

Abstract
Prediction of driver genes (tumor suppressors and oncogenes) is an essential step in understanding cancer development and discovering potential novel treatments. We recently proposed Moonlight as a bioinformatics framework to predict driver genes and analyze them in a system-biology-oriented manner based on -omics integration. Moonlight uses gene expression as a primary data source and combines it with patterns related to cancer hallmarks and regulatory networks to identify oncogenic mediators. Once the oncogenic mediators are identified, it is important to include extra levels of evidence, called mechanistic indicators, to identify driver genes and to link the observed gene expression changes to the underlying alteration that promotes them. Such a mechanistic indicator could be for example a mutation in the regulatory regions for the candidate gene. Here, we developed new functionalities and released Moonlight2 to provide the user with a mutation-based mechanistic indicator as a second layer of evidence. These functionalities analyze mutations in a cancer cohort to classify them into driver and passenger mutations. Those oncogenic mediators with at least one driver mutation are retained as the final set of driver genes. We applied Moonlight2 to the basal-like breast cancer subtype, lung adenocarcinoma and thyroid carcinoma using data from The Cancer Genome Atlas. For example, in basal-like breast cancer, we found four oncogenes (COPZ2, SF3B4, KRTCAP2 and POLR2J) and nine tumor suppressor genes (KIR2DL4, KIF26B, ARL15, ARHGAP25, EMCN, GMFG, TPK1, NR5A2 and TEK) containing a driver mutation in their promoter region, possibly explaining their deregulation. Moonlight2R is available at https://github.com/ELELAB/Moonlight2R.
Keywords: driver genes, driver mutations, basal-like, breast cancer, oncogenes, tumor suppressors
INTRODUCTION
Cancer is a well-known and widespread disease and can, in many cases, lead to premature death. In 2020, it is estimated that 19 million people were diagnosed with cancer and almost 10 million people died because of cancer [1]. Today, in many (especially, developed) countries, it is the leading cause of premature deaths.
At the molecular level, different hallmarks of cancer have been identified [2–4]. They are related to the deregulation of certain cellular functions, including increased cell proliferation, evasion of cell death, invasion or escape of immune response. Cancer driver genes, which play important roles in connection to cancer hallmarks, are altered due to the accumulation of genomic alterations. They are known as tumor-promoting (oncogenes, OGs) or tumor suppressor genes (TSGs) [5]. Mutations that activate OGs or inactivate TSGs drive tumor progression. Cancer driver genes can vary in cancer (sub)types, making them elusive to discover and annotate in a specific way. Even tumors with the same tissue of origin can be associated with different driver genes, complicating tumor stratification, accurate diagnosis and targeted treatments [6]. In addition, a new group of genes has emerged in the last decade, known as ‘dual role’ driver genes or ‘double agents’. For example, Shen et al. [7] found that out of 12 cancer types, breast cancer had the second highest occurrence of dual role genes such as ARHGEF12, CBFA2T3, CDKN1B, DDB2 and FOXA1. Dual role driver genes can exhibit both oncogenic and tumor suppressor patterns depending on the cellular context [7–9].
Advances in cancer genomics and sequencing provided a multitude of data on profiling cancer samples, including data on gene expression, mutations, methylation, etc. To cite an example, The Cancer Genome Atlas (TCGA) accounts for more than 20,000 adult tumors [10, 11]. Moreover, the Genomic Data Commons has been developed as a portal for deposition and access to different cancer -omics data [12]. These data provide a precious source for the investigation and prediction of driver genes and their classifications.
As stated above, efforts in the discovery of driver genes are important not only for a fundamental understanding of cancer mechanisms. They have applicative interest since they can be investigated as drug targets [13, 14] or they can be used as biomarkers to distinguish subtypes [15], which can increase the precision of prognosis [16]. Moreover, the knowledge of their dual role can help identify the most suitable treatment for a patient.
Many tools have been proposed to identify driver genes [17] but not all of them focus on the classification in TSGs or OGs [17]. In 2020, we contributed to this challenge by developing Moonlight, and its accompanying Bioconductor package, MoonlightR [9] which takes the biological context, the gene function and its regulatory network into account. Moonlight is not solely based on changes in gene expression because this might be a poor indicator for driver genes [18]. The Moonlight framework requires at least an additional layer of evidence to link the changes in expression and regulation to what has been defined as a ‘mechanistic indicator’ [9]. Mechanistic indicators should help to understand the underlying reasons for a driver pattern (named oncogenic mediator) identified by MoonlightR. The evidence used can for example be chromatin accessibility, copy number variations, DNA methylation, and mutations [9]. The integration of these data allows for covering both genetic and epigenetic alterations that explain the changes in gene expression. However, in the original version of MoonlightR the step of definition of mechanistic indicators is left to the user and no specific protocols are provided. To tackle this challenge, streamline the process and provide a proper workflow for the identification of mechanistic indicators we devised Moonlight2R. In details, we provided a solution to the identification of mechanistic indicators based on mutation data, along with its implementation in a set of functions to streamline and automate the analysis. The functions are released within a new version of the package, Moonlight2R (https://github.com/ELELAB/Moonlight2R).
DESIGN AND IMPLEMENTATION
Overview of Moonlight
For the sake of clarity, we will here give a brief overview of Moonlight as originally conceived (Figure 1). The tool is designed to predict driver genes based on differentially expressed genes (DEGs) and additional layers of mechanistic indicators described in the original publication [9]. The first step in the Moonlight pipeline is a functional enrichment analysis (FEA) which determines if any of Moonlight’s 101 cancer-related biological processes are overrepresented among the DEGs. The next step is a gene regulatory network analysis (GRN) which considers the network of genes that the DEGs are a part of through mutual information. Following GRN, the Moonlight pipeline diverges into two modes: a machine-learning approach and an expert-based approach. The next step is an upstream regulatory analysis (URA) which will in both modes calculate the effect of the DEGs on either user-specified biological processes (the expert-based approach) or in all of Moonlight’s 101 biological processes (the machine learning approach). The final step is a pattern recognition analysis (PRA) which identifies the oncogenic mediators and divides them into putative OGs and putative TSGs. In the expert-based approach, this is done using patterns of the effect of DEGs on two biological processes with opposite effects on cancer. In the machine learning approach, the prediction of oncogenic mediators is carried out using a random forest classifier.
Figure 1.
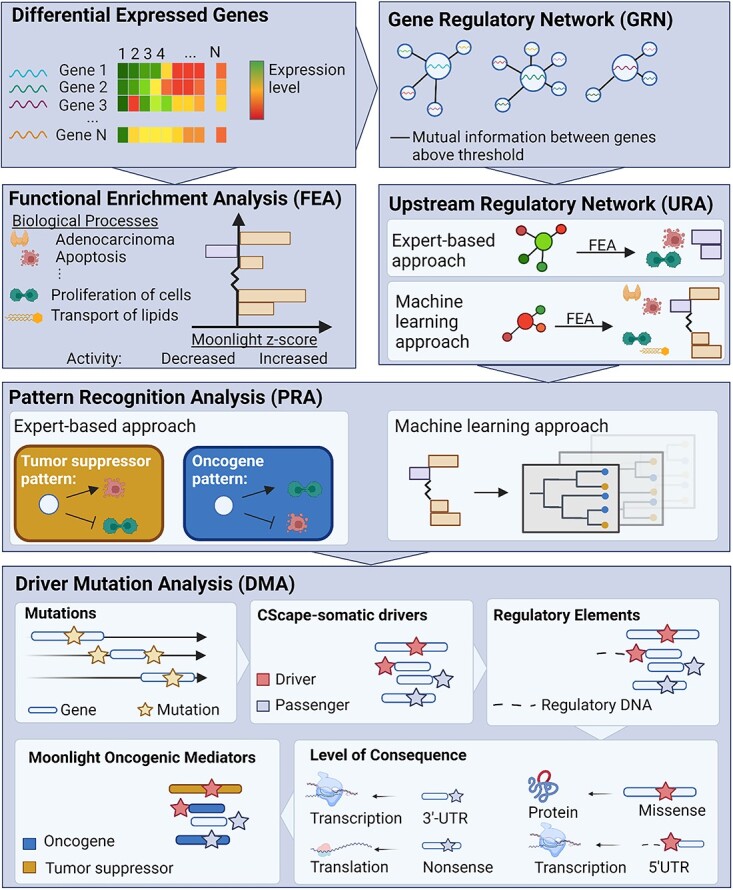
The Moonlight pipeline. The first step in Moonlight is a FEA which determines if any of Moonlight’s 101 cancer-related biological processes are enriched among an input set of differentially expressed genes (DEGs). This is done through Fisher’s exact tests and Moonlight Process Z-scores. The Moonlight Process Z-scores indicate if the activity of the process is increased or decreased based on literature reportings and gene expression levels. The next step, a gene regulatory network analysis (GRN), creates gene networks for each DEG by calculating the mutual information between all pairs of DEGs. Following GRN, the Moonlight pipeline diverges into an expert-based and a machine learning approach. The next step, an upstream regulatory analysis (URA), then evaluates the effect of each DEG on the biological processes through Moonlight Gene Z-scores. If the expert-based approach is selected, the Moonlight Gene Z-scores will only be calculated for chosen biological processes. If the machine learning approach is selected, the Moonlight Gene Z-scores will be calculated for all of Moonlight’s 101 biological processes. In the expert-based approach, pattern recognition analysis (PRA) then identifies the oncogenic mediators which fit an oncogene or tumor suppressor pattern based on the Moonlight Gene Z-scores. The two chosen biological processes must have opposite effects on cancer (growing/blocking), e.g. proliferation of cells and apoptosis. In the machine learning approach, the prediction of oncogenic mediators is done using a random forest classifier. Following Moonlight’s primary layer, a secondary mutational layer is applied through the DMA step which identifies driver mutations. First, the mutations are divided into passengers and drivers by CScape-somatic. Then, regulatory elements from ENCODE are added. The consequence of the type of mutation is annotated to either the protein’s structure, the level of transcription or level of translation. Finally, the data is cross-referenced with the Moonlight driver genes.
Design of the driver mutation analysis
In this paper, we present a new functionality to the Moonlight pipeline which allows for a mechanistic explanation of the predicted oncogenic mediators from Moonlight’s primary layer. The new function is called driver mutation analysis (DMA) and must be used subsequently to the PRA step in the Moonlight pipeline. The function can distinguish between relevant and irrelevant mutations and help filter and prioritize the mutations found in a cohort of patients with the same cancer (sub)type. The function produces a summary of the oncogenic mediators and the assessed mutations, thereby strengthening the evidence for the Moonlight prediction of driver genes. The assessment by the function is visualized in Figure 1 and has several internal steps. It removes all mutations from the Mutation Annotation Format (MAF) file that do not belong to any of the DEGs, and the remaining mutations are then classified as either drivers or passengers with CScape-somatic [19], then additional annotations are added on both mutation and gene levels. The details are provided in the next sections.
The function needs three inputs: the DEGs, the predicted oncogenic mediators, and a MAF file. The DMA function outputs the following: (i) a list of oncogenic mediators with at least one driver mutation, now predicted as driver genes, (ii) a table containing all annotations including CScape-somatic scores found to every DEG on both gene and mutational level, (iii) a summary of the mutations found in the oncogenic mediators, and finally (iv) a table containing the CScape-somatic file as if it was run outside of the DMA function.
CScape-somatic is a driver mutation predictor based on gradient-boosted decision trees. CScape-somatic defines a driver as a disease enabler that includes gain-of-function, loss-of-function, or both simultaneously [19]. CScape-somatic classifies somatic Single Nucleotide Polymorphisms (SNPs) on autosomes and scores each mutation with a number between zero and one, where one represents a highly likely driver mutation and zero represents a passenger mutation. We selected CScape-somatic for multiple reasons. First, it has the advantage of scoring mutations in both coding and non-coding regions of the genome. The possibility to cover driver variants in non-coding regions is central to DMA because the current Moonlight2 framework is still relying on gene expression data and thus the mutations in the non-coding regions are the most interesting to unveil as mechanistic indicators. Second, CScape-somatic provides data on mutations at a low computational cost since the machine-learning model has already scored the entire human genome and we do not need to include training/testing into our pipeline. In addition, CScape-somatic discriminates between germline neutral variants and somatic passenger mutations, which is not common to many available tools. We also considered the fact that the model has been trained on cancer samples and not any other disease, and as such should be more cancer-specific, which is an advantage according to a recent benchmark study [20]. The main limitations to consider are that it does not cover the X and Y chromosomes and that it annotates SNPs only.
In our DMA workflow, we set the threshold to define a driver mutation for the CScape-somatic score to 0.5 (Table 1) as suggested in the original publication [19]. This means that all mutations with a score >0.5 are denoted as driver mutations, and all mutations with a score ≤0.5 are denoted as passenger mutations. A threshold of 0.89 denotes driver mutations with high confidence, and so driver mutations with a score between 0.5 and 0.89 are labeled with low confidence. Thus, the driver genes are selected as those oncogenic mediators containing at least one mutation with a CScape-somatic score > 0.5. The CScape-somatic scores of all mutations in all DEGs together with thresholds and corresponding confidence labels are retained in the output of DMA. In this way, the user can easily filter mutations and genes based on various criteria of interest (see Supplementary Text S1 for more details).
Table 1.
Thresholds of CScape-somatic scores annotating a mutation as a driver or passenger including the confidence of the mutation as a driver mutation
| CScape-somatic score of mutation | Annotation of mutation by CScape-somatic | Confidence of mutation by CScape-somatic |
|---|---|---|
| ≥0.89 (coding) | Driver | High confidence |
| ≥0.7 (non-coding) | Driver | High confidence |
| >0.5 | Driver | Low confidence |
| ≤0.5 | Passenger | Neutral |
Predicting the level of consequence of mutations in DMA
The purpose of adding a mechanistic layer into Moonlight is to validate and explain the oncogenic patterns based on differential expression. For the mutations, it means that although a mutation might be a driver, it is not necessarily influencing the up- or downregulation of genes. We addressed this issue by categorizing mutations based on their position (e.g. 5’flank, 5’UTR, missense) and their type (e.g. SNP, insertion, deletion) into three categories which we call level of consequence: (i) mutations which can influence the level (rate or amount) of transcription, (ii) mutations which can influence the level of translation, and finally, (iii) mutations which can influence a protein’s structure or function (see more details in Supplementary Table S1 and Supplementary Text S2). The binary values in the level of consequence tables denote a presumed effect (1) or no presumed effect (0) of the given mutation type on either the transcription, translation, or protein structure/function level. NA values represent cases where the mutation type (e.g. missense, in frame deletion, silent) and variant type (e.g. SNP, INS, DEL) are not in accordance with each other. For example, as a missense mutation per definition is a SNP, the combination between this mutation type, i.e. a missense mutation, and variant types INS and DEL cannot occur. Consequently, such effects are not evaluated in the tables, resulting in NA. For instance, a mutation at the 5’flank of a gene can be inside a promoter. If the promoter is mutated, its corresponding transcription factor might have a stronger or weaker binding with it, thereby causing a change in the expression level of the corresponding gene, resulting in a presumed effect (1) of a 5′ flank mutation on the transcription level. On the other hand, a mutation at the translational start site (TSS) will not affect the transcription of the candidate gene but will influence the translation of the gene. This is represented as a 1 for a TSS mutation in the translational level of consequence table, but as a 0 in the transcriptional level of consequence table.
Besides annotating the predicted level of consequence of mutations, experimentally found promoter regions from the ENCODE consortium [21, 22] are integrated in the DMA function. We downloaded the dataset from ENCODE (https://www.encodeproject.org/) with the following ENCODE identifier: ENCSR294YNI. If a mutation falls within a promoter region, we have added a column with the promoter start and end position to the output table containing a complete overview of all annotations belonging to the DEGs.
Comparing driver genes with the Network of Cancer Genes
Finally, we incorporated data from the Network of Cancer Genes (NCG) [23] into DMA. From this, it is possible to cross-reference the oncogenic mediator annotation from Moonlight2 with findings from other cancer studies. In NCG, two categories of cancer genes are included. The first one contains known cancer genes with associated experimental support. When a gene has been reported by either Vogelstein et al. 2013 [5], Saito et al. 2020 [24] and the Cancer Gene Census [25], the driver type (OG or TSG) found by the respective study is stated. The second category contains candidate cancer genes which have somatic alterations that are predicted to have cancer driver roles but without any experimental validation. The PubMed PMID identifier and the associated cancer types for each gene are also listed.
PubMed literature search of driver genes and mutations
To explore the available information in literature on our set of predicted driver genes and mutations, we implemented a new function called Gene Literature Search (GLS) using the easyPubMed R package [26]. This function takes a user-supplied gene list and a string that constitutes part of a PubMed search query. The gene list can for instance be the predicted driver genes or other genes of interest. The string is expected to use general keywords such as ‘driver’, ‘cancer’ etc. that can work with each gene of the list. Standard PubMed syntax can be used in the query string. For each gene in the input list, a final PubMed query string is constructed by concatenating the user-supplied query string and the gene name; this is then used to query PubMed and retrieve information on identified publications, up to a user-specified number. GLS generates a table containing PubMed PMID identifier, doi, title, abstract, year of publication, keywords, and total number of PubMed publications for each of the genes supplied in the input. This allows for a quick and easy overview of the current state-of-the-art of genes of interest.
Visualizing results of the DMA function
Additionally, we have added two plotting functions to Moonlight to visualize the results of the DMA function: plotDMA() which creates heatmaps of the driver classifications of mutations for the oncogenic mediators (Figure 2) and plotMoonlight() which visualizes the effect of genes on the biological processes calculated in the URA step (Figure 3).
Figure 2.
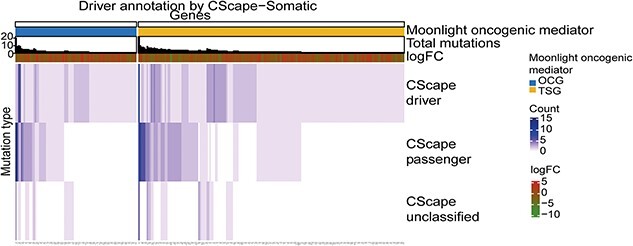
Classification of mutations in the 278 driver genes predicted in basal-like breast cancer by Moonlight. The 278 driver genes contain at least one driver mutation. Genes are in the columns while the mutation type classified by CScape-somatic is in the rows. The values in the heatmap indicate the number of driver, passenger, and unclassified mutations. The heatmap is divided into predicted oncogenes (OGs) and predicted tumor suppressor genes (TSGs). The total number of mutations and log2FC values of the driver genes are included in the heatmap. This plot was created with the plotDMA function where the input data was filtered to contain only the driver genes.
Figure 3.
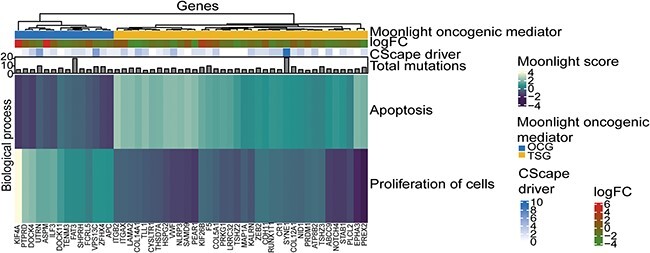
The top 50 oncogenic mediators with the highest total number of mutations in basal-like breast cancer. The columns are genes and the rows are the Moonlight Gene Z-scores for the two biological processes selected in the expert-based approach: apoptosis and proliferation of cells. The genes are divided into predicted oncogenes (OGs) and predicted tumor suppressor genes (TSGs). Number of driver mutations, log2FC values, and total number of mutations of the oncogenic mediators are included in the heatmap. This plot was created with the plotMoonlight function.
RESULTS
Case study: discovering driver genes in basal-like breast cancer with Moonlight
To demonstrate the new functionalities in Moonlight2R, we conducted a case study on basal-like breast cancer using data from TCGA. Gene expression, mutational and clinical data from the TCGA-BRCA project were retrieved via TCGAbiolinks [27, 28] and further curated to include only the subtype basal-like using the classification provided by the PanCancerAltas_subtype() function of TCGAbiolinks [29]. We performed a differential expression analysis (DEA) between the basal-like subtype and normal samples to generate the input to Moonlight2. We implemented the steps in the Moonlight2 framework as outlined above (Supplementary Text S3). Finally, we used the resulting tables from the DMA to select genes and mutations of interest which we investigated in the literature and other databases such as COSMIC [30] and TRRUST [31]. We predicted the driver genes in the context of the two biological processes apoptosis and proliferation of cells as these are well-known cancer hallmarks as done in previous publications with Moonlight [9, 32]. We also performed enrichment analyses of the driver genes with the R package EnrichR [33, 34] using the databases GO Molecular Function 2021, GO Biological Process 2021, and KEGG 2021 Human (Supplementary Text S3).
Applying the Moonlight pipeline on basal-like breast cancer
From the DEA, we identified 9300 DEGs between basal-like breast cancer and normal samples which we then input to Moonlight2’s primary layer. This resulted in the prediction of 852 oncogenic mediators divided into 260 putative OGs and 592 putative TSGs. We then applied Moonlight2’s secondary mutational layer, implemented in the DMA function presented here, to allow one potential mechanistic explanation of the predicted DEGs. This resulted in the classification of 4125 driver mutations, 4543 passenger mutations, and 1557 unclassified mutations. On the gene level, we found that 278 oncogenic mediators contained at least one driver mutation, resulting in our final set of driver genes. Of these 278 driver genes, 87 and 191 were predicted as OGs and TSGs, respectively (Table 2 and Supplementary Tables S2 and S3).
Table 2.
Number of mutations and genes in basal-like breast cancer; the upper part of the table contains the number of genes in different categories while the lower part contains the number of mutations; these categories are DEGs, oncogenic mediators, driver genes, and driver genes with transcriptional mutation(s); the numbers in brackets contain the mutations for OGs and TSGs, respectively; note that the numbers are identical for driver mutations for oncogenic mediators and driver genes as this is the basis on which driver genes are chosen
| DEGs | Oncogenic mediators | Driver genes | Driver genes with transcriptional mutation(s) | |
|---|---|---|---|---|
| Genes | ||||
| OGs | - | 260 | 87 | 32 |
| TSGs | - | 592 | 191 | 61 |
| Total | 9300 | 852 | 278 | 93 |
| Genes without mutations [OG/TSG] | 4539 | 364 [114/250] | - | - |
| Mutations [OG/TSG] | ||||
| Driver mutations | 4125 | 394 [119/275] | 394 [119/275] | 154 [51/103] |
| Passenger mutations | 4543 | 537 [157/380] | 262 [84/178] | 76 [23/53] |
| Unclassified mutations | 1557 | 151 [55/96] | 59 [19/40] | 23 [5/18] |
| Total | 10,225 | 1082 | 715 | 253 |
Abbreviations: DEGs, differentially expressed genes; OGs, oncogenes; TSGs, tumor suppressor genes.
We visualized the classification of the mutations of the 278 driver genes in a heatmap with the function plotDMA() (Figure 2). From Figure 2, we can notice that if an oncogenic mediator has a driver mutation, in about half of the cases, it will also have a passenger mutation. Moreover, we observe that most drivers only carry one driver mutation. We find one OG (FAT3) and one TSG (SYNE1) which have around 20 total associated mutations whereas the rest of the predicted driver genes have 10 or less total mutations.
Exploration of oncogenic mediators and driver genes in terms of driver and passenger mutations
First, we explored the oncogenic mediators with a high number of mutations. In Figure 3, the top 50 oncogenic mediators with the highest number of total mutations are visualized in a heatmap, created with plotMoonlight(). It is clear that not all highly mutated oncogenic mediators contain few or any driver mutations e.g. SAMD9, FAT3 and SYNE1. This clearly showcases the need to assess the mutations individually because the total number of mutations does not necessarily reflect driver status of the gene.
CScape-somatic has defined driver mutations as ‘disease-enablers’ [19], however, not all of them are placed such that they can directly interfere with the gene expression level. We therefore decided to focus on mutations with a transcriptional level of consequence. We found 93 driver genes containing in total 154 driver mutations which potentially affect the transcriptional level (Table 2). Of these transcriptional driver mutations, mutated promoter regions are of special interest because mutations in these sites can alter transcription factor binding and thereby change the transcription level (Figure 4 and Table 3). Of the 93 predicted basal-like driver genes that had a driver mutation with a potential transcriptional level of consequence, we found that four OGs and nine TSGs had one driver mutation located within an experimentally validated promoter region from ENCODE [21, 22]. Of the driver mutations in these 13 genes (Table 3), COPZ2’s mutation scored the highest, suggesting high confidence of this driver mutation. These specific mutations are candidates for further structural studies and experimental investigation.
Figure 4.

Oncogenes and tumor suppressors with a driver mutation in an experimentally validated promoter region in basal-like breast cancer. Four oncogenes (OGs) and nine tumor suppressor (TSGs) have a driver mutation located within an experimentally validated promoter region from ENCODE. The columns are genes and the rows are the Moonlight Gene Z-scores for the two biological processes selected in the expert-based approach: apoptosis and proliferation of cells. The genes are divided into predicted OGs and predicted TSGs. Number of driver mutations, log2FC values, and total number of mutations of the driver genes are included in the heatmap.
Table 3.
Summary of driver mutations in basal-like breast cancer which are located in an experimentally validated promoter region from ENCODE; these mutations can therefore potentially alter the transcription level of the genes; the table includes which chromosome the mutation is placed on, Moonlight2 prediction of driver type (TSG/OG), SNP position in the chromosome according to grch38, mutation type, CScape-somatic score of mutation, and log2FC value of gene; all mutations were predicted as non-coding driver mutations by CScape-somatic
| Chromosome | Gene | Driver type predicted by Moonlight2 | SNP Position | Classification of mutations | CScape-somatic score | Log2FC |
|---|---|---|---|---|---|---|
| chr1 | KIF26B | TSG | 245,155,300 | 5’UTR | 0.746 | 3.03 |
| chr1 | SF3B4 | OG | 149,927,847 | 5’UTR | 0.682 | 1.12 |
| chr1 | NR5A2 | TSG | 200,027,802 | 5’UTR | 0.594 | -2.10 |
| chr1 | KRTCAP2 | OG | 155,173,311 | 5’UTR | 0.683 | 1.27 |
| chr2 | ARHGAP25 | TSG | 68,735,192 | 5’UTR | 0.589 | -0.536 |
| chr4 | EMCN | TSG | 100,517,922 | 5’UTR | 0.532 | -2.67 |
| chr5 | ARL15 | TSG | 54,310,486 | 5’UTR | 0.528 | -1.11 |
| chr17 | COPZ2 | OG | 48,037,182 | Intron | 0.837 | -1.58 |
| chr7 | POLR2J | OG | 102,478,861 | 5’UTR | 0.815 | 0.770 |
| chr7 | TPK1 | TSG | 144,835,605 | 5’UTR | 0.524 | -0.712 |
| chr9 | TEK | TSG | 27,109,485 | 5’UTR | 0.697 | -2.66 |
| chr19 | KIR2DL4 | TSG | 54,803,614 | 5’Flank | 0.542 | 2.98 |
| chr19 | GMFG | TSG | 39,335,532 | Splice_Site | 0.599 | -0.642 |
Abbreviations: OG, oncogenes; TSG, tumor suppressor gene; log2FC, log2 fold change; chr, chromosome; SNP, single nucleotide polymorphisms; UTR, untranslated region.
On the flip side of the regulatory mechanisms lie the transcription factors (TFs) which bind to promoter regions and regulate transcription levels. Thus, we next sought to investigate the presence of any TFs in the 278 predicted basal-like driver genes using the TRRUST database [31]. We found four and nine OGs and TSGs, respectively, which are TFs with a known mode of regulation (Table 4). Transcription factors can be of particular interest because they, like mutated promoters, can alter the transcription level. Though in this case it can be necessary to investigate all levels of consequences. For instance, if a mutation is placed in the coding region, the TF could have altered binding ability to the promoter, thereby changing the target’s expression level. The TF could also have its own transcription lowered, indirectly causing lowered transcription of the target. Such patterns might show up through further investigation of TF-target interactions.
Table 4.
Basal-like driver genes predicted by Moonlight2 that are annotated as TFs in the TRRUST database with a known mode of action; four and nine predicted OGs and TSGs are annotated as TFs in TRRUST, respectively; the log2FC value, number of driver mutations including type of driver mutations and which genes the TF has been found to regulate are shown; for the targets, the letters in parenthesis indicate mode of regulation: A = activation and R = repression
| Gene | Moonlight2-predicted basal-like driver gene type | log2FC | Number of driver mutations | Target including mode of regulation by TF |
|---|---|---|---|---|
| APC | OG | -0.752 | 1 | AKT1 (R), AMHR2 (R), DNMT1 (R), MYC (R), NOS2 (A), ODC1 (R), PTGS2 (R), SGK1 (R) |
| EPAS1 | TSG | -2.26 | 3 | CA9 (A), CCR7 (A), COL10A1 (A), FLT1 (A), MMP14 (A), MSC (A), SERPINE1 (A), VEGFA (A) |
| ERG | TSG | -2.29 | 1 | ADAMTS1 (A), CXCL8 (R), CXCR4 (A), ENG (A), EPB41L3 (R), EPB41L4B (A), ERG (A), FGF2 (R), ICAM1 (R), ILK (R), PIM1 (A), SPP1 (A), TDRD1 (A,R), VIM (A), VWF (A), WNT11 (A) |
| FOXM1 | OG | 4.96 | 2 | AR (A), BTG2 (A), CCNB1 (A), CDC25B (A), CDC6 (A), CDKN2A (R), FGB (R), MYC (A), SFTPB (A) |
| ILF3 | OG | 0.569 | 2 | ACRV1 (A), BIRC5 (A), HLA-DRA (R), IL2 (A), PLAU (A) |
| KLF11 | OG | -0.722 | 2 | INS (A) |
| MITF | TSG | -1.40 | 1 | ACP5 (A), BCL2A1 (R), BEST1 (A), CTSK (A), DCT (A), FOS (A), GPNMB (A), HIF1A (A), KIT (A), OCA2 (A), PPARGC1A (A), SERPINF1 (A), TRAP (A), TRPM1 (A), TYR (A), TYRP1 (A) |
| NR5A2 | TSG | -2.10 | 2 | ABCG5 (A), ABCG8 (A), CETP (A), CYP11B1 (A), HSD3B2 (A), NR5A2 (A), STAR (A) |
| PGR | TSG | -5.18 | 1 | BCL2 (A), CYP19A1 (R), DUSP1 (A), E2F1 (A), ERBB2 (R), ESR1 (A), FOXP3 (R), HLTF (A), IL10 (R), KLK4 (A), PTGS2 (R), RELA (R) |
| PRDM1 | TSG | 0.562 | 1 | CIITA (R), GCSAM (R), LMO2 (R), MYC (R) |
| TAL1 | TSG | -2.39 | 1 | ALDH1A2 (A), CD34 (R), ERG (A), MYB (A), MYCN (A), NFKB1 (R) |
| TCF4 | TSG | -1.23 | 1 | ABCB1 (A), CCND1 (A), CLEC4C (A), CNTNAP2 (A), HECA (R), MITF (R), MYC (A), NOTUM (A), NRXN1 (A), PLD1 (A), PTEN (R), VEGFA (A), VIM (A) |
| ZEB2 | TSG | -1.47 | 2 | CDH1 (R), CXADR (R), ITGA5 (A), MEOX2 (R), POU5F1 (A), VIM (A) |
Abbreviations: TFs, transcription factors; OGs, oncogenes; TSGs, tumor suppressor genes; log2FC, log2 fold change; A, activation; R, repression.
Similarities and differences between TSGs and OGs
In the literature, it has been reported that OGs and TSGs generally promote and limit cell growth, respectively [35, 36]. We sought to investigate possible differences between the two predicted classes of driver genes through an enrichment analysis on the 191 predicted TSGs and 87 predicted OGs (Figure 5).
Figure 5.
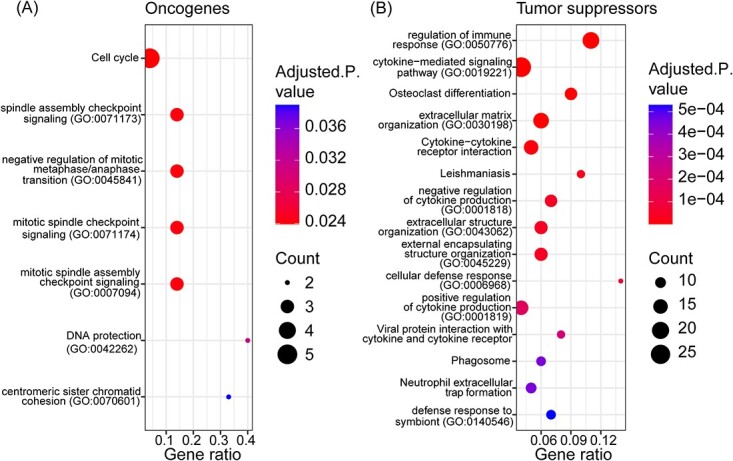
Enrichment analysis of 87 and 191 predicted oncogenes and tumor suppressors, respectively, in basal-like breast cancer. The top 15 most significantly enriched terms (adjusted P-value <0.05) within the GO Molecular Function 2021, GO Biological Process 2021 and KEGG 2021 human databases among the (A) 87 predicted oncogenes (OGs) and (B) 191 predicted tumor suppressor genes (TSGs). The enriched terms are sorted on the adjusted P-value. The gene ratio refers to the ratio between the number of predicted TSGs or OGs intersecting with genes annotated in the given process and the total number of genes annotated in the given process. The sizes of the points represent the number of TSGs or OGs participating in the given process.
The enrichment analysis reveals expected oncogenic roles of the OGs. The top enriched terms among the OGs are related to DNA replication, spindle checkpoints, and cell cycle. Since the OGs are upregulated in basal-like breast cancer compared to normal tissue, the activity of the enriched biological processes related to DNA replication and cell cycle is increased, potentially driving cancer progression. In contrast, the enriched terms among the TSGs are for the most part associated with immune response and regulation, with both positive and negative impacts, indicating immunological roles of the predicted TSGs. The enriched terms among the OGs and TSGs are related to the cancer hallmarks. For example, processes related to DNA replication and cell cycle are manifested in the Sustaining proliferative signaling hallmark. Similarly, the immunological processes can play a role in the Avoiding immune destruction and Tumor-promoting inflammation hallmarks [2–4]. Collectively, these results indicate that Moonlight2, thanks to the integration of the core functions and the DMA step, is capable of finding two distinct classes of driver genes which both have strong associations to cancer related pathways. Moreover, no enriched terms were shared between the TSGs and OGs when comparing the significantly enriched terms (adjusted P-value <0.05), indicating that the two driver gene classes are highly distinct in terms of functional roles.
Besides investigating the functional profile of the predicted OGs and TSGs, we explored the difference between these two driver gene classes in terms of driver mutation types (Supplementary Figure S1). We noticed no distinct difference in the types of driver mutations between the OGs and TSGs, suggesting that it is not possible to classify the driver gene type based on driver mutation type alone.
Comparison of predicted basal-like driver genes with other findings and annotations
With the aim of investigating the novelty and consistency of Moonlight2’s driver gene annotations, we compared Moonlight2’s predictions with the NCG database (Figure 6). Most driver genes (i.e. 62 OGs and 123 TSGs) determined by Moonlight2 were not reported in the NCG database. The comparison between genes predicted as either OG or TSG with Moonlight2 and in NCG is listed in Supplementary Table S4. The small overlap between Moonlight2’s predicted driver genes and NCG genes may also be attributed to NCG being a collection of genes across multiple cancer types and not only breast cancer. It could also be due to the lack of functional studies on some of the candidate genes with regards to their oncogenic or tumor-suppressor potential. Nevertheless, our predicted candidates would still need further investigation to support their driver signature and constitute an interesting dataset for the research community working with breast cancer subtypes.
Figure 6.
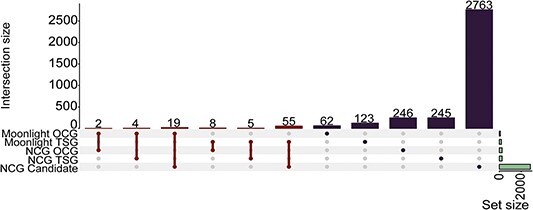
Comparison between Moonlight’s predicted driver genes in basal-like breast cancer and genes in the Network of Cancer Genes database. The first six intersections represent genes in common between two groups and the next five intersections represent genes that are specific to one group. The horizontal bar to the right represents the total number of genes in the five sets.
One of the strengths of Moonlight is its classification of driver genes into TSGs and OGs which allows for the prediction of dual role genes–genes that are predicted as TSGs in one biological context but as OGs in another context [7, 9]. We found 12 possible dual role driver genes which Moonlight2 has predicted as a TSG or an OG in the context of basal-like breast cancer, whereas NCG may have reported the gene functioning with the opposite driver gene type in possibly another cancer type. However, we found that none of the potential dual role genes had a reported cancer type by NCG. Thus, we could not establish the biological context of the dual role genes.
Finally, we compared Moonlight’s basal-like driver genes with breast cancer genes reported in NCG to explore validity and consistency of our results. NCG contains 147 driver genes associated with breast cancer which are all marked as candidates. Comparing these 147 breast cancer genes with our 278 predicted driver genes revealed an overlap of three driver genes (SYNE1, CTSS, and STAB1) which are all predicted to be TSGs by Moonlight. Above, we also described SYNE1 as the Moonlight2 predicted driver gene with the most driver mutations (Figure 3). Additionally, the two other genes (CTSS and STAB1) have both been documented to be recurrently mutated across breast cancer cohorts [37–39]. Following the investigation of breast cancer related driver genes, we more specifically examined triple-negative breast cancer genes from NCG. A total of 13 (CDKN2B, DNMT3B, EPHB1, IGF1R, MCL1, NOTCH3, PIK3CD, AURKA, DIS3, TBK1, SHQ1, RPTOR, and EPHA6) of the 147 breast cancer candidates in NCG are specifically associated with triple-negative breast cancer. These genes were also not identified by Moonlight2 as oncogenic mediators except for AURKA. Additionally, we only found five out of the 13 genes to be differentially expressed between basal-like and normal samples used in our study, namely DNMT3B, EPHB1, IGF1R, AURKA, and EPHA6, meaning the other eight genes were not input to Moonlight, and consequently, we could not evaluate their driver gene potential. The fact that Moonlight2 with DMA did not predict any of these five genes as driver genes suggests that other mechanisms not dependent on changes in expression or mutations in non-coding regulatory elements could be at play, or features that are common only to a subset of patients and not identified in the TCGA dataset. To further investigate this direction, we retrieved the literature cited by NCG for the five genes and evaluated the expected driver alteration associated with them. DNMT3B, EPHB1, EPHA6 as for AURKA were annotated in NCG from the same study [40] but the results seem to mostly rely on predictions of sparse missense mutations or splice site and not clear experimental validation of the tumorigenic potential of these variants. Moreover, one study discovered EPHA6 as a driver pan-cancer using the tool OncodriveFML [41] and another study reported EPHA6 as a significantly mutated gene in ampullary carcinomas [42]. Furthermore, EPHB1 was found as a driver in thyroid carcinoma by OncodriveFML [41] and mutations in EPHB1 were also found to be involved in chronic lymphocytic leukemia [43]. Finally, Weisman et al. 2016 reported an in-frame indel in IGF1R which was predicted as likely pathogenic, deleterious by PROVEAN, and disease-causing by MutationTaster [40]. Additionally, IGF1R was found as a novel driver in breast cancer [44] and glioblastoma [45].
Few other tools besides Moonlight classifies driver genes as TSGs and OGs. We compared Moonlight basal-like predicted driver genes with another driver gene prediction tool, GUST [46]. We selected GUST due to the availability of precomputed publicly available results on breast cancer (https://liliulab.shinyapps.io/gust/). These predictions from GUST were however not basal-like specific. Compared to our results, we found a very small overlap of predicted driver genes. Only two genes (IFFO1 and HERC5) were predicted as driver genes with both Moonlight2 and GUST (Supplementary Table S5). These two genes are not reported in NCG.
Case studies: discovering driver genes in lung adenocarcinoma and thyroid carcinoma with Moonlight
To further showcase the applicability of Moonlight2 to cancer types of different mutation burdens, we applied Moonlight2 on lung adenocarcinoma (LUAD) and thyroid carcinoma (THCA). LUAD and THCA have previously been reported to have high and low tumor mutation burdens, respectively [47–49]. These case studies were conducted similarly to the basal-like breast cancer case study, except considering their cancer types in the whole and using paired samples only (Supplementary Text S3).
Among 15,457 DEGs between LUAD and normal samples, Moonlight’s primary layer predicted 610 oncogenic mediators (399 putative OGs and 211 putative TSGs). Applying Moonlight’s secondary mutational layer resulted in a final prediction of 131 driver genes categorized into 91 OGs and 40 TSGs (Figure 7A and Supplementary Tables S2 and S3). Similarly, for THCA, we found 885 oncogenic mediators (676 putative OGs and 209 putative TSGs) from 12,015 DEGs. Moonlight’s secondary mutational layer predicted 12 driver genes divided into nine OGs and three TSGs in THCA (Figure 7B and Supplementary Tables S2 and S3). The THCA driver genes all had one driver mutation in contrast to driver genes discovered in LUAD and basal-like breast cancer where the number of driver mutations in the driver genes ranged between 1–4 and 1–9, respectively. The lower number of driver genes and mutations in THCA is consistent with its lower mutation burden compared to LUAD and basal-like breast cancer.
Figure 7.
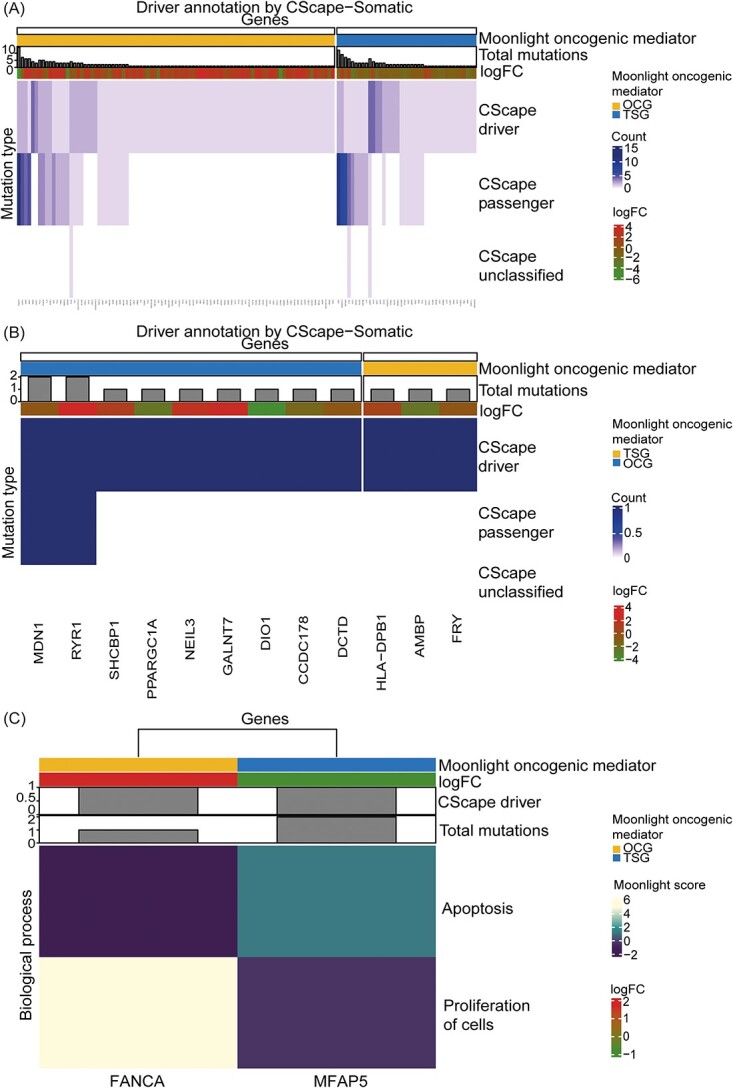
Classification of mutations in predicted driver genes in lung adenocarcinoma and thyroid carcinoma by Moonlight. (A) Classification of mutations in the 131 driver genes predicted in lung adenocarcinoma. (B) Classification of mutations in the 12 driver genes predicted in thyroid carcinoma. In (A) and (B), genes are in the columns while the mutation type classified by CScape-somatic is in the rows. The values in the heatmap indicate the number of driver, passenger, and unclassified mutations. The total number of mutations and log2FC values of the driver genes are shown in the heatmap. (C) Driver genes (one oncogene and one tumor suppressor) in lung adenocarcinoma with a driver mutation in an experimentally validated promoter region from ENCODE. The columns are genes and the rows are the Moonlight Gene Z-scores for the two biological processes selected in the expert-based approach: apoptosis and proliferation of cells. The number of driver mutations, the number of total mutations and the log2FC values of the driver genes are included.
Subsequently, we investigated more closely predicted driver genes with driver mutation(s) affecting the transcriptional level and located in the promoter regions. In THCA and LUAD, we found zero and two driver genes, respectively, fulfilling these criteria. In LUAD, these two genes are an OG, FANCA, and a TSG, MFAP5. The CScape-somatic non-coding scores of these two driver mutations were 0.77 and 0.58, respectively, suggesting high confidence of the driver mutation located in the promoter region in FANCA (Figure 7C). These driver genes highlight interesting candidates for further studies.
To examine consistency and validity of Moonlight’s predicted driver genes in LUAD and THCA, we compared these genes with NCG (Supplementary Table S4) and GUST (Supplementary Table S5). Like the basal-like breast cancer case study, we here found a low overlap between Moonlight’s predicted driver genes, NCG, and GUST. Thus, to investigate novelty of the predicted driver genes in LUAD and THCA and explore current state-of-the-art, we performed literature searches of these genes using Moonlight’s GLS function presented above (Supplementary Table S6). Given the potential of FANCA in LUAD as an interesting candidate for further studies, it is worth highlighting some of the literature results of this gene. We found that four PubMed records match the query ‘FANCA AND cancer AND driver’ [50–53]. For instance, Ognibene et al. [50] found high expression of FANCA to be associated with low survival in patients with neuroblastoma. Genetic alterations including mutations in FANCA were also frequently discovered in several cancer types including LUAD [52]. Moreover, studies have reported a role of FANCA in the molecular pathogenesis of LUAD and high expression of FANCA was significantly associated with poor prognosis of patients with LUAD [54–56].
FUTURE DIRECTIONS
In this study, we mainly focused on driver mutations located in the promoter region as these mutations are the ones that most likely can explain the observed patterns of deregulated expression. Nevertheless, other types of mutations in both the coding and non-coding regions of the driver genes are of interest. For instance, missense mutations in genes involved with mRNA degradation could also be essential targets for further studies. Additionally, we envision an inclusion of additional -omics layers such as DNA methylation, copy number variation, and chromatin accessibility in future updates of Moonlight. Moreover, we envision complementing the promoter annotations by including other regions such as silencers and enhancers.
Key Points
Discovery of cancer driver genes, tumor suppressors and oncogenes, is essential for understanding cancer development and ultimately for discovery of novel treatment strategies
We have presented new functionalities in our previously developed bioinformatics framework, Moonlight, to produce Moonlight2 which aims at predicting tumor suppressors and oncogenes
The new functionalities in Moonlight called Driver Mutation Analysis (DMA) and Gene Literature Search (GLS) classify mutations in a cancer cohort into driver and passenger mutations and perform literature searches of candidate driver genes, respectively
DMA provides one potential mechanistic explanation of deregulated genes in a cancer cohort
In basal-like breast cancer, we found four oncogenes and nine tumor suppressors which contain a driver mutation in the promoter region. In lung adenocarcinoma and thyroid carcinoma, we found two (one oncogene and one tumor suppressor) and zero driver genes with a driver mutation in the promoter region, respectively
Supplementary Material
ACKNOWLEDGEMENTS
The results published here are in whole or in part based upon data generated by The Cancer Genome Atlas (TCGA) Research Network: https://www.cancer.gov/tcga.
Mona Nourbakhsh is a PhD student in the Cancer Systems Biology group (Department of Health and Technology, Technical University of Denmark, DTU, Lyngby, Denmark). Her work focuses on discovering cancer driver genes in cancer cohorts using -omics data.
Astrid Brix Saksager was a research assistant in the Cancer Systems Biology group (Department of Health and Technology, Technical University of Denmark, DTU, Lyngby, Denmark). Her work focused on predicting driver genes and mutations. She now works as a PhD student in the Immunoinformatics and Machine Learning group (Department of Health and Technology, Technical University of Denmark, DTU, Lyngby, Denmark).
Nikola Tom worked as a postdoc in the Cancer Systems Biology group (Department of Health and Technology, Technical University of Denmark, DTU, Lyngby, Denmark) establishing workflows for analysis of -omics data. He is currently a postdoc in the Lipidomics Core Facility at the Danish Cancer Society (Danish Cancer Institute, DCI, Copenhagen, Denmark).
Xi (Steven) Chen, PhD is Professor of Biostatistics in the Department of Public Health Sciences at the University of Miami Miller School of Medicine. He is also the Director of the Biostatistics and Bioinformatics at Sylvester Comprehensive Cancer Center.
Antonio Colaprico is currently an Associate Scientist at the Department of Public Health Sciences and the Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL, USA. His research activities are focused on the development of innovative integrated bioinformatics methods and applications with the aim of modeling complex systems in biology and improving molecular diagnosis.
Catharina Olsen obtained her PhD in computer science from the Université libre de Bruxelles (ULB). She currently works as bioinformatician at the UZ Brussels and as a 10% assistant professor at the Vrije Universiteit Brussel (VUB). Her research focuses on omics analysis applied to rare diseases and oncology.
Matteo Tiberti is a staff scientist at the Cancer Structural Biology group, at the Danish Cancer Institute (DCI) in Copenhagen, Denmark. His primary research interests revolve around cancer bioinformatics and the investigation of protein variants with a focus on software development for these studies.
Elena Papaleo is an Associate Professor and group leader of the Cancer Systems Biology group at DTU and the Cancer Structural Biology group at the Danish Cancer Institute (DCI). Her group uses integrative -omics approaches to understand cancer drivers. Additionally, they employ structural methods to analyze the impact of protein variants. To enhance the computational findings, they complement their work with experimental validation using in vitro and cellularassays.
Contributor Information
Mona Nourbakhsh, Cancer Systems Biology, Section for Bioinformatics, Department of Health and Technology, Technical University of Denmark, Lyngby, Denmark.
Astrid Saksager, Cancer Systems Biology, Section for Bioinformatics, Department of Health and Technology, Technical University of Denmark, Lyngby, Denmark.
Nikola Tom, Cancer Systems Biology, Section for Bioinformatics, Department of Health and Technology, Technical University of Denmark, Lyngby, Denmark.
Xi Steven Chen, Department of Public Health Sciences, University of Miami Miller School of Medicine, Miami, FL 33136, USA; Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Antonio Colaprico, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Catharina Olsen, Vrije Universiteit Brussel (VUB), Universitair Ziekenhuis Brussel (UZ Brussel), Clinical Sciences, Reproduction and Genetics; Brussels Interuniversity Genomics High Throughput core (BRIGHTcore), VUB-ULB, Brussels 1090, Belgium; Interuniversity Institute of Bioinformatics in Brussels (IB)2, Brussels 1050, Belgium.
Matteo Tiberti, Cancer Structural Biology, Danish Cancer Institute, Copenhagen, Denmark.
Elena Papaleo, Cancer Systems Biology, Section for Bioinformatics, Department of Health and Technology, Technical University of Denmark, Lyngby, Denmark; Cancer Structural Biology, Danish Cancer Institute, Copenhagen, Denmark.
FUNDING
EP group’s research has been supported by Interregional Childhood Oncology Precision Medicine Exploration (iCOPE), a cross-Oresund collaboration between University Hospital Copenhagen, Rigshospitalet, Lund University, Region Skane, and Technical University Denmark (DTU); Hartmanns Fond (R241-A33877 to E.P. group’s research); Danmarks Grundforskningsfond (DNRF125 to E.P. group’s research); NCI (R01CA200987, R01CA158472 and U24CA210954 to A.C. and X.C.).
DATA AVAILABILITY
The new functionality of Moonlight2 called DMA is available on GitHub (https://github.com/ELELAB/Moonlight2R). The input, data, and code for the case studies are also available on GitHub (https://github.com/ELELAB/Moonlight2_case_studies). Once the Moonlight2R R package is loaded, example data of input and output files for all functions in Moonlight2R are provided.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov 2022;12:31–46. [DOI] [PubMed] [Google Scholar]
- 5. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 1979;2013(340):1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porta-Pardo E, Valencia A, Godzik A. Understanding oncogenicity of cancer driver genes and mutations in the cancer genomics era. FEBS Lett 2020;594:4233–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen L, Shi Q, Wang W. Double agents: genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stepanenko AA, Vassetzky YS, Kavsan VM. Antagonistic functional duality of cancer genes. Gene 2013;529:199–207. [DOI] [PubMed] [Google Scholar]
- 9. Colaprico A, Olsen C, Bailey MH, et al. Interpreting pathways to discover cancer driver genes with Moonlight. Nat Commun 2020;11:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hutter C, Zenklusen JC. The cancer genome atlas: creating lasting value beyond its data. Cell 2018;173:283–5. [DOI] [PubMed] [Google Scholar]
- 11. Ganini C, Amelio I, Bertolo R, et al. Global mapping of cancers: the cancer genome atlas and beyond. Mol Oncol 2021;15:2823–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grossman RL, Heath AP, Ferretti V, et al. Toward a shared vision for cancer genomic data. N Eng J Med 2016;375:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y, Hu X, Han C, et al. Targeting tumor suppressor genes for cancer therapy. Bioessays 2015;37:1277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep 2015;16:280–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao L, Lee VHF, Ng MK, et al. Molecular subtyping of cancer: current status and moving toward clinical applications. Brief Bioinform 2019;20:572–84. [DOI] [PubMed] [Google Scholar]
- 16. Jackson SE, Chester JD. Personalised cancer medicine. Int J Cancer 2015;137:262–6. [DOI] [PubMed] [Google Scholar]
- 17. Shi X, Teng H, Shi L, et al. Comprehensive evaluation of computational methods for predicting cancer driver genes. Brief Bioinform 2022;23:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pham VVH, Liu L, Bracken C, et al. Computational methods for cancer driver discovery: a survey. Theranostics 2021;11:5553–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rogers MF, Gaunt TR, Campbell C. CScape-somatic: distinguishing driver and passenger point mutations in the cancer genome. Bioinformatics 2020;36:3637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen H, Li J, Wang Y, et al. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol 2020;21:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo Y, Hitz BC, Gabdank I, et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res 2020;48:D882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Repana D, Nulsen J, Dressler L, et al. The Network of Cancer Genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol 2019;20:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saito Y, Koya J, Araki M, et al. Landscape and function of multiple mutations within individual oncogenes. Nature 2020;582:95–9. [DOI] [PubMed] [Google Scholar]
- 25. Sondka Z, Bamford S, Cole CG, et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 2018;18:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fantini D. easyPubMed: Search and Retrieve Scientific Publication Records from PubMed. R package version 2.13 2019. https://CRAN.R-project.org/package=easyPubMed.
- 27. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silva TC, Colaprico A, Olsen C, et al. TCGA workflow: analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res 2016;5:1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mounir M, Lucchetta M, Silva TC, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol 2019;15:e1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 2019;47:D941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han H, Cho JW, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 2018;46:D380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lucchetta M, da Piedade I, Mounir M, et al. Distinct signatures of lung cancer types: aberrant mucin O-glycosylation and compromised immune response. BMC Cancer 2019;19:824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 2013;14:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Z, Bailey A, Kuleshov MV, et al. Gene set knowledge discovery with Enrichr. Curr Protoc 2021;1:1–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Croce CM. Oncogenes and cancer. N Engl J Med 2008;358:502–11. [DOI] [PubMed] [Google Scholar]
- 36. Wang LH, Wu CF, Rajasekaran N, Shin YK. Loss of tumor suppressor gene function in human cancer: an overview. Cell Physiol Biochem 2018;51:2647–93. [DOI] [PubMed] [Google Scholar]
- 37. Ciriello G, Ciriello G, Gatza ML, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015;163:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Encinas G, Sabelnykova VY, de Lyra EC, et al. Somatic mutations in early onset luminal breast cancer. Oncotarget 2018;9:22460–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berger AC, Korkut A, Kanchi RS, et al. A comprehensive pan-cancer molecular study of Gynecologic and breast cancers. Cancer Cell 2018;33:690–705.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weisman PS, Ng CKY, Brogi E, et al. Genetic alterations of triple negative breast cancer by targeted next-generation sequencing and correlation with tumor morphology. Mod Pathol 2016;29:476–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mularoni L, Sabarinathan R, Deu-Pons J, et al. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol 2016;17:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yachida S, Wood LD, Suzuki M, et al. Genomic sequencing identifies ELF3 as a driver of Ampullary carcinoma. Cancer Cell 2016;29:229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2012;44:47–52. [DOI] [PubMed] [Google Scholar]
- 44. Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet 2016;48:768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chandrashekar P, Ahmadinejad N, Wang J, et al. Somatic selection distinguishes oncogenes and tumor suppressor genes. Bioinformatics 2020;36:1712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu H-X, Wang Z-X, Zhao Q, et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann Transl Med 2019;7:640–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niknafs N, Balan A, Cherry C, et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat Med 2023;29:440–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ognibene M, De Marco P, Parodi S, et al. Genomic analysis made it possible to identify gene-driver alterations covering the time window between diagnosis of neuroblastoma 4S and the progression to stage 4. Int J Mol Sci 2022;23:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li X, Chatla S, Wilson AF, et al. Persistent DNA damage and oncogenic stress-induced Trem1 promotes leukemia in mice. Haematologica 2022;107:2576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beltran H, Eng K, Mosquera JM, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol 2015;1:466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Severson PL, Vrba L, Stampfer MR, Futscher BW. Exome-wide mutation profile in benzo[a]pyrene-derived post-stasis and immortal human mammary epithelial cells. Mutat Res Genet Toxicol Environ Mutagen 2014;775-776:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanada H, Seki N, Mizuno K, et al. Involvement of dual strands of miR-143 (miR-143-5p and miR-143-3p) and their target oncogenes in the molecular pathogenesis of lung adenocarcinoma. Int J Mol Sci 2019;20:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu X, Zhao J, Yang L, et al. Next-generation sequencing reveals age-dependent genetic underpinnings in lung adenocarcinoma. J Cancer 2022;13:1565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao D, Li H, Mambetsariev I, et al. Molecular and clinical features of hospital admissions in patients with thoracic malignancies on immune checkpoint inhibitors. Cancers (Basel) 2021;13(11):2653. 10.3390/cancers13112653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The new functionality of Moonlight2 called DMA is available on GitHub (https://github.com/ELELAB/Moonlight2R). The input, data, and code for the case studies are also available on GitHub (https://github.com/ELELAB/Moonlight2_case_studies). Once the Moonlight2R R package is loaded, example data of input and output files for all functions in Moonlight2R are provided.