

Abstract

Targeting the colchicine binding site on tubulin is a promising approach for cancer treatment to overcome the limitations of current tubulin polymerization inhibitors. New classes of colchicine binding site inhibitors (CBSIs) are continually being uncovered; however, balancing metabolic stability and cellular potency remains an issue that needs to be resolved. Therefore, we designed and synthesized a series of novel fused imidazopyridine and -pyrazine CBSIs and evaluated their cellular activity, metabolic stability, and tubulin-binding properties. Evidence shows that the imidazo[1,2-a]pyrazine series are effective against neuroblastoma cell lines marked by MYCN amplification. Further assessment shows that a combination of an imidazo[1,2-a]pyrazine core with a trimethoxyphenyl ring D results in the highest cellular activity and binding characteristics compared with a dichloromethoxyphenyl or difluoromethoxyphenyl ring D. However, the metabolic stability of compounds with a dichloromethoxyphenyl or difluoromethoxyphenyl ring D is significantly higher than that of those containing a trimethoxyphenyl ring D, suggesting that improved metabolic stability is achieved with a moderate impact on potency.
Keywords: colchicine, tubulin polymerization inhibitor, neuroblastoma, microtubules, MYCN
α- and β-tubulin dimers make up the extremely dynamic intracellular structures known as microtubules (MTs), which are essential to many basic cellular functions such spindle pole formation, cell division, and cytoskeleton maintenance.1−4 The polymerization and depolymerization of tubulin dimers are necessary for the construction and deconstruction of MTs.5−7 Given that MTs are intimately involved in proper cell division, disruption of MT dynamics has been a well-established approach in anticancer therapy.8−11 Taxanes, vinca alkaloids, and epothilones are three primary types of tubulin inhibitors that have been widely utilized to treat cancer clinically.6,7,12 However, the effectiveness of these medications in the clinic is frequently constrained by the emergence of multidrug resistance (MDR), peripheral neuropathy, and constrictive therapeutic indices.13−19 As such, targeting of the colchicine binding site, located at the interface of the α- and β-tubulin dimers, has gained attention as a novel tubulin inhibition approach that may overcome these challenges.
The small molecule named colchicine and other small compounds that bind to the colchicine site prevent tubulin dimers from polymerizing and forming useful microtubules.3,20 Colchicine binding site inhibitors (CBSIs) consequently show significant cytotoxicity in a variety of experiments.21−23 Importantly, unlike colchicine, small-molecule CBSIs are known to be less susceptible to drug resistance mechanisms, including efflux transporters and β3 tubulin overexpression-mediated MDR.4,20 However, the significant adverse effects against normal cells, low solubility, and low oral bioavailability of small-molecule CBSIs have limited their practical applicability.24,25 Thus, in recent years, there has been extensive research on developing new classes of CBSIs with a wider therapeutic window, including phenyl-amino-thiazole (PAT),26 SAIs27 (substituted 2-aryl-imidazoles), ABIs (2-aryl-4-benzoyl-imidazoles),28 SMARTs (4-substituted methoxybenzoyl-aryl-thiazoles),29 and verubulin9,20 (Figure 1A). In human melanoma and prostate cancer xenograft models, the SMART, ABI, and PAT analogues demonstrate extremely potent in vivo effectiveness.29 However, it has been observed that the carbonyl group linking the B and C rings in ABI and SMART counterparts makes them metabolically unstable.30,31 New fused 1H-imidazo[4,5-c]pyridine analogues (Figure 1B) were reported to improve metabolic stability with moderate t1/2 values (45–56 min) against human liver microsome.30 Thus, in an attempt to derive new colchicine-site tubulin polymerization inhibitors with improved metabolic stability while enhancing or retaining potency, we designed a series of fused imidazo[1,2-a]pyridine and -pyrazine analogues (Figure 1B). Imidazo[1,2-a]pyridine and -pyrazine scaffolds are known to have a broad range of applications in medicinal chemistry, including anticancer, antiviral, and antimicrobial activities.32 Therefore, these new series of compounds were tested for their efficacy against a panel of two neuroblastoma cell lines with MYCN amplification. Neuroblastoma is one of the most prevalent extracranial solid cancers in children and infants.33 Amplification of the MYCN gene, which encodes the N-MYC oncoprotein transcription factor, is associated with high-risk cases and metastasis, resulting in poor prognosis and a higher fatality rate.34 Because of this, MYCN amplification is associated with advanced disease and failure to respond to chemotherapy. As it relates to this study, tubulin inhibitors such as vincristine are commonly used as chemotherapy agents to treat neuroblastoma.35,36 Additional preclinical studies are also actively being performed using CBSIs.37 Unfortunately, vincristine resistance is common in neuroblastoma patients,36,38 and not all tubulin inhibitors are yet effective against neuroblastoma. Although novel therapies to treat neuroblastoma have been proposed or are progressing to phase I and/or phase II clinical trials, their true impact is yet to be determined.39−41 Thus, there remains a demand to improve current therapeutics used to treat this childhood cancer. Herein, we report the design, synthesis, and biological evaluation of new fused imidazopyridine and -pyrazine CBSIs against two MYCN-amplified neuroblastoma cell lines. Our findings demonstrate that fused imidazopyrazine analogues inhibit both neuroblastoma cell viability and in vitro tubulin polymerization. Additional experimental evidence suggests that these analogues can also block cell division and disrupt cytoskeletal structures within cells, suggesting that at least some of these analogues can act as bona fide tubulin inhibitors within cells.
Figure 1.
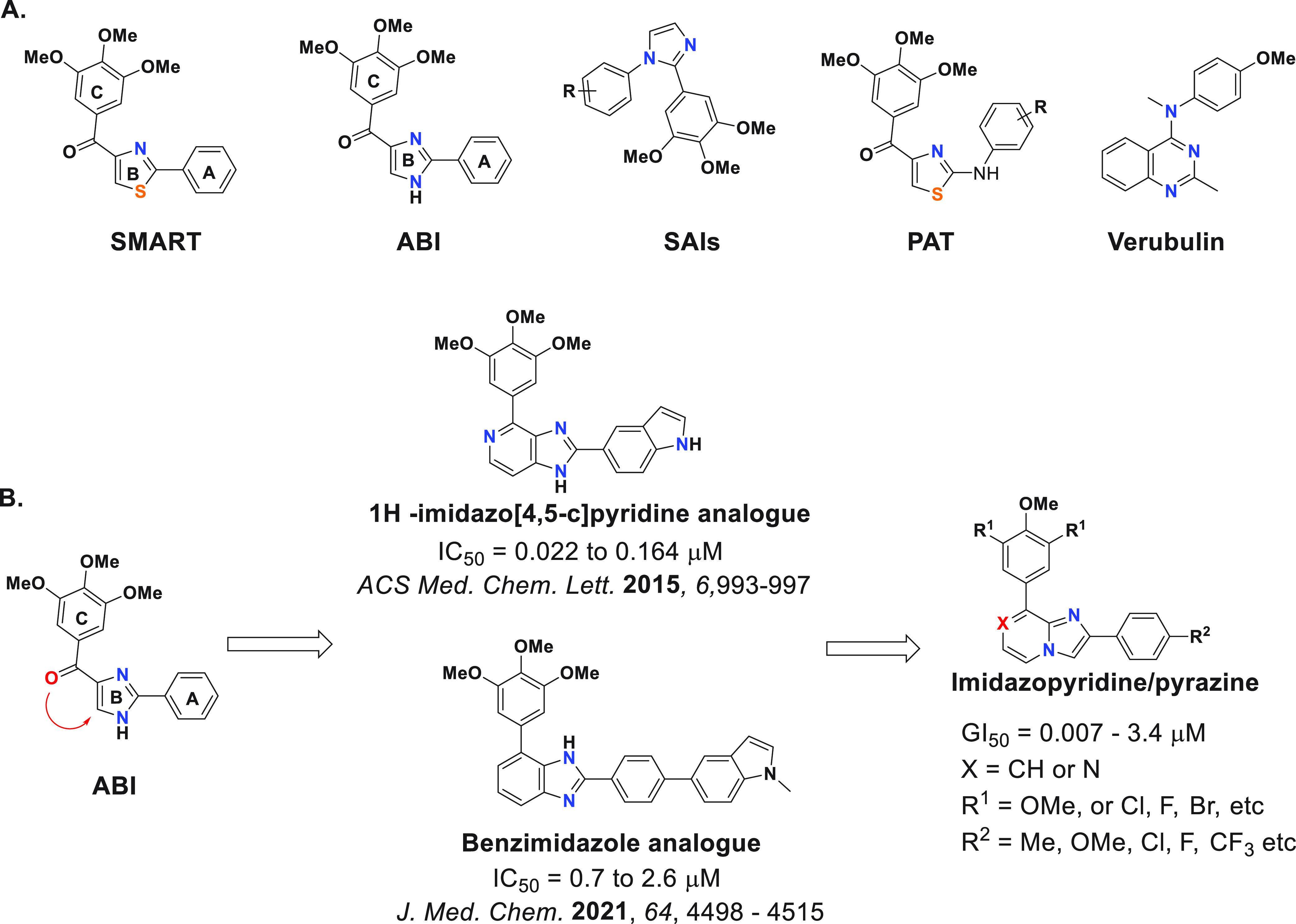
(A) Chemical structures of different classes of reported CBSIs. (B) Design and structure–activity relationship (SAR) of target compounds.
First, we synthesized six fused imidazo[1,2-a]pyridine analogues with fused A and B rings, as demonstrated in Scheme 1. We started with commercially available para-substituted α-bromoacetophenone (1) and allowed it to react with commercially available 3-bromo-5-substituted-pyridine-2-amine derivatives (2a, 2b) in the presence of scandium(III) triflate catalyst and acetonitrile as a solvent to obtain the imidazo[1,2-a]pyridine core (3a–3f) with a para-substituted ring C. Finally, the 3,4,5-trimethoxyphenyl ring (ring D) was installed using Suzuki coupling to achieve the desired imidazo[1,2-a]pyridine derivatives. These first compounds were evaluated for their ability to decrease cell viability in the Kelly and CHP-134 MYCN-amplified neuroblastoma cell lines over the course of 3 days. Commercially available colchicine was included as a comparison. The sensitivity of cells to each compound, including colchicine, was assessed by calculating the GI50 across the cell lines monitored. As reported in Table 1, the para-methyl substituent on the ring C was most tolerated, making 4a the most potent (GI50 ≈ 200 nM) compared to 4b–4f. Neither the activating methoxy substituent nor the deactivating halogen substituents (Cl or F) at the para position of ring C were tolerated, resulting in deterioration in GI50. Compound 4f, with a methyl substituent on the 6-position, lost cellular potency, however not to the same extent as 4b, 4c, and 4d.
Scheme 1. Synthesis of Imidazo[1,2-a]pyridine Analogues.
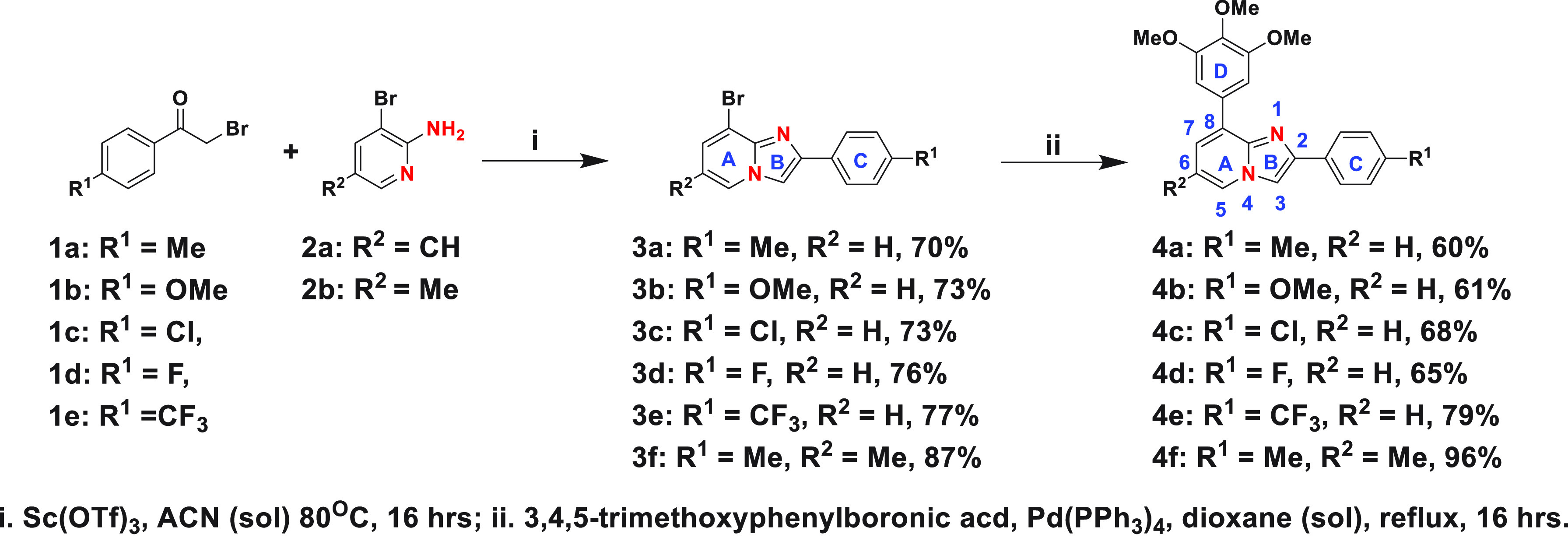
Table 1. Antiproliferative Activities of Fused Imidazopyridine and Imidazopyrazine Analogues in Neuroblastoma Cancer Cell Linesa.
| GI50 (μM) |
||
|---|---|---|
| compd | Chp-134 | Kelly |
| 4a | 0.152 ± 0.043 | 0.217 ± 0.046 |
| 4b | 0.511 ± 0.119 | 1.371 ± 0.291 |
| 4c | 1.623 ± 0.310 | 3.420 ± 0.450 |
| 4d | 1.611 ± 0.365 | 2.995 ± 0.415 |
| 4e | 2.088 ± 0.483 | >5.000 |
| 4f | 0.381 ± 0.089 | 0.717 ± 0.136 |
| 4g | 3.320 ± 0.865 | 4.915 ± 1.276 |
| 4h | 0.007 ± 0.002 | 0.012 ± 0.002 |
| 4i | 0.474 ± 0.108 | 0.438 ± 0.081 |
| 4j | 0.175 ± 0.038 | 0.402 ± 0.102 |
| 4k | 0.079 ± 0.017 | 0.165 ± 0.048 |
| 4l | 0.469 ± 0.101 | 0.504 ± 0.129 |
| 4m | >2.500 | >2.500 |
| colchicine | 0.008 ± 0.002 | 0.009 ± 0.002 |
The MYCN-amplified Kelly and CHP-134 neuroblastoma cells were treated with a nine-point serial dilution of each compound for 3 days. Cell viability was determined by the CellTiter-Glo assay. All values were normalized to DMSO-treated cells and then used to calculate a GI50 value. GI50 values are shown with the standard error of the mean. The values >5.00 and >2.50 mean that this was the highest dose (μM) tested and this dose did not decrease cell viability below 50%.
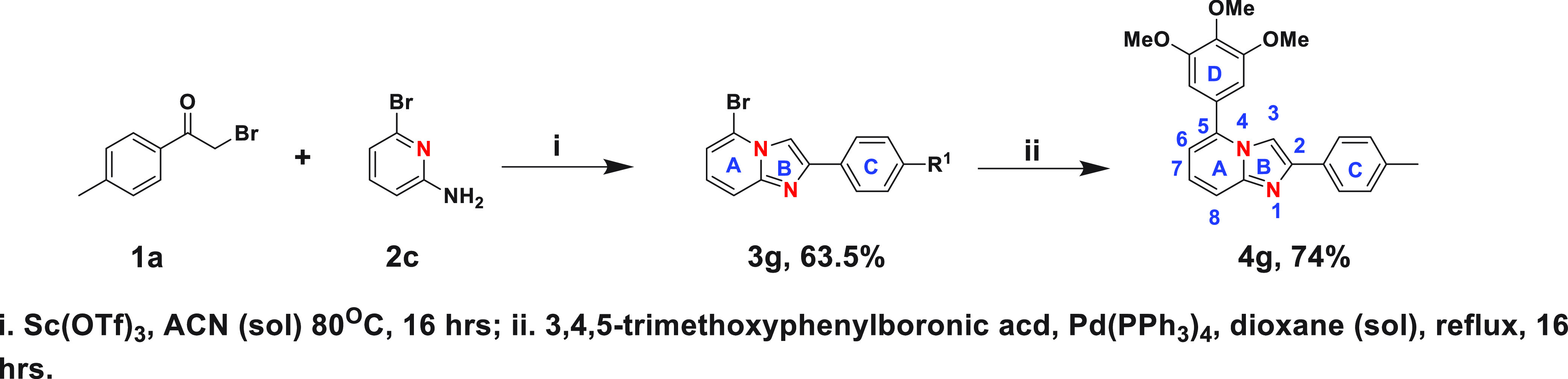
Next, we synthesized the reverse imidazo-pyridine analogue 4g following Scheme 2 to assess the effect of the location of nitrogen atoms in the imidazopyridine core on cellular activity. Cell viability screening demonstrates that the location of nitrogen atoms in the core is crucial, and having the N-1 nitrogen on the same side as the trimethoxy-phenyl ring is imperative for biological efficacy (Table 1). Finally, we synthesized six imidazo[1,2-a]pyrazine analogues, following Scheme 3, for comparison.
Scheme 2. Synthesis of Reverse Imidazopyridine Derivative.
Scheme 3. Synthesis of Imidazo[1,2-a]pyrazine Derivatives.
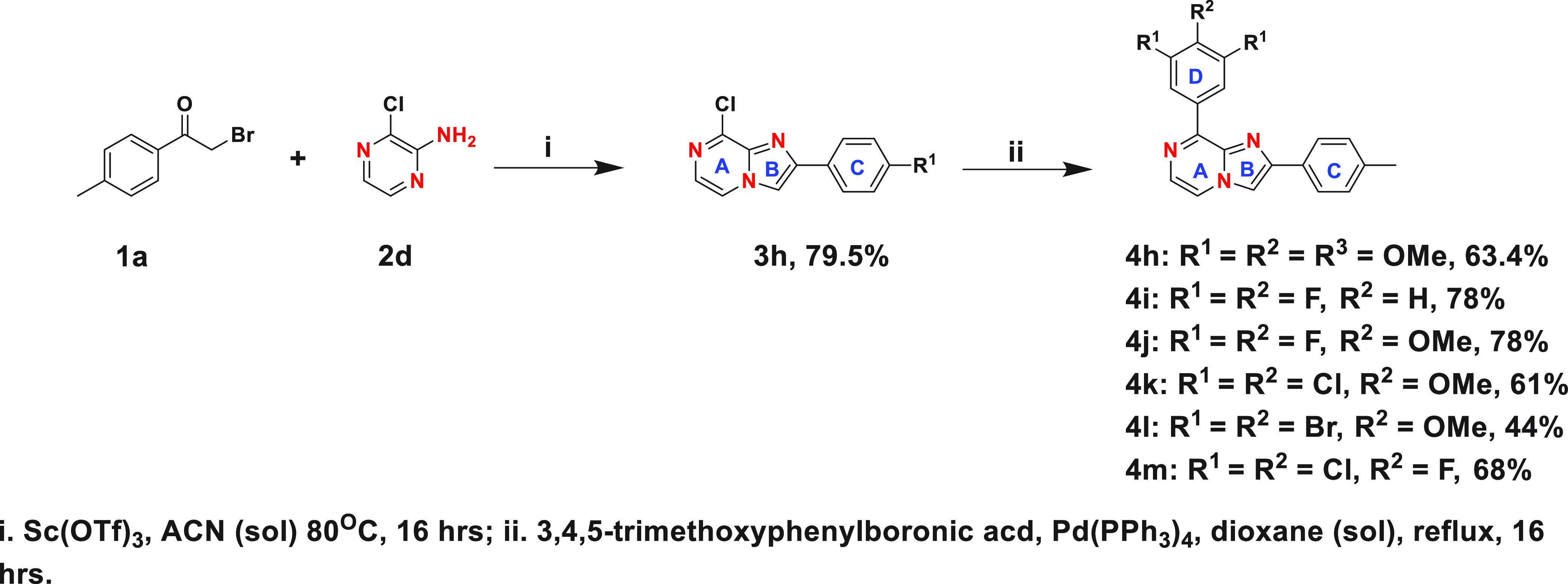
Our goal in the comparison was to assess the effect of the N-7 nitrogen of the imidazopyrazine core as well as different substituents at the 3, 4, and 5 positions of ring D on the biological efficacy. Cell viability screening showed that the N-7 nitrogen on the imidazopyrazine core and the 3,4,5-trimethoxyphenyl ring D enhanced cellular potency, and compound 4h was as potent as colchicine in decreasing cell viability, with an average GI50 of ∼10 nM (Table 1). Additional structure–activity relationship results indicate that the dichloromethoxyphenyl ring D was well-tolerated, resulting in compound 4k having a double-digit nanomolar efficacy against the CHP-134 cell line, and that at least one methoxy group at the 4-position of ring D is important, making 4j significantly more active than 4i. Surprisingly, 4l, with a bulkier dibromomethoxyphenyl ring D, was found to be detrimental to the antiproliferative activity. These findings indicate that the combination of the imidazo[1,2-a]pyrazine core with a trimethoxyphenyl ring D (4h) is the best, followed by dichloromethoxyphenyl (4k), difluoromethoxyphenyl (4j), and difluorophenyl (4i) ring D.
To determine if the compounds with the lowest GI50 values across both cell lines were capable of inhibiting tubulin polymerization, we tested compounds 4a, 4h, 4j, and 4k in an in vitro tubulin polymerization assay using purified tubulin. Colchicine was used as a control to inhibit and destabilize microtubule formation, while paclitaxel was used as a microtubule enhancer control. All four compounds sampled inhibited tubulin polymerization comparably and were as effective as colchicine in destabilizing microtubule formation (Figure 2). The overall cell viability and in vitro tubulin polymerization results suggest that many of the imidazo[1,2-a]pyrazine classes of compounds (4h, 4j, 4k, and 4l) may be capable of tubulin inhibition as their mechanism of action. To specifically compare their ability to act as tubulin inhibitors within neuroblastoma cells, we tested whether compound treatment inhibits the process of mitosis, a cellular activity dependent on active microtubule dynamics, by performing cell cycle phase distribution analysis. In Kelly cells, a 24 h treatment with 100 nM compound 4h revealed that this compound mirrors colchicine in its ability to block cell cycle progression and promote an accumulation of cells in the G2/M phase of the cell cycle (Figure 3A,B). Interestingly, compound 4j did not cause any overt changes in the cell cycle phase distribution, even at a 500 nM dose (Figure 3C,D). In CHP-134 cells, compound 4h was also as effective as colchicine (Figure 3E,F), and the overall effect was similar to that of Kelly cells, suggesting that 4h acts as an antimitotic in these neuroblastoma cells. As a second approach to compare the impact on cell cycle progression, we analyzed the level of two proteins that increase in cells during mitosis—cyclin B1 and histone 3 phosphorylated at serine residue 10 (p-S10 H3)—and therefore can act as mitotic markers.42,43 In both Kelly and CHP-134 cells, compound 4h mirrored colchicine in its ability to induce increases in the levels of both cyclin B1 and p-S10 H3 compared with the dimethyl sulfoxide (DMSO) vehicle control (Figure 4), indicative of these two compounds causing an enrichment of the cell population in the mitotic phase. In line with cell cycle phase distribution analysis (Figure 3C,D), protein levels of both mitotic markers following treatment with compound 4j were not overtly different in comparison with those of DMSO-treated cells (Figure 4). However, in both cell lines, treatment with compound 4k resulted in a moderate increase in the level of both mitotic markers, suggesting that compound 4k may function as a weaker mitotic inhibitor than compound 4h and colchicine.
Figure 2.

Compounds inhibit tubulin polymerization in vitro. In vitro tubulin polymerization was monitored in the absence of any compound (“standard”) and with the addition of 5 μM paclitaxel, 5 μM colchicine, or 5 μM of the indicated compound 4a, 4h, 4j, or 4k. The same curves generated from the standard, paclitaxel, and colchicine reactions are superimposed on all four graphs for comparison with each of the indicated compounds. All compounds tested inhibit microtubule formation as compared with the standard and colchicine reactions.
Figure 3.

Cell cycle progression is impaired with compound 4h. (A) Representative cell cycle profile images of Kelly cells treated with DMSO, 100 nM colchicine, or 100 nM compound 4h for 24 h. (B) Quantification of cell cycle phase distributions for cells treated as in (A) (n = 3 biological replicates, error bars are standard error, ****P < 0.0001, *P = 0.018, **P < 0.002 using unpaired t test, two-tailed). (C) Representative cell cycle profile images of Kelly cells treated with DMSO or 500 nM compound 4j for 24 h. (D) Quantification of cell cycle phase distributions for cells treated as in (C) (n = 3 biological replicates, error bars are standard error). (E) Representative cell cycle profile images of CHP-134 cells treated with DMSO, 100 nM colchicine, or 100 nM compound 4h for 24 h. (F) Quantification of cell cycle phase distributions for cells treated as in (E) (n = 3 biological replicates, error bars are standard error, **P = 0.0089, *P < 0.019 using unpaired t test, two-tailed).
Figure 4.

Compounds cause induction of mitotic protein markers. Western blots of protein lysates collected following treatment of indicated cells with DMSO, 100 nM colchicine, 100 nM compound 4h, or 500 nM compounds 4j and 4k for 24 h. GAPDH is used as a loading control.
Microtubule dynamics are also important in maintaining cytoskeletal structures within cells grown in culture. Therefore, we used immunofluorescence to examine the ability of compounds 4h, 4j, and 4k to disrupt the microtubule network. We first focused these experiments on Kelly cells due to their larger cytoplasmic area for visualization. DMSO treatment, followed by immunostaining for α-tubulin, revealed an intact microtubule network that extended from the cell nuclei, which were costained with DAPI. Colchicine treatment, as expected, caused a dramatic disruption to the microtubule network, with tubulin compacting around the nuclei of cells, indicative of the rounded morphology cells acquire when microtubule dynamics are impacted.44 Compound 4h was as effective as colchicine in promoting the same effect, indicating again that compound 4h mirrors the effects of colchicine (Figure 5). Consistent with the cell cycle phase distribution assay and mitotic marker analysis, compound 4k showed a moderate effect on disruption of the microtubule network, and compound 4j appeared closer to DMSO-treated cells than those treated with colchicine. As a second point of confirmation, we also visualized tubulin in another MYCN-amplified cell line, Be(2)C, which also shows high cellular sensitivity to 4h, 4j, and 4k (Table S1) and has a large cytoplasmic area for visualization. Compared to Kelly cells, the response of Be(2)C cells was very similar, and tubulin compaction was evident with 4h and 4k, compared to colchicine (Figure S1). Overall, these results indicate that compound 4h functions as a potent tubulin inhibitor in MYCN-amplified neuroblastoma cells. In addition, these results suggest that other compounds containing an imidazo[1,2-a]pyrazine scaffold structure, such as compound 4k (Table 1), may function as less potent but effective inhibitors of tubulin. To determine if compounds 4h, 4j, and 4k differ in their metabolic stability, we evaluated each in a human liver microsomal assay. As shown in Table 2, compounds 4k and 4j have improved stabilities (t1/2 ≈ 52 and 36 min, respectively) compared to compound 4h (t1/2 ≈ 26 min), indicating that addition of the 3,4,5-trimethoxyphenyl ring D improves cellular potency but reduces metabolic stability.
Figure 5.

Impact of compound treatment on intracellular tubulin. Representative immunostaining images of Kelly cells treated with DMSO, 100 nM colchicine, 100 nM compound 4h, or 500 nM compounds 4j and 4k for 24 h. Nuclei are stained with DAPI in blue, and α-tubulin is stained with Alexa-488 in green. Scale bar = 20 μm.
Table 2. In Vitro Human Liver Microsomal Stabilitya.
| compd | t1/2 (min) |
|---|---|
| 4h | 25.99 ± 0.4 |
| 4j | 36.1 ± 1.0 |
| 4k | 51.96 ± 1.15 |
| colchicine | >60 |
| verapamil | 26.3 ± 1.35 |
The half-lives of indicated compounds in human liver microsomes are shown with the standard deviation of the mean. The value >60 means that this was the longest time point (min) tested and the sample was almost intact.
To gain further insight into structure–activity relationships of the biologically evaluated hits, a molecular docking analysis was performed to envisage binding interactions within the colchicine binding site. As demonstrated in Figure 6, compound 4b (imidazopyridine) binds nicely at the colchicine binding while overlapping well with the native ligand 5m (PDB: 6X1F, Figure 6A). It is evident from Figure 6A,B that 4b occupies deep within the hydrophobic β-tubulin pocket consisting of amino acid residues Cys239, Val236, Leu240, Ala314, Ala352, Ile316, Met257, Leu253, Leu240, and Leu246. The imidazopyridine core is located close to the α-Thr179 and α-Val181 residues, suggesting that the molecule is binding at the colchicine binding site, which is at the interface of the α- and β-tubulin monomers of the α- and β-tubulin heterodimer. Figure 6C,D shows that the N-7 nitrogen of the imidazo[1,2-a]pyrazine cores of 4k and 4h forms hydrogen bonds (2.29 and 2.23 Å, respectively) with the β-Asp249 NH backbone. However, this hydrogen bonding is not observed with 4j (Figure 6E). Taken together, the docking results suggest that 4h and 4k may bind at the colchicine binding site with higher binding affinity, resulting in an enhanced inhibitory effect against tubulin polymerization.
Figure 6.

Molecular docking. (A) Docking of 4b (brown) at the colchicine binding site of tubulin (PDB: 6X1F). The α-tubulin monomer is shown in cyan and the β-tubulin monomer in green. Native ligand (SB-216/5m) is shown in pink. (B) Amino acid residues within 5 Å of the docked ligand 4b. (C) Docked pose of 4k (yellow) at the colchicine binding site. (D) Docked pose of 4h (gold) at the colchicine binding site. (E) Docked pose of 4j (purple) at the colchicine binding site. (F) Binding surface (hydrophobic) around the ligand 4k. Hydrogen bonding is shown by purple dotted lines.
We also carried out molecular dynamics (MD) simulations of the top two compounds, 4h and 4k, in complex with tubulin to assess their binding stabilities. The root-mean-square deviation (RMSD) of protein and ligand movement was analyzed (Figure 7A). The protein structure’s conformational flexibility is represented by the RMSD, and higher deviations denote a more flexible protein structure. The MD simulation of the tubulin–4h system (red) shows a sudden increase in the RMSD value for the backbone that eventually stabilizes at an average of 2.03 Å around 50 ns. The tubulin–4k complex demonstrates a gradual increase in RMSD value and finally stabilizes at 1.59 Å around 50 ns. The backbone RMSD calculations of the tubulin–ligand complexes suggest that ligand binding seems to decrease the backbone flexibility of tubulin. We also analyzed ligand movement RMSD values for 4h (green, Figure 7A) and 4k (orange, Figure 7A). As evident from Figure 7A, both ligands maintain closer proximity with the binding site over the course of the simulation. Binding free energy calculations demonstrate that 4h has higher average binding energy (red, Figure 7B, 153.44 ± 5.8 kJ/mol) than 4k (purple, Figure 7B, 135.46 ± 3.7 kJ/mol), suggesting stronger binding of 4h to the colchicine binding site.
Figure 7.

RMSD and binding energy vs time plots for 4h and 4k. (A) RMSD vs simulation time plot for protein backbone atoms with 4h as ligand in red, movement of 4h in its binding site in green, protein backbone atoms with 4k as ligand in purple, and movement of 4k in its binding site in orange. (B) Binding energy of protein–ligand interactions of the colchicine binding site of tubulin with 4h and 4k vs time. Binding energy was calculated by analyzing the MD trajectory using the BoundaryFast method in YASARA. Based on the YASARA algorithm, higher binding energy values mean stronger binding.
In summary, we designed and synthesized a series of novel fused imidazopyridine and -pyrazine CBSIs and evaluated their efficacy against two neuroblastoma cell lines with increased levels of N-MYC, a potent transcription factor implicated in diverse types of cancers.45 Among this series, the imidazo[1,2-a]pyrazine class of compounds showed the best potency in cell viability assays (compounds 4h, 4j, 4k, and 4l). Mechanism of action studies support that this class of compounds can inhibit tubulin polymerization in vitro, and 4h and 4k have the ability to impair microtubule-dependent processes in cells. Interestingly, although 4h, 4k, and 4j can each inhibit tubulin polymerization in vitro (Figure 2), 4j fails to promote robust tubulin inhibition in cells. This discrepancy may be due to the use of purified tubulin in the in vitro assay, a form of tubulin that most likely does not fully resemble tubulin in vivo, which may be bound by other proteins or cargoes or have access restricted depending on the state of the cell. Additional evidence from molecular docking, MD simulation, and metabolic stability assays helps to refine the structure–activity relationship, showing that a combination of imidazo[1,2-a]pyrazine core with trimethoxyphenyl ring D (4h) results in the highest cellular activity and stronger binding to the colchicine binding site compared to combination with a dichloromethoxyphenyl (4k) or difluoromethoxyphenyl (4j) ring D. Importantly, however, while 4k and 4j show less potency and weaker binding, they have significantly improved metabolic half-lives. Future studies that aim to optimize these lead compounds should prove useful in identifying novel compounds that have improved metabolic stability while retaining cellular potency.
Acknowledgments
We thank Dr. Jason Jessen of the Molecular Biosciences graduate program for critical support of this project.
Glossary
Abbreviations
- CBSI
colchicine binding site inhibitor
- MT
microtubule
- MDR
multi drug resistance
- GI50
50% growth inhibition
- GADPH
glyceraldehyde-3-phosphate dehydrogenase
- t1/2
half-life
- RMSD
root-mean-square deviation
- PDB
Protein Data Bank
- nM
nanomolar
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00298.
Synthetic and biology experimental procedures, cell viability against Be(2)C cell line in Table S1, immunostaining data in Figure S1, NMR spectra, HRMS spectra, and HPLC chromatograms (PDF)
Author Contributions
# J.T. and K.B.C. contributed equally to this work.
Financial support for this study was provided by a MTSU URECA award to G.E.T., a NIH/NCI grant CA263868 to A.M.W., and generous start-up funding from the Middle Tennessee State University to S.B.
The authors declare no competing financial interest.
Supplementary Material
References
- Honore S.; Pasquier E.; Braguer D. Understanding microtubule dynamics for improved cancer therapy. Cell. Mol. Life Sci. 2005, 62 (24), 3039–3056. 10.1007/s00018-005-5330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspari R.; Prota A. E.; Bargsten K.; Cavalli A.; Steinmetz M. O. Structural Basis of cis- and trans-Combretastatin Binding to Tubulin. Chem 2017, 2 (1), 102–113. 10.1016/j.chempr.2016.12.005. [DOI] [Google Scholar]
- Arnst K. E.; Banerjee S.; Chen H.; Deng S.; Hwang D.-J.; Li W.; Miller D. D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39 (4), 1398–1426. 10.1002/med.21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnst K. E.; Banerjee S.; Wang Y.; Chen H.; Li Y.; Yang L.; Li W.; Miller D. D.; Li W. X-ray Crystal Structure Guided Discovery and Antitumor Efficacy of Dihydroquinoxalinone as Potent Tubulin Polymerization Inhibitors. ACS Chem. Biol. 2019, 14 (12), 2810–2821. 10.1021/acschembio.9b00696. [DOI] [PubMed] [Google Scholar]
- Cui M.-T.; Jiang L.; Goto M.; Hsu P.-L.; Li L.; Zhang Q.; Wei L.; Yuan S.-J.; Hamel E.; Morris-Natschke S. L.; Lee K. H.; Xie L. In Vivo and Mechanistic Studies on Antitumor Lead 7-Methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one and Its Modification as a Novel Class of Tubulin-Binding Tumor-Vascular Disrupting Agents. J. Med. Chem. 2017, 60 (13), 5586–5598. 10.1021/acs.jmedchem.7b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur R.; Kaur G.; Gill R. K.; Soni R.; Bariwal J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. 10.1016/j.ejmech.2014.09.051. [DOI] [PubMed] [Google Scholar]
- Haider K.; Rahaman S.; Yar M. S.; Kamal A. Tubulin inhibitors as novel anticancer agents: an overview on patents (2013–2018). Expert Opin. Ther. Pat. 2019, 29 (8), 623–641. 10.1080/13543776.2019.1648433. [DOI] [PubMed] [Google Scholar]
- Deng S.; Banerjee S.; Chen H.; Pochampally S.; Wang Y.; Yun M.-K.; White S. W.; Parmar K.; Meibohm B.; Hartman K. L.; Wu Z.; Miller D. D.; Li W. SB226, an inhibitor of tubulin polymerization, inhibits paclitaxel-resistant melanoma growth and spontaneous metastasis. Cancer Lett. 2023, 555, 216046. 10.1016/j.canlet.2022.216046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Arnst K. E.; Wang Y.; Kumar G.; Deng S.; Yang L.; Li G.-b.; Yang J.; White S. W.; Li W.; Miller D. D. Heterocyclic-Fused Pyrimidines as Novel Tubulin Polymerization Inhibitors Targeting the Colchicine Binding Site: Structural Basis and Antitumor Efficacy. J. Med. Chem. 2018, 61 (4), 1704–1718. 10.1021/acs.jmedchem.7b01858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Ren Y.; Pan W.; Liu J.; Chen J. Discovery of Novel Acridane-Based Tubulin Polymerization Inhibitors with Anticancer and Potential Immunomodulatory Effects. J. Med. Chem. 2023, 66 (1), 627–640. 10.1021/acs.jmedchem.2c01566. [DOI] [PubMed] [Google Scholar]
- Leng J.; Zhao Y.; Sheng P.; Xia Y.; Chen T.; Zhao S.; Xie S.; Yan X.; Wang X.; Yin Y.; Kong L. Discovery of Novel N-Heterocyclic-Fused Deoxypodophyllotoxin Analogues as Tubulin Polymerization Inhibitors Targeting the Colchicine-Binding Site for Cancer Treatment. J. Med. Chem. 2022, 65 (24), 16774–16800. 10.1021/acs.jmedchem.2c01595. [DOI] [PubMed] [Google Scholar]
- Banerjee S.; Hwang D. J.; Li W.; Miller D. D. Current Advances of Tubulin Inhibitors in Nanoparticle Drug Delivery and Vascular Disruption/Angiogenesis. Molecules 2016, 21 (11), 1468. 10.3390/molecules21111468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanale D.; Bronte G.; Passiglia F.; Calò V.; Castiglia M.; Di Piazza F.; Barraco N.; Cangemi A.; Catarella M. T.; Insalaco L.; Listi A.; Maragliano R.; Massihnia D.; Perez A.; Toia F.; Cicero G.; Bazan V. Stabilizing versus Destabilizing the Microtubules: A Double-Edge Sword for an Effective Cancer Treatment Option?. Anal. Cell. Pathol. 2015, 2015, 690916. 10.1155/2015/690916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Chen J.; Wang J.; Ahn S.; Li C.-M.; Lu Y.; Loveless V. S.; Dalton J. T.; Miller D. D.; Li W. Novel Tubulin Polymerization Inhibitors Overcome Multidrug Resistance and Reduce Melanoma Lung Metastasis. Pharm. Res. 2012, 29 (11), 3040–3052. 10.1007/s11095-012-0726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Chen J.; Miller D. D.; Li W. Synergistic Combination of Novel Tubulin Inhibitor ABI-274 and Vemurafenib Overcomes Vemurafenib Acquired Resistance in BRAFV600E Melanoma. Mol. Cancer Ther. 2014, 13 (1), 16–26. 10.1158/1535-7163.MCT-13-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini K. M.; Huang S.-M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L.R.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; Hoffmaster K. A.; Ishikawa T.; Keppler D.; Kim R. B.; Lee C. A.; Niemi M.; Polli J. W.; Sugiyama Y.; Swaan P. W.; Ware J. A.; Wright S. H.; Wah Yee S.; Zamek-Gliszczynski M. J.; Zhang L. Membrane transporters in drug development. Nat. Rev. Drug Discovery 2010, 9 (3), 215–236. 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10 (3), 194–204. 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yang S.-H.; Guo X.-L. New insights into Vinca alkaloids resistance mechanism and circumvention in lung cancer. Biomed. Pharmacother. 2017, 96, 659–666. 10.1016/j.biopha.2017.10.041. [DOI] [PubMed] [Google Scholar]
- Ganguly A.; Cabral F. New insights into mechanisms of resistance to microtubule inhibitors. Biochim. Biophys. Acta 2011, 1816 (2), 164–171. 10.1016/j.bbcan.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Mahmud F.; Deng S.; Ma L.; Yun M.-K.; Fakayode S. O.; Arnst K. E.; Yang L.; Chen H.; Wu Z.; Lukka P. B.; Parmar K.; Meibohm B.; White S. W.; Wang Y.; Li W.; Miller D. D. X-ray Crystallography-Guided Design, Antitumor Efficacy, and QSAR Analysis of Metabolically Stable Cyclopenta-Pyrimidinyl Dihydroquinoxalinone as a Potent Tubulin Polymerization Inhibitor. J. Med. Chem. 2021, 64 (17), 13072–13095. 10.1021/acs.jmedchem.1c01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin E. C.; O’Boyle N. M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13 (1), 8. 10.3390/ph13010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y.; Liu W.; Tian L.; Mao Y.; Song C. Targeting Tubulin-colchicine Site for Cancer Therapy: Inhibitors, Antibody- Drug Conjugates and Degradation Agents. Curr. Top. Med. Chem. 2019, 19 (15), 1289–1304. 10.2174/1568026619666190618130008. [DOI] [PubMed] [Google Scholar]
- Song J.; Wang S.-H.; Song C.-H.; Zhang W.-X.; Zhu J.-X.; Tian X.-Y.; Fu X.-J.; Xu Y.; Jin C.-Y.; Zhang S.-Y. Discovery of N-benzylarylamide derivatives as novel tubulin polymerization inhibitors capable of activating the Hippo pathway. Eur. J. Med. Chem. 2022, 240, 114583. 10.1016/j.ejmech.2022.114583. [DOI] [PubMed] [Google Scholar]
- Mundra V.; Lu Y.; Danquah M.; Li W.; Miller D. D.; Mahato R. I. Formulation and Characterization of Polyester/Polycarbonate Nanoparticles for Delivery of a Novel Microtubule Destabilizing Agent. Pharm. Res. 2012, 29 (11), 3064–3074. 10.1007/s11095-012-0881-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai W.; Wang G.; Zhang Y.; Bu F.; Zhang S.; Miller D. D.; Li W.; Ouyang L.; Wang Y. Recent Progress on Tubulin Inhibitors with Dual Targeting Capabilities for Cancer Therapy. J. Med. Chem. 2021, 64 (12), 7963–7990. 10.1021/acs.jmedchem.1c00100. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Wang J.; Li C.-M.; Ahn S.; Barrett C. M.; Dalton J. T.; Li W.; Miller D. D. Design, Synthesis, and Biological Evaluation of Stable Colchicine Binding Site Tubulin Inhibitors as Potential Anticancer Agents. J. Med. Chem. 2014, 57 (17), 7355–7366. 10.1021/jm500764v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Quan D.; Chen J.; Ding J.; Zhao J.; Lv L.; Chen J. Design, synthesis, and biological evaluation of 1-substituted −2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. 10.1016/j.ejmech.2019.111732. [DOI] [PubMed] [Google Scholar]
- Chen J.; Ahn S.; Wang J.; Lu Y.; Dalton J. T.; Miller D. D.; Li W. Discovery of Novel 2-Aryl-4-benzoyl-imidazole (ABI-III) Analogues Targeting Tubulin Polymerization As Antiproliferative Agents. J. Med. Chem. 2012, 55 (16), 7285–7289. 10.1021/jm300564b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Li C.-M.; Wang Z.; Chen J.; Mohler M. L.; Li W.; Dalton J. T.; Miller D. D. Design, Synthesis, and SAR Studies of 4-Substituted Methoxylbenzoyl-aryl-thiazoles Analogues as Potent and Orally Bioavailable Anticancer Agents. J. Med. Chem. 2011, 54 (13), 4678–4693. 10.1021/jm2003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D.-J.; Wang J.; Li W.; Miller D. D. Structural Optimization of Indole Derivatives Acting at Colchicine Binding Site as Potential Anticancer Agents. ACS Med. Chem. Lett. 2015, 6 (9), 993–997. 10.1021/acsmedchemlett.5b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y.; Wang Y.; Li G.; Zhang Z.; Ma L.; Cheng B.; Chen J. Discovery of Novel Benzimidazole and Indazole Analogues as Tubulin Polymerization Inhibitors with Potent Anticancer Activities. J. Med. Chem. 2021, 64 (8), 4498–4515. 10.1021/acs.jmedchem.0c01837. [DOI] [PubMed] [Google Scholar]
- Deep A.; Bhatia R. K.; Kaur R.; Kumar S.; Jain U. K.; Singh H.; Batra S.; Kaushik D.; Deb P. K. Imidazo[1,2-a]pyridine Scaffold as Prospective Therapeutic Agents. Curr. Top. Med. Chem. 2016, 17 (2), 238–250. 10.2174/1568026616666160530153233. [DOI] [PubMed] [Google Scholar]
- Costa R. A.; Seuánez H. N. Investigation of major genetic alterations in neuroblastoma. Mol. Biol. Rep. 2018, 45 (3), 287–295. 10.1007/s11033-018-4161-4. [DOI] [PubMed] [Google Scholar]
- Swift C. C.; Eklund M. J.; Kraveka J. M.; Alazraki A. L. Updates in Diagnosis, Management, and Treatment of Neuroblastoma. Radiographics 2018, 38 (2), 566–580. 10.1148/rg.2018170132. [DOI] [PubMed] [Google Scholar]
- Don S.; Verrills N. M.; Liaw T. Y.; Liu M. L.; Norris M. D.; Haber M.; Kavallaris M. Neuronal-associated microtubule proteins class III beta-tubulin and MAP2c in neuroblastoma: role in resistance to microtubule-targeted drugs. Mol. Cancer Ther. 2004, 3 (9), 1137–1146. 10.1158/1535-7163.1137.3.9. [DOI] [PubMed] [Google Scholar]
- Kotchetkov R.; Cinatl J.; Blaheta R.; Vogel J.-U.; Karaskova J.; Squire J.; Hernáiz Driever P.; Klingebiel T.; Cinatl Jr J. Development of resistance to vincristine and doxorubicin in neuroblastoma alters malignant properties and induces additional karyotype changes: A preclinical model. Int. J. Cancer 2003, 104 (1), 36–43. 10.1002/ijc.10917. [DOI] [PubMed] [Google Scholar]
- Meany H. J.; Sackett D. L.; Maris J. M.; Ward Y.; Krivoshik A.; Cohn S. L.; Steinberg S. M.; Balis F. M.; Fox E. Clinical outcome in children with recurrent neuroblastoma treated with ABT-751 and effect of ABT-751 on proliferation of neuroblastoma cell lines and on tubulin polymerization in vitro. Pediatr. Blood Cancer 2010, 54 (1), 47–54. 10.1002/pbc.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommann K.; Appl B.; Hundsdoerfer P.; Reinshagen K.; Eschenburg G. Vincristine resistance in relapsed neuroblastoma can be efficiently overcome by Smac mimetic LCL161 treatment. J. Pediatr. Surg. 2018, 53 (10), 2059–2064. 10.1016/j.jpedsurg.2018.01.012. [DOI] [PubMed] [Google Scholar]
- Park J. R.; Bagatell R.; London W. B.; Maris J. M.; Cohn S. L.; Mattay K. K.; Hogarty M. Children’s Oncology Group’s 2013 blueprint for research: neuroblastoma. Pediatr. Blood Cancer 2013, 60 (6), 985–993. 10.1002/pbc.24433. [DOI] [PubMed] [Google Scholar]
- Aho E. R.; Weissmiller A. M.; Fesik S. W.; Tansey W. P. Targeting WDR5: A WINning Anti-Cancer Strategy?. Epigenet. Insights 2019, 12, 251686571986528. 10.1177/2516865719865282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan A. F.; Wang J.; Howard G. C.; Guarnaccia A. D.; Woodley C. M.; Aho E. R.; Rellinger E. J.; Matlock B. K.; Flaherty D. K.; Lorey S. L.; Chung D. H.; Fesik S. W.; Liu Qi.; Weissmiller A. M.; Tansey W. P. WDR5 is a conserved regulator of protein synthesis gene expression. Nucleic Acids Res. 2020, 48, 2924. 10.1093/nar/gkaa051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong D.; Ferrell J. E. Jr. The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol. Biol. Cell 2010, 21 (18), 3149–3161. 10.1091/mbc.e10-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley L. R.; D’Anna J. A.; Barham S. S.; Deaven L. L.; Tobey R. A. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur. J. Biochem. 1978, 84 (1), 1–15. 10.1111/j.1432-1033.1978.tb12135.x. [DOI] [PubMed] [Google Scholar]
- Taubenberger A. V.; Baum B.; Matthews H. K. The Mechanics of Mitotic Cell Rounding. Front. Cell Dev. Biol. 2020, 8, 687. 10.3389/fcell.2020.00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. 2014, 12 (6), 815–822. 10.1158/1541-7786.MCR-13-0536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.