

Significance
Despite the encouraging clinical responses generated by immunotherapies, cancer innate or acquired resistance to immunotherapy remains an important barrier to treatment effectiveness. Our research describes tumor-intrinsic fatty acid oxidation as a mechanism involved in cancer adaptive resistance to cytolytic immune cells. Our research also highlights a mode of immune editing triggered by a dynamic interplay between immune effector cells and cancer targets. Furthermore, targeting this tumor-intrinsic metabolic pathway represents a strategy for improving cancer susceptibility to T cell-based immunotherapies including human CAR T therapy.
Keywords: fatty acid oxidation, carnitine palmitoyltransferase 1A, cancer metabolism, therapeutic resistance, cellular immunotherapy
Abstract
Although tumor-intrinsic fatty acid β-oxidation (FAO) is implicated in multiple aspects of tumorigenesis and progression, the impact of this metabolic pathway on cancer cell susceptibility to immunotherapy remains unknown. Here, we report that cytotoxicity of killer T cells induces activation of FAO and upregulation of carnitine palmitoyltransferase 1A (CPT1A), the rate-limiting enzyme of FAO in cancer cells. The repression of CPT1A activity or expression renders cancer cells more susceptible to destruction by cytotoxic T lymphocytes. Our mechanistic studies reveal that FAO deficiency abrogates the prosurvival signaling in cancer cells under immune cytolytic stress. Furthermore, we identify T cell–derived IFN-γ as a major factor responsible for induction of CPT1A and FAO in an AMPK-dependent manner, indicating a dynamic interplay between immune effector cells and tumor targets. While cancer growth in the absence of CPT1A remains largely unaffected, established tumors upon FAO inhibition become significantly more responsive to cellular immunotherapies including chimeric antigen receptor-engineered human T cells. Together, these findings uncover a mode of cancer resistance and immune editing that can facilitate immune escape and limit the benefits of immunotherapies.
The encouraging clinical responses to cancer immunotherapies have now established immune-oncology as a standard treatment modality for multiple types of cancer (1, 2). Eradication of malignancies by the host immune system mainly relies on the cytolytic activity of tumor-reactive immune effector cells, particularly cytotoxic T cells (CTLs). Immune checkpoint therapy reinvigorates the antitumor function of T cells by antagonizing the negative immune regulatory proteins, e.g., cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed cell death protein-1 (PD-1) or its ligand (3). Chimeric antigen receptor (CAR) T cell therapy involves genetically engineering T cells to specially recognize cancer targets for directed and robust immune destruction (4). However, innate or acquired resistance to immunotherapy is an important barrier to treatment effectiveness (5, 6). A significant proportion of patients experience a poor response or relapse after these treatments. While cellular immunotherapy using CAR T cells has shown remarkable success in the treatment of several hematologic malignancies, its therapeutic benefit against solid tumors remains limited (7).
Unresponsiveness or reduced sensitivity to T cell–mediated cytotoxicity results from a number of tumor-intrinsic or extrinsic factors that are believed to collectively contribute to immune escape of cancer cells. These include lack of tumor antigens, impaired major histocompatibility complex (MHC)–restricted antigen presentation, mutational landscape and oncogenic signaling, defects in interferon signaling, overexpression of inhibitory checkpoint molecules, immunosuppressive cells (e.g., myeloid derived suppressor cells, M2-like macrophages, and regulatory T cells), or cytokines in the tumor microenvironment (5, 6, 8, 9). Given that the basis of therapeutic resistance is not fully understood, identifying the pathways employed by tumors to develop resistance to immune-mediated killing may lead to approaches to overcoming resistance and broadening the clinical benefits of cancer immunotherapies.
Dysregulated metabolism in cancer cells is a hallmark of malignant transformation (10). The metabolic alterations in cancer cells exert profound impacts on disease development, progression, and patient outcomes (11, 12). Fatty acid β-oxidation (FAO) is a multistep process that breaks down long-chain fatty acids to acetyl-CoA, which will be fully oxidized through the Krebs cycle and the electron transport chain to produce ATP (13). While FAO is known for its bioenergetic support of cell growth, accumulating evidence shows that FAO is abnormally activated in cancer cells in connection with oncogenes (e.g., c-Myc and c-Src) and contributes to the different aspects of malignancy (13–17). Additionally, FAO activation has been implicated in cancer resistance to certain chemotherapeutic or endocrinal drugs (18–21). However, the potential involvement of this metabolic pathway in tumor responsiveness to cytolytic immune effector cells (e.g., T cells) has yet to be determined.
Carnitine palmitoyltransferase 1 (CPT1), particularly its liver isoform CPT1A, is the rate-limiting enzyme of FAO in many tissues that catalyzes the transfer of long-chain acyl group of acyl-CoA ester to carnitine, thereby shuttling long-chain fatty acids into the mitochondrial matrix through the carnitine transporter for β-oxidation (22). Several studies, including our own, showed that CPT1A-driven FAO can facilitate cancer cell proliferation and survival as well as tumor invasion (15, 23, 24). In the current study, we examine the role of CPT1A-mediated FAO in murine or human cancer cell response to cytolytic immune cells including CTLs and CAR T cells. We demonstrate that cytotoxic stress from killer T cells induces CPT1A expression and FAO activity in cancer cells, which is mainly driven by IFN-γ in an AMP-activated protein kinase (AMPK)-dependent manner. Further, abrogating the FAO pathway using CTP1A-targeted genetic and pharmacologic approaches renders cancer cells more susceptible to cellular immune cytotoxicity. Our findings establish CPT1A-mediated FAO as an important tumor-intrinsic determinant of cancer cell response or resistance to the cytolysis of immune effector cells, which is critical for successful execution of the cancer-immunity cycle to eliminate tumor targets.
Results
FAO Is Elevated in Cancer Cells upon Interaction with Tumor-Reactive T Cells.
Given that FAO activation has been implicated in cancer therapeutic response to certain drugs (18–21), we first examined FAO rates in cancer cells with or without immunolytic pressure of cytotoxic T cells. Using the Seahorse assay to monitor the oxygen consumption rate (OCR) from oxidation of palmitic acid (Fig. 1A), we showed that the FAO activity in gp100-positive murine melanoma B16 cells was consistently elevated when coculturing with the melanoma antigen gp100–specific CD8+ cells derived from Pmel mice (25). We subsequently performed immunoblotting analysis to identify potential molecular changes associated with this metabolic pathway. Among the CPT1 family members, CPT1A exhibited the most significant upregulation in B16 melanoma cells following coculture with Pmel cells (Fig. 1B). The upregulation of CPT1A was similarly seen in murine prostate cancer RM1 cells that have been transfected with the model antigen ovalbumin (OVA), i.e., RM1-OVA, in a cytolytic assay using OVA-specific CD8+ T cells, i.e., OT-I cells (Fig. 1C), suggesting a potential biological function of CPT1A-mediated FAO in cancer cell response to T cell-based immunotherapy. Additionally, coculturing cancer cells (i.e., B16 melanoma) with antigen-nonspecific T cells (i.e., OT-I cells) failed to induce elevation of FAO activity or expression of CPT1A (SI Appendix, Fig. S1), suggesting that tumor recognition by T cells is required for hyperactivation of cancer cell–intrinsic FAO.
Fig. 1.

T cell–mediated cytotoxicity induces FAO elevation in cancer cells. B16 melanoma cells were cocultured with Pmel cells for 6 h. The FAO rate in cancer cells was determined by measuring the oxygen consumption rate (OCR) using Seahorse assays following extensive wash to remove T cells (A). The expression of CPT1 enzymes in B16 melanoma (B) or RM1-OVA prostate cancer cells (C) was examined by immunoblotting. Densitometry analysis of immunoblot replicates for CPT1A expression is shown. Data are representative of at least three independent experiments. *P < 0.05.
CPT1A Deficiency Sensitizes Cancer Cells to T Cell–Mediated Killing.
To examine the impact of the up-regulated FAO on tumor responsiveness to cytolytic immune cells, we established murine B16 melanoma lines, in which CPT1A was deleted using the CRISPR-Cas9 approach. Knockout of CPT1A was confirmed by immunoblotting (Fig. 2A) and RT-qPCR analysis (SI Appendix, Fig. S2A). Expression of other CPT1 family members was not altered in the absence of CPT1A (Fig. 2A and SI Appendix, Fig. S2A). As expected, the Seahorse assay showed that the loss of CPT1A resulted in a markedly decreased FAO rate (Fig. 2B). This reduction of FAO was verified with an FAO measurement method that we developed to directly analyze 3H-palmitic acid conversion to 3H2O (SI Appendix, Fig. S2B) (26). However, the lack of CPT1A did not significantly change the growth of these highly aggressive mouse tumor cells in culture (SI Appendix, Fig. S2C).
Fig. 2.

CPT1A deficiency sensitizes cancer cells to T cell killing. (A) Deletion of CPT1A in B16 with CRISPR-Cas9 was confirmed by immunoblotting. (B) Loss of CPT1A impairs FAO in B16 cells, indicated by OCR reduction in B16CPT1A KO cells. (C) LDH assays of B16WT or B16CPT1A KO cells following coculture with Pmel cells. (D) B16 cells were treated with Etomoxir for 16 h prior to coculture with Pmel T cells and then subjected to cytotoxicity assays. (E) Immunoblotting validation of CPT1A deletion in CRISPR-Cas9-transfected RM1-OVA cells. RM1-OVAWT or RM1-OVACPT1A KO cells were exposed to OVA-specific OT-I cells at a ratio of 1:20 for 6 h, followed by immunoblotting analyses of cell death (F) and proapoptotic or prosurvival signaling (G). Cells collected before coculture with OT-I cells were used as controls (0 h). Data are representative of at least three independent experiments. *P < 0.05. **P < 0.01. ***P < 0.001.
We next performed cytolytic assays by coculturing wild-type (WT) B16 melanoma cells or CPT1A-deficient B16 cells with gp100-specific Pmel cells. The loss of CPT1A indeed rendered B16 tumor cells more prone to killing by antigen-specific CD8+ T cells (Fig. 2C). Additionally, treatment of B16 cells with Etomoxir, a CPT1A inhibitor (26), increased tumor sensitivity to T cell–mediated cytolysis in a dose-dependent manner (Fig. 2D), further confirming the involvement of this metabolic pathway in cancer cell susceptibility to cytotoxic T cells. Similarly, OVA-expressing RM1 (RM1-OVA) prostate cancer cells that lack CPT1A were also significantly more sensitive to the cytotoxicity of OT-I cells (Fig. 2E), even though they showed a comparable growth rate and baseline LDH release in culture as compared to mock transduced counterparts (SI Appendix, Fig. S2 C–E).
FAO Deficiency in Cancer Cells Does Not Alter Antigen Processing and Presentation.
To understand the molecular basis of this improved T cell killing, we asked whether CPT1A loss in cancer cells might result in enhanced activation of CD8+ T cells. However, there was no significant difference in the activation phenotype of T cells cultured with WT or CPT1A deficient cancer cells, indicated by similar expression of the effector cytokine IFN-γ or cell degranulation marker CD107a (SI Appendix, Fig. S3). These T cells also expressed similar levels of immune checkpoint molecules including PD-1 (SI Appendix, Fig. S3), suggesting that the activation and exhaustion status of T cells are not affected by the loss of CPT1A in cancer cells. Instead, CPT1A-mediated FAO may serve as a tumor-intrinsic regulator of cancer response to T cell cytolysis.
A defect in antigen-processing machinery and downregulation of surface MHC-I expression in cancer cells can cause immunotherapeutic resistance (27). The CPT1A loss or inhibition with Etomoxir did not alter MHC-I (i.e., H-2Kb/H-2Db) expression on cancer cells stimulated with IFN-γ (SI Appendix, Fig. S4 A and B). While IFN-γ exposure up-regulated the molecular components of antigen-processing machinery, e.g., transporter associated with antigen processing 1 (TAP1) or TAP2, their overall levels in cancer cells with or without CPT1A remained unchanged (SI Appendix, Fig. S4C). Similar levels of induction of these antigen-processing and presenting molecules in the presence or absence of CPT1A suggest the presence of intact IFN-γ receptor signaling in cancer cells, which has been shown to modulate tumor immunogenicity and resistance to immunotherapy (28, 29). Last, we directly examined processing and presenting of antigen OVA using a monoclonal antibody that selectively reacts with the OVA-derived peptide (i.e., OVA257-264 or SIINFEKL) bound to H-2Kb of MHC class I. Interestingly, RM1-OVA cells lacking CPT1A expressed a lower level of the H-2Kb-OVA257-264 complexes on cell surface relative to their WT counterparts (SI Appendix, Fig. S4D), further indicating that CPT1A absence–sensitized cancer cell killing is less likely to be caused by tumor cell–intrinsic antigen processing and presenting pathways.
CPT1A Loss Enhances Proapoptotic Signaling in Cancer Cells.
The increased cancer cell susceptibility to T cell killing prompted us to examine the molecular mechanism involved in cell death pathways. Immunoblotting analyses showed that loss of CPT1A resulted in increased cleavage of poly-ADP ribose polymerase (PARP), caspase 3 (Cas3), caspase (Cas8), or caspase (Cas9) in RM1 prostate cancer cells under cytolytic stress (Fig. 2F), which was consistent with the data from cytotoxic assays. We also examined prosurvival or proapoptotic proteins of the Bcl-2 family that act as core regulators of the intrinsic pathways of apoptosis (30). Evidently, T cell–mediated cytolytic pressure caused elevation in the prosurvival molecules Bcl-2 and Bcl-Xl (31), which was significantly diminished in CPT1A-deficient RM1 tumor cells (Fig. 2G). Notably, loss of CPT1A resulted in increased expression of Bax, a proapoptotic molecule that promotes mitochondria permeabilization (32, 33) (Fig. 2G). Similarly, we also observed the enhanced proapoptotic signaling in the absence of CPT1A in B16 melanoma cells upon exposure to antigen-specific Pmel T cells (SI Appendix, Fig. S5), suggesting that an imbalance between prosurvival and proapoptotic Bcl-2 protein family members is involved in the enhanced sensitivity of FAO-deficient cells to CTLs.
Lack of CPT1A Renders Metastatic Melanoma Susceptible to T Cell Therapy.
We next sought to examine the role of CPT1A-dependent FAO in therapeutic resistance to T cell therapy using mice established subcutaneously (s.c.) with B16 melanomas that are highly refractory to therapies (34). WT or CPT1A-knockout (KO) B16 tumors grew similarly in vivo without treatment. However, adoptive transfer of gp100-specific Pmel T cells was more efficacious in controlling CPT1A-KO tumors than their WT counterparts (Fig. 3A), which consequently resulted in significantly prolonged lifespan of the mice bearing CPT1A KO B16 tumors (Fig. 3B). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) analysis of tumor sections showed significantly increased cell death in CPT1A-KO tumors following T cell therapy (Fig. 3C). Analyses of tumor infiltration by adoptively transferred Pmel T cells showed that the frequencies in IFN-γ+ or IFN-γ+TNF-α+ Pmel T cells were significantly higher in CPT1A KO B16 melanomas than in WT counterparts (Fig. 3D). Since CPT1A loss in cancer cells does not appear to alter the activation status of T cells in cytolytic coculture assays in vitro, these data suggest that the absence of CPT1A in tumors modulates the tumor microenvironment resulting in improved immune functionality in vivo. The enhanced activation phenotype was also shown by the endogenous CD8+ T cells that were infiltrating CPT1A-KO B16 tumors (Fig. 3E and SI Appendix, Fig. S6A).
Fig. 3.
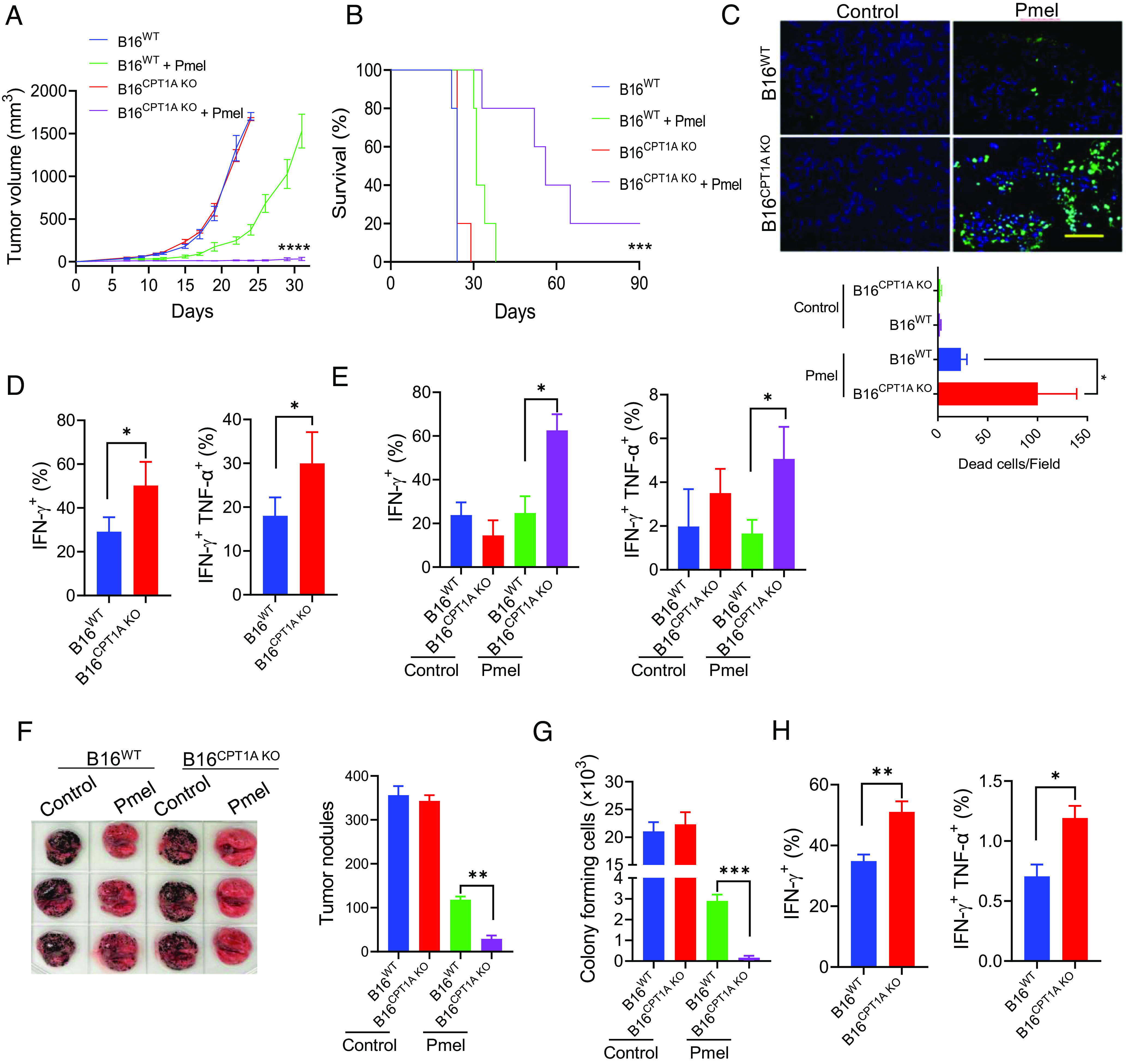
Lack of CPT1A renders metastatic melanoma susceptible to T cell therapy. C57BL/6 mice (n = 5) were injected with B16WT or B16CPT1A KO tumor cells (2 × 105, s.c.) on day 0. Mice received two doses of Pmel T cells on days 4 and 7 (107 cells, i.v.) or left untreated. Tumor growth (A) and animal survival (B) were followed. (C) Tumor-bearing mice were treated with Pmel T cells on day 17 post-tumor inoculation; tumor tissues were collected on day 21 and subjected to TUNEL assays. The frequencies of IFN-γ+ or IFN-γ+ TNF-α+ Pmel cells (D) or IFN-γ+ or IFN-γ+ TNF-α+ endogenous CD8+ T cells (E) in tumors were assessed by intracellular cytokine staining and flow cytometry analysis (gating on total cells in tumor tissue). (F) C57BL/6 mice (n = 5) were established with experimental lung metastases by inoculating B16WT or B16CPT1A KO tumor cells (1 × 105 cells, i.v.) on day 0. Mice received Pmel T cells (107 cells, i.v.) on day 8 or left untreated. Tumor nodules in the lungs were quantified on day 19. (G) Colony-formation assays were performed using lung cell suspensions to examine the frequency of metastatic cells. (H) The frequency of IFN-γ+ and IFN-γ+TNF-α+ CD8+ T cells in the lungs was analyzed by flow cytometry analysis (gating on total cells in lung tissues). Data are representative of three independent experiments. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
We next investigated the effect of CPT1A status on tumor responsiveness to T cell therapy using an experimental melanoma metastasis model (35). The pulmonary metastatic burdens from WT B16 cells and from CPT1A-KO B16 cells were comparable in untreated mice. However, T cell therapy eradicated lung tumor nodules derived from CPT1A-KO B16 cells more efficiently compared to those from WT counterparts (Fig. 3F). The results were further confirmed by in vitro colony-forming assays using lung cell suspensions of mice (Fig. 3G). Consistent with the observations in the s.c. B16 melanoma model, gp100-specific Pmel cells (Fig. 3H and SI Appendix, Fig. S6B) infiltrating the lungs also displayed an increased activation phenotype in mice carrying metastases derived from CPT1A-KO B16 cells, suggesting that lack of CPT1A in tumors may help augment immune activation in vivo during immune attack.
CPT1A-Targeted Inhibition of FAO Improves the Efficacy of T Cell Therapy.
We further examined the generality of FAO-facilitated therapeutic resistance in mice established with RM1-OVA prostate tumors. The RM1-OVA tumors that lack CPT1A were more responsive to T cell therapy using OVA-specific OT-I T cells, as evidenced by significantly enhanced inhibition of tumor growth (Fig. 4A). Increased cytotoxicity shown in CPT1A-KO RM1-OVA tumors compared to WT counterparts following T cell therapy was also confirmed by TUNEL staining (Fig. 4B). Additionally, CPT1A loss in RM1-OVA tumors facilitated the expansion of OVA-specific OT-I T cells, indicated by a higher frequency of Vα2+Vβ5+ CD8+IFN-γ+ OT-I cells in CPT1A-KO RM1-OVA tumors when compared with similarly treated WT tumors (Fig. 4C).
Fig. 4.
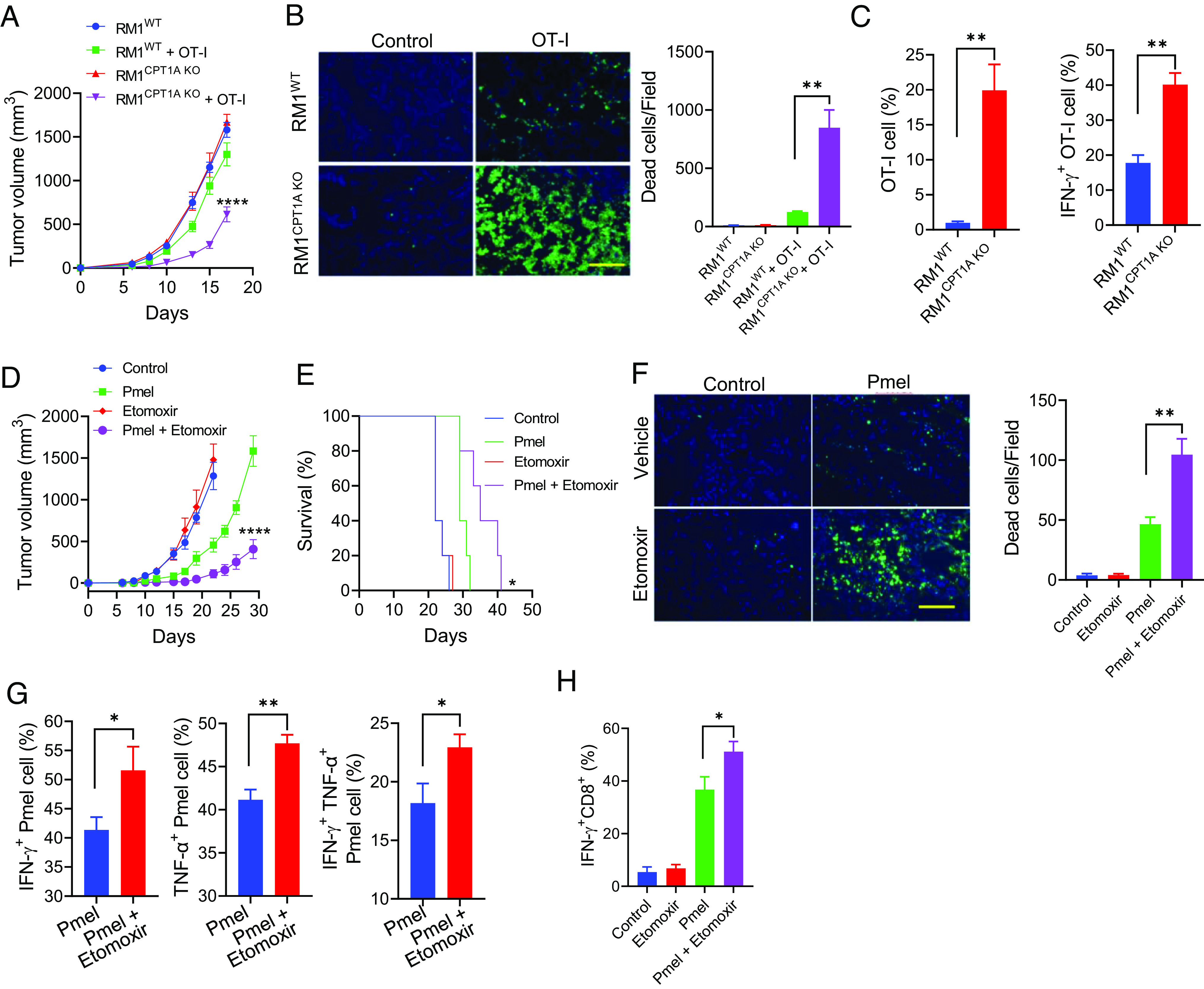
Pharmaceutical inhibition of FAO improves the antitumor efficacy of T cell therapy. Male mice with OVA-expressing RM1WT or RM1CPT1A KO prostate tumors received OT-I cells on days 2, 4, and 7 after tumor implantation (A). Prostate tumor-bearing mice (n = 5) received OT-I cells on days 9 and 12 following tumor implantation. Tumor tissues were collected 2 d after treatment for TUNEL assays (B). Infiltration and activation of OT-I cells in the tumors were assessed (C). Mice (n = 5) with B16WT or B16CPT1A KO melanomas were treated with Pmel T cells on days 4 and 8 post-tumor implantation (day 0). Etomoxir (30 mg/kg, i.p.) was administered daily starting from day 3. Tumor growth (D) and animal survival (E) were followed. (F) Melanoma-bearing mice received Pmel T cells when tumor sizes have reached 8 to 10 mm in diameters. Etomoxir treatment was initiated 2 d before T cell therapy and given daily for a total of seven doses. Tumors were collected 4 d after T cell therapy for TUNEL assays. Intracellular staining and flow cytometry analyses were performed to evaluate the activation of adoptively transferred Pmel T cells (G) or endogenous CD8+ T cells (H) in the tumors. Data are representative of three independent experiments. *P < 0.05. **P < 0.01. ****P < 0.0001.
In addition to these genetic approaches, we tested the role of FAO in tumor response to T cell therapy by pharmacologic inhibition of CPT1A. B16 tumor-bearing mice received gp100-specific T cells with or without treatment with Etomoxir. While Etomoxir alone had little effect on tumor growth, it profoundly enhanced tumor inhibition by Pmel T cells, as indicated by reduced tumor burden (Fig. 4D) and improved host survival (Fig. 4E). The enhanced tumor suppression was also supported by increased cancer cell death in Etomoxir-treated tumors as shown by TUNEL staining (Fig. 4F). Similar to CPT1A-KO tumors, Etomoxir treatment enhanced the activation of adoptively transferred Pmel T cells (Fig. 4G) and endogenous CD8+ T cells (Fig. 4H) in tumor-bearing mice. Together, these results support CPT1A-mediated FAO as a tumor-intrinsic mechanism that can impede cancer responsiveness to T cell cytolysis.
IFN-γ Induces AMPK-Dependent Upregulation of CPT1A and FAO.
Given the fact that IFN-γ is a major cytokine produced by cytotoxic immune effector cells (e.g., CTLs) that exhibits antiproliferative, proapoptotic, and antitumor activities, we sought to examine its effect on CTP1A expression and FAO activity in cancer cells. Surprisingly, IFN-γ treatment highly increased the FAO rate in B16 melanoma cells, confirmed by both FAO diffusion assay (Fig. 5A) and Seahorse assay of OCR (SI Appendix, Fig. S7A). The elevation of FAO was associated with dose-dependent induction of CPT1A and, to a lesser degree, CPT1B or CPT1C (Fig. 5B). The upregulation of CPT1A protein expression also correlated with modest but significant transcriptional activation of the cpt1a gene (SI Appendix, Fig. S7B). Additionally, the presence of IFN-γ also induced phosphorylation of AMPK and acetyl-CoA carboxylase (ACC) (Fig. 5C), which are consistent with their documented roles in regulation of FAO (13, 36).
Fig. 5.
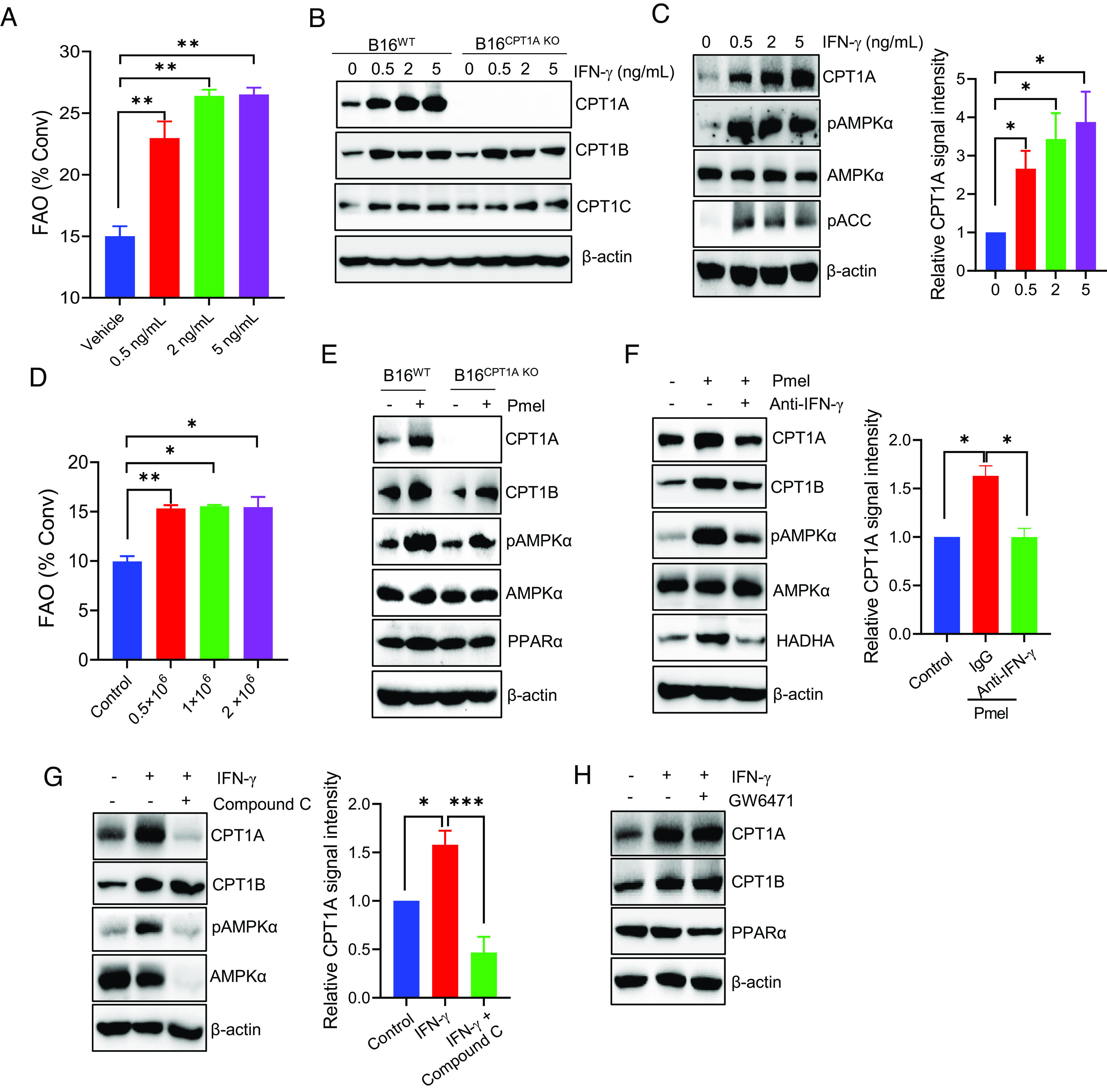
T cell–derived IFN-γ up-regulates CPT1A-mediated FAO in an AMPK-dependent manner. (A) B16 tumor cells were stimulated with IFN-γ at indicated concentrations for 24 h. The FAO activity was determined by measuring conversion of 3H-palmitic acid to 3H2O. (B) The expression of CPT1 enzymes in IFN-γ-treated B16WT or B16CPT1A KO cells was examined by immunoblotting. (C) Analysis of CPT1A, AMPKα, or phosphorylation of AMPKα and ACC in B16 cells after treatment with IFN-γ. (D) B16 cells and Pmel cells were plated into the lower and upper chambers of the transwells, respectively. The FAO activity in B16 cells was determined by diffusion assays after coculture. (E) Immunoblotting analysis of the levels of CPT1A, phosphorylated AMPKα, AMPKα, and PPARα in B16WT or B16CPT1A KO cells after coculture with T cells in transwells plates. (F) B16 cells were cocultured with activated T cells in transwell assays in the presence or absence of IFN-γ-neutralizing antibodies, followed by immunoblotting analysis of CPT1A, pAMPKα, or HADHA in tumor cells. (G and H) B16 tumor cells were treated with IFN-γ (5 ng/mL) in the presence of the AMPKα inhibitor Compound C (G) or PPAR inhibitor GW6471 (H) for 24 h. Immunoblotting was performed to determine the levels of CPT1 enzymes or pAMPKα. Data are representative of three independent experiments. *P < 0.05. **P < 0.01. ***P < 0.001.
Since IFN-γ is a major cytokine produced by activated T cells, we performed transwell assays to further test the effect of soluble factors derived from activated T cells on CPT1A-FAO in cancer cells. Indeed, activated Pmel cells cocultured with B16 melanoma cells in transwell plates efficiently triggered significant elevation of FAO (Fig. 5D) and upregulation of CPT1A in cancer cells (Fig. 5E), indicating that T cell–driven activation of FAO in tumor cells does not require cell–cell contact. To understand the molecular mechanism underlying CPT1A induction, we examined expression of PPARα and phosphorylation status of AMPKα, both of which are known to regulate FAO enzymes including CPT1A (26, 37). Coculture with activated T cells in the transwells strongly induced phosphorylation of AMPKα while having little effect on PPARα expression (Fig. 5E). Similarly, upregulation of CPT1A and activation of AMPKα were also seen in RM1-OVA prostate cancer cells after coculture with OVA-reactive OT-I T cells (SI Appendix, Fig. S8).
We next performed antibody neutralization to validate the critical role of IFN-γ in T cell-induced CPT1A elevation. Neutralization of IFN-γ significantly reduced expression of CPT1A, phosphorylation of AMPKα, as well as expression of HADHA, another enzyme of the FAO pathway (Fig. 5F), supporting IFN-γ as a major soluble factor responsible for inducing CPT1A and FAO in cancer cells upon interaction with cytolytic T cells. The inclusion of AMPK inhibitor Compound C during IFN-γ treatment abrogated induction of CPT1A (Fig. 5G), whereas PPARα inhibition had little effect on CPT1A expression (Fig. 5H), suggesting that AMPK acts as a primary mediator of IFN-γ-triggered activation of CPT1A.
Inhibition of FAO Improves Human Breast Cancer Response to CAR T Cells.
Based on publicly available gene expression databases, all subtypes of breast cancer overexpress CPT1A mRNA (SI Appendix, Fig. S9). To understand the clinical relevance of CPT1A overexpression, we analyzed RNAseq data downloaded from the Tumor Cancer Genome Atlas (TCGA) breast cancer dataset. Patients whose tumor expression of CPT1A was one normalized SD above the mean of CPT1A were grouped as CPT1AHigh, whereas those with one normalized SD below the mean were grouped as CPT1ALow. Kaplan–Meier survival and the Gehan–Breslow–Wilcoxon test showed that CPT1ALow patients had a statistically significant longer overall survival than that of CPT1AHigh patients (P = 0.0002) (Fig. 6A), suggesting that CPT1A is not only overexpressed but also associated with poor prognosis in human breast cancer. A similar correlation was also seen in patients with pancreatic cancer, ovarian cancer, and bladder carcinoma (SI Appendix, Fig. S10). Consistent with our observations in murine cancer cells, IFN-γ treatment similarly up-regulated the expression of CPT1A and phosphorylation of AMPKα (Fig. 6B) in human breast cancer MDA-MB-231 cells, which was associated with the elevation of FAO activity (Fig. 6C). In addition to the immune signal IFN-γ, we found that treatment with chemotherapeutic drugs also elevated FAO in human breast cancer cell lines (SI Appendix, Fig. S11), suggesting that FAO may represent a general prosurvival mechanism contributing to therapeutic resistance.
Fig. 6.
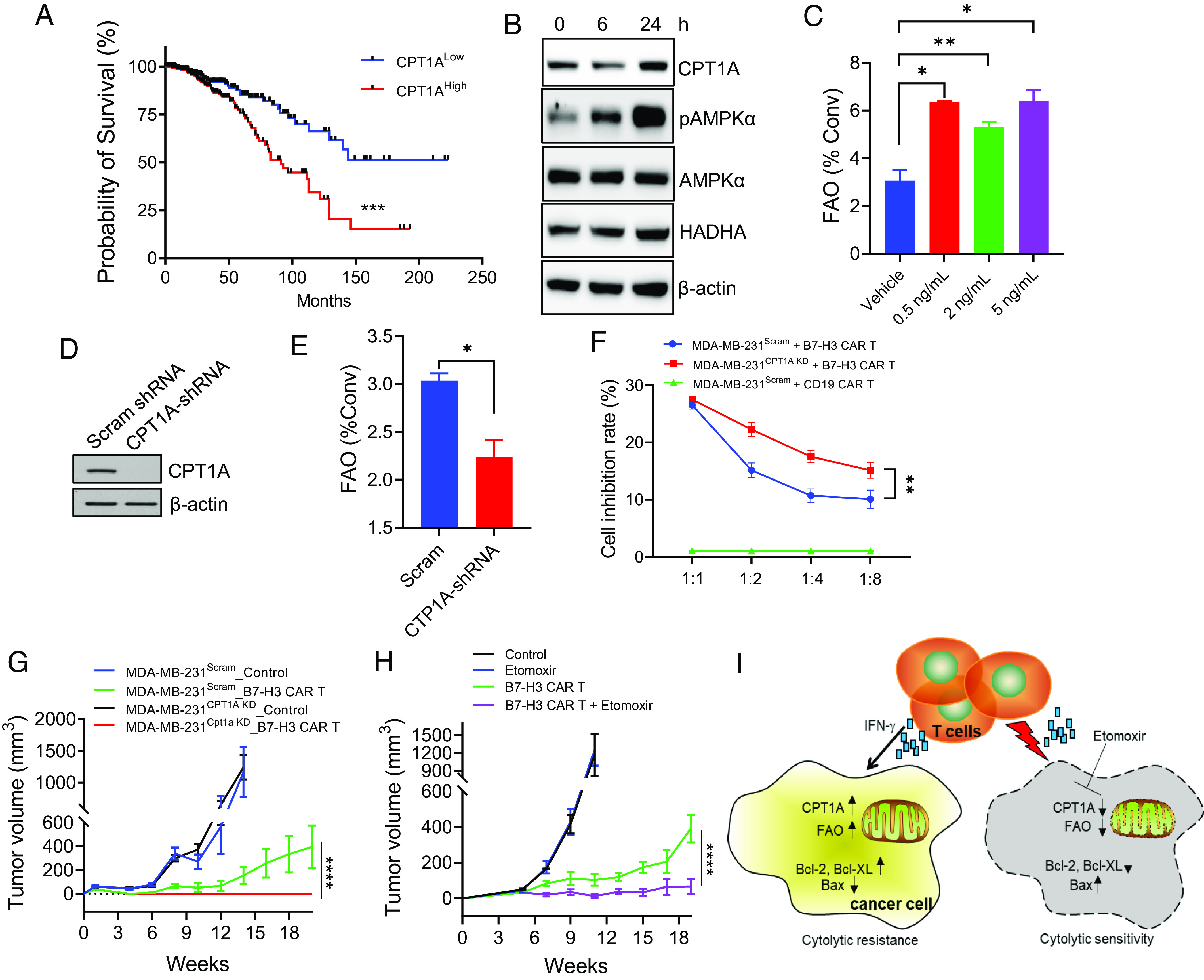
Blockade of FAO improves human breast cancer responsiveness to CAR T therapy. (A) The publicly available data on Oncomine were used to analyze the correlation of CPT1A mRNA expression and survival in patients with breast cancer. Kaplan–Meier survival and Gehan–Breslow–Wilcoxon test of two groups were performed using GraphPad PRISM software. (B) Human breast cancer MDA-MB-231 cells were stimulated with IFN-γ and analyzed for expression of CPT1 enzymes, pAMPKα, PPARα, or HADHA. (C) FAO analysis of MDA-MB-231 cells after IFN-γ stimulation at different concentrations. (D) Knockdown (KD) of CPT1A in MDA-MB-231 cells by short hairpin RNA (shRNA) was confirmed in comparison with those transduced with scramble shRNA. (E) FAO activity in MDA-MB-231Scram or MDA-MB-231CPT1A KD cells was measured by quantifying the conversion of 3H-palmitic acid to 3H2O. (F) MDA-MB-231 cells with or without CPT1A KD were cocultured with B7-H3 CAR T cells for 48 h followed by MTT assays. (G) NSG mice (n = 5) with MDA-MB-231 tumors with or without CPT1A KD were treated with B7-H3 CAR T cells (2 × 106 cells, i.v.) 1 wk after tumor implantation. (H) NSG mice with MDA-MB-231 tumors were treated with B7-H3 CAR T cells 5 wk after tumor implantations. Etomoxir treatment was initiated 1 d before CAR T therapy and administered daily during the course of study. Data are representative of three independent experiments. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001. (I) Schematic illustration of tumor resistance to immune cytolysis by IFN-γ-induced FAO in cancer cells. Upon recognition and destruction of tumor targets, IFN-γ-derived from activated T cells results in FAO elevation in cancer cells by inducing CPT1A expression via AMPKα activation. Activation of this metabolic pathway confers cancer cell resistance to immune effector cells by promoting prosurvival signaling, e.g., upregulation of Bcl-2 and Bcl-XL and downregulation of Bax. Targeted inhibition of CPT1A or blockade of FAO (e.g., Etomoxir) can enhance proapoptotic signaling, thereby potentiating tumor sensitivity to immune killing.
To investigate whether CPT1A overexpression in breast cancer is involved in resistance to immune-mediated cytolysis, we used shRNA to knock down CPT1A in human breast cancer MDA-MB-231 line (Fig. 6D), which represents highly aggressive, invasive, and poorly differentiated triple-negative breast cancer. As expected, CPT1A downregulation markedly reduced the FAO rate in MDA-MB-231 cells, assessed by the FAO diffusion assay (Fig. 6E) or Seahorse assay (SI Appendix, Fig. S12A). Since MDA-MB-231 cells highly express B7-H3 (SI Appendix, Fig. S12B), an immune checkpoint molecule that is aberrantly expressed in multiple solid cancers but rarely in normal tissues, we next examined the sensitivity of MDA-MB-231 cells with or without CPT1A to B7-H3 CAR T cells that have shown potent antitumor activity (38, 39). Consistent with our results from murine cancer models, silencing of CPT1A rendered MDA-MB-231 cells more susceptible to killing by B7-H3 CAR T cells (Fig. 6F). Since oncogenic signaling has been shown to regulate cancer cell resistance to immunotherapy (40), we also examined the potential effect of CPT1A ablation on the epidermal growth factor receptor (EGFR) pathway. The EGFR signaling appeared to be unchanged in WT or CPT1A knockdown MDA-MB-231 cells following stimulation with an EGFR ligand (SI Appendix, Fig. S12C), excluding the involvement of oncogenic EGFR signaling in FAO conferred cancer cell resistance to CAR T cell killing. Increased immune sensitivity upon CPT1A downregulation was also confirmed using another human breast cancer cell line, MCF-7. Intriguingly, MCF-7 cells with CPT1A downregulation were more sensitive than their WT counterparts or mock-treated cells to the killing by natural killer (NK) cells (SI Appendix, Fig. S13), the innate immune effector cells which use a similar lytic granule release mechanism for target destruction like CTLs (41). This further suggests that the CPT1A-FAO pathway may be broadly involved in immunolytic resistance in cancer cells.
Last, we evaluated the in vivo responsiveness of MDA-MB-231 tumors with or without CPT1A to cellular immunotherapy using B7-H3 CAR T cells. While B7-H3 CAR T cells exhibited robust therapeutic activity in treating both established tumors, CPT1A knockdown MDA-MB-231 tumors were significantly more responsive to CAR T therapy than their WT counterparts (Fig. 6G). Additionally, we showed that treatment of MDA-MB-231 tumor-bearing NSG mice with Etomoxir potentiated their response to B7-H3 CAR T cells in a similar manner (Fig. 6H), further underscoring a critical role of the CPT1A-FAO pathway in immunolytic resistance. Adoptively transferred CAR T cells showed similar activation phenotypes (i.e., IFN-γ+Granzyme B+) in mice bearing WT or CPT1A knockdown tumors as well as those treated with Etomoxir (SI Appendix, Fig. S14), suggesting that inhibition of FAO does not alter activation status of CAR T cells in NSG mice.
Discussion
Despite the remarkable success of cancer immunotherapy in the clinical management of a wide array of malignancies, the limited responses in the majority of patients indicate a need for better understanding of the molecular basis of immune resistance. Many tumor-intrinsic factors including poor antigenicity, oncogenic signaling, impaired interferon signaling, and overexpression of inhibitory checkpoint molecules have been recognized for their contributions to resistance to immunotherapies. In the present study, we have identified CPT1A-dependent FAO as a tumor autonomous factor that can define cancer cell sensitivity to killing by immune cytolytic cells (i.e., CTLs, CAR T). Given that CTLs are primarily responsible for eradicating tumor targets in the last phase of the cancer-immunity cycle, our findings have important implications for understanding tumor immune escape from the current T cell-based therapies and immunotherapies in general.
Metabolic reprogramming is a well-established hallmark of cancer, allowing tumor cells to fuel unrestrained proliferation(10). In addition to energy production for supporting tumor growth, FAO activation has been implicated in cancer resistance to certain drugs (18–21). In the current work, we have established CPT1A-mediated FAO activation as an important mechanism employed by cancer cells to resist cytolysis by CTLs or CAR T cells. First, murine tumor lines (i.e., melanoma, prostate cancer) lacking CPT1A become highly susceptible to killing by antigen-specific CD8 T cells (i.e., Pmel, OT-I) when compared to their WT counterparts. Although CPT1A downregulation was previously reported to inhibit cancer cell growth (15, 23, 24), the aggressive growth of tumor models used in our study (i.e., B16, RM1) did not seem to be affected by the CPT1A deletion. Their similar growth rates in culture and in vivo provide us a suitable system that allows reliably measuring their sensitivities to cytotoxic T cells. Second, we have demonstrated FAO conferred resistance to T cell therapy in mice bearing subcutaneous tumors or experimental metastases, indicated by significantly reduced tumor volumes or metastatic burdens as well as prolonged survival upon tumor depletion of CPT1A. Third, CPT1A stable knockdown also renders human breast cancer MDA-MB-231 prone to destruction by B7-H3 CAR T cells both in vitro and in vivo. Last, these observations are further supported by the experiments using the FAO pharmacological inhibitor Etomoxir, which similarly sensitizes these tumors to antigen-specific T cell therapy or CAR T therapy. Intriguingly, CPT1A downregulation or FAO blockade also enables human breast cancer cells more responsive to NK cell–mediated cytotoxicity, suggesting that FAO-driven immune resistance may be universal toward both innate and adaptive immune surveillance.
FAO flux is up-regulated in cancer cells both under attack by tumor-reactive CD8+ T cells and treated with chemotherapeutic drugs (SI Appendix, Fig. S11), suggesting that activation of FAO provides a general survival advantage against different types of therapeutic stress and therefore has broad implications in cancer therapies. Indeed, tumor biopsies from metastatic stage IV melanoma receiving mitogen-activated protein kinase inhibitors also exhibited increased levels of CPT1A-dependent FAO (42). Most previous studies of cancer bioenergetics have focused on the Warburg effect whereby cancer cells up-regulate glycolysis to provide quick ATP and biosynthetic intermediates favoring rapidly proliferating tumor cells. Unlike mitochondrial oxidative phosphorylation, glycolysis is poorly efficient in ATP generation. We speculate that the tumor microenvironmental stress and therapeutic pressures caused by CTLs could trigger metabolic reprograming in cancer cells by shifting from glycolysis to FAO as alternative bioenergetic and metabolic sources. Consistent with this, many types of cancer, particularly breast cancer, are characterized with CPT1A overexpression (SI Appendix, Fig. S9) and a hyperactive FAO phenotype (16, 43). In an integrated genomics study, CPT1A was identified as one of eight genes essential for the luminal subtype of breast cancer (44) and CPT1A overexpression correlates strongly with poor survival of breast cancer patients (Fig. 6A). It is possible that this metabolic reprograming in cancer cells contributes to adaptive resistance as well as immunoediting (45, 46), culminating in the selection and escape of immune-resistant cancer cells during immunotherapies that are designed to mobilize cytolytic immune cells such as CTLs or NK cells.
Our mechanistic studies provide molecular insights into elevation of FAO in cancer cells under immune cytolytic pressure. We have identified IFN-γ as a major signal that can induce expression of CPT1A and other CPT1 family members, resulting in increased FAO in cancer cells. Moreover, the elevation in CPT1A and FAO triggered by IFN-γ depends mainly on AMPK. AMPK regulates metabolic homeostasis by shutting down energy-consuming biosynthesis and activating energy-yielding catabolic processes such as FAO (36). It is known that AMPK stimulates FAO through phosphorylation and inactivation of ACC, which synthesizes malonyl-CoA, an allosteric inhibitor of CPT1 to control FAO rates in physiological conditions(13, 36). We indeed observed elevation of ACC phosphorylation in association with AMPK activation in cancer cells responding to IFN-γ stimulation (Fig. 5C). Given the significant upregulation of CPT1A expression, it is apparent that AMPK exerts diverse effects on the FAO pathway beyond its regulation of ACC. It remains to be determined how exactly T cell–derived IFN-γ signal triggers activation of the AMPK–CPT1A-FAO axis. Nonetheless, our work has unraveled an immune-driven metabolic alteration in cancer cells during their interactions with killer T cells, which can define the fate of cancer cells as well as the outcome of immunotherapy. Our finding also adds another layer of complexity to the dynamic interplay between immune effector cells and their targets, which contributes to the current understanding of the mechanisms underlying cancer resistance to immunotherapy.
CPT1A-mediated FAO represents a distinct tumor-intrinsic pathway that confers resistance to immune cytolytic activity because disruption of CPT1A does not appear to alter tumor-intrinsic factors known to modulate immune resistance such as antigen-processing and presentation machinery, interferon signaling, or expression of immune checkpoint molecules. Given that FAO is often up-regulated in multiple cancers, our findings highlight a connection of deregulated cancer metabolism with immune evasion that occurs during tumor attack by cytolytic immune cells. Our molecular studies reveal that CPT1A-mediated FAO is required for prosurvival signaling in cancer cells under cytolytic immune pressure. This is indicated by impaired induction of the prosurvival molecules (i.e., Bcl-2 and Bcl-XL) and concomitant upregulation of the proapoptotic molecule (i.e., Bax). It has been well recognized that the interplay between positive and negative regulators of the Bcl-2 family members establishes a highly sophisticated network that can control cell apoptosis (47). In support of our observations, the overexpression of antiapoptotic proteins such as Bcl-2 has been shown to promote tumor resistance to immunotherapies (48). Additionally, the Bcl-2 family protein Mcl-1 was shown to promote FAO by interacting with long-chain acyl-CoA dehydrogenase (49), and FAO can regulate the Bak-dependent mitochondrial permeability in human leukemia cells (14). While the biochemical basis remains unclear, the prosurvival effect of FAO has also been linked to maintenance of NADPH homeostasis or antagonization of oxidative stress (13). Given that evasion of apoptosis is a hallmark of cancer, tipping the proapoptotic and antiapoptotic balance in favor of survival by CPT1A-mediated FAO may represent a potentially unifying mechanism that operates under these cytotoxic stresses induced by killer T cells or drugs.
Although impairing FAO by CPT1A depletion potentiates cancer cell sensitivity to immune cytotoxicity, this enhanced tumor killing shown in cytolytic assays is not due to altered activation status of tumor-reactive T cells, which is supported by their similar production of the effector cytokine IFN-γ or surface expression of coinhibitory molecules. However, there was a striking increase in immune activation within the tumors that either lack CPT1A or have been treated with the FAO inhibitor, evidenced by high frequencies of IFN-γ+ and/or TNF-α+ tumor-infiltrating CD8+ T cells following T cell therapy. This phenotype of enhanced activation can be seen not only in those adoptively transferred T cells but also in the endogenous CD8+ T cells, suggesting that CPT1A-targeted FAO blockade in tumor may reprogram the immune compartment of the tumor in vivo in responding to T cell therapy. This discrepancy in immune phenotypes may be explained by the distinct impact of tumor-intrinsic FAO inhibition on cancer-immune interplay within the tumor microenvironment. It is possible that T cell attack triggers immunogenic cell death when CPT1A-mediated FAO is impaired in cancer cells, which can amplify antitumor immune responses by re-engaging the multiple phases of cancer-immunity cycle, i.e., cross-presentation of tumor-released antigens, expansion of tumor-reactive T cells. Under cytolytic immune pressure, inhibition of CPT1A-driven FAO also leads to robust activation of caspase cascades in cancer cells, which have been reported to define the immunogenicity of cancer cell death and consequent protective CTL response (50, 51). Alternatively, tumor-intrinsic FAO may impact on metabolic cues and immune environment in vivo. It has been recognized that tumor-intrinsic factors can also indirectly contribute to tumor-extrinsic mechanisms of therapeutic resistance by affecting the interplay between the tumor and the immune environment (52). This scenario is supported by a recent study showing that inhibiting tumor mitochondrial respiration with the mitochondrial complex 1 inhibitor metformin enhanced tumor response to anti-PD-1 treatment through alleviating oxygen tension within the tumor microenvironment to improve T cell activation and function (53, 54). Nonetheless, additional studies are necessary to confirm the intriguing roles of tumor-intrinsic FAO in shaping tumor immunogenicity and possibly the immune microenvironment.
CAR T cell therapy has produced impressive clinical responses in patients with hematologic malignancies but generated disappointing results in the treatment of solid cancers. The lack of efficacy is likely caused by the immune escape mechanisms utilized by targeted cancer cells, including innate or adaptive immune resistance. B7-H3 is an immune inhibitory molecule overexpressed in a large number of solid cancers, and recent preclinical studies have demonstrated remarkable therapeutic effects of B7-H3 CAR T cells against solid tumors (39, 55). Currently, there are multiple ongoing clinical trials evaluating B7-H3 CAR T cell therapy in patients with solid tumors (e.g., glioblastoma, melanoma, sarcoma, ovarian cancer). In this study, we have provided experimental evidence supporting the use of B7-H3 CAR T cells to eradicate human breast cancer. Additionally, CPT1A knockdown or Etomoxir treatment can significantly improve the sensitivity of human breast cancer to B7-H3 CAR T cells in vitro and in vivo, further underscoring the important role of tumor-intrinsic FAO in immune resistance. Since FAO has also been implicated in regulation regulatory T cells or myeloid-derived suppressive cells that are known for their T cell inhibitory activity (56, 57), the pharmacological approaches targeting FAO strategically may be exploited to broaden the impact of cancer immunotherapies including CAR T cell therapy.
Accumulating preclinical and clinical research has now recognized that a number of cancer-cell-autonomous cues together with tumor-microenvironmental factors account for the heterogeneous responses and failures often encountered during immunotherapies. Our work has established tumor-intrinsic FAO as a mechanism supporting tumor survival and potentially facilitating its escape from direct immune attack (Fig. 6I). Considering that cytolytic destruction of cancer cells is the most critical step of the cancer-immunity cycle, a better understanding of this metabolic pathway may provide opportunities to overcome tumor resistance to immunotherapies for improved outcomes.
Materials and Methods
Mice and Cell Lines.
OT-I mice and Pmel mice were purchased from the Jackson Laboratory (Bar Harbor, ME). C57BL/6 mice were purchased from Envigo (Somerset, NJ). NOD.CB17-Prkdcscid Il2rgtm1Wjl/SzJ NSG mice were provided by the Virginia Commonwealth University (VCU) Cancer Mouse Models Core. The mouse melanoma B16 cell line and human breast cancer cell line MD-MBA-231 were purchased from ATCC (Manassas, VA). OVA-expressing mouse prostate cancer RM1 cells were created by transfecting RM1 cells with a plasmid encoding ovalbumin as we previously described (58). Tumor cells were maintained in Dulbecco’s Modified Eagles’ Media supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin (Hyclone, Logan, UT). All experimental procedures were conducted according to the protocols approved by the VCU Institutional Animal Care and Use Committee.
Preparation and Expansion of Murine T Cells.
Ex vivo expansion of antigen-specific T cells was performed as we previously described (35). Briefly, single-cell suspensions from the spleen and lymph nodes of OT-I mice or Pmel mice (2 × 106 cells/mL) were cultured in complete RPMI1640 media containing OVA257 to 264 or gp10025 to 33 (1 μg/mL) peptides and IL-2 (80 IU/mL) for 16 to 18 h (day 0). On day 1, cells were washed three times with warm medium and cultured at 106 cells/mL in RPMI1640 media containing IL-7 and IL-15 (10 ng/mL each). Medium were replaced and cells split every 2 d. T cells on day 5 were harvested and used for experiments.
Generation of B7-H3 CAR T Cells.
B7-H3 CAR T cells were prepared as we previously described (55). Deidentified peripheral blood mononuclear cells from normal human donor blood (1 × 106 cells/well) were activated with 1 μg/mL anti-CD3 (clone OKT3; Miltenyi Biotec, Germany) and 3 μg/mL anti-CD28 antibodies (clone CD28.2; BD Biosciences, CA). T cells were expanded by addition of 10 ng/mL of IL-7 and 5 ng/mL IL-15 on day 1. Cells were transduced on day 2 with retroviral particles of the B7-H3 CAR construct using RetroNectin (Takara Bio Inc., Japan) precoated 24-well plates (39). CAR T cells were collected and stored on days 13 to 14.
Metabolic Flux Assays.
Cellular FAO activity was quantified by measuring OCR using XF-24 Extracellular Flux Analyzers (Agilent Technologies, Santa Clara, CA) following the manufacturer’s instructions. Alternatively, FAO activity was measured directly by quantifying the conversion of 3H-palmitate to 3H2O as we previously described (26).
In Vitro Cytotoxicity Assays.
Cancer cells were plated in U-bottom 96-well plates and cocultured with T cells at indicated effector to target ratios for 24 or 48 h. Supernatants were collected for lactate dehydrogenase analysis using the CytoTox 96® Cytotoxicity Assay kit from Promega (Madison, WI). Alternatively, the viability of cancer cells was quantified by the MTT assay after removing T cells.
Tumor Studies.
Mice with s.c. tumors or experimental lung metastases received 4.5-Gy whole-body irradiation followed by intravenous (i.v.) injection of antigen-specific T cells at the indicated time points. Analysis of tumor-infiltrating leukocytes was performed using intracellular cytokine staining and/or flow cytometry as previously described (59).
Statistical Analysis.
Data are expressed as mean ± SEM values. Statistical differences between groups within experiments were determined by the Student t test. The two-way repeated-measures ANOVA test was performed to compare the tumor growth rate of tumor-bearing mice. The log-rank test was used to compare survival of tumor-bearing mice between experimental groups. Values of P < 0.05 were considered to be statistically significant.
For more method details, see SI Appendix, Supplementary Materials.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported in part by CA229812, CA240004, CA259599, CA226981, and W81XWH2110433, and the Virginia Commonwealth University (VCU) Massey Cancer Center Research Development Funds. Services in support of the research project were provided by the VCU Massey Cancer Center Flow Cytometry Core and Cancer Mouse Models Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Author contributions
X.F., C.G., and X.-Y.W. designed research; Z.L., W.L., W.W., Y.M., Y.W., D.L.D., J.C., H.B., and C.G. performed research; Z.L., W.L., W.W., Y.M., Y.W., D.L.D., J.C., H.B., C.G., and X.-Y.W. analyzed data; and Z.L., E.L., S.S., P.B.F., X.W., X.F., C.G., and X.-Y.W. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. G.K. is a guest editor invited by the Editorial Board.
Contributor Information
Xianjun Fang, Email: xianjun.fang@vcuhealth.org.
Chunqing Guo, Email: chunqing.guo@vcuhealth.org.
Xiang-Yang Wang, Email: xiang-yang.wang@vcuhealth.org.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Mellman I., Coukos G., Dranoff G., Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldman A. D., Fritz J. M., Lenardo M. J., A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ribas A., Wolchok J. D., Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim W. A., June C. H., The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Donnell J. S., Teng M. W. L., Smyth M. J., Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finck A. V., Blanchard T., Roselle C. P., Golinelli G., June C. H., Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 28, 678–689 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunn G. P., Old L. J., Schreiber R. D., The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Kalbasi A., Ribas A., Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Galluzzi L., Kepp O., Vander Heiden M. G., Kroemer G., Metabolic targets for cancer therapy. Nat. Rev. Drug. Discov. 12, 829–846 (2013). [DOI] [PubMed] [Google Scholar]
- 12.DeBerardinis R. J., Thompson C. B., Cellular metabolism and disease: What do metabolic outliers teach us? Cell 148, 1132–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y., et al. , Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 435, 92–100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samudio I., et al. , Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Invest. 120, 142–156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao H., et al. , Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 7, 3832–3846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camarda R., et al. , Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 22, 427–432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T., et al. , JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27, 1357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tung S., et al. , PPARalpha and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood 122, 969–980 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Shinohara H., et al. , Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett. 371, 1–11 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Farge T., et al. , Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 7, 716–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitajima S., et al. , The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 36, 5145–5157 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Houten S. M., Violante S., Ventura F. V., Wanders R. J., The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu. Rev. Physiol. 78, 23–44 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Park J. H., et al. , Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 14, 2154–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y. N., et al. , CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 37, 6025–6040 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Abad J. D., et al. , T-cell receptor gene therapy of established tumors in a murine melanoma model. J. Immunother. 31, 1–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y., et al. , Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci. Rep. 10, 1450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Restifo N. P., et al. , Identification of human cancers deficient in antigen processing. J. Exp. Med. 177, 265–272 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J., et al. , Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404.e399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sucker A., et al. , Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat. Commun. 8, 15440 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh R., Letai A., Sarosiek K., Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng E. H., et al. , BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Hsu Y. T., Wolter K. G., Youle R. J., Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. U.S.A. 94, 3668–3672 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lovell J. F., et al. , Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Ribas A., Kirkwood J. M., Flaherty K. T., Anti-PD-1 antibody treatment for melanoma. Lancet Oncol. 19, e219 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Liu Z., et al. , Engineering T cells to express tumoricidal MDA-7/IL24 enhances cancer immunotherapy. Cancer Res. 81, 2429–2441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie D. G., AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Djouadi F., Bastin J., Mitochondrial genetic disorders: Cell signaling and pharmacological therapies. Cells 8, 289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Picarda E., Ohaegbulam K. C., Zang X., Molecular pathways: Targeting B7–H3 (CD276) for human cancer immunotherapy. Clin. Cancer Res. 22, 3425–3431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du H., et al. , Antitumor responses in the absence of toxicity in solid tumors by targeting B7–H3 via chimeric antigen receptor T cells. Cancer Cell 35, 221–237.e228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai R., et al. , Mechanisms of cancer resistance to immunotherapy. Front. Oncol. 10, 1290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiang S. C., et al. , Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 121, 1345–1356 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Aloia A., et al. , A fatty acid oxidation-dependent metabolic shift regulates the adaptation of BRAF-mutated melanoma to MAPK inhibitors. Clin. Cancer Res. 25, 6852–6867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park J. H., et al. , Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 14, 2154–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gatza M. L., Silva G. O., Parker J. S., Fan C., Perou C. M., An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 46, 1051–1059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeda K., et al. , IFN-gamma is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat. Commun. 8, 14607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreiber R. D., Old L. J., Smyth M. J., Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Pena-Blanco A., Garcia-Saez A. J., Bax, Bak and beyond - mitochondrial performance in apoptosis. FEBS J. 285, 416–431 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Lickliter J. D., et al. , Small-molecule Bcl-2 inhibitors sensitise tumour cells to immune-mediated destruction. Br J. Cancer 96, 600–608 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Escudero S., et al. , Dynamic regulation of long-chain fatty acid oxidation by a noncanonical interaction between the MCL-1 BH3 helix and VLCAD. Mol. Cell 69, 729–743.e727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casares N., et al. , Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaime-Sanchez P., et al. , Cell death induced by cytotoxic CD8(+) T cells is immunogenic and primes caspase-3-dependent spread immunity against endogenous tumor antigens. J. Immunother. Cancer 8, e000528 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitt J. M., et al. , Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity 44, 1255–1269 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Scharping N. E., Menk A. V., Whetstone R. D., Zeng X., Delgoffe G. M., Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 5, 9–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Najjar Y. G., et al. , Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 4, e124989 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y., et al. , Targeting radiation-resistant prostate cancer stem cells by B7–H3 CAR T cells. Mol. Cancer Ther. 20, 577–588 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michalek R. D., et al. , Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hossain F., et al. , Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 3, 1236–1247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo C., et al. , Absence of scavenger receptor A promotes dendritic cell-mediated cross-presentation of cell-associated antigen and antitumor immune response. Immunol. Cell Biol. 90, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yi H., et al. , Targeting the immunoregulator SRA/CD204 potentiates specific dendritic cell vaccine-induced T-cell response and antitumor immunity. Cancer Res. 71, 6611–6620 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.