

Abstract
INTRODUCTION:
Genetic associations with Alzheimer’s disease (AD) age at onset (AAO) could reveal genetic variants with therapeutic applications. We present a large Colombian kindred with autosomal dominant AD (ADAD) as a unique opportunity to discover AAO genetic associations.
METHODS:
A genetic association study was conducted for ADAD dementia AAO in 340 individuals with the PSEN1 E280A mutation via TOPMed array imputation. Replication was assessed in two ADAD cohorts, one sporadic EOAD study, and four late onset AD studies.
RESULTS:
13 variants had p<1x10−7 or p<1x10−5 with replication including three independent loci with candidate associations with clusterin including near CLU. Other suggestive associations were identified in or near HS3ST1, HSPG2, ACE, LRP1B, TSPAN10, and TSPAN14.
DISCUSSION:
Variants with suggestive associations with AAO were associated with biological processes including clusterin, heparin sulfate and amyloid processing. The detection of these effects in the presence of a strong mutation for ADAD reinforce their potentially impactful role.
Keywords: alzheimer, PSEN1, E280A, age at onset, age of onset, AAO, AOO, CLU, clusterin, SIAH3, ZC3H13, HS3ST1, HSPG2, ACE, LRP1B, TSPAN10, TSPAN14, tetraspanin, heparin sulfate, amyloid
Background
Complex genetic, environmental, and lifestyle risk factors confounded by the aging process underlie risk for late onset Alzheimer’s disease (LOAD). Autosomal dominant Alzheimer’s disease (ADAD) closely resembles the clinical and neuropathological features of LOAD, but without the confound of aging, and thus provides a less heterogeneous view of underlying AD-associated processes. ADAD accounts for less than 1% of all cases of AD and mutations in PSEN1 account for 80% of this monogenic group (reviewed in [1]).
There is a strong correlation between age at onset (AAO) and a particular ADAD mutation (r2 = 0.52) [2], but there still remains substantial unexplained variability. Large ADAD families such as the kindred harboring the Colombian PSEN1 NM_000021:c.839A>C, p.(Glu280Ala) (canonically known as PSEN1 E280A) mutation, the world’s largest ADAD founder population with a comprehensive family tree of thousands of individuals [3], provide an opportunity to assess the contribution of genetic variation to unexplained variability in age of dementia onset. PSEN1 E280A mutation carriers typically develop mild cognitive impairment (MCI) at a median age of 44 years (95% CI, 43–45) and dementia at age of 49 years (95% CI, 49–50) [4]. The value of this family for the nomination of genetic variants that delay the onset of AD was recently affirmed by the report of a PSEN1 E280A carrier who developed MCI nearly three decades after the kindred’s median age at clinical onset [5] (this individual is also included in this study). This individual was homozygous for the rare APOE ε3 Christchurch variant (APOE NM_000041:c.460C>A, p.(R154S), rs121918393) and had an exceptionally high amyloid-β plaque burden, but limited neurofibrillary tau burden. In addition to this case report, several studies have explored genetic associations with AAO in PSEN1 E280A carriers [6–9], but all with substantially lower numbers of cases (at most 72 individuals) [6]. To expand on the valuable insights gained from these previous studies, we conducted the most comprehensive search to date for genetic variants associated with age at dementia onset in this founder population by assessing 340 individuals, which is the current snapshot of all individuals from this cohort, that currently have high quality genotypic and phenotypic information available.
Methods
Patient Recruitment
A cohort of 368 patients was selected from the Neuroscience Group of Antioquia (GNA) database of the PSEN1 E280A family. After all quality control steps, 340 individuals remained for analysis. Selection criteria included being a PSEN1 E280A carrier with diagnosis of dementia, having adequate medical and neuropsychological evaluations and follow-up for a confident age determination of clinical age at dementia onset, and having a DNA sample. Participants were evaluated following a standard protocol including physical and neurological examination, as well as population-validated neuropsychological assessment [10, 11]. Dementia was diagnosed according to most recent DSM criteria at the time of diagnosis. Collected data were stored in medical records software (SISNE v2.0). Family history was obtained from the patients and their relatives, and genealogical data from baptism and death certificates was gathered from local parishes and was incorporated into the pedigree reconstruction. Blood samples from each individual were obtained through standard phlebotomy and collected in EDTA tubes. Genomic DNA was purified from peripheral blood leukocytes using a modified salting-out technique (Gentra Puregene Blood Kit, Qiagen). All individuals were genotyped for PSEN1 E280A using a restriction length fragment polymorphism assay.
Genotyping Arrays
1,923,394 variants were genotyped using the Illumina Multi-Ethnic Genotyping Array plus Neuro consortium content (catalog #WG-316-1014, beadchip #20028352). Data were annotated with build hg38 and processed and analyzed using PLINK v1.90b5.2, PLINK v2.00aLM [12], and GEMMA [13] (GEMMA was used for the main association analysis, see Results for details). Genetic relatedness was assessed using KING 2.2 [14]. Imputation was conducted using the TOPMed Imputation Panel and Server (version 1.3.3), which includes 97,256 references samples and 308,107,085 variants and uses Minimac4 for imputation. Imputation methods and quality control are described in detail in the Supplemental Methods.
Replication sets
Seven cohorts were selected for replication. For ADAD, we used the Dominantly Inherited Alzheimer’s Network (DIAN) cohort, with 116 mutation carrier cases (96 European ancestry and 20 Native American ancestry) with age of dementia onset as the phenotype as in the main cohort analyzed. The DIAN cohort was analyzed using GEMMA on TOPMed-imputed genotyping array data with an allele frequency cutoff of 1% for all variants considered. Fixed effect covariates were the parental age at onset, the gene, including considering PSEN1 before and after codon 200 as separate “genes” given more deleterious effects of PSEN1 variants after codon 200 [15], and the first three principal components.
As a second dominant AD replication cohort, we used the Alzheimer’s Disease in Adults with Down Syndrome (ADDS) cohort, which was obtained from the Synapse AD Knowledge portal (Synapse ID: syn25871263) and imputed using TOPMed. After quality control for missingness, heterozygosity, and relatedness, 222 individuals remained for analysis. We used the available phenotype if the individuals had converted to MCI or AD (105 not yet converted, 58 MCI, and 59 AD) weighted as 0, 0.5, and 1 respectively for the phenotype. We performed the GEMMA analysis in the same manner as our cohort, with sex and PCs 1-10 included in the model. For this cohort, chromosome 21 was not considered for replication.
Given the limited sample sizes for dominant AD, we also evaluated sporadic AD cohorts. For EOAD, we evaluated the largest sporadic early onset AD cohort aggregated to date, an ADGC EOAD study cohort currently in analysis with 6,282 European ancestry early onset AD cases and 13,386 European ancestry controls (European ancestry is the largest admixture component in our cohort). For this cohort, all Single variant analyses were performed with Plink v2.0 GLM function with the following model: Status~SNP+SEX+PC1–10. For LOAD, we selected an AD age at onset study (9,162 cases) [16], a study of AD age at onset survival (14,406 cases and 25,849 controls) [17], a genome wide association study (GWAS) meta-analysis for AD (21,982 AD vs. 41,944 controls) [18], and the latest meta-analysis of AD and AD by proxy (111,326 cases and 677,663 controls) [19]. See supplemental methods for discussion of International Genomics of Alzheimer’s Project (IGAP) replication data.
Role of the funding source
The study sponsors were not involved in study design, the collection, analysis, and interpretation of data, the writing of the report, or the decision to submit the paper for publication.
Results
Cohort demographics
The final cohort had a mean age of dementia onset of 49.3 years (median: 48, range: 37–75, 10th–90th percentile: 43–56). 198 of the patients were genetically female (58.2%). The patients had extensive follow up data; the mean number of medical evaluations was 6.7 (1–27), and 4.8 (1–18) for neuropsychological evaluations. A partial pedigree of enrolled individuals annotated with age at dementia onset is presented in Supplemental Figure 1.
Association analysis
Association analysis was conducted using age at dementia onset as a quantitative outcome for 340 individuals passing QC. We employed GEMMA, a package that performs a likelihood ratio test using a linear mixed model to adjust for relatedness between individuals. We adjusted for genetic sex, the first ten principal components (calculated from the set of 540,753 high quality variants used as imputation input using PLINK v2.00aLM) because this was an admixed population, and batch. The chip heritability calculated by GEMMA was 0.74+/−0.14 with a Vg estimate of of 24.6 and Ve estimate of 8.5.
Top nominally significant loci of interest
To determine if any hits observed statistically deviated from random chance, we generated a QQ plot (Supplemental Figure 2). No variants deviated detectably from a uniform distribution’s expected error range except for from modest inflation (genomic inflation factor of 1.05), but this was not surprising given the small size of the cohort, and the modest level of inflation reflects that GEMMA’s kinship matrix adjustment works well for this familial cohort. Because of this, the variants presented throughout should be viewed as speculative, particularly variants where a small number of alleles account for the association (including 6 out of the 13 loci presented in Table 1 where allele count ranges from 3–7). To add evidence for possible biological significance, we relied primarily on replication. First, we compared the number of variants with p<1x10−5 that exhibit nominal replication (p<0.05) in one of the seven replication cohorts. Second, we used a stricter threshold (p<1x10−7) where we did not require replication. The result of these filtering conditions is shown in Table 1 and includes three variants at different loci associated with clusterin biology, rs138295139, rs35980966 and rs4942482. LocusZoom plots and single nucleus multiomics linkages (correlations between single nucleus RNA-seq and ATAC-seq from the same nuclei [20]) for these variants are presented in Figure 1. In addition, the age of each individual harboring each variant in Table 1 is illustrated in Supplemental Figure 3 by variant to illustrate the spread of the variants across the cohort by zygosity. The Manhattan plot for the study is shown in Supplemental Figure 4. Unannotated summary statistics for all variants are provided in Supplemental Table 1. Annotated summary statistics including replication information from described cohorts for variants with p<1x10−5, coding variants with p<0.05, APOE coding variants, and variants that are index variants for previous GWAS are provided in Supplemental Table 2. This table also includes imputation quality (R2) for variants presented in all tables, including those in Table 1 (average +/− standard deviation = 0.95 +/− 0.09), with only one variant with an R2 < 0.85 (rs11705431, R2=0.671)). As an additional quality control measure, note that we also only considered variants called in the genomes of the 26 individuals sequenced at HudsonAlpha (see methods). Variants with p<1x10−5 that overlap with a single nucleus multiomics linkage between ATAC-seq and RNA-seq in the same nuclei from a recent study [20] are shown in Supplemental Table 3 along with more detailed information including which cell types are implicated in each multiomics linkage. LocusZoom plots of all regions with p<1x10−5 are presented in Supplemental File 1.
Table 1:
Variants that both met a suggestive threshold (p<1x10−5) and exhibited one or both of replication at p=0.05 and consistent effect direction in at least one cohort and/or presence in a locus within 500 kb of a GWAS variant previously linked to an AD-associated phenotype (less closely related phenotypes are shown in gray). β is the effect in years on age at dementia onset (i.e., positive β values indicate an association with later age of onset). Replication cohorts varied in design but required a consistent effect direction with the matched effect allele. Naj, 2014 is an age of onset study where a positive β indicates later age of onset. Huang, 2017 is an age of onset survival design where a hazard ratio less than 1 indicates a protective correlation. The DIAN cohort was analyzed in the same manner as the main cohort. Bellenguez, 2022, Kunkle, 2019, and ADDS are case/control designs where negative betas indicate a protective correlation. Full summary statistics are presented in Supplemental Table 1 and All variants with p<1x10−5 along with more detailed information are presented in Supplemental Table 2.
| Chrom: Pos | Variant–Effect Allele | p | β (SE) | Cohort AF | Replication Information | snATAC-seq + snRNA-seq linked gene(s) and H3K27Ac | Nearby (+/− 500 kb of 1.0x10−5) NHGRI-EBI Catalog Hits |
|---|---|---|---|---|---|---|---|
| 1:80.5 | rs77107089–A | 5.2x10−6 | 5.0 (1.1) | 0.044 | Huang, 2017: p=0.01, HR=0.91 | Aging [rs11162963], Stroke [rs1937787] | |
| 1:194.4 | rs138295139–C | 5.8x10−8 | 17.4 (3.1) | 0.006 | LOAD [rs6678275], Plasma clusterin [rs4428865], WM hyper. [rs71642944] | ||
| 1:213.4 | rs466632–A | 3.7x10−6 | 10.6 (2.3) | 0.009 | DIAN: p=0.02, β=4.2 | AC096639.1, AL592402.1, RPS6KC1 (excitatory neurons) | AD [rs340849] |
| 4:208.8 | rs80136406–A | 3.0x10−6 | 8.0 (1.7) | 0.018 | ADGC EOAD: p=0.047, OR=0.83 | PD and Lewy body path. [rs141863958] | |
| 6:140.4 | rs76268851–A | 9.6x10−6 | 6.3 (1.5) | 0.024 | Bellenguez, 2022: p=0.045, β= −0.07 | AD and age of onset [rs17069431] | |
| 8:27.6 | rs35980966–G | 5.5x10−8 | 16.7 (3.0) | 0.004 |
Kunkle, 2019: p=0.02, β= −0.28 Bellenguez, 2022: p=2.1x10−5, β= −0.26 ADGC EOAD: p=0.016, OR=0.65 |
AC013643.2, CLU, EPHX2, SCARA3, STMN4 (astrocytes, inhibitory neurons; H3K27Ac in astrocytes and neurons; r2=1 with rs538480041: PTK2B (multiple cell types); H3K27Ac in astrocytes, microglia, oligodendrocytes | Cingulate cortical Aβ [rs4625043], LOAD [rs28834970, rs1532278, rs9331896], APOE ε4-AD [rs2271920, rs2279590], AD [rs11136000, rs569214], APOE ε4+ AD [rs9331896], Plasma clusterin [rs4545046], CBD [rs643472], Cog. Performance [rs2978263] |
| 10:0.6 | rs75279020–C | 7.8x10−6 | 13.0 (2.8) | 0.007 | ADDS: p=0.03, β= −0.28 | Psychosis in AD [rs11252926] | |
| 13:46.1 | rs4942482–T | 2.7x10−6 | 2.1 (0.5) | 0.484 |
Huang, 2017: p=0.037, HR=0.95 Bellenguez, 2022: p=0.002, β= −0.03 ADGC EOAD: p=0.028, OR=0.95 |
CPB2, CPB2-AS1, LRCH1, LRRC63, SIAH3 (AD-specific), ZC3H13 (astrocytes, excitatory and inhibitory neurons, microglia, oligodendrocytes, OPCs); CPB2-AS1, LRCH1 and ZC3H13: H3K27Ac in Oligodendrocytes [54] | ZC3H13/SIAH3: CSF clusterin [rs741668], SIAH3: Rate of ventricular enlargement [rs11620312, rs79174114] [42] |
| 14:75.4 14:77.8 14:78.4 |
rs145134226–T rs72688827-C rs184135706–G |
1.1x10−7 2.6x10−6 2.6x10−6 |
10.8 (2.0) 11.4 (2.4) 11.4 (2.4) |
0.010 0.007 0.007 |
ADGC EOAD: p=0.046, OR=0.84 Naj, 2014: p=0.0497, β=1.2 Kunkle, 2019: p=0.01, β= −0.21 |
NRXN3 (excitatory and inhibitory neurons) (link is to rs78560216 which is r2=1 with rs184135706, rs184135706 is r2=0.71 with rs145134226); H3K27Ac: neurons; POMT2 and SPTLC2 linked to rs72688827; Others: see Supplemental Table 3. | Temporal lobe volume [rs7155434], Cognitive performance [rs6574433], ALS age of onset [rs7147705] |
| 16:0.2 | rs10153124–C | 2.2x10−6 | 3.7 (0.8) | 0.090 | Huang, 2017: p=0.047, HR=0.95 | r2=1 with rs9972706: RHBDL1 (excitatory and inhibitory neurons); H3K27Ac: neurons | |
| 18:59.9 | rs76020589–A | 9.9x10−6 | 2.7 (0.6) | 0.187 | Huang, 2017: p=0.04, HR=0.95 | AD and age of onset [rs142176337] | |
| 21:22.3 | rs17592663–T | 3.7x10−8 | 5.7 (1.0) | 0.040 | APOE ε4+ AD [rs721146], Longitudinal change in brain amyloid plaque burden [rs8129913], CSF Aβ1-42 [rs239713] | ||
| 22:48.5 | rs11705431–A | 3.4x10−6 | 16.6 (3.5) | 0.004 | DIAN: p=0.005, β=6.2 | TAFA5, Z84468.2 [promoter] (astrocytes, excitatory and inhibitory neurons, microglia oligodendrocytes, OPCs); H3K27Ac: astrocytes, neurons | AD and age of onset [rs150927461], LOAD [rs1034435] |
Abbreviations: Chrom:Pos – Chromosome: build hg38 position, Cohort AF – Cohort allele frequency, DIAN – Dominantly Inherited Alzheimer’s Network cohort, ADDS – Alzheimer’s Disease in Adults with Down Syndrome cohort, LOAD – late onset Alzheimer’s disease, AD – Alzheimer’s disease, Aβ – amyloid beta.
Figure 1:
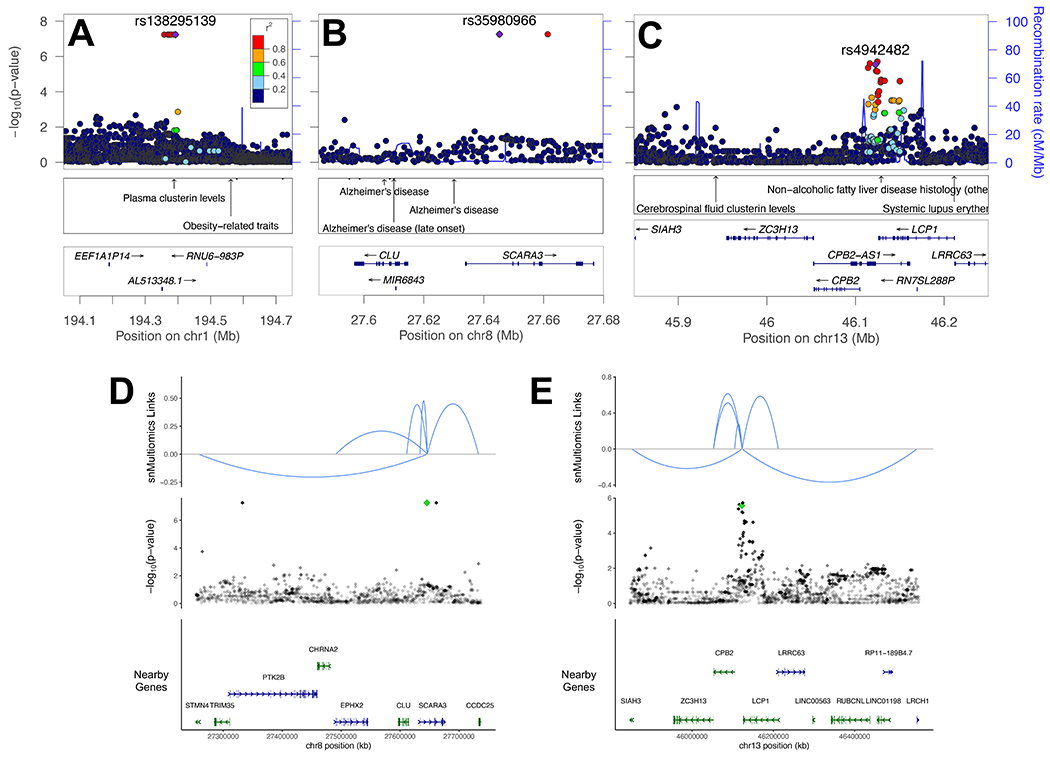
Plots of loci linked to clusterin or clusterin related phenotypes meeting criteria in Table 1. (A–C) LocusZoom plots of loci with index variants indicated with a purple diamond. Nearby NHGRI-EBI GWAS hits are indicated. (A) Note nearby variant previously linked to plasma clusterin levels. (B) Note several nearby variants previously linked to AD near CLU. (C) Note nearby variant previously linked to CSF clusterin levels between SIAH3 and ZC3H13. (D,E) Single nucleus multiomic (snMultiomics) links (RNA-seq–ATAC-seq correlations from the same nuclei) indicated for hits on chromosomes 8 and 13. Strength of the link is indicated by height, and direction indicates direction of correlation. Index variants are indicated with a green diamond. (D) Note link to CLU. (E) Note links to SIAH3 and ZC3H13.
Results at key APOE variants
Effects of previously established APOE variants important for AD association in LOAD are in the expected direction based on previous studies, but modest in magnitude (Table 2). Overall, the observations are consistent with previously reported observations including a protective effect of APOE ε2 in the Colombian E280A population (β=8.2, 95% CI=4.5–12.0, p=3.8x10−5) [21], a deleterious effect of APOE ε4 in the Colombian E280A population in one study (hazard ratio 2.1, 95% CI 1.1–4.0, p=0.03) [22] but an inability to detect an effect of APOE ε4 in three other studies in this population [21, 23, 24], and a non-significant trend towards an APOE ε2 > APOE ε3 > APOE ε4 age-of-onset in dominant AD families with a variety of mutations [2].
Table 2:
Assessment of previously implicated variants in APOE. These variants were directly genotyped by qPCR given their previously implicated role in disease. β is the effect in years on age at dementia onset (i.e., positive β values indicate an association with later age of onset). One other APOE coding variant, rs440446–G, was imputed by TOPMed but did not exhibit a meaningful signal (β= −0.57, p=0.16, see Supplemental Table 2 for details). TOPMed Bravo Freeze 8 was used for Population Allele Frequency.
| Variant–Effect Allele | Description | β (SE) | p | # Homozygous | Cohort Allele Frequency | Population Allele Frequency | Transcript ENST00000252486.9 Coding Change | CADD version 1.6 |
|---|---|---|---|---|---|---|---|---|
| rs429358–C | APOE ε4 (when rs7412 ref.) | −2.0 (0.6) | 0.0025 | 8 | 0.138 | 0.155 | p.(Cys130Arg) | 16.7 |
| rs121918393–A | APOE Christchurch | 3.1 (1.6) | 0.0496 | 1 | 0.020 | 1.9x10−5 | p.(Arg154Ser) | 25.3 |
| rs7412–T | APOE ε2 (when rs429358 ref.) | 2.0 (0.9) | 0.0891 | 0 | 0.068 | 0.078 | p.(Arg176Cys) | 26 |
A recent case report implicated the APOE Christchurch variant (rs121918393) [5]. That individual was also enrolled in this study, and while we do observe a nominally significant effect on age at onset of this variant, we note that the effect size is modest, which could be because our model does not consider homozygosity effects. No other coding variants in APOE beyond those described in Table 2 were observed in either the imputed set or the subset of cases with genomes available.
Replication at known AD-associated loci
We evaluated 17 AD GWAS, including the largest case/control studies for AD in European ancestry populations [18, 19, 25–27], studies in non-European ancestry populations [28–32], age at onset modifier studies [16, 17], and endophenotype studies [33–37]. These studies identified 108 loci (at least 500kb between unique loci) and 184 index variants within these loci with high confidence associations for AD and endophenotypes (Supplemental Table 4 (an expansion from a table put forward by [38])). Of these variants, 151 were genotyped in this cohort, nine with p<0.05, but only six of these were in a consistent direction. Replication of hits with genome-wide significance for AD-associated phenotypes with nominal significance (p<0.05) with consistent effect direction in this cohort are shown in Table 3. This table should be interpreted with caution, as it is close to the number of variants that would be expected based on random sampling of this set of GWAS hits (six observed versus ~four expected), however the variants identified do share some nearby genes or pathways with variants from other nomination approaches (see Discussion).
Table 3:
Hits with genome-wide significance for AD-associated phenotypes from previous studies with p<0.05 and consistent direction of effect in this cohort. β is the effect in years on age at dementia onset (i.e., positive β values indicate an association with later age of onset). For previous studies, all effects are meta-analysis OR (95% CI) except Beecham et al., 2014 and Deming et al., 2017, which are β (SE).
| Variant | Locus | Key Nearby Gene(s) | p | β (SE) | Cohort AF | Population AF | Previous Study | Previous Study p | Previous Study Effect (Error) | Previous Study Outcome | Other Replication |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs6448453-G | 4p16.1 | CLNK/HS3ST1 | 0.010 | 1.4 (0.5) | 0.75 | 0.77 | Jansen, 2019 | 1.9x10−09 | 0.99 (0.98–0.99) | AD Risk |
Huang, 2017
Kunkle, 2019 Bellenguez, 2022 |
| rs62341097-A | 4q34.1 | GALNT7 | 0.035 | 2.2 (1.2) | 0.05 | 0.03 | Beecham, 2014 | 6.0x10−09 | −1.147 (0.198) | Neuritic Plaque | |
| rs316341-A | 6p25 | SERPINB1 | 0.015 | −1.3 (0.5) | 0.69 | 0.71 | Deming, 2017 | 1.8x10−08 | −0.025 (0.004) | CSF Aβ42 | |
| rs6586028-T | 10q23.1 | TSPAN14 | 0.035 | −1.4 (0.7) | 0.86 | 0.87 | Bellenguez, 2022 | 2.0x10−19 | 1.08 (1.06–1.10) | AD Risk |
Huang, 2017 Kunkle, 2019 DIAN |
| rs1140239-T | 16p11.2 | DOC2A | 0.026 | 1.0 (0.5) | 0.39 | 0.31 | Bellenguez, 2022 | 2.6x10−13 | 0.94 (0.93–0.96) | AD Risk |
Huang, 2017
Kunkle, 2019 |
| rs138190086-A | 17q23.3 | ACE | 0.021 | −3.0 (1.3) | 0.03 | 0.01 |
Marioni, 2018
Kunkle, 2019 |
1.9x10−09 5.3x10−09 |
1.25 (1.16–1.35) 1.30 (1.19–1.42) |
AD Risk AD Risk |
Bellenguez, 2022 |
Abbreviations: Cohort AF indicates minor allele frequency in this cohort. Population AF indicates the population allele frequency (TOPMed Bravo Freeze 8). CADD – Combined Annotation Dependent Depletion score.
In addition to testing known LOAD risk loci individually, we also evaluated the effect of LOAD variants combined using a LOAD polygenic risk score (Figure 2). Polygenic risk score both without (Figure 2A) and with APOE ε allele–defining variants rs429358 and rs7412 (Figure 2B) exhibited a significant correlation with age at dementia onset in the expected direction (later age of onset associated with a lower polygenic risk score).
Figure 2:
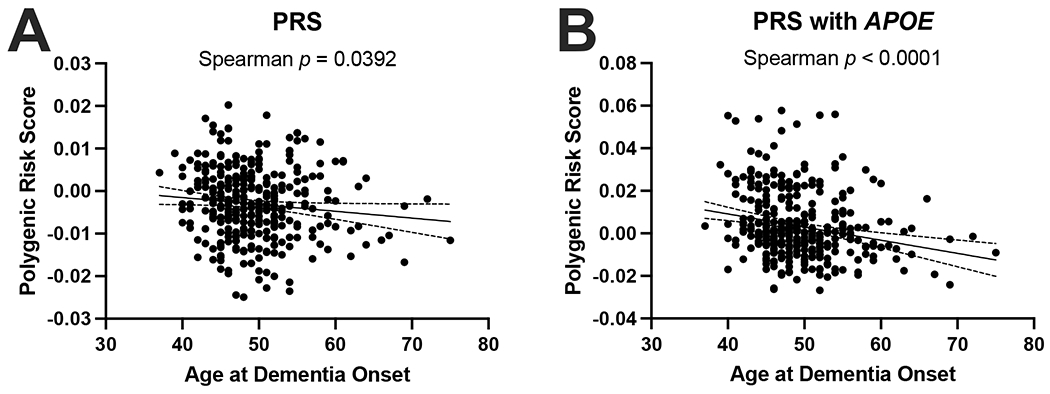
Late onset AD polygenic risk score applied to the PSEN1 E280A cohort. (A) LOAD polygenic risk score with APOE excluded (Spearman p = 0.0392). (B) LOAD polygenic risk score with APOE included (Spearman p < 0.00001).
Coding variants of interest
We next asked if any coding variants speculatively associate with age of dementia onset (Table 4). We chose four conditions: p<1x10−5; p<0.01, Combined Annotation Dependent Depletion (CADD) phred score [39] >20 and replication in more than 1 study; p<0.01, population allele frequency < 2%, CADD>20, and replication in at least 1 study; and coding variants in high priority AD genes with p<0.05 including APP, PSEN1, PSEN2, MAPT, APOE (not shown because in Table 2), ABCA7, SORL1, TREM2, and recently implicated GWAS loci with signal for coding variation in a recent exome meta-analysis [40] including ATP8B4, ABCA1, ADAM10, CLU, ZCWPW1, and ACE.
Table 4:
Coding variants of speculative interest. Conditions used to filter to variant categories highlighted in this table are noted. β is the effect in years on age at dementia onset (i.e., positive β values indicate an association with later age of onset). Replication cohorts varied in design but required a consistent effect direction with the matched effect allele. Huang, 2017 is an age of onset survival design where a hazard ratio less than 1 indicates a protective correlation. Naj, 2014 is an age of onset design with the same directional effect as this cohort. Kunkle, 2019 and Bellenguez, 2022 are case/control designs where negative betas indicate a protective correlation. All coding variants with p<0.05 along with more detailed information (including transcript for coding changes) are presented in Supplemental Table 2.
| Category | Chrom: b38Pos | Variant–Effect Allele | p | β (SE) | Cohort AF | Population AF | Gene | Protein Change | CADD v1.6 | Replication Information |
|---|---|---|---|---|---|---|---|---|---|---|
| p<1x10−5 | 15:72.3 | rs758150448–C | 7.5x10−6 | −5.8 (1.3) | 0.022 | 6.8x10−5 | CELF6 | p.(His361Arg) | 23.8 | |
|
p<0.01, CADD >20, multiple replications |
4:3.5 | rs9684786–A | 0.001 | −1.6 (0.5) | 0.254 | 0.185 | DOK7 | p.(Gly461Asp) | 21.8 |
Huang, 2017: p=0.032, HR= 1.03 Naj, 2014: p=0.012, β= −0.34 |
| 14:73.3 | rs4903104–T | 0.003 | −2.8 (0.9) | 0.059 | 0.125 | PAPLN | p.(Thr1201Met) | 26.0 |
Huang, 2017: p=0.004, HR= 1.05 Kunkle, 2019: p=0.031, β= 0.04 |
|
| 17:81.6 | rs7210026–C | 0.007 | −1.4 (0.5) | 0.604 | 0.483 | TSPAN10 | p.(Val172Ala) | 24.5 |
Huang, 2017: p=0.011, HR= 1.03 Naj, 2014: p=0.037, β= −0.22 |
|
| 22:20.4 | rs361566–A | 0.008 | 1.8 (0.7) | 0.140 | 0.129 | SCARF2 | p.(Pro174Ser) | 28.6 |
Huang, 2017: p=0.015, HR= 0.96 Kunkle, 2019: p=0.048, β= −0.04 |
|
| p<0.01, PopAF <2%, CADD>20, replication | 6:7.6 | rs28763967–T | 0.002 | −4.8 (1.7) | 0.024 | 0.009 | DSP | p.(Arg1537Cys) | 25.8 | Naj, 2014: p=0.039, β= −0.78 |
| 1:21.8 | rs143543800–T | 0.002 | 9.6 (3.0) | 0.006 | 0.004 | HSPG2 | p.(Val3500Met) | 24.6 | Bellenguez, 2022: p=0.017, β= −0.17 | |
| 16:67.2 | rs150417999–A | 0.005 | 11.3 (4.1) | 0.004 | 0.004 | EXOC3L1 | p.(Pro44Leu) | 27.0 | Bellenguez, 2022: p=0.049, β= −0.15 | |
| 2:140.4 | rs76554185–G | 0.009 | 6.5 (2.6) | 0.007 | 0.019 | LRP1B | p.(Gly3615Ala) | 21.6 | Kunkle, 2019: p=0.008, β= −0.12 | |
| High Priority AD-associated gene | 21:25.9 | rs112263157–C | 3.7x10−4 | −5.3 (1.6) | 0.022 | 0.005 | APP | p.(Ser614Gly) | 21.1 | |
| 17:63.5 | rs3730025–G | 0.011 | −3.5 (1.5) | 0.025 | 0.009 | ACE | p.(Tyr244Cys) | 26.6 |
Kunkle, 2019: p=9.9x10−5, β= 0.25 Bellenguez, 2022: p=1.1x10−7, β= 0.17 |
Abbreviations: Chrom: b38 Pos – Chromosome: build hg38 position, Cohort AF – Cohort allele frequency, Population AF indicates the population allele frequency (TOPMed Bravo Freeze 8), CADD v1.6 – Combined Annotation Dependent Depletion score version 1.6.
Shared pathways between previous GWAS and coding variants of interest
Several pathways emerged with variants in both the previous GWAS replication set and the coding variants of interest set. First, TSPAN14 and TSPAN10 are involved in scaffolding ADAM10 and had GWAS and coding variants respectively. Second, ACE had a GWAS and coding variant. Third, HS3ST1 had a GWAS variant, and HSPG2 had a coding variant, with both involved in heparin sulfate biology.
Discussion
Genetic association studies for LOAD are limited by heterogeneity of cases and unknown levels of contribution from environmental sources. This study addresses these limitations by employing a well-described phenotype in a geographically isolated population with a monogenic form of AD [3]. While environmental influences will always be present, this population has a relatively homogeneous set of environmental influences.
We identified 13 loci with p<1x10−5 and replication or p<1x10−7 with a nearby GWAS hit associated with AD phenotypes as well as more speculative signals when considering replication of previous GWAS in this cohort or important coding variants. This study nominates several important biological processes and pathways for consideration including clusterin, heparin sulfate and amyloid processing.
One of the most significant variants was rs35980966 (p=5.5x10−8), which is a rare variant (gnomAD v3.1.2 MAF=0.35%) that tags the CLU locus on chromosome 8 and exhibits replication in three studies [18, 19] and ADGC EOAD study in progress. The variant falls within a single nucleus multiomics linkage [20] to CLU. In addition, rs138295139 on chromosome 1 is only 4.4kb from a variant previously associated with plasma clusterin, rs4428865 [41], though these variants are not in LD, which could be explained by the rarity of rs138295139. Finally, rs4942482 on chromosome 13 is near a variant previously associated with CSF clusterin [41] and replicates in three studies [17, 19] and ADGC EOAD study in progress. This variant is linked via single nucleus multiomics measurements to nearby genes including ZC3H13 and SIAH3 (the linkage to SIAH3 is particularly interesting as it is AD-specific). The variant previously associated with CSF clusterin levels [41] falls between these genes. In addition, SIAH3 has been associated through another GWAS to rate of ventricular enlargement in the ADNI cohort [42], an association that has also been separately observed with variants near CLU [43]. Taken together, these observations, along with evidence for diverse contributions of clusterin in LOAD (recently review in [44]), suggest that further investigation of the role of clusterin and processes that may influence the effects of clusterin in ADAD is warranted.
Two variants were identified in or near heparin sulfate associated genes including rs6448453, a common variant near HS3ST1, and rs143543800, a rare variant in HSPG2. Heparin sulfate has been implicated in cell-to-cell spread of tau [45] as well as other AD-associated processes [46], pointing to potential importance of this pathway for dominant AD.
Variants in genes associated with amyloid processing were also identified in this study. A common variant in TSPAN14, rs6586028 (recently newly implicated in LOAD [19]), replicated in this cohort, and we also identified a coding variant in TSPAN10 (Table 4). These two genes code for tetraspanins that are a part of the TspanC8 subgroup of tetraspanins which promote ADAM10 maturation [47]. Given ADAM10’s established role as an α-secretase promoting non-amyloidogenic processing of Aβ [48] as well as its ability to cleave TREM2 (reviewed in [49]) and the recent association of genetic variation in or near ADAM10 with AD risk by GWAS [18, 25, 27] along with a candidate study of mutations [50], the basis of the observed association between age of dementia onset and these variants in TSPAN14 and TSPAN10 (both with a deleterious correlation) may result from disruption of a protective role of ADAM10.
As the largest age at onset modifier study in ADAD to date (to our knowledge), this study has nominated several new candidate genetic associations with age of dementia onset in ADAD. The most important limitation of this study is the small sample size (despite being the largest available sample size for this population) which precluded variants passing multiple corrections adjusted genome-wide significance. However, because we analyzed the three largest ADAD datasets available in the field, it is not possible to further increase sample size or replication in ADAD, and we therefore present these findings in light of replication with these available ADAD cohorts as well as sporadic EOAD and LOAD cohorts. Still, we recognize the speculative nature of the nominal associations identified in this study. Recruitment of more patients with early onset and/or dominant dementias from South American countries will help to overcome this limitation in future studies [51].
An important overarching theme from this analysis is that while age at dementia onset in ADAD has a strong heritable component, it is likely that, as with LOAD, there are many different genetic contributors that sum to determine an individual’s age at dementia onset for ADAD. Indeed, previous studies have suggested that further study of these types of genetic contributions is warranted [52]. Based on the unique demography of this population as a tri-continental admixture that passed through a narrow bottleneck [53], we conducted this study with the hypothesis that rare variants with a large effect size, i.e., the APOE Christchurch mutation [5], could account for much of the difference in age at dementia onset. Indeed, we identified many genetic variants of a similar rarity in this study that are candidates for having a large effect on age at dementia onset. However, we note that due to the nature of the analysis, it is possible for the presence of alleles in a small number of individuals with a particularly late age at onset to result in a low p value and large effect size (“winner’s curse”), therefore large effect sizes in this study should be interpreted with caution. In particular, 6 of the 13 associations highlighted as top candidate associations are observed with an allele count of between 3–7, and thus these associations are driven by a small number of individuals with a late age of onset. Further functional analysis in future studies could help to clarify the possible role of these rare variants on biological processes that may affect AD age at onset.
Importantly, we also detected common and/or lower effect size variation associated with age of dementia onset in pathways and biological processes including clusterin, heparin sulfate and amyloid processing. Because many of these variants replicate or were identified in non-admixed European populations, it suggests that the associations for many of these variants are robust to ancestral background. The identified variants in this study occur in the presence of a very strong causative mutation for ADAD, emphasizing the importance of the association signals observed for these variants and the need for more investigation of these variants in future studies.
Supplementary Material
Acknowledgements
We thank the patients and their caregivers for their generous contributions to this study. We thank other members of the GNA for their contributions to study of this cohort which made this study possible. We thank Nithesh Perumal for assisting with pedigree reconstruction. Replication statistics were obtained from publicly available data for NIAGADS project NG00075 [18] and controlled access data from NIAGADS projects NG00048 [16] and NG00058 [17] and the Synapse AD Knowledge portal (Synapse ID: syn25871263). More extensive acknowledgement details for DIAN, IGAP, ADDS, and ADGC are provided in the supplemental acknowledgements.
Funding
Funding was provided by the HudsonAlpha Memory and Mobility Program, the Larry L. Hillblom Foundation, the Rainwater Foundation, NIH grant 4R00AG068271 (Cochran), NIA, the Alzheimer’s Association, and the Michael J. Fox Foundation (Cruchaga), 1R56AG062479-01 and 5U54NS100717-04 (Kosik), and the Sustainability Program of the CODI University of Antioquia (Lopera).
Disclosures
Grant and other financial support for this study overall and for particular investigators related to this study is detailed in the Funding section. J. Nicholas Cochran has consulted for Caraway Therapeutics. Richard M. Myers is a member of the board of directors for the Coalition for the Life Sciences and is a science advisor for Decheng Capital, Accuragen, iRepertoire, IMIDomics, Inc., iCubate, and Sentieon. Carlos Cruchaga has consulted for Circular Genomics and Alector, has received travel support from Somalogic, has a leadership or fiduciary role and holds stock or stock options in Vivid Genetics and Circular Genomics, and has received support not related to this study from GSK, Alector, Eisai, an anonymous foundation, Biogen, and Parabon. All other authors declared no relevant competing interests.
Footnotes
Consent Statement
All human subjects enrolled in this study provided informed consent.
References
- [1].Neuner SM, Tcw J, Goate AM. Genetic architecture of Alzheimer’s disease. Neurobiol Dis. 2020;143:104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lopera F, Ardilla A, Martinez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793–9. [PubMed] [Google Scholar]
- [4].Acosta-Baena N, Sepulveda-Falla D, Lopera-Gomez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet neurology. 2011;10:213–20. [DOI] [PubMed] [Google Scholar]
- [5].Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lalli MA, Bettcher BM, Arcila ML, Garcia G, Guzman C, Madrigal L, et al. Whole-genome sequencing suggests a chemokine gene cluster that modifies age at onset in familial Alzheimer’s disease. Mol Psychiatry. 2015;20:1294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Velez JI, Lopera F, Silva CT, Villegas A, Espinosa LG, Vidal OM, et al. Familial Alzheimer’s Disease and Recessive Modifiers. Mol Neurobiol. 2020;57:1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Velez JI, Rivera D, Mastronardi CA, Patel HR, Tobon C, Villegas A, et al. A Mutation in DAOA Modifies the Age of Onset in PSEN1 E280A Alzheimer’s Disease. Neural Plast. 2016;2016:9760314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lalli MA, Garcia G, Madrigal L, Arcos-Burgos M, Arcila ML, Kosik KS, et al. Exploratory data from complete genomes of familial alzheimer disease age-at-onset outliers. Hum Mutat. 2012;33:1630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aguirre-Acevedo DC, Gomez RD, Moreno S, Henao-Arboleda E, Motta M, Munoz C, et al. [Validity and reliability of the CERAD-Col neuropsychological battery]. Rev Neurol. 2007;45:655–60. [PubMed] [Google Scholar]
- [11].Ardila A, Lopera F, Rosselli M, Moreno S, Madrigal L, Arango-Lasprilla JC, et al. Neuropsychological profile of a large kindred with familial Alzheimer’s disease caused by the E280A single presenilin-1 mutation. Arch Clin Neuropsychol. 2000;15:515–28. [PubMed] [Google Scholar]
- [12].Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T, Members of the Familial Alzheimer’s Disease Pathology Study G. Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am J Pathol. 2001;158:2165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Naj AC, Jun G, Reitz C, Kunkle BW, Perry W, Park YS, et al. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol. 2014;71:1394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Anderson AG, Rogers BB, Loupe JM, Rodriguez-Nunez I, Roberts SC, White LM, et al. Single nucleus multiomics identifies ZEB1 and MAFB as candidate regulators of Alzheimer’s disease-specific cis regulatory elements. bioRxiv. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Velez JI, Lopera F, Sepulveda-Falla D, Patel HR, Johar AS, Chuah A, et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol Psychiatry. 2016;21:916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G, et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54:163–9. [DOI] [PubMed] [Google Scholar]
- [23].Aguirre-Acevedo DC, Lopera F, Henao E, Tirado V, Munoz C, Giraldo M, et al. Cognitive Decline in a Colombian Kindred With Autosomal Dominant Alzheimer Disease: A Retrospective Cohort Study. JAMA Neurol. 2016;73:431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lendon CL, Martinez A, Behrens IM, Kosik KS, Madrigal L, Norton J, et al. E280A PS-1 mutation causes Alzheimer’s disease but age of onset is not modified by ApoE alleles. Hum Mutat. 1997;10:186–95. [DOI] [PubMed] [Google Scholar]
- [25].Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry. 2018;8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jun GR, Chung J, Mez J, Barber R, Beecham GW, Bennett DA, et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 2017;13:727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mez J, Chung J, Jun G, Kriegel J, Bourlas AP, Sherva R, et al. Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimers Dement. 2017;13:119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309:1483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhou X, Chen Y, Mok KY, Zhao Q, Chen K, Chen Y, et al. Identification of genetic risk factors in the Chinese population implicates a role of immune system in Alzheimer’s disease pathogenesis. Proc Natl Acad Sci USA. 2018;115:1697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kunkle BW, Schmidt M, Klein HU, Naj AC, Hamilton-Nelson KL, Larson EB, et al. Novel Alzheimer Disease Risk Loci and Pathways in African American Individuals Using the African Genome Resources Panel: A Meta-analysis. JAMA Neurol. 2021;78:102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014;10:e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78:256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Deming Y, Filipello F, Cignarella F, Cantoni C, Hsu S, Mikesell R, et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nelson PT, Estus S, Abner EL, Parikh I, Malik M, Neltner JH, et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. 2014;127:825–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Andrews SJ, Fulton-Howard B, Goate A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet neurology. 2020;19:326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Holstege H, Hulsman M, Charbonnier C, Grenier-Boley B, Quenez O, Grozeva D, et al. Exome sequencing identifies rare damaging variants in the ATP8B4 and ABCA1 genes as novel risk factors for Alzheimer’s Disease. medRxiv. 2021:2020.07.22.20159251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Deming Y, Xia J, Cai Y, Lord J, Holmans P, Bertelsen S, et al. A potential endophenotype for Alzheimer’s disease: cerebrospinal fluid clusterin. Neurobiol Aging. 2016;37:208 e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li X, Chu SG, Shen XN, Hou XH, Xu W, Ou YN, et al. Genome-wide association study identifies SIAH3 locus influencing the rate of ventricular enlargement in non-demented elders. Aging (Albany NY). 2019;11:9862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roussotte FF, Gutman BA, Madsen SK, Colby JB, Thompson PM, Alzheimer’s Disease Neuroimaging I. Combined effects of Alzheimer risk variants in the CLU and ApoE genes on ventricular expansion patterns in the elderly. J Neurosci. 2014;34:6537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yuste-Checa P, Bracher A, Hartl FU. The chaperone Clusterin in neurodegeneration-friend or foe? Bioessays. 2022;44:e2100287. [DOI] [PubMed] [Google Scholar]
- [45].Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Snow AD, Cummings JA, Lake T. The Unifying Hypothesis of Alzheimer’s Disease: Heparan Sulfate Proteoglycans/Glycosaminoglycans Are Key as First Hypothesized Over 30 Years Ago. Front Aging Neurosci. 2021;13:710683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Haining EJ, Yang J, Bailey RL, Khan K, Collier R, Tsai S, et al. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J Biol Chem. 2012;287:39753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Peron R, Vatanabe IP, Manzine PR, Camins A, Cominetti MR. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals (Basel, Switzerland). 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lichtenthaler SF, Tschirner SK, Steiner H. Secretases in Alzheimer’s disease: Novel insights into proteolysis of APP and TREM2. Curr Opin Neurobiol. 2022;72:101–10. [DOI] [PubMed] [Google Scholar]
- [50].Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18:3987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ibanez A, Yokoyama JS, Possin KL, Matallana D, Lopera F, Nitrini R, et al. The Multi-Partner Consortium to Expand Dementia Research in Latin America (ReDLat): Driving Multicentric Research and Implementation Science. Front Neurol. 2021;12:631722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Day GS, Cruchaga C, Wingo T, Schindler SE, Coble D, Morris JC. Association of Acquired and Heritable Factors With Intergenerational Differences in Age at Symptomatic Onset of Alzheimer Disease Between Offspring and Parents With Dementia. JAMA Netw Open. 2019;2:e1913491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mooney JA, Huber CD, Service S, Sul JH, Marsden CD, Zhang Z, et al. Understanding the Hidden Complexity of Latin American Population Isolates. Am J Hum Genet. 2018;103:707–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, et al. Brain cell type-specific enhancer-promoter interactome maps and disease risk association. Science (New York, NY). 2019;366:1134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.