

Summary
The National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) investigates tumors from a proteogenomic perspective, creating rich multi-omics datasets connecting genomic aberrations to cancer phenotypes. To facilitate pan-cancer investigations, we have generated harmonized genomic, transcriptomic, proteomic, and clinical data for >1000 tumors in 10 cohorts to create a cohesive and powerful dataset for scientific discovery. We outline efforts by the CPTAC pan-cancer working group in data harmonization, data dissemination, and computational resources for aiding biological discoveries. We also discuss challenges for multi-omics data integration and analysis, specifically the unique challenges of working with both nucleotide sequencing and mass spectrometry proteomics data.
Keywords: pan-cancer, proteogenomics, data harmonization, multi-omics, open data, CPTAC
eTOC Blurb
Li et al. presents a data compendium from the Clinical Proteomic Tumor Analysis Consortium (CPTAC), covering tumor cohorts from 10 cancer types. This pan-cancer resource provides comprehensive molecular characterization of tumors, with genomic and proteomic data to connect genomic aberrations to cancer phenotypes.
Introduction
Comprehensive molecular profiling is radically changing cancer research. Genomic catalogs of tens of thousands of tumors generated by The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) add immense depth to our understanding of mutations that drive tumorigenesis1. As sequencing on individual tumor cohorts are published, the next wave of manuscripts from these consortia examine patterns across cancer types to elucidate the context-dependent nature of mutations and their impacts2. One limitation of these sequencing-centric efforts is the paucity of data for proteins and their modifications. A few select proteins were monitored through antibody-based approaches such as reverse phase protein arrays (RPPA), but broad and unbiased proteomics data were not generated. As proteins represent the primary molecules responsible for metabolism, signaling and structure, comprehensive and quantitative protein measurements are an essential part of phenotypic characterization. To connect genotype to phenotype, a true proteogenomic approach is needed3.
Proteogenomics analysis is a powerful method for discovering the next generation of precision treatments for cancer as it explicitly links genomic mutations to their impact on cellular physiology 4–6. Early work by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) demonstrated extensive proteome coverage with TCGA samples7, but also identified that sample collection protocols for TCGA allowed significant ischemia prior to tissue freezing. Thus the phosphorylation data measured in these tumors represented a mix of cancer-related and ischemia-related signaling8. As aberrant cellular signaling is an important hallmark of cancer dysfunction and ischemia activates several of the same pathways (e.g. MAPK signaling and apoptosis), it is necessary to create proteogenomic data from freshly acquired tumors with protocols designed to avoid ischemic artifacts 9,10.
The CPTAC dataset currently includes 10 cancer cohorts of prospectively collected tumors analyzed with genomics, transcriptomics, proteomics and phosphoproteomics (Figure 1). Molecular classifications derived from these primary data types are also available, e.g. HLA typing, immune cell decomposition, and ancestry prediction. Other protein post-translational modification (PTM) data such as acetylomics and glycoproteomics were generated for select cancer types. Standard clinical/demographic data and histology images have also been made available. Distributions of sex, age, tumor grade, tumor stage, smoking history, and recurrence status are illustrated in Figure 2. Detailed information of sample provenance is given in Tables S1 and S2. In the original publications investigating a single cancer cohort 11–20, data were processed and analyzed by disease-specific working groups using customized genomics and proteomics data analysis pipelines. Therefore, to enable pan-cancer integrative analysis, and for consistency and reproducibility, we created a compendium of datasets where all proteogenomic data has been re-processed and harmonized.
Fig. 1 -. Tumor types and data types of the CPTAC pan-cancer dataset.

Overview of the available molecular data types for the CPTAC pan-cancer cohort (n=1072, see Table S1 for list of excluded cases and reasons for exclusion from the original data sets). Whole exome, whole genome, transcriptome, proteome, and phosphoproteome data are available for all ten cancer types. Normal samples are available for a subset of tumor types, see Table S1 and S2.
Fig. 2 -. Demographics of the CPTAC dataset.
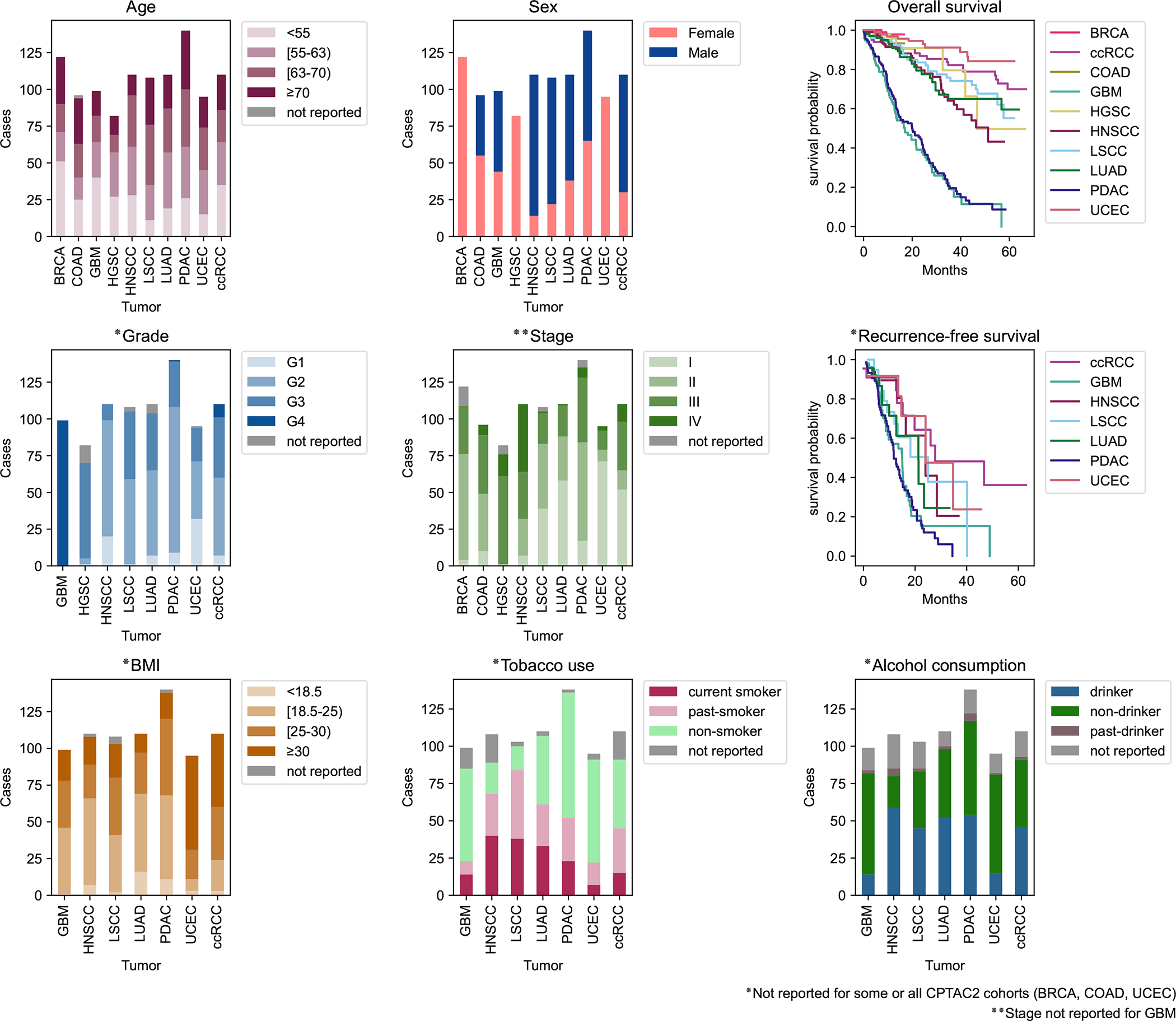
Distributions of selected clinical features among the pan-cancer cohort illustrated in Fig 1. Age is stratified by quartiles. Grade information is not available for BRCA and COAD cohorts. Stage information is not available for the GBM cohort. BMI, Tobacco use, and Alcohol use data is not available for BRCA, COAD, and HGSC cohorts. For survival plots, time starts at diagnosis. Additional clinical features, such as race and ethnicity, are available for exploration on the ProTrack pan-cancer sample dashboard.
Concurrent with this manuscript detailing the data processing and dissemination, CPTAC investigators have pursued biologically motivated pan-cancer analyses to illuminate mechanisms of cancer development. Pan-cancer investigation of protein post-translational modifications identified a subset of tumors with significant changes to cellular regulation, including dysregulated DNA repair, altered metabolic regulation associated with immune response, and patterns of acetylation that affect kinase specificity21. An integration of somatic driver mutations and proteomics data across tumor types resolves distinct cancer hallmark patterns22. Analysis groups continue to conduct thematic studies using the Pan Cancer dataset described here, according to five identified themes: Oncogenic drivers and pathways; DNA Damage Response; Cell of origin; Tumor microenvironment and immunotherapy; and Clinical imaging, biomarkers, and actionable targets.
CPTAC datasets are generated as a resource for cancer research, and community-driven re-analysis is a positive and anticipated outcome from the program. Indeed, numerous groups have already begun re-examining the data23,24. They powerfully use proteogenomic data to reveal new molecular subtypes25–27, prognostic markers28–30, novel protein variants from alternative splicing and RNA editing31–33, and extensive post-translational regulation for protein complexes34,35. To facilitate an increased data reuse and serve the broad audience of cancer data stakeholders, we present our computational methodology for data harmonization and multiple dissemination mechanisms to share both the raw and processed data.
NCI’s Data Commons
The Genomic Data Commons (GDC, https://portal.gdc.cancer.gov) and Proteomic Data Commons (PDC, https://pdc.cancer.gov) are NCI Cloud resources that coordinate storage and analysis of genomics and proteomics data for cancer research. The proteogenomic data generated by the CPTAC program is publicly disseminated through GDC and PDC, which host raw and processed data according to their in-house pipelines. As components of NCI Cloud resource, the GDC and PDC are fully integrated with other NCI Research Data Commons resources, e.g., the Cancer Imaging Archive (TCIA, https://www.cancerimagingarchive.net/), facilitating cloud-based analysis of proteomic, genomic and imaging data. Driven primarily by the CPTAC projects, PDC organizes the data through a robust data model to maintain consistency and integrity of both data and associated metadata, and provides an interface to filter, query, search and visualize proteogenomic data. A direct link to the harmonized data tables stored at the Proteome Data Commons is https://pdc.cancer.gov/pdc/cptac-pancancer.
Finally, in addition to thematic repositories, NCI’s Cancer Research Data Commons contains a data type-agnostic resource, the Cancer Data Service. CPTAC has placed the processed and curated data files into the Cancer Data Service (CDS; https://datacommons.cancer.gov/). The CPTAC data stored in the CDS includes all the harmonized proteogenomic data for our pan-cancer analyses, including: mutation calls, RNA and protein quantification tables, clinical and demographic data, and derived molecular data such as HLA typing, immune cell decomposition, and ancestry prediction. The CPTAC pan-cancer data hosted in CDS is controlled data. Access to controlled access data on CDS is through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC), accessible with a queryable web portal through the Seven Bridges Cancer Genomic Cloud with dbGaP Study Accession, phs001287.v16.p6.
Data from Multiple Pipelines
Proteomic and genomic data analysis methods are continually evolving, and a variety of software tools exist for processing raw data into variant calls and quantifications (e.g., RNA or protein abundance matrices) that can be used for downstream analyses. As CPTAC consists of multiple groups with expertise in each data type, we have often analyzed data with multiple pipelines. Applying different tools to the same set of data may lead to different results and sometimes different conclusions. Therefore, benchmarking is important for tool assessment and selection. For somatic mutation calling, results from the ICGC-TCGA DREAM Somatic Mutation Calling Challenge show that different algorithms have characteristic error profiles, and an ensemble of pipelines always outperforms the best individual pipeline36. Based on this observation, and leveraging our team members’ experience from the Multi-Center Mutation Calling in Multiple Cancers (MC3) project37, somatic mutation calling in our harmonized dataset was based on integrated results from the Broad Institute and Washington University in St. Louis pipelines, which each included multiple algorithms. RNA-Seq data processing pipelines are now relatively mature with much overlap between widely-used pipelines (e.g., https://nf-co.re/rnaseq). The major difference between the three pipelines used in this project is that the pipeline from Baylor College of Medicine includes circular RNAs in addition to linear RNAs. Quantifications for the vast majority of genes are not affected by circular RNAs and show very high correlation among the three pipelines. To compare different pipelines for proteomics data quantification, we have developed OmicsEV38, which uses more than a dozen evaluation metrics to comprehensively assess data depth, data normalization, batch effect, biological signal, platform reproducibility, and multi-omics concordance. Among the publicly available tools used by the CPTAC centers, the FragPipe pipeline usually provides higher data depth while maintaining similar or better performance for other metrics. Using three deep learning-derived features as evaluation metrics (predicted phosphosite probability, absolute retention time (RT) difference between observed and predicted RTs, and Pearson’s correlation coefficient between observed and predicted spectra), we further found that FragPipe achieved higher sensitivity and quality for phosphopeptide identification and phosphosite localization compared with the other tested pipelines39. Based on these evaluation results, we provide one non-redundant, harmonized version with data across all cancer types and omics data types (see BCM pipeline for pan-cancer multi-omics data harmonization in Data S1 for details). However, we would like to emphasize that benchmarking is usually complicated by the lack of absolute ground truth, and thus more efforts should be put towards this important but challenging task. We have therefore also included results from multiple data processing pipelines in the data compendium. Users are encouraged to read the method description associated with each pipeline; explicit details can be found in the Data S1.
Computational API
Simplifying data access can significantly remove barriers to community use and improve transparency and reproducibility. Therefore, CPTAC has created a software package that streams final quantitative data tables directly into a programming environment as dataframe variables (Figure 3). The Python API40, which originally streamed data from the individual cancer type publications, has been updated to provide access to the harmonized pan-cancer datasets described above. Because data is streamed in native pandas dataframes, it is easily integrated with common machine learning and visualization packages such as SciKit-learn, PyTorch, plotly, seaborn, etc. Additionally, access to this API is also straightforward within R using the reticulate package for Python/R interconversion.
Figure 3 -. Streaming data with APIs.
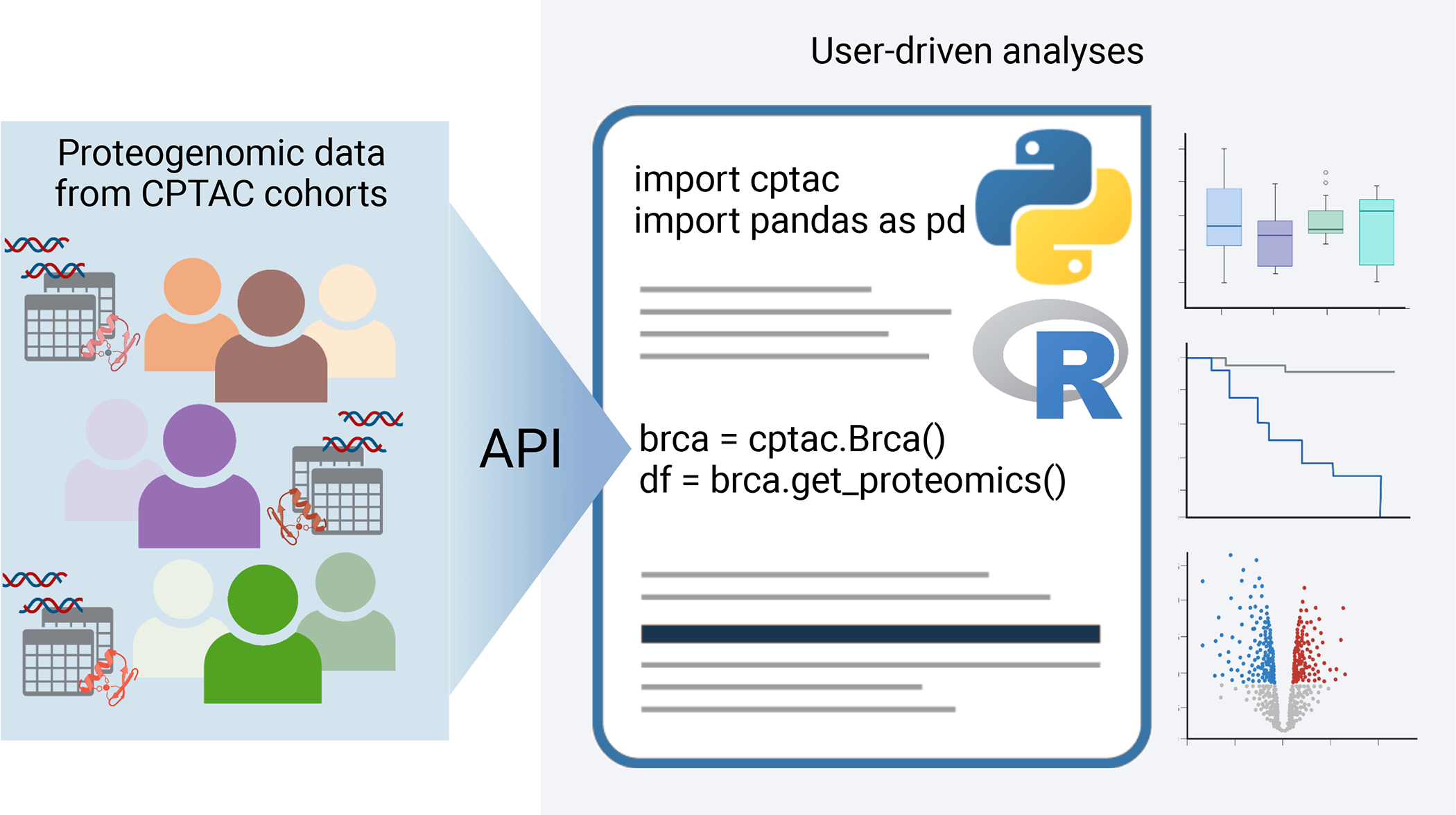
Programmatic access to CPTAC proteogenomic data across all cohorts is provided by both a Python and R API.
Computational APIs also extend the utility of CPTAC proteogenomic data by connecting them to other large public datasets41. We have recently expanded our popular R/Bioconductor tool, TCGAbiolinks42, to stream CPTAC pan-cancer data. In addition to leveraging the numerous software tools available within Bioconductor, TCGAbiolinks facilitates access to molecular data from TCGA, GENIE, MET500, GTEx, GEO, and IHEC. With TCGAbiolinks internal functions to harmonize data from diverse consortia, end-users can explore and validate hypotheses on a comprehensive library of reference datasets using sharable and reproducible codes43. See http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html for tutorials and instructions.
Web Portals for Data Visualization and Analysis
CPTAC teams have created several web portals for visualization and exploration of pan-cancer proteogenomics data (Figure 4). Each of these websites draws from the data compendium the appropriate datasets for pan-cancer analyses.
Figure 4 -. Web portals to CPTAC data.
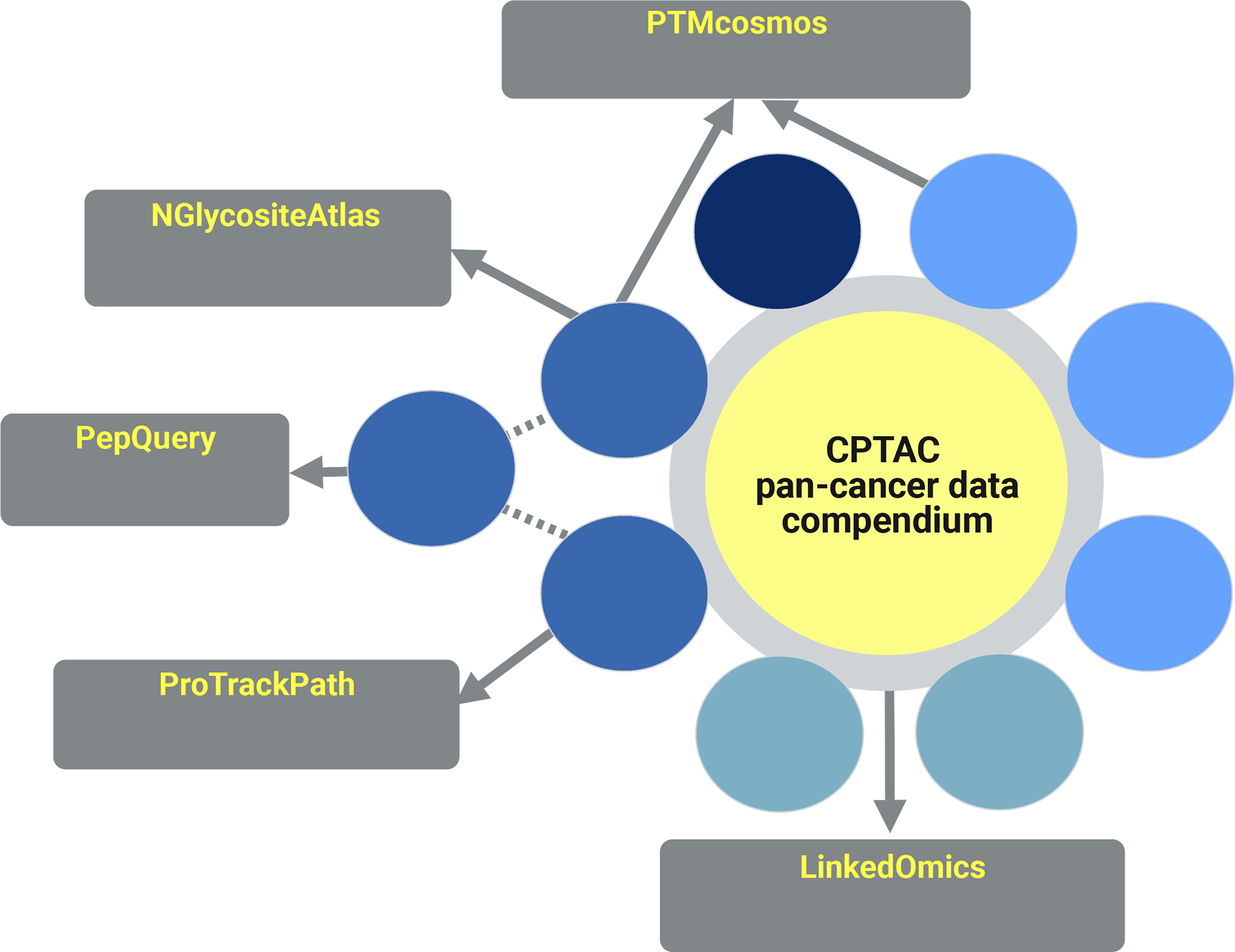
Multiple websites present CPTAC’s proteogenomic data for visual exploration.
PepQuery.
Cancer genomic studies have identified many genomic aberrations that may give rise to abnormal proteins, which are promising candidates for cancer biomarkers, drug targets, and neoantigens. Validation of their expression at the protein level is a critical step toward the clinical translation of these findings. PepQuery (http://www.pepquery.org) allows quick and easy proteomic validation of genomic aberrations, such as single nucleotide variants (SNVs), insertions and deletions (INDELs), RNA editing sites, novel junctions, fusions, and novel transcription regions, using MS/MS data44,45. We have recently recently introduced a new data indexing algorithm in to improve the search speed and have expanded the dataset collection in the PepQuery web server to include MS/MS data from all 10 CPTAC studies, which increased the total number of MS/MS spectra to more than one billion46. Through the PepQuery web server and a mirror site at PDC (https://pdc.cancer.gov), users can directly query CPTAC and other MS/MS data with a novel peptide or DNA sequence of interest to look for supporting peptide spectrum matches (PSMs). For each PSM, annotated spectra are provided for manual evaluation. Moreover, the stand-alone version and the implementation of PepQuery in the Galaxy Proteomics platform (https://proteomics.usegalaxy.eu/) support batch analysis and user-provided MS/MS data, and the identification results can be visualized using PDV47.
LinkedOmics and LinkedOmicsKB.
LinkedOmics (http://www.linkedomics.org) is a data analysis portal that allows the characterization of any clinical or molecular feature of interest (e.g., survival, BRAF_V600E mutation, miR200c expression, or CHEK2-S422 phosphorylation) using cancer multi-omics data from TCGA and CPTAC48. We now provide the pan-cancer harmonized datasets described in this paper for all CPTAC cohorts in LinkedOmics. For each CPTAC study, the database stores data for >500,000 attributes including clinical attributes, mutations at site and gene levels, copy number alterations at region and gene levels, methylations at site and gene levels, mRNA expression, miRNA expression, protein expression, and PTM at site and protein levels. Using three analytical modules, including LinkFinder, LinkCompare, and LinkInterpretor, these data can be mined to reveal the consequences of genetic aberrations, characterize functions of genes and PTMs, and uncover molecular basis of cancer phenotypes.
The on-the-fly, user-defined data queries in LinkedOmics provide a high level of flexibility for analyzing CPTAC data, but performing data analysis on-the-fly is time consuming, and integrating and co-visualizing results from multiple cancer types and multiple omics data types remains challenging. To address these challenges, we further developed LinkedOmicsKB, a new knowledge portal that makes precomputed results for individual genes and phenotypes readily available through a single query49. All results for a query gene or phenotype are presented on a single page with user-friendly visualization to facilitate easy comprehension. The knowledge portal is available at https://kb.linkedomics.org.
PTMcosmos.
PTMcosmos is an interactive web portal designed to catalog and visualize PTMs in humans. As a key regulator of protein activity, PTMs play an essential role in our understanding of cancer and dysregulated cellular states. The PTM sites detected across all CPTAC studies were harmonized using protein sequences from UniProt’s reviewed proteome, allowing for the integration of extensive annotations from many established databases including the UniProt Knowledge Base, PhosphoSitePlus, and protein 3D structures. In total, we harmonized 210,112 PTM sites and annotated them with 11,265 publications. Additionally, to investigate the relationship between genetic alterations found in cancer and PTMs that are in close spatial proximity, we included cancer somatic mutations detected in the samples of CPTAC and the Cancer Genome Atlas (TCGA). Finally, we developed interactive visualization tools to allow researchers to explore the existing literature on a PTM site, the difference in abundance between tumor and normal samples, and the PTM-mutation clusters on protein structures. PTMcosmos portal is publicly available at https://ptmcosmos.wustl.edu/.
ProTrackPath: Pan-Cancer Portal.
We have developed a web application for accessing pathway enrichment scores across the pan-cancer cohorts. While previous ProTrack applications allow users to visualize normalized raw data for individual cancers50–52, the ProTrackPath pan-cancer portal presents pathway enrichment scores across cancer types, calculated with a single sample gene set enrichment analysis (ssGSEA)53. The user specifies a pathway database such as Hallmark54, KEGG55, or Reactome56, then selects a set of pathways to visualize. An interactive heatmap is then generated, which users can customize by sorting according to any given track or toggling categorical variables on and off. Additionally, the portal includes a sample dashboard view, which allows for viewing clinical characteristics. This allows users to explore the distributions of the cancer types along with various demographic and clinical features as bar graphs. Users can filter samples by toggling features in each bar graph’s interactive legend, then populate the heatmap with their custom-generated cohort. The portal is available to the public at http://pancan.cptac-data-view.org/.
NGlycositeAtlas Portal.
N-linked glycosylation is one of the most abundant protein modifications and is highly relevant to disease progression in cancer57. With the advances in experimental and computational approaches, glycoproteomics has provided comprehensive characterization of glycosite-specific glycosylation of glycoproteins and valuable insights into their biological functions in cancer58–62. However, there is still a lack of the integration of large-scale characterization of glycoproteomic data from different cancer types for pan-cancer research. We identified intact N-linked glycopeptides (see Data S1) to create a database resource termed NGlycositeAtlas 2.0, which contains more than 90,629 intact N-linked glycopeptides (representing 5,665 N-linked glycosite-containing peptides) of over 2,000 glycoproteins from CPTAC data. The NGlycositeAtlas database and consensus MS/MS spectra are available at https://www.biomarkercenter.org/nglycositeatlas.
Analytical challenges for pan-cancer multi-omics
With the rapid development of molecular measurement technologies, cancer datasets have become multi-modal. CPTAC has created rich proteogenomic datasets that measure DNA, RNA, and protein molecules within tumors and adjacent normal tissues (NATs). This diversity of data catalogs a comprehensive map of cellular state, providing researchers the opportunity to understand the subtle regulatory interplay between DNA mutation events that give rise to dysregulated signaling networks and the ultimate cellular phenotype. This large and comprehensive dataset presents several challenges in data integration and interpretation. In this section, we outline several important considerations for the re-use and re-analysis of proteogenomic data.
The first challenge in a proteogenomic dataset is to ensure that identifiers are harmonized. The following examples demonstrate the challenge. Many genes have multiple protein isoforms due to alternative splicing, including a noted change in splicing patterns in cancers63–65. Each isoform may have a unique function and combining all data into a single ‘gene level’ measurement could obscure these differences. Suppose that mRNA data identifies two distinct transcripts. The transcriptomics data table, therefore, reports two database identifiers each with a separate quantitative value. If the proteomics data does not identify peptides that differentiate the two isoforms, which protein identifier should be used? To which transcript data should the protein abundance be compared? As orthogonal data types, proteomics and transcriptomics frequently identify different isoforms. This situation is equally complex when integrating PTMs, mutations, or epigenetics. If a phosphorylation or a coding mutation is observed, which protein isoform should it be associated with? Which transcript/protein should be used in comparison with methylation data? Mapping PTMs and coding mutations to different protein isoforms will make it difficult to study the impact of somatic mutations on PTMs. Thus, for a large multi-omics harmonization task such as presented here, we recommend careful consideration and transparency in reporting analytical methods. As potential solutions to mitigate the above challenges, we suggest the following: 1) using the same versions of genome assembly and gene annotation for the processing of data from all omics platforms and all cancer types; 2) reporting gene-level quantification when isoform level analysis is unrealistic; 3) applying a consistent and transparent rule for representative isoform selection when representative isoform selection is needed but the data is isoform agnostic, e.g., phosphosite localization annotation. A second challenge is embracing the full proteogenomic landscape as the molecular characterization of cells and tissues becomes more complete. We emphasize that each data type provides unique value and helps to clarify complex phenotypes. For example, the proteome and the transcriptome are distinct, and each provides a meaningful view of cellular processes. A rich body of research demonstrates that the mRNA and protein abundances frequently have a poorer correlation than expected66–70, a consequence of both translational and post-translational regulation71–74. As cancers are often characterized by regulatory dysfunction, exploring the source of this dysfunction can be best understood by combining transcriptomics and proteomics75. Similarly, the consequence of somatic mutation in kinases is best observed by combining genomics and phosphoproteomics. Indeed, many biological hypotheses can be best addressed by a fruitful combination of data types. To understand the consequence of genomic copy number variation, Gonçalves et al., combined genomics and proteomics and discovered widespread post-transcriptional attenuation in protein abundance mitigating the impact of gene amplification, especially to preserve stoichiometry in protein complexes34. The search for novel amino acid variants76 and cancer neo-antigens77–79 is inherently a proteogenomic investigation, as is the discovery of tumor-specific splice isoforms80,81 and fusion proteins82. Combining all the proteogenomics levels into a single analysis is challenging, but the NMF methodology is frequently used for integrative clustering to highlight the unique contribution of each data type83.
Despite the great effort to harmonize the multi-omics datasets across different cancer studies, we want to emphasize that “batch” effects between different cancer types could still remain in the pan-cancer datasets due to both technical factors, as omics experiments of different cancer types were carried out by different labs and/or using different platforms, and biological factors, as different organs and cancer types have intrinsically different biology. Thus, when analyzing the pan-cancer data, one needs to carefully adjust for these batch effects across different cancer types. For example, when fitting a regression model to study the dependence of molecular abundances on other attributes, one can include cancer type indicators as covariates to account for cancer-type specific mean values of molecules. Other analysis techniques, such as meta-analysis framework, could also be used to perform pan-cancer level inferences.
Finally, we focus on a challenge specific to post-translational modifications (PTMs). In the CPTAC data, we report quantitative measurement of phosphorylation and selected datasets also have data for acetylation, and glycosylation. Although missing values are a regular part of all omics data, they are more pronounced in PTM data. One place where this is particularly problematic is pan-cancer analysis. If a PTM site is well quantified in one cancer type (e.g., EGFR tyrosine 1172), it may have many missing values in another, which would complicate a pan-cancer comparison of protein activation. One might be tempted to roll together all PTMs in a protein into a single measurement - e.g., the average phosphorylation state of EGFR. However, we advise against this, as PTMs at each site in a protein can be functionally independent and may not correlate across samples. Both experimental and computational approaches are being developed to improve PTM peptide identification, which will help alleviate the missing value problem in PTM proteomics 84.
Conclusion
Pan-cancer proteogenomic data analysis requires a consistent data set processed with a unified pipeline across all samples. Several groups have created proteogenomic datasets on cancer cohorts, exploring diverse genetic backgrounds for common cancers85–88, pediatric tumors51 or understudied tumor types89,90. For pan-cancer analyses it is important that individual datasets follow similar SOPs and process data in a consistent manner. Therefore, we have re-processed the data from CPTAC’s 10 cancer cohorts to create a pan-cancer proteogenomic dataset. We presented the description of methods used to create this data compendium, methods of data access, as well as key considerations for pan-cancer multi-omics data analysis. This resource has been used within CPTAC for biological discoveries under various themes. We hope this also serves as a resource for the broader cancer research community to advance cancer diagnosis and treatment.
Supplementary Material
Table S1 – Clinical and demographic information, related to Figure 1.
Table S2 – Clinical information related to tumor and normal samples, related to Figure 1.
Table S3 – Representative isoforms for each gene in the BCM pipelines, related to Data S1.
Acknowledgements
The Clinical Proteomic Tumor Analysis Consortium (CPTAC) is supported by the National Cancer Institute of the National Institutes of Health under award numbers U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, and U01CA214125. Additional funding support was provided by NIH awards R33CA263705, T32CA203690, T32GM136542 and Leidos Biomed contract 20X042F01/TO01 and the Simmons Center for Cancer Research. Figure 1 representing the data overview for this manuscript was created using BioRender.com. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. 75N91019D00024, Task Order No. 7591029F00029, and Contract No. 75N91019D00024, Task Order 75N91020F00029. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Footnotes
Declaration of Interest
F.A. is an inventor on a patent application related to SignatureAnalyzer-GPU filed by the Broad Institute and is an employee and shareholder of Illumina Inc. since 8 November 2021.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ding L, Bailey MH, Porta-Pardo E, Thorsson V, Colaprico A, Bertrand D, Gibbs DL, Weerasinghe A, Huang K-L, Tokheim C, et al. (2018). Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 173, 305–320.e10. 10.1016/j.cell.2018.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alfaro JA, Sinha A, Kislinger T, and Boutros PC (2014). Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods 11, 1107–1113. 10.1038/nmeth.3138. [DOI] [PubMed] [Google Scholar]
- 4.Mani DR, Krug K, Zhang B, Satpathy S, Clauser KR, Ding L, Ellis M, Gillette MA, and Carr SA (2022). Cancer proteogenomics: current impact and future prospects. Nat Rev Cancer 22, 298–313. 10.1038/s41568-022-00446-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez H, Zenklusen JC, Staudt LM, Doroshow JH, and Lowy DR (2021). The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 184, 1661–1670. 10.1016/j.cell.2021.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, and Paulovich AG (2019). Clinical potential of mass spectrometry-based proteogenomics. Nat Rev Clin Oncol 16, 256–268. 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. (2014). Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, et al. (2014). Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics 13, 1690–1704. 10.1074/mcp.M113.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, Huang C, Li J, Dong X, Zhou Y, et al. (2019). Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 179, 561–577.e22. 10.1016/j.cell.2019.08.052. [DOI] [PubMed] [Google Scholar]
- 10.Mun D-G, Bhin J, Kim S, Kim H, Jung JH, Jung Y, Jang YE, Park JM, Kim H, Jung Y, et al. (2019). Proteogenomic Characterization of Human Early-Onset Gastric Cancer. Cancer Cell 35, 111–124.e10. 10.1016/j.ccell.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih T-SM, Chang H-Y, et al. (2020). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 180, 207. 10.1016/j.cell.2019.12.026. [DOI] [PubMed] [Google Scholar]
- 12.Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 183, 1436–1456.e31. 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049.e19. 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L-B, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 39, 509–528.e20. 10.1016/j.ccell.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. 10.1016/j.ccell.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e40. 10.1016/j.cell.2021.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang W-W, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. 10.1016/j.cell.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDermott JE, Arshad OA, Petyuk VA, Fu Y, Gritsenko MA, Clauss TR, Moore RJ, Schepmoes AA, Zhao R, Monroe ME, et al. (2020). Proteogenomic Characterization of Ovarian HGSC Implicates Mitotic Kinases, Replication Stress in Observed Chromosomal Instability. Cell Rep Med 1, 100004. 10.1016/j.xcrm.2020.100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. 10.1016/j.cell.2021.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748.e26. 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geffen Y (2023). Patterns and regulation of post-translational modifications in cancer. Cell. CELL-D-22–02032 [Google Scholar]
- 22.Li Yize (2023). Pan-Cancer Proteogenomic Impacts of Oncogenic Drivers. Cell. CELL-D-22–01960 [Google Scholar]
- 23.Wu P, Heins ZJ, Muller JT, Katsnelson L, de Bruijn I, Abeshouse AA, Schultz N, Fenyö D, and Gao J (2019). Integration and Analysis of CPTAC Proteomics Data in the Context of Cancer Genomics in the cBioPortal. Mol Cell Proteomics 18, 1893–1898. 10.1074/mcp.TIR119.001673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhan X, Cheng J, Huang Z, Han Z, Helm B, Liu X, Zhang J, Wang T-F, Ni D, and Huang K (2019). Correlation Analysis of Histopathology and Proteogenomics Data for Breast Cancer. Molecular & Cellular Proteomics 18, S37–S51. 10.1074/mcp.RA118.001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen F, Chandrashekar DS, Varambally S, and Creighton CJ (2019). Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat Commun 10, 5679. 10.1038/s41467-019-13528-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tong M, Yu C, Zhan D, Zhang M, Zhen B, Zhu W, Wang Y, Wu C, He F, Qin J, et al. (2019). Molecular subtyping of cancer and nomination of kinase candidates for inhibition with phosphoproteomics: Reanalysis of CPTAC ovarian cancer. EBioMedicine 40, 305–317. 10.1016/j.ebiom.2018.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Chen F, Chandrashekar DS, Varambally S, and Creighton CJ (2022). Proteogenomic characterization of 2002 human cancers reveals pan-cancer molecular subtypes and associated pathways. Nat Commun 13, 2669. 10.1038/s41467-022-30342-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang W, Chen J, Weng W, Xiang Y, Shi H, and Shan Y (2020). Development of cancer prognostic signature based on pan-cancer proteomics. Bioengineered 11, 1368–1381. 10.1080/21655979.2020.1847398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao J, Cheng M, Gai J, Zhang R, Du T, and Li Q (2020). SPOCK2 Serves as a Potential Prognostic Marker and Correlates With Immune Infiltration in Lung Adenocarcinoma. Front Genet 11, 588499. 10.3389/fgene.2020.588499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Z-H, and Yang D-L (2020). Identification of a protein signature for predicting overall survival of hepatocellular carcinoma: a study based on data mining. BMC Cancer 20, 720. 10.1186/s12885-020-07229-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kahles A, Lehmann K-V, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research Network, et al. (2018). Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224.e6. 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han L, Zhou Z, Mojumdar K, Jeong KJ, Labrie M, et al. (2018). A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 33, 817–828.e7. 10.1016/j.ccell.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prakash A, Taylor L, Varkey M, Hoxie N, Mohammed Y, Goo YA, Peterman S, Moghekar A, Yuan Y, Glaros T, et al. (2021). Reinspection of a Clinical Proteomics Tumor Analysis Consortium (CPTAC) Dataset with Cloud Computing Reveals Abundant Post-Translational Modifications and Protein Sequence Variants. Cancers (Basel) 13, 5034. 10.3390/cancers13205034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonçalves E, Fragoulis A, Garcia-Alonso L, Cramer T, Saez-Rodriguez J, and Beltrao P (2017). Widespread Post-transcriptional Attenuation of Genomic Copy-Number Variation in Cancer. Cell Syst 5, 386–398.e4. 10.1016/j.cels.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan CJ, Kennedy S, Bajrami I, Matallanas D, and Lord CJ (2017). A Compendium of Co-regulated Protein Complexes in Breast Cancer Reveals Collateral Loss Events. Cell Syst 5, 399–409.e5. 10.1016/j.cels.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ewing AD, Houlahan KE, Hu Y, Ellrott K, Caloian C, Yamaguchi TN, Bare JC, P’ng C, Waggott D, Sabelnykova VY, et al. (2015). Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat Methods 12, 623–630. 10.1038/nmeth.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6, 271–281.e7. 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen B, Jaehnig EJ, and Zhang B (2022). OmicsEV: a tool for comprehensive quality evaluation of omics data tables. Bioinformatics, btac698. 10.1093/bioinformatics/btac698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang W, Wen B, Li K, Zeng W-F, da Veiga Leprevost F, Moon J, Petyuk VA, Edwards NJ, Liu T, Nesvizhskii AI, et al. (2021). Deep-Learning-Derived Evaluation Metrics Enable Effective Benchmarking of Computational Tools for Phosphopeptide Identification. Mol Cell Proteomics 20, 100171. 10.1016/j.mcpro.2021.100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindgren CM, Adams DW, Kimball B, Boekweg H, Tayler S, Pugh SL, and Payne SH (2021). Simplified and Unified Access to Cancer Proteogenomic Data. J Proteome Res 20, 1902–1910. 10.1021/acs.jproteome.0c00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colaprico A, Olsen C, Bailey MH, Odom GJ, Terkelsen T, Silva TC, Olsen AV, Cantini L, Zinovyev A, Barillot E, et al. (2020). Interpreting pathways to discover cancer driver genes with Moonlight. Nat Commun 11, 69. 10.1038/s41467-019-13803-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71. 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehmann BD, Colaprico A, Silva TC, Chen J, An H, Ban Y, Huang H, Wang L, James JL, Balko JM, et al. (2021). Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat Commun 12, 6276. 10.1038/s41467-021-26502-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen B, Li K, Zhang Y, and Zhang B (2020). Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat Commun 11, 1759. 10.1038/s41467-020-15456-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen B, Wang X, and Zhang B (2019). PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res 29, 485–493. 10.1101/gr.235028.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen B, and Zhang B (2023). PepQuery2 democratizes public MS proteomics data for rapid peptide searching. Nat Commun 14, 2213. 10.1038/s41467-023-37462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li K, Vaudel M, Zhang B, Ren Y, and Wen B (2019). PDV: an integrative proteomics data viewer. Bioinformatics 35, 1249–1251. 10.1093/bioinformatics/bty770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vasaikar SV, Straub P, Wang J, and Zhang B (2018). LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res 46, D956–D963. 10.1093/nar/gkx1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao A proteogenomics data-driven knowledge base of human cancer. Cell Systems. CELL-SYSTEMS-D-23–00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calinawan AP, Song X, Ji J, Dhanasekaran SM, Petralia F, Wang P, and Reva B (2020). ProTrack: An Interactive Multi-Omics Data Browser for Proteogenomic Studies. Proteomics 20, e1900359. 10.1002/pmic.201900359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov D, Krek A, Ma W, Zhu Y, Ji J, et al. (2020). Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell 183, 1962–1985.e31. 10.1016/j.cell.2020.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang D, Chowdhury S, Wang H, Savage SR, Ivey RG, Kennedy JJ, Whiteaker JR, Lin C, Hou X, Oberg AL, et al. (2021). Multiomic analysis identifies CPT1A as a potential therapeutic target in platinum-refractory, high-grade serous ovarian cancer. Cell Rep Med 2, 100471. 10.1016/j.xcrm.2021.100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, D353–D361. 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Res 48, D498–D503. 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinho SS, and Reis CA (2015). Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15, 540–555. 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 58.Dong M, Lih TM, Chen S-Y, Cho K-C, Eguez RV, Höti N, Zhou Y, Yang W, Mangold L, Chan DW, et al. (2020). Urinary glycoproteins associated with aggressive prostate cancer. Theranostics 10, 11892–11907. 10.7150/thno.47066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu Y, Pan J, Shah P, Ao M, Thomas SN, Liu Y, Chen L, Schnaubelt M, Clark DJ, Rodriguez H, et al. (2020). Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep 33, 108276. 10.1016/j.celrep.2020.108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan J, Hu Y, Sun S, Chen L, Schnaubelt M, Clark D, Ao M, Zhang Z, Chan D, Qian J, et al. (2020). Glycoproteomics-based signatures for tumor subtyping and clinical outcome prediction of high-grade serous ovarian cancer. Nat Commun 11, 6139. 10.1038/s41467-020-19976-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabang DN, Ford M, and Li L (2021). Recent Advances in Mass Spectrometry-Based Glycomic and Glycoproteomic Studies of Pancreatic Diseases. Front Chem 9, 707387. 10.3389/fchem.2021.707387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Jiao J, Yang P, and Lu H (2014). Mass spectrometry-based N-glycoproteomics for cancer biomarker discovery. Clin Proteomics 11, 18. 10.1186/1559-0275-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Climente-González H, Porta-Pardo E, Godzik A, and Eyras E (2017). The Functional Impact of Alternative Splicing in Cancer. Cell Rep 20, 2215–2226. 10.1016/j.celrep.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 64.Venables JP (2004). Aberrant and alternative splicing in cancer. Cancer Res 64, 7647–7654. 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 65.Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, et al. (2009). Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 16, 670–676. 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 66.Fortelny N, Overall CM, Pavlidis P, and Freue GVC (2017). Can we predict protein from mRNA levels? Nature 547, E19–E20. 10.1038/nature22293. [DOI] [PubMed] [Google Scholar]
- 67.McManus J, Cheng Z, and Vogel C (2015). Next-generation analysis of gene expression regulation--comparing the roles of synthesis and degradation. Mol Biosyst 11, 2680–2689. 10.1039/c5mb00310e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, and Mann M (2011). Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7, 548. 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Payne SH (2015). The utility of protein and mRNA correlation. Trends Biochem Sci 40, 1–3. 10.1016/j.tibs.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13, 227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aviner R, Shenoy A, Elroy-Stein O, and Geiger T (2015). Uncovering Hidden Layers of Cell Cycle Regulation through Integrative Multi-omic Analysis. PLoS Genet 11, e1005554. 10.1371/journal.pgen.1005554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cai Y, Yu X, Hu S, and Yu J (2009). A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinformatics 7, 147–154. 10.1016/S1672-0229(08)60044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grzmil M, and Hemmings BA (2012). Translation regulation as a therapeutic target in cancer. Cancer Res 72, 3891–3900. 10.1158/0008-5472.CAN-12-0026. [DOI] [PubMed] [Google Scholar]
- 74.He R-Z, Luo D-X, and Mo Y-Y (2019). Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis 6, 6–15. 10.1016/j.gendis.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang W, Zhou M, Dorsey TH, Prieto DA, Wang XW, Ruppin E, Veenstra TD, and Ambs S (2018). Integrated proteotranscriptomics of breast cancer reveals globally increased protein-mRNA concordance associated with subtypes and survival. Genome Med 10, 94. 10.1186/s13073-018-0602-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Da Cunha LM, Terrematte P, Fiuza TDS, Silva VLD, Kroll JE, De Souza SJ, and De Souza GA (2022). dbPepVar: A Novel Cancer Proteogenomics Database. IEEE Access 10, 90982–90994. 10.1109/ACCESS.2022.3201897. [DOI] [Google Scholar]
- 77.Cleyle J, Hardy M-P, Minati R, Courcelles M, Durette C, Lanoix J, Laverdure J-P, Vincent K, Perreault C, and Thibault P (2022). Immunopeptidomic analyses of colorectal cancers with and without microsatellite instability. Mol Cell Proteomics 21, 100228. 10.1016/j.mcpro.2022.100228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Polyakova A, Kuznetsova K, and Moshkovskii S (2015). Proteogenomics meets cancer immunology: mass spectrometric discovery and analysis of neoantigens. Expert Rev Proteomics 12, 533–541. 10.1586/14789450.2015.1070100. [DOI] [PubMed] [Google Scholar]
- 79.Xiang R, Ma L, Yang M, Zheng Z, Chen X, Jia F, Xie F, Zhou Y, Li F, Wu K, et al. (2021). Increased expression of peptides from non-coding genes in cancer proteomics datasets suggests potential tumor neoantigens. Commun Biol 4, 496. 10.1038/s42003-021-02007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miller RM, Jordan BT, Mehlferber MM, Jeffery ED, Chatzipantsiou C, Kaur S, Millikin RJ, Dai Y, Tiberi S, Castaldi PJ, et al. (2022). Enhanced protein isoform characterization through long-read proteogenomics. Genome Biol 23, 69. 10.1186/s13059-022-02624-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hatakeyama K, Ohshima K, Fukuda Y, Ogura S, Terashima M, Yamaguchi K, and Mochizuki T (2011). Identification of a novel protein isoform derived from cancer-related splicing variants using combined analysis of transcriptome and proteome. Proteomics 11, 2275–2282. 10.1002/pmic.201100016. [DOI] [PubMed] [Google Scholar]
- 82.Kim C-Y, Na K, Park S, Jeong S-K, Cho J-Y, Shin H, Lee MJ, Han G, and Paik Y-K (2019). FusionPro, a Versatile Proteogenomic Tool for Identification of Novel Fusion Transcripts and Their Potential Translation Products in Cancer Cells. Mol Cell Proteomics 18, 1651–1668. 10.1074/mcp.RA119.001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mani DR, Maynard M, Kothadia R, Krug K, Christianson KE, Heiman D, Clauser KR, Birger C, Getz G, and Carr SA (2021). PANOPLY: a cloud-based platform for automated and reproducible proteogenomic data analysis. Nat Methods 18, 580–582. 10.1038/s41592-021-01176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bekker-Jensen DB, Bernhardt OM, Hogrebe A, Martinez-Val A, Verbeke L, Gandhi T, Kelstrup CD, Reiter L, and Olsen JV (2020). Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat Commun 11, 787. 10.1038/s41467-020-14609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y-J, Roumeliotis TI, Chang Y-H, Chen C-T, Han C-L, Lin M-H, Chen H-W, Chang G-C, Chang Y-L, Wu C-T, et al. (2020). Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell 182, 226–244.e17. 10.1016/j.cell.2020.06.012. [DOI] [PubMed] [Google Scholar]
- 86.Lehtiö J, Arslan T, Siavelis I, Pan Y, Socciarelli F, Berkovska O, Umer HM, Mermelekas G, Pirmoradian M, Jönsson M, et al. (2021). Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune evasion mechanisms. Nat Cancer 2, 1224–1242. 10.1038/s43018-021-00259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu J-Y, Zhang C, Wang X, Zhai L, Ma Y, Mao Y, Qian K, Sun C, Liu Z, Jiang S, et al. (2020). Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 182, 245–261.e17. 10.1016/j.cell.2020.05.043. [DOI] [PubMed] [Google Scholar]
- 88.Qu Y, Feng J, Wu X, Bai L, Xu W, Zhu L, Liu Y, Xu F, Zhang X, Yang G, et al. (2022). A proteogenomic analysis of clear cell renal cell carcinoma in a Chinese population. Nat Commun 13, 2052. 10.1038/s41467-022-29577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi X, Sun Y, Shen C, Zhang Y, Shi R, Zhang F, Liao T, Lv G, Zhu Z, Jiao L, et al. (2022). Integrated proteogenomic characterization of medullary thyroid carcinoma. Cell Discov 8, 120. 10.1038/s41421-022-00479-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dong L, Lu D, Chen R, Lin Y, Zhu H, Zhang Z, Cai S, Cui P, Song G, Rao D, et al. (2022). Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 40, 70–87.e15. 10.1016/j.ccell.2021.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – Clinical and demographic information, related to Figure 1.
Table S2 – Clinical information related to tumor and normal samples, related to Figure 1.
Table S3 – Representative isoforms for each gene in the BCM pipelines, related to Data S1.