

Abstract
Integrating single-cell multi-omics data is a challenging task that has led to new insights into complex cellular systems. Various computational methods have been proposed to effectively integrate these rapidly accumulating datasets, including deep learning. However, despite the proven success of deep learning in integrating multi-omics data and its better performance over classical computational methods, there has been no systematic study of its application to single-cell multi-omics data integration. To fill this gap, we conducted a literature review to explore the use of multimodal deep learning techniques in single-cell multi-omics data integration, taking into account recent studies from multiple perspectives. Specifically, we first summarized different modalities found in single-cell multi-omics data. We then reviewed current deep learning techniques for processing multimodal data and categorized deep learning-based integration methods for single-cell multi-omics data according to data modality, deep learning architecture, fusion strategy, key tasks and downstream analysis. Finally, we provided insights into using these deep learning models to integrate multi-omics data and better understand single-cell biological mechanisms.
Keywords: multi-omics, single-cell, deep learning, data integration
INTRODUCTION
Single-cell technologies have revolutionized how biologists study cellular systems, enabling biologists to examine the molecular characteristics of individual cells within a population [1]. Single-cell omics data generated by single-cell technologies have numerous applications, such as novel cell type discovery and regulatory network identification. Recent biotechnology advancements have generated various types of omics data, including genomics, transcriptomics, epigenomics and proteomics. Integrating these multi-omics datasets can enhance our biological understanding, which would have been unachievable using omics data of a single modality [2, 3].
Recently, there has been a surge in the development of multimodal deep learning (MDL) approaches for integrating single-cell multi-omics data. They have demonstrated impressive predictive power [4] and offered several advantages over existing computational methods [5, 6]. For example, MDL techniques can uncover complex patterns and provide a more comprehensive understanding of the molecular characteristics of individual cells [7]. Unlike existing computational methods like matrix factorization [8, 9] and correlation-based approaches [5, 6] that require manually extracted features for each modality, MDL can automatically learn a hierarchical representation for each modality by extracting meaningful features through a multilayer neural network model. MDL can better manage high-dimensional data by mapping features from different modalities into a smaller, unified subspace [10]. This is especially true when non-linear feature relationships are expected [11] because each model layer involves non-linear feature mapping [12].
While MDL approaches for single-cell data integration hold great potential, several challenges need to be addressed. For example, overfitting can be a problem during MDL model training, especially for high-dimensional, imbalanced single-cell multi-omics data [12]. In addition, the sparsity of the data can also be an issue, as single-cell multi-omics data often contain numerous missing values [13]. Furthermore, there is a lack of consensus on the best methods for integrating single-cell multi-omics data using MDL techniques, making comparing results across different studies challenging. Nonetheless, MDL is an active area of research, and ongoing efforts are to improve these methods and make them more accessible to researchers.
There is a lack of systematic investigation into the application of MDL approaches for single-cell multi-omics data integration. There are reviews on computational strategies for single-cell multi-omics integration [10, 14]. However, these reviews focused on computational methods other than MDL approaches and did not include more recently published works on MDL. Considering the advancement of deep learning methods and the development of next-generation sequencing data, this study aims to review various modalities of single-cell multi-omics data and the current state-of-the-art MDL models for their integration. We categorize the published work based on MDL model architecture, fusion strategy, key integration tasks and downstream biological analysis.
The paper is structured as follows: Overview of Single-Cell Multi-omics Data Modalities section provides an overview of the different modalities of single-cell omics data. Overview of MDL Techniques section describes the deep learning techniques commonly used for multimodal data analysis. MDL Models for Single-Cell Data Integration section discusses the current state-of-the-art MDL models for single-cell data integration. Finally, Discussion and Conclusion section focuses on the limitations of the current approaches, future research directions and conclusions.
OVERVIEW OF SINGLE-CELL MULTI-OMICS DATA MODALITIES
Single-cell technologies aim to comprehensively measure biological molecules like RNAs, proteins and chromatin structures at a single-cell resolution [15]. The major data modalities for single-cell multi-omics data are summarized in Table 1.
Table 1.
Single-cell multi-omics data modalities
| Single-cell omics type | Data modality | Example |
|---|---|---|
| Genomics | scDNA-seq | DOP-PCR, MDA, MALBAC |
| Transcriptomics | scRNA-seq | Full-length transcript: scNaUmi-seq, MATQ-seq, Smart-seq, Smart-seq2 3′ transcript: 10x Chromium, CEL-seq2, Drop-seq, InDrop, MARS-seq 5′ transcript: STRT-seq |
| Epigenomics | DNA methylation | scBS-seq |
| Histone modification | ChIP-seq | |
| Chromatin accessibility | ATAC-seq, DNase-seq | |
| Chromosome conformation | Hi-C | |
| Proteomics | Protein expression | CyTOF, FACS |
| Multi-omics | DNA methylation data and transcriptomic data | scM&T-seq, scMT-seq, scTrio-seq, and snmCT-seq |
| Transcriptome and chromatin accessibility | Paired-seq and SNARE-seq |
(i) Single-cell genomics data
Single-cell DNA sequencing (scDNA-seq) has proven effective in identifying copy number aberrations [16], somatic mutations [17] and tracking cell lineage [18]. It has found extensive use in cancer research, where it helps track the growth of different cell clones and understand tumor development [19–21]. It also enables the identification of rare cell types that may be missed in conventional bulk analysis methods [22]. Various scDNA-seq whole genome amplification techniques exist, such as degenerate oligonucleotide-primed polymerase chain reaction (DOP-PCR), multiple displacement amplification (MDA), and multiple annealing and looping–based amplification cycles (MALBAC) [23].
(ii) Single-cell transcriptomics data
scRNA-seq, also known as single-cell transcriptomics or gene expression data, is a powerful method for measuring the expression levels of genes in individual cells, enabling scientists to characterize cellular diversity and heterogeneity at a high resolution [24]. Various scRNA-seq protocols are available that differ in the extent of transcript analysis. While some analyze the entire transcript, others examine only the 3′ or 5′ end [25]. Examples of such protocols include 10x Chromium, CEL-seq2 and MARS-seq, among others. Full-length transcript methods can detect allele-specific expression, low expressive genes and alternative splicing occurrences. In contrast, partial-length methods can analyze a bulk amount of single cells, but cannot detect allele-specified expression [26, 27].
(iii) Single-cell epigenomics data
Epigenomics measures genome-wide epigenomic modifications, such as DNA methylation, histone modifications and chromosome accessibilities [28–30]. Recent developments in single-cell epigenomic approaches have led to the creation of methods such as single-cell bisulfite sequencing (scBS-seq) and single-cell reduced representation bisulfite sequencing (scRRBS-seq) for the single-base resolution mapping of DNA methylation in individual cells [31–33]. Other techniques, such as TET-assisted bisulfite sequencing and Aba-seq, have also been used to study hydroxymethylated cytosine (5hmC) in bulk samples and have the potential to be adapted for single-cell analysis [34–37]. Histone modification can be detected using chromatin immunoprecipitation sequencing (ChIP-seq) [38]. Finally, high-throughput sequencing techniques, such as transposase-accessible chromatin with sequencing (ATAC-seq) and DNase I hypersensitive site sequencing (DNase-seq), can identify genome regions open for transcription and thus measure chromatin accessibility in single cells [39, 40].
(iv) Single-cell proteomics data
Single-cell proteomics investigates individual cells’ protein content, analyzing their roles and interactions [41]. This technique is especially valuable when studying cells with distinct functions or at varying stages of development. Various methods are employed in single-cell proteomics, such as fluorescence-activated cell sorting (FACS), single-cell mass spectrometry (CyTOF) and microfluidics-based techniques.
(v) Joint-modality single-cell multi-omics data
New techniques have allowed for the simultaneous measurement of multiple modalities, resulting in joint-modality data that provides a more comprehensive understanding of the molecular and cellular processes involved in tissue and organ function [42, 43]. Several techniques allow researchers to simultaneously measure the DNA methylation and transcriptomic data in individual cells, such as scM&T-seq [44], scMT-seq [45], scTrio-seq [46] and snmCT-seq. Perturb-seq [47, 48] and CRISP-seq [49] are techniques that measure CRISPR-based transcriptional interference and high-throughput scRNA-seq. Similar techniques, such as Paired-seq [50] and SNARE-seq [51], investigate transcriptome and chromatin accessibility in single cells or nuclei. The data generated by these techniques allow multimodal omics analysis at the single-cell level.
OVERVIEW OF MDL TECHNIQUES
Recent studies have investigated the potential of deep learning models in tackling complex and multimodal biological challenges [52] with encouraging outcomes. The main goal of deep learning is to train models that can learn high-level features of input data by processing them through a series of layers. In this process, earlier layers learn simpler data abstractions, which are combined in deeper layers to form more informative and complex representations relevant to the task at hand [53]. Deep learning can capture non-linear and cross-modal relationships, making it a powerful tool for addressing multimodal biological problems.
FCNN: A fully connected neural network (FCNN) connects all the nodes of one layer to the subsequent layer’s nodes. Researchers have employed FCNN in various studies to solve different problems. For example, Park et al. [54] used FCNN to predict Alzheimer’s disease (AD) by concatenating gene expression and DNA methylation data. Huang et al. [55] utilized FCNN to integrate mRNA and miRNA data to predict cancer patient survival. Dent et al. [56] trained four FCNNs with four types of drug features and integrated their predictions by taking the mean of the probabilities of the 65 targets. Huang et al. [57] fused clinical and imaging data using FCNN during the final decision stage.
CNN: Convolutional neural network (CNN) is a model that comprises three layers: the convolutional layer, the pooling layer and the fully connected layer. It has demonstrated impressive performance in dealing with image and audio data. Chang et al. [58] combined genomic profiles of 787 human cancer cell lines and structural profiles of 244 drugs and applied CNN to predict drug effectiveness. Similarly, Islam et al. [59] used a deep CNN model to combine copy number alteration and gene expression data to classify molecular subtypes of breast cancer. Spasov et al. [60] utilized a CNN to fuse magnetic resonance images (MRI) and clinical data to predict AD.
RNN: A recurrent neural network (RNN) maintains a state vector that encodes information from past time steps and updates it at each time step. It is particularly effective for analyzing temporal data. For instance, Bichindaritz et al. [61] utilized long short-term memory techniques to predict the survival rate for breast cancer by integrating gene expression and DNA methylation data. Lee et al. [62] used gated recurrent units to learn marginal representation from multi-omics data to predict AD progression.
AE: Autoencoder (AE) is a neural network architecture that consists of an encoder and a decoder, and it is commonly used for representation learning. The encoder learns a compressed latent representation of the input data, and the decoder aims to reconstruct the input data from the latent representation. To tackle multimodal biological problems, AE and its variations have been widely used. For instance, Guo et al. [63] applied denoising autoencoders to multi-omics ovarian cancer data to identify cancer subtypes. Islam et al. [59] employed a stacked autoencoder to predict breast cancer subtypes using early fused copy number alteration and gene expression data. Ronen et al. [64] utilized a stacked variational AE (VAE) to measure the similarity between colorectal tumors and cancer cell lines.
DBN: A deep belief network (DBN) comprises multiple Boltzmann machines arranged in a specific order, with lower computational complexity than deep neural networks. Suk et al. [65] adopted a multimodal DBN to predict AD by aggregating positron emission tomography scans and MRI. To predict disease–gene associations, Luo et al. [52] utilized two DBNs to learn latent representation from protein–protein interaction networks and gene ontology. A joint representation was then learned from that latent representation using another DBN.
Heterogenous model: Zhang et al. [66] developed fusion models based on CNNs and RNNs to learn patient representation by combining sequential clinical notes, static demographic and admission data. In another study, Lin et al. [67] utilized three separate encoder networks to learn marginal representations of mRNA, DNA methylation and copy number variation data for breast cancer subtype prediction. These marginal representations were concatenated and fed into a classification subnetwork to learn a joint representation. More recently, sciCAN [68] combined generative adversarial networks (GAN) and encoder models for integrating single-cell multi-omics data.
MDL MODELS FOR SINGLE-CELL DATA INTEGRATION
We were able to identify a total of 21 studies published by 2022. Figure 1 shows the workflow of integrating single-cell multi-omics data using MDL techniques. We first analyze the single-cell multi-omics data (Table 2) and the proposed models (Table 3). Then, we categorized the studies based on fusion strategy, data type, key task and downstream analysis.
Figure 1.
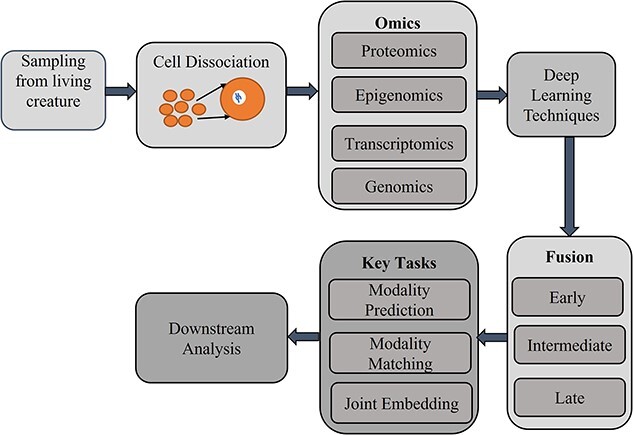
Workflow of deep learning-based single-cell multi-omics data integration.
Table 2.
Data description of current works of MDL-based single-cell multi-omics data integration
| Year | Papers | Cell type | Integration type | Data sample | Technology | Modality |
|---|---|---|---|---|---|---|
| 2018 | MAGAN [69] | Paired | Vertical | Human bone marrow [70] | CyTOF, scRNA-seq | FACS-sorted/scRNA-seq |
| 2019 | k-Coupled AE [71] | Paired | Vertical | Single neurons [72] | Patch-seq | scRNA-seq/electrophysiological profiles |
| 2020 | SCIM [73] | Unpaired | Diagonal | Melanoma tumor from the Tumor Profiler project [74], human bone marrow sample [75] | scRNA-seq/CyTOF | scRNA-seq/CyTOF |
| 2021 | scMM [76] | Paired | Vertical | Human PBMC [2] and BMNC [5] | CITE-seq | scRNA-seq/surface protein |
| Mouse skin [3] | SHARE-seq | scRNA-seq/scATAC-seq | ||||
| Cobolt [77] | Paired | Vertical | Adult mouse cerebral cortices [51] | SNARE-seq | mRNA-seq/scATAC-seq | |
| Mop [78] | scRNA-seq, scATAC-seq | scRNA-seq/scATAC-seq | ||||
| Human PBMC [79–82] | 10X Multiome | scRNA-seq/scATAC-seq | ||||
| BABEL [83] | Paired | Vertical | Human PBMC [84], DM and HSR cells (GSE160148) | 10X Multiome | scRNA-seq/scATAC-seq (single nuclei) | |
| Adult mouse cerebral cortex [51] (GSE126074) | SNARE-seq | |||||
| Mouse skin [3] (GSE140203) | SHARE-seq | scRNA-seq/scATAC-seq | ||||
| Human bone marrow [5] (GSE128639) | CITE-seq | scRNA-seq/protein epitope | ||||
| scMVAE [85] | Paired | Vertical | Human cell lines mixture [51] | SNARE-seq | scRNA-seq/scATAC-seq | |
| Adult mouse cerebral cortex [86] | scCAT-seq | |||||
| totalVI [87] | Paired | Vertical | Human PBMC10k [88] and PBMC5k [89], MALT [90], SLN111-D1, SLN111-D2, SLN208-D1, SLN208-D2 (GSE150599) | CITE-seq | scRNA-seq/surface protein | |
| Crossmodal-AE [91] | Paired and unpaired | Vertical and diagonal | Human lung [92] (GSE117089), human PBMCs [93] | scRNA-seq, scATAC-seq | scRNA-seq/scATAC-seq | |
| Human PBMC [94], Zenodo [91] | – | Chromatin image data | ||||
| SMILE [95] | Paired | Vertical | Mouse kidney [92] (GSE117089) | sci-CAR | scRNA-seq/scATAC-seq | |
| Mixed cell lines [51] (GSE126074) | SNARE-seq | |||||
| Mouse brain and skin [3] (GSE140203) | SHARE-seq | |||||
| Human prefrontal cortex [96] (GSE130711) | snm3c-seq | Single-cell DNA methylation/ Hi-C | ||||
| Mouse brain [97] (GSE152020) | Paired-tag | scRNA-seq/4 histone mark data (H3K4me1, H3K9me3, H3K27me3, H3K27ac) | ||||
| 2022 | scMoGNN [98] | Paired | Vertical | Hallmark genesets [99] from MsigDB | – | GEX (scRNA-seq) to ADT (protein), and GEX (scRNA-seq) to scATAC-seq |
| SAILERX [100] | Paired | Vertical | Human PBMC [2] | 10X Multiome | scRNA-seq/scATAC-seq | |
| Mouse skin [3] | SHARE-seq | |||||
| Mouse cortex [51] | SNARE-seq | |||||
| GLUE [101] | Unpaired | Diagonal | Mouse cortex [51] (GSE126074) | SNARE-seq | scRNA-seq/scATAC-seq | |
| Mouse skin [3] (GSE140203) | SHARE-seq | |||||
| Human PBMC [84, 102] | 10X Multiome | |||||
| Mouse nephron [103] (GSE151302) and Mop [78] | scRNA-seq, scATAC-seq | |||||
| Mop [104] | snmC-seq | DNA methylation | ||||
| scMVP [105] | Paired | Vertical | Mouse cerebral cortex (GSE126074) [51] | SNARE-seq | scRNA-seq/scATAC-seq | |
| Human and mouse (GSM3271040, GSM3271040) [92] | sci-CAR | |||||
| Mouse (GSE130399) [50] | Paired-seq | |||||
| Mouse skin (GSE140203) [3] | SHARE-seq | |||||
| Human PBMC and lymph node [106] | 10X Multiome | |||||
| scJoint [107] | Paired and unpaired | Vertical and diagonal | Mouse atlas [108, 109], human hematopoiesis [110] | scRNA-seq, scATAC-seq | scRNA-seq/scATAC-seq | |
| Human fetal atlas [111, 112] | scRNA-seq (GSE156793), scATAC-seq (GSE149683) | |||||
| Adult mouse cerebral cortex [51] (GSE126074) | SNARE-seq | |||||
| Human PBMC T-cell stimulation experiment [113] (GSE156478) | CITE-seq [114] and ASAP-seq [113] | Gene expression levels (scRNA-seq) or chromatin accessibility (scATAC-seq) simultaneously with surface protein levels | ||||
| sciCAN [68] | Unpaired | Diagonal | Mixed cell lines [51] (GSE126074) | SNARE-seq | scRNA-seq/scATAC-seq | |
| Mouse skin [3] (GSE140203) | SHARE-seq | |||||
| Human PBMC [115] | 10X Multiome | |||||
| Human hematopoiesis [110] (GSE139369), human lung [116] (GSE161383) and mouse kidney [117] (GSE157079) | scRNA-seq, scATAC-seq | |||||
| CRISPR-perturbed single-cell K562 | Perturb-seq [47] (GSE90063) | scRNA-seq | ||||
| Spear-ATAC [118] (GSE168851) | scATAC-seq | |||||
| scDART [119] | Unpaired | Diagonal | Mouse neonatal brain cortex [51] (GSE126074) | SNARE-seq | scRNA-seq/scATAC-seq | |
| Mouse endothelial cells [120] (GSE137117) | scRNA-seq, scATAC-seq | |||||
| Human hematopoiesis | scRNA-seq [121] (GSE117498), scATAC-seq [122] (GSE96772) | |||||
| Portal [123] | Paired and unpaired | Vertical and diagonal | Mouse brain atlas [124] | SPLiT-seq | snRNA-seq | |
| Mouse brain atlas [125, 126] | Drop-seq and 10X | scRNA-seq | ||||
| Human PBMC [127] | scRNA-seq | scRNA-seq | ||||
| Human brain [128, 129] | snRNA-seq | snRNA-seq | ||||
| Human PBMC [113] | CITE-seq | scRNA-seq | ||||
| ASAP-seq | scATAC-seq | |||||
| MIRA [130] | Paired | Vertical | Mouse skin [3] | SHARE-seq | scRNA-seq/scATAC-seq | |
| Mouse embryonic brain [115] | 10X Multiome | |||||
| SCALEX [131] | Paired | Vertical | Human PBMC [102, 132] | 10X Multiome | scRNA-seq/scATAC-seq | |
| scMDC [133] | Paired | Vertical | BMNC (GSE128639), CBMN (GSE100866), PBMC, SLN111_D1, SLN111_D2, SLN208_D1 and SLN208_D2 (GSE150599) | CITE-seq | scRNA-seq/surface protein (ADT) | |
| Mouse brain E18, PBMC10K, PBMC3K [134] | SMAGE-seq (SNARE-seq and 10X Multiome) | scRNA-seq/scATAC-seq | ||||
| STACI [135] | Unpaired | Diagonal | Mouse brain (STARmap PLUS dataset) | STARmap | Spatial RNA (gene expression, cell adjacency matrix) and chromatin imaging |
Note: Abbreviations. PBMC: peripheral blood mononuclear cells. BMNC: bone marrow mononuclear cells. Mop: mouse primary motor cortex. DM: colon adenocarcinoma COLO-320 cells. HSR: colorectal adenocarcinoma COLO-320 cells. MALT: mucosa-associated lymphoid tissue. SLN111-D1, SLN111-D2, SLN208-D1 and SLN208-D2: the murine spleen and lymph node data. MSigDB: Molecular Signatures Database. CBMN: cord blood mononuclear cells.
Table 3.
Current work on deep learning-based single-cell multi-omics data integration
| Year | Papers | Methods | Fusion | Key task | Evaluation |
|---|---|---|---|---|---|
| 2018 | MAGAN [69] | GAN | Intermediate (marginal homogeneous) | ● Modality prediction | ● No downstream task |
| 2019 | k-Coupled AE [71] | AE | Late | ● Modality prediction | ● Cell type discovery |
| 2020 | SCIM [73] | VAE (2 encoders + 2 decoders + 1 discriminator) | Intermediate (joint homogeneous) | ● Joint embedding | ● Cell matching ● Cell type discovery |
| 2021 | scMM [76] | VAE (2 encoders + 2 decoders) | Intermediate (joint homogeneous) | ● Joint embedding ● Modality prediction |
● Cell type discovery ● cis-Regulatory analysis |
| Cobolt [77] | VAE (2 encoders + 2 decoders) | Intermediate (joint homogeneous) | ● Joint embedding | ● DE ● Cell type discovery |
|
| BABEL [83] | AE | Intermediate (joint homogeneous) | ● Joint embedding ● Modality prediction |
● Cell type discovery | |
| scMVAE [85] | VAE | Early and intermediate (joint homogeneous) | ● Joint embedding | ● Cell type discovery ● cis-Regulatory analysis |
|
| totalVI [87] | VAE | Early | ● Joint embedding ● Modality prediction |
● Protein identification and correction ● DE ● Archetypal analysis |
|
| Crossmodal-AE [91] | AE | Intermediate (joint heterogeneous) | ● Joint embedding ● Modality prediction |
● DE ● Cell matching |
|
| SMILE [95] | Encoder | Intermediate (marginal homogeneous) | ● Joint embedding ● Modality matching |
● DE | |
| 2022 | scMoGNN [98] | GCNN | Intermediate (marginal and joint homogeneous) | ● Joint embedding ● Modality prediction ● Modality matching |
● No downstream analysis |
| SAILERX [100] | VAE (1 encoder + 1 decoder) | Intermediate (joint heterogeneous) | ● Joint embedding | ● cis-Regulatory analysis ● Cell type discovery |
|
| GLUE [101] | VAE (3 encoders + 3 decoders) | Intermediate (marginal homogeneous) | ● Joint embedding | ● cis-Regulatory analysis ● Cell type discovery |
|
| scMVP [105] | VAE (3 encoders +2 decoders) | Intermediate (joint homogeneous) | ● Joint embedding | ● Cell type discovery ● cis-Regulatory analysis ● Trajectory inference |
|
| scJoint [107] | Encoder | Intermediate (joint homogeneous) | ● Joint embedding ● Modality prediction |
● Cell type discovery ● DE |
|
| sciCAN [68] | Encoder + GAN | Intermediate (joint heterogeneous) | ● Joint embedding ● Modality prediction |
● Trajectory inference analysis ● Cellular response analysis to genetic perturbation |
|
| scDART [119] | FCNN | Intermediate (joint homogeneous) | ● Joint embedding ● Modality prediction |
● Trajectory inference analysis ● DE ● Cell matching |
|
| Portal [123] | GAN | Intermediate (joint homogeneous) | ● Joint embedding | ● Cell type discovery | |
| MIRA [130] | VAE | Late | ● Joint embedding | ● Trajectory inference analysis ● cis-Regulatory analysis |
|
| SCALEX [131] | VAE | Early | ● Joint embedding | ● DE ● Cell type discovery |
|
| scMDC [133] | AE | Early | ● Joint embedding ● Modality matching |
● DE | |
| STACI [135] | Over-parameterized AE | Early | ● Joint embedding ● Modality prediction |
● DE ● Amyloid plaques prediction ● Identification of spatio-temporal changes in AD |
Data description
The input datasets of the studies (Table 2) are classified as ‘paired’ or ‘unpaired’. ‘Paired’ means the same cells or the same type of cells are selected from different integrating modalities and used as anchors for the integration task. ‘Unpaired’ means the cells are not matched between different modalities, and no anchor is defined.
MDL integration methods can be classified into three types, horizontal, vertical and diagonal data integration, depending on pairing and anchor information [136]. Horizontal integration uses shared features like genes to link data from different modalities, while vertical integration uses paired cells as anchors. In contrast, diagonal integration methods perform integration without using paired cells or shared features as anchors. These methods aim to build a simplified representation of the relationships between data modalities, assuming an underlying low-dimensional structure links them. The current review excludes studies in the horizontal category because they often use only one data modality from different sources. As shown in Table 2, only a few papers have used unpaired cells of different modalities to perform diagonal integration. SCIM, crossmodal-AE, STACI, GLUE, scJoint, sciCAN, scDART and Portal are examples of diagonal integration methods. Crossmodal-AE, scJoint and Portal perform both vertical and diagonal integration on paired or unpaired cell types.
Table 2 also describes the datasets based on data sources, technology or platform used for sequencing the data and comparing modalities. Most studies performed the integration task between scRNA-seq and scATAC-seq [68, 76, 77, 83, 85, 91, 95, 100, 101, 105, 107, 119, 123, 130, 131]. Some studies performed integration of multiple modality pairs like scMM [76], Cobolt [77], SMILE [95], Portal [123], scMoGNN and scMDC [133]. For example, besides integrating scRNA-seq and scATAC-seq data, scMM and totalVI also integrated scRNA-seq and surface protein profiling data. Crossmodal-AE is the only study that performed integration between chromatin image and scRNA-seq data. STACI [135] integrated single-cell spatial transcriptomics data with chromatin images. Some studies go beyond modality pairs and can integrate multiple data modalities. For example, GLUE integrated three different omics data modalities and named it triple omics integration. SMILE also integrated three modalities using a combination of the two model variants.
Model architecture
Table 3 summarizes the recent MDL models developed for single-cell multi-omics data integration. All the models used two-dimensional numerical matrices to represent the input data. We categorize these models into seven groups, VAE, AE, encoders, GAN, FCNN, GNN and heterogenous models, detailed as follows.
VAE: Most studies we surveyed utilized VAE. For instance, SCIM employs VAE to integrate scRNA-seq and CyTOF modalities. In SCIM, each modality is modeled by an encoder-decoder network, and a discriminator is incorporated to identify a specific source modality from the latent representation of other modalities. Through adversarial training, SCIM can generate an integrated latent space. However, SCIM cannot predict one modality from another and thus cannot accomplish cross-modal translation tasks.
scMM, similar to SCIM, aims to integrate multiple modalities into a shared space. However, scMM has the additional capability of cross-modal translation. scMM uses a VAE to integrate two modalities. The encoder-decoder network first takes the feature vectors for each modality as input. An encoder is trained to generate a low-dimensional joint variational posterior that can be factorized by a ‘mixture of experts model’ (MOE) [137]. This joint representation is then used to train decoders that reconstruct the underlying data distribution in each modality. The MOE factorization enables the separation of each modality from the joint representations, which helps scMM to perform cross-modality predictions.
Cobolt adopts an approach similar to scMM in projecting different modalities into a shared latent space. However, Cobolt distinguishes itself from scMM in its attempt to integrate joint-modality data with single-modality data. Given the current prevalence of single-modality data over joint-modality data in both quality and quantity [77], there is a strong interest in integrating both types of multi-omics data. Cobolt employs three encoders to learn the latent feature distributions of the input modalities: one for scRNA-seq, one for joint-modality data (scRNA-seq + scATAC-seq) and another for scATAC-seq. Each encoder learns separate latent embeddings and posterior distributions of latent variables. Cobolt then projects the modalities into a shared latent space by taking the posterior mean of these distributions. Finally, three separate decoders learn from the shared latent embeddings.
The MDL models described earlier can generate a shared feature representation (joint embedding) that preserves modality-specific information [105]. However, when significant noise or sparsity exists in joint-modality data, the resulting joint embedding may not accurately capture the biological variation, causing difficulties in downstream analysis and interpretation [105]. To address this issue, scMVAE and scMVP were developed. scMVAE utilizes one multimodal encoder, two single-modal encoders and two single-modal decoders. The multimodal encoder models scRNA-seq and scATAC-seq data with three joint-learning techniques. One is to estimate a joint posterior from the product of posteriors of each modality. One is to learn a joint-learning space using a neural network, and another is to obtain a concatenation of the original features of each modality. Meanwhile, the single-modal encoders play the roles of data normalization, denoising and imputation of the input modalities. scMVP has the same architecture as scMVAE, but scMVP’s joint-modality encoder only uses a neural network to learn the joint-learning space of the scRNA-seq and scATAC-seq modalities. scMVP improves joint embedding by connecting its two decoders through a cell-type-guided attention module that captures the correlation between the two modalities. Furthermore, scMVP’s single-modal encoders are connected to the joint embedding with two extra modules that ensure clustering consistency.
The assumption that all data sources are equally valuable and follow the same distribution is not always true. For example, in scATAC-seq experiments, the amount of data per cell is typically lower and more variable than in scRNA-Seq experiments from the same cell [100]. Directly combining data using neural networks from such imbalanced modalities can lead to overfitting [100]. SAILERX was introduced to address these challenges by only learning scATAC-seq data with a VAE, and for scRNA-seq, using pre-trained scRNA-seq embeddings. SAILERX also enforces similarity between the latent space of both modalities through regularization, which preserves local cell structure across modalities. The goal is to avoid overfitting and allow hybrid integration of joint profiling data with single-modality data, similar to Cobolt.
Combining RNA and protein data to create a unified representation of cell state is challenging due to technical biases and inherent noise in each data modality. In particular, protein data presents a unique challenge due to background noise from ambient or nonspecifically bound antibodies [87]. The VAE-based totalVI model provides a solution for integrating scRNA-seq and protein data while correcting for protein background noise. To achieve this, totalVI takes matrices of scRNA-seq and protein count data as input, along with categorical covariates such as experimental batch or donor information. The encoder then generates a joint latent representation of both modalities, which helps to control modality-specific noise properties and batch effects. Finally, the decoder estimates the parameters of the underlying distributions of both modalities from the joint latent representation while correcting for protein background noise.
Integrating unpaired multi-omics data can be difficult since each modality has unique feature spaces. To address this issue, GLUE was developed to integrate unpaired multi-omics data via graph-guided embeddings. GLUE uses a separate VAE to model each data modality. To merge modality-specific feature spaces, GLUE constructed a knowledge-based graph using cross-modality regulatory interactions, with vertices representing the features of different omics data modalities and edges representing regulatory interactions. A variational graph AE (VGAE) is adopted to create graph embeddings from the knowledge-based graph. The VGAE is then connected with the modality-specific decoders to help integrate these unpaired multi-omics data.
The above-discussed MDL techniques were not designed specifically for online integration tasks [131]. As a solution, SCALEX was developed to continuously integrate new single-cell multi-omics data without recalculating all previous integrations [131]. The VAE-based SCALEX model aims to create a generalized encoder for data projection without retraining, and it achieves this through three key design elements.
AE: Several studies have adopted AEs for multimodal data integration. One such approach is the k-coupled AE, which uses a multi-agent AE approach [71], where each AE agent learns a modality separately. These agent AEs are coupled by an overall cost function that measures the dissimilarity among the representations learned by the agent AEs. All agent AEs minimize this overall cost function during training to produce a better integrated latent representation. The k-coupled AE is useful for cross-modal translation. Another study, BABEL, uses two AE-based architecture to generate cross-modal translation. Two separate encoders learn two modalities separately and project them into a shared latent space, which is learned by two decoders to create the original modalities. Another AE-based model, scMDC, focuses on accurately clustering single-cell data. It uses a multimodal AE to learn a joint latent representation from the concatenated modalities of scRNA-seq and (CITE-seq or scATAC-sesq). Unlike the previous studies focusing on sequencing data only, crossmodal-AE integrates image and sequencing data. The approach of the crossmodal-AE is similar to BABEL. However, the AEs are customized according to specific modalities in crossmodal-AE. For example, to project single-cell image data, scRNA-seq, scATAC-seq and single-cell Hi-C data into the shared latent space, AEs corresponding to CNN, FCNN and GNN are designed, respectively. STACI extended the work of crossmodal-AE, which used AE with over-parameterization. Over-parameterization means extending the size of hidden layers to be greater than the input feature space. The model used separate decoders to obtain each modality from the latent embedding.
Encoders: The encoder-based SMILE model integrates single-cell multi-omics data using contrastive learning. There are two variants of SMILE: pSMILE and mpSMILE. The former has two encoders corresponding to two input modalities. One-layer multilayer perceptrons (MLPs) are applied to each encoder to reduce the dimensionality of its output. The outputs are then subjected to noise contrastive estimation to maximize mutual information in the shared latent space. mpSMILE uses two encoders as well but has one encoder duplicated to give more weight to the corresponding data modality. Doing so can improve discriminative representations [107, 138]. In another study, scJoint presents an encoder-based transfer learning method that learns a joint embedding space from scRNA-seq and scATAC-seq data. Since unpaired multi-omics data integration is challenging due to distinct feature spaces, scJoint uses two loss functions to identify orthogonal latent features and maximize the alignment of different modalities. In addition to a cross-entropy loss for cell-type prediction, the joint embedding space can be used by a k-nearest neighbor approach to transfer cell labels from scRNA-seq cells to ATAC-seq cells, further improving the joint embedding space.
GAN: MAGAN is a model that addresses the challenge of integrating unpaired data using a manifold alignment strategy. MAGAN uses a dual GAN framework consisting of two GANs, which align manifolds from two modalities. One GAN is responsible for creating a mapping from the first modality to the second modality, while the other GAN learns the mapping from the second to the first modality.
FCNN: Existing methods for integrating unpaired data into a single latent space primarily focus on cells that form clusters rather than continuous cells that follow trajectories [8, 9, 139]. scDART addresses this limitation by utilizing a FCNN to project data into a shared latent space while preserving cell trajectories. The scDART model consists of two parts: the gene activity function, which generates a ‘pseudo-scRNA-seq’ count matrix from scATAC-seq data; and the projection module, which takes both the original scRNA-seq data and the ‘pseudo-scRNA-seq’ data as input to produce a shared latent space. The diffusion/random-walk-based distances between cells along the trajectory manifold in the original and the latent space are considered in the overall loss function to preserve the cell trajectory structure.
Graph neural network (GNN): Integration studies like BABEL, scMM and Cobolt often treat each cell as an independent input, which can overlook important high-order interactions between cells and modalities. Such interactions are critical for effective learning from single-cell data’s high-dimensional and sparse cell features. scMoGNN leverages GNNs to integrate single-cell modalities while preserving high-order structural information to address this limitation. Specifically, scMoGNN first constructs a cell-feature graph from a given modality and applies a graph CNN (GCNN) to obtain latent embeddings of cells. These cell embeddings are then fed into a task-specific head for downstream tasks such as modality prediction, matching and joint embedding.
Heterogenous model: The SMILE model, mentioned earlier, requires cell anchors and can only be applied when corresponding cells are known across multiple modalities. sciCAN was developed to address this limitation. The sciCAN model consists of representation learning using an encoder and modality alignment using a GAN. The encoder employs noise contrastive estimation as its loss function to learn a joint low-dimensional representation. The GAN component includes two discriminators: one identifies source domains represented by a latent representation, and the other generates one data modality from the other, such as chromatin accessibility data from gene expression data. sciCAN differentiates itself from similar models like single-cell GAN (scGAN) [140] and AD-AE by using one additional cycle-consistent adversarial network, which introduces cycle-consistent loss to learn the connections between two modalities.
With the emergence of Atlas-level scRNA-seq datasets, there is a growing need for integration techniques that can handle large numbers of cell populations and are computationally scalable. In response, the Portal model was developed. To align single-cell datasets, the Portal model employs a domain translation network of two encoders, generators and discriminators. The encoders learn latent embeddings for input modalities, the generators generate one modality data from the latent embedding of the other modality and the discriminators identify non-aligned data points to improve network training further.
Key tasks
Single-cell data integration can be divided into three key tasks: modality prediction, matching and joint embedding [98]. In the modality prediction task, one modality is predicted given the other modality. Among the surveyed papers, MAGAN and k-coupled AE both perform modality prediction tasks, as shown in Table 3. MAGAN predicts scRNA-seq from CyTOF or vice versa, while k-coupled AE predicts neuron morphological data from scRNA-seq. No studies have focused solely on modality matching. SCIM, Cobolt, scMVAE, SAILERX, GLUE, scMVP, Portal, MIRA and SCALEX all perform joint embedding tasks exclusively. In contrast, scMM performs joint embedding and modality prediction simultaneously. First, scRNA-seq and scATAC-seq modalities are jointly embedded, then used by decoders to predict one modality given another. Similarly, BABEL, scJoint, sciCAN, totalVI, crossmodal-AE, STACI and scDART perform both joint embedding and modality prediction tasks on various single-cell omics data modalities. SMILE performs joint embedding and modality matching tasks with scRNA-seq, scATAC-seq, DNA methylation and Hi-C modalities. scMoGNN performs all three tasks (joint embedding, modality prediction and modality matching) with mRNA, scATAC-seq and ADT modalities.
Fusion methods
Methods for integrating data from multiple modalities in an MDL model architecture are called fusion methods. Three types of fusion strategies have been identified: early fusion, intermediate fusion and late fusion [52] (as shown in Figure 2). The fusion strategies of the various studies are detailed as follows (Table 4).
Figure 2.
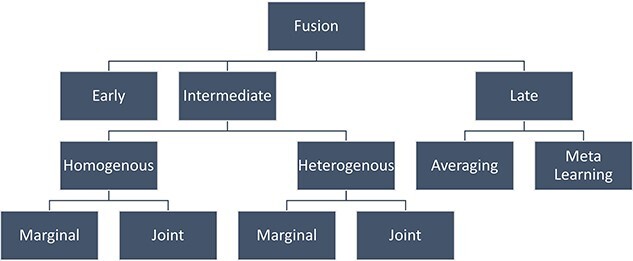
Classification of fusion strategy.
Table 4.
Classification of single-cell multimodal studies according to fusion strategy
| Fusion | Category 1 | Category 2 | Papers |
|---|---|---|---|
| Early | scMVAE (with direct learning), totalVI and SCALEX, STACI | ||
| Intermediate | Homogeneous | Marginal | MAGAN, SMILE, scMoGNN (with modality prediction and modality matching tasks) and GLUE |
| Joint | SCIM, scMM, Cobolt, BABEL, scMVAE, scMoGNN (with joint embedding task), scMVP, scJoint, scDART and Portal | ||
| Heterogeneous | Marginal | – | |
| Joint | Crossmodal-AE, SAILERX and sciCAN | ||
| Late | k-Coupled AE and MIRA |
Early fusion: The strategy of ‘early fusion’ involves concatenating input features from different modalities to serve as the input of a deep learning model (Figure 3A). scMVAE, totalVI, STACI and SCALEX are among the studies that utilized the early fusion strategy. These studies aggregated modalities first and then utilized VAE to learn the joint latent embedding.
Figure 3.
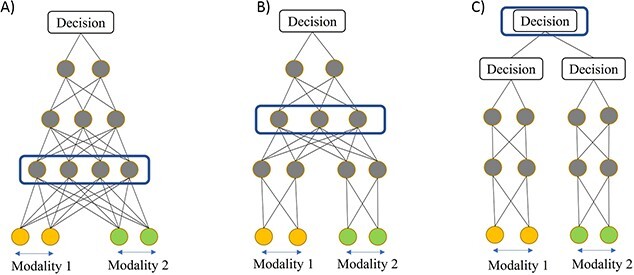
(A) Early fusion strategy. (B) Intermediate fusion strategy. (C) Late fusion strategy. Layers enclosed in blue rounded rectangle are shared between modalities and used to integrate modalities [52].
Intermediate fusion: The majority of studies we surveyed have used intermediate fusion, where the modalities are learned first and fused later inside the MDL model. For instance, in Figure 3B, the marginal representation of modality 1 and 2 are learned first and integrated later inside the neural network layer. Intermediate fusion can be further classified into two categories: homogeneous and heterogeneous. Homogeneous fusion is used when the modalities are learned through the same type of neural network. In contrast, heterogeneous fusion is used when the modalities are learned through different types of neural networks. Moreover, based on representation, both homogeneous and heterogeneous fusion can be divided into marginal and joint types. The marginal representation uses features to represent latent components based on a single modality, while joint representation encodes information from several modalities.
In joint homogeneous fusion, marginal representations are first concatenated, and then a joint representation is learned from that concatenated marginal representation. For example, SCIM assumes joint homogeneous fusion since modalities are learned by two separate VAEs and then imposed into a shared latent space. Joint heterogeneous intermediate fusion is a good strategy for learning informative cross-modality interactions, where different modalities are learned through different types of neural networks concatenated. Then, a joint representation is learned from that concatenated marginal representation using a separate neural network. For example, crossmodal-AE, SAILERX and sciCAN follow the joint heterogeneous fusion strategy.
The marginal homogeneous fusion strategy involves using the same type of neural network to learn marginal feature representation from different modalities, which are later merged into the decision function. For example, SMILE adopted this strategy by using two separate encoders to learn two modalities and create two latent embeddings, followed by two MLPs that learned from both embeddings to minimize the model’s loss.
Late fusion: Late fusion integrates the decisions of separate models to make a final decision (Figure 3C). k-Coupled AE and MIRA are examples of models that use the late fusion strategy. In k-coupled AE, two modalities are learned through two separate coupled AE, and the latent representations are later aggregated. In MIRA, two modalities are learned through two separate VAEs, and the latent representations are combined later to make the final decision.
Downstream analysis
The integration of single-cell modalities using MDL provides great assistance for downstream analysis. Following integration, the MDL models are assessed through downstream analysis and utilized for such purposes. Commonly practiced downstream analysis includes cell type discovery, differential expression analysis (DE), cell trajectory inference, cell matching and cis-regulatory analysis, as detailed below (Table 3).
Cell type discovery: The goal of cell type discovery is to identify the different types of cells present within a sample. Most tools performed cell type discovery to validate their results and illustrate their usage. For example, SCIM recovered T cells by integrating scRNA and CyTOF modalities. scJoint found 19 cell types common between scRNA-seq and scATAC-seq data. However, the detected cell types often vary across the studies. For instance, in the crossmodal-AE study, scRNA-seq data from human peripheral blood mononuclear cells (PBMCs) [93] was analyzed and clustered, revealing the presence of four types of T cells. In comparison, SAILERX clustered the PBMC 10k dataset [2] and identified 29 cell types. Similarly, scMM performed clustering on the same PBMC dataset [2] and identified 54 cell types. Cell type discovery accuracy is often validated by comparing the consistency of the detected cell types with the cell labels annotated in the original data. For example, scMM compared their discovered cell types with the cell-type annotation of PBMC dataset [2]. Some studies such as SCALEX validated cell type discovery by assessing through quantitative clustering metrics such as ARI (adjusted Rand index), NMI (normalized mutual information) and silhouette scores. Crossmodal-AE performed protein immunofluorescence staining to validate the discovered cell types. In this experiment, they selected two genes, CORO1A and RPL10A, that were predicted to be strongly upregulated in specific subpopulations of naive T cells with distinct patterns of chromatin density. They then analyzed immunofluorescence staining data of the two proteins along with the chromatin images. They demonstrated that the tool effectively aligns gene expression with the image features, allowing for the characterization of distinct subpopulations of naive T cells.
DE: The process of DE involves identifying genes that are expressed differently across distinct cell types. This analysis provides insights into how gene expression changes in response to various biological conditions [141]. The reviewed studies utilized DE analysis on several single-cell multi-omics datasets to investigate alterations in gene expression. For example, in a study conducted by Cobolt, DE analysis showed distinct expression levels of Adarb2 and Sox6 genes in scRNA-seq and scATAC-seq clusters, which are known markers distinguishing between CGE and Pvalb clusters. The scDART, SCALEX, STACI and SMILE also performed DE on their data and made discoveries supported by the literature. Most studies validated their DE analysis results utilizing the existing evidence. For instance, scDART compared their DE analysis findings with existing evidence [51]. On the other hand, totalVI performed Welch’s t-test and Wilcoxon rank-sum test to validate their DE analysis findings [87].
Trajectory inference: Trajectory inference aims to identify the progression of a cellular dynamic process and organize cells based on their movement through the process. scMVP, sciCAN, scDART and MIRA all performed trajectory inference. For example, scMVP conducted trajectory inference analysis on growing bulge cells of the SHARE-seq mouse skin dataset and identified two paths from αhigh CD34+ bulge to new bulge cells. sciCAN conducted co-trajectory analysis to investigate the hematopoietic hierarchy. The scDART algorithm was evaluated by trajectory inference analysis on a neonatal mouse brain cortex dataset. Moreover, MIRA investigated hair follicle maintenance and differentiation, revealed the hierarchy of different follicle lineages and recreated the true layout of the follicle. All studies validated the trajectory inference results by comparing with the existing evidence [3].
Cell matching: Cell matching has been used as an assessment technique to evaluate the effectiveness of joint latent embedding. For example, SCIM employed this strategy to match cells between scRNA and CyTOF modalities in a melanoma tumor sample. Crossmodal-AE employed the cell-matching strategy on the human lung adenocarcinoma dataset [92] to match samples between RNA-seq and ATAC-seq modalities. In addition, scDART evaluated the cell matching capability using the mouse neonatal brain cortex dataset [51]. To evaluate cell matching accuracy, several metrics such as k-nearest neighbors’ accuracy, neighborhood overlapping score and cosine similarity score were utilized. For instance, scDART utilized neighborhood overlapping and cosine similarity scores to evaluate their cell matching accuracy and achieved the neighborhood overlapping scores of 0.6 and the cosine similarity scores of 0.712 in the mouse neonatal brain cortex dataset [51].
cis-Regulatory analysis: cis-Regulatory analysis studies various cis-acting DNA sequences that modulate gene transcription. It includes identifying distal and proximal gene regulatory regions such as enhancers and promoters, transcription factor binding sites and their binding patterns called motifs in regulatory regions, as well as the grammars orchestrated by these binding elements and motifs. Several tools, including scMM, SAILERX, MIRA, GLUE and scMVP, have performed cis-regulatory analysis as a downstream analysis of multi-omics data integration. For example, scMM identified enriched regulatory motifs in genes and peaks associated with latent dimensions, while SAILERX determined the top motifs that were most enriched in individual cell types. MIRA used topic modeling of cell states and the regulatory-potential modeling of individual gene loci to identify enriched motifs, while GLUE identified distal gene regulatory regions based on the cosine similarity between feature embeddings. All studies evaluated cis-regulatory analysis findings by comparing their predictions with the existing evidence. For example, scMVP compared their cis-regulatory analysis findings with a previous study [51] and found a higher enrichment of H3K27ac and H3K4me1 in the translation start site (TSS) distal peaks and H3K4me3 in the TSS proximal regions.
DISCUSSION AND CONCLUSION
Several limitations and challenges must be addressed to effectively integrate single-cell multi-omics data using deep learning models. Firstly, data preprocessing is crucial for efficiently integrating multi-omics data, but defining a unified pipeline for data preprocessing tasks is challenging. Although many deep learning models use one-hot encoding to standardize two data modalities, other preprocessing tasks like gene finding, cell labeling, filtering, scaling, normalization and data formatting are not standardized. As a result, there is a disparity in results, even for the same input data, making it difficult to explain the differences.
Secondly, more data information is always needed. For example, studies such as SMILE and BABEL require paired data as cell anchors that are often not readily available. scJoint requires cell annotations for scRNA-seq data. However, paired information or annotated cell anchors are not present in all datasets. Although the diagonal integration method addresses this limitation, as no pairing information is required for this type of integration, it is not easy to find common ground to perform the integration among different modalities without using any anchors. Besides, when multiple encoders are involved in the AE-based models, it can be difficult to train them at once without linking information from various modalities. In addition, as the data become more complex, it becomes harder to find a shared embedding space. Moreover, as different studies used different datasets, it is difficult to compare the methodologies fairly. Furthermore, MDL models need a large amount of data for training and testing. For example, scMM faces limitations in generating cell populations due to insufficient large-scale atlas data for training. Cobolt needs a large amount of single-modality data for cross-modality prediction. An imbalance in data for the input modalities may create overfitting issues.
The third challenge in integrating multiple modalities is the lack of interpretability of modality-specific information from the joint latent embedding. To address this challenge, scMM created pseudocells and used Spearman correlation to associate each latent dimension with the features of each modality. However, interpreting the latent dimensions limits the model to specific data, and predicting cell populations not included in the training data could be challenging.
Another challenge in DL methods is the selection of appropriate hyperparameters. These models require extensive tuning of various parameters, such as the number of hidden nodes, layers, training epochs and batch size, among others, which can be time consuming and computationally intensive. While some models have specific parameters to tune, such as scJoint’s cosine similarity loss, which requires tuning the fraction of data pairs to achieve optimal results, other models require more general tuning. Furthermore, DL models suffer from a lack of generalizability, and customizations in the architecture are often required for new modalities of data. For instance, crossmodal-AE requires changing the model architecture for each data modality. As a result, there are still numerous areas to investigate and significant potential for growth in the field.
Our study shows that MDL is gaining popularity in single-cell multi-omics data integration. As the technology continues to improve and data increases in different modalities (e.g. single-cell imaging), the model performance has room for improvement in single-cell multi-omics data integration. Although deep learning methods demonstrate their influence in single-cell multi-omics integration, no benchmark pipeline has been defined for dataset selection, data preprocessing, architecture design, etc. So, future research can be conducted to obtain a benchmark for specific tasks. The current studies not only perform single-cell multi-omics data integration, but also explore various downstream analyses. However, the downstream analysis tasks are validated mostly with the existing evidence. To show the power of the integration methods and generate new biological knowledge, future research can be conducted to validate the downstream analysis findings by performing experiments such as multiplex FISH [142].
MDL is a crucial area of research that aims to enhance our understanding of single-cell data and potentially reveal novel biological insights [143]. With a growing body of published research, new and innovative MDL architectures are expected to emerge. This review does not provide specific recommendations for architecture design, as it depends on the problem being addressed. However, the insights presented in this review can serve as a valuable reference for future research and help advance the field more coherently.
Key Points
Surveyed 22 recent studies on single-cell MDL
Analyzed these studies from five aspects
No unified pipeline for data processing in MDL studies
Interpreting of modality-specific information is still challenging
Author Biographies
Tasbiraha Athayaa is a graduate student from Department of Computer Science, University of Central Florida. She mainly works on multi-omics data integration.
Rony Chowdhury Ripana is a graduate student from the Department of Computer Science, University of Central Florida. He mainly works on single-cell data analysis.
Haiyan Hu is a professor from the Department of Computer Science, University of Central Florida. She works on miRNAs, epigenomics and gene transcriptional regulation.
Xiaoman Li is a professor from Burnett School of Biomedical Science, University of Central Florida. He works on chromatin interactions and metagenomics.
Contributor Information
Tasbiraha Athaya, Department of Computer Science, University of Central Florida, Orlando, Florida, United States of America.
Rony Chowdhury Ripan, Department of Computer Science, University of Central Florida, Orlando, Florida, United States of America.
Xiaoman Li, Burnett School of Biomedical Science, College of Medicine, University of Central Florida, Orlando, Florida, United States of America.
Haiyan Hu, Department of Computer Science, University of Central Florida, Orlando, Florida, United States of America.
FUNDING
The National Science Foundation (grants 2015838, 1661414, and 2120907).
AUTHORS’ CONTRIBUTION
H.H. and X.L. conceived the idea. A.T. and R.R.C. implemented the idea and generated results. A.T., R. R.C., X.L. and H.H. analyzed the results and wrote the manuscript. All authors reviewed the manuscript.
DATA AVAILABILITY
All data underlying this article are available in the articleand its tables.
REFERENCES
- 1. Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet 2019;20:631–56. [DOI] [PubMed] [Google Scholar]
- 2. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma S, Zhang B, LaFave LM, et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 2020;183:1103–1116.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramachandram D, Taylor GW. Deep multimodal learning: a survey on recent advances and trends. IEEE Signal Process Mag 2017;34:96–108. [Google Scholar]
- 5. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu B, Chen S, Bai Y, et al. Robust single-cell matching and multimodal analysis using shared and distinct features. Nat Methods 2023;20:304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Welch JD, Kozareva V, Ferreira A, et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 2019;177:1873–1887.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duren Z, Chen X, Zamanighomi M, et al. Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc Natl Acad Sci 2018;115:7723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adossa N, Khan S, Rytkönen KT, Elo LL. Computational strategies for single-cell multi-omics integration. Comput Struct Biotechnol J 2021;19:2588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 12. Kang M, Ko E, Mersha TB. A roadmap for multi-omics data integration using deep learning. Brief Bioinform 2022;23:bbab454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song M, Greenbaum J, Luttrell J, et al. A review of integrative imputation for multi-omics datasets. Front Genet 2020;11:570255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stanojevic S, Li Y, Ristivojevic A, Garmire LX. Computational methods for single-cell multi-omics integration and alignment. Genomics Proteomics Bioinformatics 2022;20:836–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Subedi P, Moertl S, Azimzadeh O. Omics in radiation biology: surprised but not disappointed. Radiation 2022;2:124–9. [Google Scholar]
- 16. Mallory XF, Edrisi M, Navin N, Nakhleh L. Methods for copy number aberration detection from single-cell DNA-sequencing data. Genome Biol 2020;21:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luquette LJ, Bohrson CL, Sherman MA, Park PJ. Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat Commun 2019;10:3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woodworth MB, Girskis KM, Walsh CA. Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat Rev Genet 2017;18:230–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kester L, van Oudenaarden A. Single-cell transcriptomics meets lineage tracing. Cell Stem Cell 2018;23:166–79. [DOI] [PubMed] [Google Scholar]
- 20. Evrony GD, Hinch AG, Luo C. Applications of single-cell DNA sequencing. Annu Rev Genomics Hum Genet 2021;22:171–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kashima Y, Sakamoto Y, Kaneko K, et al. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med 2020;52:1419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J, Adhav R, Xu X. Current progresses of single cell DNA sequencing in breast cancer research. Int J Biol Sci 2017;13:949–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang L, Ma F, Chapman A, et al. Single-cell whole-genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet 2015;16:79–102. [DOI] [PubMed] [Google Scholar]
- 24. Adil A, Kumar V, Jan AT, Asger M. Single-cell transcriptomics: current methods and challenges in data acquisition and analysis. Front Neurosci 2021;15:591122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen G, Ning B, Shi T. Single-cell RNA-Seq technologies and related computational data analysis. Front Genet 2019;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kulkarni A, Anderson AG, Merullo DP, Konopka G. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol 2019;58:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 2018;50:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007;128:669–81. [DOI] [PubMed] [Google Scholar]
- 29. Stein CM, Weiskirchen R, Damm F, Strzelecka PM. Single-cell omics: overview, analysis, and application in biomedical science. J Cell Biochem 2021;122:1571–8. [DOI] [PubMed] [Google Scholar]
- 30. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature 2019;571:489–99. [DOI] [PubMed] [Google Scholar]
- 31. Smallwood SA, Lee HJ, Angermueller C, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 2014;11:817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo H, Zhu P, Guo F, et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc 2015;10:645–59. [DOI] [PubMed] [Google Scholar]
- 33. Gu H, Raman AT, Wang X, et al. Smart-RRBS for single-cell methylome and transcriptome analysis. Nat Protoc 2021;16:4004–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clark SJ, Lee HJ, Smallwood SA, et al. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol 2016;17:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu M, Hon GC, Szulwach KE, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012;149:1368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Booth MJ, Branco MR, Ficz G, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012;336:934–7. [DOI] [PubMed] [Google Scholar]
- 37. Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011;473:398–402. [DOI] [PubMed] [Google Scholar]
- 38. Rotem A, Ram O, Shoresh N, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 2015;33:1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Buenrostro JD, Wu B, Litzenburger UM, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015;523:486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin W, Tang Q, Wan M, et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015;528:142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vistain LF, Tay S. Single-cell proteomics. Trends Biochem Sci 2021;46:661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu C, Preissl S, Ren B. Single-cell multimodal omics: the power of many. Nat Methods 2020;17:11–4. [DOI] [PubMed] [Google Scholar]
- 43. Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med 2020;52:1428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Angermueller C, Clark SJ, Lee HJ, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 2016;13:229–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu Y, Huang K, An Q, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 2016;17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hou Y, Guo H, Cao C, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 2016;26:304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dixit A, Parnas O, Li B, et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 2016;167:1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adamson B, Norman TM, Jost M, et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 2016;167:1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaitin DA, Weiner A, Yofe I, et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-Seq. Cell 2016;167:1883–1896.e15. [DOI] [PubMed] [Google Scholar]
- 50. Zhu C, Yu M, Huang H, et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol 2019;26:1063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen S, Lake BB, Zhang K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol 2019;37:1452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stahlschmidt SR, Ulfenborg B, Synnergren J. Multimodal deep learning for biomedical data fusion: a review. Brief Bioinform 2022;23:bbab569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bengio Y, Courville A, Vincent P. Representation learning: a review and new perspectives. IEEE Trans Pattern Anal Mach Intell 2013;35:1798–828. [DOI] [PubMed] [Google Scholar]
- 54. Park C, Ha J, Park S. Prediction of Alzheimer’s disease based on deep neural network by integrating gene expression and DNA methylation dataset. Expert Syst Appl 2020;140:112873. [Google Scholar]
- 55. Huang Z, Zhan X, Xiang S, et al. SALMON: survival analysis learning with multi-omics neural networks on breast cancer. Front Genet 2019;10:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Deng Y, Xu X, Qiu Y, et al. A multimodal deep learning framework for predicting drug–drug interaction events. Bioinformatics 2020;36:4316–22. [DOI] [PubMed] [Google Scholar]
- 57. Huang S-C, Pareek A, Zamanian R, et al. Multimodal fusion with deep neural networks for leveraging CT imaging and electronic health record: a case-study in pulmonary embolism detection. Sci Rep 2020;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chang Y, Park H, Yang H-J, et al. Cancer drug response profile scan (CDRscan): a deep learning model that predicts drug effectiveness from cancer genomic signature. Sci Rep 2018;8:8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mohaiminul Islam M, Huang S, Ajwad R, et al. An integrative deep learning framework for classifying molecular subtypes of breast cancer. Comput Struct Biotechnol J 2020;18:2185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spasov SE, Passamonti L, Duggento A, et al. A multi-modal convolutional neural network framework for the prediction of Alzheimer’s disease. Annu Int Conf IEEE Eng Med Biol Soc 2018;2018:1271–4. [DOI] [PubMed] [Google Scholar]
- 61. Bichindaritz I, Liu G, Bartlett C. Integrative survival analysis of breast cancer with gene expression and DNA methylation data. Bioinformatics 2021;37:2601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee G, Nho K, Kang B, et al. Predicting Alzheimer’s disease progression using multi-modal deep learning approach. Sci Rep 2019;9:1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guo L-Y, Wu A-H, Wang Y, et al. Deep learning-based ovarian cancer subtypes identification using multi-omics data. BioData Min 2020;13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ronen J, Hayat S, Akalin A. Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci Alliance 2019;2:e201900517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Suk H-I, Lee S-W, Shen D. Hierarchical feature representation and multimodal fusion with deep learning for AD/MCI diagnosis. Neuroimage 2014;101:569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang D, Yin C, Zeng J, et al. Combining structured and unstructured data for predictive models: a deep learning approach. BMC Med Inform Decis Mak 2020;20:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lin Y, Zhang W, Cao H, et al. Classifying breast cancer subtypes using deep neural networks based on multi-omics data. Genes 2020;11:888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu Y, Begoli E, McCord RP. sciCAN: single-cell chromatin accessibility and gene expression data integration via cycle-consistent adversarial network. NPJ Syst Biol Appl 2022;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Amodio M, Krishnaswamy S. MAGAN: aligning biological manifolds. In: Dy J, Krause A (Ed). Proceedings of the 35th International Conference on Machine Learning. Stockholm, Sweden: Proceedings of Machine Learning Research, 2018, 215–23.
- 70. Velten L, Haas SF, Raffel S, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 2017;19:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gala R, Gouwens N, Yao Z, et al. A coupled autoencoder approach for multi-modal analysis of cell types. In: Wallach H, Larochelle H, Beygelzimer A, d'Alché-Buc F, Fox E, and Garnett R (Ed). Advances in Neural Information Processing Systems Vancouver, CANADA. Proceedings of the 33rd International Conference on Neural Information Processing Systems. 2019, 9267–76.
- 72. Cadwell CR, Palasantza A, Jiang X, et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat Biotechnol 2016;34:199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stark SG, Ficek J, Locatello F, et al. SCIM: universal single-cell matching with unpaired feature sets. Bioinformatics 2020;36:i919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Irmisch A, Bonilla X, Chevrier S, et al. The tumor profiler study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell 2021;39:288–93. [DOI] [PubMed] [Google Scholar]
- 75. Oetjen KA, Lindblad KE, Goswami M, et al. Human bone marrow assessment by single-cell RNA sequencing, mass cytometry, and flow cytometry. JCI Insight 2018;3:e124928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Minoura K, Abe K, Nam H, et al. A mixture-of-experts deep generative model for integrated analysis of single-cell multiomics data. Cell Rep Methods 2021;1:100071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gong B, Zhou Y, Purdom E. Cobolt: integrative analysis of multimodal single-cell sequencing data. Genome Biol 2021;22:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yao Z, Liu H, Xie F, et al. A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 2021;598:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.[dataset] 2021, Pbmcs from human (no cell sorting, chromium next gem), single cell multiome atac + gene expression dataset by cell ranger arc 2.0.0, 10x Genomics, https://www.10xgenomics.com/resources/datasets/pbmc-from-a-healthy-donor-no-cell-sorting-10-k-1-standard-2-0-0 and https://www.10xgenomics.com/resources/datasets/pbmc-from-a-healthy-donor-no-cell-sorting-3-k-1-standard-2-0-0.
- 80. [dataset] 2021, Pbmcs from human (multiome v1.0, chromium x), single cell multiome atac + gene expression dataset by cell ranger arc 2.0.0, 10x Genomics, https://www.10xgenomics.com/resources/datasets/10-k-human-pbm-cs-multiome-v-1-0-chromium-x-1-standard-2-0-0.
- 81.[dataset] 2021, Pbmcs from human (3′ ht v3.1, chromium x), single cell gene expression dataset by cell ranger 6.1.0, 10x Genomics, https://www.10xgenomics.com/resources/datasets/20-k-human-pbm-cs-3-ht-v-3-1-chromium-x-3-1-high-6-1-0.
- 82.[dataset] 2021, Pbmcs from human (atac v1.1, chromium x), single cell atac dataset by cell ranger atac 2.0.0, 10x Genomics, https://www.10xgenomics.com/resources/datasets/10-k-human-pbm-cs-atac-v-1-1-chromium-x-1-1-standard-2-0-0.
- 83. Wu KE, Yost KE, Chang HY, Zou J. BABEL enables cross-modality translation between multiomic profiles at single-cell resolution. Proc Natl Acad Sci 2021;118:e2023070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.[dataset] 2020, PBMC from a healthy donor - granulocytes removed through cell sorting (10k), Single Cell Multiome ATAC + Gene Exp. Dataset by Cell Ranger ARC 1.0.0, 10x Genomics, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160148.
- 85. Zuo C, Chen L. Deep-joint-learning analysis model of single cell transcriptome and open chromatin accessibility data. Brief Bioinform 2021;22:bbaa287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu L, Liu C, Quintero A, et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun 2019;10:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gayoso A, Steier Z, Lopez R, et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat Methods 2021;18:272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.[dataset] 2018, 10k PBMCs from a healthy donor—gene expression and cell surface protein, 10x Genomics, https://www.10xgenomics.com/resources/datasets/10-k-pbm-cs-from-a-healthy-donor-gene-expression-and-cell-surface-protein-3-standard-3-0-0.
- 89.[dataset] 2019, 5k Peripheral blood mononuclear cells (PBMCs) from a healthy donor with cell surface proteins (v3 chemistry), 10x Genomics, https://www.10xgenomics.com/resources/datasets/5-k-peripheral-blood-mononuclear-cells-pbm-cs-from-a-healthy-donor-with-cell-surface-proteins-v-3-chemistry-3-1-standard-3-1-0.
- 90.[dataset] 2018, 10k Cells from a MALT tumor—gene expression and cell surface protein, 10x Genomics, https://www.10xgenomics.com/resources/datasets/10-k-cells-from-a-malt-tumor-gene-expression-and-cell-surface-protein-3-standard-3-0-0.
- 91. Yang KD, Belyaeva A, Venkatachalapathy S, et al. Multi-domain translation between single-cell imaging and sequencing data using autoencoders. Nat Commun 2021;12:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cao J, Cusanovich DA, Ramani V, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018;361:1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Satpathy AT, Granja JM, Yost KE, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 2019;37:925–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.[dataset] 2017, 8k PBMCs from a Healthy Donor, Single Cell Gene Expression Dataset by Cell Ranger 2.1.0, 10x Genomics, https://www.10xgenomics.com/resources/datasets/8-k-pbm-cs-from-a-healthy-donor-2-standard-2-1-0.
- 95. Xu Y, Das P, McCord RP. SMILE: mutual information learning for integration of single-cell omics data. Bioinformatics 2022;38:476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee D-S, Luo C, Zhou J, et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat Methods 2019;16:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhu C, Zhang Y, Li YE, et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods 2021;18:283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wen H, Ding J, Jin W, et al. Graph neural networks for multimodal single-cell data integration. In: Doerr A (Ed). Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining. London: Springer Nature, 2022, 4153–63.
- 99. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The Molecular Signatures Database hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cao Y, Fu L, Wu J, et al. Integrated analysis of multimodal single-cell data with structural similarity. Nucleic Acids Res 2022;50:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cao Z-J, Gao G. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nat Biotechnol 2022;40:1458–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. [dataset] 2018, 10k Peripheral blood mononuclear cells (PBMCs) from a healthy donor, Single Cell ATAC Dataset by Cell Ranger 1.0.1, 10x Genomics, https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/1.0.0/pbmc_granulocyte_sorted_10.
- 103. Muto Y, Wilson PC, Ledru N, et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun 2021;12:2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Luo C, Keown CL, Kurihara L, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017;357:600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Li G, Fu S, Wang S, et al. A deep generative model for multi-view profiling of single-cell RNA-seq and ATAC-seq data. Genome Biol 2022;23:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. 10x Genomics, Datasets. https://www.10xgenomics.com/resources/datasets (24 February 2023, date last accessed).
- 107. Lin Y, Wu T-Y, Wan S, et al. scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning. Nat Biotechnol 2022;40:703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schaum N, Karkanias J, Neff NF, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cusanovich DA, Hill AJ, Aghamirzaie D, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 2018;174:1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Granja JM, Klemm S, McGinnis LM, et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 2019;37:1458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cao J, O’Day DR, Pliner HA, et al. A human cell atlas of fetal gene expression. Science 2020;370:eaba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Domcke S, Hill AJ, Daza RM, et al. A human cell atlas of fetal chromatin accessibility. Science 2020;370:eaba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mimitou EP, Lareau CA, Chen KY, et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat Biotechnol 2021;39:1246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017;14:865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. 10x Genomics, Single Cell Multiome ATAC + Gene Expression. https://www.10xgenomics.com/resources/datasets (24 February 2023, date last accessed).
- 116. Wang A, Chiou J, Poirion OB, et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. Elife 2020;9:e62522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Miao Z, Balzer MS, Ma Z, et al. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat Commun 2021;12:2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Pierce SE, Granja JM, Greenleaf WJ. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat Commun 2021;12:2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhang Z, Yang C, Zhang X. scDART: integrating unmatched scRNA-seq and scATAC-seq data and learning cross-modality relationship simultaneously. Genome Biol 2022;23:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhu Q, Gao P, Tober J, et al. Developmental trajectory of prehematopoietic stem cell formation from endothelium. Blood 2020;136:845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pellin D, Loperfido M, Baricordi C, et al. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat Commun 2019;10:2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Buenrostro JD, Corces MR, Lareau CA, et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 2018;173:1535–1548.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhao J, Wang G, Ming J, et al. Adversarial domain translation networks for integrating large-scale atlas-level single-cell datasets. Nat Comput Sci 2022;2:317–30. [DOI] [PubMed] [Google Scholar]
- 124. Rosenberg AB, Roco CM, Muscat RA, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018;360:176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Saunders A, Macosko EZ, Wysoker A, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018;174:1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zeisel A, Hochgerner H, Lönnerberg P, et al. Molecular architecture of the mouse nervous system. Cell 2018;174:999–1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.[dataset] 2016, 3k PBMCs from a Healthy Donor, Single Cell Gene Expression Dataset by Cell Ranger 1.1.0, 10x Genomics, https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k.
- 128. Fullard JF, Lee H-C, Voloudakis G, et al. Single-nucleus transcriptome analysis of human brain immune response in patients with severe COVID-19. Genome Med 2021;13:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tran MN, Maynard KR, Spangler A, et al. Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 2021;109:3088–3103.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lynch AW, Theodoris CV, Long HW, et al. MIRA: joint regulatory modeling of multimodal expression and chromatin accessibility in single cells. Nat Methods 2022;19:1097–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Xiong L, Tian K, Li Y, et al. Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space. Nat Commun 2022;13:6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.[dataset] 2018, 10k PBMCs from a Healthy Donor (v3 chemistry), Single Cell Gene Expression Dataset by Cell Ranger 3.0.0., 10x Genomics, https://www.10xgenomics.com/resources/datasets/10-k-pbm-cs-from-a-healthy-donor-v-3-chemistry-3-standard-3-0-0.
- 133. Lin X, Tian T, Wei Z, Hakonarson H. Clustering of single-cell multi-omics data with a multimodal deep learning method. Nat Commun 2022;13:7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. 10x Genomics, Single Cell Gene Expression. https://www.10xgenomics.com/resources/datasets (24 February 2023, date last accessed).
- 135. Zhang X, Wang X, Shivashankar GV, Uhler C. Graph-based autoencoder integrates spatial transcriptomics with chromatin images and identifies joint biomarkers for Alzheimer’s disease. Nat Commun 2022;13:7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Argelaguet R, Cuomo ASE, Stegle O, Marioni JC. Computational principles and challenges in single-cell data integration. Nat Biotechnol 2021;39:1202–15. [DOI] [PubMed] [Google Scholar]
- 137. Shi Y, Siddharth N, Paige B, et al. Variational mixture-of-experts autoencoders for multi-modal deep generative models. Adv Neural Inf Process Syst 2019;32:15718–29. [Google Scholar]
- 138. Jain MS, Polanski K, Conde CD, et al. MultiMAP: dimensionality reduction and integration of multimodal data. Genome Biol 2021;22:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Argelaguet R, Arnol D, Bredikhin D, et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol 2020;21:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bahrami M, Maitra M, Nagy C, et al. Deep feature extraction of single-cell transcriptomes by generative adversarial network. Bioinformatics 2021;37:1345–51. [DOI] [PubMed] [Google Scholar]
- 141. Westwood JT. Chapter 12—Using transcriptomics to study behavior. In: van den Berghe V (Ed). Molecular-Genetic and Statistical Techniques for Behavioral and Neural Research. United Kingdom: BioMed Central Ltd., 2018, 267–88.
- 142. Kearney L. Multiplex-FISH (M-FISH): technique, developments and applications. Cytogenet Genome Res 2006;114:189–98. [DOI] [PubMed] [Google Scholar]
- 143. Liu J, Fan Z, Zhao W, Zhou X. Machine intelligence in single-cell data analysis: advances and new challenges. Front Genet 2021;12:655536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data underlying this article are available in the articleand its tables.