

Abstract
The genetic information of human cells is stored in the context of chromatin, which is subjected to DNA methylation and various histone modifications. Such a ‘language’ of chromatin modification constitutes a fundamental means for gene and (epi)genome regulation, underlying a myriad of cellular and developmental processes. In recent years, mounting evidence has demonstrated that mis-writing, mis-reading or mis-erasing of the modification language embedded in chromatin represents a common, sometimes early and pivotal, event across a wide range of human cancers, contributing to oncogenesis through the induction of epigenetic, transcriptomic and phenotypic alterations. It is increasingly clear that cancer-related metabolic perturbations and oncohistone mutations also directly impact chromatin modification, thereby promoting cancerous transformation. Phase separation-based deregulation of chromatin modulators and chromatin structure also is emerging to be an important underpinning of tumorigenesis. Understanding the various molecular pathways that underscore a mis-regulated chromatin language in cancer, together with discovery and development of more effective drugs to target these chromatin-related tumor vulnerabilities, will enhance treatment of human malignancies.
ToC blurb:
Deregulation of chromatin modification underlies a myriad of oncogenic processes. This review synthesizes the many connections between chromatin modifications and cancer, discussing recent advances and highlighting options for therapeutic targeting.
Introduction
In eukaryotes, chromatin is organized in the repeated units of nucleosomes, each composed of a histone octamer and a piece of surrounding DNA. Histone posttranslational modifications (PTMs) and DNA methylation create chromatin variations1,2. Besides the core replicative histones (i.e., H2A, H2B, H3 and H4), histone variants also exist and are assembled into nucleosomes in a replication-independent manner. Some of the better studied histone lysine methylations occur predominantly on histones H3 and H4, including histone H3 lysine 4 (H3K4), lysine 9 (H3K9), lysine 27 (H3K27), lysine 36 (H3K36), lysine 79 (H3K79) and histone H4 lysine 20 (H4K20). Each of these lysines can exist in four methyl states - unmodified, monomethylated (Kme1), dimethylated (Kme2) and trimethylated lysine (Kme3); alternatively, they can exist in acetylated or other acylated forms yielding staggering complexity, although the abundance and relative weight of various PTMs in terms of function can vary (FIG. S1a). In mammalian cells, DNA modifications are mainly 5-methylcytosine (5mC) and its oxidative derivatives, which all play a role in gene or genome regulation3,4. These chromatin variations establish a fundamental means for regulating essentially all of the DNA-templated processes such as gene transcription, DNA replication, DNA damage repair and DNA recombination.
Histone PTMs and DNA methylation, established and removed by antagonizing enzymes of writers and erasers, respectively (FIG. 1a), regulate chromatin-based processes both in cis and in trans. In cis, histone PTMs change structural or physical properties of nucleosomes, for example increasing DNA accessibility or neutralizing the negative charge of DNA via histone charge-altering modifications (e.g., acetylation and phosphorylation). In trans, chromatin modification serves as a context-dependent docking site for recruiting readers or other effectors1. It is noteworthy to mention that a good number of writers and erasers also harbor a chromatin reader module, thereby opening up various possibilities for potential cross-talk among chromatin modifiers including self-propagation, cooperation, competition or antagonism. Writers, erasers and readers can also potentially target nonhistone proteins to regulate their respective functions5–8.
Figure 1. The misregulated ‘language’ of chromatin modification in cancer.
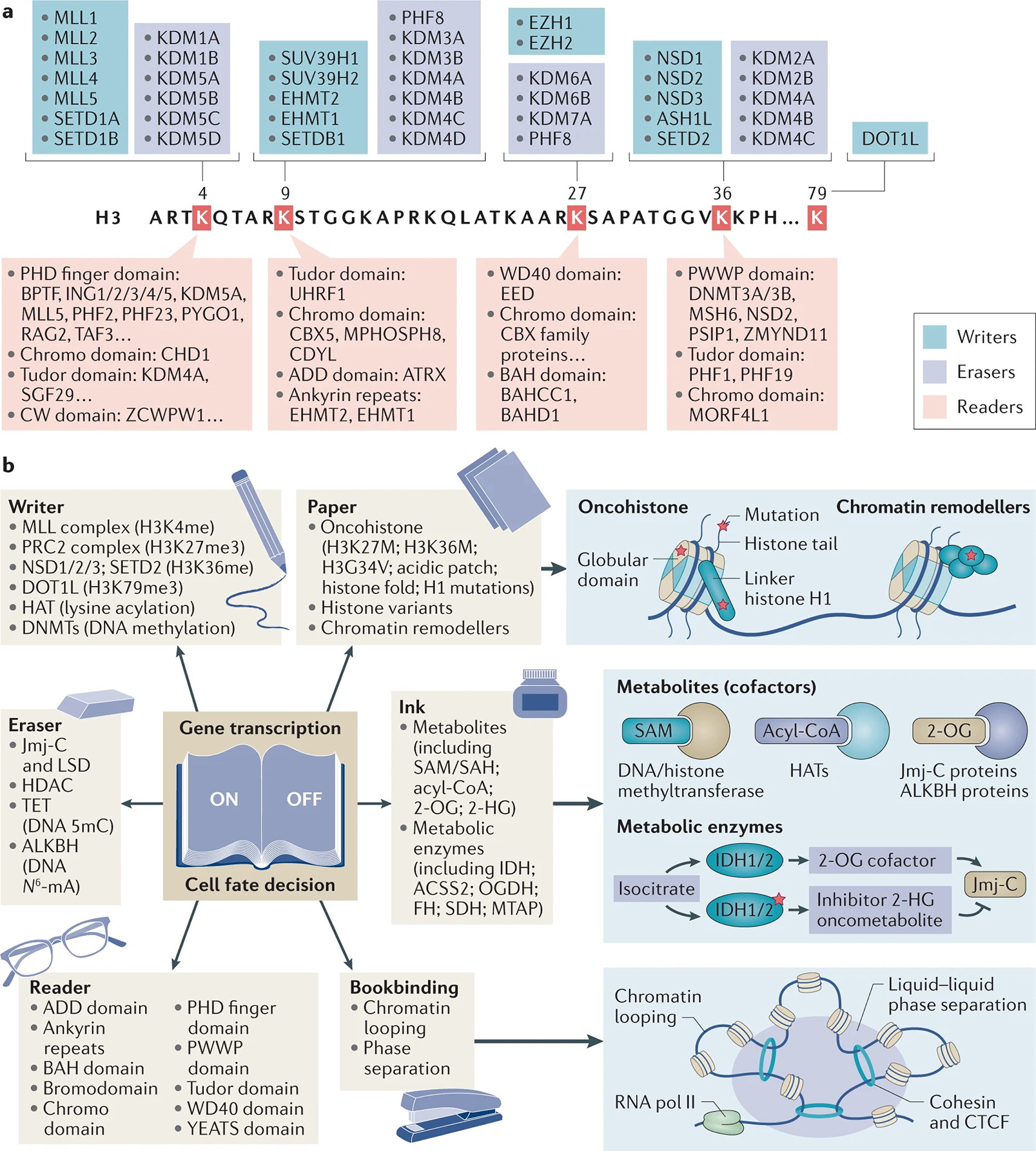
a. Overview of key writers, erasers and readers of histone H3 methylations at K4, K9, K27, K36 and K79. Note that this list is not exclusive; many other modifications such as acetylation, phosphorylation, ubiquitylation and arginine methylation also occur in all histones261, which are not shown in the figure.
b. A mis-regulated ‘language’ of chromatin modification underlies oncogenesis. In addition to cancer-related mutations of chromatin modification writers, erasers or readers, the oncohistone and chromatin remodeler mutations alter numerous fundamental aspects of chromatin, as illustrated by the ‘paper’ that the ‘language’ of chromatin modification operates on. Moreover, cellular metabolites and oncometabolites form essential cofactors for erasers, and also represent the ‘paint and ink’ used by writers (and in some cases erasers) to modify histones and DNA. A versatile set of readers allow cells to recognise and engage various chromatin marks such as lysine methylation, acetylation, crotonylation, and more. Similar to a process of ‘stapling and bookbinding’, chromatin looping and phase separation-based regulation are involved in the high-order organization of chromatin and regulation of (epi)-genome, processes frequently altered in cancers. Altogether, a collection of deregulated mechanisms converge, resulting in a mis-regulated chromatin ‘language’.
A wealth of evidence now supports an intimate relationship between chromatin misregulation and cancer. In this review, we discuss recent advances that illustrate how a range of mechanisms lead to a misregulated ‘language’ of chromatin modifications, profoundly impacting on cancer initiation and progression. Due to the prevalence and impact of chromatin deregulation, a mechanistic understanding of this process in cancer will ideally lead to the development of promising therapeutics.
A concept of chromatin misregulation in cancers
As an extension to our previous review of the topic9, we here wish to further develop and expand the notion of a misregulated chromatin ‘language’ by first covering cancer-related alterations of DNA methylation and cell metabolism (FIG. 1b). Much evidence shows that various metabolic pathways generate metabolites such as acetyl-CoA, S-adenosylmethionine and lactate, which serve as cofactors for chromatin writer enzymes to deposit their respective chemical tags onto the chromatin10,11. In addition, metabolites such as α-ketoglutarate and NAD+ are essential cofactors for certain chromatin erasers10,11. As a hallmark of cancer, altered cell metabolism can therefore cause perturbations in the chromatin state leading to deregulation of gene expression10,12–14. Furthermore, it is increasingly clear that a versatile set of reader domains evolve as a ‘toolkit’ for cells to sense and recognize the previously less-studied chromatin PTMs such as lysine crotonylation (Kcr) or benzoylation15–17. Therefore, in addition to the fact that cancers frequently carry mutations of chromatin writers, readers, or erasers, an excess or lack of metabolites and/or their misuse can equally contribute to the mis-regulation of chromatin modification (FIG. 1b), thereby profoundly shaping cancer pathogenesis10,12–14.
An unexpected finding of studies based on data from The Cancer Genome Atlas is the identification of recurrent oncogenic somatic mutation of histones, also known as ‘oncohistones’18 (Box 1), across cancer types including glioma19, sarcoma20 and lymphoma21. Studies of the most common oncohistones have convincingly shown that histone mutations alter epigenomic patterning, affecting DNA-templated processes such as gene transcription and DNA damage repair. A common theme of classic H3 tail oncohistones is the functional inhibition of cognate histone writers to which oncohistones bind, leading to perturbation of epigenetic and transcriptomic states. H3 oncohistones have been reviewed in detail elsewhere18–20,22,23.
BOX 1. Oncohistones.
Histone missense mutations were recurrently detected in cancers and thus named oncohistones. Certain oncohistones including H3K27M, H3K36M and H3G34V/R/W/L mutations are common in pediatric cancers262. Recently, oncohistone sites have been expanded from histone tails to globular domains and linker histones18. For example, while the mutations of H2B (E71Q or E76Q) and H4 (D68Y/N/H and R92T) are predicted to disrupt the H2B-H4 binding interface, those of histone H2A (E56Q, E92D/K) and H2B (E113K/Q) occur in the ‘acidic patch’, a regulatory interface on the histone ‘disk’ surface essential for binding of chromatin regulators263. Approximately 60 nonsynonymous mutations of linker histone H1 were detected in human lymphomas264. Recently, two studies have shown that depletion of linker H1 histones in mice (which mimics their LOF mutations found in lymphoma patients) caused chromatin decompaction and an increase in H3K36me2 levels with concurrent decrease in H3K27me3 levels, ultimately leading to transcriptional activation of lymphoma-related oncogenes21,33 (Figure). Many oncohistones are tumor type-specific, influencing histone modification patterns globally. Although it only accounts for 3–17% of total H3, H3K27M has a dominant effect265.
The mechanisms underlying oncohistone-induced oncogenesis are complex. A simple model is that oncohistones influence histone methyltransferases in trans or in cis262. H3K9M, H3K27M and H3K36M were reported to inhibit their corresponding lysine methyltransferases in trans. This explains the globally decreased H3K27me3 or H3K36me3 seen in the H3K27M or H3K36M-mutant cells. Dominant effects of H3K27M and H3K36M also support the in trans inhibition model. Histone H3G34 mutations inhibit H3K36me2/3 methyltransferases in cis: only nucleosomes containing H3G34 mutations exhibit the decreased H3K36me2/3265. In addition, the H3.3G34R/V mutations found primarily in glioma also lead to globally elevated levels of H3K27me3, which impede neuronal differentiation; an aberrant chromatin looping brings a cis-regulatory element of GSX2 (hs687) in proximity to the promoter of PDGFRA, thereby facilitating the expression of mutant PDGFRA during gliomagenesis (Figure)34,266–268.
Effects of oncohistones are complex and content-dependent. For example, although global H3K27me3 loss is observed in H3K27M-mutant cells, a striking gain of H3K27me3 was identified at certain genomic loci269,270. Elevated H3K27ac and global DNA hypomethylation are observed in H3K27M-mutant tumors265,269,270, indicating a complex crosstalk among different chromatin modifications.
We hypothesize that ‘oncohistones’ at core and linker histones cause chromatin deregulation, which involves the perturbed functionality of writers, readers or erasers, de-compaction of chromatin and structural alteration of nucleosomes.
Mutations within chromatin-remodeling complexes represent another class of cancer-related lesions which are estimated to affect about 10–20% of all cancers24. Similar to oncohistones, deregulation of chromatin remodelers is known to alter the accessibility of chromatin, leading to chromatin ‘openness’ versus ‘compaction’, and may additionally crosstalk with other chromatin modifiers, as exemplified by antagonism between the Polycomb repressive complex 2 (PRC2) and the SWI/SNF chromatin remodeling complex25,26. Thus, recurrent oncohistone and chromatin remodeler mutations constitute an important mechanism by which tumors alter the normal process of chromatin-based gene and genome regulations, in order to gain growth advantages.
Emerging evidence from in vitro studies now demonstrates that phase separation, a phenomenon of molecule compartmentalization without sub-cellular membranes (Box 2), is critically involved in assembly of chromatin itself and chromatin-associated factors; this phase separation-based regulation is proposed to be vital not only for establishment of appropriate chromatin modification patterning but also three-dimensional (3D) organization of chromatin 27–31. In addition, chromatin loops can connect together the spatially distal enhancer and promoter thereby modulating transcription (FIG. 2a). It is conceivable that mis-regulations of chromatin looping and 3D organization represent vital oncogenic pathways leading to epigenomic and gene deregulation. In support of this, an oncogenic gain-of-function (GOF) mutation of enhancer of Zeste homologue 2 (EZH2), an H3K27me3 writer, was recently reported to induce structural changes in 3D chromatin structure and long-range chromatin interactions32. Furthermore, while loss-of-function (LOF) mutation of histone H1 was shown to alter the 3D chromatin architecture during lymphomagenesis21,33, the H3.3-G34R/V oncohistone establishes aberrant chromatin looping and a permissive chromatin environment for transcriptional activation of platelet derived growth factor receptor alpha (PDGFRA), an oncogene frequently mutated in glioma34. Collectively, these examples highlight the connection among histone modifications, oncohistones and chromatin high-order structures.
BOX 2. Chromatin phase separation.
Liquid-liquid phase separation (LLPS) compartmentalizes biochemical reactions without subcellular membranes. LLPS is driven by multivalent nonspecific ionic and hydrophobic interactions, and relies on the polymeric nature and intrinsically disordered regions of macromolecules30. Chromatin is composed of repeated nucleosomes with disordered histone tails. Recent studies demonstrate that the chromatin itself can form liquid droplets in vitro and in the nucleus30. Other transcription machineries such as RNA polymerase II, mediators such as MED1 and transcription factors also have intrinsically disordered regions and are regulated by phase separation271,272.
The phase separation behavior of chromatin is regulated by many factors such as histone H1, histone tails, linker DNA and nucleosome dynamics30. Histone modifications and reader proteins also directly regulate chromatin phase separation. Histone acetylation itself antagonizes chromatin LLPS30, while multi-bromodomain proteins form multivalent interactions with acetylated chromatin and facilitate LLPS of chromatin30. Transcriptional coactivators BRD4 and MED1 mediate LLPS at super-enhancer regions273. Heterochromatin regions also form nuclear puncta mediated by H3K9me3 and its reader, chromobox protein homolog 5 (CBX5; also known as HP1α)28,31. CBX5 binds H3K9me3 and recruits the H3K9me writer SUV39H1 and scaffold protein TRIM28274. This multi-subunit complex forms condensed macromolecule-enriched droplets and can exclude DNase or general transcription factors274.
In recent years, more and more epigenetic factors were reported to form separated phases and this property is directly related with human diseases. For example, mutations of ENL promote its phase separation behavior and activate oncogene expression in Wilms tumour168.
Figure 2. Miswriting of chromatin modification promotes oncogenic development.

a. Active enhancer is marked by H3K4me1 and H3K27ac, which are generated by mixed lineage leukemia 3 (MLL3) and MLL4, and histone acetyltransferases such as CREBBP and EP300. Enhancers are bound by transcription factors (TF), mediators and transcription coactivators such as BRD4, which activate RNA polymerase II (Pol II) for mediating productive transcription from promoters and generating enhancer RNA (eRNA) to facilitate gene activation. Enhancer-promoter looping underlies activation of gene transcription. Loss or inactivation mutation of CREBBP or EP300 and/or MLL3 or MLL4 is characteristic of cancers such as B-cell lymphoma, resulting in decreased H3K27ac and/or H3K4me1 at enhancers and reduced expression of genes related to tumor suppression, cell differentiation and/or antitumor immunity.
b. Wildtype MLL1 uses a N-terminal region for interacting with chromatin-binding cofactors, menin and PSIP1. MLL1 or its partial tandem duplication (PTD) results in elevated H3K4me3 at oncogenes such as HOX, promoting acute leukaemogenesis. MLL1 fusion oncoprotein gains a C-terminal segment from its fusion partner, such as AF9, ENL or AF4, which recruits DOT1L complex (DotCom) for catalyzing H3K79 methylation and/or the super elongation complex (SEC) for catalyzing serine 2 phosphorylation (Ser2ph) of the C-terminal domain (CTD) of RNA Polymerase II (Pol II). H3K79me and Pol-II CTD Ser2ph, possibly with other activators such as PAF1, promote expression of oncogenes such as those of the HOX family.
c. A collective action of wildtype EZH2 and its gain-of-function mutation, Y646X (X= F, C, H, S or N), causes abnormal elevation of H3K27me3 in lymphoma, leading to downregulation of transcripts related to cell cycle control and B cell differentiation.
d. Regulatory roles of H3K36me2/3 modifications at gene body, intergenic regions and CpG islands. First, intergenic H3K36me2, installed by NSD family proteins, and SETD2-mediated H3K36me3 at gene body both antagonizes H3K27me3. Meanwhile, H3K36me2/3 serves as a docking site of the DNMT3A/3B PWWP domain, resulting in co-localization of H3K36me2/3 with DNA 5mC at gene body and intergenic regions. Additionally, recognition of H3K36me3/2 by the Tudor domain of PHD finger protein 1 (PHF1) or PHF19 provides a possible mechanism for PRC2 complex to establish de novo H3K27me3. Deregulation of NSD family proteins, SETD2, PRC2, PHF1/19 and DNMT3A is frequent among various human tumors.
e. In breast cancer, overexpressed DOT1L interacts with MYC and EP300 to antagonize histone deacetylases (HDACs) and DNMT1, leading to the elevated H3K79me and H3Kac levels at epithelial-to-mesenchymal transition (EMT)-promoting oncogenes such as SNAIL and ZEB1.
Phenotypic and functional heterogeneity of human cancers is well documented and intratumoral epigenomic heterogeneity leads to considerable variation among tumor cell subpopulations35. In addition, epigenetic plasticity results in tumor cell adaption and resistance to therapeutic treatment, which represents a huge challenge in the clinic36,37. Indeed, mis-regulation of DNA and histone modifications can be as frequent as, if not more than, gene mutations, contributing to cancer heterogenicity36–38.
‘Miswriting’ of chromatin modifications
Recent studies have gained important insights, partly via cryogenic electron microscopy structures, into how multi-subunit writer machineries deposit their respective modification39,40. Although chromatin modification potentially affects numerous gene targets, a common theme is that miswritten chromatin modifications exert their oncogenic effects more heavily by deregulating one or a few sets of critical nodes, notably those genes that regulate stemness, cell cycle, and antitumor immunity, amongst others41. In addition to the more well-studied PTMs, several newly identified ones such as lysine lactylation (FIG. S1a) have emerged, providing new connections between metabolic malfunction and chromatin deregulation42.
Deregulation of histone acylations, beyond acetylation
Histone lysine acetylation (Kac) positively regulates gene transcription by promoting DNA accessibility and, importantly, creating a docking site for reader proteins. Histone acetylation can be catalyzed by three major families of histone acetyltransferases (HATs), namely the GCN5-related N-acetyltransferase (GNAT), EP300 and related CREB binding protein (CREBBP; also known as CBP), and MYST families. Because of a close relationship between histone acetylation and transcriptional activation, malfunction of HATs often perturbs appropriate gene-expression programs, leading to development of disease43–45.
Numerous reported cases exist in the literature illustrating how deregulation of histone acetylation writers contributes to oncogenesis. For example, the MYST family proteins, KAT8 (also known as MOF) and KAT7 (also known as HBO1), were recently demonstrated to be oncoproteins in the setting of acute leukaemias, with their in vivo leukemogenic functions dependent on the acetyl-‘writing’ activity harbored within HAT domain46,47. MacPherson et al showed that KAT7 and associated complex components are essential for inducing histone H3 lysine 14 acetylation (H3K14ac) in leukaemia-initiating stem cells (LSCs) and that H3K14ac facilitates the processivity of RNA Polymerase II to maintain the high expression of proto-oncogenes such as HOXA9 and HOXA10, an event that can be targeted by inhibitor of KAT747.
Another prominent example of pro-oncogenic deregulation by chromatin writers comes from inactivating mutations of CREBBP or EP300, which together account for approximately 60–70% of follicular lymphoma (FL) and 25–30% of diffuse large B cell lymphoma (DLBCL) cases48,49 and represent a common, early event during pathogenesis of these lymphomas (FIG. 2a). Aside from hematological cancers, mutations of HATs such as EP300 were also reported in patients with solid cancers such as glioma and melanoma44,45. Somatic mutations within EP300 or CREBBP are predominantly truncation and missense substitutions clustered within the HAT domain, which impair acetyl-‘writing’ onto chromatin. Studies in mouse models demonstrated that CREBBP deletion alone results in merely hyperplasia but is able to cooperate with deregulated apoptosis regulator BCL2 to cause a spectrum of fully penetrant tumor phenotypes that resemble human diseases50–52. These findings establish CREBBP as a bona fide tumor suppressor in lymphomagenesis49–53. Mechanistically, CREBBP mutation in germinal center (GC) B cells causes a range of epigenetic, transcriptomic and cell microenvironment perturbations. These include aberrant silencing of genes involved in cell differentiation, cell cycle control and immune response, mediated partly by an onco-repressor complex of HDAC3-BCL6-NCOR2 (also known as SMRT); the enhanced MYC node and immune evasion were both observed in these lymphoma animal models, contributing to tumorigenesis 50–55. In lymphoma, a large portion of perturbations that involve CREBBP or EP300 loss occur at gene enhancers resulting in unopposed histone deacetylation, and CREBBP mutation often coexists with inactivation of mixed lineage leukemia 4 (MLL4; also known as KTM2D) or MLL3 (also known as KTM2C) (FIG. 2a), another class of histone writers functioning at enhancer chromatin56,57. Together, this indicates synergy between the two lesions in causing epigenetic perturbation.
HATs may also cooperate with oncoproteins. For example, the histone acetyltransferases KAT2A (also known as GCN5) and KAT5 (also known as TIP60) both act as coactivators of MYC and are critically involved in MYC-driven transcriptional activation and oncogenesis8,58–62. Additionally, direct acetylation of the MYC protein by HATs can regulate its protein stability60.
Advances in high-sensitivity mass spectrometry permit identification of short-chain acylations of histone lysines, such as crotonylation and 2-hydroxyisobutyrylation15 (FIG. S1a). In cells, a combination of acetylation and acylations, rather than a single type, are often used by HATs to ‘mark’ target genes15. Non-acetyl histone acylations are generally correlated with gene activation but can be functionally distinctive from acetylation15. Moreover, histone lysine lactylation, generated from either exogenous or endogenous lactate, was identified as a new acylation that regulates the expression of homeostatic genes42. Cancer cells generate energy predominantly through anaerobic glycolysis followed by lactic acid fermentation, a metabolic alteration known as the Warburg effect63. It is therefore conceivable that miswriting of these histone lysine lactylation may link the Warburg effect with gene expression alterations in cancer, which merits further investigation.
MLL mutation and deregulation of H3K4
The MLL family of lysine methyltransferases (also known as KMT2 family) activate transcription partly via ‘writing’ H3K4 methylation at gene promoters and enhancers. The N-terminus of MLL1 (also known as KMT2A) physically interacts with cofactors required for genomic targeting such as menin and PC4 and SFRS1-interacting protein (PSIP1; also known as LEDGF)64,65, whereas the C-terminal SET domain of MLL1 associates with WDR5, RBBP5 and ASH2L to establish a highly ordered catalysis center to mediate histone methylation66,67 (FIG. 2b). Structures of MLL1 complexes and their interactors were recently solved (FIG. S1b–c), which revealed that binding of RBBP5 and ASH2L stabilizes an active conformation of MLL1 and that the subsequent MLL1 binding to histone H3 substrates induces a conformational change of the catalytic center, presenting H3K4 to the catalytic pocket68. The MLL1 complex engages mononucleosomes by interacting with both nucleosomal DNA and histone H4 (FIG. S1b)69,70. Overall, the activity and chromatin targeting of MLL1 methyltransferase is delicately regulated by its physical interaction and crosstalk with cofactors, as well as the chromatin context.
Rearrangement of the MLL1 gene in acute myelogenous leukemia (AML) or acute lymphoblastic leukaemia (ALL) provides a classic example of how miswriting of histone methylation causes oncogenesis71–73. As a common form of MLL1 rearrangement, MLL1 fusion essentially replaces the MLL1 C-terminal segment with one of the fusion partners, many of which recruit gene-activating machineries such as Super Elongation Complex (SEC) and/or the H3K79 methyltransferase, DOT1L. More than 100 MLL1 fusion partners have been reported to date, with the most frequent being AF4, ENL, AF9, AF10 and ELL71,72,74. Recruitment of DOT1L, SEC and other coactivators (such as wildtype MLL1 and PAF1) by the MLL1 fusion protein establishes a self-reinforcing gene-activation network, resulting in abnormal potent transcription of leukaemia-related oncogenes such as HOX family genes and MEIS171,72,75 (FIG. 2b). Partial tandem duplication of MLL1, the other type of MLL1 rearrangement occurring in approximately 5–10% of AMLs, generates in-frame partial duplication of an N-terminal regulatory region of MLL1, resulting in aberrant elevation of H3K4me3 and histone acetylation at oncogenes such as HOXA9 76,77. While MLL1 rearrangement acts as a GOF mutation to promote acute leukemias, its family members often function as tumor suppressors in other cancers73. Moreover, mutations of MLL family genes also exist widely among solid cancers such as lung cancer and breast cancer73,78.
LOF mutation of MLL3 (also known as KMT2C) and MLL4 (also known as KMT2D), the two main writers of enhancer-associated H3K4me179, is frequent in a range of cancers such as FL, DLBCL, lung cancer and melanoma57,78,80–82. In normal cells, MLL3 and MLL4 assemble complexes for establishing H3K4me1 at target enhancers or superenhancers, promoting transcription of genes related to differentiation or tumor suppression such as tumor necrosis factor alpha-induced protein 3 (TNFAIP3) and suppressor of cytokine signaling 3 (SOCS3)78,81,83. Besides core subunits (WDR5, ASH2L and RBBP5), MLL3 also interacts with BAP1, a histone H2A deubiquitinase and tumor suppressor that is required for recruiting MLL3 to enhancers84. Mutational hotspots have been reported to occur within the sequence encoding the Plant HomeoDomain (PHD) finger of MLL3 which mediates BAP1 interaction. Mutations in this region disrupted BAP1 binding, resulting in a defective recruitment of MLL3 to enhancers of tumor suppressor genes84. Additionally, the MLL3 complex interacts with KDM6A (also known as UTX), an H3K27me3-specifc demethylase frequently mutated in cancer57,85. Thus, MLL3 inactivation induces an increase in H3K27me3 at tumor suppressors84. Altogether, MLL3 and MLL4 mutations provide an example of enhancer-specific histone modification leading to enhancer malfunction and, subsequently, cancerous transformation.
Mutation of PRC2 and the miswritten pattern of H3K27me3
Trimethylation of H3K27 is catalyzed by PRC2, which is composed of EZH2, polycomb proteins SUZ12 and EED, and histone-binding proteins RBBP7 or RBBP4. Deregulation of EZH2, the catalytic subunit of PRC2, is intimately associated with cancer development86,87. Intriguingly, EZH2 has both oncogenic and tumor suppressive functions under different contexts, highlighting a multifaceted function86,87.
Approximately 20% of B cell lymphoma (FL and DLBCL) and 3% of melanoma patients harbor a heterozygous hotspot mutation of EZH2 within the catalytic domain residue Tyr646 (Y646X, where X is F, C, H, S, or N)88–90. In wildtype cells, activity of PRC2 is tightly regulated and inversely correlated to the degree of methylation at H3K27; the catalytic efficiency of PRC2 is greatest in converting H3K27me0 to H3K27me1, and dramatically diminished in subsequent methylation reactions86,89. However, lymphomas additionally produce a PRC2 that contains mutant EZH2-Y646X, which displays an altered substrate preference. PRC2 with EZH2-Y646X shows a greatly enhanced catalysis in converting H3K27me1 or H3K27me2 to higher methylation states86,89. As a result, the two forms of PRC2 carrying wildtype EZH2 or EZH2-Y646X act in concert to increase global H3K27me3 in cancer86,89(FIG. 2c). Two relatively rare EZH2 mutations, A677G and A687V found in 1–3% of lymphomas, have a similar GOF effect to EZH2-Y646X 86. In mouse models, EZH2-Y646F promotes pathogenesis of lymphoma91 or melanoma90 in the presence of a coexisting oncogene, such as an activating mutation in BCL2 or BRAF. In lymphoma, wildtype and mutant EZH2 together repress transcription of cell cycle inhibitors (such as CDKN2A and CDKN1A) and transcription factors required for post-GC B cell development (such as IRF4 and PRDM1)91–93.
EZH2 also controls genes related to tumor immunity. In particular, wildtype EZH2 and the induced H3K27me3 repress expression of CXC-chemokine ligand 9 (CXCL9) and CXCL10 within tumours94, as well as transcripts related to major histocompatibility complex class 1 antigen processing95, uncovering a role for EZH2 in tumour immune evasion. Additionally, EZH2-Y646F perturbs the interaction between GC B cells and immune cells (such as T cell help) in the animal model and helps establish a niche for promoting malignant transformation of GC B cells96. Thus, EZH2 regulates a battery of transcripts related to cell cycle control, cell differentiation, antitumor immunity and tumor cell microenvironment.
In addition to the oncogenic GOF mutations in EZH2 that are seen in lymphoma and melanoma, LOF mutations of PRC2 components such as EZH2, EED or SUZ12 are prevalent among many other cancers including myeloid neoplasms, T-cell ALL (T-ALL) and malignant peripheral nerve sheath tumors97–99. A tumor suppressive role of PRC2 was indeed substantiated by recent investigations of mouse knockout models in which abolishment of PRC2 function promoted progression of cancers such as leukemia and malignant peripheral nerve sheath tumour 100,101. Structural characterization of PRC2 provides important molecular explanations of how certain missense mutations detected in cancers influence PRC2 activity102–104. EZH2, EED and SUZ12 form a ‘core’ complex with wide-range interactions102,103 (FIG. S1d). It shall be noted that EED contains a ‘reader’ domain to bind H3K27me3 or trimethylation of JARID2 (a PRC2 cofactor), an event that mediates allosteric activation of PRC2 (FIG. S1d)103. Interestingly, a number of cancer-related mutations of PRC2 precisely target those residues involved in either the H3K27me3-‘reading’ domain of EED or the stimulatory responsive motif of EZH2, all of which interfere with allosteric activation of PRC2104,105. Together, an exquisite regulation of PRC2 and ‘writing’ of H3K27me3 are critically involved in appropriate control of gene expression and tumor-microenvironment interaction, two processes frequently deregulated during oncogenesis.
H3K36me2 and H3K36me3 miswriting underscores development of certain cancers
Deregulation of H3K36me2 and H3K36me3 is now appreciated to be pivotal in a wide range of cancer types106,107. Although they differ by just one methyl group, H3K36me2 and H3K36me3 have distinct genomic localizations, writers and biological functions106. While H3K36me3 is predominantly located within the body of actively transcribed genes, H3K36me2 is enriched in intergenic regions106. The histone-lysine N-methyltransferase SETD2 (also known as KMT3A) is the only identified enzyme catalyzing H3K36me3 whereas H3K36me2 can be generated by a diverse set of writers including the histone lysine methyltransferases NSD1, NSD2, NSD3 and ASH1L39. Under different contexts, H3K36 methyltransferases can be oncogenic or tumor suppressive. For example, while SETD2 acts as tumor suppressor in acute leukaemia, T-cell lymphoma, lung cancer and renal cancer106, gene rearrangement and/or GOF mutation of NSD2 occurs frequently in multiple myeloma and pediatric ALLs, resulting in globally elevated H3K36me2 levels and enhanced tumorigenicity in vitro and in vivo106–109. In a subset of AMLs, abnormal fusion between NUP98, a nucleoporin protein, and a C-terminal SET domain-containing segment of NSD1 or NSD3 is sufficient to drive AML development by activating proto-oncogenes such as those of the Hox gene family110. Furthermore, inactivating mutations in NSD1 were found at a frequency of 5–15% in several types of epithelial cell cancers106,111,112, suggesting a general relevance of H3K36 miswriting in oncogenesis.
Mechanistically, H3K36 di- or tri-methylation (H3K36me2/3) modulates various processes, such as mRNA splicing, DNA damage repair and gene transcription, and profoundly shapes epigenomic landscapes of cancer cells, notably H3K27me3 and DNA methylation106 (FIG. 2d). An antagonistic effect of H3K36me2/3 on PRC2 restricts deposition and/or spreading of H3K27me3113–117. Additionally, H3K36me2/3 is recognized by a Pro-Trp-Trp-Pro (PWWP) ‘reader’ module harbored within the DNA methyltransferases DNMT3A and DNMT3B, thereby regulating the chromatin localization of these methyltransferases and resultant de novo DNA methylation118–120. In support of this, cancers with NSD1 or NSD2 deregulation are characterised by DNA hypomethylation and/or a miswritten H3K27me3 pattern108,111,113,115,121. Furthermore, recent studies have illustrated the structural basis of NSD3 engagement with the nucleosomal substrate and also showed that overexpression and/or GOF mutation of NSD3 drives lung squamous cell carcinoma (LUSC) in both genetically engineered mouse models and patient derived xenograft (PDX) models 122,123. Specifically, NSD3 interacts extensively with nucleosomal DNA, histone H3 and histone H2A (FIG. S1e), which facilitates the active conformation of NSD3122. GOF mutation of NSD3 (such as T1232A) enhances its H3K36me2-writing activity, significantly contributing to development and progression of LUSC in vivo123. Mechanistically, NSD3 GOF mutation potentiates the oncogenic gene-expression program, notably MYC targets and mTOR signaling, in a catalytic activity-dependent manner123.
Deregulation of DOT1L and H3K79 methylation influences histone acetylation.
DOT1L is the only methyltransferase known to ‘write’ H3K79 methylation. In addition to its oncogenic role whereby it interacts with MLL fusion proteins, DOT1L also promotes oncogenesis by antagonizing histone deacetylation. In breast cancer, DOT1L overexpression is correlated to poorer prognosis, and a complex of DOT1L, MYC, CREBBP and EP300 activates genes related to epithelial-mesenchymal transition (EMT)124. Mechanistically, recruitment of DOT1L and resultant H3K79 methylation repel histone deacetylase 1 (HDAC1) and DNMT1 from EMT-regulatory genes, thereby promoting breast cancer progression124 (FIG. 2e). Likewise, DOT1L antagonizes the histone deacetylase sirtuin 1 (SIRT1) in MLL-rearranged leukaemia and HDAC1 in thymic lymphoma125,126. However, the molecular underpinnings for such an antagonism and acetylation state switch remain to be further determined.
Structural studies of DOT1L in complex with nucleosome (FIG. S1f) revealed how DOT1L activity is (de)regulated127–131. DOT1L binds nucleosome via multivalent interactions with the histone H4, nucleosome acidic patch, nucleosomal DNA and mono-ubiquitinated histone H2B lysine 120, which together stabilize active conformation of DOT1L and augment its enzymatic activity. DOT1L binding also induces a conformational change of histone H3, reorienting H3K79 from an inner position into the catalytic center130. A recent work further delineated a role for histone H4 lysine 16 acetylation (H4K16ac), a histone mark of decondensed, transcriptionally permissive chromatin, in direct stimulation of Dot1, the yeast homologue methyltransferase of DOT1L132. Structural studies also reveal how MLL fusion recruits DOT1L— the leucine zipper of AF10, a common MLL fusion partner, is complexed with a coiled-coil region of DOT1L, forming a parallel coiled-coil dimer through various hydrophobic interactions127 (FIG. S1g). Mutations at the AF10-DOT1L interaction interface abrogated leukaemogenicity of MLL1-AF10 in cell models127. MLL also forms oligomers with AF10 through coiled-coil interactions and potentiates leukemogenenesis133 (FIG. S1g). Thus, in addition to directly targeting DOT1L with available catalytic inhibitors, inhibiting DOT1L binding to nucleosome or cofactors may represent an alternative therapeutic strategy.
DNA methylation is frequently miswritten in cancer
DNA methyltransferases (DNMTs) establish 5mC, which modulates characteristics such as gene expression, mRNA splicing and genome stability4. A well-known characteristic of almost all tumors is global hypomethylation and concurrent abnormal hypermethylation at localized sites such as CpG islands 134. While the mechanisms underlying this deregulated 5mC pattern in cancer are likely complex and remain elusive, recent studies suggested an involvement of Ubiquitin Like With PHD And Ring Finger Domains 1 (UHRF1), a multifaceted E3 ligase that not only interacts with DNMT1, a writer of maintenance DNA methylation, but also ‘reads’ various chromatin modifications including hemi-methylated-CpG, H3K9me3, and H3K18 and H3K23 ubiquitination at replication forks135–138. In colon and liver cancers, UHRF1 overexpression is a predictor of poor clinical outcomes and, intriguingly, exerts dual effects on DNA methylation patterning136. Specifically, while UHRF1 overexpression in the zebrafish liver was sufficient to induce global DNA hypomethylation136, presumably via reduction of DNMT1 levels, UHRF1 also sustained aberrant promoter hypermethylation at tumor suppressor genes and promoted the in vivo tumorigenesis in the xenografted tumor models of liver and colon cancers 136,137. Interplay among UHRF1, DNMT1, replication fork machinery and chromatin are complicated, warranting further investigation136–139.
Another recently appreciated example that highlights a close relationship between 5mC miswriting and oncogenesis comes from DNMT3A, a de novo DNA methyltransferase frequently mutated in hematological malignancies such as AML and premalignant disorders such as clonal hematopoiesis140–144. Somatic mutations of DNMT3A in hematopoietic stem or progenitor cells represent an early premalignant event; mutations are clustered at DNMT3A functional domains including the catalytic domain and protein-protein interaction interfaces, with a noted mutational hotspot at Arg882. In hematopoietic stem or progenitor cells, loss of DNMT3A or introduction of a hotspot mutation (such as R882H in human or R878H in mouse DNMT3A) predominantly causes CpG hypomethylation at gene-regulatory elements, such as enhancers, leading to potentiation of gene-expression programs related to stemness145–153. Acquisition of additional oncogenic mutations, such as those of a proliferative kinase and/or nucleophosmin (NPM1), is required for progression into full-blown cancer145–153.
DNMT3A uses its homo-dimeric interface, a target recognition domain (TRD) and a catalytic loop for mediating multivalent interactions with CpG-containing DNA duplex154,155 (FIG. S1h, S1i). A set of hematological cancer-related DNMT3A mutations are spatially clustered at the substrate binding interface of DNMT3A as exemplified by V716D, R792H and K841E, which interfere with activity of wildtype DNMT3A in a dominant-negative fashion154. Intriguingly, structural analyses permit valuable insights into the mutational hotspot of DNMT3A— its R882 residue forms interactions with both DNA substrate and the TRD loop (FIG. S1i), a DNMT3A motif critically involved in engaging CpG dinucleotides154. A hotspot mutation such as R882H enhances dynamics of the TRD loop and compromises both the activity and CpG specificity of DNMT3A154,156,157; conceivably, non-CpG methylation by R882H-mutated DNMT3A can potentially be immediately eliminated during replication, since DNMT1, the enzyme responsible for maintenance DNA methylation, shows strict CpG specificity156. R882-mutated DNMT3A also displays altered preference for CpG-flanking sequences156,158,159. Together, this suggests that diseased hematological cells harboring the R882-mutated DNMT3A predominantly display a CpG hypomethylation phenotype and that the effect of DNMT3A hotspot mutation on CpG methylation is hypomorphic.
Chromatin modifications mis-interpreted in cancers
Specific histone modifications are recognized by specific readers. For example, bromodomains are classic histone acetylation readers and anticancer drug targets160. Following the discovery of bromodomains, double PHD finger domain and YEATS domains were also identified as histone acetylation readers161,162. In this section, we discuss recent advances which illustrate how improper interpretation of chromatin marks due to deregulation of chromatin readers represents a pivotal pathway leading to oncogenesis, indicative of attractive drug targets for therapeutics.
YEATS domain proteins recognize and bind histone acetylation and crotonylation
In humans, there are four YEATS domain-containing proteins, AF9, ENL, YEATS2 and GAS41 (also known as YEATS4). The YEATS domain of AF9 was initially identified as a histone acetylation reader, with a noted preference towards H3K9ac162; likewise, a YEATS domain harbored within ENL binds H3K27ac and H3K9ac163,164. Recognition of H3K27ac and H3K9ac by the YEATS modules of AF9 and ENL helps to recruit and/or stabilize transcriptional (co)activator machineries (including SEC, DOT1L, RNA Polymerase II and others) onto targets, mediating active transcription162–164 (FIG. 3a). In acute leukaemias, AF9 and ENL are frequently fused with MLL, generating an abnormal chimeric oncoprotein of MLL-AF9 and MLL-ENL respectively, which retains capability for SEC and DOT1L recruitment. Two independent studies demonstrated that ENL, instead of AF9, is required for tumorigenicity of MLL-rearranged AML in xenografted tumor models163,164. In AML, the H3ac-binding-defective mutation of the ENL YEATS domain largely phenocopied ENL depletion, impaired recruitment of SEC and RNA Polymerase II, and rendered AML sensitive to inhibitors of bromodomain-containing proteins and DOT1L. Combining inhibitors of YEATS domain, bromodomain and DOT1L thus provides a promising therapeutic strategy. In support of this, a peptide-based inhibitor selective for the YEATS domain of ENL was recently developed, which acts synergistically with bromodomain and DOT1L inhibitors in killing leukaemia cells165.
Figure 3. Misinterpretation of histone modification in cancer.

a. The YEATS domain of ENL recognizes acetylated lysine (Kac). ENL, which interacts with the mixed lineage leukemia 1 (MLL1) fusion oncoprotein, recruits the Super Elongation Complex (SEC) or DotCom into target oncogenes, maintaining a potently activated state in leukaemia cells. The ENL gain-of-function mutations facilitate self-aggregation and form phase separated puncta. The concentrated ENL mutant proteins recruit more SEC for activation of oncogenes in Wilms tumor.
b. The histone acetyltransferase Ada-two-A-containing (ATAC) complex contains a subunit YEATS2, recognizing H3K27ac and a subunit SAGA-associated factor 29 (SGF29), recognizing H3K4me3. The catalytic subunit consisting of histone acetyltransferase KAT2A results in elevated H3K9ac and activates expression of oncogenes in non-small cell lung cancer. The YEATS domain of GAS41 recognizes H3K27ac and H3K14ac at promoter regions. GAS41 is a subunit of chromatin remodeling complexes SRCAP and TIP60-EP400. The remodeling complex SRCAP substitutes histone H2A with histone variant H2A.Z and thus activates gene expression.
c. BAH and coiled-coil domain-containing protein 1 (BAHCC1) binds to H3K27me3-marked chromatin regions through a conserved BAH domain. BAHCC1 interacts with corepressors including histone deacetylases (HDACs) and SAP30 binding protein (SAP30BP) to silence tumor suppressor genes and lineage-specification transcription factors in acute leukaemias.
d. Histone modifications H3.3K36me3 and phosphorylated serine 31 on histone H3.3 (H3.3S31ph) influence chromatin localization of zinc finger MYND domain-containing protein 11 (ZMYND11). ZMYND11 specifically recognizes H3.3K36me3 at gene bodies and functions as a transcriptional corepressor by recruiting the NCOR2-HDAC3 complex. H3.3S31ph leads to the ‘ejection’ of ZMYND11 from its binding sites. A ZMYND11-MBTD1 fusion protein was identified in a subset of acute myeloid leukemia patients. The PWWP domain of ZMYND11 binds to H3K36me3 and the fusion partner (MBTD1) recruits the nucleosome acetyltransferase of H4 (NUA4)-TIP60 histone acetyltransferase complex. Elevated histone acetylation maintains the high expression of pro-leukemic genes in leukemia stem cells.
e. Under normal conditions, the tumor suppressor RACK7 recognizes the H3 tail carrying lysine acetylation and/or H3K4me1 and recruits the H3K4me3 erasers KDM5C and KDM5D onto enhancer regions. Loss of the RACK7-KDM5C/5D complex fails to demethylate H3K4me3 and results in overexpression of oncogenes (such as S100A) and metastasis-linked genes (such as SLUG and VEGFA) in breast and prostate tumor cells.
Recurrent mutations of the ENL YEATS domain also exist in Wilms tumours; these mutations are characterized by small in-frame amino acid insertions or deletions that are distant from the Kac-binding pocket of the YEATS domain166,167. While these YEATS-mutant ENL proteins bind acetylated histones with similar affinities and also occupy similar genomic loci when compared with wildtype ENL, they exhibit stronger self-association and the enhanced binding to chromatin targets168. Consistently, the intrinsically disordered regions harbored within ENL were recently shown to promote multivalent phase separation of SEC, allowing rapid, robust transcriptional activation168,169. Thus, a phase separation-like behavior seen with the ENL YEATS mutants most likely contributes to the increased recruitment and targeting of SEC and phosphorylated RNA Polymerase II onto target genes for enforcing oncogenic programs (FIG. 3a, BOX 2). Such a GOF form of ENL enforces aberrant activation of ENL targets such as the development-associated HOX family genes, thereby impairing cell-fate regulation during development of Wilms tumour168. Likewise, MLL-ENL oncoproteins also boosted phase separation-like properties of SEC, suggesting a role of abnormal SEC phase separation in leukaemogenesis169. These examples collectively illustrate how cancer-related proteins ‘interpret’ and engage specific histone modification by assembling downstream co-activator complexes at chromatin targets, at least partly via phase-separated protein condensates, to induce oncogenesis.
Two other YEATS-containing proteins, YEATS2 and GAS41, were also shown to be oncogenic170,171. YEATS2, a subunit of histone acetyltranferase Ada-two-A-containing (ATAC) complex, is amplified in non-small cell lung cancer (NSCLC). Recognition and binding of H3K27ac by the YEATS domain of YEATS2 facilitates subsequent ATAC-mediated deposition of H3K9ac, reinforcing target gene activation170 (FIG. 3b). Similarly, the YEATS domain contained within GAS41 binds H3K27ac and H3K14ac171. In NSCLC, GAS41 co-localizes with H3K27ac and H3K14ac, an event required for genome-wide deposition of histone variant H2A.Z at target promoters171 (FIG. 3b). Disrupting the YEATS reader-mediated interaction of YEATS2 or GAS41 with histone acetyl marks slowed NSCLC growth and suppressed transformation in the tumor xenograft models170,171.
Compared to the bromodomains, YEATS domain generally has a longer and flatter binding channel, which accommodates longer and bulkier non-acetyl acylation (FIG. S1j)16. In vitro binding assays indeed showed histone Kcr bound strongest to the YEATS domain16,172–174. Due to a π-electron conjugation, the crotonylamide group of Kcr is planar, forming a π-π-π stacking with aromatic residues of YEATS16,172–174 (FIG. S1j). AF9 also co-localizes with H3K18cr, activating gene expression in macrophages16. H3K18cr can be induced at the de novo activated gene-regulatory elements upon lipopolysaccharide (LPS) stimulation of macrophages175, and knocking down AF9 significantly reduced the LPS-induced gene-stimulation effect, which cannot be rescued by the YEATS mutant of AF916.
A new class of H3K27me3-specific reader: The BAH module
H3K27me3 regulates gene silencing and cell differentiation, misregulation of which is a frequent event in tumorigenesis176. Recently, Fan et al reported that an evolutionarily conserved Bromo-Adjacent Homology (BAH) module harbored within BAH and coiled-coil domain-containing protein 1 (BAHCC1), a chromatin regulator significantly overexpressed in acute leukemia patients, recognizes and binds H3K27me3 and enforces silencing of Polycomb target genes177 (FIG. S1k). BAHCC1 interacts with corepressors such as HDACs and SAP30 binding protein (SAP30BP), thus providing a molecular basis for Polycomb gene silencing177. In acute leukemias, depletion of BAHCC1, or disruption of its BAH-mediated ‘readout’ of H3K27me3, caused derepression of H3K27me3-demarcated genes that are involved in tumor suppression and cell differentiation, which in turn suppressed tumorigenesis in the xenografted leukemia models (FIG. 3c). This study thereby unveils a novel H3K27me3-directed transduction pathway in mammalian cells that relies on a conserved BAH reader, deregulation of which contributes to oncogenesis.
PWWP domains and ‘readout’ of H3K36me2/3
Evidence suggests PWWP as a multivalent reader of H3K36me2/3 and nucleosomal DNA178. Recognition of H3K36me2/3 by PWWP-containing proteins is crucial to a range of processes including DNA methylation, transcriptional activation and elongation, pre-mRNA processing and DNA damage repair106.
Notably, DNMT3A and related DNMT3B contain a PWWP domain involved in the binding of H3K36me2/3. In vitro and in vivo binding studies show that DNMT3B is recruited by H3K36me3, whereas DNMT3A is more preferentially recruited by H3K36me2118,120,179,180. PWWP is one of the recurrently mutated domains of DNMT3A in patients with clonal hematopoiesis, hematological cancers and paraganglioma142,180. Misinterpretation of H3K36me2/3 due to mutation of the DNMT3A PWWP domain was also recently reported to be responsible for microcephalic dwarfism181. Thus, H3K36me2/3 ‘misreading’ mechanisms frequently underlie deregulation of DNA methylation during oncogenesis and pathogenesis.
The tumor suppressor and transcriptional corepressor zinc finger MYND domain-containing protein 11 (ZMYND11; also known as BS69) harbors a PHD-bromo-PWWP combination cassette that functions as a histone variant H3.3-specific reader of H3K36me3182,183 (FIG. S1l). Low expression of ZMYND11 is associated with poor outcome in patients with triple-negative breast cancer. Via its PHD-bromo-PWWP module, ZMYND11 interacts with H3.3K36me3 located within gene body regions and suppresses the transition of paused RNA Polymerase II to transcriptional elongation, leading to downregulation of gene-expression programs related to tumor growth182; recently, Armache et al further showed that phosphorylation of H3.3 at Ser31 leads to ‘ejection’ of ZMYND11 from chromatin during rapid stimulation and activation of genes, indicating a dynamic nature of this pathway184 (FIG. 3d). Interestingly, the H3.3K36me3-‘reading’ module of ZMYND11 was previously reported to be involved in an aberrant gene fusion termed ZMYND11-MBTD1 among a subset of AML patients106,185. A recent study further demonstrated that the H3.3K36me3-‘reading’ activity of ZMYND11-MBTD1 is essential for recruiting and stabilizing this chimeric oncoprotein and associated HATs onto proto-oncogenes such as Hoxa9 and Meis1, which then maintains the high expression of oncogenes in LSCs and induces AML development in mouse models186 (FIG. 3d). Altogether, by engaging gene-activation-related H3K36me3, ZMYND11 impacts on tumorigenicity in various ways.
Recognition and engagement of H3K36me3 by PWWP domains is part of the DNA damage repair process. MSH6, a DNA mismatch repair protein frequently mutated in tumors such as colorectal and uterine cancer, contains a PWWP domain for binding H3K36me3187. Both H3K36me3 and the MSH6 PWWP domain were found to be essential for chromatin localization of heterodimeric MSH2-MSH6 complex187. In addition, interaction between PSIP1, another PWWP domain-containing protein, and H3K36me3 helps recruit the DNA endonuclease RBBP8 (also known as CtIP) to DNA damage sites188. Proper ‘interpretation’ of H3K36me3 by a set of damage repair-related PWWP domain proteins therefore provides an important safeguard mechanism for ensuring genome stability, a process frequently deregulated in cancer.
PHD finger domains recognise various histone tail PTMs
Different subclasses of PHD finger domains ‘sense’ the status of H3K4 PTMs, either highly methylated or unmethylated. Previously, the PHD finger of KDM5A (also known as JARID1A or RBP2) or PHF23 was found aberrantly fused to the nuclear pore complex protein NUP98 among a subset of AMLs189,190. The resultant NUP98-KDM5A and NUP98-PHF23 PHD finger chimeras were found to bind H3K4me3 and were associated with stemness and expression of leukemogenic genes such as HOX family genes and MEIS1, as well as the perturbed chromatin dynamics and cell differentiation189–191. Recent studies further showed that the NUP98-KDM5A fusion is a driver mutation present in ~10% of acute megakaryoblastic leukemias192,193 and a subset of such lethal pediatric cancers also harbor a similar chimera between NUP98 and the BPTF PHD finger194, suggesting commonality among these NUP98-PHD chimeric oncoproteins and a druggable PHD finger target190,191.
More broadly, ‘misreading’ of H3K4me3 by PHD finger domain proteins such as inhibitor of growth (ING) proteins 1–5, PHF23, pygopus (PYGO) and PHF20, have been shown to exert either oncogenic or tumor suppressive effects via improper modulation of target gene expression41,195. Recent studies of the tumor suppressor protein kinase C-binding protein 1 (also known as RACK7 or ZMYND8) showed that its PHD finger forms a tandem cassette together with adjacent bromo and PWWP domains for recognition of a dually modified histone H3 tail, consisting of H3K4me1K14ac196,197. In the human breast and prostate cancer models, RACK7 binds gene enhancers and suppresses transcription of oncogenes (such as S100A family genes) and metastasis-linked genes (such as SLUG and VEGFA) via recruitment of the H3K4me3 erasers, KDM5C and KDM5D 196,197 (FIG. 3e). Loss of RACK7 results in abnormally elevated H3K4me3 at enhancers, leading to onco-target overactivation in tumor cells and enhanced oncogenesis in the tumor xenograft models196,197. Also, H3.3G34R, an oncohistone detected in pediatric glioblastoma (BOX 1), can create a docking site for RACK7, which then acts to suppress genes related to major histocompatibility complex, a known mediator of anti-tumor immunity198. Thus, via various misreading mechanisms, RACK7 contributes to tumorigenesis among a wide range of cancers.
The Double PHD finger (DPF) domain harbored within DPF3, a BAF chromatin-remodeling complex subunit, and MOZ (also known as KAT6A) or MORF (also known as KAT6B), two closely related HATs involved in AML tumorigenicity, also emerges as a combinational reader for both N-terminus and acetylation of histone H3161,199,200. Additionally, the DPF domain of MOZ or DPF2, a DPF3-related BAF subunit, can bind various histone acylations, such as crotonylation201. Engaging of histone ligands by the DPF module is critical for development because DPF2 mutations that abolish histone binding were found responsible for a neurodevelopmental disorder, Coffin-Siris Syndrome202, as well as the affected myeloid differentiation of hematopoietic stem or progenitor cells203. Overall, the bromo, DPF and YEATS domains provide a ‘toolkit’ for cells to recognize the subtlety and unique features of various histone acylations15–17,161,162. The detailed interplays of histone PTM readers with the epigenomic, transcriptomic and metabolic alterations during oncogenesis warrant further investigation.
Chromatin modifications mis-erased in cancers
HDACs, the eraser of Kac, have long been implicated in oncogenesis, via either histone or nonhistone substrate regulation204. Recently, Vlaming et al reported that inactivation of HDAC1 in thymocytes results in enhanced DOT1L activity and lymphomagenesis in mice126. Like the more labile Kac, histone Kme and DNA 5mC modifications can also be reversible, a process executed by enzymes or enzyme complexes that collectively have been referred to as ‘erasers’ (FIG. 1a). In particular, Jumonji C (Jmj-C) domain-containing proteins form a large family of KDMs205 and DNA 5mC is ‘erased’ by TET family proteins through successive oxidizations3. Both Jmj-C domain proteins and TET family proteins are Fe (II) and 2-oxoglutarate (2-OG) dependent enzymes. Cellular metabolites and hypoxic conditions influence the Fe (II) and 2-OG level, thereby affecting the enzymatic activities of Jmj-C domain-containing and TET proteins206.
Deregulation of Jumonji C domain causes mis-erasing of histone methylation
While non-Jmj-C proteins KDM1A (also known as LSD1) and KDM1B (also known as LSD2) are FAD-dependent monoamine oxidases catalyzing demethylation of histone H3K4me1 and H3K4me2207, a larger family of Jmj-C domain proteins are Fe (II) and 2-OG dependent hydroxylases that catalyze demethylation of various histone sites and of all methylation states (FIG. S1m)205.
Malfunction of histone lysine demethylases (KDMs) is a common theme among cancers205. Depending on cancer types and contexts, Jmj-C domain proteins have both oncogenic and tumor suppressive functions. For example, while the KDM5 family proteins act as tumor suppressors in MLL-rearranged AML and clear cell renal cell carcinoma by transcriptionally repressing oncogenes208,209, KDM5 is generally regarded as oncogene in other tumors such as breast cancer and melanoma, in which KDM5A is frequently overexpressed210–213. For example, KDM5B suppresses a stimulator of interferon genes (STING)-related pathway of antitumor immunity210 and can additionally promote drug resistance in multiple cancer types such as melanoma, breast tumor and lung cancer211–213. These oncogenic effects of KDM5B can be reversed by inhibitors that target its demethylating function, demonstrating an attractive means of targeting ‘mis-erased’ H3K4me3 or H3K4me2 PTMs which are required for tumorigenicity212.
Different erasers can mediate opposing effects on tumour growth, even when acting on the same histone mark. For instance, both KDM6A and KDM6B (also known as JMJD3) are H3K27me3 and H3K27me2 erasers. In murine models of T-ALL, KDM6B was reported to activate the NOTCH1-related axis by eliminating oncogene-associated H3K27me3, thus promoting T-ALL oncogenesis; meanwhile, KDM6A activated tumor suppressor genes thereby inhibiting T-ALL development214 (FIG. 4a). Furthermore, LOF mutations in KDM6A are frequently present in various solid cancers, further supporting a tumor suppressive role85.
Figure 4. Mis-erasing of chromatin modification is critically involved in cancer initiation and progression.
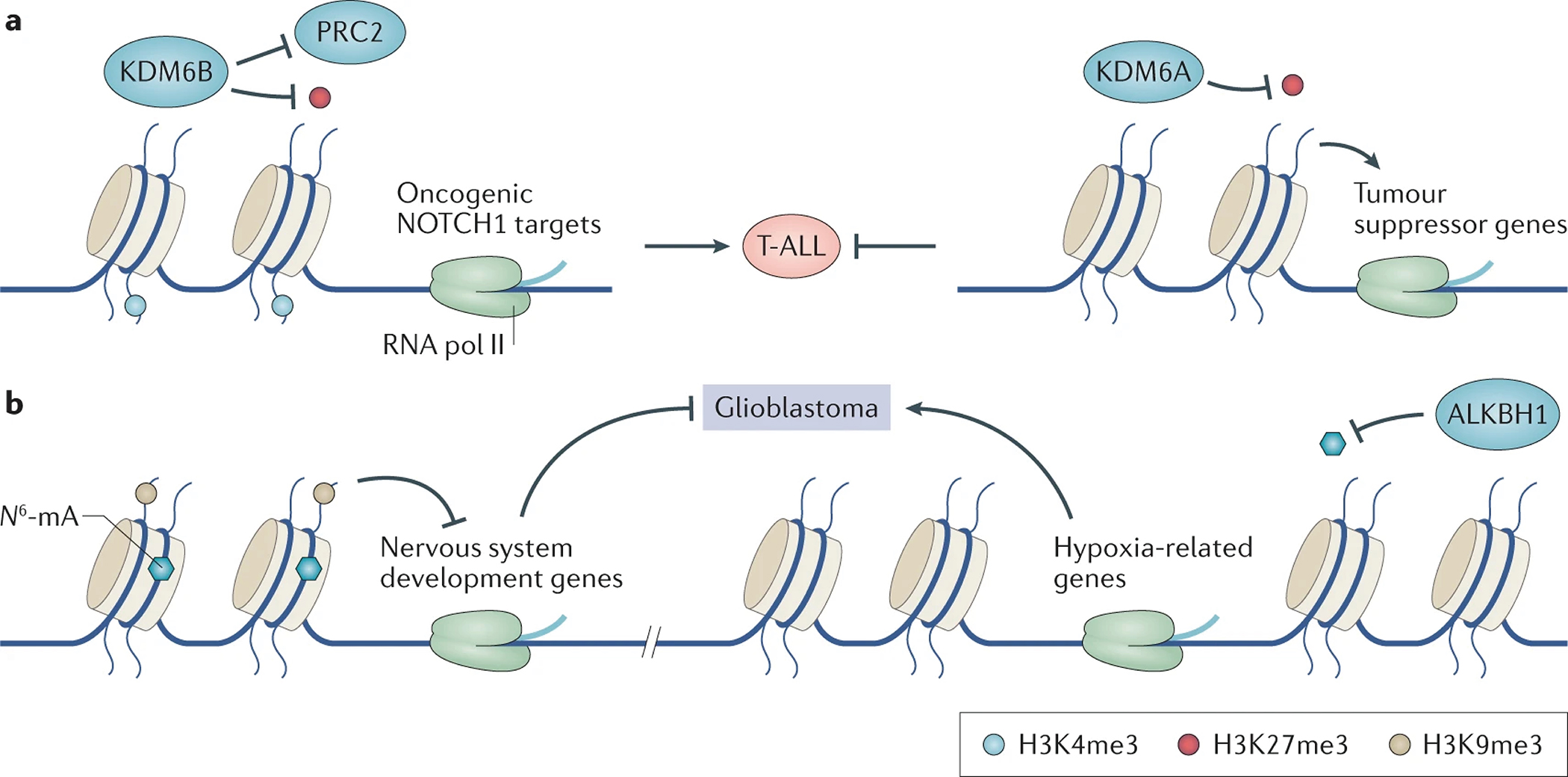
a. In T-ALL, the expression level of KDM6B increases while the (KDM6A level decreases. Both KDM6A and KDM6B are erasers of H3K27me3. KDM6B binds to oncogenic NOTCH1 target genes, catalyzes the demethylation of H3K27me3 and antagonizes the polycomb repressor complex 2 (PRC2), an H3K27me3 writer. Decreased H3K27me3 and increased H3K4me3 facilitate the expression of oncogenic genes. In contrast, KDM6A binds to tumor suppressor genes and facilitates their expression. KDM6A thus functions as tumor suppressor in T-ALL.
b. In glioblastoma stem cells (GSC), DNA adenine methylation (N6-mA) co-exists with H3K9me3, suppressing the neuronal differentiation-related gene-expression program. Depletion of nucleic acid dioxygenase ALKBH1 in glioblastoma facilitates silencing of oncogenic genes and thus decreases GSC proliferation.
The eraser activity of the Jmj-C domain requires 2-OG, which can be competitively inhibited by its analog 2-hydroxyglutarate (2-HG). Normally, 2-OG is generated from glutamine in cells. However, the core region of solid tumors features glutamine deficiency and a decreased 2-OG level, which leads to elevated H3K9me3 and H3K27me3215,216. Low glutamine-induced histone hypermethylation, notably H3K27me3, induced cancer cell dedifferentiation and drug resistance in xenografted melanoma models, impacting on both tumour heterogeneity and therapeutic response215. Moreover, the isocitrate dehydrogenase (IDH) metabolic enzymes convert isocitrate into 2-OG217. IDH1 is cytosolic while IDH2 is a mitochondrial protein217. IDH1 and IDH2 mutations are somatic, predominantly heterozygous and mutually exclusive218. Tumours carrying a GOF mutation of IDH1 or IDH2 produce an astonishingly high level of D-2-HG, a so-called oncometabolite that competes with 2-OG219,220. 2-HG is a chiral metabolite that has D-(R)-enantiomer and L-(S)-enantiomeric conformations. IDH mutants specifically generate D-2-HG while L-2-HG can be generated under hypoxic conditions221. Both D-2-HG and L-2-HG inhibit ‘erasing’ function of Jmj-C domain proteins and impair histone demethylation220–223.
TET inactivation causes mis-erasing of DNA cytosine methylation
5mC in DNA (FIG. S1a) is erased through successive oxidizations by TET1, TET2 or TET3 224–226. Malfunctions of TET family proteins are responsible for a wide range of hematological cancers and certain solid tumours227. TET2 mutations, as detected in myeloid cancers, are both truncation mutations and missense substitutions centered within the region encoding the catalytic domain, all of which impair the 5mC ‘erasing’ activity of TET2228 (FIG. S1n).
TET family proteins are regulated by various proteins and metabolites. For example, a WNT pathway protein, CXXC-type zinc finger protein 4 (CXXC4, also known as IDAX), downregulates TET2 by caspase activation229. Like Jmj-C domain proteins, activity of TET family proteins also relies on 2-OG as a cofactor and hence they are sensitive to levels of the oncometabolite 2-HG220,230. IDH1, IDH2 and TET2 mutations in cancers are mutually exclusive230. IDH1 and IDH2 mutations inhibit TET2 function and result in DNA hypermethylation in cancers230. Molecular oxygen, ascorbic acid and reduced Fe (II) are additional cofactors of TET family proteins. Consequently, tumor-associated hypoxia can reduce TET activity, causing DNA hypermethylation at promoters of tumor suppressor genes231. On the other hand, Vitamin C, an antioxidant regulating Fe (II) reduction, potentiates TET-mediated demethylation. It has been shown that Vitamin C levels can influence leukemogenesis by regulating TET family proteins232–234. Since haploinsufficiency of TET family proteins is a frequent event in hematological cancer, treatment with high-dose Vitamin C may restore TET2 deficiency and represents a possible therapeutic strategy, a topic under active investigation234.
Altogether, deregulation of TET family proteins, the m5C erasers, exerts a plethora of context-dependent effects on cancer evolution and progression, stem cell aging and clonal hematopoiesis, immunity, and cellular responses to environmental cues.
ALKBH family demethylases ‘erase’ DNA adenine methylation
The abundance of DNA adenine methylation (N6-mA) (FIG. S1a) in normal mammalian cells is very low, typically below 20 parts per million235–237. However, a study reported that the N6-mA level in glioblastoma stem cells (GSCs) from cancer patient samples can be elevated by more than 100-fold, compared to normal astrocytes; this correlated with levels of H3K9me3 and functioned as a repressive mark238. Increased levels of N6-mA were also observed in other central nervous system cancers, such as diffuse intrinsic pontine glioma, meningioma and medulloblastoma238.
The Fe (II)- and 2-OG-dependent nucleic acid dioxygenase ALKBH1 is an N6-mA eraser235,238,239. In the PDX models of glioblastoma, knockdown of ALKBH1 increased the level of N6-mA and induced transcriptional silencing of hypoxia-related oncogenes through decreased chromatin accessibility, thereby inhibiting glioblastoma growth238 (FIG. 4b). Under hypoxic stress, levels of N6-mA in mitochondrial DNA are also significantly elevated, indicating its relationship with cell stress240. ALKBH1 favors a “bubble” DNA structure instead of B-form duplex241,242 (FIG. S1o), consistent with the discovery that N6-mA is more enriched at regions with stress-induced DNA double helix destabilization and/or R-loop243–245. Other potential regulators of DNA N6-mA modification were recently proposed, such as the methyltransferase METTL4 (on mitochondrial DNA240) and the METTL3-METTL14 complex (on single-strand and unpaired DNA246), the DNA-binding protein SATB1 (binding of which is repelled by N6-mA243), and YTH domain-containing protein 1 (YTHDC1, which binds to N6-mA on single-strand DNA247). These molecular players are likely to regulate DNA and/or RNA, in a context-dependent manner. However, additional verification and functional characterizations are warranted237,248. In particular, a reliable high-resolution sequencing method needs to be developed for evaluating the function of N6-mA modification in cancer.
Future directions
Chromatin writers, readers and erasers are promising targets for pharmacological manipulation. The first-generation epigenetic drugs targeting DNMTs and HDACs have been approved for clinical use, for example in treatment of T cell lymphoma and multiple myeloma. The next-generation small-molecules targeting various chromatin modulators are being developed with improved selectivity and potency, some of which are currently under clinical evalution249. Chromatin modulator targets that proved to be ‘druggable’ include writers (such as EZH2, DOT1L and EP300), readers (such as bromodomain-containing proteins) and erasers (such as KDM5 and HDACs). Aside from those against epigenetic modulators per se, inhibitors of cancer-related metabolic enzymes, such as GOF IDH1 or IDH2 mutant proteins, show promising results as well, reversing their pro-tumour epigenetic effects249. For detailed progress in this area, we recommend recent reviews134,249.
Chromatin modulators are also suitable for new drug development strategies such as heterobifunctional PROteolysis TArgeting Chimeras (PROTACs)250, which have several advantages. First, instead of targeting one aspect of multifunctional chromatin modulators, PROTACs bring about target protein degradation, thus temporally eliminating all functions of targets to exert strong antitumor effects. Second, PROTACs can be more potent; due to their catalytic mechanism of action, PROTACs can be reused for repeated cycles of target depletion, potentially reducing the need for high residence time and continuous drug exposure relative to small-molecule inhibitors that typically rely on receptor occupancy pharmacology. Thirdly, PROTACs can potentially improve drug specificity by introducing more interactions between the PROTAC, the target protein and the E3-ligase ternary complex251. PROTACs may therefore have the potential to improve specificity and efficacy of the existing inhibitors available for targeting chromatin regulators, may effectively suppress target chromatin-regulatory oncoproteins that are often multifunctional in cancer, and may address drug resistance252. In support of this, PROTACs targeting BRD4253, HDACs254 and EZH2255 are being developed and show promising effects in vitro and in vivo.
Lastly, epigenetic variations contribute to tumor heterogeneity36–38. With recent advances in single cell-based chromatin modification profiling technologies256,257, we shall anticipate more comprehensive mapping of cancer epigenomes, at both spatial and temporal levels (4D) during the dynamic course of cancer evolution36. Tumor cell adaptation and therapeutic resistance frequently occur; therefore, future studies should explore strategies to address this, such as epigenetic synthetic lethality and combination therapy, for (epi)targeting of human cancers. For example, treatment with a bromodomain inhibitor leads to accumulation of DNA damage in tumors and thus a high demand for DNA damage repair machinery, which renders tumors hypersensitive to PARP inhibitors258,259. Likewise, combining epigenetic therapies with immunotherapies represents another attractive means for enhancing treatment success260. Further dissection of potential crosstalk between epigenetic players and other cancer-related pathways shall continue to impact on cancer therapeutics and medicine.
Conclusions
The molecular understanding of chromatin deregulation, one of the central mechanisms underlying oncogenesis, together with development of potent and specific pharmacological agents, will aid in development of effective treatments for human cancers. We look forward to further advances along these lines in years to come.
Supplementary Material
Acknowledgements
This work was supported by NIH grants (R01-CA215284 and R01-CA218600 to G.G.W.). G.G.W. is an American Cancer Society Research Scholar and a Leukemia and Lymphoma Society Scholar. The authors apologize to colleagues whose works are not cited in this Review due to space limitations.
Glossary:
- Polycomb repressive complex 2 (PRC2)
The PRC2 complex, consisting of core subunits EZH2 or EZH1, EED, SUZ12 and RbAp46 or RbAp48, catalyzes the methylation of histone H3K27
- SWI/SNF chromatin remodeling complex
The SWI/SNF chromatin remodeling complex, comprising approximately 15 subunits, utilizes the energy from ATP hydrolysis to mobilize nucleosomes
- Germinal center (GC) B cells
B cells residing in the GC sites of secondary lymphoid organs such as spleen and lymph nodes where B cells proliferate, differentiate, and mutate the antibody-encoding genes (through somatic hypermutation) to generate antibodies of higher affinity during the immune response
- CpG islands
CpG islands (CGIs) are genomic regions (typically 300–3000 bps) that contain highly enriched CpG dinucleotides and are usually lack of DNA methylation
- Hypomorphic
Hypomorphic mutation causes a partial loss of gene function (such as reduced enzymatic activity
- intrinsically disordered regions
Intrinsically disordered regions (IDRs), which are hotspot regions mediating phase separation, are flexible linkers/loops within a protein and form no secondary structure
- π-π-π stacking
π-π-π stacking is the noncovalent interaction between aromatic rings
- Dioxygenase
Dioxygenases refer to the oxidoreductase enzymes that incorporate both atoms of O2 into the substrate
- Oncohistone
Onco’-histone is a term used to describe the cancer-associated recurrent mutations of histones that can dominantly drive the malignant development
- acidic patch
The acidic patch is a negatively charged region in nucleosome formed by six residues from H2A and H2B
- phase separation
Phase separation is a physical phenomenon created by forming two distinct phases from one homogeneous mixture
- chromatin loop
Stretches of chromosome regions can be brought together in close physical proximity, forming a structure called chromatin loop
- Acylation
Acylation is a chemical process of transferring an acyl group to the substrate. It includes but not limited to acetylation
- super-enhancers
Super-enhancers are chromatin regions that comprise multiple enhancers and enriched in transcription factors and mediators
- maintenance DNA methylation vs de novo DNA methylation
Maintenance DNA methylation is created based on the existing template DNA methylation while de novo DNA methylation is created at previously unmethylated sites
- clonal hematopoiesis
Clonal hematopoiesis is a phenomenon of the expansion of a clonal blood cell population with the same genetic mutation
- premalignant disorder
Premalignant disorder is a condition that abnormal cells have a high risk of developing into cancer
- Oncometabolite
Oncometabolite is the metabolite whose quantity is significantly elevated in tumors
- R-loop
The R-loop is a three-stranded nucleic acid structure formed by one DNA-RNA duplex and one associated non-template single strand DNA
Footnotes
Competing interests
The authors declare no competing interests
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s415XX-XXX-XXXX-X
References
- 1.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Jenuwein T & Allis CD Translating the histone code. Science (New York, N.Y 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Wu X & Zhang Y TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18, 517–534, doi: 10.1038/nrg.2017.33 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Ren R, Horton JR, Zhang X, Blumenthal RM & Cheng X Detecting and interpreting DNA methylation marks. Curr Opin Struct Biol 53, 88–99, doi: 10.1016/j.sbi.2018.06.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson SM & Gozani O Nonhistone Lysine Methylation in the Regulation of Cancer Pathways. Cold Spring Harb Perspect Med 6, doi: 10.1101/cshperspect.a026435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biggar KK & Li SS Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol 16, 5–17, doi: 10.1038/nrm3915 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Narita T, Weinert BT & Choudhary C Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 20, 156–174, doi: 10.1038/s41580-018-0081-3 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Farria A, Li W & Dent SY KATs in cancer: functions and therapies. Oncogene 34, 4901–4913, doi: 10.1038/onc.2014.453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chi P, Allis CD & Wang GG Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10, 457–469, doi: 10.1038/nrc2876 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai Z, Ramesh V & Locasale JW The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet, doi: 10.1038/s41576-020-0270-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J & Thompson CB Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20, 436–450, doi: 10.1038/s41580-019-0123-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gut P & Verdin E The nexus of chromatin regulation and intermediary metabolism. Nature 502, 489–498, doi: 10.1038/nature12752 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Reid MA, Dai Z & Locasale JW The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 19, 1298–1306, doi: 10.1038/ncb3629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suganuma T & Workman JL Chromatin and Metabolism. Annu Rev Biochem 87, 27–49, doi: 10.1146/annurev-biochem-062917-012634 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Sabari BR, Zhang D, Allis CD & Zhao YM Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Bio 18, 90–101, doi: 10.1038/nrm.2016.140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Molecular cell 62, 181–193, doi: 10.1016/j.molcel.2016.03.028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren X et al. Histone benzoylation serves as an epigenetic mark for DPF and YEATS family proteins. Nucleic Acids Res 49, 114–126, doi: 10.1093/nar/gkaa1130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nacev BA et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–478, doi: 10.1038/s41586-019-1038-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips RE, Soshnev AA & Allis CD Epigenomic Reprogramming as a Driver of Malignant Glioma. Cancer cell 38, 647–660, doi: 10.1016/j.ccell.2020.08.008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nacev BA et al. The epigenomics of sarcoma. Nat Rev Cancer 20, 608–623, doi: 10.1038/s41568-020-0288-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yusufova N et al. Histone H1 loss drives lymphoma by disrupting 3D chro0matin architecture. Nature 589, 299–305, doi: 10.1038/s41586-020-3017-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flaus A, Downs JA & Owen-Hughes T Histone isoforms and the oncohistone code. Current opinion in genetics & development 67, 61–66, doi: 10.1016/j.gde.2020.11.003 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Ghiraldini FG, Filipescu D & Bernstein E Solid tumours hijack the histone variant network. Nat Rev Cancer, doi: 10.1038/s41568-020-00330-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kadoch C & Crabtree GR Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1, e1500447, doi: 10.1126/sciadv.1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.St Pierre R & Kadoch C Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev 42, 56–67, doi: 10.1016/j.gde.2017.02.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodges C, Kirkland JG & Crabtree GR The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med 6, doi: 10.1101/cshperspect.a026930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nair SJ et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nature structural & molecular biology 26, 193–203, doi: 10.1038/s41594-019-0190-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strom AR et al. Phase separation drives heterochromatin domain formation. Nature 547, 241–245, doi: 10.1038/nature22989 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin Y et al. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 175, 1481–1491.e1413, doi: 10.1016/j.cell.2018.10.057 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson BA et al. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421, doi: 10.1016/j.cell.2019.08.037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson AG et al. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240, doi: 10.1038/nature22822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donaldson-Collier MC et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nature genetics 51, 517–528, doi: 10.1038/s41588-018-0338-y (2019). [DOI] [PubMed] [Google Scholar]
- 33.Willcockson MA et al. H1 histones control the epigenetic landscape by local chromatin compaction. Nature 589, 293–298, doi: 10.1038/s41586-020-3032-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen CCL et al. Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 183, 1617–1633 e1622, doi: 10.1016/j.cell.2020.11.012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazor T, Pankov A, Song JS & Costello JF Intratumoral Heterogeneity of the Epigenome. Cancer Cell 29, 440–451, doi: 10.1016/j.ccell.2016.03.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marine JC, Dawson SJ & Dawson MA Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer 20, 743–756, doi: 10.1038/s41568-020-00302-4 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Flavahan WA, Gaskell E & Bernstein BE Epigenetic plasticity and the hallmarks of cancer. Science 357, doi: 10.1126/science.aal2380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Easwaran H, Tsai HC & Baylin SB Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 54, 716–727, doi: 10.1016/j.molcel.2014.05.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husmann D & Gozani O Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol 26, 880–889, doi: 10.1038/s41594-019-0298-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jang S & Song JJ The big picture of chromatin biology by cryo-EM. Curr Opin Struct Biol 58, 76–87, doi: 10.1016/j.sbi.2019.05.017 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Chi P, Allis CD & Wang GG Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10, 457–469, doi: 10.1038/nrc2876 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580, doi: 10.1038/s41586-019-1678-1 (2019). This paper reported a new histone acylation type and its relationship with metabolism.
- 43.Attar N & Kurdistani SK Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb Perspect Med 7, doi: 10.1101/cshperspect.a026534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Martile M, Del Bufalo D & Trisciuoglio D The multifaceted role of lysine acetylation in cancer: prognostic biomarker and therapeutic target. Oncotarget 7, 55789–55810, doi: 10.18632/oncotarget.10048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheikh BN & Akhtar A The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat Rev Genet 20, 7–23, doi: 10.1038/s41576-018-0072-4 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Valerio DG et al. Histone Acetyltransferase Activity of MOF Is Required for MLL-AF9 Leukemogenesis. Cancer Res 77, 1753–1762, doi: 10.1158/0008-5472.CAN-16-2374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacPherson L et al. HBO1 is required for the maintenance of leukaemia stem cells. Nature 577, 266–270, doi: 10.1038/s41586-019-1835-6 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Pasqualucci L et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195, doi: 10.1038/nature09730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer SN et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity 51, 535–547 e539, doi: 10.1016/j.immuni.2019.08.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov 7, 322–337, doi: 10.1158/2159-8290.CD-16-1417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Y et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov 7, 38–53, doi: 10.1158/2159-8290.CD-16-0975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Ramirez I et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood 129, 2645–2656, doi: 10.1182/blood-2016-08-733469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashwah H et al. Inactivation of CREBBP expands the germinal center B cell compartment, down-regulates MHCII expression and promotes DLBCL growth. Proc Natl Acad Sci U S A 114, 9701–9706, doi: 10.1073/pnas.1619555114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mondello P et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov 10, 440–459, doi: 10.1158/2159-8290.CD-19-0116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mlynarczyk C, Fontan L & Melnick A Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev 288, 214–239, doi: 10.1111/imr.12755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bakhshi TJ & Georgel PT Genetic and epigenetic determinants of diffuse large B-cell lymphoma. Blood Cancer J 10, 123, doi: 10.1038/s41408-020-00389-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sze CC & Shilatifard A MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb Perspect Med 6, doi: 10.1101/cshperspect.a026427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flinn EM et al. Recruitment of Gcn5-containing complexes during c-Myc-dependent gene activation. Structure and function aspects. J Biol Chem 277, 23399–23406, doi: 10.1074/jbc.M201704200 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Liu X, Tesfai J, Evrard YA, Dent SY & Martinez E c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J Biol Chem 278, 20405–20412, doi: 10.1074/jbc.M211795200 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel JH et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol 24, 10826–10834, doi: 10.1128/MCB.24.24.10826-10834.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer N & Penn LZ Reflecting on 25 years with MYC. Nat Rev Cancer 8, 976–990, doi: 10.1038/nrc2231 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Farria AT et al. Transcriptional Activation of MYC-Induced Genes by GCN5 Promotes B-cell Lymphomagenesis. Cancer research 80, 5543–5553, doi: 10.1158/0008-5472.CAN-20-2379 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cairns RA, Harris IS & Mak TW Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95, doi: 10.1038/nrc2981 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Yokoyama A & Cleary ML Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer cell 14, 36–46 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang J et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 482, 542–546, doi: 10.1038/nature10806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dou Y et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121, 873–885, doi: 10.1016/j.cell.2005.04.031 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Dou Y et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nature structural & molecular biology 13, 713–719 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Li Y et al. Structural basis for activity regulation of MLL family methyltransferases. Nature 530, 447–452, doi: 10.1038/nature16952 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xue H et al. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature 573, 445–449, doi: 10.1038/s41586-019-1528-1 (2019). This study revealed the structural basis of MLL binding to nucleosome.
- 70. Park SH et al. Cryo-EM structure of the human MLL1 core complex bound to the nucleosome. Nat Commun 10, 5540, doi: 10.1038/s41467-019-13550-2 (2019). This study revealed the structural basis of MLL binding to nucleosome.
- 71.Krivtsov AV & Armstrong SA MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7, 823–833, doi: 10.1038/nrc2253 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Slany RK MLL fusion proteins and transcriptional control. Biochim Biophys Acta Gene Regul Mech 1863, 194503, doi: 10.1016/j.bbagrm.2020.194503 (2020). [DOI] [PubMed] [Google Scholar]
- 73.Rao RC & Dou Y Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15, 334–346, doi: 10.1038/nrc3929 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meyer C et al. The MLL recombinome of acute leukemias in 2017. Leukemia 32, 273–284, doi: 10.1038/leu.2017.213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Milne TA et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Molecular cell 38, 853–863, doi: 10.1016/j.molcel.2010.05.011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dorrance AM et al. The Mll partial tandem duplication: differential, tissue-specific activity in the presence or absence of the wild-type allele. Blood 112, 2508–2511 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dorrance AM et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. The Journal of clinical investigation 116, 2707–2716 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alam H et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37, 599–617 e597, doi: 10.1016/j.ccell.2020.03.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heintzman ND et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39, 311–318, doi: 10.1038/ng1966 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Zhang J et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21, 1190–1198, doi: 10.1038/nm.3940 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ortega-Molina A et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21, 1199–1208, doi: 10.1038/nm.3943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maitituoheti M et al. Enhancer Reprogramming Confers Dependence on Glycolysis and IGF Signaling in KMT2D Mutant Melanoma. Cell Rep 33, 108293, doi: 10.1016/j.celrep.2020.108293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dhar SS et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Molecular cell 70, 825–841 e826, doi: 10.1016/j.molcel.2018.04.028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang L et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nature medicine 24, 758–769, doi: 10.1038/s41591-018-0034-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tran N, Broun A & Ge K Lysine Demethylase KDM6A in Differentiation, Development, and Cancer. Mol Cell Biol 40, doi: 10.1128/MCB.00341-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu B, Konze KD, Jin J & Wang GG Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp Hematol 43, 698–712, doi: 10.1016/j.exphem.2015.05.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu JR, Lee CH, Oksuz O, Stafford JM & Reinberg D PRC2 is high maintenance. Genes & development 33, 903–935, doi: 10.1101/gad.325050.119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morin RD et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature genetics 42, 181–185, doi: 10.1038/ng.518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sneeringer CJ et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proceedings of the National Academy of Sciences of the United States of America 107, 20980–20985, doi: 10.1073/pnas.1012525107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Souroullas GP et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med 22, 632–640, doi: 10.1038/nm.4092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beguelin W et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer cell 23, 677–692, doi: 10.1016/j.ccr.2013.04.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caganova M et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. The Journal of clinical investigation 123, 5009–5022, doi: 10.1172/JCI70626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Velichutina I et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255, doi:- 10.1182/blood-2010-04280149 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253, doi: 10.1038/nature15520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burr ML et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer cell 36, 385–401 e388, doi: 10.1016/j.ccell.2019.08.008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beguelin W et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 37, 655–673 e611, doi: 10.1016/j.ccell.2020.04.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nikoloski G et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetics 42, 665–667, doi: 10.1038/ng.620 (2010). [DOI] [PubMed] [Google Scholar]
- 98.Ntziachristos P et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nature medicine 18, 298–301, doi: 10.1038/nm.2651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee W et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nature genetics 46, 1227–1232, doi: 10.1038/ng.3095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Raedt T et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514, 247–251, doi: 10.1038/nature13561 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Gu Z et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov 9, 1228–1247, doi: 10.1158/2159-8290.CD-19-0152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jiao L & Liu X Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350, aac4383, doi: 10.1126/science.aac4383 (2015). This study provided the structural basis of core PRC2 complex.
- 103. Justin N et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 7, 11316, doi: 10.1038/ncomms11316 (2016). This study provided the structural basis of core PRC2 complex.
- 104.Lee CH et al. Allosteric Activation Dictates PRC2 Activity Independent of Its Recruitment to Chromatin. Molecular cell 70, 422–434 e426, doi: 10.1016/j.molcel.2018.03.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ueda T et al. Propagation of trimethylated H3K27 regulated by polycomb protein EED is required for embryogenesis, hematopoietic maintenance, and tumor suppression. Proc Natl Acad Sci U S A 113, 10370–10375, doi: 10.1073/pnas.1600070113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li J, Ahn JH & Wang GG Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell Mol Life Sci 76, 2899–2916, doi: 10.1007/s00018-019-03144-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bennett RL, Swaroop A, Troche C & Licht JD The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb Perspect Med 7, doi: 10.1101/cshperspect.a026708 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jaffe JD et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nature genetics 45, 1386–1391, doi: 10.1038/ng.2777 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oyer JA et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 28, 198–201, doi: 10.1038/leu.2013.204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang GG, Cai L, Pasillas MP & Kamps MP NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol 9, 804–812, doi: 10.1038/ncb1608 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Papillon-Cavanagh S et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet 49, 180–185, doi: 10.1038/ng.3757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brennan K et al. NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma. Sci Rep 7, 17064, doi: 10.1038/s41598-017-17298-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng Y et al. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proceedings of the National Academy of Sciences of the United States of America 109, 13549–13554, doi: 10.1073/pnas.1205707109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miyazaki H et al. Ash1l methylates Lys36 of histone H3 independently of transcriptional elongation to counteract polycomb silencing. PLoS Genet 9, e1003897, doi: 10.1371/journal.pgen.1003897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Popovic R et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet 10, e1004566, doi: 10.1371/journal.pgen.1004566 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lu C et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849, doi: 10.1126/science.aac7272 (2016). This paper reported the functional roles of oncohistone H3K36M in cancer.
- 117. Fang D et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352, 1344–1348, doi: 10.1126/science.aae0065 (2016). This paper reported the functional roles of oncohistone H3K36M in cancer.
- 118.Weinberg DN et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286, doi: 10.1038/s41586-019-1534-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shirane K, Miura F, Ito T & Lorincz MC NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nature genetics 52, 1088–1098, doi: 10.1038/s41588-020-0689-z (2020). [DOI] [PubMed] [Google Scholar]
- 120.Xu W et al. DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein Cell 11, 150–154, doi: 10.1007/s13238-019-00672-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Swaroop A et al. An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene 38, 671–686, doi: 10.1038/s41388-018-0474-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Li W et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 590, 498–503, doi: 10.1038/s41586-020-03069-8 (2021). This study illustrated the structural basis of NSD family proteins catalyzing nucleosomal H3K36me and the role of NSD3 GOF mutation in lung cancer.
- 123. Yuan G et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature 590, 504–508, doi: 10.1038/s41586-020-03170-y (2021). This study illustrated the structural basis of NSD family proteins catalyzing nucleosomal H3K36me and the role of NSD3 GOF mutation in lung cancer.
- 124.Cho MH et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun 6, 7821, doi: 10.1038/ncomms8821 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen CW et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nature medicine 21, 335–343, doi: 10.1038/nm.3832 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vlaming H et al. Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L-dose dependency in HDAC1-deficient thymic lymphoma. EMBO J 38, e101564, doi: 10.15252/embj.2019101564 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang H et al. Structural and functional analysis of the DOT1L-AF10 complex reveals mechanistic insights into MLL-AF10-associated leukemogenesis. Genes Dev 32, 341–346, doi: 10.1101/gad.311639.118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jang S et al. Structural basis of recognition and destabilization of the histone H2B ubiquitinated nucleosome by the DOT1L histone H3 Lys79 methyltransferase. Genes Dev 33, 620–625, doi: 10.1101/gad.323790.118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Valencia-Sanchez MI et al. Structural Basis of Dot1L Stimulation by Histone H2B Lysine 120 Ubiquitination. Mol Cell 74, 1010–1019 e1016, doi: 10.1016/j.molcel.2019.03.029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Worden EJ, Hoffmann NA, Hicks CW & Wolberger C Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell 176, 1490–1501 e1412, doi: 10.1016/j.cell.2019.02.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Anderson CJ et al. Structural Basis for Recognition of Ubiquitylated Nucleosome by Dot1L Methyltransferase. Cell Rep 26, 1681–1690 e1685, doi: 10.1016/j.celrep.2019.01.058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Valencia-Sanchez MI et al. Regulation of the Dot1 histone H3K79 methyltransferase by histone H4K16 acetylation. Science 371, doi: 10.1126/science.abc6663 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Song X et al. A higher-order configuration of the heterodimeric DOT1L-AF10 coiled-coil domains potentiates their leukemogenenic activity. Proc Natl Acad Sci U S A 116, 19917–19923, doi: 10.1073/pnas.1904672116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheng Y et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 4, 62, doi: 10.1038/s41392-019-0095-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nishiyama A et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 502, 249–253, doi: 10.1038/nature12488 (2013). [DOI] [PubMed] [Google Scholar]
- 136.Mudbhary R et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 25, 196–209, doi: 10.1016/j.ccr.2014.01.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kong X et al. Defining UHRF1 Domains that Support Maintenance of Human Colon Cancer DNA Methylation and Oncogenic Properties. Cancer Cell 35, 633–648 e637, doi: 10.1016/j.ccell.2019.03.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bronner C, Alhosin M, Hamiche A & Mousli M Coordinated Dialogue between UHRF1 and DNMT1 to Ensure Faithful Inheritance of Methylated DNA Patterns. Genes (Basel) 10, doi: 10.3390/genes10010065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang ZM et al. An Allosteric Interaction Links USP7 to Deubiquitination and Chromatin Targeting of UHRF1. Cell Rep 12, 1400–1406, doi: 10.1016/j.celrep.2015.07.046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ley TJ et al. DNMT3A mutations in acute myeloid leukemia. The New England journal of medicine 363, 2424–2433, doi: 10.1056/NEJMoa1005143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang L, Rau R & Goodell MA DNMT3A in haematological malignancies. Nat Rev Cancer 15, 152–165, doi: 10.1038/nrc3895 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brunetti L, Gundry MC & Goodell MA DNMT3A in Leukemia. Cold Spring Harb Perspect Med 7, doi: 10.1101/cshperspect.a030320 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kaner J et al. Clonal Hematopoiesis and Premalignant Diseases. Cold Spring Harb Perspect Med 10, doi: 10.1101/cshperspect.a035675 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu R & Wang GG Pharmacologic Targeting of Chromatin Modulators As Therapeutics of Acute Myeloid Leukemia. Front Oncol 7, 241, doi: 10.3389/fonc.2017.00241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Challen GA et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44, 23–31, doi: 10.1038/ng.1009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Russler-Germain DA et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer cell 25, 442–454, doi: 10.1016/j.ccr.2014.02.010 (2014). This study illustrated the mechanism by which the DNMT3A R882H mutation contributed to development of acute leukemia.
- 147.Yang L et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell 30, 363–365, doi: 10.1016/j.ccell.2016.07.015 (2016). [DOI] [PubMed] [Google Scholar]
- 148. Lu R et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer cell 30, 92–107, doi: 10.1016/j.ccell.2016.05.008 (2016). This study illustrated the mechanism by which the DNMT3A R882H mutation contributed to development of acute leukemia.
- 149.Meyer SE et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discov 6, 501–515, doi: 10.1158/2159-8290.CD-16-0008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guryanova OA et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nature medicine 22, 1488–1495, doi: 10.1038/nm.4210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ferreira HJ et al. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene 36, 4233, doi: 10.1038/onc.2017.57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lu R et al. A Model System for Studying the DNMT3A Hotspot Mutation (DNMT3A(R882)) Demonstrates a Causal Relationship between Its Dominant-Negative Effect and Leukemogenesis. Cancer research 79, 3583–3594, doi: 10.1158/0008-5472.CAN-18-3275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Uckelmann HJ et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 367, 586–590, doi: 10.1126/science.aax5863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zhang ZM et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391, doi: 10.1038/nature25477 (2018). This study provided the structural basis of DNMT3A in complex with substrate DNA and in complex with substrate nucleosome.
- 155.Gao L et al. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat Commun 11, 3355, doi: 10.1038/s41467-020-17109-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Anteneh H, Fang J & Song J Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat Commun 11, 2294, doi: 10.1038/s41467-020-16213-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Norvil AB et al. Dnmt3b Methylates DNA by a Noncooperative Mechanism, and Its Activity Is Unaffected by Manipulations at the Predicted Dimer Interface. Biochemistry 57, 4312–4324, doi: 10.1021/acs.biochem.6b00964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Emperle M et al. Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res 47, 11355–11367, doi: 10.1093/nar/gkz911 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Norvil AB et al. The acute myeloid leukemia variant DNMT3A Arg882His is a DNMT3B-like enzyme. Nucleic acids research 48, 3761–3775, doi: 10.1093/nar/gkaa139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zaware N & Zhou MM Bromodomain biology and drug discovery. Nat Struct Mol Biol 26, 870–879, doi: 10.1038/s41594-019-0309-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zeng L et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 466, 258–262, doi: 10.1038/nature09139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Li Y et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571, doi: 10.1016/j.cell.2014.09.049 (2014). This paper identified the YEATS domain as a new reader of histone acetylation.
- 163. Wan L et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269, doi: 10.1038/nature21687 (2017). This paper illustrated the functional roles of ENL YEATS domain in leukaemia.
- 164. Erb MA et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274, doi: 10.1038/nature21688 (2017). This paper illustrated the functional roles of ENL YEATS domain in leukaemia.
- 165.Li X et al. Structure-guided development of YEATS domain inhibitors by targeting pi-pi-pi stacking. Nat Chem Biol 14, 1140–1149, doi: 10.1038/s41589-018-0144-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Perlman EJ et al. MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6, 10013, doi: 10.1038/ncomms10013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gadd S et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49, 1487–1494, doi: 10.1038/ng.3940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wan L et al. Impaired cell fate through gain-of-function mutations in a chromatin reader. Nature 577, 121–126, doi: 10.1038/s41586-019-1842-7 (2020). This paper reported that gain-of-function mutations of ENL YEATS domain resulted in enhanced self-association and contributed to pathogenesis of Wilms tumour.
- 169.Guo C et al. ENL initiates multivalent phase separation of the super elongation complex (SEC) in controlling rapid transcriptional activation. Sci Adv 6, eaay4858, doi: 10.1126/sciadv.aay4858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mi W et al. YEATS2 links histone acetylation to tumorigenesis of non-small cell lung cancer. Nat Commun 8, 1088, doi: 10.1038/s41467-017-01173-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hsu CC et al. Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z deposition in non-small cell lung cancer. Genes Dev 32, 58–69, doi: 10.1101/gad.303784.117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Andrews FH et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol 12, 396–398, doi: 10.1038/nchembio.2065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Klein BJ et al. Structural insights into the pi-pi-pi stacking mechanism and DNA-binding activity of the YEATS domain. Nat Commun 9, 4574, doi: 10.1038/s41467-018-07072-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang Q et al. Structural Insights into Histone Crotonyl-Lysine Recognition by the AF9 YEATS Domain. Structure 24, 1606–1612, doi: 10.1016/j.str.2016.05.023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sabari BR et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell 58, 203–215, doi: 10.1016/j.molcel.2015.02.029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Margueron R & Reinberg D The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349, doi: 10.1038/nature09784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Fan H et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat Genet, doi: 10.1038/s41588-020-00729-3 (2020). This paper reported the BAH domain within BAHCC1 as a new H3K27me3 reader in mammalian cells, significantly contributing to Polycomb gene silencing and oncogenesis.
- 178.Wang H, Farnung L, Dienemann C & Cramer P Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat Struct Mol Biol 27, 8–13, doi: 10.1038/s41594-019-0345-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Baubec T et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247, doi: 10.1038/nature14176 (2015). [DOI] [PubMed] [Google Scholar]
- 180.Dukatz M et al. H3K36me2/3 Binding and DNA Binding of the DNA Methyltransferase DNMT3A PWWP Domain Both Contribute to its Chromatin Interaction. J Mol Biol 431, 5063–5074, doi: 10.1016/j.jmb.2019.09.006 (2019). [DOI] [PubMed] [Google Scholar]
- 181.Heyn P et al. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet 51, 96–105, doi: 10.1038/s41588-018-0274-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Wen H et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268, doi: 10.1038/nature13045 (2014). This paper reported ZMYND11 as a histone variant H3.3K36me3 specific reader and its roles in tumour suppression.
- 183.Guo R et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell 56, 298–310, doi: 10.1016/j.molcel.2014.08.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Armache A et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 583, 852–857, doi: 10.1038/s41586-020-2533-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.De Braekeleer E et al. Recurrent translocation (10;17)(p15;q21) in acute poorly differentiated myeloid leukemia likely results in ZMYND11-MBTD1 fusion. Leuk Lymphoma 55, 1189–1190, doi: 10.3109/10428194.2013.820292 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Li J et al. ZMYND11-MBTD1 induces leukemogenesis through hijacking NuA4/TIP60 acetyltransferase complex and a PWWP-mediated chromatin association mechanism. Nat Commun 12, 1045, doi: 10.1038/s41467-021-21357-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li F et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153, 590–600, doi: 10.1016/j.cell.2013.03.025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Daugaard M et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol 19, 803–810, doi: 10.1038/nsmb.2314 (2012). [DOI] [PubMed] [Google Scholar]
- 189.Wang GG et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851, doi: 10.1038/nature08036 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gough SM et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov 4, 564–577, doi: 10.1158/2159-8290.CD-13-0419 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang Y et al. Mechanistic insights into chromatin targeting by leukemic NUP98-PHF23 fusion. Nat Commun 11, 3339, doi: 10.1038/s41467-020-17098-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.de Rooij JD et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27, 2280–2288, doi: 10.1038/leu.2013.87 (2013). [DOI] [PubMed] [Google Scholar]
- 193.Cardin S et al. Human models of NUP98-KDM5A megakaryocytic leukemia in mice contribute to uncovering new biomarkers and therapeutic vulnerabilities. Blood Adv 3, 3307–3321, doi: 10.1182/bloodadvances.2019030981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Roussy M et al. NUP98-BPTF gene fusion identified in primary refractory acute megakaryoblastic leukemia of infancy. Genes Chromosomes Cancer 57, 311–319, doi: 10.1002/gcc.22532 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Klein BJ et al. PHF20 Readers Link Methylation of Histone H3K4 and p53 with H4K16 Acetylation. Cell Rep 17, 1158–1170, doi: 10.1016/j.celrep.2016.09.056 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li N et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol Cell 63, 470–484, doi: 10.1016/j.molcel.2016.06.035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shen H et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell 165, 331–342, doi: 10.1016/j.cell.2016.02.064 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jiao F et al. RACK7 recognizes H3.3G34R mutation to suppress expression of MHC class II complex components and their delivery pathway in pediatric glioblastoma. Sci Adv 6, eaba2113, doi: 10.1126/sciadv.aba2113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Qiu Y et al. Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev 26, 1376–1391, doi: 10.1101/gad.188359.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Klein BJ et al. Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat Commun 10, 4724, doi: 10.1038/s41467-019-12551-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xiong X et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat Chem Biol 12, 1111–1118, doi: 10.1038/nchembio.2218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vasileiou G et al. Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome. Am J Hum Genet 102, 468–479, doi: 10.1016/j.ajhg.2018.01.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Huber FM et al. Histone-binding of DPF2 mediates its repressive role in myeloid differentiation. Proc Natl Acad Sci U S A 114, 6016–6021, doi: 10.1073/pnas.1700328114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Li Y & Seto E HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med 6, doi: 10.1101/cshperspect.a026831 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Klose RJ, Kallin EM & Zhang Y JmjC-domain-containing proteins and histone demethylation. Nature reviews 7, 715–727 (2006). [DOI] [PubMed] [Google Scholar]
- 206.Losman JA, Koivunen P & Kaelin WG Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer 20, 710–726, doi: 10.1038/s41568-020-00303-3 (2020). [DOI] [PubMed] [Google Scholar]
- 207.Shi Y Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet 8, 829–833, doi: 10.1038/nrg2218 (2007). [DOI] [PubMed] [Google Scholar]
- 208.Niu X et al. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 31, 776–786, doi: 10.1038/onc.2011.266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wong SH et al. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 28, 198–209, doi: 10.1016/j.ccell.2015.06.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wu L et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol 16, e2006134, doi: 10.1371/journal.pbio.2006134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hinohara K et al. KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell 34, 939–953 e939, doi: 10.1016/j.ccell.2018.10.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Vinogradova M et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat Chem Biol 12, 531–538, doi: 10.1038/nchembio.2085 (2016). [DOI] [PubMed] [Google Scholar]
- 213.Roesch A et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer cell 23, 811–825, doi: 10.1016/j.ccr.2013.05.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ntziachristos P et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514, 513–517, doi: 10.1038/nature13605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pan M et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18, 1090–1101, doi: 10.1038/ncb3410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Carey BW, Finley LW, Cross JR, Allis CD & Thompson CB Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416, doi: 10.1038/nature13981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tommasini-Ghelfi S et al. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci Adv 5, eaaw4543, doi: 10.1126/sciadv.aaw4543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ye D, Guan KL & Xiong Y Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer 4, 151–165, doi: 10.1016/j.trecan.2017.12.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Dang L et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744, doi: 10.1038/nature08617 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Xu W et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30, doi: 10.1016/j.ccr.2010.12.014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Intlekofer AM et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab 22, 304–311, doi: 10.1016/j.cmet.2015.06.023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Chowdhury R et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. Embo Rep 12, 463–469, doi: 10.1038/embor.2011.43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lu C et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478, doi: 10.1038/nature10860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Tahiliani M et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935, doi: 10.1126/science.1170116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ito S et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303, doi: 10.1126/science.1210597 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.He YF et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307, doi: 10.1126/science.1210944 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Huang Y & Rao A Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet 30, 464–474, doi: 10.1016/j.tig.2014.07.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hu L et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155, 1545–1555, doi: 10.1016/j.cell.2013.11.020 (2013). [DOI] [PubMed] [Google Scholar]
- 229.Ko M et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497, 122–126, doi: 10.1038/nature12052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Figueroa ME et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567, doi: 10.1016/j.ccr.2010.11.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Thienpont B et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63–68, doi: 10.1038/nature19081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Agathocleous M et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481, doi: 10.1038/nature23876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cimmino L et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095 e1020, doi: 10.1016/j.cell.2017.07.032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Yue X & Rao A TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood 136, 1394–1401, doi: 10.1182/blood.2019004158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wu TP et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 532, 329–333, doi: 10.1038/nature17640 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Koziol MJ et al. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat Struct Mol Biol 23, 24–30, doi: 10.1038/nsmb.3145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Schiffers S et al. Quantitative LC-MS Provides No Evidence for m(6) dA or m(4) dC in the Genome of Mouse Embryonic Stem Cells and Tissues. Angew Chem Int Ed Engl 56, 11268–11271, doi: 10.1002/anie.201700424 (2017). [DOI] [PubMed] [Google Scholar]
- 238. Xie Q et al. N(6)-methyladenine DNA Modification in Glioblastoma. Cell 175, 1228–1243 e1220, doi: 10.1016/j.cell.2018.10.006 (2018). This paper linked abnormal levels of DNA N6mA modification to glioblastoma.
- 239.Xiao CL et al. N(6)-Methyladenine DNA Modification in the Human Genome. Mol Cell 71, 306–318 e307, doi: 10.1016/j.molcel.2018.06.015 (2018). [DOI] [PubMed] [Google Scholar]
- 240.Hao Z et al. N(6)-Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol Cell 78, 382–395 e388, doi: 10.1016/j.molcel.2020.02.018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhang M et al. Mammalian ALKBH1 serves as an N(6)-mA demethylase of unpairing DNA. Cell Res 30, 197–210, doi: 10.1038/s41422-019-0237-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tian LF et al. Structural basis of nucleic acid recognition and 6mA demethylation by human ALKBH1. Cell Res 30, 272–275, doi: 10.1038/s41422-019-0233-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Li Z et al. N(6)-methyladenine in DNA antagonizes SATB1 in early development. Nature 583, 625–630, doi: 10.1038/s41586-020-2500-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Yang X et al. m(6)A promotes R-loop formation to facilitate transcription termination. Cell Res 29, 1035–1038, doi: 10.1038/s41422-019-0235-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Abakir A et al. N(6)-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat Genet 52, 48–55, doi: 10.1038/s41588-019-0549-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Woodcock CB et al. Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov 5, 63, doi: 10.1038/s41421-019-0136-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Woodcock CB et al. Biochemical and structural basis for YTH domain of human YTHDC1 binding to methylated adenine in DNA. Nucleic Acids Res 48, 10329–10341, doi: 10.1093/nar/gkaa604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Douvlataniotis K, Bensberg M, Lentini A, Gylemo B & Nestor CE No evidence for DNA N (6)-methyladenine in mammals. Sci Adv 6, eaay3335, doi: 10.1126/sciadv.aay3335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mohammad HP, Barbash O & Creasy CL Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med 25, 403–418, doi: 10.1038/s41591-019-0376-8 (2019). [DOI] [PubMed] [Google Scholar]
- 250.Burslem GM & Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114, doi: 10.1016/j.cell.2019.11.031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Vogelmann A, Robaa D, Sippl W & Jung M Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr Opin Chem Biol 57, 8–16, doi: 10.1016/j.cbpa.2020.01.010 (2020). [DOI] [PubMed] [Google Scholar]
- 252.Pisa R & Kapoor TM Chemical strategies to overcome resistance against targeted anticancer therapeutics. Nat Chem Biol 16, 817–825, doi: 10.1038/s41589-020-0596-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Winter GE et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (New York, N.Y 348, 1376–1381, doi: 10.1126/science.aab1433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yang K et al. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg Med Chem Lett 28, 2493–2497, doi: 10.1016/j.bmcl.2018.05.057 (2018). [DOI] [PubMed] [Google Scholar]
- 255.Ma A et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol 16, 214–222, doi: 10.1038/s41589-019-0421-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ludwig CH & Bintu L Mapping chromatin modifications at the single cell level. Development 146, doi: 10.1242/dev.170217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Shema E, Bernstein BE & Buenrostro JD Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nature genetics 51, 19–25, doi: 10.1038/s41588-018-0290-x (2019). [DOI] [PubMed] [Google Scholar]
- 258.Yang L et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med 9, doi: 10.1126/scitranslmed.aal1645 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sun C et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer cell 33, 401–416 e408, doi: 10.1016/j.ccell.2018.01.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hogg SJ, Beavis PA, Dawson MA & Johnstone RW Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov 19, 776–800, doi: 10.1038/s41573-020-0077-5 (2020). [DOI] [PubMed] [Google Scholar]
- 261.Huang H, Sabari BR, Garcia BA, Allis CD & Zhao Y SnapShot: histone modifications. Cell 159, 458–458 e451, doi: 10.1016/j.cell.2014.09.037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Weinberg DN, Allis CD & Lu C Oncogenic Mechanisms of Histone H3 Mutations. Cold Spring Harb Perspect Med 7, doi: 10.1101/cshperspect.a026443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.McGinty RK & Tan S Recognition of the nucleosome by chromatin factors and enzymes. Curr Opin Struct Biol 37, 54–61, doi: 10.1016/j.sbi.2015.11.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lohr JG et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proceedings of the National Academy of Sciences of the United States of America 109, 3879–3884, doi: 10.1073/pnas.1121343109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (New York, N.Y 340, 857–861, doi: 10.1126/science.1232245 (2013). This paper illustrated the mechanism of oncohistone H3K27M in pediatric glioblastoma.
- 266.Jain SU et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc Natl Acad Sci U S A 117, 27354–27364, doi: 10.1073/pnas.2006076117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Huang TY et al. Effects of H3.3G34V mutation on genomic H3K36 and H3K27 methylation patterns in isogenic pediatric glioma cells. Acta Neuropathol Commun 8, 219, doi: 10.1186/s40478-020-01092-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Shi L, Shi J, Shi X, Li W & Wen H Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. Journal of molecular biology 430, 1562–1565, doi: 10.1016/j.jmb.2018.04.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Chan KM et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & development 27, 985–990, doi: 10.1101/gad.217778.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bender S et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672, doi: 10.1016/j.ccr.2013.10.006 (2013). [DOI] [PubMed] [Google Scholar]
- 271.Boija A et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855 e1816, doi: 10.1016/j.cell.2018.10.042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Cho WK et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415, doi: 10.1126/science.aar4199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, doi: 10.1126/science.aar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wang L et al. Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol Cell 76, 646–659 e646, doi: 10.1016/j.molcel.2019.08.019 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.