

Abstract
Background
AUTO1 is a fast off-rate CD19-targeting chimeric antigen receptor (CAR), which has been successfully tested in adult lymphoblastic leukemia. Tscm/Tcm-enriched CAR-T populations confer the best expansion and persistence, but Tscm/Tcm numbers are poor in heavily pretreated adult patients. To improve this, we evaluate the use of AKT inhibitor (VIII) with the aim of uncoupling T-cell expansion from differentiation, to enrich Tscm/Tcm subsets.
Methods
VIII was incorporated into the AUTO1 manufacturing process based on the semiautomated the CliniMACS Prodigy platform at both small and cGMP scale.
Results
AUTO1 manufactured with VIII showed Tscm/Tcm enrichment, improved expansion and cytotoxicity in vitro and superior antitumor activity in vivo. Further, VIII induced AUTO1 Th1/Th17 skewing, increased polyfunctionality, and conferred a unique metabolic profile and a novel signature for autophagy to support enhanced expansion and cytotoxicity. We show that VIII-cultured AUTO1 products from B-ALL patients on the ALLCAR19 study possess superior phenotype, metabolism, and function than parallel control products and that VIII-based manufacture is scalable to cGMP.
Conclusion
Ultimately, AUTO1 generated with VIII may begin to overcome the product specific factors contributing to CD19+relapse.
Keywords: Immunotherapy, Translational Medical Research, T-Lymphocytes, Cytokines
WHAT IS ALREADY KNOWN ON THIS TOPIC
AUTO1 is a fast off-rate CD19 chimeric antigen receptor (CAR) with a 41BBz endodomain which has been successfully tested against B-cell acute lymphoblastic leukemia. Like many CAR T-cell studies, a proportion of patients relapsed with CD19+ disease, often contributed by impaired T-cell ‘fitness’. AKT inhibitor VIII (VIII) has been previously tested and shown to limit T-cell differentiation, enhancing T-cell ‘fitness’ and improving adoptive cell therapy products.
WHAT THIS STUDY ADDS
To date, VIII is not used in clinical scale CAR T-cell manufacturing or tested in the 41BBz-CAR setting. Here, we show that AUTO1 products manufactured with VIII show enhanced expansion, cytotoxicity and polyfunctionality in vitro accompanied by superior antitumor activity in vivo. Most notably with the ability to enhance in vitro/in vivo functionality in T-cells from patients that have previously suffered from CD19+ relapse on trial. We further show that VIII-based manufacture can be successfully scaled to clinical scale GMP manufacturing.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
VIII-based manufacturing has the potential to overcome impaired CAR T-cell ‘fitness’ and holds potential in overcoming the product-related factors contributing to CAR T-cell failure and patient relapse.
Introduction
AUTO1 is a fast off-rate CD19-targeting chimeric antigen receptor (CD19CAR-T) with a 41BBz endodomain, designed to reduce CAR-T immunotoxicity and improve engraftment. AUTO1 was tested in pediatric and adult B-cell acute lymphoblastic leukemia (B-ALL) on the CARPALL (NCT02443831) and ALLCAR19 (NCT02935257) clinical studies. While clinical response rates were excellent, a proportion of patients relapsed with CD19+disease.
Outcome data shows that CAR-T products yielding poor expansion and persistence in vivo contribute to an increased risk of CD19+ relapse and CAR-T treatment failure.1 2 By contrast, products enriched for naïve/stem-cell memory/central memory T-cells (Tn/Tscm/Tcm) can display better engraftment, expansion, and persistence in vivo, more often leading to complete and durable clinical responses.3 4 Efforts to generate Tn/Tscm/Tcm enriched patient products are challenging due to inherently poor T-cell fitness in patients, contributed to by age-related T-cell senescence, chemotherapy exposure, underlying cancer diagnosis and manufacturing protocols that prioritize expansion over differentiation phenotype.3 5 6
Mechanistically, T-cell expansion and differentiation are coupled through MAPK and PI3K/AKT/mTOR T-cell signaling pathways.7 8 Pharmacological inhibitors to MAPK/PI3K/AKT/mTOR have been tested ex vivo with the aim of uncoupling T-cell expansion from differentiation. PI3K inhibitors, such as LY294002 (PI3K α/β/δ),9 Idelalisib (CAL-101) (PI3Kδ),10 Duvelisib (IPI-145) (PI3Kδ/γ)11 and Akt inhibitors, including MK2206,12 Ipatasertib (GDC-0088),13 Sirolimus (Rapamycin)14 and VIII15–17 remain popular for use in ex vivo T-cell manufacture. Such inhibitors each in their own capacity have demonstrated preservation of less differentiated T-cell subsets and enhanced anti-tumor activity.18 With a focus on AKT inhibitor VIII (VIII), Klebanoff et al showed that inhibition of AKT signaling during manufacture of FMC63 CD19CAR products incorporating CD28z endodomains could generate large numbers of early memory T-cells with enhanced antitumor efficacy, driven through the transcription factor FOXO1.15 Mousset et al compared several pharmacological AKT inhibitors in vitro and concluded that the allosteric serine/threonine kinase AKT1/2/3 inhibitor VIII outperformed most compounds with respect to enhancing cell therapy memory phenotypes and CD8+T cell polyfunctionality.13 However, further analysis cautioned against routine integration of VIII into mixed CD4/CD8 cultures, as this was shown to induce immunomodulatory Th2 skewing in the final product.19 This poses a significant problem for CAR-T manufacture where mixed CD4/CD8 cultures are standard practice, and where an immunomodulatory Th2 profile would be considered disadvantageous, potentially compromising anti-tumor activity.
Overall, there is compelling evidence that VIII can substantially improve T-cell biology and has the potential to benefit CAR-T manufacture, but several questions remain. First, are the potential benefits of VIII on T-cell phenotype and function outweighed by the negative implications of a Th2 skewed CAR-T product. Second, are the potential benefits of VIII on CAR-T phenotype/function unique to CD28z endodomain CAR-T constructs as previously described,15 16 or can the impact of VIII be realized in the setting of 41BBz endodomains where there is less dependence on AKT signaling. Third, can VIII potentially rescue product phenotype and function in patients where CAR-T engraftment fails to persist. Lastly, what is the feasibility of a VIII-focussed cGMP CAR-T manufacture process for patients. To our knowledge, there are no current CAR-T clinical trials using VIII-cultured products.
Here, we addressed these questions with a focus on AUTO1 and the ALLCAR19 clinical study in adult B-ALL and have conducted a series of experiments to definitively evaluate VIII in CD4/CD8 mixed cultures. This has been done at small and cGMP scale as a strategy to enhance CAR-T functionality, and in the context of the 41BBz AUTO1 CAR-T endodomain. We compared the effect of VIII CAR-T at the phenotypic, transcriptomic, and metabolic level, comparing healthy donor and adult B-ALL patient material from the ALLCAR19 study. We assess how VIII affects CAR-T functionality in vivo and in vitro using a CAR-T ‘stress’ model designed to replicate chronic antigen exposure, and polyfunctionality using the Isoplexis single-cell proteomic platform. Further, we retrospectively evaluate ALLCAR19 patient trial products to assess whether VIII could have positively impacted product phenotype and function in patients who experienced CD19+ relapse.
Finally, we review the integration of cGMP VIII into the clinical scale Miltenyi CliniMACS Prodigy process, comparing trial products with matched scale-ups incorporating VIII, toward a modified AUTO1 manufacture process and better CAR-T products for future patients.
Methods
Cell lines
Cell lines were obtained from the American Type Culture Collection. HEK-293T cells were cultured in complete Iscove’s modified Dulbecco’s medium (Sigma Aldrich) supplemented with 10% FCS and 2 mM Glutamax (Gibco) (cIMDM). RAJI and NALM6 were maintained in complete RPMI medium (Sigma Aldrich) supplemented with 10% FCS and 2 mM Glutamax (Gibco) (cRPMI). RAJI CD19 wild type (RAJI-19WT) cells were transduced with SFG vector for enhanced green fluorescence protein (eGFP) (RAJI-19GFP), and RAJI CD19 knockout (RAJI-19KO) cells were edited using CRISPR-Cas9. NALM6 FLUC cells were transduced with SFG vector to express firefly luciferin and a lipid anchored hemagglutinin (HA-GPI) tag.
T-cell isolation, activation, and lentiviral transduction
Healthy donor-derived enriched CD4+/8+ T cells were isolated from leukocyte cones using SepMate, RosetteSep reagent (Stemcell Technologies) and Ficoll Paque density grade centrifugation. Excess cryopreserved leukapheresis from B-ALL patients on the ALLCAR19 trial (NCT02935257) were thawed, rested overnight in TexMACS (Miltenyi Biotec) supplemented with 3% human serum (Sigma Aldrich, Life Science Production) and enriched for CD4+/8+T cells using a microbead-based pan T-cell isolation kit (Miltenyi Biotec). Activation/transduction at small-scale was designed to mimic the cGMP Miltenyi CliniMACS Prodigy-based CAR-T manufacture process on ALLCAR19.20 Selected T-cells were maintained in TexMACS supplemented with 3% human serum and 10 ng/mL IL7/IL15 (cTexMACS). Cells were activated with TransAct (Miltenyi Biotec) and transduced with concentrated lentivirus on retronectin-coated plates at a multiplicity of infection of 5, cells were maintained for a further 4 days for a total manufacturing time of 8 days. AKT inhibitor VIII (Merck Millipore/Ardena) was added at T-cell activation at a concentration of 1–5 µM and maintained throughout the culture period, with a media change every 48 hours.
Scale-up runs on the CliniMACS Prodigy
ALLCAR19 uses the Miltenyi CliniMACS Prodigy manufacture platform as previously described.20 To assess the feasibility of incorporating VIII into the AUTO1 manufacture process, three scale-ups were performed in parallel with ALLCAR19 patient manufactures, comparing VIII-exposed and trial products. GMP-grade VIII was manufactured by Ardena.
Functional assays
CAR-T co-culture assay was set up with 1:1 culture of non-transduced (NT)/CAR-T with RAJI-19WT or RAJI-19KO target cell lines (irradiated, 60 Gy) in 3% human serum supplemented TexMACS media, without cytokines/VIII, for 7 days. On day 3, 500 µL of cell supernatant was harvested and frozen for cytokine analysis. At completion of co-culture, CountBright beads (Thermofisher Scientific) were added to each sample during flowcytometry to determine absolute cell numbers. Cytotoxicity was determined in a flowcytometry-based killing assay (FBK). NT or CAR-T effectors were serially diluted 2-fold in a 96 well plate. RAJI-19GFP target cells were added toward effector to target (E:T) ratios of 1:1, 1:2, 1:4 and 1:8, and incubated for 72 hours in 3% human serum supplemented TexMACS media, without cytokines/VIII. Cytotoxicity was determined by the number of remaining viable RAJI-19GFP targets by flowcytometry and CountBright beads were added to determine absolute cell numbers. A ‘stress’ model was developed by rechallenging tumor-stimulated CAR-T in a FBK assay. CAR T-cells from the expansion co-culture harvested on day 7 were restimulated in a repeat FBK.
Cytokine bead array
Cytokine bead array (CBA) was performed on day 3 cell culture supernatant from the co-culture assay. IL-2, IFN-γ, TNF-α, IL-4, IL-10, IL-6, IL-22, IL-17A and IL-17F cytokine concentrations were determined by LEGENDplex Human Th Cytokine CBA kit (BioLegend). Fluorescence intensities were measured using the BD LSRFortessa, and concentrations determined using LEGENDplex software.
Isoplexis
CAR transduced cells were violet membrane-stained, and co-cultured with RAJI-19WT targets at a 1:2 E:T ratio for 20 hours. Cells were harvested, selected for CD4+and CD8+ subsets, and stained for CD4/8 Alexa Fluor 647. 30,000 total cells were loaded onto each Isoplexis chip, measuring 31-plex cytokines grouped as effector, chemoattractive, regulatory and inflammatory. Data were analyzed using the Isospeak V.2.9.0 software.
Flowcytometry, including intracellular staining for cytokines/transcription factors
Cells were stained in PBS for 30 min at 4°C. A bespoke anti-idiotype antibody and secondary fluorochrome-labeled anti-rabbit IgG antibody (Jackson ImmunoResearch) was used to detect the AUTO1 CAR. Fixable viability dye eFluor 780 (Invitrogen), was used to discriminate live/dead cells. Anti-human CD4 (RPA-T4), CD8 (RPA-T8), CD45RA (HI100), CD62L (DREG-56), CCR7 (G025H7), CD27 (M-T271), CD28 (CD28.2), CD95 (DX2), CCR4 (L291H4), CCR6 (G034E3), CXCR3 (G025H7), CD127 (A019D5), CD25 (BC69), CD45 (HI30) and HA tag (16B12) were obtained from BioLegend.
Prior to intracellular stains, CAR-T were stimulated overnight with irradiated RAJI-19WT using GolgiStop Protein Transport Inhibitor (BD Bioscience). Following cell-surface stains, cells were fixed/permeabilised (BD Cytofix/Cytoperm kit, or the BD Pharmingen Transcription-Factor Buffer kit) and stained with intracellular antibodies. Anti-human IL-2 (MQ1-17H12), IFN-γ (B27) (BD), TNF-α (Mab-11), GranzymeB (GB11) (BD), T-bet (4B10) (Invitrogen), IFN-γ (B27) (BD), GATA3 (16E10A23), IL-4 (8D4-8) (BD), RORgt (AFKJS-9) (Invitrogen), IL-17A (VL168), FoxP3 (PCH101) (Invitrogen) and IL-10 (JES3-19F1) (BD), remaining antibodies were purchased from BioLegend. Samples were analyzed on the BD LSRFortessa, or Agilent Novocyte flow cytometers. Analysis was performed using FlowJo V.10 software. For intracellular cytokine assessments, Cytokine Strength Index was calculated as per Polyfunctionality Strength Index (PSI) measured by the Isoplexis platform by multiplying the mean fluorescence intensity (MFI) of secreted cytokines with the % of cells secreting each.
CytoID, Mitrotracker and MitoProbe staining
Autophagy was measured using the CYTO-ID Autophagy Detection Kit 2.0 (Enzo Life Sciences) diluted 1:1000 in assay buffer. This kit contains a 488 nm excitable green, fluorescent dye that can selectively label autophagic vacuoles in live cells. Total mitochondria were measured using MitoTracker Green FM (Invitrogen). Cells were stained at 100 nM in PBS. Mitochondrial membrane potential was measured using the MitoProbe JC-1 Assay Kit (Invitrogen). Cells were stained in cTexMACS at a concentration of 2 µM. Samples were analyzed on the BD LSRFortessa and analysis was performed using FlowJo V.10 software.
Seahorse metabolic profiling
Seahorse measurements were carried out on 2×105 selected CD8 T-cells. Mitochondrial Stress and Long Chain Fatty Acid Oxidation Stress was measured using the Seahorse XF Long Chain Fatty Acid Oxidation Test Kit (Agilent). Long Chain Fatty Acid Assay drugs used include, 4 µM Etomoxir, 2 µM Oligomycin, 0.5 µM FCCP and 0.5 µM Rotenone/Antimycin A. As a control, cells were run in parallel without the addition of Etomoxir which was further used to inform Mitochondrial Stress. Glycolysis was measured using the XF Glycolysis Stress Kit (Agilent), drugs used include, 10 mM Glucose, 2 µM Oligomycin and 50 mM 2-DG. Plates were run on the Seahorse XFe96 Analyzer where oxygen consumption (OCR) and extracellular acidification rates were measured. Results were analyzed using Agilent Seahorse Analytics and GraphPad Prism V.9.3.1 software.
RNA sequencing
RNA was extracted from sorted CD4+ and CD8+ CAR T at the end of manufacture, day 8. Library preparation and sequencing was carried out by the UCL Great Ormond Street Institute of Child Health Genomics Facility. cDNA was synthesized and library preparation were done using the KAPA mRNA HyperPrep Kit (Roche). Single-end sequencing was performed on the Illumina NextSeq 500 using the high output 75 Cycle Kit. Approximately 33 million reads were generated per sample. Output FASTQ files were mapped to the target genome and differential gene analysis was carried out using the DESeq2-based and EdgeR-based R pipeline, SARTools.21 To identify enriched pathways, count-based gene enrichment tests were performed on differential gene data using the topGO package mapped against Gene Ontology Pathway Database.
In vivo modelling
All protocols were performed in accordance with a UK Home Office approved project licence. Systemic leukemia was established in NOD/SCID γ (NSG) mice aged 8–12 weeks, housed in individually ventilated cages, via intravenous injection of 5×105 NALM6-FLUC followed by 1×106 NT or CAR T-cells 4 days later. Tumor burden was measured bi-weekly via bioluminescent imaging (BLI) using the IVIS spectrum in vivo imaging system (Perkin Elmer) following intraperitoneal (IP) injection of 2 mg D-luciferin in 200 µl PBS. Photon emission from NALM6 cells was measured as photons/s/cm2/steradian.
Statistics
Statistical tests used are described in the figure legends. Statistical analysis was performed on GraphPad Prism V.9.3.1. Statistical analysis for transcriptome data was carried out on R. Transcripts were filtered, where those with a mean count below 5 were removed. Remaining genes were fit in a negative binomial model for differential expression. A Wald test was used to identify significantly differentially expressed genes subsequently corrected for multiple testing using the Benjamin-Hochberg false discovery rate (FDR) method. The FDR rate was set to 5%. A Fisher’s exact test was used to identify significantly overrepresented gene fractions in each pathway at a p<0.05 cut off.
Results
AKTi VIII produces Tscm and Tcm-enriched AUTO1 CAR-T products
VIII concentrations of 1–18 µM have been tested in other T-cell expansion studies.13 15–17 Here, GMP-grade VIII (Ardena) and research-grade VIII (Merck Millipore) were titrated in healthy donor T-cells at small-scale. Total expansion, CD8 CAR-T Tcm subset enrichment, and functional analysis were comparable between GMP and research grade VIII, and 2.5 µM was defined as the optimal concentration for downstream use based on superior cytotoxicity (online supplemental figure 1A–E).
jitc-2023-007002supp001.pdf (3.1MB, pdf)
AUTO1 was manufactured with VIII (VIII-CAR) and without VIII /untreated (UT-CAR) at small-scale from healthy donor T-cells in a mock-up of the GMP CliniMACS Prodigy process employed on ALLCAR1920 (figure 1A). VIII was associated with reduced T-cell expansion (figure 1B), but increased transduction efficiency (figure 1C). VIII-CAR was enriched for Tcm (but not effector/terminal (Te/Tte)) (figure 1D) and Tscm subsets (figure 1E) at the end of manufacture on day 8. Of note, the beneficial impact of VIII on Tscm populations was apparent as early as day 4 and throughout the manufacture period. This suggests that VIII has the potential to optimize even shortened CAR-T manufacture protocols. The greatest phenotypic benefit of VIII was observed in CD8 CAR-T cells, and over the manufacture period, VIII-CAR showed preserved CCR7 and increased Tcm populations over UT-CAR (figure 1F). A summary illustration of CAR-T phenotype for all donors throughout manufacture is outlined in online supplemental figure 2.
Figure 1.

VIII produces Tscm and Tcm-enriched AUTO1 CAR-T products (A) Schema of CAR T-cell manufacturing process. (B) Total T-cell fold expansion throughout manufacture, ±SEM. (C) AUTO1 CAR T-cell transduction at end of manufacture day 8, ±SD/individual data points. (D) Graphical representation of Tn (CCR7+/CD45RA+), Tcm (CCR7+/CD45RA−), Te (CCR7−/CD45RA−) and Tte (CCR7−/CD45RA+) subsets in CD4 and CD8 CAR T-cells determined by flowcytometry, ±SD at end of manufacture day 8. (E) Percentage CD95+ positive, sub gated from Tn population throughout manufacture, depicting individual data points. (F) Representative flowcytometry plots from one donor demonstrating CCR7/CD45RA staining in T-cell starting material and subsequently in CAR CD8s throughout manufacture. (G) Percentage CD27+/CD28+ and IL2+/Granzyme− in CD4/8 CAR T-cells at end of manufacture, determined by flowcytometry, graphs depicting individual data points. (B-E/G) n=6, Two tailed Mann-Whitney U test, ns p>0.05. *p<0.05 and **p<0.01. CAR, chimeric antigen receptor.
Previous reports suggest that Tcm subsets produce high levels of IL-2, but not cytotoxins such as Perforin and Granzyme B (GZMB).22 VIII-CAR had significantly greater proportions of CD4+ and CD8+ CAR T producing IL-2 (but not GZMB) consistent with Tcm enrichment (figure 1G).
Loss of CD27/CD28 expression is commonly observed in patients following chemotherapy and is associated with T-cell senescence and failure to proliferate following stimulation.23 In this analysis, VIII led to increased CD27/CD28 double positive CD4+ and CD8+ CAR T subsets, more marked in the CD8+ compartment (figure 1G).
Together these data show that VIII in small-scale experiments in healthy donor T-cells leads to a less differentiated CAR-T profile, consistent with the desirable phenotypes associated with effective T-cell therapies.4 24 25
AKTi VIII enhances CAR-T proliferation, cytotoxicity, and cytokine secretion
Following co-culture with tumor cells, VIII-CAR CD8+ (but not VIII-CAR CD4+) showed a statistically significant expansion over UT-CAR (22.4-fold increase against RAJI-19KO and 57.9-fold increase against RAJI-19WT, figure 2A). Cytotoxicity at the end of manufacture showed CD19-specific and comparable killing between UT-CAR and VIII-CAR. To uncover functional differences, we performed a rechallenge ‘stress’ test, designed to replicate protracted, chronic antigen exposure in vivo by rechallenging CAR-T cells in a cytotoxicity assay following a 7-day co-culture with RAJI-19WT targets. In these experiments, VIII-CAR displayed significantly improved cytotoxicity at all E:T ratios (figure 2B). After primary co-culture with RAJI-19WT targets, VIII-CAR produced significantly more effector cytokines (IL-2, IFN-γ, TNF-α) than UT-CAR. Regulatory/inflammatory cytokines showed a significant increase in IL-6, IL-22, IL-17A and IL-4. IL-4 was found at low concentrations and no differences in IL-10 or IL-17F were observed, (figure 2C).
Figure 2.

VIII enhances CAR-T proliferation, cytotoxicity, and cytokine secretion in vitro (A) CD4/CD8 CAR fold expansion following a 7-day co-culture with irradiated RAJI-19KO or RAJI-19WT target cell lines, ±SD/induvial data points. (B) Graphs depicting % killing of RAJI-19GFP target by NT or CAR T-cells in a 72-hour killing assay at the end of manufacture or post rechallenge where NT or CAR-T co-cultured with RAJI-19WT targets for 7 days prior. Results from all conditions were normalized to the untreated NT condition at each E:T ratio, graphs show individual data points. (C) Cytokine concentrations measured by CBA from day 3 of the 7- day co-culture with RAJI-19WT targets, graphs show individual data points. (A–C) n=6, Two-tailed Mann-Whitney U test, ns p>0.05. *p<0.05 and **p<0.01. CBA, cytokine bead array; NT, non-transduced.
Similarly, in vivo assessments in NSG mice-bearing systemic B-ALL, treated with 5×105 NT/UT-CAR/VIII-CAR healthy donor T-cells revealed significantly lower tumor burden in VIII-treated mice vs UT-CAR and NT groups. Spleen/BM analysis on day 21 showed persisting T-cells, a virtual absence of tumor and a trend toward the presence of less differentiated CAR-T populations in VIII-treated mice (online supplemental figure 3A–E).
AKTi VIII promotes Th1/Th17 polarization and enhances T-cell polyfunctionality
Mousset et al observed that VIII, in the context of a non-CAR engineered mixed CD4+/CD8+ culture, drives CD4 Th2 differentiation.19 Here, we characterized CD4+CAR T helper subsets (Th1, Th2, Th17, Treg) following stimulation with RAJI-19WT. We found AUTO1 production in the presence of VIII leads to an inflammatory/stimulatory profile, with a 2.2-fold increase in Th1 subsets, a 3.7-fold increase in Th17 subsets, and no change in Th2 or Tregs (figure 3A). IL-6 was significantly increased in VIII-CAR co-culture (figure 2C) potentially driving the Th17 skew. IL-6 is recognized to play a role in Th17 differentiation and maintenance26 and has been shown to correlate with therapeutic response in CAR-T treated chronic lymphocytic leukemia patients.24
Figure 3.
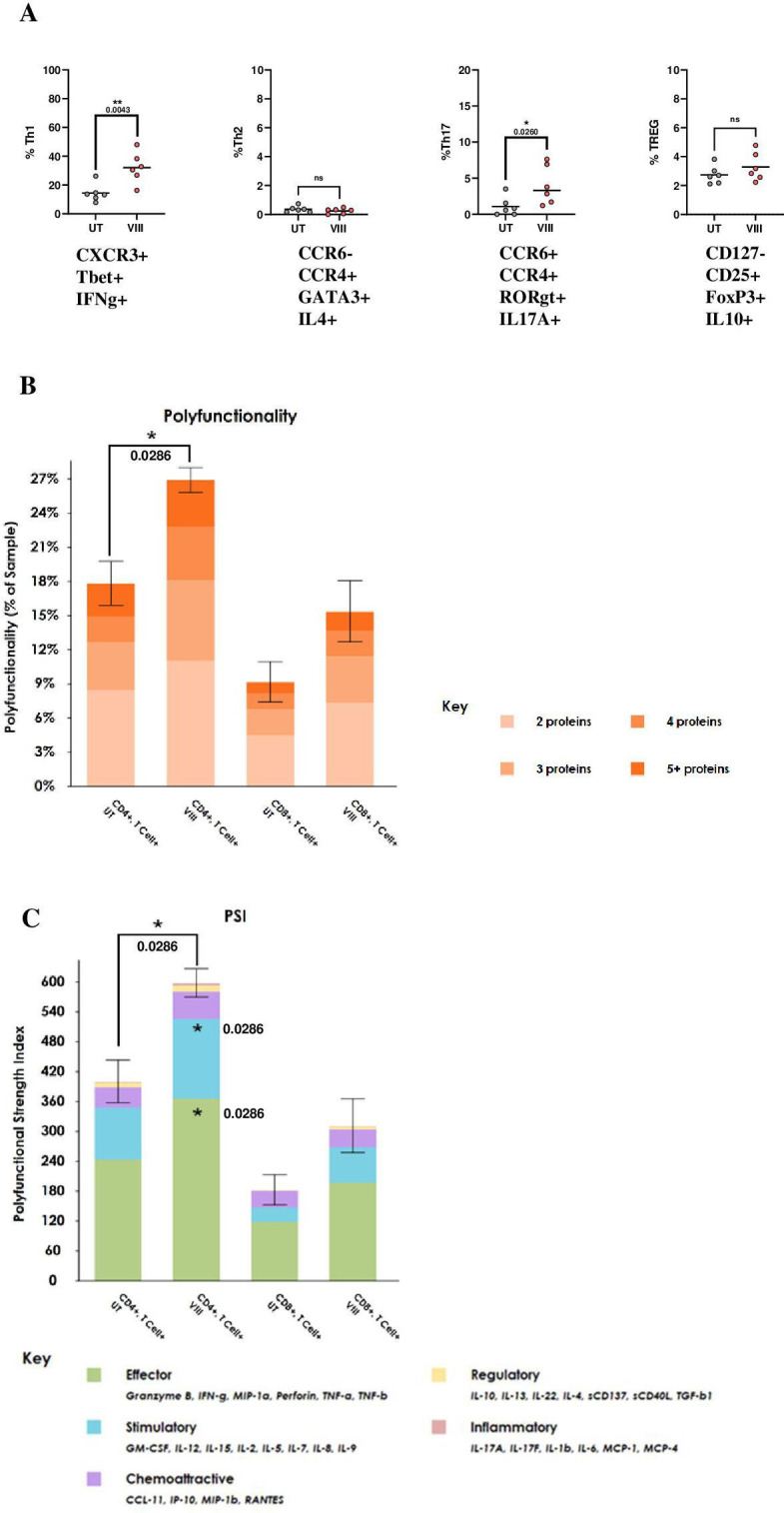
VIII inhibition promotes Th1/Th17 polarization and enhances T-cell polyfunctionality (A) Graphs representing percentage of CAR CD4 Th1, Th2, Th17 and TREG subsets determined by flowcytometry post overnight stimulation at 1:1 with RAJI-19WT targets, graphs show individual data points. (B) Total percentage of polyfunctional cells per sample±SD. Statistical comparisons were made against paired UT samples (C) Polyfunctionality Strength Index (PSI) calculated by multiplying the total percentage of polyfunctional cells with the secretion intensity of each cytokine grouped into effector, stimulatory, chemoattractive, regulatory and inflammatory subsets, ±SD. All statistical comparisons were made against paired UT sample in each subset. (A) n=6, (B, C) determined using Isoplexis platform, following 20-hour 2:1 RAJI-19WT:CAR stimulation, n=4. (A–C) Two-tailed Mann-Whitney U test, ns p>0.05. *p<0.05 and **p<0.01. CAR, chimeric antigen receptor.
Polyfunctionality is the ability of a single T-cell to simultaneously produce multiple cytokines, chemokines and chemotoxins, and can be measured using the Isoplexis single-cell proteomic analysis platform. Readouts include percentage of polyfunctional cells and PSI. In other studies, CAR T-cell polyfunctionality has been reported to correlate with clinical response.27 Here, VIII-CAR products show enhanced polyfunctionality, with a 1.6-fold increase in PSI, reflecting increased effector/stimulatory cytokine secretion (including GM-CSF, IL-2, TNF-α and TNF-β) from both CD4+ and CD8+ subsets (figure 3B), without a parallel increase in regulatory cytokines (figure 3C). This is further illustrated by principal component analysis (PCA) (online supplemental figure 4).
AKTi VIII regulates the CAR-T cell transcriptome and metabolic signature
CAR CD4+and CD8+ subset transcriptome analysis revealed 417 (CD4+) and 609 (CD8+) significantly differentially expressed transcripts. Count-based enrichment testing on differential genes mapped to the Gene Ontology Pathway Database showed that leukocyte migration, activation, lymphocyte differentiation and proliferation signatures were downregulated in VIII-CAR CD4+ (figure 4A). In contrast, leukocyte migration, T-cell activation, and autophagy signatures were upregulated in VIII-CAR CD8 (figure 4B). AKT signaling has different biological effects in CD4 and CD8 T-cells, namely controlling effector vs memory CD8 T-cell differentiation, and promoting naïve CD4 Th cell differentiation,28 this may explain the differential effect of VIII inhibition on both subsets. Further analysis of VIII-CAR CD8+ at the individual gene level confirmed a T-cell activation signature with upregulation of CD28 and ICOS, and downregulation of granzyme and FASLG transcripts. These transcript-level findings are supported by flowcytometry (online supplemental figure 5A).
Figure 4.

VIII inhibition regulates the CAR-T cell transcriptome and metabolic signature. (A, B) Dot plots of cellular pathways mapped to Gene Ontology database CAR CD4 (A) and CAR CD8 (B) Size of dots represents the number of genes contributing to a pathway, dot color represents adjusted p values and x-axis shows the fraction of mapped genes in each pathway. (C) Heat map representation of autophagy signature in CAR CD8 T-cells. Color represents low to high transcript expression. Rows are specific to each gene and columns highlights expression in each donor following UT/VIII manufacture. Only significantly differentially expressed pathways and genes are included in figures at a p<0.05 cut off. (A–C) n=3. (D) Histogram and MFI representation of CytoID staining for autophagic vesicles in total CAR and CD8 CAR T-cells. (E) Mitochondrial mass determined using Mitrotracker Green measured by MFI in total CAR and CAR CD8 T-cells. Oxygen consumption rates (OCR) in CD8 T-cells treated with etomoxir or media control. Graphs representing change in maximal respiration following etomoxir treatment compared with media alone and mitochondrial spare respiratory capacity (SCR). (F) ΔΨm of total CAR and CAR CD8 T-cells using the JC-1 dye, ratio of aggregate (red) to monomer (green) fluorescence. Extracellular acidification rates (ECAR) in CD8 T-cells. Graph representing basal and maximal glycolysis. (D–F) n=6, depicting individual data points, seahorse experiments represent pooled data from two independent experiments, n=6 each ±SEM. Two-tailed Mann-Whitney U test, ns p>0.05 and **p<0.01. CAR, chimeric antigen receptor.
FOXO1 is recognized to be important for CD4 and CD8 T-cell homing to secondary lymphoid organs through regulated of expression of L-selectin (SELL), sphingosine-1-phosphate receptor 1 (S1PR1) and CCR7.29 VIII-CAR CD8+ showed a FOXO1 transcriptomic signature, with upregulation of SELL (CD62L), IL7R, KLF2 and S1PR1, consistent with other studies.15 30 31 VIII-CAR CD4+ also showed upregulation of FOXO1-dependent SELL and IL7R transcripts, and additionally CD28 and ICOS transcripts (online supplemental figure 5A,B). These signatures correlate with enrichment for Tn/Tcm subsets, which are associated with persistence.30 31
Autophagy is a process through which cells maintain homeostasis via lysosome-mediated degradation of damaged organelles/proteins and pathogens that is associated with the generation and maintenance of memory T-cells.32 Autophagy is inhibited during PI3K/AKT signaling through mammalian target of rapamycin (mTOR). Conversely, when mTOR is inhibited by rapamycin, autophagy is ‘switched on’.33 In this study, we show that VIII-CAR CD8+ possess a transcriptomic profile for increased autophagy (figure 4C). As a phenotypic measure of autophagy, we used CYTO-ID MFI to detect preautophagosomes, autophagosomes, and autolysosomic vesicles and showed increases of 61% and 46% in VIII-treated total CAR and CD8+CAR subsets, respectively (figure 4D).
Autophagy contributes to improved T-cell survival through degradation of dysfunctional mitochondria (mitophagy)33 and enhanced fatty acid oxidation (FAO).34 Using MitoTracker Green, we showed a slight reduction in mitochondrial mass in VIII-treated total CAR (−9%) and CD8+CAR T cell subsets (−7%) (figure 4E). To characterize FAO by OCR etomoxir, a long chain FAO inhibitor of carnitine palmitoyltransferase-1, was used to inhibit maximal respiration in FAO-dependent cultures. Spare respiratory capacity (SCR) is a measure of mitochondrial respiration and is commonly increased in Tcm compared with Tn/Te subsets.35 Here, we showed a 2.8-fold reduction in maximal respiration following etomoxir in VIII-treated CAR-T (figure 4E), but no overt change in SCR, despite Tcm enrichment (figure 4E). Together, these data support the hypothesis of VIII-induced autophagy.
Research suggests that metabolic CAR-T fitness correlates with anti-tumor efficacy.36 In T-cells, low mitochondrial membrane potential (low-ΔΨm) has been associated with superior persistence,37 however, high-ΔΨm represents energy stored that can ultimately be used to produce ATP and has been shown to enhance CD8 effector function.38 The mTOR inhibitor rapamycin has been shown to increase ΔΨm,39 but to date this has not been described in VIII-treated CAR-T cells. Using the JC-1-dye assay, we showed a 4/4.9-fold increase in ΔΨm in VIII-treated total CAR and CAR CD8s, respectively (figure 4F), representing an increased capacity to produce ATP. Glycolysis, as a means to produce ATP, can be regulated by AKT40 but the impact of VIII is unclear.13 15 We observed no changes in glycolytic capacity between postmanufacture VIII-treated CAR CD8s compared with UT-CAR (figure 4F).
AKTi VIII enhances phenotype and function of CAR-T cells derived from B-ALL patients
We next sought to determine if manufacturing AUTO1 with VIII using excess leukapheresis from 6 B-ALL ALLCAR19 patients, three with long-term remission and three with CD19+relapse, would yield similar outcomes to previous assessments on healthy donors.
Compared with healthy donors, B-ALL patient leukapheresis material had lower Tn subsets, higher Te/Tte subsets, and a lower proportion of non-senescent CD27+/CD28+T cells (online supplemental figure 6A,B). This pattern has also been seen in diffuse large B-cell lymphoma patients following chemotherapy23 and represents more challenging starting material for CAR-T manufacture.
Despite the more differentiated profile of patient leukapheresis, postmanufacture VIII-CAR from B-ALL patients revealed similar phenotypic and functional benefits to those observed with healthy donor material. Tscm/Tcm subsets were increased, Te/Tte subsets were reduced (figure 5A), and on extended analysis, CD27+/CD28+ subsets productive of IL-2 (but not GZMB) were increased (figure 5B). While phenotypic analysis failed to reach significance in all subsets due to variability among B-ALL patients, a trend similar to healthy donors was observed, accompanied by improved function. This suggests that VIII may enrich poor quality starting material for desirable cell subsets through the course of CAR-T manufacture.
Figure 5.

VIII enhances phenotype and function of CAR-T cells derived from B-ALL patients. (A) Graphical representation of Tn (CCR7+/CD45RA+), Tcm (CCR7+/CD45RA−), Te (CCR7−/CD45RA−) and Tte (CCR7−/CD45RA+) subsets in CD4 and CD8 CAR T-cells determined by flowcytometry, ±SD, and percentage CD95+ positive cells, subgated from the Tn population, ±SD/individual data points. (B) Percentage CD27+/CD28+and IL2+/Granzyme− in CD4/8 CAR T-cells at end of manufacture, determined by flowcytometry, graphs depicting individual data points. (C) CD4/CD8 CAR fold expansion following a 7-day co-culture with irradiated RAJI-19KO or RAJI-19WT target cell lines. ±SD/induvial data points. (D) Graphs depicting % killing of RAJI-19GFP target by NT or CAR T-cells in a 72-hour killing assay at the end of manufacture or post rechallenge where NT or CAR-T co-cultured with RAJI-19WT targets for 7 days prior. Results from all conditions were normalized to the untreated NT condition at each E:T ratio, graphs show individual data points. (E) Cytokine concentrations measured by CBA from day 3 of the 7- day co-culture with RAJI-19WT targets, graphs show individual data points. (F) Cytokine Strength Index, calculated by multiplying the MFI of each cytokine with the percentage of cells producing each, grouped into effector (IFN-γ/TNF-α) and stimulatory (IL-2) in end of manufacture CD4/8 CAR-T derived from B-ALL patients in small-scale assessments, ±SEM. (G) Percentage of CAR CD4 Th1, Th2, Th17 and TREG subsets determined by flowcytometry post overnight stimulation at 1:1 with RAJI-19WT targets, graphs show individual data points. (H) CytoID, Mitrotracker Green and JC-1 staining of CAR and CAR CD8 T-cells for autophagy, mitochondrial mass and ΔΨm, aggregate (red) to monomer (green) fluorescence assessments, depicting individual data points. (A–H) n=6, Two tailed Mann-Whitney U test, ns p>0.05. *p<0.05 and **p<0.01. (A–E, G, H) Circle symbols on graphs represent patients in remission and square symbols represent patients with CD19+ relapse. MFI, mean fluorescence intensity.
B-ALL patient derived VIII-CAR in vitro showed improved survival and expansion with and without target (figure 5C). Contrary to what was observed in healthy donors, end of manufacture patient-derived UT-CAR vs VIII-CAR showed significantly higher cytotoxicity and GZMB production (83% vs 42%, respectively). A less significant decrease in GZMB production was seen in VIII-CAR vs UT-CAR in healthy donors and may explain the comparable cytotoxicity observed at the end of manufacture here but not in patients (online supplemental figure 7B). Nonetheless, similarly to what was observed in healthy donors, VIII-CAR demonstrated significantly improved cytotoxicity at rechallenge ‘stress’ test across all E:T ratios, even in ALLCAR19 ‘non-responder’ patients (figure 5D) and retained higher production of GZMB (online supplemental figure 7B). Importantly, this was associated with improved cytokine secretion following RAJI-19WT co-culture, with increases in IL-2, IFN-γ, TNF-α and IL-6. IL-22 and IL-17A levels were increased in some (but not all) B-ALL patient samples (figure 5E), and while significant increases in IL-4 and IL-10 were observed, overall concentrations were low.
In healthy donors, we showed that VIII positively impacts polyfunctionality, predominantly through increased production of the effector cytokines IL-2, IFN-γ, and TNF-α (figure 3C and online supplemental figure 4). In B-ALL patient-derived CAR-T products, we measured cytokine secretion following overnight stimulation with RAJI-19WT targets using intracellular flowcytometric analysis. Results revealed a significant increase in the levels of the effector cytokines IL-2, IFN-γ, TNF-α, in both VIII-CAR CD4 and CD8 conditions (figure 5F and online supplemental figure 7A). While some patient-derived VIII-CAR products demonstrated an increase in Th1 and Th17 subsets (akin to healthy donors), this was variable between patients. No enrichment for Th2 or TREG subsets was observed in patient-derived VIII-CAR products (figure 5G).
Analysis of autophagy, mitochondrial mass and ΔΨm in patient-derived VIII-CAR showed a significant increase in autophagic vesicles by CYTO-ID, a significant reduction in mitochondrial mass by MitoTracker Green, and a marked increase in ΔΨm by JC-1 assessment (figure 5H), demonstrating that a profile for metabolic fitness and autophagy can be induced even in poor quality patient material.
Together, this suggests that modification of manufacture to incorporate VIII has the capability to functionally enhance patient products that are otherwise at risk of CD19+relapse.
AKTi VIII can be scaled to produce superior AUTO1 patient products on the CliniMACS Prodigy
To test whether the desirable phenotypic and functional profiles of VIII-CAR in vitro and in vivo can be reproduced at clinical scale, we used surplus leukapheresis from three B-ALL patients on ALLCAR19 and manufactured VIII-CAR in parallel with the conventional trial manufacture process using the CliniMACS Prodigy. Despite a reduction in T-cell expansion with VIII, scaled manufacture comfortably generated the 410×106 AUTO1 target dose specified on study. VIII-treatment showed similar transduction efficiency to the standard manufacture process, but with non-significant trends toward enrichment for Tscm/Tcm subsets, increased effector cytokines IL-2, IFN-γ, TNF-α (figure 6A–C) and a reduction in GZMB which increases in VIII-CAR at rechallenge (online supplemental figure 7C,D). To assess function in vivo, we tested products manufactured from a trial patient with CD19+relapse in an NSG NALM6 mouse model in three cohorts: 1×106 NT/UT-CAR/VIII-CAR. Bi-weekly BLI revealed improved tumor control (figure 6D,E) and survival (figure 6F) in all mice treated with VIII-CAR over UT-CAR, suggesting that VIII in clinical-scale manufacture can restore functionality and improve efficacy even in heavily pretreated patients.
Figure 6.

VIII can be scaled to produce superior AUTO1 patient products on the CliniMACS Prodigy. (A) Total number of T-cells and CAT CAR transduction percentage at the end of scaled manufactures with (VIII) or without (UT), individual data points±SD. (B) Graphical representation of Tn (CCR7+/CD45RA+), Tcm (CCR7+/CD45RA−), Te (CCR7−/CD45RA−) and Tte (CCR7−/CD45RA+) subsets in CD4 and CD8 CAR T-cells determined by flowcytometry, ±SD and percentage CD95+ positive, subgated from Tn population, ±SD/induvial data points. (C) Cytokine Strength Index of effector (IFN-γ/TNF-α) and stimulatory (IL-2) cytokines at the end of manufacture in CD4/8 CAR-T derived from B-ALL patients in prodigy scale assessments, ±SEM and percentage of cells producing GZMB in total CAR-T at end of manufacture and rechallenge ±SD. (A–C) n=3, Two-tailed Mann-Whitney U test, ns p>0.05. (D) Tumor burden measured by bioluminescent imagining (BLI) in NALM6 tumor established mice treated with NT T-cells or CAR T-cells manufactured with (VIII) or without (UT) Akt VIII ±SEM. (E) Overall survival of mice. (F) Representative BLI images of 4 mice pre NT/CAR treatment (day 4) and post (day 10/17). (D–F) Cells derived from one ALL patient, n=5 mice per group. Two-way ANOVA corrected for multiple comparisons by Tukey’s test on log transformed data, ns p>0.05, ***p<0.001 and ****p<0.0001. Differences in survival were determined using Mantel Cox test. CAR, chimeric antigen receptor. ANOVA, analysis of variance.
Discussion
Preclinical studies of adoptive T-cell immunotherapy show that Tscm/Tcm subsets provide greater proliferative capacity, enhanced metabolic fitness and superior anti-tumor responses than Te/Tte subsets in patients.4 25 41 Thus, to improve CAR-T products, several studies have sought to modify manufacture to promote Tscm/Tcm skewing, through the inclusion of small molecule inhibitors targeting T-cell differentiation,11 15 16 23 42 43 where the PI3K inhibitor BB007 has been used in CAR-T manufacture for a multiple myeloma trial (NTC03274219).44
VIII-driven AKT inhibition during FMC63-CD28z CD19CAR manufacture has been shown to generate early memory T-cells with enhanced antitumor efficacy.15 Mousset et al cautioned against its use in mixed CD4/CD8 cultures due to the risk of a Th2-skewed final product.19 In contrast, we observed a VIII-associated skew toward Th1 and Th17 polarization in AUTO1, without a signal for Th2/TREGs. The different results may be explained by variations in experimental conditions including choice of effector cells (CAR-T vs unmodified T-cells), strength in T-cell activation protocols and the VIII concentrations tested.19
Th1/Th17 polarization is desirable for adoptive cell therapy. Th1 subsets secrete IFNγ, enhance CD8 expansion and improve antitumor activity.45 46 Th17 subsets have similar antitumor properties to Th1, but with a less differentiated phenotype, harboring plasticity under conditions of repetitive stimulation.47 Previous studies implicate ICOS in the induction and regulation of Th1/Th2 and Th17 responses.48 49 Here, transcriptomic analysis of VIII-CAR CD4 T-cells showed enrichment for ICOS, supporting this hypothesis. Together, this data strongly favors the integration of VIII into mixed CD4/CD8 cultures for the generation of AUTO1. A fundamental question for this project was whether the benefits conferred by VIII on CAR-T phenotype/function were unique to CD28z endodomain CAR-T constructs, as the majority of testing has been conducted in CAR-T models bearing CD28z endodomains, manufactured at small scale with IL-2 supplementation and dynabead/soluble CD3 antibody protocols.11 15 16 23 CD28 signaling is more heavily dependent on AKT than 41BB signaling, such that the impact of VIII on 4-1BBz endodomain CAR-T models cannot be directly inferred from CD28z data.36 50 Thus, for biological relevance to the AUTO1 program, where the CAR construct is designed with a 41BBz endodomain, and where manufacture incorporates TransAct activation and IL-7/IL-15, we have tested GMP-grade VIII ex vivo with a view toward enhanced clinical scale CAR-T manufacture.
Consistent with the CD28z-CAR preclinical data, VIII-treated AUTO1 products showed transcriptional dependence on FOXO1,31 Tscm/Tcm enrichment, superior in vitro expansion, enhanced cytotoxicity and superior tumor control in vivo.15 16 This commonality of readout despite differing CD19-binding scFv moieties and different endodomains suggests a broadly applicable mechanism of action, agnostic to CAR structure. Of note, discrimination of the functional benefits of VIII was not obvious here in vitro by conventional cytotoxicity assay, but instead by our novel rechallenge stress test and the readouts from the Isoplexis polyfunctionality platform. Together, these assays delineated the biological advantages of VIII in AUTO1 manufacture and highlight the importance of potency assessment when investigating potential changes in CAR-T manufacturing protocols.
In our analysis, VIII was associated with an autophagy signature in CAR T-cells. Autophagy plays an important role in the generation and maintenance of memory T-cell during contraction.51 52 This is likely to be important for CAR-T efficacy, as high circulating Tn/Tcm and CCR7+CD27+ populations positively correlate with antitumor response.24 53 Here, VIII-treated cells also displayed hybrid features of longevity (Tcm), with an increase in FAO and effector (Te) function, with high-ΔΨm (ie, high stored energy contributing to enhanced effector function38)but without an overt change in glycolysis or SRC.54 These findings identify multiple active metabolic pathways required to support the energy requirements of rapid activation, expansion, and longevity in VIII-CAR CD8s. Together, these data support the integration of VIII into the AUTO1(41BBz) CAR-T manufacture process.
A focus for this project was whether VIII could potentially rescue product phenotype and function in ALLCAR19 study patients where AUTO1 on trial did not confer durable persistence/response. To this end, we evaluated leukapheresis material from patients on the ALLCAR19 study with and without durable remissions. Despite a paucity of Tn and CD27+/CD28+ subsets in B-ALL patient leukapheresis material compared with healthy donor material, VIII exposure ex vivo appeared to enrich for desirable Tscm/Tcm subsets and yielded functionally superior AUTO1 products in vitro and in vivo compared with UT-CAR. This was particularly notable in patients with CD19+ relapse on study. These data further support the integration of VIII into the AUTO1 CAR-T manufacture process and may be particularly valuable for patients with impaired T-cell fitness who are potentially at higher risk of relapse with conventionally manufactured CAR-T.
There are no current CAR-T clinical trials using VIII-cultured products, which may in part be due to a lack of access to cGMP grade VIII. Through an academic collaboration with Professor Harry Dolstra at the Radboud University Medical Center, we have been able to access cGMP grade VIII for scale-ups and process development work. We identified 2.5 μM VIII as the optimal concentration for functional benefits. The higher concentration affected expansion more so than 1 µM used in other CAR-T studies,15 16 however, CAR-T doses as stipulated for the ALLCAR19 trial were reached for all patient products manufactured at scale with 2.5μM-VIII. Expansion is recognized to drive differentiation55; in limiting excessive and unnecessary expansion ex vivo, we may protect AUTO1 products from terminal differentiation. This data illustrates that VIII can be integrated into the cGMP AUTO1 CAR-T manufacture process without compromising yield, even in poor quality patient starting material.
While in this study we demonstrate that addition of VIII enhances phenotype and performance of AUTO1 products, we continue to question how VIII-based culture compares with other CAR-T manufacturing advances such as shortened manufacture protocols (1–5 days),43 55 some without a T-cell activation step,49. Biotech companies such as Novartis (Tcharge),56 and Gracell Biotechnologies (F-CAR-T)57 have developed a shortened manufacture process to 1–4 days in vitro. This approach has potential advantages of less time in cGMP with subsequent impacts on lowering costs of staff/room hire per product and a reduction in vein-to-vein time, critical for patients with rapidly progressive chemo-refractory disease. From a product perspective, shorter ex vivo expansion in the absence of CD3/CD28 activation is proposed to preserve T-cell stemness in the final product.56 57
As the technology stands, shorter manufacture protocols possess several challenges which may impede their broad deliverability into the clinic. First, much higher T-cell numbers are required at baseline for a non-expansion protocol, and this may be difficult to obtain from lymphopenic patient leukapheresis. Second, high viral vector volumes are required to modify large cell numbers ex vivo, thus negatively impacting the cost per patient product, as vector is the most expensive component of current CAR-T manufacture processes. Third, Tn/Tscm subsets are often under-represented in patient leukapheresis, particularly in heavily pretreated, older patients.5 6 23
By contrast, VIII-based manufacture does not require higher starting T-cell numbers, nor higher vector volumes, and consistently yields Tn/Tscm/Tcm enrichment, even in heavily pretreated patient starting material. In our hands, AUTO1 characterization as early as day 4 of the manufacture process shows a significant difference in Tscm populations between VIII and UT cultures, suggesting a tangible benefit from VIII, even in short duration manufacture, and the integration of VIII in shortened manufacture protocols is worthy of further investigation.
In summary, we have shown that VIII can improve the phenotype and functionality of AUTO1 in both healthy donor and B-ALL patient material, with gains observed in patients who experienced CD19+ relapse on the ALLCAR19 study. It is possible that VIII-based manufacture may help to prevent CD19+ relapse and CAR-T failure for future patients. Based on this data, we have developed a cGMP manufacture process incorporating VIII which is currently being tested in a UCL CAR-T trial of multiple myeloma (NCT04795882) with plans to integrate this into other CAR-T trials.
Acknowledgments
We thank the NIHR Blood and Transplant Research Unit (BTRU) for funding this project. We thank patient’s part of the ALLCAR19 trial for consent to use surplus cellular material. We thank Dr P. Niola and T. Brooks at the UCL Great Ormond Street Institute of Child Health Genomics Facility for their assistance conducting RNA sequencing and differential gene expression. We acknowledge, M.Abbasian, M.Vaughan, V.M.C.Pereira and L.Bosshard-Carter for assistance conducting large scale CAR T-cell manufactures. We thank H. Roddy for assistance harvesting and processing tissue from in vivo models. We thank Dr Lydia Lee for reviewing the manuscript.
Footnotes
Contributors: VM designed and conducted all experiments, analysed data and wrote the manuscript. GA assisted with B-ALL patient, IsoplexisTM, in vivo assessments and edited the manuscript. JDAP provided surplus trial patient material and conducted large scale CAR-T cell manufactures. MSS conducted analysis and data interpretation of RNA sequencing. ACG and LG conducted large scale CAR-T cell manufactures. AH assisted with designing and performing in vivo experiments. FAV provided access to Isoplexis platform and assisted with data interpretation. ABvdW and HD provided GMP-VIII drug used in this study and reviewed the manuscript. MAP conceived the project, provided AUTO1 constructs and reviewed the manuscript. KSP reviewed the manuscript and CR guarantor, conceived the project and edited the manuscript.
Funding: NIHR NHSBT BTRU (IS-BTU-0214-10074). All animal studies were performed in accordance to the UK Home Office approved project licence PP8379762.
Competing interests: MAP owns stock in and is employed Autolus Therapeutics; an inventor on patents licensed to Autolus Therapeutics to which he receives a share of revenues. KSP share holder and consultant of Autolus Therapeutics. CR speaker fees and advisory boards for Novartis, Kite/Gilead, Amgen and BMS/Celgene.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data available on reasonable request
Ethics statements
Patient consent for publication
Not applicable.
References
- 1.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell Lymphoblastic leukemia. N Engl J Med 2018;378:439–48. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult Relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med 2019;380:45–56. 10.1056/NEJMoa1804980 [DOI] [PubMed] [Google Scholar]
- 3.Fraietta JA, Lacey SF, Orlando EJ, et al. Author correction: determinants of response and resistance to Cd19 Chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2021;27:561. 10.1038/s41591-021-01248-2 [DOI] [PubMed] [Google Scholar]
- 4.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full Effector function in vitro paradoxically impairs the in vivo antitumor efficacy of Adoptively transferred Cd8+ T cells. J Clin Invest 2005;115:1616–26. 10.1172/JCI24480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das RK, Storm J, Barrett DM. T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit Chimeric antigen receptor potential. Cancer Res 2018;78:1631. 10.1158/1538-7445.AM2018-1631 [DOI] [Google Scholar]
- 6.Tu W, Rao S. Mechanisms underlying T cell Immunosenescence: aging and cytomegalovirus infection. Front Microbiol 2016;7:2111. 10.3389/fmicb.2016.02111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han JM, Patterson SJ, Levings MK. The role of the Pi3K signaling pathway in Cd4(+) T cell differentiation and function. Front Immunol 2012;3:245. 10.3389/fimmu.2012.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim EH, Suresh M. Role of Pi3K/AKT signaling in memory Cd8 T cell differentiation. Front Immunol 2013;4:20. 10.3389/fimmu.2013.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng W, O’Hear CE, Alli R, et al. Pi3K orchestration of the in vivo persistence of Chimeric antigen receptor-modified T cells. Leukemia 2018;32:1157–67. 10.1038/s41375-017-0008-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stock S, Übelhart R, Schubert M-L, et al. Idelalisib for Optimized Cd19-specific Chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int J Cancer 2019;145:1312–24. 10.1002/ijc.32201 [DOI] [PubMed] [Google Scholar]
- 11.Funk CR, Wang S, Chen KZ, et al. Pi3Kδ/Γ inhibition promotes human CART cell epigenetic and metabolic Reprogramming to enhance antitumor cytotoxicity. Blood 2022;139:523–37. 10.1182/blood.2021011597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Ding J, Sun S, et al. Akt inhibition at the initial stage of CAR-T preparation enhances the CAR-positive expression rate, memory phenotype and in vivo efficacy. Am J Cancer Res 2019;9:2379–96. [PMC free article] [PubMed] [Google Scholar]
- 13.Mousset CM, Hobo W, Ji Y, et al. Ex vivo AKT-inhibition facilitates generation of Polyfunctional stem cell memory-like Cd8(+) T cells for adoptive Immunotherapy. Oncoimmunology 2018;7:e1488565. 10.1080/2162402X.2018.1488565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nian Z, Zheng X, Dou Y, et al. Rapamycin pretreatment Rescues the bone marrow AML cell elimination capacity of CAR-T cells. Clin Cancer Res 2021;27:6026–38. 10.1158/1078-0432.CCR-21-0452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klebanoff CA, Crompton JG, Leonardi AJ, et al. Inhibition of AKT signaling Uncouples T cell differentiation from expansion for receptor-engineered adoptive Immunotherapy. JCI Insight 2017;2:e95103. 10.1172/jci.insight.95103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urak R, Walter M, Lim L, et al. Ex vivo AKT inhibition promotes the generation of potent Cd19Car T cells for adoptive Immunotherapy. J Immunother Cancer 2017;5:26. 10.1186/s40425-017-0227-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Waart AB, van de Weem NMP, Maas F, et al. Inhibition of AKT signaling promotes the generation of superior tumor-reactive T cells for adoptive Immunotherapy. Blood 2014;124:3490–500. 10.1182/blood-2014-05-578583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Passang T, Ravindranathan S, et al. The magic of small-molecule drugs during ex vivo expansion in adoptive cell therapy. Front Immunol 2023;14:1154566. 10.3389/fimmu.2023.1154566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mousset CM, Hobo W, de Ligt A, et al. Cell composition and expansion strategy can reduce the beneficial effect of AKT-inhibition on Functionality of Cd8(+) T cells. Cancer Immunol Immunother 2020;69:2259–73. 10.1007/s00262-020-02612-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roddie C, Dias J, O’Reilly MA, et al. Durable responses and low toxicity after fast off-rate Cd19 Chimeric antigen receptor-T therapy in adults with Relapsed or refractory B-cell acute Lymphoblastic leukemia. J Clin Oncol 2021;39:3352–63. 10.1200/JCO.21.00917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varet H, Brillet-Guéguen L, Coppée J-Y, et al. Sartools: A Deseq2- and Edger-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One 2016;11:e0157022. 10.1371/journal.pone.0157022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahnke YD, Brodie TM, Sallusto F, et al. The who’s who of T-cell differentiation: human memory T-cell Subsets. Eur J Immunol 2013;43:2797–809. 10.1002/eji.201343751 [DOI] [PubMed] [Google Scholar]
- 23.Petersen CT, Hassan M, Morris AB, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with Pi3Kδ inhibitors and VIP antagonists. Blood Adv 2018;2:210–23. 10.1182/bloodadvances.2017011254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to Cd19 Chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24:563–71. 10.1038/s41591-018-0010-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinrichs CS, Borman ZA, Gattinoni L, et al. Human Effector Cd8+ T cells derived from naive rather than memory Subsets possess superior traits for adoptive Immunotherapy. Blood 2011;117:808–14. 10.1182/blood-2010-05-286286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn T, Hiltensperger M. Role of IL-6 in the commitment of T cell Subsets. Cytokine 2021;146. 10.1016/j.cyto.2021.155654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi J, Paczkowski P, Shen Y-W, et al. Preinfusion Polyfunctional anti-Cd19 Chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018;132:804–14. 10.1182/blood-2018-01-828343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdullah L, Hills LB, Winter EB, et al. Diverse roles of AKT in T cells. Immunometabolism 2021;3:e210007. 10.20900/immunometab20210007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cabrera-Ortega AA, Feinberg D, Liang Y, et al. The role of Forkhead box 1 (Foxo1) in the immune system: Dendritic cells, T cells, B cells, and hematopoietic stem cells. Crit Rev Immunol 2017;37:1–13. 10.1615/CritRevImmunol.2017019636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerdiles YM, Beisner DR, Tinoco R, et al. Foxo1 links homing and survival of naive T cells by regulating L-Selectin, Ccr7 and interleukin 7 receptor. Nat Immunol 2009;10:176–84. 10.1038/ni.1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macintyre AN, Finlay D, Preston G, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is Dispensable for T cell metabolism. Immunity 2011;34:224–36. 10.1016/j.immuni.2011.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabatino M, Choi K, Chiruvolu V, et al. Production of anti-Cd19 CAR T cells for ZUMA-3 and -4: phase 1/2 multicenter studies evaluating KTE-C19 in patients with Relapsed/refractory B-precursor acute Lymphoblastic leukemia (R/R ALL). Blood 2016;128:1227. 10.1182/blood.V128.22.1227.1227 [DOI] [Google Scholar]
- 33.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3–12. 10.1002/path.2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Sullivan D, van der Windt GJW, Huang S-C, et al. Memory Cd8(+) T cells use cell-intrinsic Lipolysis to support the metabolic programming necessary for development. Immunity 2014;41:75–88. 10.1016/j.immuni.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Windt GJW, Everts B, Chang C-H, et al. Mitochondrial respiratory capacity is a critical regulator of Cd8+ T cell memory development. Immunity 2012;36:68–78. 10.1016/j.immuni.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawalekar OU, O’ Connor RS, Fraietta JA, et al. Distinct signaling of Coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 2016;44. 10.1016/j.immuni.2016.02.023 [DOI] [PubMed] [Google Scholar]
- 37.Sukumar M, Liu J, Mehta GU, et al. Mitochondrial membrane potential identifies cells with enhanced Stemness for cellular therapy. Cell Metab 2016;23:63–76. 10.1016/j.cmet.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amitrano AM, Berry BJ, Lim K, et al. Optical control of Cd8+ T cell metabolism and Effector functions. Front Immunol 2021;12. 10.3389/fimmu.2021.666231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felizardo TC, Foley J, Steed K, et al. Harnessing Autophagy for cell fate control gene therapy. Autophagy 2013;9:1069–79. 10.4161/auto.24639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via Phosphatidylinositol-3 kinase/AKT regulation of Glut1 activity and trafficking. Mol Biol Cell 2007;18:1437–46. 10.1091/mbc.e06-07-0593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graef P, Buchholz VR, Stemberger C, et al. Serial transfer of single-cell-derived Immunocompetence reveals Stemness of Cd8(+) central memory T cells. Immunity 2014;41:116–26. 10.1016/j.immuni.2014.05.018 [DOI] [PubMed] [Google Scholar]
- 42.Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic induction of Cd8+ T cell memory: better living through chemistry. Sci Transl Med 2009;1:11ps12. 10.1126/scitranslmed.3000302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghassemi S, Durgin JS, Nunez-Cruz S, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat Biomed Eng 2022;6:118–28. 10.1038/s41551-021-00842-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raje NS, Shah N, Jagannath S, et al. Updated clinical and correlative results from the phase I CRB-402 study of the BCMA-targeted CAR T cell therapy Bb21217 in patients with Relapsed and refractory multiple myeloma. Blood 2021;138:548. 10.1182/blood-2021-146518 [DOI] [Google Scholar]
- 45.Nishimura T, Nakui M, Sato M, et al. The critical role of Th1-dominant immunity in tumor Immunology. Cancer Chemother Pharmacol 2000;46 Suppl:S52–61. 10.1007/pl00014051 [DOI] [PubMed] [Google Scholar]
- 46.Wilde S, Sommermeyer D, Leisegang M, et al. Ls producing Th1 Polycytokines show superior antigen sensitivity and tumor recognition. J Immunol 2012;189:598–605. 10.4049/jimmunol.1102165 [DOI] [PubMed] [Google Scholar]
- 47.Muranski P, Borman ZA, Kerkar SP, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 2011;35:972–85. 10.1016/j.immuni.2011.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guedan S, Chen X, Madar A, et al. ICOS-based Chimeric antigen receptors program bipolar Th17/Th1 cells. Blood 2014;124:1070–80. 10.1182/blood-2013-10-535245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson EH, Zaph C, Mohrs M, et al. B7Rp-1-ICOS interactions are required for optimal infection-induced expansion of Cd4 Th1 and Th2 responses. J Immunol 2006;177:2365–72. 10.4049/jimmunol.177.4.2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salter AI, Ivey RG, Kennedy JJ, et al. Phosphoproteomic analysis of Chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal 2018;11:eaat6753. 10.1126/scisignal.aat6753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor Immunotherapy. Cell Metab 2017;26:94–109. 10.1016/j.cmet.2017.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu X, Araki K, Li S, et al. Autophagy is essential for Effector Cd8(+) T cell survival and memory formation. Nat Immunol 2014;15:1152–61. 10.1038/ni.3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng Q, Han G, Puebla-Osorio N, et al. Characteristics of anti-Cd19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell Lymphomas. Nat Med 2020;26:1878–87. 10.1038/s41591-020-1061-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang M, Jin X, Sun R, et al. Optimization of metabolism to improve efficacy during CAR-T cell manufacturing. J Transl Med 2021;19:499. 10.1186/s12967-021-03165-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghassemi S, Nunez-Cruz S, O’Connor RS, et al. Reducing ex vivo culture improves the Antileukemic activity of Chimeric antigen receptor (CAR) T cells. Cancer Immunol Res 2018;6:1100–9. 10.1158/2326-6066.CIR-17-0405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Novartis . Novartis Press Release: Novartis announces T-Charge. 2022. [Google Scholar]
- 57.Yang J, He J, Zhang X, et al. Next-day manufacture of a novel anti-Cd19 CAR-T therapy for B-cell acute Lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J 2022;12:104. 10.1038/s41408-022-00694-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2023-007002supp001.pdf (3.1MB, pdf)
Data Availability Statement
Data available on reasonable request