

Abstract
Heterotopic ossification (HO) is a common, potentially debilitating pathology that is instigated by tissue damage or other insults and involves inflammation followed by chondrogenesis, osteogenesis, and extraskeletal bone accumulation. Current remedies are not very effective and have side effects, including the risk of triggering additional HO. Activin A is a member of the transforming growth factor–β (TGF-β) superfamily that is produced by activated macrophages and other inflammatory cells and signals through the activation of the canonical intracellular effectors SMAD2 and SMAD3 (SMAD2/3). Because HO starts with inflammation and because SMAD2/3 activation is chondrogenic, we tested whether activin A stimulated HO development. Using standard mouse models of acquired intramuscular and subdermal HO, we found that blockage of endogenous activin A by systemic administration of a neutralizing antibody markedly reduced HO development and bone accumulation. Single-cell RNAseq and developmental trajectories showed that the antibody treatment sharply reduced the number of Sox9+ skeletal progenitors, many of which also expressed the gene encoding activin A (Inhba), that were recruited to the HO site. In line with the latter finding, gain-of-function assays showed that activin A enhanced the chondrogenic differentiation of the progenitor cells and did so through SMAD2/3 signaling, and inclusion of activin A to HO-inducing in vivo implants enhanced HO development. Together, our data reveal that activin A is a critical upstream signaling stimulator of acquired HO in mice and could represent an effective therapeutic target against forms of this pathology in patients.
INTRODUCTION
Heterotopic ossification (HO) is a common, potentially debilitating, acquired pathology in which masses of ectopic endochondral bone form and accumulate within or around connective tissues, muscles, joints, blood vessels, and other sites (1–3). HO can be instigated by tissue damage, invasive surgeries, protracted immobilization, or spinal cord injury (4–7). It is particularly common amongst severely wounded soldiers (8, 9). The HO masses can impede routine daily functions and mobility, and can cause other health problems including nerve and blood vessel impingement, pressure ulcers, joint ankylosis, prosthesis fitting problems, and chronic pain (1, 4, 10, 11). The exact pathogenesis of HO remains unclear (2); however, evidence indicates that following injury or other events, local activities of macrophages, monocytes, and other cells at the prospective HO site result in substantial increases in inflammatory cytokines, including tumor necrosis factor (TNF), interleukin-1β (IL-1β), transforming growth factor–β (TGF-β), IL-6, and monocyte chemoattractant protein 1 (MCP-1) (2, 12, 13). This in turn leads to mobilization and recruitment of certain stem cell populations (14–16) and local stimulation of pro-skeletogenic signaling mechanisms (17–19), ultimately inducing chondrogenesis, osteogenesis, and ectopic bone tissue formation and accumulation.
Current clinical treatments for HO include local low-dose radiation and systemic administration of non-steroidal anti-inflammatory drugs (NSAIDs) (20, 21). Radiation is most commonly used with patients undergoing total hip arthroplasty and also in other groups at risk, including spinal cord injury patients (5, 22). This treatment affects local cell progenitors, reducing their functioning and viability and dampening their potential for ectopic skeletogenic cell differentiation (20, 23, 24). NSAIDs interfere with prostaglandin production (25), and animals treated with the NSAID indomethacin display more resistance to HO provoked by tissue injury or intramuscular injection of recombinant bone morphogenetic proteins (BMPs) and reduced differentiation of local skeletal cell progenitors (26, 27). Clinically, the above treatment modalities are associated with lower incidence of HO, but are neither ideal nor wholly effective, do not target specific skeletogenic mechanisms, and cannot be given to certain patient populations (28). NSAIDs can cause side effects that are potentially serious or lead to reduced patient compliance, such as gastrointestinal pain and ulceration, low platelet function, and renal toxicity (20, 29). NSAIDs that are selective cyclooxygenase 2 (COX-2) inhibitors, such as celecoxib, are used to reduce side effects (30). The HO masses can also be removed by surgery (31). This intervention is used within six months from onset of HO and when the HO masses are deemed mature. However, surgery is prone to complications and could potentially trigger another round of HO if accompanied by extensive tissue damage or if the preexisting HO masses are not resected completely (32). In sum, although diverse clinical treatment options are available, there is an urgent need for safer and more effective ones.
Activin A is a member of the TGF-β superfamily that includes BMPs, TGF-βs, and growth and differentiation factors (GDFs) (33, 34). Although Activin A was originally isolated from gonadal fluids and found to stimulate the secretion of pituitary follicle stimulating hormone (35, 36), it is now well established that the protein acts locally and systemically to regulate a host of important physiologic processes, including immune responses and wound healing as well as pathologic responses including inflammation and fibrosis (37, 38). Activin A systemic abundance rapidly increase during inflammation, and its overall roles are modulated at the tissue level by interactions with natural inhibitors, such as follistatin, and through the formation of diffusion gradients (39–41). At the cellular level, one mechanism of activin A action involves its interaction with the cell surface type II kinase receptors ACVR2A and ACVR2B in association with the type I receptor ALK4 or ALK7 and signaling through the phosphorylation of the canonical effectors SMAD2 and SMAD3 (SMAD2/3), which modulate target gene expression to regulate cellular responses (33, 42). Of particular relevance here is the fact that activin A is produced by inflammatory macrophages and other activated immune cells and stimulates the release of inflammatory cytokines such as IL-1β and TNF and the recruitment of mast cells (40, 43, 44), processes usually seen during the initial phases of HO. In addition, studies have indicated that activin A promotes bone fracture repair, a process that closely resembles HO, and can stimulate bone accrual when delivered locally (45, 46). As importantly, canonical signaling through phosphorylated SMAD2/3 (pSMAD2/3) elicited by cytokines such as TGF-β has long been known to stimulate chondrogenic cell differentiation (47, 48), and this property appears to be shared by activin A (49). When considered together, the above studies raise the possibility that activin A could actually promote the development of HO and, if so, could represent an effective target for attenuation. To test that thesis, we used standard subdermal and intramuscular BMP implant mouse models of acquired HO, and our data provide clear evidence in its support.
RESULTS
Systemic administration of activin A neutralizing antibody inhibits subcutaneous HO development
To test whether activin A has a role in acquired HO, we asked whether systemic administration of an activin A neutralizing antibody would mitigate its development. To this end, we used standard HO mouse models in which a matrix scaffold containing a skeletogenic protein is implanted ectopically (50–52). Provoked by the matrix-protein implant, host inflammatory cells readily surround and infiltrate the scaffold and are followed by mesenchymal progenitors that undergo heterotopic chondrogenesis, osteogenesis, and endochondral bone formation over time (52). Accordingly, adult 2 months-old female mice were implanted at two subcutaneous abdominal sites with 250 μl Matrigel aliquots containing recombinant human BMP2 (rhBMP2) or rhBMP6. We tested two BMPs to minimize biological biases. Mice were then given biweekly injections of a neutralizing monoclonal antibody (IgG2b isotype) against mouse activin A (10 mg/kg/injection), a dose comparable to that used in a previous study (53). Effectiveness of this antibody was verified by its ability to inhibit recombinant activin A–induced pSMAD2/3 signaling in cultured cells; specificity was verified by its inability to interfere with activin B–induced pSMAD2/3 accumulation (fig. S1, A and B). Control BMP-Matrigel–implanted mice received pre-immune mouse IgG2b isotype antibody obtained from the same manufacturer and given at identical dose, route, and frequency. Micro-computed tomography (μCT) imaging and quantification on day 14 from implantation showed that very large HO masses were present in control, pre-immune IgG-treated mice receiving rhBMP2-Matrigel or rhBMP4-Matrigel implants (Fig. 1, A and B). Strikingly, systemic administration of activin A antibody potently inhibited HO development (Fig. 1, A and B), amounting to an inhibition of about 70% based on bone volume/total volume (BV/TV) measurement and regardless of whether HO had been promoted by rhBMP2 or rhBMP6 (Fig. 1, C and D). Histochemistry and in situ hybridization with samples from rhBMP2-implanted mice showed that, compared to controls, activin A antibody treatment markedly reduced the formation of both safranin O–positive cartilage and alizarin red–positive endochondral bone and reduced the expression of the cartilage maturation and hypertrophy and bone markers Collagen type II alpha 1 (Col2a1), Collagen type X alpha 1 (Col10a1), and Collagen type I alpha 1 (Col1a1) (Fig. 2). Similar outcomes were observed in HO masses retrieved from control vs neutralizing antibody–treated rhBMP6-implanted mice (fig. S2).
Fig. 1. Systemic administration of activin A neutralizing antibody inhibits subcutaneous HO development.
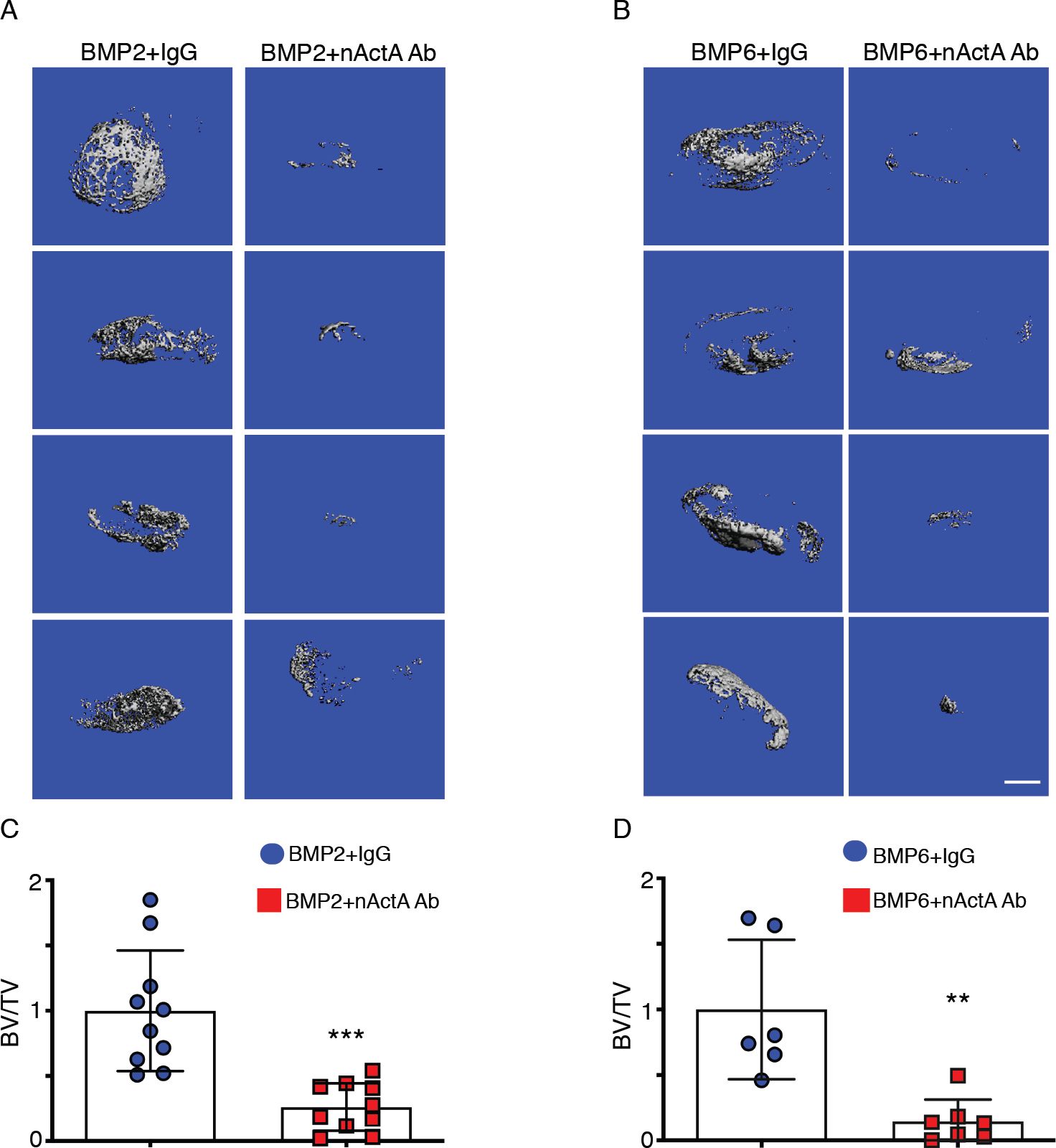
(A) Images of μCT scans of ectopic mineralized tissue masses present in mice implanted with Matrigel-rhBMP2 mixtures and then treated twice per week with control pre-immune IgGb2 mouse monoclonal antibody (BMP2 + IgG) or neutralizing activin A mouse monoclonal IgGb2 antibody (BMP2 + nActA.Ab). Samples were harvested and processed for analyses on day 14 after Matrigel implantation. Images show mineralized masses from 4 different mice in each treatment group. (B) Images of μCT scans of ectopic masses on day 14 in mice implanted with Matrigel-rhBMP6 mixtures and administered pre-immune or neutralizing activin A antibody as in (A). (C and D) Quantification of bone volume relative to total volume (BV/TV) in ectopic masses retrieved from the mice implanted with Matrigel-rhBMP2 (C) and Matrigel-BMP6 (D) and treated with pre-immune or neutralizing activin A antibody. Data are presented a mean ± S.D. N = 6–10 ectopic masses per experimental group, 2 masses per mouse, obtained from 3 independent experiments. Student’s t tests were used in all experiments to determine statistical significance. **P<0.01; ****P<0.0001. Scale bar for all panels, 1 mm.
Fig. 2. Activin A antibody administration inhibits heterotopic cartilage and bone formation.
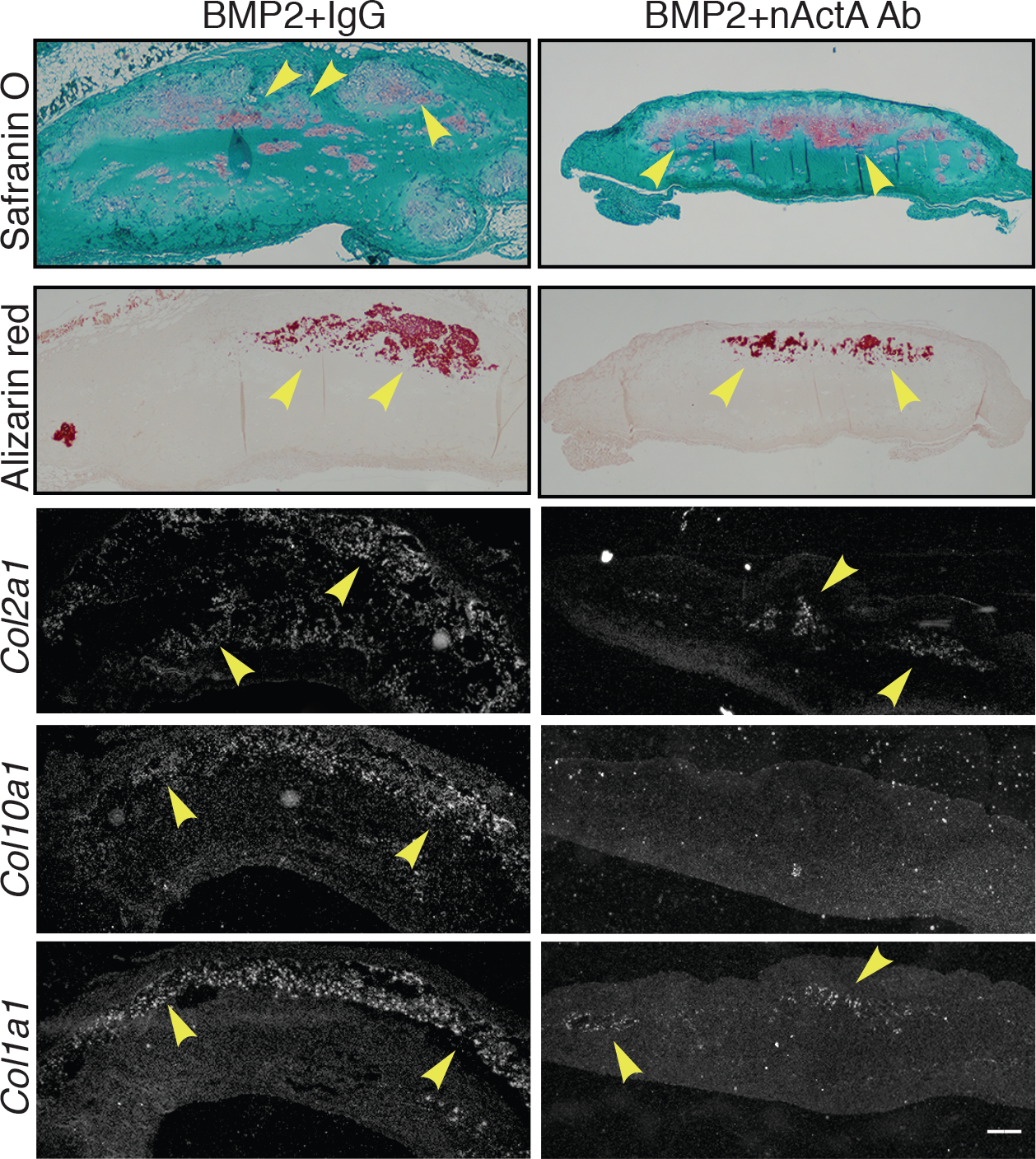
Serial sections of HO samples harvested on day 14 from rhBMP2-implanted mice treated systemically with control IgG (BMP2 + IgG) or activin A neutralizing antibody (BMP2 + nActA.Ab) were stained with safranin O to reveal cartilage and alizarin red to reveal mineralized bone. Sections were also processed for in situ hybridization to analyze gene expression of the endochondral ossification markers Col2a1 (immature cartilage), Col10a1 (hypertrophic cartilage), and Col1a1 (bone). Arrowheads in each panel point to prominent sites of cartilage and bone formation and expression of tissue-specific markers. Images are representative of N = 4 ectopic masses in each of 3 independent experiments. Scale bar, 200 μm.
Single-cell RNA-seq identifies inflammatory and chondrogenic cells recruited into developing HO lesions
To gain insights into how systemic administration of activin A antibody inhibited HO, we carried out single-cell RNAseq (scRNA-seq) analyses of cell populations recruited into developing heterotopic tissue masses. Mice implanted subcutaneously with rhBMP2-Matrigel mixtures were injected with pre-immune antibody (referred to as BMP2/IgG) or neutralizing activin A antibody (BMP2/nActA.Ab) on day 1 and day 3 from implantation, and tissue masses were harvested on day 5. By this time point, numerous host inflammatory and mesenchymal cells were already recruited to the ectopic site in Matrigel and Matrigel/BMP-implanted mice (52) (fig. S3, A to C), and many of the latter were invading the matrix and undergoing skeletogenic cell differentiation (52). As an additional control, tissue masses were also harvested on day 5 from companion mice that had been implanted with Matrigel only. Cells were liberated by enzymatic digestion and directly processed for scRNA-seq analysis (about 10,000 from each sample) (54). Libraries of cells were generated by Chromium controller (10X Genomics, Inc), barcoded, and sequenced on an Illumina HiSeq platform, and data were processed with Cell Ranger to obtain unique molecular identifiers (UMIs). We used the Seurat analysis package V3 (55) for filtering, variable gene selection, and dimensionality reduction and for eliciting t-distributed stochastic neighbor embedding (t-SNE) and graph-based clustering. Total cell numbers in the datasets shown here were as follows: 8,728 cells for Matrigel only, 5,476 cells for BMP2/IgG, and 5,657 cells for BMP2/nActA.Ab.
To delineate cluster composition, we superimposed datasets from Matrigel-only samples with those from BMP2/IgG and BMP2/nActA.Ab samples (Fig. 3A). On the basis of differential expression of representative marker genes, we identified ten major cell population clusters (Fig. 3, B to D). Two of them—clusters 2 and 4—constituted skeletogenic and mesenchymal lineage cells expressing markers such as Platelet-derived growth factor receptor alpha (Pdgfra) and Paired-related homeobox 1 (Prrx1) (56, 57) (Fig. 3, C and D). The several remaining clusters represented hematopoietic and inflammatory cells expressing Protein tyrosine phosphatase receptor type C (Ptprc) (58) (Fig. 3B), reflecting the typical preponderance of inflammatory cells at early HO stages (52). We first focused on the skeletogenic and mesenchymal cell populations in clusters 2 and 4. The Matrigel-only samples displayed a large number of Prrx1-expressing cells in both clusters (Fig. 3, D and E), confirming that the Matrigel implant itself was sufficient to trigger a host’s response and simulate cell recruitment (fig. S3). The samples displayed also a small number of cells expressing the chondrogenic master regulator Sox9 (Fig. 3F), likely representing committed skeletogenic cells (59). Sox9-expressing cells as well as chondrogenic cells expressing Aggrecan (Acan) and Col2a1 were evident and abundant in BMP2/IgG samples and were largely confined to cluster 2 (Fig. 3, F to H). This was confirmed by gene ontology analyses (fig. S4, A–D). In agreement with the anti-HO action of activin A antibody administration (Fig. 1 above), there was a sharp reduction in the number of Sox9-, Acan- and Col2a1-expressing cells in BMP2/nActA.Ab samples, visually apparent in the t-SNE plots and quantified by violin plots (Fig. 3, F to H). KEGG Pathway Mapper (60, 61) suggested that nActA.Ab effects against HO included an increase in the expression of genes encoding members of the differential screening–selected gene in neuroblastoma (DAN) family, which are well known for their BMP antagonism (fig. S5) (62).
Fig. 3. Single-cell RNA-seq analysis of populations recruited into developing HO masses.
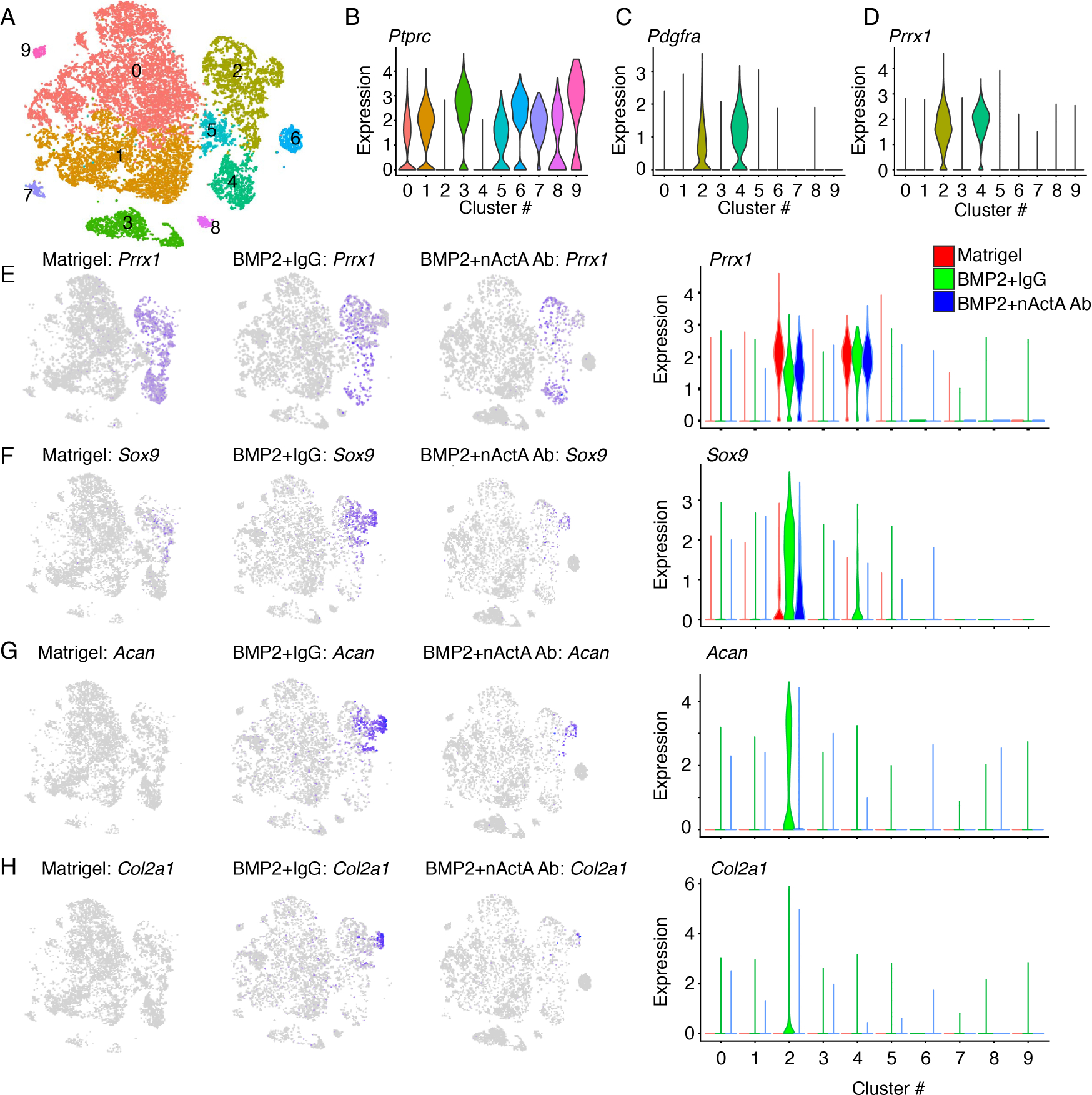
Heterotopic tissue masses were harvested on day 5 from mice implanted with Matrigel alone or Matrigel+rhBMP2 mixture, with the latter group of mice treated systemically with either pre-immune control IgG or activin A neutralizing antibody (identified in the panels as BMP2/IgG and BMP2/nAct A.Ab, respectively). (A) Sample contribution to cell type clusters after superimposition of t-SNE data from all samples, yielding 10 major cell clusters (0 to 9). (B to D) Violin plots of selected genes identifying eight hematopoietic and inflammatory cell clusters (clusters 1, 3, and 5–9) expressing Ptprc (B) and skeletogenic and mesenchymal cell clusters (clusters 2 and 4, respectively) expressing Pdgfra and/or Prrx1 (C and D). (E to H) t-SNE plots and violin plots showing Prrx1 expression that characterizes cells in clusters 2 and 4 (E) and expression of Sox9 (F), Acan (G) and Col2a1 (H) in cluster 2 cells. N = 6 ectopic masses per each experimental group pooled for scRNA-seq analysis.
To establish pseudotime and reveal trajectories of developmental progression from a mesenchymal to a chondrogenic cell phenotype, we utilized Monocle (63) and examined the effects of antibody treatment on such progression, using an unsupervised approach. Monocle identified the expression of differentially expressed genes characteristic of mesenchymal cells and chondrogenic cells, establishing pseudo-temporal patterns of most significantly expressed genes (Fig. 4A). Using the top 1,000 genes, Monocle ordered cells retrieved from Matrigel-only samples in a continuous two-dimensional pseudotime trajectory in which the cells occupied the early and medial portions only (Fig. 4B). In comparison, cells present in BMP2/IgG samples more prominently occupied the terminal portion of the trajectory, with fewer present in the early portions (Fig. 4C). The number of cells reaching the terminal portion of the trajectory was clearly reduced in BMP2/nActA.Ab samples (Fig. 4D). These trends were confirmed using single marker gene analysis. In Matrigel-only samples, cells expressing such mesenchymal genes as Cd34 and Ly6a were limited to early and medial trajectory segments (Fig. 4, B, E and F), whereas Sox9-expressing committed skeletogenic cells were mostly located within the medial portion, confirming their more advanced developmental status (Fig. 4, B and G). Acan- and Col2a1-expressing chondrogenic cells in BMP/IgG samples occupied the terminal portion of the trajectory (Fig. 4, C, H and I), but were far fewer in number in BMP/nActA.Ab samples (Fig. 4, D, H and I) (371 cells in BMP/nActA.Ab versus 1,248 in BMP/IgG).
Fig. 4. Pseudotime trajectories of mesenchymal-to-chondrogenic developmental cell progression.
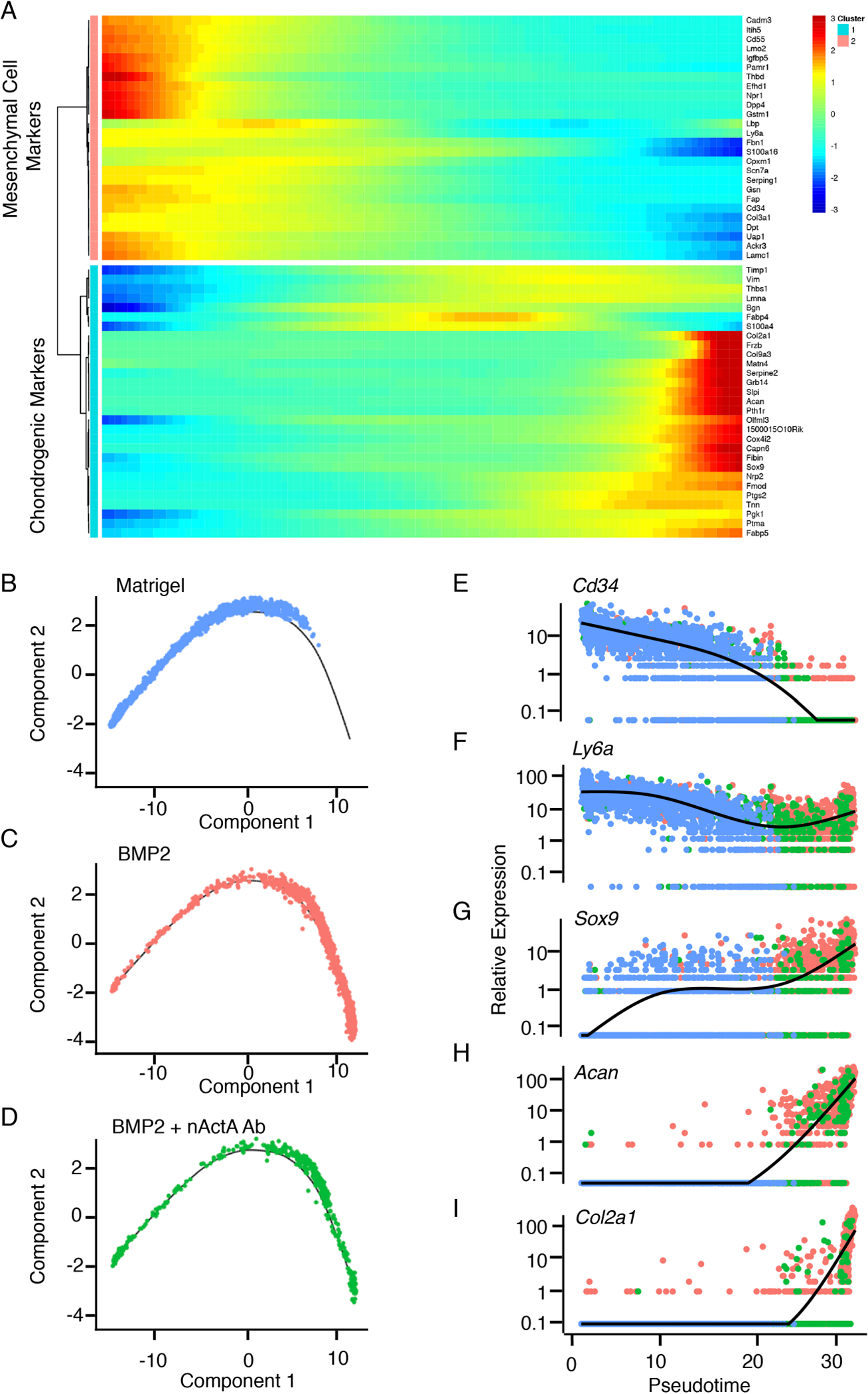
(A) Heat maps of expression levels of genes characteristic of mesenchymal cells and chondrogenic cells within clusters 2 and 4 over developmental pseudotime. (B to D) Monocle-defined two-dimensional order of progress toward differentiation of cells retrieved on day 5 HO masses from mice implanted with Matrigel only and not treated (B), implanted with Matrigel + rhBMP2 and treated with pre-immune serum (BMP2, C), or implanted with Matrigel + rhBMP2 and treated with activin A neutralizing antibody (BMP2 + nAct A Ab, D). (E to I) Pseudotime cell developmental trajectories based on the indicated individual genes in cells from Matrigel-only (blue), BMP2 (red) and BMP2 + nAct A Ab (green) samples. Comparison of early mesenchymal markers Cd34 and Ly6a with skeletogenic (Sox9) and chondrogenic (Acan and Col2a1) markers effectively decomposes the mesenchyme to chondrogenic progression. N = 6 ectopic masses per each experimental group.
Next, we examined the inflammatory and hematopoietic cell clusters (Fig. 3, A and B) and asked whether and which cell population(s) may be endowed with endogenous Inhba expression and thus could be a source of activin A. Based on differential expression of marker genes (fig. S6, A to F), the clusters mainly represented macrophages, mast cells, neutrophils, and B and T cells (Fig. 5A). The macrophages were the most conspicuous and occupied different clusters (clusters 0, 1 and 5 in Fig. 5, A and B), as also observed in an injury HO mouse model study (64). Expression of Inhba mainly characterized macrophages in clusters 0 and 1, and there was a small but appreciable number of Inhba-expressing cells in other clusters such as mast cells (Fig. 5C). The overall number of Inhba-expressing macrophages was largely comparable in BMP2/IgG versus BMP2/nActA.Ab samples, but was far higher than that in matrigel-only samples (Fig. 5D), indicating that BMP2 had served to attract the cells (65). Further analysis showed that many cluster 0 and 1 macrophages in BMP2/IgG and BMP2/nActA.Ab samples displayed an activated phenotype, indicated by Arginase 1 (Arg1) and Rnase2a expression, whereas there were fewer such cells in Matrigel-only samples (fig. S7, A and B). The former samples also displayed considerable numbers of cells in cluster 0 expressing matrix metallopeptidase 14 (Mmp14) and Ccl7 and Ccl24, which encode chemoattractant proteins (fig. S7C). Gene Set Enrichment Analysis (GSEA) indicated that compared to Matrigel-only cells, macrophages in the BMP2/IgG and BMP2/nActA.Ab samples were phenotypically enriched in inflammatory and immune system responses, and those in the BMP2/IgG samples displayed traits of skeletal and endochondral bone development (Table S1), reiterating the importance of inflammation in HO formation.
Fig. 5. Inhba is expressed by macrophages and mesenchymal progenitor cells in developing HO masses.
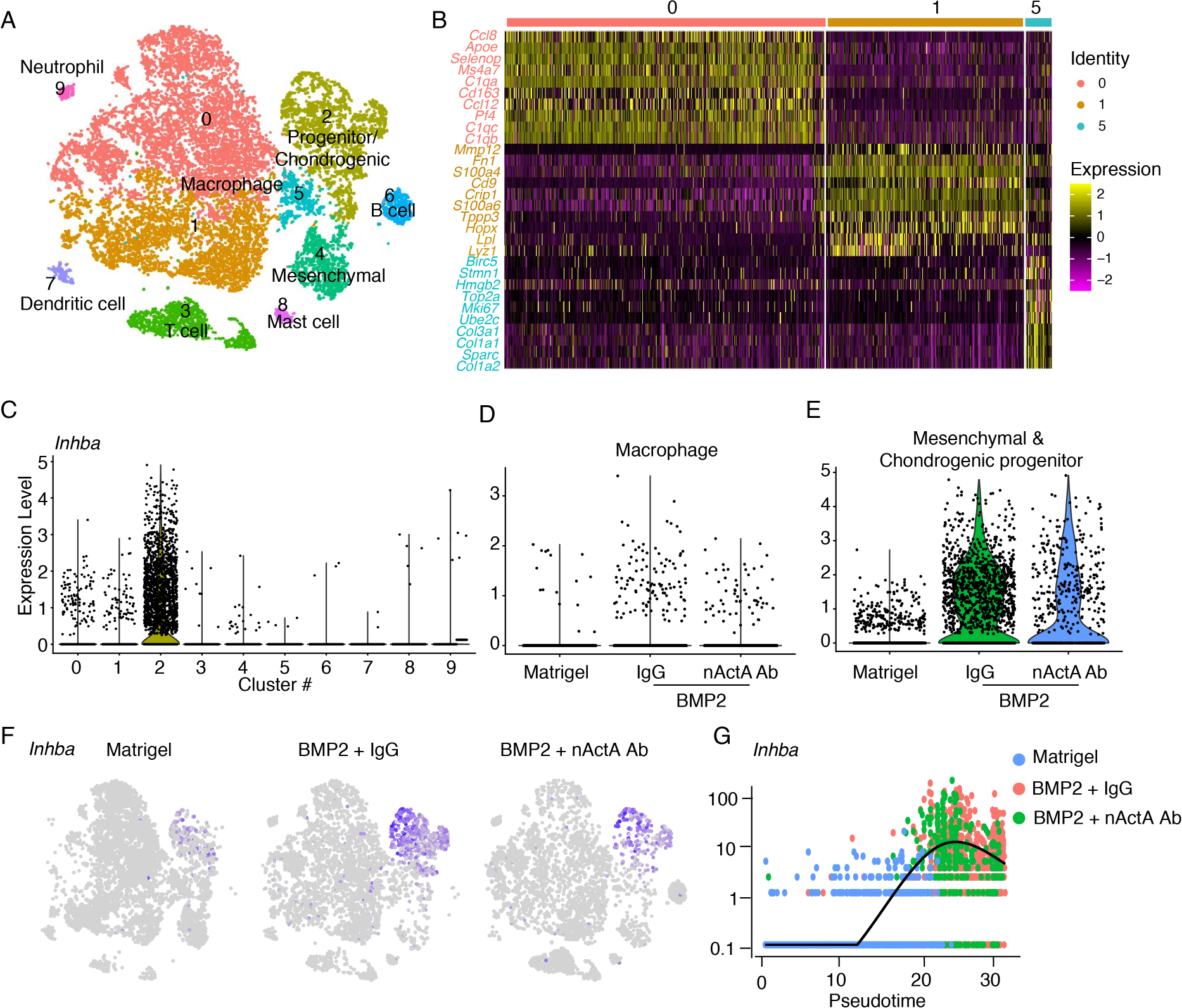
Single-cell RNA-seq datasets were further analyzed to discern Inhba expression amongst the population clusters depicted in superimposed t-SNE plots of Matrigel-only, BMP2/IgG, and BMP2/nActA.Ab samples. (A) Clusters identified by differential gene expression. Clusters 0, 1 and 5, macrophages; cluster 2, progenitor and chondrogenic cells; cluster 3, T cells; cluster 4, mesenchymal cells; cluster 6, B cells; cluster 7, dendritic cells; cluster 8, mast cells; cluster 9, neutrophils. (B) Heat map of differentially expressed genes depicts the phenotypic traits of the 3 main macrophage clusters 0, 1 and 5. (C to E) Violin plots of Inhba expression in cells from all 10 clusters across all samples (C), in macrophage populations (clusters 0, 1, and 5) from Matrigel-only, BMP2/IgG, and BMP2/nActA.Ab samples (D), and in all mesenchymal and chondrogenic progenitors (clusters 2 and 4) in Matrigel-only, BMP2/IgG and BMP2/nActA.Ab samples (E). (F) t-SNE plots showing the number of Inhba-expressing cells in the Matrigel-only, BMP2/IgG, and BMP2/nActA.Ab samples. (G) Pseudotime developmental trajectory of Inhba-expressing cells in the different samples. N = 6 ectopic masses per each experimental group.
The scRNA-seq analyses revealed that 1502 out of 2768 progenitor cells in cluster 2 expressed Inhba (Fig. 5C). The cells were abundant in the BMP/IgG samples, but their number was much lower in the BMP/nActA.Ab samples and lower yet in the Matrigel-only samples (Fig. 5, E and F). Out of 1247 cells in BMP/IgG sample cluster 2, 882 of them co-expressed Inhba and Sox9, and 350 of them co-expressed Inhba, Sox9, Acan, and Col2a1. These cell sub-cohorts decreased proportionally in the BMP2/nActA.Ab and Matrigel-only samples. Monocle-based developmental pseudotime analysis confirmed that Inhba-expressing progenitors present in the BMP2/IgG and BMP2/nActA.Ab samples occupied the later portion of the trajectory, whereas those from Matrigel-only samples were confined to the early portion (Fig. 5G). To envision possible regulators of Inhba expression, we used CellphoneDB and NicheNet to predict cell-cell interactions and ligand-receptor interactions (66–68). We found that the progenitors would mainly be influenced by macrophages and mast cells (fig. S8, A to C) and their expression of Inhba would be regulated by inflammatory cytokines such as IL-1β and TNF (fig. S8, D to F).
Because the above series of scRNA-seq data stemmed from a single set of experiments, we carried out additional studies to verify the findings. Flow cytometry was used with antibodies recognizing CD45 to monitor immune cell populations and with antibodies against CD34 to monitor chondrogenic and mesenchymal cells (69, 70) during HO development. The data indicated that the overall numbers of CD45+ populations did not vary markedly in day 5 HO masses retrieved from untreated versus nActA.Ab-treated mice (fig. S9, A to C), whereas there were notable changes in the proportion of mesenchymal and chondrogenic populations, with the latter decreasing in treated mice (fig. S9D) in agreement with the scRNA-seq data.
Activin A neutralizing antibody treatment inhibits intramuscular HO development
Because each animal model of HO has inherent limitations (51, 71), we used a muscle model of HO to test whether the activin A immunotherapy was effective in this system as well (72). Accordingly, 50 μl Matrigel aliquots containing rhBMP2 or rhBMP6 were micro-injected into the central portion of the gastrocnemius muscle of 2 months-old female mice, where each aliquot elicited an immediate and palpable intramuscular mass upon matrix solidification. Half of the mice received biweekly injections of control pre-immune IgG2b isotype antibody, whereas the remainder received the neutralizing activin A antibody (20 mg/kg/injection). As shown by μCT imaging, very large intramuscular HO masses had formed by day 14 in mice receiving pre-immune antibody and regardless of which BMP was used (Fig. 6, A and B). The masses were on average much larger than those forming subcutaneously (Fig. 1, A and B). Systemic administration of activin A antibody markedly reduced the development of intramuscular HO in both rhBMP2- and rhBMP6-implanted mice (Fig. 6, A and B). Inhibition was over 80%, as indicated by μCT imaging quantification (Fig. 6, C and D). Histochemical analyses showed that compared to controls, the administration of neutralizing antibody reduced cartilage formation (fig. S10).
Fig. 6. Systemic administration of activin A neutralizing antibody inhibits intramuscular HO development.
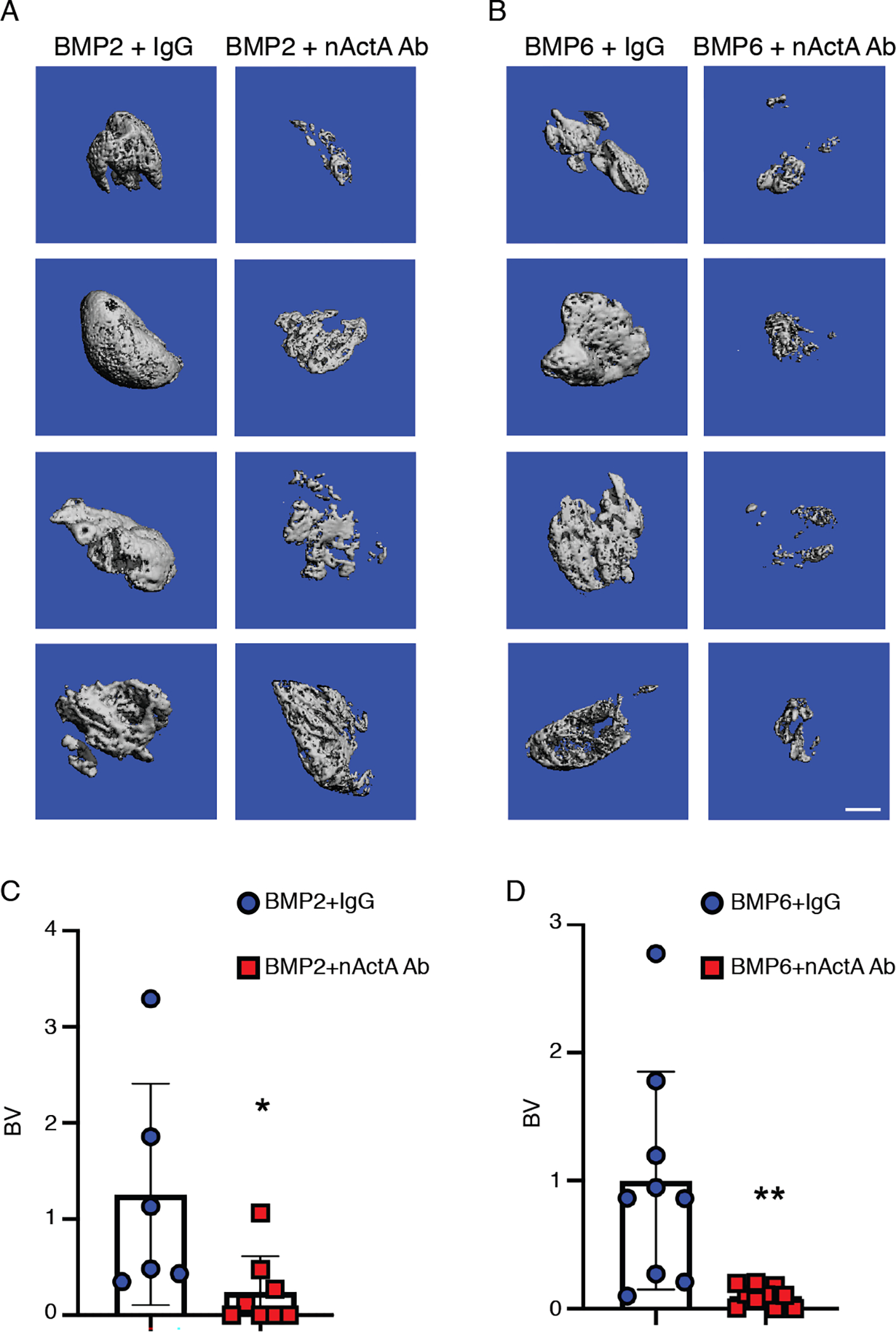
(A and B) μCT images of intramuscular mineralized tissue masses in mice implanted with Matrigel-rhBMP2 (A) or Matrigel-rhBMP6 (B) mixtures and then treated twice per week with control pre-immune mouse monoclonal IgG (IgG) or neutralizing activin A mouse monoclonal antibody (nActA.Ab) as indicated. Samples were harvested and processed for analyses on day 14 from implantation. (C and D) Quantification of bone volume (BV) within the ectopic masses retrieved from the indicated mouse groups. Data are from indicated numbers of mice (2 data point per mouse) from a minimum of 3 independent experiments and are plotted as mean ± S.D. N = 6–10 ectopic masses, 2 masses per mouse, per experimental group. Student’s t tests were used in all experiments to determine statistical significance. *P<0.05; **P<0.01. Scale bar, 1 mm
Next, we analyzed whether endogenous activin A was present in developing HO masses and what major cell population(s) were characterized by it. Using day 7 HO masses to capture the chondrogenic phase, we found that the protein was readily apparent in chondrogenic cells and chondrocytes within the interstitial area of muscle (fig. S11). By day 14, activin A strongly characterized residual chondrogenic cells and also bone lining cells and osteoblasts (fig. S11). The overall amounts of immunologically detectable activin A were markedly lower in the marginal HO masses present in nActA.Ab-treated mice at either time point (fig. S11).
Exogenous activin A stimulates HO development and chondrogenic cell differentiation
Because administration of activin A neutralizing antibody inhibited HO, we reasoned that the opposite should occur if exogenous activin A were to be provided. To test this possibility, adult female mice were implanted with 250 μl Matrigel aliquots containing rhBMP2 or rhBMP6 without or with inclusion of recombinant activin A, up to 4-fold w/w excess. Although rhBMP2 and rhBMP6 each induced prominent HO by day 14, as expected (Fig. 7A and fig. S12A), inclusion of activin A boosted HO formation (Fig. 7, B and C and fig. S12, B and C), amounting to a 2- to 3-fold increase by BV quantification (Fig. 7D and fig. S12D). HO masses from mice receiving rhBMP6 alone and rhBMP6 plus activin A all contained large amounts of safranin O–positive maturing cartilage and alizarin red–positive endochondral bone, but there was more endochondral bone accumulation in HO masses from mice receiving rhBMP6 and activin A combination (fig. S12, E and F).
Fig. 7. Exogenous recombinant activin A stimulates HO development.
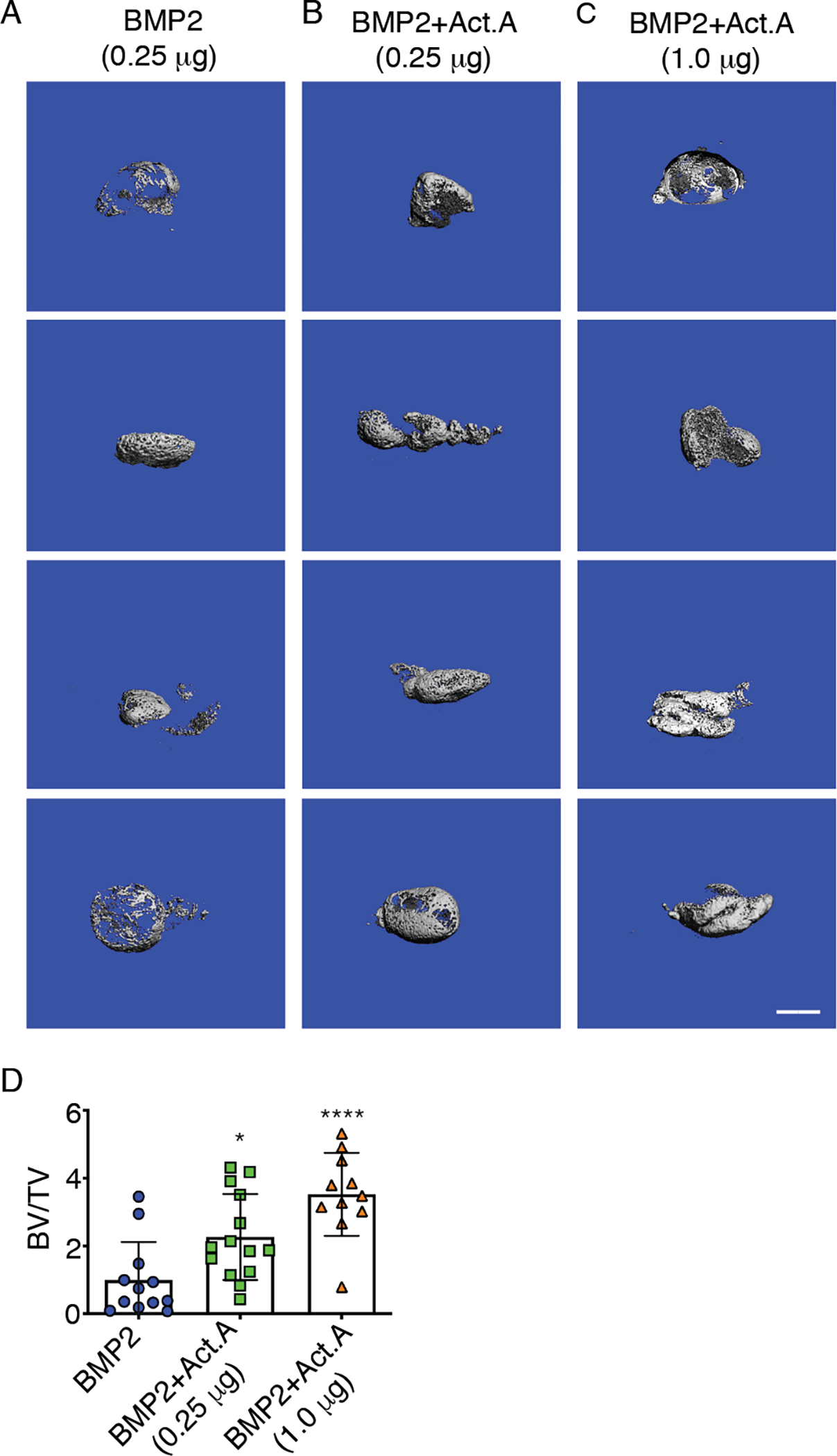
(A to C) μCT images of ectopic mineralized tissue masses present on day 14 in mice implanted subcutaneously with Matrigel-rhBMP2 (A), Matrigel-rhBMP2 plus recombinant activin A at 1:1 w/w ratio (B), or Matrigel-rhBMP2 plus recombinant activin A at 1:4 w/w ratio (C). Images show mineralized masses from 4 different mice in each treatment group. (D) Quantification of bone volume relative to total volume (BV/TV) in ectopic masses retrieved from all the mouse groups in (A–C). N = 11–14 mice per experimental group, 2 masses per mouse. Data was compiled from 3 independent experiments and presented as mean ± S.D. Student’s t tests were used in all experiments to determine statistical significance. *P<0.05; ****P<0.0001. Scale bar, 1 mm
Activin A could possibly stimulate HO by various means given its multiple properties and functions (38). Our single-cell transcriptome data showed that endogenous Inhba expression characterized mesenchymal and chondrogenic progenitors present in developing HO masses, raising the possibility that a role of activin A signaling could be to further enhance the differentiation of committed chondrogenic cells. To test this thesis, we used preskeletal progenitor cell populations isolated from E11.5 mouse embryo limb buds (73). Upon seeding in micromass culture, these committed progenitors resume their development, undergo aggregation and condensation, differentiate, and produce cartilage nodules over time in vitro (73, 74). Starting about 2 hrs after seeding, the limb bud cell cultures were continuously treated with different doses of recombinant activin A in the absence or presence of SB431542, a potent pSMAD2/3 signaling inhibitor (75). As negative and positive controls, companion cultures received an equal volume of vehicle or were treated with rhTGF-β1, which promotes chondrogenesis through activation of pSMAD2/3 (42, 47, 48). Control cultures receiving vehicle displayed an appreciable and stereotypic degree of chondrogenic cell differentiation over culture time, but activin A treatment (100 ng/ml) greatly enhanced chondrogenesis as measured by alcian blue staining and quantification (Fig. 8A) and the expression of such chondrogenic markers as Sox9, Acan, and Col2a1 (Fig. 8B). Activin A treatment also increased pSMAD2 abundance (Fig. 8C), and increases in both chondrogenesis and pSMAD2 signaling were prevented by 10 μM SB431542 co-treatment (Fig. 8, A and C). As expected, rhTGF-β1 treatment (5 ng/ml) stimulated both chondrogenesis and pSMAD2 abundance, responses that were both prevented by SB431542 co-treatment (Fig. 8, A and C).
Fig. 8. Activin A stimulates chondrogenic cell differentiation.
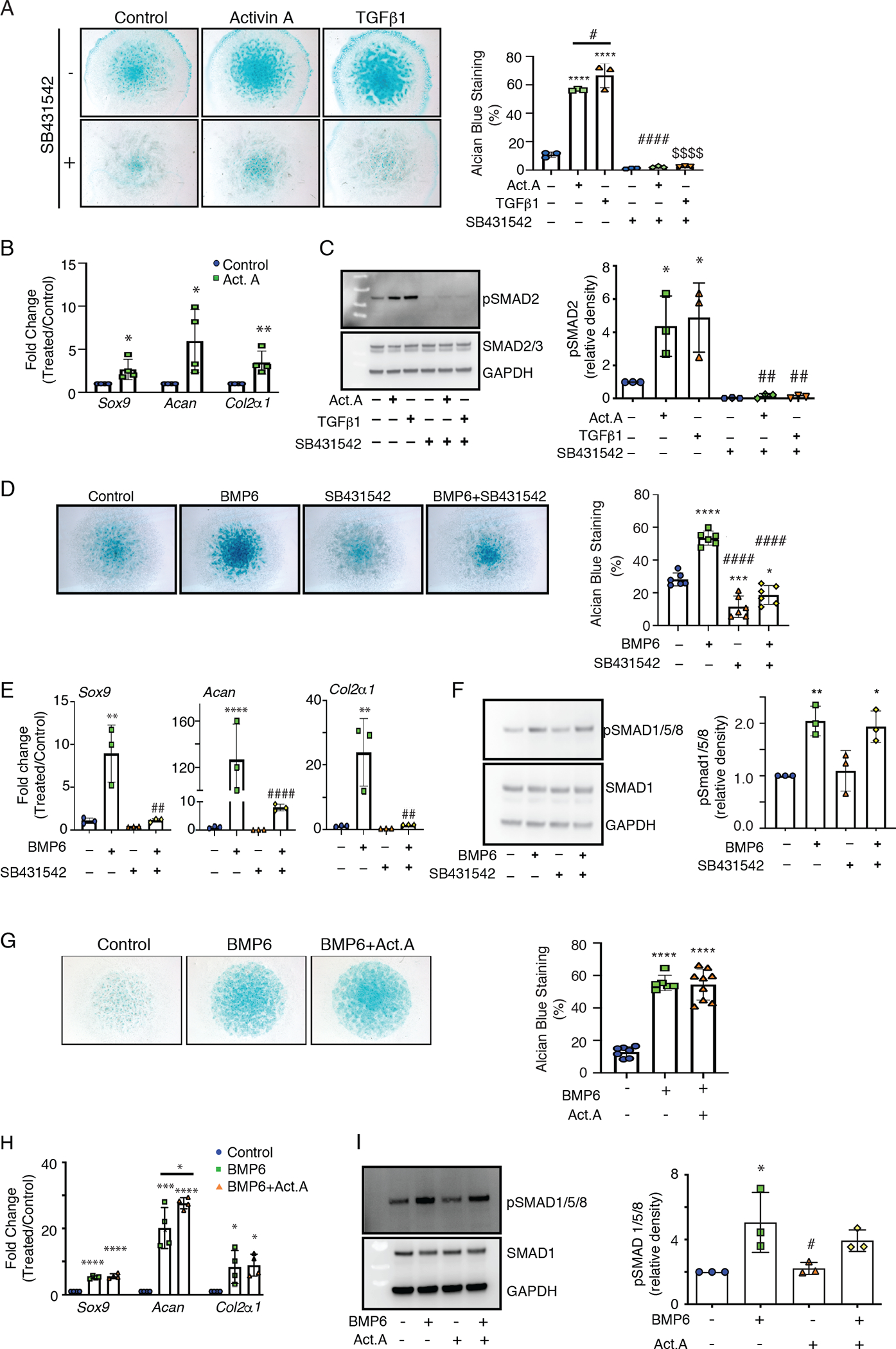
(A) Images and quantification of alcian blue staining of E11.5 limb bud mesenchymal cells in micromass cultures treated with recombinant activin A (100 ng/ml) or rhTGF-β1 (5 ng/ml) in the absence (−) or presence (+) of the pSMAD2/3 signaling antagonist SB431542 (10 μM), starting about 2 hrs after plating. Control cultures received vehicle only. Alcian blue staining was quantified at day 3 using ImageJ and Particle Analysis software. (B) Expression of chondrogenic genes Sox9, Acan, and Col2a1 in day 3 control versus activin A–treated cultures in (A). (C) Immunoblot analysis and quantification of phosphorylated SMAD2 (pSMAD2) and SMAD2/3 in limb bud mesenchymal cell cultures that were acutely treated for 1 hr with activin A, TGF-β1, and the pSMAD2/3 inhibitor SB431542 as indicated. Molecular weight markers are in the left-most lane. Protein loading consistency was assessed by re-processing the blots with antibodies specific for nonphosphorylated SMAD2/3 and GAPDH. (D) Images and quantification of alcian blue staining of micromass cultures treated with rhBMP6, SB431542, or both and processed on day 3. (E) RT-PCR quantification of Sox9, Acan, and Col2a1 expression in day 3 control versus treated and co-treated cultures in (D). (F) Immunoblot analysis and quantification of pSMAD1/5/8 in micromass cultures after 1 hr treatment with rhBMP6 and SB431542 as indicated. Molecular weight markers are in the left-most lane. Loading was assessed by re-blotting for SMAD1 and GAPDH. (G) Images and quantification of alcian blue staining in micromass cultures on day 3 treated with rhBMP6 (50 ng/ml) alone or in combination with activin A (100 ng/ml) starting about 2 hrs from plating. (H) RT-PCR quantification of Sox9, Acan, and Col2a1 expression in day 3 control versus BMP6-treated and BMP6 and activin A co-treated cultures. (I) Immunoblot analysis and quantification of pSMAD1/5/8 in micromass cultures treated for 1 hr with rhBMP6 and activin A as indicated. Molecular weight markers are in the left-most lane. Loading was assessed by re-blotting for SMAD1 and GAPDH. Statistical significance was determined by Student’s t tests and one-way ANOVA in all experiments. *P<0.05; **P<0.01; ****P<0.0001; #P<0.05; ##P<0.01; ####P<0.0001; $ $<0.01. * indicates a comparison to the control; # indicates a comparison to BMP6 or Activin A; and $ indicates a comparison to TGF-β1. N = 3 to 5 independent experiments with 3 repeats per sample, and data are presented as mean ± S.D.
Given the relationship between pSMAD2/3 activation and chondrogenesis, we asked whether this pathway could be part of the overall cellular mechanisms through which BMPs normally promote chondrogenesis. Thus, we treated micromass cultures with rhBMP6 in the absence and presence of SB431542. As assessed by alcian blue staining and expression of chondrogenic markers, treatment with rhBMP6 alone stimulated chondrogenesis compared to control cultures with no treatment, as expected (Fig. 8, D and E). SB431542 inhibited baseline chondrogenesis on its own and inhibited rhBMP6-induced chondrogenesis (Fig. 8, D and E). The specificity of drug action was confirmed by the fact that the drug had no obvious effects on pSMAD1/5/8 abundance (Fig. 8F).
Previous studies with cultured human kidney and myeloma cells showed that activin A can antagonize the function of BMP6 and its canonical signaling occurring through phosphorylation of SMAD1, SMAD5, and SMAD8 (SMAD1/5/8) (33), though this antagonism was not observed toward BMP2 (76). Because the combination of rhBMP6 plus activin A further stimulated HO development as our data show, it would appear that activin A does not antagonize BMP6 in the context of skeletogenic cell differentiation. To test this prediction, we treated mouse limb bud cells in micromass cultures as above with rhBMP6 in the absence or presence of activin A (up to 2-fold excess). At the treatment dose used, rhBMP6 maximally stimulated chondrogenesis over control cultures treated with vehicle only (Fig. 8G), in line with the known pro-chondrogenic properties of this BMP (77). We observed no obvious inhibitory effects of activin A on rhBMP6-stimulated chondrogenesis as revealed by alcian blue staining and quantification (Fig. 8G) and chondrogenic gene expression (Fig. 8H). rhBMP6 treatment increased SMAD1/5/8 phosphorylation compared to the control (Fig. 8I) but again, activin A did not interfere with this increase in pSMAD1/5/8 (Fig. 8I). The responses to rhBMP6 alone or rhBMP6 plus activin A were not accompanied by major changes in the expression of genes encoding relevant type I and type II TGF-β superfamily surface receptors (fig. S13, A and B), indicating that increased receptor activities, rather than expression, were responsible for the increased pSMAD1/5/8 response. One exception was a decrease in Alk2 (also known as Acvr1) and Alk7 (also known as Acvr1c) expression, possibly a negative feedback response to limit excessive cellular responses to ligands. Together, the data indicate that activin A antagonism of BMP6 action and signaling does not apply to chondrogenesis and may rather be cell type and context–dependent.
DISCUSSION
Our data provide evidence that activin A is an important regulator and stimulator of acquired forms of endochondral HO. The cytokine appears to act early and upstream during HO given the ability of activin A neutralizing antibody to markedly reduce its development in the BMP-implant mouse models used. Our single-cell transcriptome data uncover the fact that in addition to macrophages and other inflammatory cells, Inhba expression prominently characterizes progenitor cell populations present around and within developing HO masses and coincides in part with the expression of Sox9 and other chondrogenic genes. This indicates that the protein has at least a dual role in the genesis and progression of HO, first as a component of the initial inflammatory response and then as part of signaling mechanisms prodding and stimulating committed cell progenitors toward chondrogenesis and heterotopic bone formation. This is supported by our data with limb bud chondrogenic progenitors in micromass cultures in which activin A greatly enhanced their baseline differentiation and did so through canonical pSMAD2/3 signaling. Because treatment with a pSMAD2/3 signaling antagonist dampened both baseline and BMP-stimulated chondrogenesis, this pathway does appear to occupy an upstream position in the regulatory hierarchy controlling chondrogenesis. Previously, endogenous Inhba expression was shown to characterize human bone marrow progenitors, and inhibition of Inhba expression by RNA interference impaired their differentiation under chondrogenic conditions in vitro (49). In addition, activin A is present in bone and facilitates bone accrual and fracture repair when applied locally in rodents (45, 46). Together, our data and previous studies offer new insights into the biology of activin A as a TGF-β superfamily member intimately involved in skeletal tissue formation in both physiological and pathological contexts.
These tantalizing conclusions relate well with those reached in recent studies on the positive roles of TGF-β itself and canonical pSMAD2/3 signaling in HO development (64, 78) and normal bone homeostasis (79, 80). With regard to HO, Wang et al. first examined surgical retrieval specimens of HO masses from surgery and trauma patients and detected a high number of pSMAD2/3-positive cells in osteoid tissues and high amounts of active TGF-β1 in their serum (78). They also observed an abundance of pSMAD2/3-positive cells—including mesenchymal progenitors—in developing HO masses in intramuscular BMP-implant and tendon injury mouse models of HO. Conditional Tgfβ1 ablation in the monocyte and macrophage lineage or systemic treatment with a TGF-β1 neutralizing antibody (1D11) attenuated HO formation. In another study, Sorkin et al. used a tendon injury model of mouse HO, scRNA-seq, and developmental trajectories to further probe HO pathogenesis (64). They uncovered dynamic functional interplays amongst TGF-β–producing monocytes and macrophages, chondrogenic progenitor cells, and HO progression, and conditional Tgfβ1 ablation in monocytes and macrophages caused a marked decrease in both cartilage formation and HO development. Considered together, these TGF-β studies and our data herein lead to a model in which canonical activin A and TGF-β signaling through pSMAD2/3 would set the HO process in motion together with other inflammatory cytokines (fig. S14). These ligands act upstream of—but strictly rely upon—BMP signaling to induce both skeletogenic commitment and differentiation of progenitor cells, and targeting activin A or TGF-β by immunotherapy elicits a marked attenuation of HO development (fig. S14).
It remains to be determined what exactly activin A and TGF-β each does with regard to progenitor cell commitment and differentiation, how their putative actions relate to those exerted by BMP signaling, and how Inhba expression is regulated in inflammatory and progenitor cells during HO development. With regard to the latter question, our predictions of cell-cell interplays and ligand-receptor interactions suggest that progenitor cells could be influenced by macrophages and mast cells, and their expression of Inhba would be regulated by such inflammatory factors as IL-1β and TNF. In addition, it remains to be determined whether activin A and TGF-β exert similar central roles in other forms of acquired HO, including spinal cord- and tendon-injury-associated HO; limb amputation–associated HO that is propelled by local bacterial infection; and intramembranous-preponderant HO that mostly involves direct bone formation without a cartilage intermediate (2, 81–83). At this regard, a just published study reported that systemic administration of an activin A antibody did not appreciably inhibit HO in a skin burn/tendon-injury mouse model of HO (84). The observed lack of response in this tendon model compared to those used here could be due to differences in: HO pathogenesis and severity; inciting cues; treatment regimens; antibody potency and specificity; or antibody bioavailability at different anatomical sites. Addressing these possibilities will require side-by-side comparisons of the different models. Lastly, it is important to highlight here exciting studies on fibrodysplasia ossificans progressiva (FOP), a rare, congenital and severe disorder characterized by aggressive endochondral HO and caused by gain-of-function mutations in the type I BMP receptor ACVR1 (also called ALK2) (85). Two groups discovered that a neomorphic function in mutant ALK2 allows it to directly respond to activin A, elicit BMP-like pSMAD1/5/8 signaling in addition to canonical pSMAD2/3 signaling, and induce ectopic skeletogenesis and HO (53, 86). In FOP then, activin A would also elicit pSMAD1/5/8 signaling and could on its own provoke and sustain HO development, offering a potent therapeutic target (53). Notably, TGF-β may represent an alternative therapeutic target for FOP because systemic administration of the TGF-β1 neutralizing antibody 1D11 effectively inhibits HO in a mouse model of aggressive FOP (78).
In cell types such as human kidney and myeloma cells, activin A can actually antagonize the biological effects and canonical pSMAD1/5/8 signaling elicited by BMP6 and BMP9, though not the effects and canonical pSMAD1/5/8 signaling of BMP2 and BMP4 (76). As pointed out above, these data indicate that in certain circumstances, activin A is able to counteract BMPs that normally signal through the type 2 receptors ACVR2A and ACVR2B in combination with type 1 receptor ALK2, but is unable to antagonize BMP2 and BMP4 that normally signal through BMPR2 in combination with ALK3 and ALK6. The antagonistic effects of activin A on BMP6 canonical signaling have been confirmed in mouse embryonic stem cells in culture (53). In our studies here, however, activin A did not interfere with, but actually potentiated, the pro-HO action of BMP6 in vivo and did not interfere with BMP6 stimulation of chondrogenesis in vitro. This indicates that the roles of—and responses to—activin A depend on the physiological context and very likely, on the specific combinations of type 1 and type 2 receptors expressed by given cell types and at different developmental stages (33, 34). It should also be pointed out that based on structural features and phylogenic considerations, (i) BMP2 and BMP4, (ii) BMP5, BMP6, and BMP7, and (iii) the activins occupy three evolutionary subgroups within the TGFβ protein superfamily that have diverse overall roles and functions (33). In recent structural studies on these three protein subgroups, we compared their Cardin-Weintraub (CW) peptide domain (87) that normally regulates their interactions with cell surface heparan sulfate proteoglycan co-receptors and their overall bio-availability and signaling activities (88–90). We found that the CW domain is structurally, stereologically, and functionally distinct in each subgroup, and such diversity is strictly conserved though evolution (91, 92). In sum, the overall functional properties of TGF-β superfamily members are likely to result from complex interactions and interplays amongst ligands, receptors, co-receptors, and other modulators and are still to be wholly deciphered, including differences that depend on the biological context. Further insights into these mechanisms should provide a better understanding of activin A functioning during HO and could uncover additional modalities for therapy.
MATERIALS AND METHODS
HO mouse models
All mouse studies were conducted after review and approval by the IACUC at our Institutions. Standard subcutaneous and intramuscular models of HO were used that we and others have shown to be effective, mild and non-invasive previously (50, 52, 72, 93, 94). Briefly, growth factor-reduced, phenol red-free Matrigel (Corning) was mixed with: recombinant human bone morphogenetic protein-2 (0.25 μg) (rhBMP2; Gene Script Corp); recombinant human BMP-6 (0.1 μg) (rhBMP6; R&D Systems); recombinant activin A (Act A; 1 μg R&D Systems); rhBMP2 (0.25 μg) plus Act A (0.25 or 1.0 μg; R&D Systems); or rhBMP6 (0.1 μg) plus Act A (0.25 or 1 μg) per 250 μl. For experiments utilizing activin A neutralizing monoclonal antibody (nActA.Ab; IgG2b isotype; Biolegend), Matrigel was pre-mixed with: rhBMP2 (0.25 μg) plus mouse pre-immune control IgG2b isotype antibody (25 μg; Biolegend); rhBMP2 (0.25 μg) plus nActA.Ab (25 μg); rhBMP6 (0.1 μg) plus pre-immune control IgG2b isotype (25 μg); or rhBMP6 (0.1 μg) plus nActA.Ab (25 μg). For the subcutaneous HO model, the above mixtures were prepared on ice, and 250 μl aliquots of each combination were injected at two ventral subcutaneous sites in 6–8 week old CD-1 female mice. For the intramuscular HO model, a 50 μl aliquot of each protein combination was injected in the right and left gastrocnemius muscle in female mice. Twice per week the mice received subcutaneous injections of 10 mg/kg (subcutaneous HO model) or 20 mg/kg (intramuscular HO model) of pre-immune mouse antibody or Act A neutralizing antibody as above. Female mice were used because they are easier to handle and elicit a consistent HO process indistinguishable from that in males (94). Subcutaneous and intramuscular implantations were carried out under anesthesia by inhalation of 1.5% isoflurane in 98.5% oxygen as prescribed by IACUC. HO samples were harvested at Day 5 or Day 14, fixed in 4% paraformaldehyde (PFA) overnight and then processed for micro computed tomography (μCT) and histological and immunohistological analyses.
Imaging and quantification by μCT
After fixation, HO samples were washed in 1X PBS and stored in 70% ethanol at 4°C. We carried out scanning and analysis using a Scanco μCT 35 instrument (52). Samples were scanned at a resolution of 21 μm, 200 ms integration time, 55kVp energy and 145 μA intensity. HO samples from the subcutaneous model were analyzed at threshold 33 to measure total tissue volume (TV) and re-analyzed at threshold 125 to measure bone volume (BV). Data were used to calculate bone volume/total volume ratios and are presented in relation to control values. HO samples from the intramuscular model were analyzed at threshold 165 to measure bone volume. These values were calculated based on averages obtained by multiple samples from at least two independent experiments.
Histochemistry, immunohistochemistry and in situ hybridization
Day 5 and Day 14 HO samples were fixed overnight and then immediately processed without decalcification. Fixed samples were embedded in paraffin and serial 5.0 μm cross sections were stained with hematoxylin and eosin for routine histology, Safranin O/Fast green to reveal cartilage and Alizarin red to reveal mineralized matrix. Representative sections were subjected to heat-antigen retrieval and processed for immunohistochemistry using 1:200 dilution of goat activin A antibodies (R&D Systems, AF338) at 4°C overnight. Companion sections were incubated with a similar concentration of pre-immune goat serum. After rinsing, sections were reacted with biotinylated secondary antibodies followed by DAB color development. Bright-field images were taken with a Nikon Eclipse Ci equipped with a Nikon camera operated with NIS Elements software. In situ hybridization was carried out on serial paraffin sections as described (95). Briefly, after paraffin removal and rehydration, serial tissue sections were pretreated with 10 μg/ml proteinase K for 10 min at room temperature, post-fixed in 4% PFA, washed with PBS containing 2 mg/ml glycine, and treated with 0.25% acetic anhydride in triethanolamine buffer. Sections were hybridized with antisense or sense 35S-labeled riboprobes (approximately 1 × 106 DPM/section) at 50°C for 16 hrs, coated with Kodak NTB-3 emulsion diluted 1:1 with water and exposed for 2 to 4 days. Slides were developed with Kodak D-19 at 20°C and stained with hematoxylin. cDNA clones used as templates for probe transcription included: collagen I alpha1 (nt.233–634; NM_007742); collagen II alpha 1 (nt. 1095–1344; X_57982),; and collagen X alpha1 (nt. 1302–1816; NM_009925). Dark-field images were taken with a Nikon Eclipse Ci equipped with a Nikon camera operated with NIS Elements software.
Preparation, treatment and analysis of micromass cultures
Primary mesenchymal cell micromass cultures were prepared from E11.5 CD-1 mouse embryo limb buds as described previously (73, 96). Briefly, limb buds were dissociated into single cell suspensions by incubation with 0.5% trypsin-EDTA at 37°C for 30 min. The cells were diluted to a concentration of 10 × 106 cells/ml in DMEM containing 3% FBS and antibiotics. Micromass cultures were initiated by spotting 15 μl of the cell suspension (1.5 × 105 cells/spot) onto the surface of 24-well tissue culture plates. After a 2 hr incubation at 37°C in a humidified CO2 incubator to allow for cell attachment, the cultures were flooded with medium containing the following reagents depending on the experiment performed: Act A (100 ng/ml); TGF-β1 (5 ng/ml; R&D Systems); TGF-β receptor inhibitor (SB431542; 10 μM; Tocris); Act A (100 ng/ml) plus SB431542 (10 μM); TGF-β1 (5 ng/ml) plus SB431542 (10 μM); rhBMP6 (50 ng/ml); or Act A (100 ng/ml) plus rhBMP6 (50 ng/ml). Fresh reagents were given with medium change daily. Equivalent amounts of vehicle (4 mM HCl or ethanol) were added to respective companion control cultures. Cultures were stained with Alcian Blue (pH 1.0) after 3 days to monitor chondrogenic cell differentiation (73, 96). Images were taken with a Nikon SMZ-U microscope equipped with a SPOT insight camera and acquired with SPOT 4.0 software. Micromass analysis was performed using ImageJ. Images were made binary under a RGB threshold and “Particle Analysis” was utilized to measure Alcian blue positive area (97).
Gene expression analysis
Total RNA was isolated from control and treated micromass cultures on day 3 using TRIzol reagent (cat# 15596–026, Life Technologies) according to the manufacturer’s protocol. RNA was quantified by Nanodrop. One microgram total RNA was reversed transcribed using the Verso cDNA kit (cat# AB1435/A, Thermo Scientific). Quantitative real-time PCR was carried out using SYBR Green PCR Master Mix in an Applied Biosystems 7500 according to manufacturer’s protocol. Gapdh was used as the endogenous control, and relative expression was calculated using the ΔΔ Ct method. Primer information is in Table S2.
Protein signaling assays
To monitor and quantify protein signaling activity in primary mouse embryo limb bud mesenchymal cells, freshly-isolated cells from E11.5 embryos were seeded at a lower density (10 × 105 cells/ml) to elicit confluent monolayers and allow ready cell surface access to exogenous proteins and reagents to be tested acutely. Cultures were maintained for 24 hrs in DMEM containing 3% FBS and antibiotics and were then treated for 1 hr with: appropriate vehicle control; Act A (100 ng/ml); rhBMP6 (50 ng/ml); Act A (100 ng/ml) plus rhBMP6 (50 ng/ml); TGF-β1 (5 ng/ml); SB431542 (10 μM); Act A (100 ng/ml) plus SB431542 (10 μM); or TGF-β1 (5 ng/ml) plus SB431542 (10 μM). Cells were immediately lysed in 1X RIPA solution with protease and phosphatase inhibitors; samples were centrifuged at 13,200 rpm at 4°C for clarification and supernatants were collected. Protein concentration for each sample was determined using the MicroBCA Protein Assay Kit (Thermo Scientific) according to the manufacturer’s protocol. Total cellular proteins (30 μg/lane) were electrophoresed on 4–12% NuPAGE Bis-Tris gel (Life Technologies) and transferred to PVDF membranes (Life Technologies). Membranes were blocked in 5% nonfat milk/1X Tris buffered saline/Tween 20 (TBST) and incubated overnight at 4°C with one of the following antibodies: phospho-SMAD1/5/9 (pSMAD1/5/9) (1:1000; Cell Signaling) or phospho-SMAD2 (1:1000; Cell Signaling). Membranes were washed in 1X TBST and incubated with anti-rabbit HRP-linked antibodies (1:2000; Cell Signaling) for 1 hr at room temperature. Antigen-antibody complexes were detected with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) chemiluminescent detection system. Membranes were re-blotted with SMAD1 (1:1000; Cell Signaling) or SMAD 2/3 (1:1000; Santa Cruz Biotechnology) antibodies for normalization. For loading control, membranes were blotted with GAPDH (1:1000; Santa Cruz Biotechnology). ImageJ was used to determine band intensities. All the above procedures were also used in experiments involving Ad-293 cells, a cell line commonly used in studies on signaling activities of TGFβ superfamily members (89). Cells were transiently transfected with full length expression plasmids encoding wild type ALK2 or mutant ALK2R206H which is the most common mutation seen in patients with FOP (85). Treatments, including the addition of Activin B (100 ng/ml; R&D Systems), cell harvesting and immunoblots were all as above.
Single-cell transcriptome analyses
Mice were implanted subcutaneously with: Matrigel alone; Matrigel containing rhBMP2 plus control preimmune mouse antibody; or rhBMP2 plus neutralizing Act A antibody, following the procedure and doses detailed above. Additional antibodies were provided by injection on day 1 and day 3. Ectopic tissue masses were harvested on day 5 from initial implantation, minced and dissociated into single cell suspension by treatment with dispase (50 U/ml in HBSS) for 2 hrs at 37°C. Cells were then suspended in 5% FBS/DMEM at a concentration of 700–1200 cells/μl. Cell suspensions were immediately delivered to the Center for Applied Genomics (Children’s Hospital of Philadelphia) for determination of cell viability and sample processing. Viability of > 95% in all samples. cDNA libraries of cells from each group were generated by Chromium controller (10X Genomics Inc.), barcoded and purified as described by the manufacturer, and sequenced using a 2×150 pair-end configuration on an Illumina HiSeq platform at a sequencing depth of ~400 million reads. Cell Ranger (version 3.0.2) was used to de-multiplex reads, followed by extraction of cell barcode and unique molecular identifiers (UMIs). The cDNA insert was aligned to a modified reference mouse genome (mm10) using STAR (98).
Seurat package V3 was used for filtering, variable gene selection, dimensionality reduction analysis and clustering standardly (55). Doublets or cells with poor quality (genes>6000, genes<200, or >5% genes mapping to mitochondrial genome) were excluded. Expression was natural log transformed and normalized for scaling the sequencing depth to a total of 1×104 molecules per cell. For the integrated dataset, anchors from different dataset were defined using the FindIntegrationAnchors function, and these anchors were then used to integrate datasets together with IntegrateData. Datasets were scaled by regressing out the number of UMIs and percent mitochondrial genes. Cell-cycle heterogeneity effect was also regressed out by using the Seurat Cell-cycle scoring function. Statistically significant principle components were selected as input for t-SNE plots. Different resolutions for clustering were used to demonstrate the robustness of clusters. In addition, differentially expressed genes within each cluster relative to the remaining clusters were identified using FindMarkers within Seurat. Sub-clustering was performed by isolating the mesenchymal and skeletogenic lineage clusters using known marker genes, followed by reanalysis as described above. Gene ontology analysis was performed using the clusterProfiler package (99).
To computationally delineate the developmental progression of HO from mesenchymal to chondrogenic cells and order them in pseudotime and trajectory, we used the algorithms of Monocle package V2 (63). A list of differentially expressed genes that change as a function of pseudotime was obtained using “dpFeature”. Top 1000 significant genes were selected as ordering genes. Heat maps to visualize pseudotime-dependent genes were generated by using a subset of top most significant differentially-expressed genes. Cell trajectories were established using genes selected by a principal-component-analysis-based method for cell ordering according to previously described methods (55, 63).
Flow cytometry
Ectopic tissue masses were harvested on day 5 from mice implanted with Matrigel alone and Matrigel/rhBMP2 mixture. The latter were sub-divided into two groups each systemically treated with preimmune antibody or Act A antibody as above. Samples were dispersed into single cell populations by dispase treatment for 1 hr, and cells were stained with fluorescent conjugated antibodies, including FITC anti-CD45 (Biolegend, 103107) and Brilliant Violet 421 anti-CD34 antibodies (BD Biosciences, 562608) for 60 minutes at 4°C. Cells washed in flow buffer (2% FBS in PBS) and analyzed on a BD Biosciences LSR II flow cytometer.
Cell-cell and ligand-receptor interaction predictions and analyses
To analyze possible cell–cell communication mediators among different cell types, we used CellPhoneDB, a repository of ligands, receptors and interaction data that relies on public information to annotate receptors and ligands.(66) Briefly, each cell type cluster defined by Seurat was used as input into CellphoneDB that is based on statistical methods and analyses to generate interaction numbers of ligand/ receptor pair amongst different groups. Network visualization was done using Cytoscape version 3.5.1.(67) The networks layout was set to force-directed layout.
Further predictive analyses of ligand-receptor pairs were carried out using NicheNet,(68) allowing identification of putative ligands produced by immune cells regulating target genes in mesenchymal and chondrogenic cell cohorts. Ligand-target links are inferred with existing knowledge on signaling and gene regulatory networks. NicheNet defines a list of potentially active ligands and predicts affected and non-affected background genes in target cells (mesenchymal cells in our analyses). Ligands were ranked according to how well they predict putative gene sets of interest compared to unrelated genes. For heatmaps presentation, we focused on top 5 ligands (out of 119 ligands) secreted by immune cells and ordered them according to Pearson correlation value and identified their corresponding receptors. Scores reflected weight of ligand/receptor interactions in the integrated weighted ligand signaling network.
Statistical methodologies
Data were analyzed using Prism 6 (GraphPad Software, Inc). Student’s t tests and one-way ANOVA was used to establish statistical significance. Threshold for significance for all tests was set as p ≤ 0.05.
Supplementary Material
Fig. S1. Activin A neutralizing antibody specifically interferes with recombinant activin A-induced pSMAD2 activation.
Fig. S3. Recruitment and migration of cells into implanted Matrigel
Fig. S2. Activin A antibody administration inhibits rhBMP6-induced subcutaneous heterotopic cartilage and bone formation
Fig. S4. GO term analysis of cellular phenotypic and developmental processes
Fig. S5. KEGG-based Pathview plot analysis of possible mode of action of anti-activin A treatment
Fig. S6. Inflammatory and hematopoietic cells are present at early stages of HO development
Fig. S7. Phenotypes of macrophages present in developing HO masses
Fig. S9. Flow cytometry analyses of cell populations present in HO masses
Fig. S8. Analyses of cell-cell communication and ligand/receptor/target interactions in mesenchymal and immune cells present in developing HO sites
Fig. S10. Activin A antibody administration inhibits intramuscular rhBMP6-induced HO
Fig. S12. Exogenous activin A stimulates rhBMP6-induced HO development
Fig. S11. Endogenous activin A is present in developing HO masses
Fig. S13. Gene expression analysis of receptors for TGF-β superfamily members
Fig. S14. Working model of activin A and TGF-β roles in acquired HO development
Table S2. Primer sequences
Table S1. GSEA of macrophage clusters
Acknowledgements:
We thank the Center for Applied Genomics at CHOP for advice and performance of single cell RNAseq reactions, the Biostatistics & Data Management Core (BMC) at CHOP for advice and guidance with statistical analyses, and the Penn Center for Musculoskeletal Disorders at the University of Pennsylvania for expert use of the μCT imaging core facility (supported by NIH P30 grant AR069619).
Funding:
This work was supported by the National Institutes of Health grant RO1AR071946 (to M.P.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
The RNA-seq data have been deposited in the NCBI at NIH with accession number GSE157679. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.McCarthy EF, Sundaram M, Heterotopic ossification: a review. Skeletal Radiol. 34, 609–619 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Meyers C, Lisiecki J, Miller S, Levin A, Fayad L, Ding C, Sono T, McCarthy E, Levi B, James AW, Heterotopic ossification: A comprehensive review. J. Bone Min. Res. Plus 3, e10172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Legosz P, Drela K, Pulik L, Sarzynska S, Maldik P, Challenges of heterotopic ossification. Molecular background and current treatmnet strategies. Clin, Exp. Pharmacol. Physiol. 45, 1229–1235 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Ahrengart L, Periarticular heterotopic ossification after total hip arthroplasty. Risk factors and consequences. Clin. Orth. Relat. Res. 263, 49–58 (1991). [PubMed] [Google Scholar]
- 5.Teasell RW, Mehta S, Aubut JL, Ashe MC, Sequeira K, Macaluso S, Tu L, Team SR, A systematic review of the therapeutic interventions for heterotopic ossification after spinal cord injury. Spinal Cord 48, 512–521 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nuovo MA, Norman A, Chumas J, Ackerman LV, Myositis ossificans with atypical clinical, radiographic or pathologic findings: a review of 23 cases. Skeletal Radiol. 21, 87–101 (1992). [DOI] [PubMed] [Google Scholar]
- 7.Bedi A, Zbeda RM, Bueno VF, Downie B, Dolan M, Kelly BT, The incidence of heterotopic ossification after hip arthroscopy. Am. J. Sports Med. 40, 854–863 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Forsberg JA, Pepek JM, Wagner S, Wilson K, Flint J, Andersen RC, Tadaki D, Gage FA, Stojadinovic A, Elster EA, Heterotopic ossification in high-energy wartime extremity injuries: prevalence and risk factors. J. Bone Joint Surg. 91, 1084–1091 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Potter BK, Burns TC, Lacap AP, Granville RR, Gajewski DA, Heterotopic ossification following traumatic and combat-related amputations: prevalence, risk factors, and preliminary results of excision. J. Bone Joint Surg. 89, 476–486 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Forsberg JA, Potter BK, Heterotopic ossification in wartime wounds. J. Surg. Orthop. Adv. 19, 54–61 (2010). [PubMed] [Google Scholar]
- 11.Garland DE, A clinical prospective on common forms of acquired heterotopic ossification. Clin. Orthop. 263, 13–29 (1991). [PubMed] [Google Scholar]
- 12.Evans KN, Forsberg JA, Potter BK, Hawksworth JS, Brown TS, Andersen R, Dunne JR, Takadi D, Elster EA, Inflammatory cytokines and chemokine expression is associated with heterotopic ossification in high-energy penetrating war injuries. J. Ortho. Trauma 26, e204–213 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Forsberg JA, Potter BK, Polfer EM, Safford SD, Elster EA, Do inflammatory markers portend heterotopic ossification and wound failure in combat wounds? Clin. Orth. Relat. Res. 472, 2845–2854 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal S, Loder SJ, Cholok D, Peterson J, Li J, Breuler C, Cameron Brownley R, Sung HH, Chung MT, Kamiya N, Li S, Zhao B, Kaartinen V, Davis TA, Qureshi AT, Schilpani E, Mishina Y, Levi B, Scleraxis-lineage cells contribute to ectopic bone formation in muscle and tendon. Stem Cells 35, 705–710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kan L, Peng CY, McGuire TL, Kessler JA, Glast-expressing progenitor cells contribute to heterotopic ossification. Bone 53, 194–203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ, Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Min. Res. 27, 1004–1017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal S, Loder S, Brownley C, Cholok D, Mangiavini L, Li J-S, Breuler C, Sung HH, Li S, Ranganathan K, Peterson J, Tompkins R, Herndon D, Xiao W, Jumlongras D, Olsen BR, Davis TA, Mishina Y, Schipani E, Levi B, Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl. Acad. Sci. USA 113, E338–E347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoeger T, Proetzel GE, Welzel H, Papadimitriou A, Dony C, Balling R, Hoffmann C, In situ gene expression analysis during BMP2-induced ectopic bone formation in mice shows simultaneous endochondral and intramembranous ossification. Growth Factors 20, 197–210 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Hannallah D, Peng H, Young B, Usas A, Gearhart B, Huard J, Retroviral delivery of Noggin inhibits the formation of heterotopic ossification induced by BMP-4, demineralized bone matrix, and trauma in an animal model. J. Bone Joint Surg, Am. 86, 80–91 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Sell S, Willms R, Jany R, Esenwein S, Gaissmaier C, Martini F, Bruhn G, Burkhardsmaier F, Bamberg M, Kusswetter W, The suppression of heterotopic ossifications: radiation versus NSAID therapy - A prospective study J. Arthroplasty 13, 854–859 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Popovic M, Agarwal A, Zhang L, Yip C, Kreder HJ, Nousiainen MT, Jenkinson R, Tsao M, Lam H, Milakovic M, Wong E, Chow E, Radiotherapy for the prophylaxis of heterotopic ossification: a systematic reviw and meta-analysis of published data. Radiother. Oncol. 113, 10–17 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Mishra MV, Austin L, Parvizi J, Ramsey M, Showalter TN, Safety and efficacy of radiation therapy as seceondary prophylaxis for heterotopic ossification of non-hip joints. J. Med. Imaging Radiat. Oncol. 55, 333–336 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Craven PL, Urist MR, Osteogenesis by radioisotope labeled cell populations in implants of bone matrix under the influence of ionizing radiation. Clin. Orth. Relat. Res. 76, 231–233 (1971). [DOI] [PubMed] [Google Scholar]
- 24.Sautter-Bihl ML, Liebermeister E, Nanassy A, Radiotherapy as a local treatment option for heterotopic ossifications in patients with spinal cord injury. Spinal Cord 38, 33–36 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Simmons DL, Botting RM, Hla T, Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56, 387–437 (2004). [DOI] [PubMed] [Google Scholar]
- 26.DiCesare PE, Nimni ME, Pen L, Yazdi M, Cheung DT, Effects of indomethacin on demineralized bone-induced heterotopic ossification in the rat. J. Orthop. Res. 9, 855–861 (1991). [DOI] [PubMed] [Google Scholar]
- 27.Moed BR, Resnick RB, Fakhouri AJ, Nallamothu B, Wagner RA, Effect of two nonsteroidal antinflammatory drugs on heterotopic bone formation in a rabbit model. J. Arthoplasty 9, 81–87 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Alfieri KA, Forsberg JA, Potter BK, Blast injuries and heterotopic ossification. Bone Joint Res. 1, 174–179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karunakar MA, Sen A, Bosse MJ, Sims SH, Goulet JA, Kellam JF, Indometacin as prophylaxis for heterotopic ossification after the operative treatment of fractures of the acetabulum. J. Bone Joint Surg. Br. 88, 1613–1617 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Vasileiadis GI, Sioutis IC, Mavrogenis AF, Vlabis K, Babis GC, Papagelopoulos PJ, COX-2 inhibitors for the prevention of heterotopic ossification after THA. Orthopaedics 34, 467 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Meiners T, Abel R, Bohm V, Gerner HJ, Resection of heterotopic ossification of the hip in spinal cord injured patients. Spinal Cord 35, 443–445 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Pavey GJ, Polfer EM, Nappo KE, Tintle SM, Forsberg JA, Potter BK, What risk factors predict recurrence of heterotopic ossification after excision of combat-related amputations? Clin. Orthop. Relat. Res. 473, 2814–2824 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salazar VS, Gamer LW, Rosen V, BMP signaling in skeletal development, disease and repair. Nat. Rev. Endocrinology 12, 203–221 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Schmierer B, Hill CS, TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Schwartz NB, Channing CP, Evidence for ovarian “inhibin”: suppression of the secondary rise in serum stimulating hormone levels in proestrous rats by injection of porcine follicular fluid. Proc. Natl. Acad. Sci. USA 74, 5721–5724 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vale W, Rivier J, Vaughan J, McClintock R, Corrigan A, Woo W, Karr D, Spiess J, Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature 321, 776–779 (1986). [DOI] [PubMed] [Google Scholar]
- 37.de Kretser DM, O’Hehir RE, Hardy CL, Hedger MP, The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell. Endocrinol. 359, 101–106 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Xia Y, Schneyer AL, The biology of activin: recent advances in structure, regulation and function. J. Endocrinol. 202, 1–12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrington AE, Morris-Triggs SA, Ruotolo BT, Robinson CV, Ohnuma S, Hyvonen M, Structural basis for the inhibtion of activin signaling by follistatin. EMBO J. 25, 1035–1045 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones KL, Brauman JN, Groome NP, de Kretser DM, Phillips DJ, Activin A release into the circulation is an early event in systemic inflammation and precedes the release of follistatin. Endocrinology 141, 1905–1908 (2000). [DOI] [PubMed] [Google Scholar]
- 41.McDowell N, Zorn AM, Crease DJ, Gurdon JB, Activin has direct long-range signaling activity and can form a concentration gradient by diffusion. Curr. Biol. 7, 671–681 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Rigueur D, Lyons KM, TFGβ signaling in cartilage development and maintenance. Birth Defects Res. (Part C) 102, 37–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, Phillips DJ, Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc. Natl. Acad. Sci. USA 104, 16239–16244 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Funaba M, Murakami M, Ikeda T, Ogawa K, Tsuchida K, Sugino H, Identification of tocopherol-associated protein as an activin/TGF-beta0inducible gene in mast cells. Biochim. Biophys. Acta 1763, 900–906 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Hirotani H, Ohtsuka-Isoya M, Mori S, Sakai R, Eto Y, Echigo S, Shinoda H, Activin A increases the bone mass of grafted bone in C3H/Hej mice. Calcif. Tissue Int. 70, 330–338 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Sakai R, Miwa K, Eto Y, Local administration of activin promotes fracture healing in the rat fibula fracture model. Bone 25, 191–196 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Seyedin SM, Thompson AY, Bentz H, Rosen DM, McPherson JM, Conti A, Siegel NR, Galluppi GR, Piez KA, Cartilage-inducing factor-A: Apparent identity with transforming growth factor-β. J. Biol. Chem. 261, 5693–5695 (1986). [PubMed] [Google Scholar]
- 48.Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ, Danielson KG, Hall DJ, Tuan RS, Transforming growth factor-β-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J. Biol. Chem. 278, 41227–41236 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Djouad F, Jackson WM, Bobick BE, Janjanin S, Song Y, Huang GT, Tuan RS, Activin A expression regulates multipotency of mesenchymal progenitor cells. Stem Cell Res. Therapy 1, 11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Connor JP, Animal models of heterotopic ossification. Clin. Orth. Relat. Res. 346, 71–80 (1998). [PubMed] [Google Scholar]
- 51.Scott MA, Levi B, Askarinam A, Nguyen A, Rackohn T, Ting K, Soo C, James AW, Brief review of models of ectopic bone formation. Stem Cells Dev. 21, 655–667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinha S, Uchibe K, Usami Y, Pacifici M, Iwamoto M, Effectiveness and mode of action of a combination therapy for heterotopic ossification with a retinoid agonist and an anti-inflammatory agent. Bone 90, 59–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatsell SJ, Idone V, Alessi Wolken DM, Huang L, Kim HJ, Wang LC, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN, ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Science Trans. Med. 7, 303ra137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW, Droplet barcoding for single cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, W. M. r. Mauck, Y. Hao, M. Stoeckius, P. Smibert, R. Satija, Comprehensive integration of single-cell data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Houlihan DD, Mabuchi Y, Morikawa S, Niibe K, Araki D, Suzuki S, Okano H, Matsuzaki Y, Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-α. Nat. Protocols 7, 2103–2111 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Martin JF, Bradley A, Olsen EN, The pair-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes & Dev. 9, 1237–1249 (1995). [DOI] [PubMed] [Google Scholar]
- 58.Alexander DR, The CD45 tyrosine phosphatase: a positive and negative regulator of immune cell function. Sem. Immunol. 12, 349–359 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Lefebvre V, Bhattaram P, Vertebrate skeletogenesis. Curr. Topics Dev. Biol. 90, 291–317 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanehisa M, Sato Y, Kawashima K, Furumichi M, Tanabe M, KEGG as a refernec resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo W, Pant G, Bhaynasi YK, Brouwer C, Pathwiew Web: user friendly pathway visualization and data integration. Nucleic Acids Res. 45(W1), W501–W508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dionne MS, Skarnes WC, Harland RM, Mutation and analysis of Dan, the founding member of the Dan family of transforming growth factor β antagonists. Mol. Cell. Biol. 21, 636–643 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trapnell C, Cacchiarelli D, Grimsby J, Pokharei P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sorkin M, Huber AK, Hwang C, Carson WFI, Menon R, Li J, Vasquez K, Pagani C, Patel N, Li S, Visser ND, Niknafs Y, Loder S, Scola M, Nycz D, gallagher K, McCauley LK, Xu J, James AW, Agarwal S, Kunkel S, Mishina Y, Levi B, Regulation of heterotopic ossification by monocytes in a mouse model of aberrant wound healing. Nat. Communications 11, 722 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei F, Zhou Y, Wang J, Liu C-J, Xiao Y, The immunomodulatory role of BMP-2 on macrophages to accelerate osteogenesis. Tissue Eng. Part A 24, 584–594 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Efremova M, Vento-Tormo M, Teichmann SA, V.-T. R., CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protocols 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: a software environment for intregrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Browaeys R, Saelens W, Saeys Y, NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Hermiston ML, Zikherman J, Zhu JW, CD45, CD148, and Lyp/Pep: Critical phosphatases regulating Src family kinase signaling networks in immune cells. Immunol. Rev. 228, 288–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sidney LE, Branch MJ, Dunphy SE, Dua HS, Hopkinson A, Concise review: Evidence for CD34 as a common marker of diverse progenitors. Stem Cells 32, 1380–1389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kan L, Kessler JA, Animals models of typical heterotopic ossification. J. Biomed. Biotechnol. 2011, 309287 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lounev VY, Ramachandran R, Wosczyna M, Yamamoto M, Maldment A, Shore E, Glaser DL, Goldhamer DJ, Kaplan FS, Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Joint Surg. 91, 652–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mundy C, Bello A, Sgariglia F, Koyama E, Pacifici M, HhAntag, a hedgehog signaling antagonist, suppresses chondrogenesis and modulates canonical and non-canonical BMP signaling. J. Cell. Physiol. 231, 1033–1044 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Solursh M, Jensen KL, Singley CT, Linsenmayer TF, Reiter RS, Two distinct regulatory steps in cartilage differentiation Dev. Biol. 94, 311–325 (1982). [DOI] [PubMed] [Google Scholar]
- 75.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS, SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like (ALK) receptors ALK4, ALK5 and ALK7. Mol. Pharmacol. 62, 65–74 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Olsen OE, Wader KF, Hella H, Mylin AK, Turesson I, Nesthus I, Waage A, Sundan A, Holien T, Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signaling 13, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Mara CS, Duarte AS, Sartori-Cintra AR, Luzo AC, Saad ST, Coimbra IB, Chondrogenesis from umbilical cord blood cells stimulated with BMP-2 and BMP-6. Rheumatol. Int. 33, 121–128 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Wang X, Li F, Xie L, Crane J, Zhen G, Mishina Y, Deng R, Gao B, Chen H, Liu S, Yang P, Gao M, Tu M, Wang Y, Wan M, Fan CM, Cao X, Inhibition of overactive TGF-β attenuates progression of heterotopic ossification in mice. Nat. Communications 9, 551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang SY, Alliston T, Regulation of postnatal bone homeostasis by TGFβ. BoneKEy Reports 2, 255 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Filvaroff EH, Erlebacher A, Ye J-Q, Gitelman SE, Lotz J, Heillman M, Derynck R, Inhibition of TGF-β receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 126, 4267–4279 (1999). [DOI] [PubMed] [Google Scholar]
- 81.Genet F, Kulina I, Vaquette C, Torossian F, Millard S, Pettit AR, Sims NA, Anginot A, Guerton B, Winkler IG, Barbier V, Lataillade J-J, Le Bousse-Kerdiles MC, Hutmacher DW, Levesque J-P, Neurological heterotopic ossification following spinal cord injury is triggered by macrophage-mediated inflammation in muscle. J. Pathol. 236, 229–240 (2015). [DOI] [PubMed] [Google Scholar]
- 82.Pavey GJ, Quareshi AT, Hope DN, Pavicek RL, Potter BK, Forsberg JA, Davis TA, Bioburden increases heterotopic ossification in an established rat model. Clin. Orth. Relat. Res. 473, 2840–2847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pavey GJ, Qureshi AT, Tomasino AM, Honnold CL, Bishop DK, Agarwal S, Loder S, Levi B, Pacifici M, Iwamoto M, Potter BK, Davis TA, Forsberg JA, Targeted stimulation of the retinoic acid receptor-γ mitigates the formation of heterotopic ossification in an established blast-related traumatic injury model. Bone 90, 159–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hwang C, Pagani CA, Das N, Marini S, Huber AK, Xie L, Jimenez J, Brydges S, Lim WK, Narrunu KC, Murphy AJ, Economides AN, Hatsell SJ, Levi B, Activin A does not drive post-traumatic heterotopic ossification. Bone 138, 115473 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Shore E, Xu M, Feldman GJ, Fenstermacher DA, Consortium TFIR, Brown MA, Kaplan FS, A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genet. 38, 525–527 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, Toguchida J, Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 112, 15438–15443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cardin AD, Weintraub HJ, Molecular modeling of protein-glycosaminoglycan interactions. Arterioschler. Thromb. Vasc. Biol. 9, 21–32 (1989). [DOI] [PubMed] [Google Scholar]
- 88.Billings PC, Pacifici M, Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: mechanisms and mysteries. Connect. Tissue Res. 56, 272–280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mundy C, Yang E, Takano H, Billings PC, Pacifici M, Heparan sulfate antagonism alters bone morphogenetic protein signaling and receptor dynamics, suggetsing a mechanism in Hereditary Multiple Exostoses. J. Biol. Chem. 293, 7703–7716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu D, Esko JD, Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Billings PC, Yang E, Mundy C, Pacifici M, Domains with highest heparan sulfate-binding affinity reside at opposite ends in BMP2/4 versus BMP5/6/7: Implications for function. J. Biol. Chem. 293, 14371–14383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang E, Mundy C, Rappaport EF, Pacifici M, Billings PC, Identification and characterization of a novel heparan sulfate-binding domain in Activin A longest variants and implications for function. PLOS One 14, e0222784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shimono K, Morrison TN, Tung W-E, Chandraratna RAS, Williams JA, Iwamoto M, Pacifici M, Inhibition of ectopic bone formation by a selective retinoic acid receptor α-agonist: a new therapy for heterotopic ossification? J. Orthop. Res. 28, 271–277 (2010). [DOI] [PubMed] [Google Scholar]
- 94.Shimono K, Tung W-E, Macolino C, Chi A, Didizian JH, Mundy C, Chandraratna RAS, Mishina Y, Enomoto-Iwamoto M, Pacifici M, Iwamoto M, Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nature Med. 17, 454–460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, Pacifici M, Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134, 2159–2169 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huegel J, Mundy C, Sgariglia F, Nygren P, Billings PC, Yamaguchi Y, Koyama E, Pacifici M, Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: A mechanism likely deranged in Hereditary Multiple Exostoses. Dev. Biol. 377, 100–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gutierrez MA, Guevara J, Barrera LA, Semi-automatic grading system in histologic and immunohistochemistry analysis to evaluate in vitro chondrogenesis. Universitas Scientiarum 17, 167–178 (2012). [Google Scholar]
- 98.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisoon M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guangchuang Y, Wang L-G, Han Y, He Q-Y, clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Activin A neutralizing antibody specifically interferes with recombinant activin A-induced pSMAD2 activation.
Fig. S3. Recruitment and migration of cells into implanted Matrigel
Fig. S2. Activin A antibody administration inhibits rhBMP6-induced subcutaneous heterotopic cartilage and bone formation
Fig. S4. GO term analysis of cellular phenotypic and developmental processes
Fig. S5. KEGG-based Pathview plot analysis of possible mode of action of anti-activin A treatment
Fig. S6. Inflammatory and hematopoietic cells are present at early stages of HO development
Fig. S7. Phenotypes of macrophages present in developing HO masses
Fig. S9. Flow cytometry analyses of cell populations present in HO masses
Fig. S8. Analyses of cell-cell communication and ligand/receptor/target interactions in mesenchymal and immune cells present in developing HO sites
Fig. S10. Activin A antibody administration inhibits intramuscular rhBMP6-induced HO
Fig. S12. Exogenous activin A stimulates rhBMP6-induced HO development
Fig. S11. Endogenous activin A is present in developing HO masses
Fig. S13. Gene expression analysis of receptors for TGF-β superfamily members
Fig. S14. Working model of activin A and TGF-β roles in acquired HO development
Table S2. Primer sequences
Table S1. GSEA of macrophage clusters
Data Availability Statement
The RNA-seq data have been deposited in the NCBI at NIH with accession number GSE157679. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.