

Significance
Vibrio cholerae colonizes the human intestine to cause the disease cholera. How V. cholerae binds to intestinal cells is still not well understood. We identified a peptide-binding domain (PBD) in the FrhA adhesin that facilitates V. cholerae binding to human cells. The FrhA PBD is functionally interchangeable with the PBD from the Antarctic bacterium Marinomonas primoryensis, which uses it to bind to diatoms. Peptide inhibitors that bind M. primoryensis PBD can disrupt V. cholerae human cell binding, biofilm formation, and intestinal colonization. These studies demonstrate how a protein domain that enhances colonization of microorganisms in the marine environment can also enhance colonization in humans and identify a peptide that can inhibit V. cholerae virulence.
Keywords: cholera, peptide-binding, adhesin
Abstract
Vibrio cholerae, the causative agent of the disease cholera, is responsible for multiple pandemics. V. cholerae binds to and colonizes the gastrointestinal tract within the human host, as well as various surfaces in the marine environment (e.g., zooplankton) during interepidemic periods. A large adhesin, the Flagellar Regulated Hemagglutinin A (FrhA), enhances binding to erythrocytes and epithelial cells and enhances intestinal colonization. We identified a peptide-binding domain (PBD) within FrhA that mediates hemagglutination, binding to epithelial cells, intestinal colonization, and facilitates biofilm formation. Intriguingly, this domain is also found in the ice-binding protein of the Antarctic bacterium Marinomonas primoryensis, where it mediates binding to diatoms. Peptide inhibitors of the M. primoryensis PBD inhibit V. cholerae binding to human cells as well as to diatoms and inhibit biofilm formation. Moreover, the M. primoryensis PBD inserted into FrhA allows V. cholerae to bind human cells and colonize the intestine and also enhances biofilm formation, demonstrating the interchangeability of the PBD from these bacteria. Importantly, peptide inhibitors of PBD reduce V. cholerae intestinal colonization in infant mice. These studies demonstrate how V. cholerae uses a PBD shared with a diatom-binding Antarctic bacterium to facilitate intestinal colonization in humans and biofilm formation in the environment.
Vibrio cholerae is a gram-negative bacterium that is naturally found in the marine environment. These bacteria cause the disease cholera by colonizing the gastrointestinal tract after ingestion and expressing cholera toxin (CT), which causes the life-threatening diarrhea that is associated with infection (1). V. cholerae O1 serogroup strains have been responsible for multiple cholera pandemics, with the sixth pandemic classical biotype strains being replaced by El Tor biotype strains during the ongoing seventh pandemic (2). A defining feature of V. cholerae is the single polar flagellum responsible for its motility. Flagellar-mediated motility and chemotaxis are integrated into the V. cholerae lifestyle and contribute to virulence as well as environmental persistence (biofilms) (3). Flagellar synthesis is regulated by a transcription hierarchy, a stepwise regulatory pathway that ensures that flagellar genes are expressed in the order they are assembled into the flagellar structure (4–6). The flagellar transcription hierarchy also controls expression of nonflagellar genes, including positively regulating a hemagglutinin, Flagellar-Regulated Hemagglutinin A (FrhA), that contributes to intestinal colonization and biofilm formation (5).
Multiple adhesins have been implicated in the attachment of V. cholerae to various surfaces. The Toxin Co-regulated Pilus (TCP) is a Type IV pilus (T4P) that is essential for V. cholerae intestinal colonization (7) but plays no role in adhesion in aquatic environments. While TCP has been visualized to mediate adhesion to intestinal cells, it appears not to be required for initial intestinal adherence and plays more dominant roles in bacterial-bacterial interactions and as the receptor for the CT-encoding bacteriophage (8, 9), indicating that other adhesins contribute to V. cholerae binding within the host. The Mannose Sensitive Hemagglutinin (MSH) is also a T4P that is required for attachment to zooplankton and other environmental surfaces where V. cholerae colonizes and forms biofilms (10–12) but plays no role in intestinal colonization. mshA mutants are still able to bind zooplankton (10), indicating that other adhesins also contribute to V. cholerae binding in the environment. A V. cholerae chitin-binding protein GbpA binds to GlcNaC residues and mediates binding to both epithelial cells and zooplankton (13), and FrhA has also been shown to mediate binding to both epithelial cells and chitin (5, 14).
FrhA is a large protein (2251 aa; 234.5 kD) encoded on the V. cholerae large chromosome and divergently transcribed from frhC, encoding the outer membrane pore required for FrhA secretion (5) (Fig. 1A). FrhA contains a C-terminal Type I (“RTX-like”) secretion domain, indicating that it is secreted from the cytoplasm via T1SS. The C termini of T1SS substrates engage with the translocase complex composed of an inner membrane ABC transporter (HlyB-like) and periplasmic membrane fusion protein (MFP; HlyD-like) (15). The translocase complex engages with an outer membrane pore to form a “tunnel” that spans the periplasmic space and allows proteins to translocate from the cytoplasm to outside the cell (16). FrhA is similar to a family of surface-localized “megaproteins” that function as adhesins in a variety of bacteria, including Pseudomonas spp (LapA) and Marinomonas primoryensis (MpIBP). (17, 18) These proteins remain tethered at the outer membrane surface because their N-terminal domain folds into a “plug” that is too large to fit through the outer membrane pore and thus remains in the periplasmic space (17, 19, 20). The periplasmic LapG protease cleaves and removes the N-terminal plug of LapA, releasing the adhesin from the bacterial surface, but this only occurs when intracellular cyclic di-GMP (c-di-GMP) levels are low. The transmembrane protein LapD sequesters LapG in the periplasm while bound to c-di-GMP within the cytoplasm, thus allowing an intracellular signal to control extracellular protein localization (21, 22) The LapDG system has also been shown to control surface localization of FrhA and another adhesin CraA in V. cholerae (14).
Fig. 1.
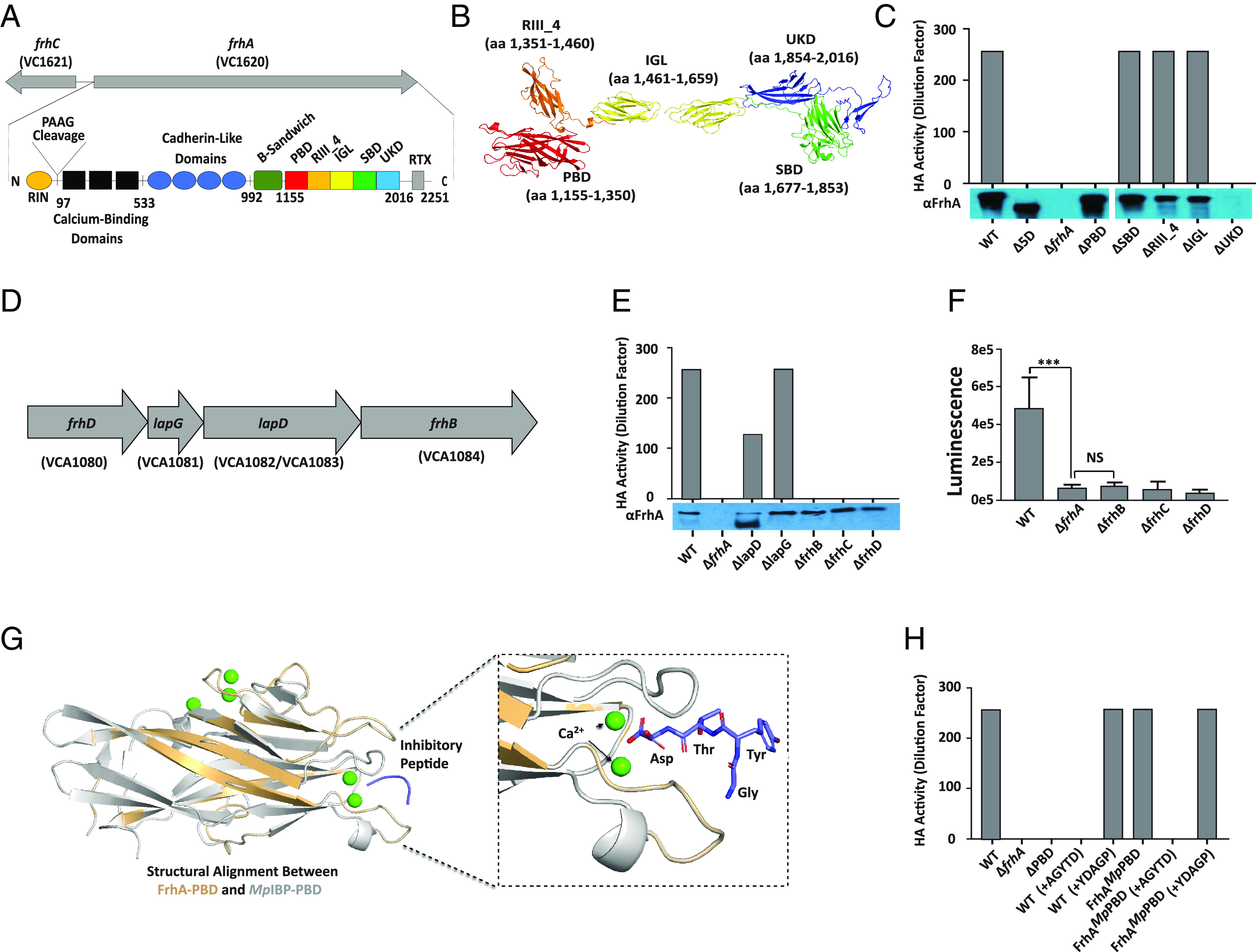
FrhA PBD is responsible for HA and a peptide inhibitor of MpIBP-PBD can prevent HA by V. cholerae. (A) Schematic of predicted FrhA architecture, described in text. (B) AlphaFold prediction of the structure of the FrhA ligand binding region. (C) Hemagglutination of human O erythrocytes by V. cholerae strains KKV598 (WT), FYVc12114 (ΔfrhA), KKV2759 (Δ5D), KKV2942 (ΔPBD), KKV2986 (ΔRIII_4), KKV2987 (ΔIGL), KKV2985 (ΔSBD), and KKV3009 (ΔUKD). Below, Western immunoblot of the same V. cholerae strains with αFrhA. (D) Schematic of frhDlapGDfhrB operon. lapD in the type strain V. cholerae N16961 contains a stop codon and is annotated as two separate ORFs (VCA1082 and VCA1083), but is a single ORF (VCO395_0160) in strain O395. (E) Hemagglutination of human O erythrocytes by V. cholerae strains KKV598 (WT), FYVc12114 (ΔfrhA), KKV2956 (ΔlapD), KKV2957 (ΔlapG), KKV3032 (ΔfrhB), KKV2075 (ΔfrhC), and KKV3033 (ΔfrhD). Results shown are representative of three independent experiments which all yielded similar values. Below, western immunoblot of the same V. cholerae strains with αFrhA. (F) Surface localization of FrhA in frhBCD mutants. αFrhA bound to the surface of V. cholerae strains KKV598 (WT), FYVc12114 (ΔfrhA), KKV3032 (ΔfrhB), KKV2075 (ΔfrhC), and KKV3033 (ΔfrhD) was detected by a chemiluminescent assay as described in Materials and Methods. *** (P < =0.001; Students paired t test), NS (not statistically significant). (G) Structural alignment between an AlphaFold Model of the FrhA-PBD and the crystal structure of MpIBP-PBD. Zoomed-in view on the right shows the ligand-binding pocket of the PBDs. Within the X-ray crystal structure of MpIBP-PBD (PDB accession: 6X5W), the peptide-binding Ca2+ ions are shown as green spheres while the peptide AGYTD (blue) is shown with the stick representation. Note the N-terminal alanine of the AGYTD peptide does not contribute to binding MpIBP-PBD and is not visible in the crystal structure. (H) Hemagglutination of human O erythrocytes by V. cholerae strains KKV598 (WT), FYVc12114 (ΔfrhA), KKV2942 (ΔPBD), and KKV3540 (FrhAMpPBD), with the addition of 100 µM peptides noted. Results for C, E, and H are representative of three independent experiments which all yielded similar values.
In the current study, we have identified a peptide-binding domain in FrhA (FrhA_PBD) that mediates HA and enhances epithelial cell binding, biofilm formation, and intestinal colonization. Surprisingly a homolog of PBD is found in a large adhesin produced by an Antarctic bacterium, M. primoryensis, where it mediates binding to diatoms. These domains are functionally interchangeable and peptide inhibitors that prevent M. primoryensis diatom binding also prevent V. cholerae HA. We suggest that PBD represents an adhesion domain for the marine environment that has been co-opted by V. cholerae to enhance interactions within the intestine.
Results
The Peptide-Binding Domain of FrhA Is Responsible for Hemagglutination.
The V. cholerae strain O395 is a classical biotype and was used for these studies because it does not express MSH, which obscures FrhA-dependent HA activity (23). While the relevance of V. cholerae binding erythrocytes during cholera infection is not clear, HA is a convenient readout of bacteria binding to host cells. The putative domain structure of FrhA was determined using a combination of BLAST (24), HHPred (25), Phyre2 (26), and AlphaFold (27) (Fig. 1A and SI Appendix, Fig. S1A). The PAAG sequence in the N-terminal region (aa 96 to 99) is where LapG cleaves between the alanine residues to release FrhA from the bacterial surface (14). Therefore, aa 1 to 97 represent the globular retention signal (RIN; SI Appendix, Fig. S2A). Within the region from aa 100 to 533, three domains are predicted with structural similarity to bacterial immunoglobulin (Big) domains of which calcium-binding domain CBD12 (pdb entry 3RB5_A) is one example. The region from aa 534 to 992 contains four cadherin-like domains (e.g., pdb entry 6C13), which are also calcium-binding domains (SI Appendix, Fig. S2A). Calcium binding to these repetitive domains as they emerge from the outer membrane through the FrhC pore is predicted to fold and rigidify the adhesin to extend it past the LPS O Antigen (28). The number of these FrhA “extender” domains varies, and there are additional BIg domains and/or cadherin-like domains in other sequenced V. cholerae strains (e.g., HE-25, MAK757, MO10, and 2740-80). The individual BIg and cadherin-like domains are predicted to fold into the same basic beta-sandwich structure (SI Appendix, Fig. S2B). The region from aa 993 to 1154 contains an additional beta-sandwich domain with some structural homology to the Ig-like domain in the β-adhesin of Streptococcus agalactiae [6V3P;(29)]. The C-terminal end of FrhA is predicted to contain the T1S sequence recognized by the T1S translocase complex. This is preceded by the “RTX repeat” domain (COG2931; aa 2073 to 2143), which is predicted to bind calcium and adopt a β roll conformation that enhances secretion (16, 28) (SI Appendix, Fig. S2A).
The sequence between aa 1155 to 2016 is the ligand-binding region of FrhA in which we identified five potential functional domains (Fig. 1B). The first putative domain in this region (aa 1155 to 1350) is a homolog of the PBD from M. primoryensis (PBD; PDB:6X5V_A; described in more detail below), followed by aa 1351 to 1460 predicted to be its companion “split” domain (PDB:6X5V_A). Amino acids 1461 to 1659 are predicted to form two tandem Ig-like domains similar to the ectodomain from e-cadherin [IGL; PDB: 3Q2V (30)] and aa 1677 to 1853 resemble a sugar-binding domain from a galactosaminidase of Bifidobacterium longum [SBD; PDB: 2ZXQ (31)], potentially allowing FrhA to bind the inhibitor fucose (32). Finally aa 1854 to 2016 is a β-sandwich from an adhesin/autotransporter of Escherichia coli with unknown function [UKD; PDB:4Q1Q (33)]. We generated in-frame deletions of each domain, as well as a deletion of all five domains (Δ5D), in the coding sequence of frhA on the V. cholerae genome. The resulting mutant V. cholerae strains were measured for HA activity with human O type erythrocytes (Fig. 1C). Deletion of the entire frhA coding sequence (ΔfrhA) or just these five domains (Δ5D) eliminated V. cholerae HA activity, indicating that HA activity is localized within the ligand binding region. Deletion of just the PBD or the UKD also eliminated HA activity, whereas deletion of any of the other three domains had no effect on HA. The V. cholerae strains were analyzed by immunoblotting for expression of stable FrhA (Fig. 1C). Deletion of the UKD led to undetectable levels of FrhA as did the deletion of the entire frhA gene, whereas deletion of any of the other four domains, as well as deletion of all five domains, resulted in stable FrhA expression at the anticipated MW. Since deletion of the PBD eliminates HA without affecting the stability of the protein, these results demonstrate that the PBD mediates FrhA-dependent HA activity. Because deletion of UKD led to unstable FrhA, we did not proceed with this mutant any further.
FrhA is the flagellar-regulated surface adhesin whose activity was identified over 40 y ago as a fucose-sensitive hemagglutinin (32). Addition of fucose inhibits V. cholerae HA (SI Appendix, Fig. S3), but deletion of the putative fucose binding domain (ΔSBD) did not (Fig. 1C), indicating that the mechanism of fucose inhibition of HA is likely complex. We thus focused in this study on the role of the PBD in adhesion.
The FrhBCD T1S Translocon Is Responsible for Secretion of FrhA.
We previously identified the outer membrane pore FrhC, which is divergently transcribed from frhA, as being required for FrhA-dependent HA activity (5), but the identity of the inner membrane T1S components was unknown. A search of the V. cholerae genome for HlyB and HlyD orthologs revealed two genes in the small chromosome, VCA1084 and VCA1080, respectively, that flank lapGD [(14); Fig. 1D]. LapGD have been shown to modulate surface localization of FrhA in response to c-di-GMP levels (14). VCA1080 shares 40% identity and 63% similarity with E. coli HlyD, and VCA1084 shares 43% identity and 65% similarity with E. coli HlyB. V. cholerae strains were created with in-frame deletions in VCA1080 and VCA1084, as well as in lapG and lapD, and assayed for HA activity (Fig. 1E).
The ΔVCA1080 and ΔVCA1084 mutants lacked HA activity, similar to ΔfrhA and ΔfrhC mutants. In contrast, the ΔlapG mutant exhibited HA activity similar to the wild-type strain, while the ΔlapD mutant exhibited reduced, but not absent, HA activity. Full-length FrhA could be detected in whole cell lysates of all these strains (Fig. 1E); the ΔlapD strain also had detectable lower molecular weight FrhA, likely due to LapG-dependent proteolysis, consistent with the role of LapD in sequestering the LapG protease (14). To determine whether VCA1080, VCA1084, and FrhC are responsible for secretion of FrhA, the surface accessibility of FrhA was measured by detecting anti-FrhA antisera adhered to intact bacterial cells (Fig. 1F). No surface-localized FrhA could be detected in ΔVCA1084, ΔfrhC, or ΔVCA1080 mutants, similar to a ΔfrhA strain, consistent with these genes encoding the T1S translocon that secretes FrhA to the bacterial surface. We propose renaming VCA1080 frhD and VCA1084 frhB.
The M. primoryensis IBP PBD Is Functionally Interchangeable with V. cholerae FrhA PBD for HA.
The V. cholerae FrhA-PBD shares sequence and structural homology with the PBD of the M. primoryensis Ice-Binding Protein (MpIBP-PBD) (65% identity, 76% similarity, Fig. 1G and SI Appendix, Fig. S1B). This Antarctic bacterium uses the 1.5-MDa MpIBP surface adhesin to simultaneously bind to diatoms and ice; this facilitates mixed-species microcolonies forming on the Antarctic ice to optimize photosynthesis and nutrient acquisition (17). The crystal structure of the MpIBP-PBD revealed that it binds to terminal amino acid residues on target molecules that include an unidentified surface protein of the Antarctic diatom Chaetoceros neogracile (17, 18). Peptide library analysis identified the pentapeptide (AGYTD) to bind strongly to the MpIBP-PBD (EC50 29 nM). The structure of MpIBP-PBD in complex with AGYTD revealed interactions between the three carboxyl-terminal amino acids in the peptide and residues within the MpIBP-PBD binding pocket (18). The critical MpIBP-PBD residues within the binding pocket that make contact with the C-terminal amino acids (YTD) are conserved in FrhA-PBD (V1242, Y1243, S1244, and N1282) [(18); Fig. 1G and SI Appendix, Fig. S1B], so we reasoned that the AGYTD peptide would also bind strongly to the FrhA-PBD and would inhibit its binding to RBCs. To test the interchangeability of these domains, a V. cholerae strain was created that expresses FrhA in which the Vc PBD has been replaced with Mp PBD (FrhAMpPBD).
The Vc strain expressing FrhAMpPBD exhibits HA activity similar to the wild-type V. cholerae strain (Fig. 1H), demonstrating that the Mp PBD is capable of mediating HA similar to the Vc PBD. Moreover, the peptide inhibitor AGYTD is able to inhibit both wild-type Vc as well as Vc expressing FrhAMpPBD for HA activity, whereas a control peptide (YDAGP), shown previously to bind poorly to MpPBD (18), had no effect on HA of either strain. The minimum inhibitory concentration of AGYTD to prevent HA was 50 μM with wild-type Vc and 25 μM with Vc expressing FrhAMPPBD under these conditions. No inhibition of HA was observed up to a concentration of 1.6 mM of control peptide YDAGP with either strain. These results demonstrate the functional interchangeability of the Vc and Mp PBD for HA and that the inhibitory peptide AGYTD can inhibit the binding of both to erythrocytes.
The FrhA-PBD Contributes to Epithelial Cell Binding.
The HA assay is a convenient measure of bacterial-erythrocyte interaction that is dependent on the FrhA adhesin on the V. cholerae surface. However, V. cholerae colonizes epithelial cells during infection, and thus, V. cholerae-epithelial cell binding is directly relevant to the pathogenic process. We measured V. cholerae interactions with human epithelial type 2 (Hep-2) cells via image flow cytometry (Fig. 2A and SI Appendix, Fig. S4 A and B). We used Hep-2 cells because these have been used previously to characterize FrhA-dependent adhesion (5, 34). V. cholerae cells expressing Red Fluorescent Protein (RFP) were allowed to bind Hep-2 cells for 1 h at an MOI of 50:1, washed, fixed, and analyzed by flow cytometry (600 to 1,200 Hep-2 cells imaged per sample) to determine the number of V. cholerae cells bound per epithelial cell (Fig. 2A and SI Appendix, Fig. S4 A and B).
Fig. 2.

FrhA PBD contributes to Epithelial cell binding and Biofilm formation. (A) V. cholerae strains KKV598 (WT), FYVc12114 (ΔfrhA), KKV2942 (ΔPBD), and KKV3540 (FrhAMpPBD) were allowed to bind to Hep-2 cells and the number of bacteria/cell was measured by flow cytometry, as described in Materials and Methods, and presented as box and whisker plot with median, upper and lower quartiles within the box. (B) V. cholerae strains KKV598 (WT) and KKV3540 (FrhAMpPBD) were allowed to bind to Hep-2 cells in the presence of 500 µM inhibitory peptide (AGYTD) or control peptide (YDAGP), and the number of bacteria/cell was measured by flow cytometry, as described in Materials and Methods. **** (P < =0.0001; Students paired t test), NS (not statistically significant). (C) V. cholerae strains KKV598 (WT), KKV3540 (FrhAMpPBD), KKV2759 (Δ5D), FYVc12114 (ΔfrhA), and KKV2942 (ΔPBD) were monitored for biofilm formation over 48 h, as described in Materials and Methods. (D) V. cholerae strain FYVc12114 (ΔfrhA) and KKV598 (WT) either without or with 100 µM AGYTD or YGATD peptides were monitored for biofilm formation over 48 h, as described in Materials and Methods. **** (P < =0.0001; Students paired t test), NS (not statistically significant).
The wild-type V. cholerae cells could be visualized and quantitated bound to the epithelial cell surface, with an average of 2.67 V. cholerae cells/Hep-2 cell under these conditions (Fig. 2A and SI Appendix, Fig. S4 A and B and Table S1). There was a 68% reduction of ΔfrhA V. cholerae bacteria bound to Hep-2 cells, and a 50% reduction of ΔPBD V. cholerae bound to Hep-2 cells, demonstrating that FrhA, and more specifically the PBD, contributes to epithelial cell binding (SI Appendix, Table S1). V. cholerae cells expressing FrhAMpPBD bound to Hep-2 cells at similar levels as those expressing the wild-type FrhA, demonstrating the interchangeability of V. cholerae and M. primoryensis PBDs for epithelial cell binding (Fig. 2A and SI Appendix, Table S1). Importantly, the addition of the inhibitory peptide AGYTD led to a 52% reduction in wild-type V. cholerae bound to Hep-2 cells and a 48% reduction in bound cells expressing FrhAMpPBD (Fig. 2B and SI Appendix, Table S1). These reductions in binding are similar to V. cholerae lacking either full-length FrhA or FrhA-PBD. The addition of the control peptide YDAGP had little effect on epithelial cell binding by either V. cholerae expressing wild-type FrhA or FrhAMpPBD. These results illustrate the functional equivalence of the PBDs from V. cholerae FrhA and M. primoryensis IBP for epithelial cell binding; both facilitate binding to epithelial cells, and both can be competed off with inhibitory peptide AGYTD.
The FrhA-PBD Contributes to Biofilm Formation.
Biofilm formation by V. cholerae strains was monitored over a 48-h time period (Fig. 2C). The wild-type V. cholerae strain forms noticeably larger biofilms during the 24 to 48 h time period. In contrast, a ΔfrhA strain is defective for biofilm formation, as has been described previously (5). V. cholerae lacking all five domains within the FrhA ligand-binding region (Δ5D) was also defective for biofilm formation, as was a strain lacking just the PBD (ΔPBD), demonstrating the involvement of the FrhA-PBD in V. cholerae biofilm formation. V. cholerae strains lacking the T1S components (FrhB, FrhC, and FrhD) were also defective for biofilm formation (SI Appendix, Fig. S5). The V. cholerae strain expressing FrhAMpPBD was restored to a large extent for biofilm formation (Fig. 2C). Biofilm formation by the wild-type strain could be disrupted by the AGYTD pentapeptide inhibitor, resembling a ΔfrhA mutant (Fig. 2D). In contrast, the control peptide YDAGP had no effect on wild-type biofilm formation. These results emphasize the functional conservation of the FrhA-PBD and MpIBP-PBD for biofilm formation and demonstrate that the peptide inhibitor AGYTD can disrupt V. cholerae biofilms.
The FrhA-PBD Facilitates Binding to Diatoms.
M. primoryensis binds to the diatom, C. neogracile, via the IBP. The purified MpPBD has previously been shown to bind to the diatom, and this binding could be inhibited by AGYTD (18). To determine whether FrhA-PBD was also able to bind to diatoms, we purified FrhA-PBD and labeled it with fluorescein isothiocyanate (FITC), then measured its binding to C. neogracile after 24 h, at which point the diatom surface was coated with FITC-labeled FrhA-PBD (Fig. 3 A and B). Importantly, addition of the peptide AGYTD (37.5 µM) led to a >20-fold reduction of FITC-FrhA-PBD bound to the diatoms. Addition of 375 µM of the control peptide AGAGD had no significant effect on FITC-FrhA-PBD binding.
Fig. 3.

The FrhA PBD binds Diatoms and contributes to intestinal colonization. (A) C. neogracile was incubated either without or with FITC-FrhA-PBD for 24 h, either without or with 37.5 µM AGYTD or 375 µM control peptide AGAGD, and imaged by brightfield and fluorescence microscopy. All images were captured with the same length of exposure. (B) Fluorescence from A quantitated, as described in Materials and Methods. (P < =0.001; Students paired t test), (C) V. cholerae strains FYVc12114 (ΔfrhA), KKV2942 (ΔPBD), and KKV3540 (FrhAMpPBD) were coinoculated with the wild-type strain O395 perorally into infant mice. (n.s. = not significant) (D) V. cholerae strains KKV598 (“WT”) and KKV2942 (ΔPBD), were coinoculated with the wild-type strain O395 perorally into infant mice, with the addition of 500 µM inhibitory peptide (AGYTD) or control peptide (YDAGP). Inocula in C and D contained a ratio of ~1:1 mutant: wild type; intestinal homogenates were recovered at 22 h after inoculation and CFU of wild-type and mutant strains determined. The competitive index is given as the output ratio of mutant:wild type divided by the input ratio of mutant:wild type; each value shown is from an individual mouse. (P < =0.01; Students paired t test).
The cold temperature required for the Antarctic C. neogracile to grow in the laboratory was incompatible with V. cholerae, so we instead visualized V. cholerae interacting with a mesophilic diatom, Extubocellulus spinifer, in room temperature seawater. V. cholerae swarmed E. spinifer and bound extensively to their surface; V. cholerae binding was reduced by the addition of the inhibitory peptide AGYTD (SI Appendix, Fig. S6A). The isolated TRITC-FrhA-PBD was also able to bind the surface of E. spinifer, similar to C. neogracile (SI Appendix, Fig. S6B). These results demonstrate that V. cholerae uses the FrhA-PBD to bind to diatoms.
The FrhA-PBD Enhances Intestinal Colonization.
V. cholerae’s ability to colonize the infant mouse small intestine is correlated with its ability to cause disease in humans (35), and a ΔfrhA V. cholerae strain has been shown to exhibit an approx. 50% reduction in intestinal colonization in this model (5). We used a competition assay in the infant mouse to determine the relative ability of the V. cholerae strain lacking only FrhA-PBD (ΔPBD) to colonize the infant mouse small intestine. This ΔPBD mutant strain showed an approximate 44% reduction in its ability to colonize (CI = 0.559), which was not significantly different to the ~59% reduction of a V. cholerae strain lacking the entire FrhA (CI = 0.411) (Fig. 3C). The replacement of VcPBD with MpPBD (FrhAMpPBD) restored wild-type levels of colonization to this strain (CI = 1.093). These results indicate that the PBD largely mediates FrhA-dependent intestinal colonization and are consistent with the approx. 50% reduction in epithelial cell binding by the ΔPBD strain seen in vitro (Fig. 2 A and B). Moreover, the MpPBD can substitute for VcPBD for intestinal colonization, emphasizing the interchangeable nature of PBD from V. cholerae and M. primoryensis.
To determine whether the AGYTD peptide could inhibit V. cholerae intestinal colonization, we again used the competition assay (Fig. 3D). We reasoned that the inhibitory peptide would decrease the colonization of the wild-type strain (PBD+) and thus increase the competitive index of the ΔPBD strain in a coinoculation experiment. As anticipated, the addition of 500 µM AGYTD to the ΔPBD:wild-type mixture increased the competitive index approximately 30% (CI = 0.798), while the competitive index of the ΔPBD:wild-type coinoculation with the addition of 500 µM of the control peptide YDAGP was unaffected (CI = 0.532). Our results demonstrate that the AGYTD peptide functions to inhibit V. cholerae PBD-dependent intestinal colonization.
Discussion
Bacterial adhesins are important for binding their hosts to various substrates, including eukaryotic cells, to initiate microcolony formation, and colonization of surfaces, as well as binding to each other to facilitate complex behaviors such as biofilm formation. All of these activities are relevant during the lifecycle of V. cholerae, which colonizes the human gastrointestinal tract during cholera pathogenesis, and which colonizes various surfaces in the marine environment during interepidemic periods. Colonization of chitinous surfaces in the marine environment also leads to the induction of T6SS-mediated killing of neighboring bacteria and uptake of their DNA through natural competence (36, 37), leading to the rapid evolution of V. cholerae through horizontal gene transfer in the marine environment (38).
The V. cholerae adhesin FrhA is a large protein that mediates binding to both human cells and various substrates, including chitin (5, 14). Similar bacterial “RTX adhesins” have a general conserved architecture, with the N terminus tethered to the membrane, a long region with multiple repeat sequences that serves to extend the protein from the cell surface, a ligand binding region that is responsible for binding to specific substrates, and a C terminus that contains the secretion signal that directs the protein to the T1SS machinery (28). We have shown here that FrhA maintains these features and moreover that a PBD within the FrhA ligand binding region mediates HA, epithelial cell binding, biofilm formation, and intestinal colonization. Interestingly, the FrhA-PBD is functionally interchangeable with the PBD of M. primoryensis IBP.
M. primoryensis are psychrophilic, motile bacteria found in Antarctic waters (39). These bacteria use their surface adhesin IBP to form mixed-species microcolonies with photosynthetic microorganisms such as diatoms that are bound to marine ice (17). The M. primoryensis IBP has the same general architecture as FrhA, with a much longer extender region. Within the IBP ligand-binding region, there is an ice-binding domain that tethers the bacteria to the underside of marine ice. Also, within the ligand binding region is the PBD, which allows M. primoryensis to bind to diatoms such as C. neogracile. M. primoryensis can be observed binding to the nonmotile C. neogracile and moving them by swimming motility to the ice surface which they colonize (17). Presumably, this interaction enhances diatom photosynthesis by moving the diatom to where the most light can be found, i.e., the underside of marine ice. The bacteria likely benefit from this interaction by using oxygen and organic compounds the diatom derives from photosynthesis.
The V. cholerae FrhA-PBD and the M. primoryensis MpIBP-PBD share 65% identity and are functionally interchangeable. We have shown that the FrhA-PBD facilitates binding to erythrocytes and epithelial cells and enhances biofilm formation, and the MpIBP-PBD can substitute for all these activities. Likewise, the MpIBP-PBD binds to diatoms, and the FrhA-PBD binds to diatoms as well. Importantly, a pentapeptide inhibitor, AGYTD, that was developed to bind with high affinity to the MpIBP-PBD (18) can also inhibit FrhA-PBD-dependent HA activity, epithelial cell binding, biofilm formation, and intestinal colonization, as well as binding to diatoms.
The M. primoryensis PBD binds to three C-terminal peptide residues. The YTD of the AGYTD pentapeptide is a particularly good match to the binding pocket of MpIBP-PBD (18), although other sequences fit the binding pocket with lower affinity. The protein(s) on the surface of erythrocytes and epithelial cells, or diatoms, to which PBD binds have not been identified. However, the efficacy with which AGYTD inhibits V. cholerae FrhA binding to erythrocytes and epithelial cells and lowers intestinal colonization is proof of FrhA-PBD’s involvement in these interactions. The role of PBDs in biofilm formation on abiotic surfaces indicates that the domain also mediates bacterial–bacterial associations, and perhaps, these interbacterial interactions also contribute to HA and intestinal colonization.
Within Vibrio spp., the PBD is only found in V. cholerae, the closely related Vibrio metoecus (40), and Vibrio vulnificus. This limited distribution within Vibrionales is suggestive of acquisition by horizontal gene transfer (HGT). Outside Vibrio spp. the PBD is found in M. primoryensis, as well as in Aeromonas and Shewanella spp. It is tempting to speculate that V. cholerae, a natural marine inhabitant, acquired the diatom-binding PBD through HGT from another bacterium associated with diatoms, perhaps even M. primoryensis. This would have not only enhanced V. cholerae’s ability to bind to diatoms and persist in the marine environment but also enhanced its ability to bind to human cells and facilitated its infectious potential in humans. The PBD appears to always be associated with large T1S surface adhesins with multiple cadherin/Ig-like extender regions, indicating that it likely functions to bind to host cells and/or substrates in these other bacteria as well. Given the conserved efficacy of the pentapeptide AGYTD in inhibiting the function of both V. cholerae FrhA-PBD and M. primoryensis MpIBP-PBD, it is likely that this peptide would also inhibit binding and biofilm formation by the other bacteria with PBD as well. Although the pentapeptide inhibitor AGYTD does not completely prevent V. cholerae intestinal colonization, its ability to reduce colonization may have value as an additive to other therapeutic measures against cholera infection. The role of FrhA in O1 El Tor intestinal colonization has not yet been evaluated, but FrhA has been shown to be important for O1 El Tor biofilm formation on chitin in a microfluidic device (14), and the LapGD system controls O1 El Tor biofilm dispersal (41), likely through FrhA. AGYTD represents a rationally designed inhibitor that interferes with both V. cholerae biofilms and adhesin-host cell interactions.
Materials and Methods
Bacterial Strains and Plasmids.
E. coli strains DH5αλpir and NEB 10β were used for cloning, and BW29427 (42) was used for conjugation. V. cholerae O1 classical strain O395, which is streptomycin resistant, and its derivatives were used for all experiments. Strains were grown in Luria broth (LB) at 37 °C overnight. Antibiotics were used with V. cholerae strains at the following concentrations: ampicillin (Ap) or carbenicillin (Cb), 100 μg/mL, streptomycin (Sm), 100 μg/mL, gentamycin (Gm) 100 μg/mL, and chloramphenicol (Cm) 2 μg/mL.
V. cholerae mutants were generated through allelic exchange using the plasmids pKEK229 (43) or pKEK2200 (44) via sacB counterselection. A complete list of primers and plasmids used is provided in SI Appendix, Tables S2 and S3. Details on plasmid and strain construction are included in SI Appendix. All plasmid constructs were verified by sequencing. A complete list of V. cholerae strains is provided in SI Appendix, Table S4. V. cholerae in-frame chromosomal deletions within frhA removed the coding sequence for the amino acids noted in text and SI Appendix, Fig. S1. All chromosomal deletions were confirmed by sequencing. rfp was inserted into the V. cholerae chromosome via Tn7 mutagenesis that inserts in the genomic region between loci VC0487 and VC0488, as described previously (45).
Hemagglutination.
The HA assay was adapted from refs. 5 and 34. V. cholerae strains from overnight cultures grown in LB at 37 °C were concentrated to an OD600 of 15 in KRT buffer. Bacterial cells were serially diluted in a round-bottomed 96-well microtiter plate. Human Type O Red Blood Cells (RBCs) (Zenbio) were resuspended in KRT buffer and washed three times. An aliquot (200 μL) of 0.7% red blood cells was added to 100 μl of bacterial suspension and monitored for HA titer at 1 h. For experiments using inhibitory peptides, peptides were suspended in KRT buffer at a concentration of 100 mM peptides and then added to the final concentration indicated. Because in our hands the HA titer is identical between biological replicates of the same Vc strain, the HA experiments were performed a minimum of three separate times, and representative results from a single experiment are shown here.
Western Immunoblot and Cell Surface Protein Expression.
Rabbit polyclonal antisera to a purified fragment of FrhA that contains a single cadherin, as well as PBD and RIII_4 (aa 884 to 1460; SI Appendix, Fig. S1), was used in immunoblots and cell surface expression assays. To detect surface localization, mid-log cells were concentrated to OD600 of 1 and then pelleted and washed 3× in Phosphate Buffered Saline (PBS). α-FrhA (10 μL) was added to 1 mL of cells and then incubated at room temperature for 1 h. The cells were then washed 3× PBS, 10 μL of anti-rabbit-HRP IgG (Southern Biotech) was added, and the cells were incubated 1 h at room temperature. The cells were washed 3× with PBS, and the cell pellet was resuspended in 200 μL SuperSignal West Pico PLUS Chemiluminescent substrate (Thermo) and measured for chemiluminescence.
HEp-2 Cell Binding Assay.
Hep-2 cells (106) were seeded in Dulbecco’s Modified Eagle’s Medium (DMEM) + 5% FBS into a 24-well plate and allowed to adhere at 37 °C and 5% CO2 for 2 h. V. cholerae strains expressing RFP were grown overnight at 37 °C in Luria Broth with 100 μg/mL streptomycin and 2 μg/mL chloramphenicol. The cells were then back diluted and grown to mid-log at 37 °C for 4 h. A total of 5 × 107 CFU of bacteria (MOI 50:1) were added to Hep-2 cells, and plates were spun at 150 × g for 5 min. The medium was replaced with KRT buffer, containing the desired concentration of inhibitor, if applicable, and allowed to incubate at 37 °C and 5% CO2 for 1 h. The plates were spun at 150 × g for 5 min and washed with KRT buffer three times to remove any unbound bacteria. The cells were then fixed with 4% paraformaldehyde and filtered twice for single cells using 20 μm cell-straining capped tubes (Falcon).
Cells were then imaged using an Amnis ImageStreamX MKII (Millipore) image flow cytometer with a 7-μm core at low flow rate and high sensitivity using INSPIRE software. RFP was excited using a 561-nm laser, and image data were collected through a 60× objective. Single cells were gated using Area/Aspect Ratio 200-1000/0.6-1, and 10,000 instances were collected per sample. Images were analyzed using IDEAS® software version 6.2 (Millipore). Machine learning was applied to count the number of RFP spots present on each cell using the spot count wizard, and histograms were derived using the histogram smoothing operation.
Biofilm Assay.
Bacterial strains were grown overnight at 37 °C in LB and then inoculated to an OD600 of 0.1 into 0.75 mL of LB in Falcon disposable plastic tubes at an OD600 of 0.1. Tubes were incubated statically at 37 °C. At the designated time points, tubes were washed with 0.5 mL of distilled water and dried for 12 h at 37 °C. Tubes were stained for 20 min with 2% crystal violet, washed 3× with 1 mL distilled water, dissolved in 1 mL dimethyl sulfoxide (DMSO) for 1 h at room temperature, and then quantitated by measuring OD570.
Diatom Binding.
FrhA-PBD was purified and labeled as described in SI Appendix. C. neogracile cells and E. spinifer cells were grown at 4 °C in F/2 media with natural sea water from the Gulf of Maine with periodic lighting at 5,000 lux (13 h ON, 11 h OFF). The cultures were passaged into new flasks every 4 wk and were used for the experiments 2 to 3 wk after passaging. C. neogracile or E. spinifer cultures (2 mL) were centrifuged at 3,000 × g, and the pellet was resuspended in 0.2 mL resuspension buffer (50 mM Tris–HCl pH 9.0, 300 mM NaCl, and 5 mM CaCl2). The resuspended diatom culture was mixed with TRITC- FrhA-PBD or FITC-FrhA-PBD and inhibitory or control peptides at the concentration indicated to a total volume of 0.4 mL in 1.5 mL centrifuge tubes. The tubes were covered and incubated at 4 °C with gentle mixing for 24 h. The samples were centrifuged at 4,500 × g for 3 min and washed with the resuspension buffer three times. The final pellet was resuspended in 20 µL of the buffer. For interaction studies with V. cholerae, E. spinifer cell cultures (20 mL), grown as indicated above, were centrifuged at 3,000 × g for 5 min and resuspended in 1 mL of culture medium and allowed to reach room temperature. An aliquot (50 µL) of a 37 °C overnight culture of V. cholerae was added to the diatoms, and the cell mixtures were incubated at room temperature for 5 h. Samples were visualized using an Olympus IX83 inverted fluorescence microscope. Images of at least 30 single diatom cells were used for quantitation, using ImageJ software. The collected data were used to calculate the Corrected Total Cell Fluorescence (CTCF) using this formula: CTCF = Integrated Density−(Area of selected cell × Mean fluorescence of background readings).
Mouse Intestinal Colonization Assay.
V. cholerae mutant strains were each mixed in a 1:1 ratio with the isogenic wild-type strain and then inoculated intragastrically into 5-d-old CD-1 suckling mice (approximately 106 mutant: 106 wild type). Infected pups were placed in a humidified 30 °C incubator. After 22 h, the mice were killed, and their small intestines were isolated and homogenized. The mutant/wild type ratios were determined by plating dilutions on LB agar containing X-Gal (5-bromo-4-chloro-3-indolyl-D-galactopyranoside). The competitive index is given as the output ratio of mutant:wild type divided by the input ratio of mutant:wild type; Prism 5.0b was used for statistical analyses using the Mann–Whitney U test.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
K.E.K. was funded by the Brown Foundation, the Kleberg Foundation, and the San Antonio Area Foundation. P.L.D. holds the Canada Research Chair in Protein Engineering and acknowledges the support of a Foundation Award from the Canadian Institutes of Health Research (CIHR). S.G. is partially supported by a CIHR fellowship. We are grateful to Mustafa Sherik for technical assistance in the preparation of VcPDB and Thomas Hanse for advice with AlphaFold modeling.
Author contributions
C.J.L., S.G., B.K., S.K.A., I.K.V., P.L.D., and K.E.K. designed research; C.J.L., S.G., B.K., H.Z., R.E., S.K.A., and K.E.K. performed research; S.G., H.Z., F.Y., I.K.V., and P.L.D. contributed new reagents/analytic tools; C.J.L., S.G., B.K., H.Z., R.E., S.K.A., F.Y., I.K.V., P.L.D., and K.E.K. analyzed data; and C.J.L., P.L.D., and K.E.K. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and SI Appendix.
Supporting Information
References
- 1.Mekalanos J. J., Cholera toxin: Genetic analysis, regulation, and role in pathogenesis. Curr. Top. Microbiol. Immunol. 118, 97–118 (1985). [DOI] [PubMed] [Google Scholar]
- 2.Deen J., Mengel M. A., Clemens J. D., Epidemiology of cholera. Vaccine 38, A31–A40 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Echazarreta M. A., Klose K. E., Vibrio flagellar synthesis. Front. Cell Infect. Microbiol. 9, 131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prouty M. G., Correa N. E., Klose K. E., The novel s54- and s28-dependent flagellar gene transcription hierarchy of Vibrio cholerae. Mol. Microbiol. 39, 1595–1609 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Syed K. A., et al. , The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol. 191, 6555–6570 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klose K. E., Mekalanos J. J., Distinct roles of an alternative sigma factor during both free-swimming and colonizing phases of the Vibrio cholerae pathogenic cycle. Mol. Microbiol. 28, 501–520 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Taylor R. K., Miller V. L., Furlong D. B., Mekalanos J. J., Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U.S.A. 84, 2833–2837 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krebs S. J., Taylor R. K., Protection and attachment of Vibrio cholerae mediated by the toxin-coregulated pilus in the infant mouse model. J. Bacteriol. 193, 5260–5270 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waldor M. K., Mekalanos J. J., Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Chiavelli D. A., Marsh J. W., Taylor R. K., The mannose-sensitive hemagglutinin of Vibrio cholerae promotes adherence to zooplankton. Appl. Environ. Microbiol. 67, 3220–3225 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watnick P. I., Kolter R., Steps in the development of a Vibrio cholerae biofilm. Mol. Microbiol. 34, 586–595 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Utada A. S., et al. , Vibrio cholerae use pili and flagella synergistically to effect motility switching and conditional surface attachment. Nat. Commun. 5, 4913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirn T. J., Jude B. A., Taylor R. K., A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438, 863–866 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Kitts G., et al. , A conserved regulatory circuit controls large adhesins in Vibrio cholerae. mBio 10, e02822-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delepelaire P., Type I secretion in gram-negative bacteria. Biochim. Biophys. Acta 1694, 149–161 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Thomas S., Holland I. B., Schmitt L., The Type 1 secretion pathway - the hemolysin system and beyond. Biochim. Biophys. Acta 1843, 1629–1641 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Guo S., et al. , Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci. Adv. 3, e1701440 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo S., et al. , Molecular basis for inhibition of adhesin-mediated bacterial-host interactions through a peptide-binding domain. Cell Rep. 37, 110002 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Smith T. J., Font M. E., Kelly C. M., Sondermann H., O’Toole G. A., An N-terminal retention module anchors the giant adhesin LapA of Pseudomonas fluorescens at the cell surface: A novel subfamily of Type I secretion systems. J. Bacteriol. 200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo S., et al. , Conserved structural features anchor biofilm-associated RTX-adhesins to the outer membrane of bacteria. FEBS J. 285, 1812–1826 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Newell P. D., Monds R. D., O’Toole G. A., LapD is a bis-(3’,5’)-cydlic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc. Natl. Acad. Sci. U.S.A. 106, 3461–3466 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newell P. D., Boyd C. D., Sondermann H., O’Toole G. A., A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol. 9, e1000587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanne L. F., Finkelstein R. A., Characterization and distribution of the hemagglutinins produced by Vibrio cholerae. Infect. Immunity 36, 209–214 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sayers E. W., et al. , Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmermann L., et al. , A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo S., Vance T. D. R., Stevens C. A., Voets I., Davies P. L., RTX adhesins are key bacterial surface megaproteins in the formation of biofilms. Trends Microbiol. 27, 453–467 (2019). [DOI] [PubMed] [Google Scholar]
- 29.van Sorge N. M., et al. , Bacterial protein domains with a novel Ig-like fold target human CEACAM receptors. EMBO J. 40, e106103 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison O. J., et al. , The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19, 244–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki H., Toyota M., Nojima M., Mori M., Imai K., SFRP, a family of new colorectal tumor suppressor candidate genes. Nihon rinsho. Japanese J. Clin. Med. 63, 707–719 (2005). [PubMed] [Google Scholar]
- 32.Jones D. H., Franklin F. C., Thomas C. M., Molecular analysis of the operon which encodes the RNA polymerase sigma factor s54 of Escherichia coli. Microbiol. 140, 1035–1043 (1994). [DOI] [PubMed] [Google Scholar]
- 33.Lu Q., et al. , An iron-containing dodecameric heptosyltransferase family modifies bacterial autotransporters in pathogenesis. Cell Host. Microbe. 16, 351–363 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Gardel C. L., Mekalanos J. J., Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect. Immun. 64, 2246–2255 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klose K. E., The suckling mouse model of cholera. Trends Microbiol. 8, 189–191 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Meibom K., Blokesch M., Dolganov N. A., Wu C. Y., Schoolnik G. K., Chitin induces natural competence in Vibrio cholerae. Science 310, 1824–1827 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Borgeaud S., Metzger L. C., Scrignari T., Blokesch M., The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science 347, 63–67 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Matthey N., et al. , Neighbor predation linked to natural competence fosters the transfer of large genomic regions in Vibrio cholerae. Elife 8, e48212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romanenko L. A., Uchino M., Mikhailov V. V., Zhukova N. V., Uchimura T., Marinomonas primoryensis sp. nov., a novel psychrophile isolated from coastal sea-ice in the Sea of Japan. Int. J. Syst. Evol. Microbiol. 53, 829–832 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Kirchberger P. C., et al. , Vibrio metoecus sp. nov., a close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int. J. Syst. Evol. Microbiol. 64, 3208–3214 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Bridges A. A., Fei C., Bassler B. L., Identification of signaling pathways, matrix-digestion enzymes, and motility components controlling Vibrio cholerae biofilm dispersal. Proc. Natl. Acad. Sci. U.S.A. 117, 32639–32647 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan C. H., Levar C. E., Zacharoff L., Badalamenti J. P., Bond D. R., Scarless genome editing and stable inducible expression vectors for geobacter sulfurreducens. Appl. Environ. Microbiol. 81, 7178–7186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Correa N. E., Lauriano C. M., McGee R., Klose K. E., Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Mol. Microbiol. 35, 743–755 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Mejia-Santana A., Lloyd C. J., Klose K. E., New cloning vectors to facilitate quick allelic exchange in gram-negative bacteria. Biotechniques 70, 116–119 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Fong J. C., Karplus K., Schoolnik G. K., Yildiz F. H., Identification and characterization of RbmA, a novel protein required for the development of rugose colony morphology and biofilm structure in Vibrio cholerae. J. Bacteriol. 188, 1049–1059 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and SI Appendix.