

Key Points
Questions
Are gray matter changes across different stages of psychosis constrained by brain network architecture and are certain regions epicenters of volume loss?
Findings
In this case-control study of 534 participants from 4 independent samples spanning different stages of psychotic illness, gray matter alterations were constrained by the underlying architecture of the brain’s axonal pathways, and the hippocampus was consistently identified as a putative source from which volume loss may spread to connected regions.
Meaning
These findings suggest that white matter fibers may act as conduits for the spread of pathology across all stages of psychotic illness, and medial temporal regions play a critical role in the origins of gray matter volume reductions.
This case-control study models the processes that explain spatial patterns of gray matter volume loss in different stages of psychotic illness and assesses whether certain brain regions act as putative epicenters from which volume loss spreads.
Abstract
Importance
Psychotic illness is associated with anatomically distributed gray matter reductions that can worsen with illness progression, but the mechanisms underlying the specific spatial patterning of these changes is unknown.
Objective
To test the hypothesis that brain network architecture constrains cross-sectional and longitudinal gray matter alterations across different stages of psychotic illness and to identify whether certain brain regions act as putative epicenters from which volume loss spreads.
Design, Settings, and Participants
This case-control study included 534 individuals from 4 cohorts, spanning early and late stages of psychotic illness. Early-stage cohorts included patients with antipsychotic-naive first-episode psychosis (n = 59) and a group of patients receiving medications within 3 years of psychosis onset (n = 121). Late-stage cohorts comprised 2 independent samples of people with established schizophrenia (n = 136). Each patient group had a corresponding matched control group (n = 218). A sample of healthy adults (n = 356) was used to derive representative structural and functional brain networks for modeling of network-based spreading processes. Longitudinal illness-related and antipsychotic-related gray matter changes over 3 and 12 months were examined using a triple-blind randomized placebo-control magnetic resonance imaging study of the antipsychotic-naive patients. All data were collected between April 29, 2008, and January 15, 2020, and analyses were performed between March 1, 2021, and January 14, 2023.
Main Outcomes and Measures
Coordinated deformation models were used to estimate the extent of gray matter volume (GMV) change in each of 332 parcellated areas by the volume changes observed in areas to which they were structurally or functionally coupled. To identify putative epicenters of volume loss, a network diffusion model was used to simulate the spread of pathology from different seed regions. Correlations between estimated and empirical spatial patterns of GMV alterations were used to quantify model performance.
Results
Of 534 included individuals, 354 (66.3%) were men, and the mean (SD) age was 28.4 (7.4) years. In both early and late stages of illness, spatial patterns of cross-sectional volume differences between patients and controls were more accurately estimated by coordinated deformation models constrained by structural, rather than functional, network architecture (r range, >0.46 to <0.57; P < .01). The same model also robustly estimated longitudinal volume changes related to illness (r ≥ 0.52; P < .001) and antipsychotic exposure (r ≥ 0.50; P < .004). Network diffusion modeling consistently identified, across all 4 data sets, the anterior hippocampus as a putative epicenter of pathological spread in psychosis. Epicenters of longitudinal GMV loss were apparent in posterior cortex early in the illness and shifted to the prefrontal cortex with illness progression.
Conclusion and Relevance
These findings highlight a central role for white matter fibers as conduits for the spread of pathology across different stages of psychotic illness, mirroring findings reported in neurodegenerative conditions. The structural connectome thus represents a fundamental constraint on brain changes in psychosis, regardless of whether these changes are caused by illness or medication. Moreover, the anterior hippocampus represents a putative epicenter of early brain pathology from which dysfunction may spread to affect connected areas.
Introduction
Psychotic disorders such as schizophrenia are characterized by anatomically distributed reductions of gray matter volume (GMV),1,2,3,4,5,6 which can progress over time and across different illness stages.7,8,9,10,11,12,13 Meta-analyses and mega-analyses identify robust cross-sectional GMV reductions in frontal, cingulate, and temporal cortices, as well as medial temporal lobe and thalamus,3,4,5,6,14,15,16,17 along with longitudinal reductions in frontal, temporal, and parietal cortices.8,10,18 However, despite a large literature describing the location and nature of these brain changes, the specific mechanisms that give rise to their characteristic spatial pattern remain unknown.
The human brain is an intricate network of functionally specialized regions linked by a complex web of axonal fibers, referred to as a connectome.19 These fibers enable the widespread coordination of neuronal dynamics and the transport of trophic and other biological molecules throughout the brain. They can also act as conduits for the spread of pathology, such that illness processes originating in one area can propagate to affect distributed systems via multiple mechanisms.20,21 Accordingly, the spread of GMV reductions in different neurodegenerative conditions is constrained by the brain’s white-matter architecture.22,23,24,25
A network-based spreading process may also be involved in psychosis. Cross-sectional regional GMV reductions in patients correlate with the microstructure of adjacent white matter,26 across spatially distributed regions,27,28,29 with normative connectome organization,30,31,32 and with reductions in structurally connected regions.33 Together, these findings support the hypothesis that the spatial patterning of GMV loss in psychotic illness is constrained by connectome architecture. However, the few studies addressing this question26,27,28,29,30,31,33 have been cross-sectional and only examined patients with chronic illness, precluding an opportunity to track how coordinated gray matter changes evolve through time and across illness stages. It thus remains unclear whether longitudinal GMV changes are actually constrained by brain network architecture, as would be expected for a network-based spreading process. Moreover, the reliance on samples of patients taking antipsychotic medication makes it difficult to determine whether coupled gray matter changes are driven by treatments for the illness or the illness process itself.13,34
Herein, we used magnetic resonance imaging (MRI) in multiple cohorts spanning different stages of psychosis to comprehensively evaluate network constraints on cross-sectional and longitudinal GMV changes. Specifically, we evaluated the capacity of different coordinated deformation model (CDM) variants, constrained by distinct aspects of connectome structure or function, to model cross-sectional GMV differences in 2 samples of patients at early illness stages and 2 samples of patients at later stages, allowing for independent replication of our findings at each stage (Figure 1). We further leveraged longitudinal scans acquired in one of the early-stage data sets during a randomized placebo-controlled trial13,35 to disentangle network constraints on brain changes related to illness from those related to antipsychotic exposure. Our approach allowed us to robustly investigate brain network constraints on diverse cross-sectional and longitudinal GMV pathology across different stages of psychosis and to identify possible focal points of early brain volume loss.
Figure 1. Analysis Workflow for the Coordinated Deformation Model (CDM).
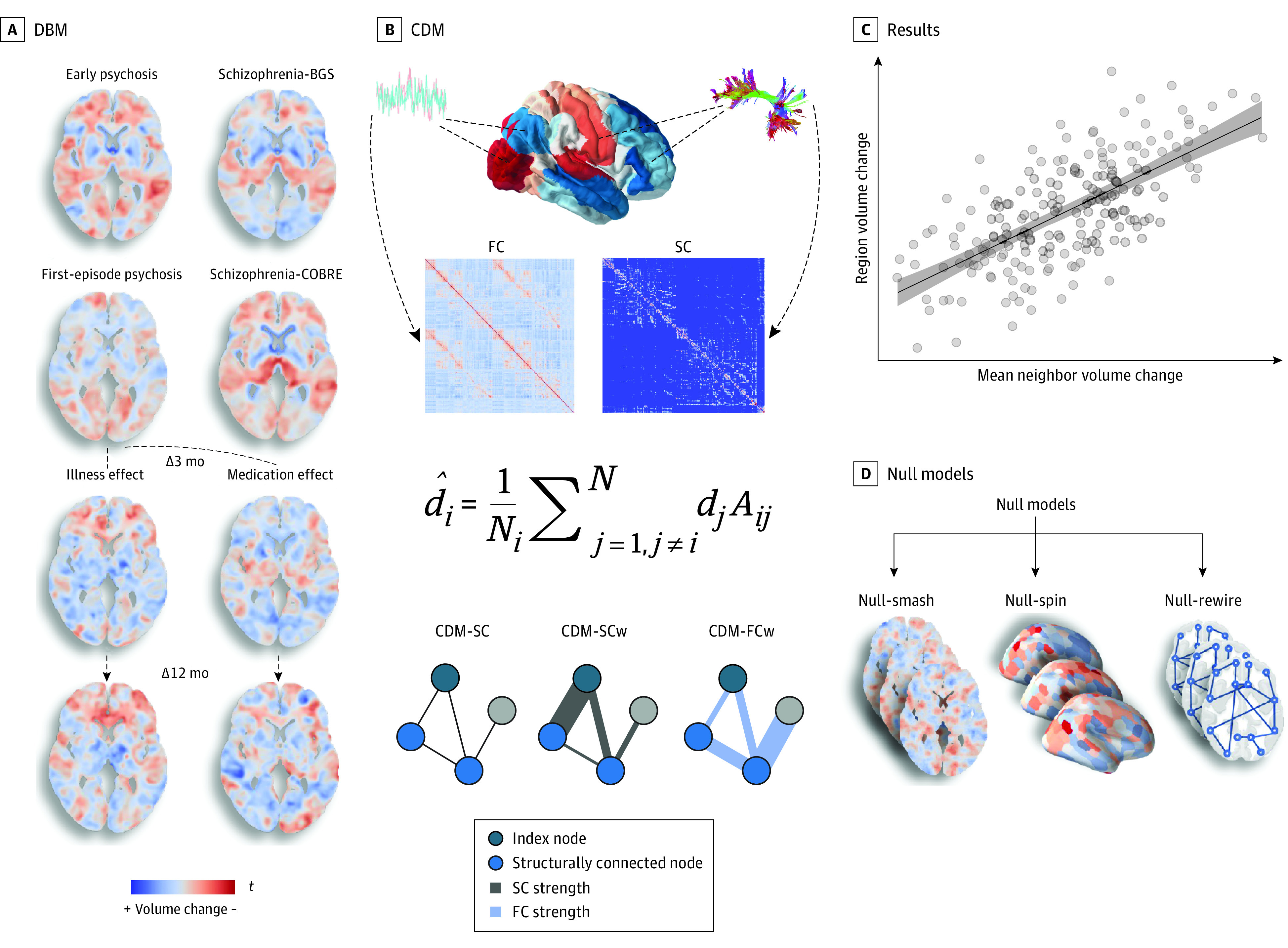
A, We derived voxelwise gray matter volume (GMV) estimates using deformation-based morphometry (DBM). Five separate contrasts were specified using a robust marginal model to infer baseline GMV differences and longitudinal GMV changes associated with illness and antipsychotic medication at 3 and 12 months. Cross-sectional contrasts were specified such that positive values (in red) in the resulting voxelwise t statistic maps indicate lower volume in patients compared with controls. For the illness-related longitudinal contrasts, positive values (in red) in the resulting voxelwise t statistic maps indicate greater longitudinal GMV decline in patients receiving placebo compared with controls. For the medication-related longitudinal contrasts, positive values in the resulting voxelwise t statistic maps indicate greater longitudinal GMV decline in patients receiving medication compared with both those receiving placebo and controls. Schizophrenia data sets included participants in the BrainGluSchi (BGS) and COBRE studies. Δ indicates change from baseline. B, The contrast statistics were mapped to a brain parcellation comprising 332 regions, and diffusion and functional magnetic resonance imaging data from an independent healthy sample were used to generate sample-averaged functional coupling (FC) and structural connectivity (SC) matrices. These matrices were used to model mean volume changes in structurally connected neighbors. Under the CDM, the estimated deformation of a node, d̂i, is modeled as a weighted sum of the deformation values observed in its structurally connected neighbors, di (shown as light blue nodes in the example graphs). The weights are given by the adjacency matrix, Aij. Three different matrices were used, yielding 3 CDM variants: (1) a model denoted as CDM-SC, in which Aij = 1 if regions i and j share a connection and Aij = 0 otherwise; (2) a model denoted as CDM-SCw in which the elements of Aij correspond precisely to the weighted SC matrix, such that the contribution of each neighbor is weighted by the strength of its structural connectivity to the index node; and (3) a model denoted CDM-FCw, in which the elements of Aij correspond precisely to the weighted FC matrix, such that the contribution of each neighbor is weighted by its FC with the index node. C, Model performance was evaluated using the product-moment correlation between regional estimates of observed and estimated GMV differences. D, We also compared model performance with 3 benchmark null models accounting for spatial autocorrelations in the deformation maps (Null-smash and Null-spin) and basic topological properties of the connectome (Null-rewire) (see Statistical Analysis subsection of the Methods section and eMethods 9 in Supplement 1).
Methods
Sample Characteristics
This case-control study used data from 4 independent data sets sampling different stages of psychotic illness: the STAGES (Staged Treatment and Acceptability Guidelines in Early Psychosis Study) clinical trial13,35 (first-episode psychosis), Human Connectome Project Early Psychosis36 (early psychosis), BrainGluSchi37 (schizophrenia), and COBRE38 (schizophrenia). Representative structural and functional connectomes were derived using an independent control sample. The Table shows sample characteristics (further details in eMethods 1 in Supplement 1). All data were collected between April 29, 2008, and January 15, 2020, and analyses were performed between March 1, 2021, and January 14, 2023. The first-episode psychosis data set received ethics approval from Melbourne Health Human Research and Ethics committee. The independent control data set received ethics approval from Monash University Human Research Ethics committee. All participants gave written informed consent after having the study fully explained to them.13,35 The remaining 3 publicly available data sets were approved by their respective committees, which ensured appropriate informed consent procedures.36,37,38 This study aligns with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Table. Sample Characteristics of Included Data Sets.
| Characteristic | First-episode psychosis (n = 86)a | Early psychosis (n = 178) | Schizophrenia (SCZ-COBRE) (n = 138) | Schizophrenia (SCZ-BGS) (n = 132) | Independent healthy controls (n = 356) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo group (n = 30) | Antipsychotic group (n = 29) | Matched controls (n = 27) | Patients (n = 121) | Matched controls (n = 57) | Patients (n = 66) | Matched controls (n = 72) | Patients (n = 70) | Matched controls (n = 62) | ||
| Baseline age, mean (SD), y | 18.8 (2.7) | 19.5 (2.9) | 21.9 (1.9) | 23.3 (3.9) | 23.4 (3.9) | 38.0 (14.1) | 35.9 (11.7) | 36.6 (13.4) | 38.2 (12.4) | 23.7 (5.4) |
| Baseline sex, No. (%) | ||||||||||
| Women | 14 (46.7) | 13 (44.8) | 17 (63.0) | 47 (38.8) | 30 (52.6) | 12 (18.2) | 23 (31.9) | 10 (14.3) | 14 (22.6) | 198 (55.6) |
| Men | 16 (53.3) | 16 (55.2) | 10 (37.0) | 74 (61.2) | 27 (47.4) | 54 (81.8) | 49 (68.1) | 60 (85.7) | 48 (77.4) | 158 (44.4) |
| Diagnosis, No.b | ||||||||||
| Major depression with psychosis | 7 | 5 | NA | 5 | NA | 0 | NA | 0 | NA | NA |
| Schizophreniform disorder | 5 | 5 | NA | 8 | NA | 0 | NA | 0 | NA | NA |
| Psychotic disorder NOS | 8 | 7 | NA | 0 | NA | 0 | NA | 0 | NA | NA |
| Substance-induced psychotic disorder | <5 | <5 | NA | 0 | NA | 0 | NA | 0 | NA | NA |
| Delusional disorder | <5 | <5 | NA | 0 | NA | 0 | NA | 0 | NA | NA |
| Schizoaffective disorder | 0 | 0 | NA | 13 | NA | 6 | NA | 0 | NA | NA |
| Schizophrenia | 5 | 5 | NA | 61 | NA | 60 | NA | 70 | NA | NA |
| Bipolar disorder with psychosis | 0 | 0 | NA | 25 | NA | 0 | NA | 0 | NA | NA |
| Missing diagnosis | 0 | <5 | NA | 0 | NA | 0 | NA | 0 | NA | NA |
| Illness duration, mean (SD), y | <0.5 | <0.5 | NA | 1.8 (1.4) | NA | 16.0 (12.2) | NA | 18.2 (12.9) | NA | NA |
| Symptom severity score, mean (SD) | BPRS total, 59.4 (9.64)c,d | BPRS total, 55.8 (10.10)c,d | NA | PANSS total, 49.7 (11.0)e | NA | PANSS total, 58.4 (13.6)e | NA | PANSS, 60.7 (16.9)e | NA | NA |
| Functional outcome score, mean (SD) | SOFAS, 52.9 (14.0)c,f | SOFAS, 51.7 (10.6)c,f | NA | GAF, 66.4 (16.6)g | NA | CGI, 3.7 (0.6)h | NA | CGI, 3.9 (0.8)h | NA | NA |
Abbreviations: BPRS, Brief Psychiatric Rating Scale, version 4; CGI, Clinical Global Impressions Scale; GAF, Global Assessment of Functioning Scale (mean of the social and occupational rating items); NA, not applicable; NOS, not otherwise specified; PANSS, Positive and Negative Syndrome Scale; SCZ-BGS, schizophrenia-BrainGluSchi study; SCZ-COBRE, schizophrenia-COBRE study; SOFAS, Social and Occupational Functioning Assessment Scale.
The placebo group included 21 patients at 3-month follow-up and 22 at 12-month follow-up; the antipsychotic group, 20 at 3-month follow-up and 14 at 12-month follow-up; and the matched controls, 21 at 3- and 12-month follow-ups.
Diagnostic groups with fewer than 5 patients are specified as less than 5 to avoid compromising identifiability.
Obtained at baseline.
Scores range from 24 to 168, with higher scores indicating worse symptoms.
Scores range from from 30 to 210, with higher scores indicating worse symptoms.
Scores range from 1 to 100, with higher scores indicating better functioning.
Scores range from 1 to 100, with higher scores indicating better functioning.
Scores range from 1 to 7, with higher scores indicating more severe illness.
Quantifying Cross-Sectional and Longitudinal Gray Matter Changes in Patients
Deformation-based morphometry was used to map regional volume changes (see eMethods 2 for MRI acquisition parameters and eMethods 3 for details on deformation-based morphometry protocol in Supplement 1). Robust marginal models implemented in the Sandwich Estimator Toolbox39 quantified spatial patterns of group-level cross-sectional and longitudinal volume change. All contrasts were adjusted for age, sex, and handedness, with site additionally included for the early psychosis data set.
Cross-sectional contrasts in each of the 4 patient data sets captured GMV differences between patients and controls (Figure 1A). The cross-sectional contrast for first-episode psychosis compared patients and controls at baseline, prior to treatment group allocation (see eMethods 1 in Supplement 1). Longitudinal GMV contrasts in first-episode psychosis (Figure 1A) isolated (1) illness-related change over time, by comparing longitudinal GMV changes in the placebo group with matched healthy controls, and (2) antipsychotic-related changes over time, by comparing longitudinal GMV changes in the medication group to both the placebo group and matched healthy controls (see eMethods 4 in Supplement 1 for further details). Longitudinal contrasts were assessed from baseline to 3 months and from baseline to 12 months, with a linear contrast used for the latter.
Given our primary goal to model the processes that explain spatial patterns of GMV loss in psychosis, rather than merely map these patterns, we converted the voxelwise, contrast-specific t maps encoding regional GMV changes to unthresholded z maps. These maps were used as inputs to our models, allowing us to model the complete spatial pattern of GMV differences across the brain and not just differences surviving a statistical threshold. Renderings of the unthresholded t maps can be found in the top rows of Figure 2 and Figure 3. False discovery rate–corrected and –uncorrected voxel-level t statistic maps for each contrast are provided in eMethods 7 and 8 in Supplement 1. The maps were then parcellated into 300 discrete cortical40 and 32 subcortical41 areas (see eMethods 4 in Supplement 1 for details) to enable analysis with respect to estimates of normative connectome architecture.
Figure 2. Baseline and Longitudinal Illness-Related Gray Matter Volume Changes Are Constrained by Connectome Anatomy.
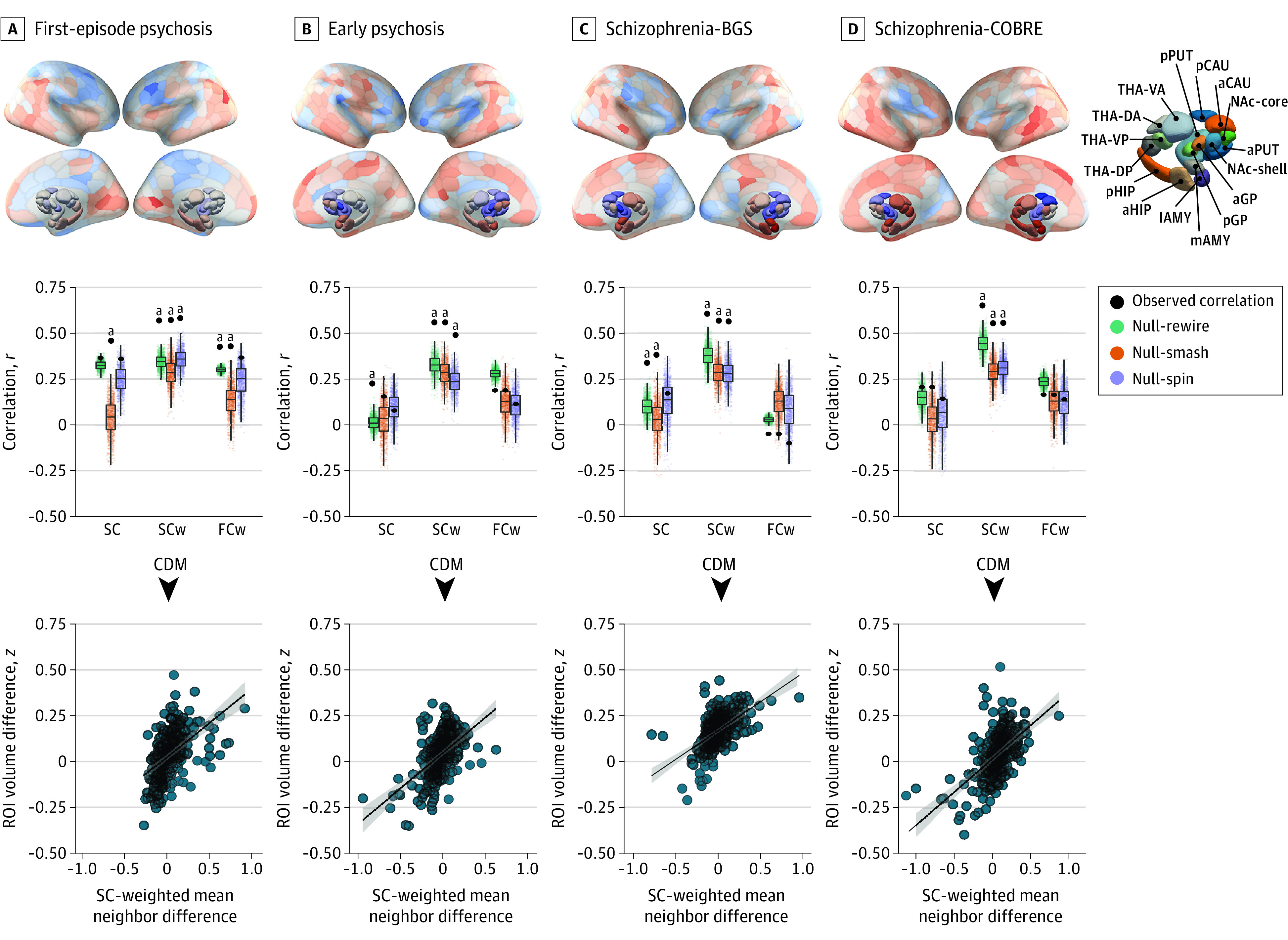
Top row, The contrast statistics for 4 cross-sectional contrasts (first-episode psychosis, early psychosis, schizophrenia-BrainGluSchi [BGS], and schizophernia-COBRE data sets) mapped to a brain parcellation comprising 332 regions. a Indicates anterior; AMY, amygdala; CAU, caudate nucleus; d, dorsal; DA, dorsoanterior; DMN, default mode network; DorsAttn, dorsal attention network; DP, dorsoposterior; FPN, frontoparietal network; GP, globus pallidus; HIP, hippocampus; l, lateral; Lim, cortical limbic network; m, medial; MTL, medial-temporal lobe (amygdala and hippocampus); NAc, nucleus accumbens; p, posterior; SomMot, somatomotor network; Stri, striatum; PUT, putamen; and THA, thalamus. Middle row, Performance of the equally weighted (SC), structural connectivity–weighted (SCw), and functional coupling–weighted (FCw) coordinated deformation models (CDMs) relative to the Null-smash, Null-spin, and Null-rewire benchmarks. Black circles indicate the observed product-moment correlations between estimated and actual regional deformation values for each model. Note that the observed value used for comparison against the Null-spin models is different because the subcortex was excluded. Bottom row, Scatterplots of the association between observed and estimated regional volume deformation values for the best-performing CDM-SCw model at each time point. ROI indicates region of interest.
aP < .016.
Figure 3. Longitudinal Illness-Related and Antipsychotic-Related Gray Matter Volume Changes Are Constrained by Connectome Anatomy.
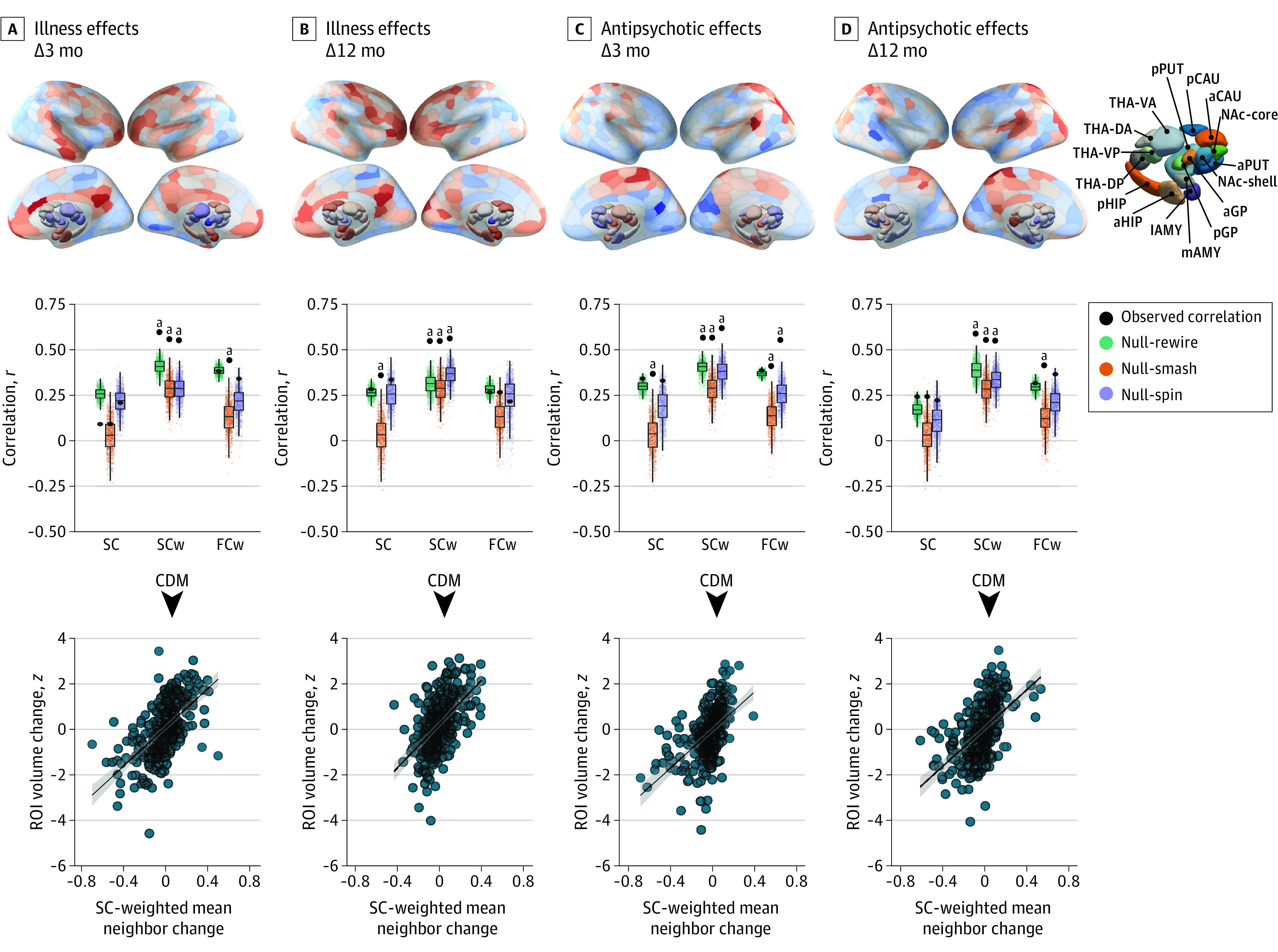
Top row, The contrast statistics for illness-related and antipsychotic-related contrasts mapped to a brain parcellation comprising 332 regions. a Indicates anterior; AMY, amygdala; CAU, caudate nucleus; d, dorsal; DA, dorsoanterior; DMN, default mode network; DorsAttn, dorsal attention network; DP, dorsoposterior; FPN, frontoparietal network; GP, globus pallidus; HIP, hippocampus; l, lateral; Lim, cortical limbic network; m, medial; MTL, medial-temporal lobe (amygdala and hippocampus); NAc, nucleus accumbens; p, posterior; SomMot, somatomotor network; Stri, striatum; PUT, putamen; and THA, thalamus. Middle row, Performance of the equally weighted (SC), structural connectivity–weighted (SCw), and functional coupling–weighted (FCw) coordinated deformation models (CDMs) relative to the Null-smash, Null-spin, and Null-rewire benchmarks. Black circles indicate the observed product-moment correlations between estimated and actual regional deformation values for each model at each time point. Note that the observed value used for comparison against the Null-spin models is different because the subcortex was excluded. Bottom row, Scatterplots of the association between observed and estimated regional deformation values for the best-performing CDM-SCw model at each time point. ROI indicates region of interest.
aP < .004.
Healthy Reference Connectomes
Our models relied on a normative estimate of connectome architecture derived from diffusion-weighted MRI data acquired in an independent sample of 356 healthy adults (Figure 1B and Table; see eMethods 5 in Supplement 1 for details on image acquisition and processing). This procedure resulted in a single 332 × 332 weighted group-average matrix encoding pairwise interregional structural connectivity. We also derived a group-level 332 × 332 weighted matrix of interregional functional coupling estimates using resting-state functional MRI data acquired in the same sample (Figure 1B; see eMethods 6 in Supplement 1 for details on image acquisition and processing).
Coordinated Deformation Models
We evaluated network constraints on cross-sectional and longitudinal GMV changes using the CDM introduced by Shafiei et al33 and defined as
 |
where d̂i is the estimated GMV change in node i, Ni is the number of structurally connected neighbors of i, dj is the deformation observed in the j-th neighbor of node i, and Aij defines the connectivity between nodes i and j. Three different matrices were substituted for Aij, yielding 3 variants of the CDM (Figure 1B). For the first model, denoted CDM-SC, Aij = 1 if nodes i and j are connected in the group-mean structural connectivity matrix and zero otherwise. Therefore, all j structurally connected neighbors make an equal contribution to estimating the deformation observed in node i.
For the second and third models, denoted CDM-SCw and CDM-FCw, Aij corresponded to the weighted structural connectivity or functional coupling matrices, respectively. Therefore, under these models, the contributions of node i’s neighbors were weighted by either interregional structural connectivity (CDM-SCw) or functional coupling (CDM-FCw) estimates, such that neighbors of node i with a more strongly weighted connection made a stronger contribution to estimating node i’s volume change (Figure 1B). In all models, only edges with a corresponding structural connection were included, and structural connectivity and functional coupling edge weights were taken from the healthy reference connectome, unless otherwise specified.
Network Diffusion Model–Epicenter Identification
The CDM evaluates the degree to which spatial patterns of GMV change are shaped by connectome properties. A close coupling between GMV change and network architecture implies that volume loss may spread through the connectome, but the CDM offers limited insight into the dynamics of the spreading process, nor is it able to identify regions from which the spreading may initiate. We therefore used a network diffusion model (NDM) to directly test whether GMV loss spreads through the brain via a process of diffusion and whether certain brain regions act as sources, or epicenters, of pathological spread through the brain (Figure 4).24 The NDM simulates the dynamic spread of pathology between the nodes of a weighted network via a process of diffusion (Figure 4A), defined as f(t) = e–aHtf0, where t is the model diffusion time, which has arbitrary units, and f(t) is a vector characterizing the amount of diffusion in each region at time t. The strength of the diffusion process is controlled by a constant (a), H is the Laplacian of the weighted structural connectivity matrix, and f0 represents the initial distribution of pathology. We repeatedly initialized the model using each of the 332 regions as the starting seed, such that the initial state was set to 1 for the seed region and 0 for all other regions. At each initialization, using a constant of a = 1, the NDM was used to estimate the diffusion at all other regions at time t = 0 to 50. In this way, we were able to determine whether a diffusion process seeded from each region resulted in a spatial distribution of volume loss that matched the empirically observed patterns. Further information about the NDM can be found in eMethods 10 in Supplement 1.
Figure 4. Regional Epicenters of Gray Matter Volume (GMV) Loss.
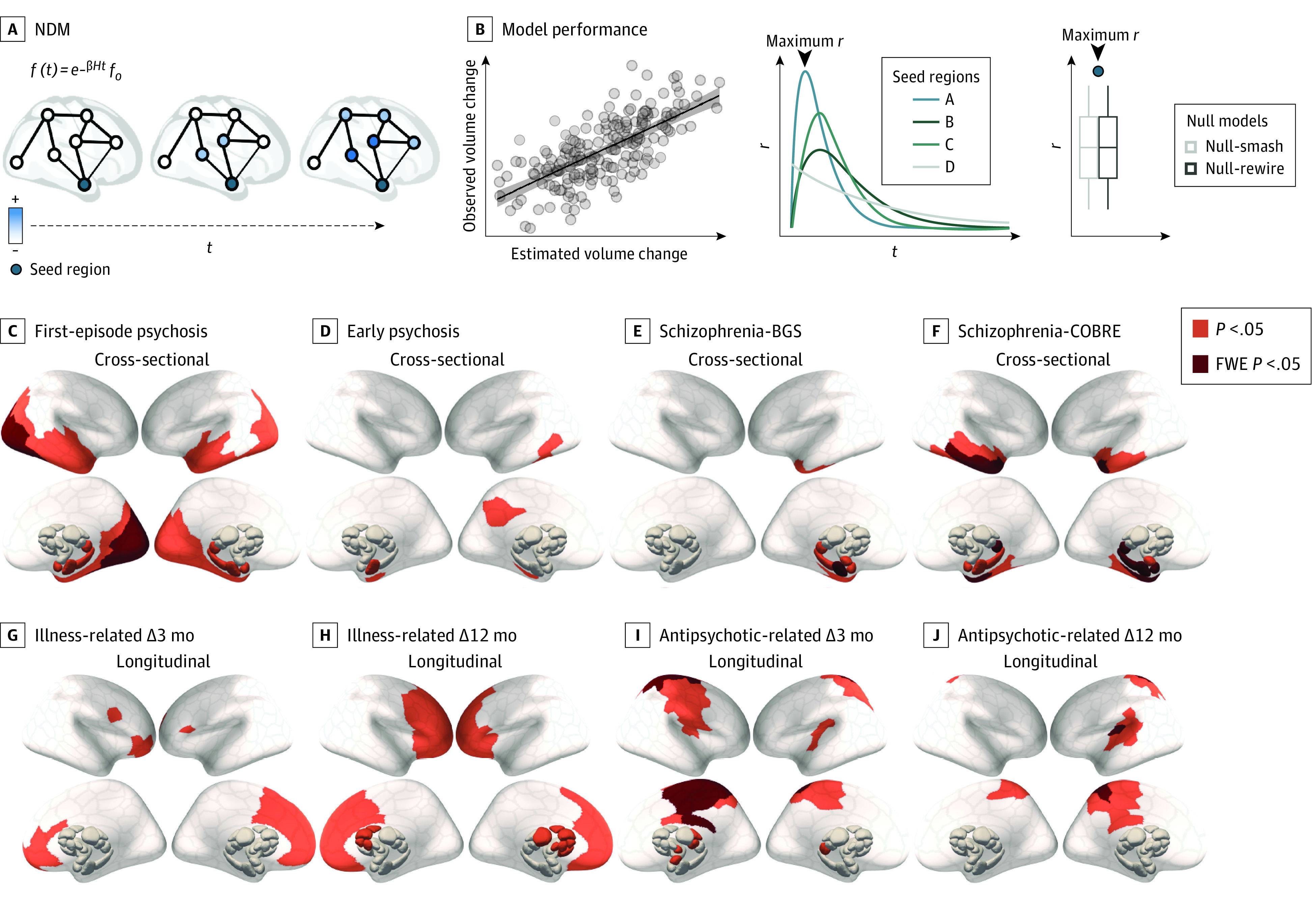
A, Epicenters were defined as potential sources of pathological volume loss from which GMV reductions spread (blue) to affect structurally connected areas. To identify such regions, we simulated a spreading process using a network diffusion model (NDM). Schizophrenia data sets included participants in the BrainGluSchi (BGS) and COBRE studies. B, Using each of the 332 parcellated regions as a seed, we retained the maximum correlation between the simulated and observed GMV abnormalities (maximum r). For each contrast, we then compared maximum r values for each region to a distribution of maximum r values from 2 benchmark null models accounting for spatial autocorrelations in the deformation maps (Null-smash) and basic topological properties of the connectome (see Statistical Analysis subsection of the Methods section [NDM]). Regional epicenters with significantly greater maximum r than a spatially constrained null model (orange indicates P < .05; red, familywise error [FWE] P < .05) are shown for cross-sectional (C-F) and longitudinal (G-J) effects. Results using Null-rewire benchmark models and scatterplots of observed and estimated volume abnormalities are provided in eFigure 3 in Supplement 1.
Statistical Analysis
We evaluated CDM performance using the product-moment correlation (r) between regionwise estimates of observed and estimated deformation (Figure 1C). We also compared the performance of the CDM-SC, CDM-SCw, and CDM-FCw variants with 3 benchmark null models: 2 models, denoted Null-smash and Null-spin, ensured our findings were not explained by spatial properties of the deformation maps and the third model, denoted Null-rewire, ensured they could not be explained by low-level topological properties of the connectome (see eMethods 9 in Supplement 1 for details). Statistical inferences on cross-sectional analyses (Figure 2) were Bonferroni-corrected for 3 models within each data set and evaluated at α = .016. Illness-related and medication-related longitudinal comparisons from the first-episode psychosis data set (Figure 3) were corrected for 3 models at each of the 4 contrasts, with α = .004.
We quantified NDM performance for each seed region as the maximum correlation between observed and estimated GMV contrast maps obtained across all time steps of the model24,42 (Figure 4A) (maximum r). Performance was compared to the same benchmark null models as in the CDM evaluation, with familywise error (FWE) correction applied to account for 332 seed regions (details in eMethods 10 in Supplement 1). Two-sided P < .05 indicated statistical significance. All statistical analyses used R, version 4.1.1 (R Program for Statistical Computing).
Results
Structural Connectivity Shapes Cross-Sectional GMV Alterations Across Illness Stages
Of 534 cases and controls included in the analysis, 354 (66.3%) were men and 180 (33.7%) were women; the mean (SD) age was 28.4 (7.4) years. Details on race and ethnicity across the 4 data sets are available in depth elsewhere.13,35,36,37,38 We first evaluated the performance of the 3 CDMs in capturing cross-sectional differences in regional GMV. In all data sets, the CDM-SCw variant yielded more accurate estimates of empirical differences (r range, >0.46 to <0.57) when compared with the CDM-SC and CDM-FCw models (all r < 0.35) (Figure 2, middle row). The performance of the CDM-SCw variant was also significantly better than that of all 3 benchmark models in all data sets (all P < .01). The CDM-SC and CDM-FCw variants generally did not surpass the performance of the benchmark models. Across samples, model performance was positively correlated with mean illness duration and symptom severity (P < .05) (eFigure 1 in Supplement 1), suggesting that gray matter differences may be more constrained by network architecture in later illness stages.
Structural Connectivity Shapes Longitudinal Illness and Medication-Related GMV Changes
Having robustly demonstrated that connectome structure shapes cross-sectional GMV differences, we next tested the implicit assumption of the CDM—that longitudinal GMV changes spread across axonal pathways—by considering longitudinal illness-related and medication-related changes in the sample with first-episode psychosis. Estimates of the CDM-SCw model for illness-related gray matter changes at 3 and 12 months were correlated with empirical changes at r = 0.58 (Figure 3A, middle row) and r = 0.52 (Figure 3B, middle row), respectively, and both correlations were significantly larger than all 3 benchmark models (all P < .001). In comparison, correlations for the CDM-SC and CDM-FCw variants did not exceed r = 0.38 and only showed significantly better performance compared with the Null-smash at 3 and 12 months, respectively (Figure 3A and B, middle row).
Estimates of the CDM-SCw model for antipsychotic-related GMV changes were correlated with the empirical maps at r = 0.51 (Figure 3C, middle row) for 3 months and r = 0.50 (Figure 3D, middle row) for 12 months. These correlations were significant when compared with all 3 null models (all P < .004) (Figure 3C and D, middle row). Associations at 3 and 12 months were smaller for the CDM-SC (r = 0.34 and r = 0.24, respectively) and CDM-FCw (r = 0.38 and r = 0.31, respectively) variants, which differed from the performance of the null benchmarks only in isolated cases (Figure 3). Thus, connectome structure represents a generic and fundamental constraint on both illness-related and medication-related longitudinal GMV changes in psychosis.
Epicenters of GMV Loss
We next used the NDM to simulate the dynamic spread of GMV loss from each individual brain region. Results using the Null-smash benchmark are presented below (Figure 4C-J). Across all cross-sectional comparisons, medial temporal lobe regions emerged as statistically significant epicenters (Figure 4C-F), with the anterior hippocampus consistently implicated across all data sets (P < .05) and surviving multiple comparison correction (FWE P < .05) in the 2 samples with schizophrenia. In first-episode psychosis, additional epicenters were identified in bilateral occipital and temporal cortex, as well as amygdala and posterior thalamic regions (Figure 4C). In the early psychosis data set, additional epicenters were identified in temporal and posterior cingulate cortex (Figure 4D). In both samples with schizophrenia, additional epicenters were identified in the temporal cortex, amygdala, and posterior thalamic regions (Figure 4E and F). Consistent results were obtained using the Null-rewire benchmark (eFigure 4 in Supplement 1). The spatial locations of our epicenters aligned with those identified through an alternative data-driven method used in past work33 (eFigure 2 in Supplement 1; see eMethods 11 in Supplement 1 for details).
In first-episode psychosis, epicenters of longitudinal illness-related loss were identified in medial frontal regions at 3 months and progressed to include much of the frontal cortex, as well as striatal and thalamic regions, by 12 months (Figure 4G and H). Comparisons with the connectome-based null benchmarks were more conservative, but also implicated prefrontal regions (eFigure 4 in Supplement 1).
Epicenters of longitudinal antipsychotic-related GMV loss in first-episode psychosis were identified in the sensorimotor, cingulate, and insula cortices, as well as the thalamic and amygdala regions at 3 months, with the same cortical epicenters also identified at 12 months (Figure 4I and J). These results were largely consistent when using the Null-rewire models (eFigure 4 in Supplement 1). Scatterplots of observed and estimated volume abnormalities for all contrasts are provided in eFigure 3 in Supplement 1.
Robustness Analyses
Our findings were robust to the use of alternative normative connectomes, modeling and data processing choices, and diagnostic subgroups. Findings are detailed in eMethods 12 and eFigures 5 through 10 in Supplement 1.
Discussion
The mechanisms driving spatially patterned GMV changes in psychotic illness have thus far been unclear. In this case-control study, we used a simple CDM to confirm that, across both early and later stages of illness, cross-sectional GMV changes are shaped by the topology and strength of structural, but not functional, connectivity between brain regions. We further found that both illness-related and antipsychotic-related longitudinal changes are constrained by structural connectivity, indicating that the temporal evolution of brain changes in the illness is also constrained by the brain’s axonal pathways. Moreover, using an NDM to simulate the spread of pathology from different brain regions, we identified the anterior hippocampus as a putative epicenter of volume loss across all illness stages and further showed a progression of epicenters of dynamic GMV loss from posterior to anterior areas, suggesting that pathological burden within temporal and prefrontal systems increases as the illness progresses.
Structural Connectivity Constrains GMV Changes in Psychotic Illness
The strength and topology of the structural connectome shaped the spatial pattern of volume abnormalities across both early and late stages of illness. Our findings in later illness stages align with previous research showing that the CDM captures the spatial pattern of cross-sectional GMV differences in people with established schizophrenia.33 This earlier result was observed using the CDM-SC variant considered in the present study. We extend this result to show that the strength of structural connectivity between regions further modulates coupled GMV differences within structurally connected neighborhoods, given that the CDM-SCw variant showed clearly superior performance to the CDM-SC and CDM-FCw variants in all data sets. This result indicates that GMV differences are more tightly coupled between areas with high structural connectivity. Critically, network constraints on cross-sectional GMV differences cannot be explained by antipsychotic medication, as our sample with first-episode psychosis was antipsychotic naive at the baseline scan. Moving beyond cross-sectional differences, our longitudinal analysis further demonstrates that both illness-related and antipsychotic-related changes in GMV are constrained by connectome architecture.
These results are in line with a spreading process in which pathology propagates across axonal connections. The precise mechanisms driving this process remain unclear. While limited evidence exists for visible deposits of aggregated pathological proteins in psychotic illness, more subtle changes in protein homeostasis43 may occur in subsets of patients and spread to synaptically connected distant brain regions.44 Alternatively, and given the commonly reported finding of functional brain alterations in psychotic disorders, dysfunction of one region may trigger abnormal activity in connected sites that, over time, may trigger structural changes as a result of aberrant neurotransmission or a loss of trophic support.21 This process may be exacerbated by a breakdown of white matter fiber integrity, which may further disrupt the interregional transport of trophic factors. Although our analyses suggest that using a structural connectome derived from a patient sample did not change the overall pattern of our findings, further work may investigate how coordinated GMV changes interact with white matter pathology in patients.
An alternative explanation for our findings is that regions sharing a strong anatomical connection have a more similar molecular and cytoarchitectonic profile, resulting in a shared vulnerability to illness-related or treatment-related changes.45,46,47 Future research should examine associations between the strength of structural connectivity and shared molecular features such as receptor profiles, gene expression, and synaptic density in patient populations.
Medial Temporal Lobe as an Epicenter of Gray Matter Differences in Psychosis
Our NDM analysis indicated that the medial temporal lobe, and the anterior hippocampus in particular, is a putative source of GMV loss in psychosis (see eMethods 11 in Supplement 1 for comparison with prior epicenter mapping findings33). The hippocampus has repeatedly been implicated in the pathogenesis of psychosis. It has been linked to early neurodevelopmental aberrations48,49,50 and often shows lower levels of messenger RNA and protein markers of synaptic and dendritic function post mortem.51,52 Recent in vivo positron emission tomography imaging studies have also identified a loss of synaptic vesicle proteins.53,54 Multiple animal models and human studies have indicated that a primary dysfunction occurring within the hippocampus,55,56 such as a loss of pyramidal cell inhibition, results in downstream brain abnormalities, including disinhibition of striatal dopamine release57,58 and aberrant corticostriatothalamic functioning.59 Other evidence suggests that dysregulation of glutamate neurotransmission beginning in the CA1 region60,61 initiates the transition to psychotic illness and triggers atrophy in other medial temporal and structurally connected regions.
Regional Epicenters of Longitudinal Gray Matter Change Dynamically Evolve With Illness Progression
While the hippocampus was robustly implicated as an epicenter for cross-sectional GMV differences at different illness stages, our analysis of longitudinal changes in the group with first-episode psychosis identified putative epicenters in striatal and prefrontal areas. This contrasts the largely posterior focus of cortical epicenters for baseline GMV differences in this group, suggesting that the most pronounced longitudinal GMV changes occurring early in the illness affect the prefrontal cortex. This finding aligns with the greater involvement of striatal and prefrontal areas at the 12-month compared with the 3-month follow-ups. Thus, while posterior and medial temporal areas appear important for understanding GMV difference near psychosis onset (ie, at baseline), prefrontal cortical areas are epicenters for dynamic GMV changes that occur shortly after psychosis onset. These findings accord with longitudinal studies in early-onset schizophrenia demonstrating a dynamic wave of volume contraction progressing from posterior to anterior regions,62,63 and other evidence of pronounced prefrontal GMV reductions in the earliest stages of illness,7,18,64,65,66,67,68 which may reflect an exaggeration of normal neurodevelopmental processes.69,70 Notably, these regional epicenters of illness-related GMV loss were distinct from epicenters of antipsychotic-related GMV loss, which were identified in somatosensory, motor, and posterior cingulate regions.
Limitations
This study has some limitations. Our findings depend on group-level summary metrics of brain volume and may not represent volume changes at the individual patient level, which can show substantial heterogeneity.71,72,73 Subsequent work could look at whether using individual-level measures of brain volume and connectivity, such as those obtained through normative modeling,74 can improve model estimates, given the increasing realization that GMV changes can be highly heterogeneous across individual patients.71,72,73 Such work will require the collection of large normative samples to adequately train the models.74,75
Conclusions
In this case-control study, we identify a robust and central role for axonal connectivity as a conduit for the spread of GMV reductions across early and late stages of psychotic illness, mirroring findings reported in neurodegenerative conditions. Our findings also align with those in animal models to suggest that medial temporal regions may play a critical role in the origins of brain pathology and indicate that the structural connectome represents a fundamental constraint on brain changes in psychosis, regardless of whether they are caused by illness or medication.
eMethods 1. Description of Each Data Set Used
eMethods 2. MRI Acquisition Parameters
eMethods 3. DBM Processing
eMethods 4. Volume Change Contrasts
eMethods 5. DWI Processing
eMethods 6. fMRI Processing
eMethods 7. FDR-Corrected and -Uncorrected Voxel-Level DBM t Statistic Maps for Each Contrast
eMethods 8. FDR-Corrected and -Uncorrected Voxel-Level VBM t Statistic Maps for Each Contrast
eMethods 9. Benchmark Null Models for the Coordinated Deformation Model (CDM)
eMethods 10. Further Information Network Diffusion Model (NDM) and Benchmark Null Models
eMethods 11. Data-Driven Epicentre Mapping Methods
eMethods 12. Robustness Analyses
eFigure 1. Sample-Level Associations Between Model Performance and Illness Duration and Severity
eFigure 2. Data-Driven Epicentre Region Identification Using Different Null Models
eFigure 3. Scatterplots of Observed and Estimated GMV Alterations Using the Best Seed Across the Whole Brain
eFigure 4. NDM Epicentres Using Null-Rewire Benchmarks (Top) and Null-Rewire Benchmarks That Do Not Preserve Distance Rules (Bottom)
eFigure 5. Replication of Results After Excluding Patients With Nonschizophrenia Diagnosis
eFigure 6. Replication of Results Using Representative Structural and Functional Connectomes From the First Episode Psychosis (STAGES) Patient Population
eFigure 7. Replication of Results Using Alterative Representative Structural and Functional Connectomes
eFigure 8. Replication of Results Using Voxel-Based Morphometry
eFigure 9. Replication of Results Applying Global Signal Regression to FC Data
eFigure 10. Replication of Results Using Representative Structural and Functional Connectomes From an Older Subset of the Independent Healthy Controls
eReferences.
Data Sharing Statement
References
- 1.Gur RE, Turetsky BI, Bilker WB, Gur RC. Reduced gray matter volume in schizophrenia. Arch Gen Psychiatry. 1999;56(10):905-911. doi: 10.1001/archpsyc.56.10.905 [DOI] [PubMed] [Google Scholar]
- 2.Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull. 2013;39(5):1129-1138. doi: 10.1093/schbul/sbs118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry. 2006;188:510-518. doi: 10.1192/bjp.188.6.510 [DOI] [PubMed] [Google Scholar]
- 4.van Erp TGM, Walton E, Hibar DP, et al. ; Karolinska Schizophrenia Project . Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biol Psychiatry. 2018;84(9):644-654. doi: 10.1016/j.biopsych.2018.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta CN, Calhoun VD, Rachakonda S, et al. Patterns of gray matter abnormalities in schizophrenia based on an international mega-analysis. Schizophr Bull. 2015;41(5):1133-1142. doi: 10.1093/schbul/sbu177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vieira S, Gong Q, Scarpazza C, et al. Neuroanatomical abnormalities in first-episode psychosis across independent samples: a multi-centre mega-analysis. Psychol Med. 2021;51(2):340-350. doi: 10.1017/S0033291719003568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pantelis C, Velakoulis D, McGorry PD, et al. Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI comparison. Lancet. 2003;361(9354):281-288. doi: 10.1016/S0140-6736(03)12323-9 [DOI] [PubMed] [Google Scholar]
- 8.Vita A, De Peri L, Deste G, Sacchetti E. Progressive loss of cortical gray matter in schizophrenia: a meta-analysis and meta-regression of longitudinal MRI studies. Transl Psychiatry. 2012;2(11):e190-e190. doi: 10.1038/tp.2012.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akudjedu TN, Tronchin G, McInerney S, et al. Progression of neuroanatomical abnormalities after first-episode of psychosis: a 3-year longitudinal sMRI study. J Psychiatr Res. 2020;130:137-151. doi: 10.1016/j.jpsychires.2020.07.034 [DOI] [PubMed] [Google Scholar]
- 10.Olabi B, Ellison-Wright I, McIntosh AM, Wood SJ, Bullmore E, Lawrie SM. Are there progressive brain changes in schizophrenia? a meta-analysis of structural magnetic resonance imaging studies. Biol Psychiatry. 2011;70(1):88-96. doi: 10.1016/j.biopsych.2011.01.032 [DOI] [PubMed] [Google Scholar]
- 11.Andreasen NC, Nopoulos P, Magnotta V, Pierson R, Ziebell S, Ho BC. Progressive brain change in schizophrenia: a prospective longitudinal study of first-episode schizophrenia. Biol Psychiatry. 2011;70(7):672-679. doi: 10.1016/j.biopsych.2011.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liloia D, Brasso C, Cauda F, et al. Updating and characterizing neuroanatomical markers in high-risk subjects, recently diagnosed and chronic patients with schizophrenia: a revised coordinate-based meta-analysis. Neurosci Biobehav Rev. 2021;123:83-103. doi: 10.1016/j.neubiorev.2021.01.010 [DOI] [PubMed] [Google Scholar]
- 13.Chopra S, Fornito A, Francey SM, et al. Differentiating the effect of antipsychotic medication and illness on brain volume reductions in first-episode psychosis: a longitudinal, randomised, triple-blind, placebo-controlled MRI study. Neuropsychopharmacology. 2021;46(8):1494-1501. doi: 10.1038/s41386-021-00980-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bora E, Fornito A, Radua J, et al. Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta-analysis and meta-regression analysis. Schizophr Res. 2011;127(1-3):46-57. doi: 10.1016/j.schres.2010.12.020 [DOI] [PubMed] [Google Scholar]
- 15.Fornito A, Yücel M, Patti J, Wood SJ, Pantelis C. Mapping grey matter reductions in schizophrenia: an anatomical likelihood estimation analysis of voxel-based morphometry studies. Schizophr Res. 2009;108(1-3):104-113. doi: 10.1016/j.schres.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 16.Del Re EC, Zeng V, Alliey-Rodriguez N, et al. Anterior-posterior axis of hippocampal subfields across psychoses: a B-SNIP study. Biomark Neuropsychiatry. 2021;5:100037. doi: 10.1016/j.bionps.2021.100037 [DOI] [Google Scholar]
- 17.Job DE, Whalley HC, Johnstone EC, Lawrie SM. Grey matter changes over time in high risk subjects developing schizophrenia. Neuroimage. 2005;25(4):1023-1030. doi: 10.1016/j.neuroimage.2005.01.006 [DOI] [PubMed] [Google Scholar]
- 18.McIntosh AM, Owens DC, Moorhead WJ, et al. Longitudinal volume reductions in people at high genetic risk of schizophrenia as they develop psychosis. Biol Psychiatry. 2011;69(10):953-958. doi: 10.1016/j.biopsych.2010.11.003 [DOI] [PubMed] [Google Scholar]
- 19.Sporns O, Tononi G, Kötter R. The human connectome: a structural description of the human brain. PLoS Comput Biol. 2005;1(4):e42. doi: 10.1371/journal.pcbi.0010042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raj A, Powell F. Models of network spread and network degeneration in brain disorders. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3(9):788-797. doi: 10.1016/j.bpsc.2018.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fornito A, Zalesky A, Breakspear M. The connectomics of brain disorders. Nat Rev Neurosci. 2015;16(3):159-172. doi: 10.1038/nrn3901 [DOI] [PubMed] [Google Scholar]
- 22.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42-52. doi: 10.1016/j.neuron.2009.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012;73(6):1216-1227. doi: 10.1016/j.neuron.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012;73(6):1204-1215. doi: 10.1016/j.neuron.2011.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shafiei G, Bazinet V, Dadar M, et al; Frontotemporal Lobar Degeneration Neuroimaging Initiative (FTLDNI); GENetic Frontotemporal dementia Initiative (GENFI). Network structure and transcriptomic vulnerability shape atrophy in frontotemporal dementia. Brain. 2023;146(1):321-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Biase MA, Cropley VL, Cocchi L, et al. Linking cortical and connectional pathology in schizophrenia. Schizophr Bull. 2019;45(4):911-923. doi: 10.1093/schbul/sby121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wannan CMJ, Cropley VL, Chakravarty MM, et al. Evidence for network-based cortical thickness reductions in schizophrenia. Am J Psychiatry. 2019;176(7):552-563. doi: 10.1176/appi.ajp.2019.18040380 [DOI] [PubMed] [Google Scholar]
- 28.Cauda F, Nani A, Costa T, et al. The morphometric co-atrophy networking of schizophrenia, autistic and obsessive spectrum disorders. Hum Brain Mapp. 2018;39(5):1898-1928. doi: 10.1002/hbm.23952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y, Wang Y, Huang H, et al. Antipsychotics effects on network-level reconfiguration of cortical morphometry in first-episode schizophrenia. Schizophr Bull. 2022;48(1):231-240. doi: 10.1093/schbul/sbab082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hettwer MD, Larivière S, Park BY, et al. ; ENIGMA ADHD Working Group; ENIGMA Autism Working Group; ENIGMA Bipolar Disorder Working Group; ENIGMA Major Depression Working Group; ENIGMA OCD Working Group; ENIGMA Schizophrenia Working Group . Coordinated cortical thickness alterations across six neurodevelopmental and psychiatric disorders. Nat Commun. 2022;13(1):6851. doi: 10.1038/s41467-022-34367-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cauda F, Nani A, Manuello J, et al. Brain structural alterations are distributed following functional, anatomic and genetic connectivity. Brain. 2018;141(11):3211-3232. doi: 10.1093/brain/awy252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgiadis F, Lariviere S, Glahn D, et al. Connectome architecture shapes large-scale cortical reorganization in schizophrenia: a worldwide ENIGMA study. bioRxiv. Preprint posted online February 13, 2023. doi: 10.1101/2023.02.12.527904 [DOI] [PMC free article] [PubMed]
- 33.Shafiei G, Markello RD, Makowski C, et al. Spatial patterning of tissue volume loss in schizophrenia reflects brain network architecture. Biol Psychiatry. 2020;87(8):727-735. doi: 10.1016/j.biopsych.2019.09.031 [DOI] [PubMed] [Google Scholar]
- 34.Chopra S, Francey SM, O’Donoghue B, et al. Functional connectivity in antipsychotic-treated and antipsychotic-naive patients with first-episode psychosis and low risk of self-harm or aggression: a secondary analysis of a randomized clinical trial. JAMA Psychiatry. 2021;78(9):994-1004. doi: 10.1001/jamapsychiatry.2021.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Francey SM, O’Donoghue B, Nelson B, et al. Psychosocial intervention with or without antipsychotic medication for first-episode psychosis: a randomized noninferiority clinical trial. Schizophr Bull Open. 2020;1(1):sgaa015. doi: 10.1093/schizbullopen/sgaa015 [DOI] [Google Scholar]
- 36.Lewandowski KE, Bouix S, Ongur D, Shenton ME. Neuroprogression across the early course of psychosis. J Psychiatr Brain Sci. 2020;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bustillo JR, Jones T, Chen H, et al. Glutamatergic and neuronal dysfunction in gray and white matter: a spectroscopic imaging study in a large schizophrenia sample. Schizophr Bull. 2017;43(3):611-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Çetin MS, Christensen F, Abbott CC, et al. Thalamus and posterior temporal lobe show greater inter-network connectivity at rest and across sensory paradigms in schizophrenia. Neuroimage. 2014;97:117-126. doi: 10.1016/j.neuroimage.2014.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillaume B, Hua X, Thompson PM, Waldorp L, Nichols TE; Alzheimer’s Disease Neuroimaging Initiative . Fast and accurate modelling of longitudinal and repeated measures neuroimaging data. Neuroimage. 2014;94:287-302. doi: 10.1016/j.neuroimage.2014.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaefer A, Kong R, Gordon EM, et al. Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb Cortex. 2018;28(9):3095-3114. doi: 10.1093/cercor/bhx179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian Y, Margulies DS, Breakspear M, Zalesky A. Topographic organization of the human subcortex unveiled with functional connectivity gradients. Nat Neurosci. 2020;23(11):1421-1432. doi: 10.1038/s41593-020-00711-6 [DOI] [PubMed] [Google Scholar]
- 42.Raj A, LoCastro E, Kuceyeski A, Tosun D, Relkin N, Weiner M; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . Network diffusion model of progression predicts longitudinal patterns of atrophy and metabolism in Alzheimer’s disease. Cell Rep. 2015;10(3):359-369. doi: 10.1016/j.celrep.2014.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bradshaw NJ, Korth C. Protein misassembly and aggregation as potential convergence points for non-genetic causes of chronic mental illness. Mol Psychiatry. 2019;24(7):936-951. doi: 10.1038/s41380-018-0133-2 [DOI] [PubMed] [Google Scholar]
- 44.Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med. 2014;20(2):130-138. doi: 10.1038/nm.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnatkeviciute A, Fulcher BD, Oldham S, et al. Genetic influences on hub connectivity of the human connectome. Nat Commun. 2021;12(1):4237. doi: 10.1038/s41467-021-24306-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fornito A, Arnatkevičiūtė A, Fulcher BD. Bridging the gap between connectome and transcriptome. Trends Cogn Sci. 2019;23(1):34-50. doi: 10.1016/j.tics.2018.10.005 [DOI] [PubMed] [Google Scholar]
- 47.Hansen JY, Shafiei G, Vogel JW, et al. Local molecular and global connectomic contributions to cross-disorder cortical abnormalities. Nat Commun. 2022;13(1):4682. doi: 10.1038/s41467-022-32420-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lipska BK, Weinberger DR. A neurodevelopmental model of schizophrenia: neonatal disconnection of the hippocampus. Neurotox Res. 2002;4(5-6):469-475. doi: 10.1080/1029842021000022089 [DOI] [PubMed] [Google Scholar]
- 49.Rosso IM, Cannon TD, Huttunen T, Huttunen MO, Lönnqvist J, Gasperoni TL. Obstetric risk factors for early-onset schizophrenia in a Finnish birth cohort. Am J Psychiatry. 2000;157(5):801-807. doi: 10.1176/appi.ajp.157.5.801 [DOI] [PubMed] [Google Scholar]
- 50.Bearden CE, Meyer SE, Loewy RL, Niendam TA, Cannon TD. The neurodevelopmental model of schizophrenia: updated. In: Cicchetti D, Cohen DJ, eds. Developmental Psychopathology: Risk, Disorder, and Adaptation. John Wiley & Sons Inc; 2015:542-569. [Google Scholar]
- 51.Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry. 2019;24(4):549-561. doi: 10.1038/s41380-018-0041-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison PJ, Eastwood SL. Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus. 2001;11(5):508-519. doi: 10.1002/hipo.1067 [DOI] [PubMed] [Google Scholar]
- 53.Radhakrishnan R, Skosnik PD, Ranganathan M, et al. In vivo evidence of lower synaptic vesicle density in schizophrenia. Mol Psychiatry. 2021;26(12):7690-7698. doi: 10.1038/s41380-021-01184-0 [DOI] [PubMed] [Google Scholar]
- 54.Onwordi EC, Halff EF, Whitehurst T, et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat Commun. 2020;11(1):246. doi: 10.1038/s41467-019-14122-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44(7):660-669. doi: 10.1001/archpsyc.1987.01800190080012 [DOI] [PubMed] [Google Scholar]
- 56.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32(9):507-513. doi: 10.1016/j.tips.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grace AA. Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology. 2012;62(3):1342-1348. doi: 10.1016/j.neuropharm.2011.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Modinos G, Allen P, Grace AA, McGuire P. Translating the MAM model of psychosis to humans. Trends Neurosci. 2015;38(3):129-138. doi: 10.1016/j.tins.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sabaroedin K, Tiego J, Fornito A. Circuit-based approaches to understanding corticostriatothalamic dysfunction across the psychosis continuum. Biol Psychiatry. 2023;93(2):113-124. doi: 10.1016/j.biopsych.2022.07.017 [DOI] [PubMed] [Google Scholar]
- 60.Lieberman JA, Girgis RR, Brucato G, et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018;23(8):1764-1772. doi: 10.1038/mp.2017.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schobel SA, Chaudhury NH, Khan UA, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78(1):81-93. doi: 10.1016/j.neuron.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vidal CN, Rapoport JL, Hayashi KM, et al. Dynamically spreading frontal and cingulate deficits mapped in adolescents with schizophrenia. Arch Gen Psychiatry. 2006;63(1):25-34. doi: 10.1001/archpsyc.63.1.25 [DOI] [PubMed] [Google Scholar]
- 63.Thompson PM, Vidal C, Giedd JN, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci U S A. 2001;98(20):11650-11655. doi: 10.1073/pnas.201243998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun D, Phillips L, Velakoulis D, et al. Progressive brain structural changes mapped as psychosis develops in “at risk” individuals. Schizophr Res. 2009;108(1-3):85-92. doi: 10.1016/j.schres.2008.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jalbrzikowski M, Hayes RA, Wood SJ, et al. ; ENIGMA Clinical High Risk for Psychosis Working Group . Association of structural magnetic resonance imaging measures with psychosis onset in individuals at clinical high risk for developing psychosis: an ENIGMA Working Group mega-analysis. JAMA Psychiatry. 2021;78(7):753-766. doi: 10.1001/jamapsychiatry.2021.0638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cannon TD, Chung Y, He G, et al. ; North American Prodrome Longitudinal Study Consortium . Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 2015;77(2):147-157. doi: 10.1016/j.biopsych.2014.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Del Re EC, Stone WS, Bouix S, et al. Baseline cortical thickness reductions in clinical high risk for psychosis: brain regions associated with conversion to psychosis versus non-conversion as assessed at one-year follow-up in the Shanghai-at-Risk-for-Psychosis (SHARP) Study. Schizophr Bull. 2021;47(2):562-574. doi: 10.1093/schbul/sbaa127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collins MA, Ji JL, Chung Y, et al. Accelerated cortical thinning precedes and predicts conversion to psychosis: The NAPLS3 longitudinal study of youth at clinical high-risk. Mol Psychiatry. 2023;28(3):1182-1189. doi: 10.1038/s41380-022-01870-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cropley VL, Klauser P, Lenroot RK, et al. Accelerated gray and white matter deterioration with age in schizophrenia. Am J Psychiatry. 2017;174(3):286-295. doi: 10.1176/appi.ajp.2016.16050610 [DOI] [PubMed] [Google Scholar]
- 70.Gogtay N, Vyas NS, Testa R, Wood SJ, Pantelis C. Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr Bull. 2011;37(3):504-513. doi: 10.1093/schbul/sbr030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolfers T, Doan NT, Kaufmann T, et al. Mapping the heterogeneous phenotype of schizophrenia and bipolar disorder using normative models. JAMA Psychiatry. 2018;75(11):1146-1155. doi: 10.1001/jamapsychiatry.2018.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lv J, Di Biase M, Cash RFH, et al. Individual deviations from normative models of brain structure in a large cross-sectional schizophrenia cohort. Mol Psychiatry. 2021;26(7):3512-3523. doi: 10.1038/s41380-020-00882-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Segal A, Parkes L, Aquino K, et al. Regional, circuit and network heterogeneity of brain abnormalities in psychiatric disorders. Nature Neurosci. Published online August 14, 2023. doi: 10.1038/s41593-023-01404-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rutherford S, Kia SM, Wolfers T, et al. The normative modeling framework for computational psychiatry. Nat Protoc. 2022;17(7):1711-1734. doi: 10.1038/s41596-022-00696-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bozek J, Griffanti L, Lau S, Jenkinson M. Normative models for neuroimaging markers: impact of model selection, sample size and evaluation criteria. Neuroimage. 2023;268:119864. doi: 10.1016/j.neuroimage.2023.119864 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods 1. Description of Each Data Set Used
eMethods 2. MRI Acquisition Parameters
eMethods 3. DBM Processing
eMethods 4. Volume Change Contrasts
eMethods 5. DWI Processing
eMethods 6. fMRI Processing
eMethods 7. FDR-Corrected and -Uncorrected Voxel-Level DBM t Statistic Maps for Each Contrast
eMethods 8. FDR-Corrected and -Uncorrected Voxel-Level VBM t Statistic Maps for Each Contrast
eMethods 9. Benchmark Null Models for the Coordinated Deformation Model (CDM)
eMethods 10. Further Information Network Diffusion Model (NDM) and Benchmark Null Models
eMethods 11. Data-Driven Epicentre Mapping Methods
eMethods 12. Robustness Analyses
eFigure 1. Sample-Level Associations Between Model Performance and Illness Duration and Severity
eFigure 2. Data-Driven Epicentre Region Identification Using Different Null Models
eFigure 3. Scatterplots of Observed and Estimated GMV Alterations Using the Best Seed Across the Whole Brain
eFigure 4. NDM Epicentres Using Null-Rewire Benchmarks (Top) and Null-Rewire Benchmarks That Do Not Preserve Distance Rules (Bottom)
eFigure 5. Replication of Results After Excluding Patients With Nonschizophrenia Diagnosis
eFigure 6. Replication of Results Using Representative Structural and Functional Connectomes From the First Episode Psychosis (STAGES) Patient Population
eFigure 7. Replication of Results Using Alterative Representative Structural and Functional Connectomes
eFigure 8. Replication of Results Using Voxel-Based Morphometry
eFigure 9. Replication of Results Applying Global Signal Regression to FC Data
eFigure 10. Replication of Results Using Representative Structural and Functional Connectomes From an Older Subset of the Independent Healthy Controls
eReferences.
Data Sharing Statement