

Abstract
Cysteine proteases comprise an important class of drug targets, especially for infectious diseases such as Chagas disease (cruzain) and COVID-19 (3CL protease, cathepsin L). Peptide aldehydes have proven to be potent inhibitors for all of these proteases. However, the intrinsic, high electrophilicity of the aldehyde group is associated with safety concerns and metabolic instability, limiting the use of aldehyde inhibitors as drugs. We have developed a novel class of self-masked aldehyde inhibitors (SMAIs) for cruzain, the major cysteine protease of the causative agent of Chagas disease—Trypanosoma cruzi. These SMAIs exerted potent, reversible inhibition of cruzain while apparently protecting the free aldehyde in cell-based assays. We synthesized prodrugs of the SMAIs that could potentially improve their pharmacokinetic properties. We also elucidated the kinetic and chemical mechanism of SMAIs and applied this strategy to the design of anti-SARS-CoV-2 inhibitors.
Graphical Abstract
INTRODUCTION
Cysteine proteases are prospective drug targets for a variety of diseases caused by viruses (e.g., picornaviruses and coronaviruses),1,2 bacteria (e.g., Salmonella and Listeria),3 and particularly, many species of parasitic protozoa.4 One of these, Trypanosoma cruzi, is the causative agent of Chagas disease, which affects an estimated 6–7 million people worldwide.5 During the life cycle of T. cruzi, its major cysteine protease, known as cruzain, plays a multifaceted role involving nutrient processing, morphological transformation, invasion into host cells, and evasion of the immune response.6 Consequently, cruzain has been widely studied and also serves as a paradigm for inhibitor design for other cysteine proteases.
Substrate–analogue peptidomimetic compounds containing a suitable electrophilic warhead comprise the most common classes of inhibitors and inactivators of cruzain, as well as other cysteine proteases.7 A notable example is K777 (or K11777), a dipeptide analogue containing a vinyl sulfone warhead, which undergoes irreversible thia-Michael addition to the active-site Cys25 of cruzain,6 and other cysteine proteases including cathepsins K, L, and B (Figure 1A).8 However, irreversible binding of small molecules to protein targets is associated with idiosyncratic immunotoxicity,9 especially for drugs which require lengthy regimens. Accordingly, an emerging trend is the development of reversible covalent inhibitors which provide potent inhibition of the desired target, but will not permanently inactivate off-target proteins.10 Among the reversible covalent warheads explored for cruzain to date, an aldehyde group often, if not always, affords outstanding inhibitory potency compared to α-keto amides, nitriles, oximes, heterocycles, and so on.11–14 On the other hand, the inherent electrophilicity of aldehydes contributes to their rapid metabolism, immunotoxicities, and untoward drug–drug interactions,15 which have all but disqualified their use in drug design. Theoretically, these issues can be mitigated by confining the exposure of the aldehyde solely to the germane protein target while eschewing off-target proteins; however, such approaches have been challenging to implement.9
Figure 1.

Inactivator and inhibitors of cysteine proteases. (A) Structure of K777 and its mechanism of covalent inactivation of cysteine proteases. (B) 1,2,4-trioxolane inhibitor PG4b undergoes Fe(II)-catalyzed fragmentation to elaborate aldehyde PG3b, a potent inhibitor of falcipain, and a radical byproduct. (C) γ-lactol inhibitor MN3 is a masked aldehyde of SJA0617. It exhibits higher transcorneal permeability and releases the free aldehyde to inhibit μ-calpain.
An alternative approach to incorporate aldehydes into drug design is to mask the aldehyde using another functional group. The antimalarial aldehyde inhibitor of falcipain PG3b was “masked” by a 1,2,4-trioxolane structure (Figure 1B), resulting in the prodrug PG4b from which the active aldehyde is liberated upon reaction with Fe(II) inside the vacuoles of the plasmodial parasite.16 Unfortunately, this clever masked aldehyde resulted in toxicity to the host cells, apparently from the action of free radical byproduct(s). Its utility is also limited by the variable availability of Fe(II) in target cells. The calpain inhibitor MN3 is another type of masked aldehyde in which a homoserine at the P1 position17 spontaneously forms a cyclic hemiacetal (Figure 1C). Compared to the potent free aldehyde SJA6017 (with Leu at P1, ), protection of the aldehyde in MN3 greatly improved transcorneal permeability despite lower potency against calpain .18,19 Herein, we describe an unprecedented class of self-masked aldehyde inhibitors (SMAIs) for cruzain, their kinetic and chemical mechanisms of reversible covalent inhibition, and their effects on trypanosomal infection in cell cultures. In addition, we developed prodrugs of these SMAIs to improve their metabolic stability to optimize their pharmacokinetic properties. We adapted this concept to develop a novel inhibitor for the main cysteine protease (3CL protease) of SARS-CoV-2.
RESULTS AND DISCUSSION
Design of SMAIs.
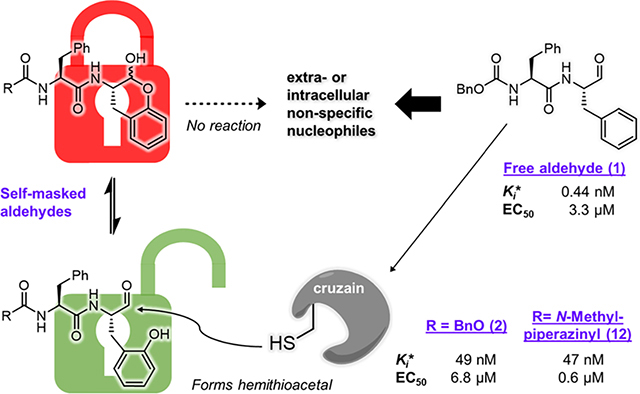
The dipeptide aldehyde 1 (Cbz-Phe-Phe-H, Figure 2A) was first prepared for use as an intermediate in the synthesis of other protease inhibitors and proved to be an extraordinarily potent inhibitor of cruzain, as discussed below. To modify its P1 structure to afford a self-masked aldehyde, we added a hydroxyl group to the 2′ position of the phenyl ring, converting the P1 group to an ortho-tyrosine (2, Cbz-Phe-o-Tyr-H). The oxygen of the phenol group is in close proximity to the aldehydic carbon permitting a nucleophilic addition reaction to occur, producing a cyclic hemiacetal (-lactol) similar to the -lactol found in calpain inhibitor MN3.19 We anticipated that a SMAI would remain “locked” in its -lactol form before binding to cruzain, after which enzyme catalysis would elaborate the free aldehyde followed by formation of a hemithioacetal adduct with cruzain (Figure 2A). If true, then the potential advantages of SMAIs include: (a) the intramolecular nature of the -lactol likely provides sustained protection of the aldehyde outside of an enzyme active site; (b) compared to the 1,2,3-trioxolane masked aldehyde,16 the cleavage of the hemiacetal does not produce reactive byproducts; and (c) the introduced hydroxyl group on the o-tyrosine sidechain is small enough so as to impose minimal perturbation of the original binding mode of the parent aldehyde 1.
Figure 2.

Rationale for the design of SMAIs. (A) In compound 2, the aldehyde group is expected to be masked by the 2′-phenol group via spontaneous formation of a lactol. It is anticipated that the SMAI will undergo enzyme-catalyzed opening of the lactol ring and subsequently form the hemithioacetal adduct with Cys25. The scheme describes a two-step inhibition mechanism in which rapid formation of an complex precedes isomerization to , which slowly converts back to . (B) Lactol form of 2 (green) noncovalently docked to cruzain. (C) Opened form of 2 covalently docked to form a hemithioacetal with Cys25. Both structures are superimposed with a covalently bound K777 (white) from the crystal structure (PDB ID: 2OZ2).20 Colored dashed lines represent corresponding cruzain–inhibitor interactions.
The first question arising from our hypothesis is: could the lactol form of 2 bind to cruzain and remain closed? To preliminarily address this question, we employed molecular modeling using docking methods conforming to the binding of compound 2 in both its noncovalent, lactol form (Figure 2B) and its free aldehyde form which was docked as a hemithioacetal adduct with the catalytic Cys25 (Figure 2C). The covalent adduct of 2 is predicted to bind in a manner similar to that of the covalent inactivator K777 as seen in the cruzain-K777 cocrystal structure.20 As expected, the phenoxy substituent of the covalently bound inhibitor 2 is well tolerated in the cruzain active site and may form hydrogen bonds with Gln19, His162, or Trp184, similar to the sulfone oxygens of K777. Although the Cbz-Phe group in Figure 2B adopts a similar orientation to the open form (Figure 2C), the lactol displays a puckered conformation in the active site. The bound lactol has a poorer binding free energy than that of the hemithioacetal , apparently owing to the bicyclic lactol moiety. One may infer that the recognition of the Cbz-Phe scaffold of 2 by cruzain assists in the binding of the lactol group and orients it for enzyme-catalyzed ring-opening to yield the high-affinity aldehyde.
Kinetic Analysis.
A series of SMAIs and related compounds 1–12 were prepared and evaluated as inhibitors of cruzain (Table 1). Time-course data for inhibition of cruzain by many of these compounds conformed to the kinetic scheme shown in Figure 2A, that is, initiation of reaction by adding enzyme to substrate and inhibitor led to curvilinear time courses (Figure 3A,C) in which reaction rates demonstrably decreased as the complex (characterized by ) progressed to the tighter complex (characterized by ). The observation of time dependence may or may not indicate that covalent bond formation has occurred, but it does reflect that , arising from either the formation of a hemithioacetal between the enzyme and inhibitor or a slow isomerization step of to not involving covalent bond formation.
Table 1.
| Compound | Structure | Cruzain Inhibition | |
|---|---|---|---|
|
|
|||
| Number | P3–P2 | P1 | (nM)a |
|
| |||
| 1 | Cbz-Phe- |
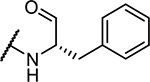
|
0.44 ± 0.02 |
| 2 | Cbz-Phe- |
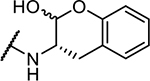
|
49 ± 2 |
| 3 b | NMePip-Phe- |
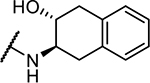
|
>100,000 |
| 4 | Cbz-Phe- |
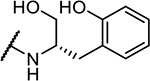
|
>10,000 |
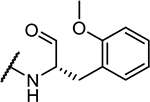
| 5 | Cbz-Phe- |
|
22 ± 2 |
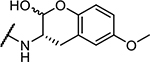
| 6 | Cbz-Phe- |
|
350 ± 30 |
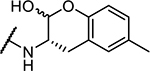
| 7 | Cbz-Phe- |
|
103 ± 5 |
| 8 | Cbz-Phe- |
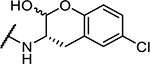
|
74 ± 10 |
| 9 | Cbz-Phe- |
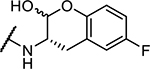
|
48 ± 2 |
| 10 | Cbz-Phe- |
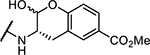
|
18 ± 0.5 |
| 11 | NMePip-Phe- |
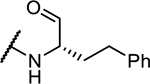
|
0.5 ± 0.2 |
| 12 | NMePip-Phe- |
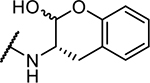
|
47 ± 2 |
| K777 | NMePip-Phe- |
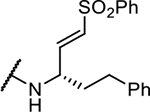
|
0.2c |
To the assay buffer (50 mM MES, 50 mM TAPSO, 1 mM CHAPS, 1 mM Na2EDTA, 5 mM DTT, 10% DMSO (v/v), and pH 7.5) containing variable concentrations of inhibitors (0.02–20 μM) and 10 μM of substrate Cbz-Phe-Arg-AMC () was added cruzain (final concentration of 0.2 nM), and changes in fluorescence were monitored for 20–60 min. Values of ) were obtained as described.
3 contains two diastereomeric anti products, and only one is shown here.
Reported as an apparent IC50 in ref 48.
Figure 3.

Kinetic analysis of inhibition of cruzain by 1, 2, and 12. (A–C) Time courses of cruzain inhibition by 1, 2, and 12. Insets for (A) and (C): the values were obtained by fitting progress curves to eq 1. Replot of vs with the line drawn through data points from fitting to eq 3.1: ; 12: . Inset for (B): the steady-state rates with and without inhibitor were obtained at min. Plot of vs [2] with the line drawn through data points from fitting to eq 4. (D) Proposed mechanism of phenoxy-assisted conversion of back to .
We first evaluated cruzain inhibition by time-course data containing fixed concentrations of substrates and variable concentrations of inhibitors for which reactions were initiated by the addition of enzyme. Inhibition of cruzain by the free aldehyde 1 was characterized by downward-concave curvilinear time courses, often referred to as “burst kinetics” (Figure 3A), indicative of time-dependent inhibition for which equilibrium between cruzain and 1 was slowly established over 30 min. Each curve was fitted to eq 1, and the resulting values of were replotted versus inhibitor concentration [1] (Figure 3A, inset). The replot was best fitted to eq 3, implying a lack of saturation of the complex by 1 for which . The unimolecular rate constant for conversion of back to was extremely slow . The low value of is likely the reason for the slow onset of inhibition and potency of aldehyde 1.
Unlike aldehyde 1, inhibition of cruzain by 2 exhibited almost linear time courses with only slight curvature observed at early stages (Figure 3B), as was also observed for SMAIs 6–10 (all containing the -Phe scaffold, Figure S1A). The nominal burst phase was only observed within the first 1–2 min, after which an apparent steady-state reaction was established, indicating a significantly faster rate (greater ) for conversion of back to than for compound 1. This difference suggests that, compared to a relatively stable hemithioacetal intermediate cruzain-1 species, the reverse reaction of this intermediate for a SMAI is likely facilitated by the attack of its phenoxy group on the hemithioacetal (Figure 3D). To characterize these inhibitors, we obtained steady-state rates of and at assay times ≥20 min. For instance, upon plotting versus [2] (Figure 3B, inset) and fitting using eq 4, we obtained a value of the overall inhibition constant of 49 ± 2 nM. For free aldehyde 11 and SMAI 12, both of which incorporated the N-methylpiperazinyl-Phe (NMePip-Phe) scaffold as found in K777, the ratio of their activities as cruzain inhibitors was almost identical to that of their Cbz-Phe-containing counterparts 1 and 2 . Similar to aldehyde 1, slow onset of inhibition was observed for 11 (Figure S1B). Surprisingly, unlike SMAIs containing the Cbz-Phe scaffold, 12 also exhibited time-dependent inhibition (Figure 3C).
To explore the reversibility of cruzain inhibition by these compounds, we first preincubated each inhibitor with cruzain followed by a 100-fold rapid dilution accompanying the addition of substrate (Figure 4A). For inhibitors 2 and 6–10 (Figure 4B,C), 91% recovery of cruzain activity was observed over the course of minutes, wherein the residual concentration of inhibitor was , capable of affording 9% inhibition, thereby demonstrating that binding of these inhibitors was fully reversible. In comparison, the activity of cruzain preincubated with aldehydes 1, 11, or SMAI 12 was restored at much slower rates (Figure 4D–F). After fitting these “lag” time courses to eq 1, the calculated residence times for compounds 2 and 6–10 ranged from 2.5 to 5.5 min, whereas 1, 11, and 12 exhibited significantly longer residence times of 52, 45, and 26 min, respectively (Table S1). While longer residence times were expected for aldehydes 1 and 11, compound 12 was the only SMAI to effect significant time-dependent inhibition as evidenced by the observation of both burst and lag kinetic time courses, and the rate of conversion of the complex to was comparable to that of aldehyde 11. Considering that compound 12 was equipotent to 2, one could infer that the peptidomimetic scaffold of 12 might also affect the adduct formation so that remained nearly unchanged for both the SMAI and the free aldehyde. Therefore, a covalent docking study was conducted for 12, which predicted a binding free energy that was close to that of compound 2 as formerly mentioned, which was in accordance with their similar values. Interestingly, an intramolecular hydrogen bond between the phenol group and carbonyl group has dragged the two moieties away from their binding subsites (Figure S2). The consequence of this conformation is that the phenol group in 12 is less likely to take part in the conversion of back to (Figure 3D), leading to slower dissociation from cruzain and its unique lag kinetics compared to 2.
Figure 4.

Rapid dilution assay for SMAIs. (A) Dilution scheme for testing the reversibility of a SMAI. Upon 100-fold dilution, the inhibitor concentration decreases from (91% inhibition) to (9% inhibition). (B, C) Cruzain activity rapidly recovered from inhibition by 2 and 6–10. (D–F) Cruzain activity recovery showed a significant lag for 1, 11, and 12. All curves were fitted to eq 1 to provide values as listed in Table S1.
Structure–Activity Relationships of SMAIs.
Inhibition constants for all compounds prepared for this study are shown in Table 1. The equipotency of SMAIs 2 and 12 at ~50 nM suggests that their different P3 sidechains have no impact on their inhibition. We prepared compound 3, an analogue of SMAI 12 in which its ether oxygen was replaced with a methylene group, and the resulting compound containing the stable tetrahydro-naphthol did not inhibit cruzain at ≤100 μM, despite its similarity to the chroman-2-ol group of the SMAIs. Substitution of the aldehyde of compound 2 with a primary alcohol also resulted in the poor inhibitor 4, which corroborates the importance of the formation of carbon–sulfur bonds in the cruzain-SMAI complexes. In the free aldehyde 5, the phenoxy group was methylated, affording an inhibitor that is 50-fold less active than aldehyde 1, implying that a bulkier 2′ substituent sterically hinders the formation of the hemithioacetal.
Another potential feature of SMAIs is the ability to tune the reactivity of the phenol group by installing substituents on the benzene ring. We introduced several substituents at the 5′ position of the ortho-tyrosine, including electron-donating groups (i.e., -OMe and -Me), weak electron-withdrawing groups (i.e., -F and -Cl), and moderate-to-strong electron withdrawing group (i.e., -CO2Me). The electron-donating substituents on 6 and 7 appear to undermine their potencies, as these inhibitors bind, respectively, 7 - and 20-fold more weakly than un-substituted SMAI 2. The stronger electron-withdrawing capability of the ester substituent (10; ) provided a 3-fold increase in potency versus 2, suggesting that the 5′-methyl ester facilitates the opening of the lactol ring. In addition to the electronic effect, a steric effect is likely operative. The methoxy group of compound 6 is bulkier than the methyl group of 7, which may contribute to its over 3-fold lower activity. Chlorine (0.79 Å, 8) has a slightly larger radius than fluorine (0.42 Å, 9), and fluorine is similar in size to a hydrogen (0.53 Å, 2) (note: the C–F is longer than the C–H bond so that, at that increased length, the fluorine is effectively the same radius as hydrogen),21 consistent with the differences between their values. It is interesting that a steric effect apparently dictates the differences in potency between compounds 8 and 9, probably because fluorine and chlorine are only weakly electron-withdrawing. The methyl ester of 10, while comprising the bulkiest substituent among this series of compounds, provided the SMAI of the highest potency in this study, apparently because the electron-withdrawing effect afforded by this large substituent overcomes any steric effect. These results suggested that appropriate substitution of the phenyl ring in SMAIs could optimize their potencies, and further, may affect the stability of enzyme-bound lactol.
Chemical Mechanism of SMAI Binding to Cruzain.
Next, we prepared compound 12 in which the aldehydic carbon was 13C-labeled, to be used in a study of 1H-13C heteronuclear single quantum correlation (HSQC) nuclear magnetic resonance (NMR) to elucidate the chemical specie(s) of 12 that are bound to cruzain. First, 0.4 mM of 13C-labeled 12 in phosphate buffer (pH 7.5) was analyzed by HSQC NMR. No discernable peak was present at , ruling out the existence of minute concentrations of free aldehyde in this aqueous solution (see the Supporting Information for a full-view spectrum).22 The two salient peaks, A and B, occurring near , were consistent with the signal of a carbon occurring in a hemiacetal (Figure 5A, red). These signals could not be assigned to an aldehyde hydrate in view of the fact that analysis by liquid chromatography–mass spectrometry (LC–MS) displayed a molecular ion peak corresponding to the hemiacetal but not the hydrate (Figure S3). We propose that peaks A and B are associated with two -lactol anomers spontaneously generated during lactol formation. The ratio of anomer A to anomer B is 1:1.32 based on peak volumes (Table S2), yet the exact stereochemistry cannot be assigned at this point.
Figure 5.

Studies on the chemical mechanism of SMAI binding to cruzain. (A) Expansion of the superposed 1H-13C HSQC NMR spectra of 13C-labeled compound 12 with (blue) and without (red) an approximately equimolar concentration of cruzain, which were obtained at 800 MHz () at 25°C. (B) Time course of phenylhydrazone formation by treating 0.2 mM 11 (aldehyde) or 12 (lactol) with 1 mM phenylhydrazine, with a control sample containing DMSO. Data for the curve with compound 11 were fitted to from which , and , while fitting the other data to this expression led to negligible values of .
To the inhibitor sample was added an approximately stoichiometric amount of cruzain, and a new spectrum was acquired after 1 h (Figure 5A, blue). The apparent hemiacetal peak B was eliminated while a trace of peak A remained, which indicates a slight preference of cruzain for anomer B over A, in consideration of the ratio of integrated volumes (A:B = 1:1.32). The fact that the cross peaks shifted considerably upon addition of cruzain is strongly indicative of hemithioacetal formation. This is because the new signals observed at 76.2 and 79.7 ppm are very similar to those observed for the formation of a hemithioacetal adduct between N-acetyl-L-phenylalanine-[2-13C]glycinal and papain ( 75.1 ppm).23 The broadening of peaks A’ and B′ also suggested a protein-bound ligand, because of reduced tumbling and the increased relaxation time of the cruzain-bound 13C-labeled 12. In addition, mass spectrometric experiments of cruzain preincubated with 12 as well as three SMAIs with the Cbz-Phe-scaffold (2, 7, and 9) clearly revealed the formation of corresponding enzyme–inhibitor adducts as indicated by the mass changes relative to apo-cruzain (Figure S4), serving as orthogonal evidence of covalent bond formation.
As phenylhydrazine readily forms phenylhydrazone adducts with aldehydes, we treated 0.2 mM of aldehyde 11 and SMAI 12 with 1 mM phenylhydrazine in the same buffer (lacking DTT) used in the NMR study (Figure 5B) to determine the fraction of free aldehyde found in 12. While aldehyde 11 was rapidly, and apparently, completely converted to phenylhydrazone, 12 remained intact after 2 h. This finding showed that even an excess amount of phenylhydrazine cannot drive the equilibrium of 12 toward the formation of open-form aldehyde, which not only corroborated the chemical stability of lactol, but also indicated that the ring-opening is likely an enzyme-catalyzed process. Therefore, we conclude that SMAI 2 predominantly maintains its lactol form in aqueous solution in the absence of cruzain, while its binding to cruzain apparently promotes opening of its ring followed by the formation of a covalent bond with Cys25.
SMAIs Display Antitrypanosomal Activity.
Subspecies of pathogenic Trypanosoma brucei have essential cysteine proteases (TbCatL, brucipain, or rhodesain) which are close homologs of cruzain and perform similar biological functions.24 We evaluated our compounds in axenic cultures of the bloodstream forms (BSFs) of Trypanosoma brucei brucei (T. b. brucei), which are found in infected mammals (Table 2, Figure S5). The most active inhibitors for BSFs, 7 and 12, exhibited respective values of 0.5 and , which are more potent than the antitrypanosomal drug, diminazene (Berenil, in T. b. brucei BSFs).25 Notably, the free aldehyde 1, despite its 100-fold higher potency versus cruzain, was equally or less potent in inhibiting T. b brucei BSFs compared to the SMAIs. Considering that aldehyde 1 is nearly four orders of magnitude less effective versus T. b. brucei than versus purified cruzain , while the average value of for all SMAIs is 74, one may speculate that the SMAIs are more accessible to its cellular target(s) than free aldehyde 1 in cell culture media. In this sense, masking of the active aldehyde within the SMAIs affords significant protection of its electrophilic group.
Table 2. Antitrypanosomal Activities of SMAIs and their Prodrugs.
| compound number | cruzain inhibition (nM) | T. b. brucei BSFs EC50 (μM)a | T. cruzi-infected cardiomyoblasts EC50 (μM)a |
|---|---|---|---|
| 1 | 0.44 ± 0.02 | 3.3 ± 2.1 | 0.5 ± 0.4 |
| 2 | 49 ± 2 | 6.8 ± 1.1 | 2.9 ± 0.2 |
| 6 | 350 ± 30 | 2.6 ± 1.1 | NDd |
| 7 | 103 ± 5 | 0.5 ± 0.2 | 5.7 ± 0.6 |
| 8 | 74 ± 10 | 5.8 ± 3.4 | ND |
| 9 | 48 ± 2 | 2.7 ± 0.2 | 3.5 ± 0.6 |
| 10 | 18 ± 0.5 | 4.0 ± 0.2 | 3.5 ± 0.3 |
| 12 | 47 ± 2 | 0.6 ± 0.1 | >20 |
| 13 b | ~1000 | 3.7 ± 0.2 | 13 ± 2 |
| 14 b | >5000 | 3.1 ± 0.2 | ND |
| 15 b | >5000 | 4.0 ± 0.2 | 10±3 |
| 16 b | >5000 | 12 ± 0.1 | ND |
| 17 b | >5000 | 14 ± 0.9 | ND |
| K777 | 0.2c | 0.09 ± 0.06 | 0.04 ± 0.01 |
Trypanocidal activities of compounds in axenic cultures of T. b. brucei BSFs and in cardiomyoblasts with T. cruzi infection were all measured as EC50 values (half maximal effective concentration), along with SEMs from at least two replicates. Typical test concentrations of inhibitors were 0.5–20 μM for T. b. brucei BSF assays and 0.02–40 μM for T. cruzi-infected cardiomyoblast assays.
Structures of 13–17 are shown in Figure 6B.
Reported as an apparent IC50 in ref 48.
ND, not determined.
Additionally, aldehyde 1, SMAIs 2, 7, 9, 10, 12, 13, and 15 (the latter two compounds are discussed in the next section) were analyzed in a murine cardiomyoblast model of T. cruzi infection in which the disease-relevant amastigote forms of the parasite were evaluated (Table 2, Figure S6). The parent aldehyde 1 was the most potent inhibitor in this series , yet it still lost most of its activity considering its subnanomolar activity against cruzain. Compounds 2, 7,9, and 10 killed T. cruzi at values ranging from , which were similar in potency to benznidazole, a first-line antichagasic drug with an value of in this assay (Figure S6). Surprisingly, 12 exhibited much weaker trypanocidal activity in this assay in contrast to its excellent activity in T. b. brucei, probably because its peptidomimetic scaffold, with higher polarity, had difficulty in penetrating the host cell membrane. Apart from 12, these SMAIs were essentially equipotent in axenic cultures of T. b. brucei BSFs and T. cruzi-infected cardiomyoblasts, suggesting that they target a cysteine protease homologue of cruzain in T. b. brucei. Additionally, they exerted no toxicity against the host cardiomyoblasts at up to 40 μM where both aldehyde 1 and K777 exhibited significant cytotoxicity (Figure S6). Accordingly, the self-masking of the aldehyde group in SMAIs provides the apparent delivery of these otherwise reactive compounds to trypanosomes harbored within mammalian cardiomyoblasts, with no apparent untoward effects on the host cells. These results encouraged progressing the SMAIs to preclinical analysis, as well as the development of prodrug forms of SMAIs.
Prodrugs of SMAIs.
The pharmacokinetic evaluation of compound 12 in mice by intravenous administration indicated a short half-life arising from apparent first-pass metabolism (data not shown). Its analogue, K777, has superior pharmacokinetic properties to 12, and it was shown that the sites of oxidative metabolism on K777 were largely confined to the N-methylpiperazine ring, common to both inhibitors, and the homophenylalanyl sidechain.26 Therefore, metabolism of the cyclic hemiacetal of 12 may be responsible for its rapid clearance in mice.
The cyclic α-keto-acetal prodrug for caspase-1, pralnacasan (VX-740), can be regarded as an SMAI, as it is rapidly converted to the active aldehyde inhibitor (VRT-18858) by plasma esterases via formation of a hemiacetal intermediate (Figure 6A).27 We designed two types of derivatization of the hemiacetal hydroxyl group, that is, via O-acylation and O-alkylation (Figure 6B). The O-acylated compounds should be hydrolyzed to compound 12 by the action of cellular esterases, which are ubiquitous in the ER lumen of mammalian tissues.28 For the O-alkylated compounds, the enzymatic oxidation is likely to occur by the action of liver cytochrome P450 enzymes, thereby elaborating a new hemiacetal that subsequently collapses to form compound 12.
Figure 6.

Development and assessment of SMAI prodrugs. (A) Pralnacasan is a SMAI prodrug for VRT-18858, an inhibitor for caspase-1. (B) Proposed metabolic routes of O-derivatized SMAIs to 12. (C) Time course of remaining 13–15 in reactions with or without addition of porcine esterase. (D) High-performance liquid chromatography (HPLC) traces of compound 15 in buffer (control). (E) HPLC traces of 15 treated with esterase.
To explore this prodrug approach, we prepared O-acylated compounds 13–15 and O-alkylated compounds 16 and 17 (structures shown in Figure 6B). These compounds exhibited negligible inhibition against cruzain, with the exception of 13, which weakly inhibited cruzain in a time-dependent fashion (, Figure S1C). Compounds 13–15 were treated with porcine esterase in buffer (pH 7.5), and the hydrolysis of these prodrugs was monitored by LC–MS (Figure 6C). In control samples without esterase, these compounds largely remained intact. Compound 13 was an exception and degraded about 10% in buffer over 3 h, which likely explains the observed inhibition of cruzain by 13 because of the formation of 12. Upon addition of the esterase, compounds 13–15 were all converted to compound 12 at variable rates of reaction with respective half-lives of 48, 24, and 16 min (by fitting curves in Figure 6C to eq 6) and displayed a trend of increased hydrolysis with the increasing steric bulk of the acyl groups. A reasonable interpretation is that the main mammalian esterase, carboxylesterase-1 (CES1), has a preference for larger acyl groups.28 Accordingly, modifying the acyl group of O-acylated compounds to obtain a suitable half-life can potentially overcome the apparent first-pass metabolism of 12. As observed for compound 15 (Figure 6D,E), there are two peaks in the chromatograph that presumably correspond to the two anomers, but the conversion of one peak (A, ) to 12 is faster than the other (B, ). Peak A is largely eliminated after 30 min, while peak B, though it constitutes a smaller proportion of untreated compound, does not decompose completely even after 3 h, suggesting that the anomer in peak A is the more specific substrate for esterase. These results provide a proof of concept that the use of O-acylated prodrugs of SMAIs will provide a means to deliver these inhibitors in vivo.
Compound 12 was also modified to yield two mixed acetals 16 and 17 (Figure 6B) to yield prodrugs that did not inhibit cruzain. Because cytochrome P450 forms 3A4, 2D6, and 2C9 together account for over 60% of drug-metabolizing P450 isoforms,29 we used these enzymes to conduct in vitro assays to determine if 16 and 17 could be transformed to compound 12 as expected. Both compounds were incubated with different CYPs in the presence of a NADPH-regenerating system for up to 4 h. Unfortunately, no transformation to 12 was observed for either compound by any of the CYPs. However, the apparent stability versus these purified P450s does not mean that (an)other microsomal oxidase(s) cannot release the active SMAIs from these prodrug forms.
These compounds were tested in cell assays (Table 2). Although less active than their parent compound 12, compounds 13–15 showed promising trypanocidal activity against axenic T. b. brucei comparable to other SMAIs (). Moreover, 13 and 15 were also tested in the T. cruzi-infected model. It is notable that they showed moderate anti-T. cruzi effects (Table 2, and 10 μM, respectively), which were more potent than 12, indicating that their acyl groups helped them enter host cells probably by increasing the hydrophobicity of these molecules. These results also demonstrated that the O-acylated prodrug forms could well be converted to 12 inside host cells or parasites by esterases. Interestingly, compounds 16 and 17 were able to kill the T. b. brucei in spite of lower activity . Because treatment with P450s did not lead to transformation of these prodrugs, the data implied the existence of either a drug-metabolizing enzyme or another potential activating enzyme in T. b. brucei.
Application of SMAIs to SARS-CoV-2 3CLpro.
The SMAI strategy is not limited to cruzain and may also find application in other cysteine proteases. To this end, we attempted to develop SMAIs for SARS-CoV-2 main protease (3CLpro), a drug target for COVID-19.30 Numerous inhibitors of the homologous cysteine proteases of SARS-CoV, MERS-CoV, and some picornaviruses have shown different levels of inhibition against SARS-CoV-2. Peptidomimetic aldehyde inhibitors of 3CLpro contain a nearly invariant 2-oxo-pyrrolidin-2-yl group (γ-lactam) as the P1 side chain. We propose that 2-pyridone can act as a surrogate for the γ-lactam (Figure 7A). Apart from their similarity in size and heteroatom substitution, the tautomerization of 2-pyridone toward 2-hydroxypyridine has been well characterized.31 Although the 2-pyridone tautomer is preferred in aqueous solution, the presence of the C-terminal aldehyde could well form a SMAI with the 2-hydroxypyridine.
Figure 7.

Compound 18 is a potential SMAI for SARS-CoV-2 3CLpro. (A) 2-Pyridone is a surrogate for the P1 γ-lactam. (B) Structure and activity of compound 18. (C) Surface representation of the 3CLpro-18 complex (PDB ID: 7M2P). The electron density map contoured at 1.0 indicated C–S bond formation. (D) Interaction between 3CLpro and 18. Dashed lines depict hydrogen bonds; the oxyanion hole is circled.
A 2-pyridone compound 18 was synthesized (Figure 7B, Scheme 4), and the proton NMR of 18 in organic solvents confirmed the apparent absence of aldehyde, suggesting that it forms a δ-lactol, though it is unclear what the exact species is in aqueous buffers. Compound 18 was such a potent inhibitor of 3CLpro that its (9 nM) was a result of apparent titration of the enzyme (40 nM used in the assay). Anti-CoV-2 activity in SARS-CoV-2-infected A549/ACE2 cells was also demonstrated (Table S3). Interestingly, 18 also inhibited human cathepsin L, which was recently shown to be essential to the penetrance of SARS-CoV-2 into mammalian cells in which it catalyzes essential cleavage of the coronaviral Spike protein.8,32 As a result, compound 18 has the potential to be a dual-acting inhibitor for two enzymes which are critical to the infection of human cells by SARS-CoV-2.
Scheme 4. Synthesis of 18a.

a(i) DBU, DCM, (ii) Pd/C, H2, MeOH, (iii) LiOH, MeOH/H2O, (iv) N,O-dimethylhydroxylamine, DIPEA, T3P, DCM, (v) TFA, DCM; Cbz-Val-Cha-OH, DIPEA, T3P, DCM, and (vi) LAH, THF, 0 °C.
We also obtained a high-resolution (1.70 Å) crystal structure of SARS-CoV-2 3CLpro in complex with 18. The well-defined electron density (Figure 7C) confirmed the formation of a hemithioacetal of 18 with active-site Cys145, and the resulting hydroxyl group of the hemithioacetal is stabilized by an oxyanion hole provided by Gly143 and Cys145 (Figure 7D). The P1 2-pyridone establishes essential binding interactions with 3CLpro analogous to that of the γ-lactam30 in which the carbonyl oxygen of the 2-pyridone accepts two hydrogen bonds from His163 and Ser144, while the amide nitrogen acts as a hydrogen bond donor to Glu166 and Phe140. These interactions clearly demonstrate that the γ-lactam can be bioisosterically replaced by the 2-pyridone moiety, which is a potential SMAI substructure. As with cruzain, 3CLpro is apparently capable of catalyzing ring-opening of the putative masked aldehyde of 18.
Chemistry.
The synthesis of SMAIs and their analogues involved the preparation of various P1 building blocks and subsequent coupling with P3-P2 scaffolds (Scheme 1). A variety of 5-substituted salicylaldehydes were coupled with N-Boc-2-phosphonoglycine trimethyl ester in Horner–Wadsworth–Emmons reactions. The formed double bond in S1a was reduced to afford the Boc-protected methyl ester of substituted ortho-tyrosine (S1b) as a mixture of enantiomers, which was then deprotected and reacted with the carboxylic acids of corresponding P3-P2 fragments, Cbz-Phe-OH or NMePip-Phe-OH. Cbz-Phe-OH was commercially available, while NMePip-Phe-OH (S1j) was simply prepared in two steps including amide coupling and hydrogenolytic cleavage of the benzyl group. The resulting S1d and subsequent intermediates were diastereomers that could be mostly separated on a silica gel column. Because the phenol group is relatively reactive that may interfere with future reactions, a methyl group was originally used for its protection and eventually resulted in compound 5. In our effort to remove the methyl group, we experienced unwanted side reactions because of harsh conditions such as the use of BBr3. Instead, a TBS group was employed as the protecting group. Next, the methyl ester of S1e was successively reduced to alcohol using NaBH4 (S1f and 4) and oxidized to aldehyde using Dess-Martin periodinane (S1g). Finally, efficient removal of TBS by TBAF exposed the phenol group toward the reaction with aldehyde, leading to in situ formation of the lactol ring (S1h). It should be noted that each SMAI was a mixture of anomers that could be observed by HPLC or NMR (see the Supporting Information). Because the spontaneous interconversion existed in between the anomers, it was neither necessary nor possible to chromatographically isolate them.
Scheme 1. Synthesis of 2–12a.

a(i) DBU, DCM, (ii) Pd/C, H2, MeOH, (iii) TFA, DCM, (iv) Cbz-Phe-OH or NMePip-Phe-OH (S1j), DIPEA, T3P, DCM, (v) TBSCl, imidazole, DCM, (vi) K2CO3, CH3I, DMF, (vii) NaBH4, MeOH, (viii) Dess-Martin periodinane, NaHCO3, DCM, 0 °C, (ix) TBAF, THF, 0 °C, (x) Et3N, THF, 0 °C, (xi) Pd/C, H2, MeOH, (xii) mCPBA, chloroform, (xiii) NaN3, MeOH/H2O, 60 °C, (xiv) Pd/C, H2, MeOH, (xv) NMePip-Phe-OH (S1j), DIPEA, T3P, DCM, (xvi) NMePip-Phe-OH (S1j), DIPEA, T3P, DCM, (xvii) NaBH4, MeOH, and (xviii) Dess-Martin periodinane, NaHCO3, DCM, 0 °C. *S1l contains two diastereomeric anti products, and only one is drawn here, so do S1m and 3.
The P1 building block of 3 was synthesized using a different strategy. 1,4-Dihydronaphthalene was subjected to epoxidation (S1k) followed by nucleophilic attack by NaN3 to produce S1l. The azide was then converted to the amino group which was coupled with NMePip-Phe-OH to yield compound 3. Notably, 3 contains two diastereomeric anti products as the P1 NH and OH are not on the same side of the ring. The preparation of free aldehyde 11 was similar to other SMAIs, except for that the latter required one more step for TBS removal.
The derivatizations of the hydroxy group of 12 are summarized in Scheme 2. Acylation was achieved by reacting 12 with corresponding acid anhydrides under basic conditions (S2a); alkylation was performed by treating 12 with boron trifluoride etherate in the presence of corresponding alcohols (S2b).
Scheme 2. Synthesis of 13–17a.

a(i) acetic/propionic/isobutyric anhydride, Et3N, DMAP, DCM and (ii) BF3OEt2, EtOH/iPrOH.
Because 13C-lableled N-Boc-2-phosphonoglycine trimethyl ester was not purchasable, we adopted a Ni(II) complex-based synthetic route for 13C-labeled 12 (Scheme 3).33 A chiral auxiliary S3a (2-(N-benzylprolyl)aminobenzophenone, BPB) was synthesized and then treated with [1-13C] glycine and nickel(II) nitrate hexahydrate under strongly alkaline conditions to form the Ni(II) complex (S3b), which could increase the acidity of α-protons in the glycine. Next, a Mitsunobu–Tsunoda reaction using CMBP was performed to introduce the side chain onto glycine with high stereoselectivity (S3c).33 We employed 8-quinolinol for the decomplexation to release the 13C-labeled benzylated o-Tyr (S3d). After protecting the amine with Boc (S3e), the carboxylic acid was converted to a Weinreb amide (S3f). The latter was deprotected and coupled to NMePip-Phe-OH (S3g) followed by reduction to aldehyde with an equimolar amount of lithium aluminum hydride (S3h) and removal of the benzyl-protecting group, resulting in 13C-labeled 12.
Scheme 3. Synthesis of 13C-labeled 12a.

a(i) 1-methylimidazole, MsCl, DCM, 45 °C, (ii) [1-13C] glycine, Ni(NO3)2·6H2O, KOH, MeOH, 60 °C, (iii) (2-(benzyloxy)phenyl)methanol, cyanomethylenetributylphosphorane (CMBP), toluene, 120 °C, (iv) 8-quinolinol, MeCN/H2O, 40 °C, (v) Et3N, Boc anhydride, dioxane/H2O, (vi) N,O-dimethylhydroxylamine, DIPEA, T3P, DCM, (vii) TFA, DCM; NMePip-Phe-OH, DIPEA, T3P, DCM, (viii) LAH, THF, 0 °C, and (ix) Pd/C, H2, MeOH.
Similar to Scheme 1, the 2-pyridone inhibitor 18 was also synthesized from 2-oxo-1,2-dihydro-3-pyridinecarbaldehyde (Scheme 4). After hydrogenation of S4a, the methyl ester of S4b was hydrolyzed (S4c) and converted to Weinreb amide S4d. The incorporation of the Cbz-Val-Cha-scaffold resulted in S4e as diastereomers that could be separated on a silica gel column. It turned out that no protection was needed for the 2-pyridone/2-hydroxypyridine, so that the LAH reduction of the Weinreb amide afforded the final product 18. Although there is no apparent aldehydic peak in NMR spectra when using CDCl3 as the solvent, the two inherent equilibria (lactol/aldehyde and pyridone/hydroxypyridine) have led to greater complexity. The exact species of 18 are dependent on multiple factors including solvents, temperature, pH, and so on, as exemplified by its HPLC–MS data (see the Supporting Information): there are several crowded peaks in HPLC, yet all of which correspond to the mass of 18 ( 552.29).
CONCLUSIONS
While the aldehyde group has frequently been employed in chemical probes in drug discovery, rarely has this reactive functionality been advanced in earnest to drug discovery campaigns. However, the masking of aldehydes in drugs and clinical agents has provided a few examples of the successful “masking” of aldehydes for drug discovery. Here, we sequestered the potent free aldehyde inhibitor 1 into an intramolecular masked aldehyde by the introduction of a 2-hydroxyl group onto the P1 benzyl group, which resulted in a surprisingly stable δ-lactol derivative that effected potent yet reversible inhibition of cruzain. The apparent stability of δ-lactol in examples of these SMAIs suggests that the observed potent inhibition of cruzain by these SMAIs most likely arises from cruzain-catalyzed ring-opening of the δ-lactol followed by formation of a hemithioacetal. Where evaluated, these self-masked aldehyde inhibitors inhibited axenic and infected cell cultures of trypanosomes with potency equaling or exceeding that of the parent aldehydes. Furthermore, the O-acylation of SMAIs yielded prodrugs that might possess improved metabolic stability and pharmacokinetic properties. Finally, we developed and characterized a novel SMAI of SARS-CoV-2 3CLpro, suggesting a broad applicability of the SMAI strategy to other cysteine proteases.
EXPERIMENTAL SECTION
General Information of Chemistry.
Unless otherwise noted, all starting materials, reagents, and solvents were obtained commercially and used without further purification or distillation. Reactions were conducted under an inert atmosphere (Ar or N2), and reaction progress was monitored by thin layer chromatography (TLC) or HPLC–MS. TLC experiments were performed with silica gel plates on aluminum foil (Sigma-Aldrich, cat. no. 60778). HPLC analysis was conducted on an UltiMate 3000 HPLC system coupled to MS analysis by an ISQ EM single quadrupole mass spectrometer. The typical settings for HPLC–MS were as follows. Column used: Phenomenex Luna 5 μm C18(2) 100 Å, 4.6 mm × 50 mm; mobile phase A: water containing 0.1% formic acid (v/v); mobile phase B: MeCN containing 0.1% formic acid (v/v); method: gradient elution at 10–100% B over 6 min, then isocratic elution at 100% B for 2 min; flow rate: 1 mL/min; MS parameters: HESI, vaporizer temperature 350 °C, source CID voltage 20 V, and gas flow: sheath = 40–50 psi, aux = sweep = 2.0 psi. Most compounds were purified by flash column chromatography (FCC) on silica gel (200–300 mesh) with different solvent systems. Some compounds were purified by semipreparative HPLC (Prep-HPLC) on the same UltiMate 3000 HPLC system, which was connected to a fraction collector. The typical settings for Prep-HPLC were as follows: Column used: Phenomenex Luna 5 μm C18(2) 100 Å, 21.2 mm × 250 mm; mobile phases A and B were the same as those for analytical HPLC; method: gradient elution at 10–100% B over 25 min, then isocratic elution at 100% B for 5 min; flow rate: 21.2 mL/min. 1H and 13C NMR spectra of compounds, except for 13C-labeled compound 12, were recorded on a Bruker AVANCE III 400 MHz using tetramethylsilane (TMS, 0.00 ppm) or residual solvent (CDCl3, 7.26 ppm; CD3OD, 3.31 ppm; (CD3)2SO, 2.50 ppm) as the internal standards. All final compounds used for testing in assays had purities that were determined to be >95% as evaluated by their NMR spectra and/or their LC–MS.
Synthetic Procedures.
Detailed procedures and characterization of compounds for all reactions in Schemes 1234 were as follows. Some of them were general procedures, which were described by representative compounds.
Methyl 2-((tert-butoxycarbonyl)amino)-3-(2-hydroxy-5-methylphenyl)acrylate (S1a, R = Me).
To a solution of (±)-Boc-α-phosphonoglycine trimethyl ester (4.22 g, 14.2 mmol, 1.2 eq) in DCM (20 mL) was added DBU (2.12 mL, 14.2 mmol, 1.2 eq) dropwise at −10 °C, and the resulting mixture was stirred for 20 min. Then 4-methylsalicylaldehyde (1.61 g, 11.8 mmol, 1.0 eq) in DCM (10 mL) was slowly added to the mixture over 10 min. The reaction mixture was stirred at room temperature overnight. The resulting mixture was concentrated under reduced pressure, diluted with EtOAc (100 mL), and washed successively with saturated aqueous NH4Cl (20 mL), saturated aqueous NaHCO3, (20 mL), and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (30–50% EtOAc in hexane, v/v) to give S1a (R = Me, 2.91 g, 80%). 1H NMR (400 MHz, CD3OD) δ 1.45 (s, 9H, Boc), 2.25 (s, 3H, PhCH3), 3.83 (s, 3H, COOCH3), 6.77 (d, J = 8.3 Hz, 1H, olefin), 7.02 (dd, J = 2.2, 8.4 Hz, 1H, Ph), 7.41 (d, J = 2.2 Hz, 1H, Ph), 7.53 (s, 1H, Ph). 13C NMR (100 MHz, CD3OD) δ 19.22, 27.18, 51.36, 79.97, 115.23, 120.46, 124.58, 128.22, 129.63, 131.02, 153.42, 154.76, 166.73. LC–MS: tR = 4.93 min; C11H14NO3+ [M + H − Boc]+, m/z calcd 208.10, found 208.08.
Methyl 2-((tert-butoxycarbonyl)amino)-3-(2-hydroxy-5-methylphenyl)propanoate (S1b, R = Me).
S1a (R = Me, 1.15 g, 3.74 mmol, 1.0 eq) was placed in a two-necked round bottom and charged with N2 gas. 10% palladium on carbon powder (Pd/C, 110 mg, cat.) was quickly added to the flask followed by addition of MeOH (20 mL). The flask was degassed and backfilled with H2 for three cycles. The reaction mixture was stirred at room temperature overnight with a balloon of H2 for replenishment. The balloon was removed, and the mixture was filtered under reduced pressure. Note that the operation should be rapid, and the Pd/C powder must be kept wet to avoid catching fire and was appropriately disposed in a water-filled, cap-closed container. The filtrate was concentrated under reduced pressure and purified by FCC (30% EtOAc in hexane, then 6% MeOH in DCM) to give S1b (R = Me, 1.1 g, 95%). 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 9H, Boc), 2.36 (s, 3H, PhCH3), 2.79–3.02 (m, 1H, βCH2), 3.07 (m, 1H, βCH2), 3.75 (s, 3H, COOCH3), 4.43 (d, J = 6.2 Hz, 1H, αCH), 5.54 (d, J = 7.4 Hz, 1H, NH), 6.65–6.73 (m, 2H, Ph), 6.73–6.79 (m, 1H, Ph). 13C NMR (100 MHz, CDCl3) δ 20.57, 28.24, 32.84, 51.98, 54.97, 79.50, 113.30, 116.22, 119.28, 127.84, 147.51, 153.95, 155.33, 172.70. LC–MS: tR = 4.98 min; C16H24NO5+ [M + H]+, m/z calcd 310.16, found 310.14.
Methyl 2-amino-3-(2-hydroxy-5-methylphenyl)propanoate TFA Salt (S1c, R = Me).
To a suspension of S1b (R = Me, 1.1 g, 3.56 mmol, 1.0 eq) in DCM (6 mL) was added TFA (3 mL) at 0 °C, and the resulting mixture was stirred for 30 min. The mixture was then concentrated with toluene three times to remove most of the residual TFA. The obtained crude product S1c (R = Me) was used without further purification.
Methyl (S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-3-phenylpropanamido)-3-(2-hydroxy-5-methylphenyl)propanoate (S1d, R = Me, R1 = BnO).
A mixed suspension of S1c (R = Me, ~1.15 g, 3.56 mmol, 1.0 eq), Cbz-Phe-OH (1.07 g, 3.56 mmol, 1.0 eq), and DIPEA (2.17 mL, 12.46 mmol, 3.5 eq) in DCM (20 mL) was cooled to 0 °C followed by dropwise addition of T3P (>50% in MeCN, 3.53 mL, 5.34 mmol, 1.5 eq). The resulting mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure, diluted with EtOAc (75 mL), and washed successively with 5% citric acid (15 mL), saturated aqueous NaHCO3 (15 mL), and brine (15 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (40% EtOAc in hexane) to give S1d (R = Me, R1 = BnO, 1.11 g, 63% for two steps). 1H NMR (400 MHz, CDCl3) δ 2.30 (s, 3H, PhCH3), 2.83–3.16 (m, 4H, 2 × βCH2), 3.69 (s, 3H, COOCH3), 4.50 (s, 1H, αCH), 4.75 (s, 1H, αCH), 4.98–5.14 (m, 2H, Cbz CH2), 5.70 (s, 1H, OH), 6.68 (t, J = 8.8 Hz, 1H, NH), 6.91–7.10 (m, 3H, Ph), 7.22 (dd, J = 20.7, 26.3 Hz, 7H, Ph), 7.33 (s, 3H, Ph), 8.08 (s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 20.66, 33.10, 38.37, 52.47, 53.19, 56.10, 67.24, 117.11, 124.49, 127.01, 127.88, 128.22, 128.32, 128.42, 128.53, 128.61, 129.25, 130.87, 135.98, 136.14, 153.57, 156.27, 171.72, 172.08. LC–MS: tR = 5.39 min; C28H31N2O6+ [M + H]+, m/z calcd 491.22, found 491.19.
Methyl (S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-3-phenylpropanamido)-3-(2-((tert-butyldimethylsilyl)oxy)-5-methylphenyl)-propanoate (S1e, R = Me, R1 = BnO, R2 = TBS).
A mixed solution of S1d (R = Me, R1 = BnO, 990 mg, 2.02 mmol, 1.0 eq), TBSCl (609 mg, 4.04 mmol, 2.0 eq), and imidazole (412 mg, 6.06 mmol, 3.0 eq) was stirred at room temperature overnight. The reaction mixture was quenched by addition of 0.5 M HCl (10 mL) and was stirred for another 15 min. The mixture was concentrated under reduced pressure and was partitioned between EtOAc (50 mL) and water (10 mL). The organic layer was washed with saturated aqueous NaHCO3 (10 mL) and brine (10 mL) and was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (20% EtOAc in hexane) to give S1e (R = Me, R1 = BnO, R2 = TBS, 897 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 0.16 (s, 6H, TBS Si(CH3)2), 0.93 (s, 9H, TBS C(CH3)3), 2.37 (s, 3H, PhCH3), 2.59–2.87 (m, 2H, βCH2), 2.89–3.05 (m, 2H, βCH2), 3.54 (d, J = 4.3 Hz, 3H, COOCH3), 4.36 (d, J = 22.6 Hz, 1H, αCH), 4.64 (q, J = 8.3 Hz, 1H, αCH), 4.95 (p, J = 12.1 Hz, 2H, Cbz CH2), 5.32–5.55 (m, 1H, NH), 6.56–6.65 (m, 1H, Ph), 6.83–7.02 (m, 3H, Ph), 7.05–7.25 (m, 9H, Ph). 13C NMR (100 MHz, CDCl3) δ −3.50, 18.27, 20.53, 25.81, 32.94, 38.57, 52.20, 52.57, 56.08, 66.96, 119.79, 125.91, 126.90, 127.89, 127.95, 128.02, 128.04, 128.07, 128.14, 128.44, 128.47, 128.57, 128.71, 129.28, 129.35, 130.82, 136.25, 136.47, 152.62, 155.90, 170.76, 171.62, 172.00. LC–MS: tR = 7.23 min; C34H45N2O6Si+ [M + H]+, m/z calcd 605.30, found 605.24.
Methyl (S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-3-phenylpropanamido)-3-(2-methoxyphenyl)propanoate (S1e, R = H, R1 = BnO, R2 = Me).
To a solution of S1d (R = H, R1 = BnO, 172 mg, 0.36 mmol, 1.0 eq) in DMF (2 mL) were successively added K2CO3 (100 mg, 0.72 mmol, 2.0 eq) and iodomethane (67 μL, 1.08 mmol, 3.0 eq) at 0 °C. The resulting mixture was stirred at room temperature for 20 h during which the system should be kept securely sealed to avoid evaporation of iodomethane. The mixture was diluted with EtOAc (50 mL) and washed with water extensively (5 × 10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (30% EtOAc in hexane) to give S1e (R = H, R1 = BnO, R2 = Me, 167 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 3.02 (qd, J = 5.7, 13.6, 16.9 Hz, 4H, 2 × βCH2), 3.61 (s, 3H, COOCH3), 3.93 (s, 3H, PhOCH3), 4.48 (s, 1H, αCH), 4.72 (s, 1H, αCH), 4.88–5.12 (m, 2H, Cbz CH2), 5.69 (d, J = 8.2 Hz, 1H, NH), 6.64–6.88 (m, 2H, Ph), 6.92–7.06 (m, 3H, Ph), 7.08–7.18 (m, 4H, Ph), 7.24 (d, J = 18.6 Hz, 5H, Ph), 7.77 (d, J = 43.8 Hz, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 32.65, 32.96, 38.40, 52.33, 52.40, 53.70, 56.04, 67.11, 115.84, 120.35, 122.73, 126.88, 126.90, 127.88, 128.16, 128.52, 128.72, 129.30, 131.30, 136.14, 154.71, 156.21, 156.23, 171.98, 172.34. LC–MS: tR = 5.71 min; C28H31N2O6+ [M + H]+, m/z calcd 491.22, found 491.33.
Benzyl ((S)-1-(((S)-1-(2-((tert-butyldimethylsilyl)oxy)-5-methylphenyl)-3-hydroxypropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (S1f, R = Me, R1 = BnO, R2 = TBS).
To a solution of S1e (R = Me, R1 = BnO, R2 = TBS, 897 mg, 1.48 mmol, 1.0 eq) in MeOH (10 mL) was added NaBH4 (1.2 g, 31.7 mmol, >20 eq) in multiple portions every 30 min. The reaction mixture was stirred at room temperature for another 2 h and then quenched by addition of saturated aqueous NH4Cl (10 mL). The mixture was concentrated under reduced pressure and diluted with EtOAc (50 mL). The organic layer was washed with saturated aqueous NaHCO3 (10 mL) and brine (10 mL) and was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (35% EtOAc in hexane) to give S1f (R = Me, R1 = BnO, R2 = TBS, 450 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 0.26 (s, 6H, TBS Si(CH3)2), 1.03 (s, 9H, TBS C(CH3)3), 2.40 (s, 3H, PhCH3), 2.74 (dt, J = 7.0, 13.1 Hz, 2H, βCH2), 2.97 (dd, J = 6.8, 40.5 Hz, 2H, βCH2), 3.25–3.60 (m, 2H, CH2OH), 4.02–4.15 (m, 1H, αCH), 4.39 (s, 1H, αCH), 4.97–5.15 (m, 2H, Cbz CH2), 5.69 (dd, J = 7.8, 66.8 Hz, 1H, NH), 6.54 (dd, J = 6.8, 121.3 Hz, 1H, Ph), 6.72–6.81 (m, 1H, Ph), 7.04–7.14 (m, 2H, Ph), 7.15–7.30 (m, 6H, Ph & NH), 7.34 (q, J = 6.2, 7.1 Hz, 4H, Ph). 13C NMR (100 MHz, CDCl3) δ −4.05, 18.27, 21.02, 25.87, 31.17, 39.00, 52.34, 56.69, 63.66, 67.03, 119.96, 126.28, 127.00, 127.56, 127.93, 128.03, 128.16, 128.47, 128.65, 129.29, 130.27, 131.00, 136.24, 136.53, 152.38, 155.94, 170.98, 171.35. LC–MS: tR = 6.90 min; C33H45N2O5Si+ [M + H]+, m/z calcd 577.31, found 577.37.
Benzyl ((S)-1-(((S)-1-(2-((tert-butyldimethylsilyl)oxy)-5-methylphenyl)-3-oxopropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-carbamate (S1g, R = Me, R1 = BnO, R2 = TBS).
To a solution of S1f (R = Me, R1 = BnO, R2 = TBS, 151 mg, .26 mmol, 1.0 eq) in DCM (8 mL) at 0 °C were added Dess-Martin periodinane (133 mg, 0.31 mmol, 1.2 eq) and NaHCO3 powder (55 mg, 0.65 mmol, 2.5 eq). The resulting mixture was stirred at 0 °C for 1 h and then quenched by addition of saturated aqueous Na2S2O3 (2 mL). The reaction mixture was concentrated under reduced pressure, diluted with EtOAc (50 mL), and washed with brine (3 × 10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (25% EtOAc in hexane) to give S1g (R = Me, R1 = BnO, R2 = TBS, 68 mg, 45%). 1H NMR (400 MHz, CDCl3) δ 0.24 (d, J = 4.6 Hz, 6H, TBS Si(CH3)2), 1.01 (s, 9H, TBS C(CH3)3), 2.24 (s, 3H, PhCH3), 2.77–3.20 (m, 4H, 2 × βCH2), 4.51 (q, J = 6.7 Hz, 1H, αCH), 5.02–5.15 (m, 2H, Cbz CH2), 5.35 (s, 1H, αCH), 6.52 (d, J = 5.5 Hz, 1H, NH), 6.71 (d, J = 8.2 Hz, 1H, Ph), 6.85 (d, J = 2.2 Hz, 1H, Ph), 6.94 (dd, J = 2.3, 8.2 Hz, 1H, Ph), 7.16 (d, J = 7.1 Hz, 2H, Ph), 7.21–7.28 (m, 3H, Ph), 7.35 (dt, J = 4.7, 6.9 Hz, 5H, Ph), 9.41 (s, 1H, CHO). 13C NMR (100 MHz, CDCl3) δ −4.14, 18.28, 20.43, 25.90, 29.92, 38.80, 56.11, 59.57, 67.01, 118.77, 125.68, 127.09, 128.01, 128.16, 128.50, 128.67, 128.91, 129.26, 130.94, 131.91, 136.13, 151.28, 170.96, 198.75. LC–MS: tR = 6.81 min; C33H43N2O5Si+ [M + H]+, m/z calcd 575.29, found 575.36.
Benzyl ((2S)-1-(((3S)-2-hydroxy-6-methylchroman-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (S1h, 7, R = Me, R1 = BnO, R2 = TBS).
To a solution of S1g (R = Me, R1 = BnO, R2 = TBS, 68 mg, 0.12 mmol, 1.0 eq) in THF (3 mL) was slowly added 1.0 M TBAF in THF (131 μL, 0.13 mmol, 1.1 eq) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL) and washed with saturated aqueous NH4Cl (10 mL) and brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (35% EtOAc in hexane) to give S1h (R = Me, R1 = BnO, R2 = TBS, i.e., compound 7, 20 mg, 36%). 1H NMR (400 MHz, CDCl3) δ 2.10–2.18 (m, 3H, PhCH3), 2.37–3.10 (m, 4H, 2 × βCH2), 4.10–4.45 (m, 2H, αCH & NH), 4.71–5.10 (m, 3H, Cbz CH2 & NH), 5.53 (dd, J = 7.2, 51.9 Hz, 1H, αCH), 5.97–6.41 (m, 1H, lactol C(O)OH), 6.59 (td, J = 10.3, 31.2, 32.4 Hz, 2H, Ph), 6.78 (dd, J = 8.2, 18.7 Hz, 1H, Ph), 6.98–7.28 (m, 10H, Ph). 13C NMR (100 MHz, CDCl3) δ 20.46, 25.96, 39.10, 46.02, 56.24, 67.20, 91.04, 91.93, 116.68, 118.34, 119.01, 119.23, 128.04, 128.12, 128.20, 128.52, 128.67, 128.77, 129.19, 129.32, 129.64, 130.50, 130.63, 136.02, 136.26, 148.52, 156.15, 171.23. LC–MS: tR = 5.32 min; C27H29N2O5+ [M + H]+, m/z calcd 461.21, found 461.27.
Benzyl (4-methylpiperazine-1-carbonyl)-L-phenylalaninate (S1i).
To a solution of 4-methylpiperazine-1-carbonyl chloride hydrochloride (1.365 g, 6.86 mmol, 1.1 eq) in THF (20 mL) at −10 °C was added Et3N (2.08 mL, 14.96 mmol, 2.4 eq) dropwise. The resulting mixture was stirred for 15 min, and then a solution of benzyl L-phenylalaninate hydrochloride (1.82 g, 6.23 mmol, 1.0 eq) was added into THF (20 mL) dropwise. The reaction mixture was stirred at room temperature overnight, quenched by addition of water (10 mL), and concentrated under reduced pressure. The residue was diluted with EtOAc (100 mL) and washed successively with saturated aqueous NH4Cl (20 mL), saturated aqueous NaHCO3 (20 mL), and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (5–10% MeOH in DCM) to give S1i (1.55 g, 60%). 1H NMR (400 MHz, CDCl3) δ 2.31 (s, 3H, NCH3), 2.37 (t, J = 5.1 Hz, 4H, 2 × piperazinyl CH2), 3.14 (d, J = 4.2 Hz, 2H, βCH2), 3.37 (q, J = 4.9 Hz, 4H, 2 × piperazinyl CH2), 4.87 (d, J = 5.1 Hz, 2H, Cbz CH2), 5.05–5.28 (m, 2H, αCH & NH), 7.02 (dd, J = 2.9, 6.5 Hz, 2H, Ph), 7.20–7.27 (m, 3H, Ph), 7.30–7.42 (m, 5H, Ph). 13C NMR (100 MHz, CDCl3) δ 38.31, 43.69, 46.09, 54.32, 54.59, 67.11, 126.93, 128.44, 128.53, 128.57, 129.38, 135.26, 136.11, 156.46, 172.45. LC–MS: tR = 3.42 min; C22H28N3O3+ [M + H]+, m/z calcd 382.21, found 382.2.
(4-methylpiperazine-1-carbonyl)-L-phenylalanine (S1j).
The compound was prepared from S1i in a similar procedure as described for S1b. The obtained crude product S1j was used without further purification.
(1aR,7aS)-1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene (S1k).
To a solution of 1,4-dihydronaphthalene (2.09 mg, 16.1 mmol, 1.0 eq) in chloroform (40 mL) was slowly added 70% mCPBA (4.74 g, 19.3 mmol, 1.2 eq) at 0 °C. The resulting mixture was stirred at room temperature overnight and quenched by addition of 2 M KOH (60 mL). The reaction mixture was concentrated under reduced pressure and diluted with EtOAc (100 mL). The organic layer was washed with brine (3 × 20 mL) and was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (10% EtOAc in hexane) to give S1k (2.03 g, 86%) 1H NMR (400 MHz, CDCl3) δ 3.05 (d, J = 17.7 Hz, 2H, CH2), 3.18 (d, J = 16.8 Hz, 2H, CH2), 3.29–3.36 (m, 2H, 2 × epoxide CH), 6.95 (dd, J = 3.5, 5.6 Hz, 2H, Ph), 7.06 (dd, J = 3.4, 5.7 Hz, 2H, Ph). 13C NMR (100 MHz, CDCl3) δ 29.84, 51.71, 126.57, 129.33, 131.74.
(2R,3R)-3-azido-1,2,3,4-tetrahydronaphthalen-2-ol and (2S,3S)-3-azido-1,2,3,4-tetrahydronaphthalen-2-ol (S1l).
To a solution of S1k (2.02 g, 13.8 mmol, 1.0 eq) in a mixture of MeOH (30 mL) and H2O (10 mL) were added NaN3 (1.8 g, 27.7 mmol, 2.0 eq) and NH4Cl (1.11 g, 20.8 mmol, 1.5 eq). The resulting mixture was heated to 60 °C and stirred overnight. The reaction mixture was cooled to room temperature, concentrated under reduced pressure, and diluted with EtOAc (100 mL). The organic layer was washed with brine (3 × 20 mL) and was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product S1l (2.22 g, 85%) was used without further purification. 1H NMR (400 MHz, CDCl3) δ 2.77–2.86 (m, 2H, CH2), 3.17 (ddd, J = 2.9, 5.8, 16.5 Hz, 2H, CH2), 3.66 (td, 1H, CH-N3), 3.87 (td, J = 5.8, 9.4 Hz, 1H, CH–OH), 7.04–7.09 (m, 2H, Ph), 7.09–7.16 (m, 2H, Ph). 13C NMR (100 MHz, CDCl3) δ 33.64, 36.57, 63.62, 70.47, 126.50, 126.68, 128.59, 128.99, 132.62, 133.41.
(2R,3R)-3-amino-1,2,3,4-tetrahydronaphthalen-2-ol and (2S,3S)-3-amino-1,2,3,4-tetrahydronaphthalen-2-ol (S1m).
The title compound was prepared from S1l in a similar procedure as described for S1b. The obtained crude product S1m was used without further purification.
N-((S)-1-(((2S,3S)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide and N-((S)-1-(((2R,3R)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide (3).
The title compound was prepared from S1j and S1m in a similar procedure as described for S1d. The obtained crude product was purified by FCC (10% MeOH in DCM) to give compound 3. 1H NMR (400 MHz, DMSO-d6) δ 2.17 (s, 3H), 2.20 (q, J = 3.3 Hz, 3H) 2.61 (ddd, J = 6.7, 16.9, 27.1 Hz, 2H), 2.83 (dd, J = 10.2, 13.6 Hz, 1H), 2.95 (ddd, J = 4.7, 8.9, 12.8 Hz, 2H), 3.04–3.33 (m, 5H), 3.79 (td, J = 4.8, 6.8 Hz, 1H), 3.93 (p, J = 7.0 Hz, 1H), 4.33 (ddd, J = 4.6, 8.3, 10.1 Hz, 1H), 6.48 (d, J = 8.3 Hz, 1H), 7.05–7.10 (m, 3H), 7.13–7.21 (m, 1H), 7.22–7.27 (m, 3H), 7.77 (d, J = 7.8 Hz, 1H), 8.15 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 32.84, 35.71, 38.13, 43.63, 45.94, 50.11, 54.62, 56.33, 67.15, 126.12, 126.24, 126.48, 128.35, 128.99, 129.38, 129.73, 134.39, 134.78, 139.14, 157.37, 163.56, 172.89.
4-Methyl-N-((S)-1-oxo-1-(((S)-1-oxo-4-phenylbutan-2-yl)amino)-3-phenylpropan-2-yl)piperazine-1-carboxamide (11).
The preparation of S1n, S1o, and 11 was similar to the procedures for S1d, S1f, and S1g, respectively. 1H NMR (400 MHz, CDCl3) δ 1.82 (dt, J = 7.6, 14.6 Hz, 1H, piperazinyl CH2), 2.09–2.17 (m, 1H, piperazinyl CH2), 2.28 (s, 3H, NCH3), 2.34 (dt, J = 3.8, 7.1 Hz, 4H, piperazinyl CH2 & homoPhe βCH2), 2.55 (dt, J = 7.9, 27.5 Hz, 2H, homoPhe γCH2), 3.11 (p, J = 5.9, 6.5 Hz, 2H, Phe βCH2), 3.29–3.39 (m, 4H, 2 × piperazinyl CH2), 4.33 (td, J = 5.3, 7.5 Hz, 1H, homoPhe αCH), 4.64 (q, J = 7.3 Hz, 1H, Phe αCH), 5.02 (d, J = 7.3 Hz, 1H, NH), 6.75 (d, J = 7.1 Hz, 1H, NH), 7.08–7.13 (m, 2H, Ph), 7.18–7.25 (m, 5H, Ph), 7.27–7.31 (m, 3H, Ph), 9.43 (s, 1H, CHO). C25H33N4O3+ [M + H]+, m/z calcd 437.25, found 481.21.
(3S)-3-((S)-2-(4-methylpiperazine-1-carboxamido)-3-phenylpropanamido)chroman-2-yl acetate (S2a, 13, R1 = Me).
To a solution of compound 12 (50 mg, 0.114 mmol, 1.0 eq) in DCM (3 mL) were added acetic anhydride (32 μL, 0.342 mmol, 3.0 eq), Et3N (48 μL, 0.342 mmol, 3.0 eq), and DMAP (2.8 mg, 0.023 mmol, 0.2 eq). The resulting mixture was stirred at room temperature overnight and concentrated under reduced pressure. The residue was diluted with EtOAc (50 mL) and washed successively with saturated aqueous NH4Cl (10 mL), saturated aqueous NaHCO3 (10 mL), and brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (10% MeOH in DCM) to give S2a (R1 = Me, i.e., compound 13, 31 mg, 57%). 1H NMR (400 MHz, CDCl3) δ 2.01 (d, J = 23.4 Hz, 3H, CH3CO), 2.29 (d, J = 3.5 Hz, 3H, NCH3), 2.30–2.38 (m, 4H, 2 × piperazinyl CH2), 2.69–2.97 (m, 2H, lactol βCH2), 2.98–3.15 (m, 2H, Phe βCH2), 3.25–3.41 (m, 4H, 2 × piperazinyl CH2), 4.49 (dt, J = 6.9, 13.8 Hz, 1H, αCH), 5.18 (dd, J = 7.4, 40.0 Hz, 1H, Phe αCH), 6.12 (dd, J = 2.5, 17.4 Hz, 1H, lactol CH), 6.31–6.54 (m, 1H, NH), 6.82–6.91 (m, 1H, Ph), 6.91–7.07 (m, 2H, Ph), 7.07–7.19 (m, 2H, Ph), 7.19 (s, 3H, Ph & NH), 7.26–7.34 (m, 2H, Ph). 13C NMR (100 MHz, CDCl3) δ 20.99, 26.61, 29.67, 38.69, 43.71, 45.99, 54.48, 56.23, 88.88, 89.33, 117.07, 119.40, 121.89, 127.09, 128.06, 128.70, 128.74, 129.15, 129.20, 136.94, 150.22, 157.02, 169.38, 171.96. LC–MS: tR = 3.29 min; C26H33N4O5+ [M + H]+, m/z calcd 481.24, found 481.3.
N-((2S)-1-(((3S)-2-ethoxychroman-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide (S2b, 16, R2 = Et).
To a solution of compound 12 (40 mg, 0.091 mmol, 1.0 eq) in EtOH (2 mL) was added BF3OEt2 (300 μL, 2.43 mmol, >20 eq) dropwise at 0 °C. The resulting mixture was stirred at room temperature overnight and quenched by addition of saturated aqueous NH4Cl (2 mL). The reaction mixture was concentrated under reduced pressure and then partitioned between DCM (20 mL) and water (20 mL). The water layer was further washed with DCM (2 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (8% MeOH in DCM) to give S2b (R2 = Et, i.e., compound 16, 18.3 mg, 43%). 1H NMR (400 MHz, CDCl3) δ 1.00 (t, 3H, OCH2CH3), 2.16–2.36 (m, 7H, NCH3 &2 × piperazinyl CH2), 2.69 (dd, J = 9.2, 14.2 Hz, 1H, βCH2), 2.83–2.95 (m, 1H, βCH2), 3.10 (dt, J = 7.2, 13.4 Hz, 1H, Phe βCH2), 3.21 (dd, J = 4.0, 6.1 Hz, 1H, Phe βCH2), 3.31 (dt, J = 6.0, 11.1 Hz, 3H, OCH2CH3 & piperazinyl CH2), 3.58–3.76 (m, 1H, piperazinyl CH2), 4.13–4.27 (m, 1H, piperazinyl CH2), 4.41 (q, J = 7.3 Hz, 1H, piperazinyl CH2), 4.68 (dd, J = 2.3, 49.7 Hz, 1H, αCH), 4.90–5.15 (m, 1H, Phe αCH), 5.78 (dd, J = 8.9, 24.8 Hz, 1H, lactol CH), 6.73 (d, J = 8.1 Hz, 1H, Ph), 6.80 (q, J = 6.8, 7.4 Hz, 1H, Ph), 6.90 (d, J = 7.2 Hz, 1H, Ph), 7.03 (t, J = 7.2 Hz, 1H, Ph), 7.07–7.30 (m, 5H, Ph). 13C NMR (100 MHz, CDCl3) δ 14.94, 14.98, 27.08, 39.59, 43.72, 45.10, 46.06, 54.58, 56.30, 64.01, 96.02, 96.34, 116.79, 120.48, 121.16, 126.99, 127.67, 128.65, 128.75, 129.20, 129.33, 136.84, 137.17, 150.58, 150.70, 156.56, 156.68, 171.26, 171.66. LC–MS: tR = 3.53 min; C26H35N4O4+ [M + H]+, m/z calcd 467.27, found 467.3.
(S)-N-(2-benzoylphenyl)-1-benzylpyrrolidine-2-carboxamide (S3a).
To a mixed solution of benzyl-L-proline (1.03 g, 5.0 mmol, 1.0 eq) and 1-methylimidazole (877 μL, 11.0 mmol, 2.2 eq) in DCM (10 mL) was added MsCl (387 μL, 5.0 mmol, 1.0 eq) dropwise at 0 °C. The resulting mixture was stirred at room temperature for 10 min, and then was added a solution of 2-aminobenzophenone (888 mg, 4.5 mmol, 0.9 eq) in DCM (10 mL). The reaction mixture was heated to 45 °C and stirred overnight. The reaction mixture was quenched by addition of NH4Cl (15 mL) and concentrated under reduced pressure. The residue was diluted with EtOAc (75 mL) and washed with saturated aqueous NaHCO3 (15 mL) and brine (15 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (15–25% EtOAc in hexane) to give S3a (1.344 g, 70%). 1H NMR (400 MHz, CDCl3) δ 1.72–1.85 (m, 2H, Pro γCH2), 1.91–2.01 (m, 1H, Pro βCH2), 2.17–2.31 (m, 1H, Pro βCH2), 2.40 (td, J = 6.8, 9.5 Hz, 1H, Pro δCH2), 3.20 (ddd, J = 2.4, 6.4, 9.1 Hz, 1H, Pro δCH2), 3.31 (dd, J = 4.8, 10.1 Hz, 1H, Pro αCH), 3.58 (d, J = 12.9 Hz, 1H, Bn CH2), 3.91 (d, J = 12.9 Hz, 1H, Bn CH2), 7.07 (td, J = 1.1, 7.6 Hz, 1H, Ph), 7.10–7.17 (m, 3H, Ph), 7.33–7.40 (m, 2H, Ph), 7.49 (qd, J = 6.5, 7.9 Hz, 4H, Ph), 7.56–7.61 (m, 1H, Ph), 7.73–7.83 (m, 2H, Ph), 8.57 (dd, J = 1.0, 8.4 Hz, 1H, Ph), 11.50 (s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 24.18, 31.03, 53.89, 59.87, 68.32, 121.53, 122.19, 125.36, 127.06, 128.16, 128.31, 129.13, 130.10, 132.45, 132.54, 133.35, 138.15, 138.59, 139.22, 174.60, 197.99. LC–MS: tR = 3.50 min; C25H25N2O2+ [M + H]+, m/z calcd 385.19, found 384.85.
(S,E)-2-(((2-(1-benzylpyrrolidine-2-carboxamido)phenyl)(phenyl)methylene)amino)acetate-1-13C in Complex with Ni(II) (S3b).
To a mixed suspension of S3a (860 mg, 2.24 mmol, 1.0 eq), [1-13C] glycine (425 mg, 5.59 mmol, 2.5 eq), and Ni(NO3)2·6H2O (1.30 g, 4.47 mmol, 2.0 eq) in MeOH (7 mL) at 45 °C was added a solution of ground KOH (752 mg, 13.4 mmol, 6.0 eq) in MeOH (3 mL). The resulting mixture was stirred at 60 °C for 1 h (note that prolonged heating might lead to racemization). The reaction mixture was neutralized by addition of acetic acid (800 μL) and diluted with water to a volume of 50 mL. The reaction mixture was concentrated under reduced pressure and extracted with DCM (75 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (20–30% acetone in DCM) to give S3b (715 mg, 64%). 1H NMR (400 MHz, CDCl3) δ 2.07 (dddd, J = 2.4, 5.6, 8.5, 15.1 Hz, 1H, Pro γCH2), 2.12–2.21 (m, 1H, Pro γCH2), 2.42 (dddd, J = 8.3, 9.5, 10.7, 13.4 Hz, 1H, Pro βCH2), 2.57 (dddd, J = 2.6, 6.3, 9.2, 14.4 Hz, 1H, Pro βCH2), 3.25–3.40 (m, 1H, Pro δCH2), 3.46 (dd, J = 5.4, 10.7 Hz, 1H, Pro δCH2), 3.61–3.74 (m, 3H, N-Bn CH2 & Pro αCH), 3.77 (dd, J = 5.5, 20.1 Hz, 1H, Gly CH2), 4.48 (d, J = 12.7 Hz, 1H, Gly CH2), 6.69 (ddd, J = 1.2, 6.9, 8.1 Hz, 1H, Ph), 6.80 (dd, J = 1.7, 8.3 Hz, 1H, Ph), 6.92–7.04 (m, 1H, Ph), 7.05–7.13 (m, 1H, Ph), 7.20 (ddd, J = 1.7, 6.9, 8.7 Hz, 1H, Ph), 7.28–7.33 (m, 1H, Ph), 7.42 (t, J = 7.6 Hz, 2H, Ph), 7.52 (ddd, J = 3.4, 7.7, 18.9 Hz, 3H, Ph), 8.01–8.12 (m, 2H, Ph), 8.31 (dd, J = 1.1, 8.6 Hz, 1H, Ph). 13C NMR (100 MHz, CDCl3) δ 23.69, 30.76, 57.56, 61.57, 63.16, 69.94, 124.26, 125.16, 126.27, 128.91, 129.11, 129.34, 129.59, 131.73, 132.22, 132.77, 133.17, 133.37, 134.67, 142.62, 143.81, 177.21, 179.27. LC–MS: tR = 3.71 min; C2613CH26N3NiO3+ [M + H]+, m/z calcd 499.14, found 498.83.
(S)-3-(2-(benzyloxy)phenyl)-2-(((E)-(2-((S)-1-benzylpyrrolidine-2-carboxamido)phenyl)(phenyl)methylene)amino)propanoate-1-13C in Complex with Ni(II) (S3c).
A mixed slurry of S3b (715 mg, 1.43 mmol, 1.0 eq), (2-(benzyloxy)phenyl)methanol (613 mg, 2.86 mmol, 2.0 eq), and CMBP (750 μL, 2.86 mmol, 2.0 eq) in toluene (5 mL) was heated to 120 °C and stirred overnight. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The obtained crude product was purified by FCC (10% acetone in DCM) to give S3c (417 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 1.48–1.59 (m, 1H, Pro γCH2), 1.99 (ddd, J = 6.5, 8.4, 11.1 Hz, 1H, Pro δCH2), 2.04–2.14 (m, 1H, Pro γCH2), 2.31 (tdd, J = 2.9, 6.7, 9.7 Hz, 2H, Pro βCH2), 2.76 (ddd, J = 1.6, 4.4, 13.6 Hz, 1H, o-Tyr βCH2), 2.90–3.05 (m, 1H, Pro δCH2), 3.22 (ddd, J = 5.0, 7.2, 13.6 Hz, 1H, o-Tyr βCH2), 3.33 (dd, J = 8.0, 9.3 Hz, 1H, Pro αH), 3.44 (d, J = 12.6 Hz, 1H, N-Bn CH2), 4.18 (q, J = 4.4 Hz, 1H, o-Tyr αCH), 4.23 (d, J = 12.6 Hz, 1H, N-Bn CH2), 4.55 (d, J = 10.8 Hz, 1H, OBn CH2), 4.90 (d, J = 10.8 Hz, 1H, OBn CH2), 6.14 (dt, J = 1.5, 7.7 Hz, 1H, Ph), 6.41 (dd, J = 1.7, 8.2 Hz, 1H, Ph), 6.63 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H, Ph), 6.83–6.94 (m, 2H, Ph), 6.96–7.22 (m, 9H, Ph), 7.28 (d, J = 7.6 Hz, 2H, Ph), 7.33–7.46 (m, 4H, Ph), 7.95–8.08 (m, 2H, Ph), 8.35 (dd, J = 1.1, 8.7 Hz, 1H, Ph). 13C NMR (100 MHz, CDCl3) δ 22.35, 25.60, 28.10, 59.19, 63.38, 68.12, 69.68, 71.86, 112.85, 122.38, 123.33, 123.36, 124.10, 127.39, 127.74, 127.80, 128.06, 128.35, 128.51, 128.73, 128.88, 129.20, 129.27, 129.61, 130.06, 130.30, 130.32, 134.10, 134.82, 137.07, 145.11, 155.82, 158.83, 170.83, 175.71. LC–MS: tR = 4.70 min; C4013CH38N3NiO4+ [M + H]+, m/z calcd 695.22, found 694.96.
(S)-2-amino-3-(2-benzyloxy)phenyl)propanoic-1-13C Acid (S3d).
A mixed solution of S3c (522 mg, 0.75 mmol, 1.0 eq) and 8-quinolinol (272 mg, 1.88 mmol, 2.5 eq) in MeCN (10 mL) and H2O (1 mL) was heated to 40 °C and stirred overnight. The reaction mixture was filtered, and the filtrate was concentrated. The residue was diluted with DCM (30 mL) and washed with water (3 × 30 mL). The organic layer containing the retrieved BPB was concentrated and saved for future use. The combined water layers were concentrated to a small volume that was subsequently purified by Prep-HPLC to give S3d (87 mg, 43%). 1H NMR (400 MHz, CDCl3) δ 2.96 (ddd, J = 2.5, 9.4, 14.3 Hz, 1H, βCH2), 3.49 (ddd, J = 2.9, 4.5, 14.3 Hz, 1H, βCH2), 3.93 (dt, J = 4.7, 9.4 Hz, 1H, αCH), 5.20 (s, 2H, OBn CH2), 6.92 (td, J = 1.1, 7.4 Hz, 1H, Ph), 7.05 (d, J = 8.1 Hz, 1H, Ph), 7.24 (dd, J = 6.7, 8.2 Hz, 2H, Ph), 7.28–7.34 (m, 1H, Ph), 7.38 (dd, J = 6.6, 8.3 Hz, 2H, Ph), 7.45–7.53 (m, 2H, Ph). 13C NMR (100 MHz, CDCl3) δ 33.52, 55.91, 71.50, 114.97, 120.74, 124.67, 127.65, 127.97, 128.07, 128.32, 130.64, 139.00, 157.44, 173.03. LC–MS: tR = 2.70 min; C1513CH18NO3+ [M + H]+, m/z calcd 273.13, found 272.75.
(S)-3-(2-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-propanoic-1-13C Acid (S3e).
To a solution of S3d (87 mg, 0.32 mmol, 1.0 eq) in dioxane (5 mL) and H2O (5 mL) were added Et3N (134 μL, 0.96 mmol, 3.0 eq) and Boc anhydride (84 mg, 0.38 mmol, 1.2 eq) at 0 °C. The resulting mixture was stirred at room temperature for 2.5 h and then concentrated under reduced pressure. The residue was acidified by addition of 0.1 M HCl to pH 2–3 and then diluted with EtOAc (50 mL). The organic layer was washed with brine (3 × 10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product S3e (113 mg, 94%) was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.28 (s, 9H, Boc), 2.74–3.06 (m, 1H, βCH2), 3.15 (d, J = 14.5 Hz, 1H, βCH2), 4.33–4.55 (m, 1H, αCH), 5.00 (d, J = 6.7 Hz, 2H, OBn CH2), 6.82 (d, J = 8.0 Hz, 2H, Ph), 7.04–7.15 (m, 2H, Ph), 7.21 (t, J = 7.3 Hz, 1H, Ph), 7.28 (d, J = 14.8 Hz, 2H, Ph), 7.36 (d, J = 7.4 Hz, 2H, Ph), 9.86 (s, 1H, COOH). 13C NMR (100 MHz, CDCl3) δ 28.30, 29.71, 32.44, 54.81, 70.26, 111.95, 121.12, 127.22, 127.93, 128.46, 128.64, 131.40, 176.93. LC–MS: tR = 4.28 min; C2013CH25NNaO5+ [M + Na]+, m/z calcd 395.17, found 394.83; C1513CH18NO3+ [M + H − Boc]+, m/z calcd 273.13, found 272.82
Tert-Butyl (S)-(3-(2-(benzyloxy)phenyl)-1-(methoxy(methyl)-amino)-1-oxopropan-2-yl-1-13C)carbamate (S3f).
The title compound was prepared from S3e (113 mg, 0.30 mmol, 1.0 eq) and N,O-dimethylhydroxylamine hydrochloride (44.4 mg, 0.45 mmol, 1.5 eq) in a similar procedure as described for S1d. The obtained crude product was purified by FCC (25% EtOAc in hexane) to give S3f (136 mg, 100%). 1H NMR (400 MHz, CDCl3) δ 1.25 (s, 9H, Boc), 2.79–2.96 (m, 2H, o-Tyr βCH2), 2.99 (d, J = 5.6 Hz, 3H, OCH3), 3.44 (s, 3H, NCH3), 4.90 (s, 1H, αCH), 4.99 (d, J = 4.1 Hz, 2H, OBn CH2), 5.21 (d J = 8.9 Hz, 1H, NH), 6.79 (t, J = 7.7 Hz, 2H, Ph), 7.00–7.14 (m, 2H, Ph), 7.18–7.32 (m, 3H, Ph), 7.38 (d, J = 7.5 Hz, 2H, Ph). 13C NMR (100 MHz, CDCl3) δ 28.30, 29.66, 32.03, 45.85, 61.26, 70.23, 79.09, 111.68, 120.70, 125.50, 127.52, 127.87, 128.15, 128.54, 131.42, 137.08, 155.20, 157.10, 172.86. LC–MS: tR = 4.60 min; C2213CH30N2NaO5+ [M + Na]+, m/z calcd 438.21, found 437.81; C1713CH22N2O5+ [M + H − Boc]+, m/z calcd 316.17, found 315.73.
N-((S)-1-(((S)-3-(2-(benzyloxy)phenyl)-1-(methoxy(methyl)-amino)-1-oxopropan-2-yl-1-13C)amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide (S3g).
The Boc group of S3f (136 mg, 0.33 mmol, 1.0 eq) was first deprotected in a similar procedure as described for S1c. Subsequently, the intermediate product was coupled to NMePip-Phe-OH (114 mg, 0.39 mmol, 1.2 eq) in a similar procedure as described for S1d. The obtained crude product was purified by FCC (8% MeOH in DCM) to give S3g (107 mg, 55%).1H MR (400 MHz, CDCl3) δ 2.27 (s, 3H, piperazinyl NCH3), 2.30 (dt, J = 3.7, 6.5 Hz, 4H, 2 × piperazinyl CH2), 2.96 (dd, J = 2.9, 6.3 Hz, 3H, OCH3), 3.07 (s, 4H, 2 x βCH2), 3.28 (ddd, J = 7.7, 13.5, 17.9 Hz, 4H, 2 × piperazinyl CH2), 3.48 (s, 3H, NCH3), 4.49 (q, J = 6.5 Hz, 1H, αCH), 4.99 (d, J = 7.0 Hz, 1H, αCH), 5.05 (s, 2H, OBn CH2), 5.19 (s, 1H, NH), 6.62 (s, 1H, NH), 6.79 (t, J = 7.3 Hz, 1H, Ph), 6.90 (dd, J = 7.3, 19.1 Hz, 2H, Ph), 7.12–7.25 (m, 6H, Ph), 7.30 (t, J = 7.3 Hz, 1H, Ph), 7.37 (t, J = 7.4 Hz, 2H, Ph), 7.46 (d, J = 7.2 Hz, 2H, Ph). 13C NMR (100 MHz, CDCl3) δ 32.03, 32.92, 38.74, 43.62, 46.03, 49.23, 54.53, 55.10, 61.22, 70.22, 111.82, 120.68, 125.09, 126.70, 127.57, 127.97, 128.30, 128.36, 128.59, 129.68, 131.43, 136.95, 137.02, 156.53, 156.99, 171.19, 171.76. LC–MS: tR = 3.72 min; C3213CH42N5O5+ [M + H]+, m/z calcd 589.32, found 589.02.
N-((S)-1-(((S)-1-(2-(benzyloxy)phenyl)-3-oxopropan-2-yl-3-13C)-amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide (S3h).
To a solution of S3g (107 mg, 0.182 mmol, 1.0 eq) in THF (8 mL) at −10 °C was added 2.0 M LAH in THF (218 μL, 0.218 mmol, 1.2 eq) dropwise. The resulting mixture was stirred at 0 °C for 1 h and then quenched using tiny pieces of ice. The reaction mixture was concentrated under reduced pressure and diluted with EtOAc (50 mL). The organic layer was washed successively with a saturated solution of Rochelle salt (15 mL), NaHCO3 (15 mL), and brine (15 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was purified by FCC (10% MeOH in DCM) to give S3h (81 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H, piperazinyl NCH3), 2.31 (t, J = 4.2 Hz, 4H, 2 × piperazinyl CH2), 2.95–3.13 (m, 4H, 2 × βCH2), 3.29 (dt, J = 5.2, 9.7 Hz, 4H, 2 × piperazinyl CH2), 4.52 (m, 2H, αCH & NH), 4.95 (d, J = 7.3 Hz, 1H, αCH), 5.04 (dd, J = 3.6, 10.5 Hz, 2H, OBn CH2), 6.82 (s, 3H, Ph & NH), 7.10–7.24 (m, 6H, Ph), 7.29–7.50 (m, 6H, Ph), 9.36 (s, 1H, CHO). 13C NMR (100 MHz, CDCl3) δ 29.66, 38.59, 43.67, 46.04, 54.52, 55.31, 59.49, 70.25, 97.62, 111.96, 121.04, 126.93, 127.55, 128.19, 128.55, 128.60, 128.73, 129.33, 129.43, 131.61, 136.52, 136.76, 156.39, 156.60, 171.89, 198.37. LC–MS: tR = 3.76 min; C3013CH36N4O4+ [M + H]+, m/z calcd 530.28, found 530.03.
N-((2S)-1-(((3S)-2-hydroxychroman-3-yl-2-13C)amino)-1-oxo-3-phenylpropan-2-yl)-4-methylpiperazine-1-carboxamide (13C-labeled 12).
The title compound was prepared from S3h (81 mg, 0.152 mmol, 1.0 eq) in a similar procedure as described for S1b. The obtained crude product was purified by Prep-HPLC to give 13C-labeled compound 12 (45 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 1.28 (s, 1H, lactol OH), 2.45 (s, 3H, NCH3), 2.65 (p, J = 6.5 Hz, 4H, 2 × piperazinyl CH2), 3.03 (dt, J = 7.6, 21.2 Hz, 2H, o-Tyr βCH2), 3.40–3.60 (m, 4H, 2 x piperazinyl CH2), 4.21–4.40 (m, 1H, Phe βCH2), 4.64 (dq, J = 7.7, 46.1 Hz, 1H, Phe βCH2), 5.38 (dd, J = 30.9, 170.9 Hz, 1H, o-Tyr αCH), 6.16 (d, J = 8.0 Hz, 1H, Phe αCH), 6.50 (dd, J = 8.0, 141.5 Hz, 1H, lactol CH), 6.86–7.08 (m, 3H, Ph), 7.13 (dd, J = 3.0, 6.9 Hz, 1H, Ph), 7.20 (dt, J = 2.3, 6.5 Hz, 2H, Ph), 7.25 (d, J = 6.7 Hz, 2H, Ph), 7.71 (s, 2H, Ph & NH), 8.27 (s, 1H, NH). 13C NMR (100 MHz, CDCl3, scanned for only a few times) δ 91.69, 92.25. LC–MS: tR = 2.77 min; C2313CH31N4O4+ [M + H]+, m/z calcd 440.24, found 439.94.
In Scheme 4, the preparation of S4a and S4b was similar to the procedures for S1a and S1b, respectively; the preparation of S4d, S4e, and S4f was similar to the procedures for S3f, S3g, and S3h, respectively. Therefore, only the preparation of S4c was described in detail as below.
2-((tert-butoxycarbonyl)amino)-3-(2-oxo-1,2-dihydropyridin-3-yl)propanoic Acid (S4c).
A mixed solution of S4b (1.18 g, 4.0 mmol, 1.0 eq) and LiOH (192 mg, 8.0 mmol, 2.0 eq) in MeOH (10 mL) and H2O (5 mL) was stirred at room temperature for 2 h. The reaction mixture was acidified by addition of 0.1 M HCl to pH 2–3 and then concentrated under reduced pressure. The residue was extracted with DCM (3 × 50 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product S4c was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 9H, Boc), 2.86 (dd, J = 8.9, 14.0 Hz, 1H, βCH2), 3.11 (dd, J = 4.3, 13.9 Hz, 1H, βCH2), 4.42 (dd, J = 4.3, 8.8 Hz, 1H, αCH), 6.32 (t, J = 6.7 Hz, 1H, pyridine), 7.30 (dd, J = 2.0, 6.6 Hz, 1H, pyridine), 7.39–7.54 (m, 1H, pyridine).
Enzyme Assays.
The preparation of cruzain and 3CLpro was described in previous publications.8,34 The evaluation of inhibitors was modified from our lab protocol and described briefly as follows.35 All enzyme assays were conducted in 96-well, clear bottom, black plates at 25 °C, and fluorescence emanating from product formation was measured using either a SpectraMax M2 (Molecular Devices) or a Synergy HTX (Biotek) microplate reader, with excitation at and emission at . For evaluation of inhibitors that exhibited time-dependent time courses (“burst kinetics),” variable concentrations of inhibitor and a fixed concentration of substrate [typically, for cruzain: 10 μM Cbz-Phe-Arg-AMC , for 3CLpro: 40 μM ACC-SAVLQSGFRK(DNP)-NH2 ] were first added into assay buffer [for cruzain: 50 mM MES (pH 7.5), 50 mM TAPSO, 1 mM CHAPS, 1 mM Na2EDTA, 5 mM DTT, and 10% DMSO (v/v); for 3CLpro: 20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 0.1 mM EDTA-Na2, 2 mM DTT, and 10% DMSO (v/v)]. Reactions were started with addition of enzyme (typically, to a final concentration of 0.2 nM for cruzain or 40 nM for 3CLpro), and the time courses were recorded for 0.5–1 h. In a rapid dilution assay, 100X concentrations of the enzyme and inhibitor were preincubated for 1–2 h and then diluted by 100-fold into assay buffer containing standard concentrations of the substrates followed by data collection.
In Vitro Cell Assays.
BSF T. b. brucei strain SM427 was grown in HM1–9 medium that included fetal bovine serum (10%) and antibiotics (100 U/mL penicillin–streptomycin, Gibco). Inhibitors dissolved in 100% (v/v) DMSO stock solutions were added at final concentrations of 0.5–20 μM (final concentrations of DMSO were <1% (v/v)). T. b. brucei were seeded at ∼1 × 106 cells at 37 °C and diluted daily to maintain a log phase of growth for up to 96 h. Treated cells were typically grown for 4 days. After each dilution, the cell cultures were supplemented with inhibitor or DMSO (control) to keep concentrations constant. Cell density was determined and graphed as previously reported.36 Trypanosomes were quantified using a Z2 Coulter Counter, and values of EC50 were determined after 3–4 days of treatment by the interpolation method of Huber and Koella.37
T. cruzi infection was evaluated in C2C12 murine cardiomyoblast cells.
Three hundred and eighty four-microwell plates containing C2C12 murine cardiomyoblast cells were infected with 103 and 104 cells of T. cruzi (Ca-I/72), to which was added 3-fold serial dilutions of inhibitor with maximal concentrations of 40 μM followed by incubation at 37 °C for 2 days. The wells were fixed with 2% PFA in PBS and stained by 5 μg/mL of DAPI. After >30 min of incubation in a dark environment, the plates were read on an ImageXpress MicroXL microscope (Molecular Devices). The images were analyzed to quantify parasites as well host cells.
Evaluation of the anti-CoV-2 activity of compound 18 using human A549 cells transduced with the ACE2 viral receptor (A549/ACE2) was as described in our recent publication.8
Evaluation of SMAI Prodrugs.
Esterase from porcine liver was obtained as an ammonium sulfate suspension (Sigma-Aldrich, E2884, 10 U/μL). Compounds 13–15 in 100% (v/v) DMSO solutions were diluted by 10-fold into cruzain assay buffer to make 1 mL samples, each containing 2 mM compound. For the experimental sample, 4 μL of esterase suspension was added to the reaction mixtures (40 U/mL), which was immediately placed on a low-speed orbital shaker in a 37 °C incubator. In the meantime, a negative control (lacking esterase) and a blank control (buffer only) were included. Aliquots (140 μL) of the reaction mixtures were removed at 0, 15, 30, 60, 120, 180, and 300 min, and reaction mixtures were quenched by the addition of MeCN (260 μL). The resulting solution was vortexed followed by centrifugation to precipitate the protein, and the resulting compounds in the supernatant were analyzed by HPLC–MS with detection at 270 nm.
Commercially available CYPs 3A4, 2D6, and 2C9 (CypExpress; Sigma-Aldrich, MTOXCE3A4, MTOXCE3D6, and MTOXCE2C9) containing glucose-6-phosphate dehydrogenase (G6PDH) and Mg2+ were used to evaluate the stability of acetal prodrugs to these oxidases. The following components were combined in 800 μL reaction mixtures: 0.5 mM 16 or 17, 5 mM sodium glucose 6-phosphate, 2 mM NADP+, cruzain assay buffer (pH 7.5), and 2.5% DMSO (v/v). For the experimental samples, 64 mg of CypExpress P450 as a lyophilized powder was added to the reaction vial (80 mg/mL), which was gently shaken to dissolve the solid, and the mixtures were stirred at 30 °C in an incubator. Sample vials were capped loosely with aluminum foil to allow aeration as well as to reduce evaporation. Additionally, a negative control lacking CypExpress P450 and a blank control containing only buffer were included. Aliquots (65 μL) were removed from mixtures at 0, 30, 60, 120, 180, and 240 min, and reaction mixtures were quenched with the addition of MeCN (65 μL). The resulting solution was vortexed followed by centrifugation to precipitate the protein, and the resulting compounds in the supernatant were analyzed by HPLC–MS with detection at 270 nm.
2D NMR Study.
Wilmad precision NMR tubes (541-PP-8, thin wall, 800 MHz, 5 mm × 8 inch) were used for this experiment. The total volume of sample in each tube was 660 μL, and the final concentrations of the components in the NMR buffer were as follows: 18 mM phosphate (K2HPO4/KH2PO4), 45 mM NaCl, 4.5 mM DTT, pH 7.5, and 10% DMSO-d6 (v/v). A 10 mM stock of 13C-labeled 12 in DMSO-d6 was diluted into the NMR buffer to specified concentrations. A control sample contained 0.6 mM of 13C-labeled 12, and an experimental sample contained 0.4 mM of 13C-labeled 12 and 0.44 mM of cruzain freshly purified by size exclusion chromatography, directly after thawing from a −80 °C stock to minimize autoproteolysis. Both samples were analyzed on a Bruker AVANCE III HD 800 MHz (18.8 Tesla) equipped with a 5 mm triple resonance cryoprobe. 1H-13C HSQC NMR spectra were acquired at 25 °C with 72 scans for the enzyme-free control sample and 128 scans for the experimental sample containing cruzain. Spectral widths were obtained at 9615 and 28,179 Hz in the 13C and 1H dimensions, respectively. Spectra were processed using NMRpipe,38 and cross-peak intensities were analyzed using Sparky39 and MestreNova.40
Detection of Aldehyde Content Using Phenylhydrazine.
In a 96-well clear microplate, compounds 11 and 12 in DMSO solutions, or pure DMSO as a control, were diluted into the above NMR buffer without addition of DTT to make 250 μL samples, each containing 0.2 mM of compounds and 10% DMSO (v/v). Upon addition of 1 M phenylhydrazine (0.25 μL), the formation of phenylhydrazone in these wells was continuously monitored for 2 h by measuring UV absorbance at a maximal wavelength of 282 nm.
X-ray Crystallography.
The procedure of crystallization for the 3CLpro-18 complex is based on the previous publication.41 The crystals were obtained at 25 °C from 0.2 M ammonium phosphate dibasic, 17% w/v PEG3350, pH 8.0, with a protein concentration of 14 mg/mL. Soaking was performed to produce 3CLpro-18 complex crystals. The crystals were washed three times with reservoir solution plus 0.5 mM inhibitor and 2% DMSO (Inhibitor was dissolved to 25 mM in 100% DMSO). The mixture was incubated at 25 °C for 48 h. The cryo-protectant solution contained mother liquor plus 30% glycerol, 0.5 mM inhibitor, and 2% DMSO. Cryo-protected crystals were fished for data collection.
The 3CLpro-18 data were collected at 1.70 Å on a Rigaku R-AXIS IV++ image plate detector. Reflections were indexed, integrated, scaled, and merged to space group I 1 2 1 using iMosflm.42 The structure was solved by molecular replacement using the Phaser module of the Phenix package using a high-resolution structure of the apo SARS-CoV-2 3CLpro (PDB accession code: 6Y2E) as the search model.43,44 The atomic coordinates and geometric restraints for the inhibitor were generated in the CCP4 suite.42 The inhibitor was built into the Fo–Fc electron density omit maps using Coot.45 Structural refinement was performed with the Real-space Refinement module of the Phenix package. Crystallographic data were summarized in Table S4. All structural figures were generated with PyMOL (https://www.pymol.org).
Molecular Modeling.
Docking studies of compound 2 binding to cruzain were carried out using the Schrödinger Suite program package. The receptor file (PDB accession code: 2OZ2) was processed using the Protein Preparation Wizard function, and the compound was energetically minimized by the LigPrep function. Noncovalent docking was executed and scored by the Glide module. For covalent docking, a cubic box of 18 Å3 was set to encompass any simulated poses with K777 as a reference ligand at its centroid. The SMARTS pattern for 2 was manually defined as [C;H1] = [O], and Cys25 was checked as the reactive residue. The binding affinity was scored, and the best three poses were visually inspected.
Kinetic Data Analysis.
All time-course data were fitted to eq 1, wherein is the fluorescence generated by formation of AMC or liberated ACC, C is the background constant, and are, respectively, initial-state and steady-state reaction rates, is time, and is the observed rate of conversion of the initial inhibited rate to the final inhibited rate.46
| (1) |
Values of vs were then replotted and fitted to eq 2, wherein and represent the respective rates of formation and dissolution of the complex as depicted in Figure 2A.
| (2) |
As described in the main text, the linearity of versus of compounds 1 and 12 arises when . Under these conditions, the concentration of inhibitor required to observe time-dependent inhibition would be much less than the value of for the complex. In this case, eq 2 reduces to eq 3, which is a linear function with a slope of and a . For the rapid dilution assay, was fixed at , and after dilution, could be estimated as according to eq 3.
| (3) |
Overall inhibition constants ( values) were obtained by fitting to eq 4, wherein and are steady-state reaction rates in the presence and absence of the inhibitor, respectively, and represent the fixed concentration of the substrate and variable inhibitor, and is the Michaelis constant of the substrate.
| (4) |
As a tight-binding inhibitor, the values of 1 for cruzain and 18 for 3CLpro were obtained by fitting data to eq 5 in which and are the initial rates obtained with or without the inhibitor, respectively, , , , , and , are, respectively, concentrations of enzyme, inhibitor, and substrate, and and are the inhibition and Michaelis constants, respectively.47
| (5) |
Rates of deacylation reactions of SMAI prodrugs 13–15 were obtained upon evaluation of the first-order loss of reagent as fitted to eq 6 in which and are the initial and final fraction of the unhydrolyzed compound, respectively, and is the pseudo-first-order rate constant from which the half-life was .
| (6) |
PDB ID Code.
The cocrystal structure of 3CLpro-18 is deposited in Protein Data Bank under the ID code 7M2P. Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for the research was provided by NIH grants R21 AI127634, R01 GM129076, and Texas A&M AgriLife Research.
ABBREVIATIONS
- ACC
7-amino-4-carbamoylmethylcoumarin
- AMC
7-amino-4-methylcoumarin
- BSF
bloodstream form
- CES
carboxylesterase
- CHAPS
3-[3-(cholamidopropyl)dimethylammonio]-1-propanesulfonate
- CMBP
cyanomethylenetributylphosphorane
- DIPEA
N,N-diisopropylethylamine
- ER
endoplasmic reticulum
- FCC
flash column chromatography
- hPhe
homophenylalanyl
- MES
2-(N-morpholino)ethanesulfonic acid
- NMePip
N-methlypiperazinyl
- SMAI
self-masked aldehyde inhibitor
- T3P
propylphosphonic anhydride
- TAPSO
3-[N-tris(hydroxymethyl)methylamino]-2-hydroxypropanesulfonic acid
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00628.
Kinetic parameters obtained from rapid dilution assays, details of peaks in 1H-13C HSQC NMR, statistic summary of the cocrystal structure of 3CLpro-18, LC-MS of 13C-labeled 12, kinetic analysis of cruzain inhibition by 11 and 13, growth curves for T. b. brucei BSFs, cell viability of T. cruzi-infected murine cardiomyoblasts, and NMR spectra of the synthesized compounds (PDF)
Molecular formula strings (CSV)
Model coordinates for 2 bound to cruzain in its free aldehyde form (PDB) and its lactol form (PDB)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c00628
Contributor Information
Linfeng Li, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Bala C. Chenna, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States
Kai S. Yang, Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States
Taylor R. Cole, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States
Zachary T. Goodall, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States
Miriam Giardini, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States.
Zahra Moghadamchargari, Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States.
Elizabeth A. Hernandez, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States
Jana Gomez, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Claudia M. Calvet, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States
Jean A. Bernatchez, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States
Drake M. Mellott, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States
Jiyun Zhu, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Andrew Rademacher, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Diane Thomas, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States.
Lauren R. Blankenship, Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States
Aleksandra Drelich, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Arthur Laganowsky, Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States.
Chien-Te K. Tseng, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, Texas 77555, United States
Wenshe R. Liu, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States.
A Joshua Wand, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Jorge Cruz-Reyes, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States.
Jair L. Siqueira-Neto, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States
Thomas D. Meek, Department of Biochemistry and Biophysics, Texas A&M University, College Station, Texas 77843, United States
REFERENCES
- (1).Bedard KM; Semler BL Regulation of picornavirus gene expression. Microbes Infect. 2004, 6, 702–713. [DOI] [PubMed] [Google Scholar]
- (2).Morse JS; Lalonde T; Xu S; Liu WR Learning from the past: Possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Steele-Mortimer O Exploitation of the ubiquitin system by invading bacteria. Traffic 2011, 12, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sajid M; McKerrow JH Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002, 120, 1–21. [DOI] [PubMed] [Google Scholar]
- (5).Rassi A Jr.; Rassi A; Marcondes de Rezende J American trypanosomiasis (Chagas disease). Infect. Dis. Clin. North Am. 2012, 26, 275–291. [DOI] [PubMed] [Google Scholar]
- (6).Branquinha MH; Oliveira SS; Sangenito LS; Sodre CL; Kneipp LF; d’Avila-Levy CM; Santos ALS Cruzipain: An update on its potential as chemotherapy target against the human pathogen trypanosoma cruzi. Curr. Med. Chem. 2015, 22, 2225–2235. [DOI] [PubMed] [Google Scholar]
- (7).Hernandez AA; Roush WR Recent advances in the synthesis, design and selection of cysteine protease inhibitors. Curr. Opin. Chem. Biol. 2002, 6, 459–465. [DOI] [PubMed] [Google Scholar]
- (8).Mellott D; Tseng CT; Drelich A; Fajtova P; Chenna BC; Kostomiris D; Hsu JC; Zhu J; Taylor Z; Tat V; Katzfuss A; Li L; Giardini MA; Skinner D; Hirata K; Beck S; Carlin AF; Clark AE; Berreta L; Maneval D; Frueh F; Hurst BL; Wang H; Kocurek KI; Raushel FM; O’Donoghue A; Siqueira-Neto JL; Meek TD; McKerrow JH A cysteine protease inhibitor blocks SARS-CoV-2 infection of human and monkey cells. bioRxiv 2020, 2020.10.23.347534, DOI: 10.1101/2020.10.23.347534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gampe C; Verma VA Curse or cure? A perspective on the developability of aldehydes as active pharmaceutical ingredients. J. Med. Chem. 2020, 63, 14357–14381. [DOI] [PubMed] [Google Scholar]
- (10).Bandyopadhyay A; Gao J Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Choe Y; Brinen LS; Price MS; Engel JC; Lange M; Grisostomi C; Weston SG; Pallai PV; Cheng H; Hardy LW; Hartsough DS; McMakin M; Tilton RF; Baldino CM; Craik CS Development of alpha-keto-based inhibitors of cruzain, a cysteine protease implicated in chagas disease. Bioorg. Med. Chem. 2005, 13, 2141–2156. [DOI] [PubMed] [Google Scholar]
- (12).Scheidt KA; Roush WR; McKerrow JH; Selzer PM; Hansell E; Rosenthal PJ Structure-based design, synthesis and evaluation of conformationally constrained cysteine protease inhibitors. Bioorg. Med. Chem. 1998, 6, 2477–2494. [DOI] [PubMed] [Google Scholar]
- (13).Silva DG; Ribeiro JFR; de Vita D; Cianni L; Franco CH; Freitas-Junior LH; Moraes CB; Rocha JR; Burtoloso ACB; Kenny PW; Leitão A; Montanari CA A comparative study of warheads for design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5031–5035. [DOI] [PubMed] [Google Scholar]
- (14).Bonatto V; Batista PHJ; Cianni L; de Vita D; Silva DG; Cedron R; Tezuka DY; de Albuquerque S; Moraes CB; Franco CH; Lameira J; Leitão A; Montanari CA On the intrinsic reactivity of highly potent trypanocidal cruzain inhibitors. RSC Med. Chem. 2020, 11, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).O’Brien PJ; Siraki AG; Shangari N Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662. [DOI] [PubMed] [Google Scholar]
- (16).Gibbons P; Verissimo E; Araujo NC; Barton V; Nixon GL; Amewu RK; Chadwick J; Stocks PA; Biagini GA; Srivastava A; Rosenthal PJ; Gut J; Guedes RC; Moreira R; Sharma R; Berry N; Cristiano ML; Shone AE; Ward SA; O’Neill PM Endoperoxide carbonyl falcipain 2/3 inhibitor hybrids: Toward combination chemotherapy of malaria through a single chemical entity. J. Med. Chem. 2010, 53, 8202–8206. [DOI] [PubMed] [Google Scholar]
- (17).Schechter I; Berger A On the size of the active site in proteases I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [DOI] [PubMed] [Google Scholar]
- (18).Cuerrier D; Moldoveanu T; Inoue J; Davies PL; Campbell RL Calpain inhibition by alpha-ketoamide and cyclic hemiacetal inhibitors revealed by X-ray crystallography. Biochemistry 2006, 45, 7446–7452. [DOI] [PubMed] [Google Scholar]
- (19).Nakamura M; Yamaguchi M; Sakai O; Inoue J Exploration of cornea permeable calpain inhibitors as anticataract agents. Bioorg. Med. Chem. 2003, 11, 1371–1379. [DOI] [PubMed] [Google Scholar]
- (20).Kerr ID; Lee JH; Farady CJ; Marion R; Rickert M; Sajid M; Pandey KC; Caffrey CR; Legac J; Hansell E; McKerrow JH; Craik CS; Rosenthal PJ; Brinen LS Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Clementi E; Raimondi DL; Reinhardt WP Atomic screening constants from SCF functions. II. Atoms with 37 to 86 electrons. J. Chem. Phys 1967, 47, 1300–1307. [Google Scholar]
- (22).Clayden J; Greeves N; Warren SG Organic chemistry. Second ed.; Oxford University Press: Oxford, 2012. [Google Scholar]
- (23).Mackenzie NE; Grant SK; Scott AI; Malthouse JP 13C NMR study of the stereospecificity of the thiohemiacetals formed on inhibition of papain by specific enantiomeric aldehydes. Biochemistry 1986, 25, 2293–2298. [DOI] [PubMed] [Google Scholar]
- (24).Abdulla MH; O’Brien T; Mackey ZB; Sajid M; Grab DJ; McKerrow JH RNA interference of trypanosoma brucei cathepsin B and L affects disease progression in a mouse model. PLoS Negl. Trop. Dis. 2008, 2, No. e298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Enanga B; Ariyanayagam MR; Stewart ML; Barrett MP Activity of megazol, a trypanocidal nitroimidazole, is associated with DNA damage. Antimicrob. Agents Chemother. 2003, 47, 3368–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jacobsen W; Christians U; Benet LZ In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab. Dispos. 2000, 28, 1343–1351. [PubMed] [Google Scholar]
- (27).Charrier JD; Durrant SJ; Studley J; Lawes L; Weber P Synthesis and evaluation of novel prodrugs of caspase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 485–488. [DOI] [PubMed] [Google Scholar]
- (28).Wang D; Zou L; Jin Q; Hou J; Ge G; Yang L Human carboxylesterases: A comprehensive review. Acta Pharm. Sin. B 2018, 8, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zanger UM; Schwab M Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [DOI] [PubMed] [Google Scholar]
- (30).Fu L; Ye F; Feng Y; Yu F; Wang Q; Wu Y; Zhao C; Sun H; Huang B; Niu P; Song H; Shi Y; Li X; Tan W; Qi J; Gao GF Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Frank J; Katritzky AR Tautomeric pyridines. Part xv. Pyridone–hydroxypyridine equilibria in solvents of differing polarity. J. Chem. Soc., Perkin Trans 2 1976, 1428–1431. [Google Scholar]
- (32).Zhao M-M; Yang W-L; Yang F-Y; Zhang L; Huang W; Hou W; Fan C; Jin R; Feng Y; Wang Y; Yang J-K Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Sig. Transduct. Target Ther. 2021, 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Noisier AFM; Harris CS; Brimble MA Novel preparation of chiral α-amino acids using the Mitsunobu–Tsunoda reaction. Chem. Commun. 2013, 49, 7744–7746. [DOI] [PubMed] [Google Scholar]
- (34).Zhai X; Meek TD Catalytic mechanism of cruzain from Trypanosoma cruzi as determined from solvent kinetic isotope effects of steady-state and pre-steady-state kinetics. Biochemistry 2018, 57, 3176–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chenna BC; Li L; Mellott DM; Zhai X; Siqueira-Neto JL; Calvet Alvarez C; Bernatchez JA; Desormeaux E; Alvarez Hernandez E; Gomez J; McKerrow JH; Cruz-Reyes J; Meek TD Peptidomimetic vinyl heterocyclic inhibitors of cruzain effect antitrypanosomal activity. J. Med. Chem. 2020, 63, 3298–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang Z; Morris JC; Drew ME; Englund PT Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 2000, 275, 40174–40179. [DOI] [PubMed] [Google Scholar]
- (37).Huber W; Koella JC A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–261. [DOI] [PubMed] [Google Scholar]
- (38).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRpipe: A multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (39).Lee W; Tonelli M; Markley JL NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Willcott MR Mestre Nova. J. Am. Chem. Soc. 2009, 131, 13180–13180. [Google Scholar]
- (41).Yang KS; Ma XR; Ma Y; Alugubelli YR; Scott DA; Vatansever EC; Drelich AK; Sankaran B; Geng ZZ; Blankenship LR; Ward HE; Sheng YJ; Hsu JC; Kratch KC; Zhao B; Hayatshahi HS; Liu J; Li P; Fierke CA; Tseng CK; Xu S; Liu WR A quick route to multiple highly potent SARS-CoV-2 Main protease inhibitors. ChemMedChem 2021, 16, 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal structure of SARS-CoV-2 Main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Morrison JF; Walsh CT The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988, 61, 201–301. [DOI] [PubMed] [Google Scholar]
- (47).Copeland RA Tight binding inhibitors. In Enzymes: A practical introduction to structure, mechanism, and data analysis, VCH: New York, 2000; 305–317. [Google Scholar]
- (48).Chen YT; Brinen LS; Kerr ID; Hansell E; Doyle PS; McKerrow JH; Roush WR In vitro and in vivo studies of the trypanocidal properties of WRR-483 against Trypanosoma cruzi. PLoS Negl. Trop. Dis 2010, 4, No. e825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.