

Alcohol consumption has increased over the COVID-19 pandemic, and this rise in alcohol consumption has led to increases in alcohol-associated liver disease (ALD), and alcohol-associated hepatitis (AH) in particular(1). The growing problem of AH has been exacerbated by the lack of suitable therapeutics to reverse AH, with many patients requiring liver transplants. The etiology of AH is complex, and current animal models of AH have not been able to replicate certain hallmarks of severe AH (sAH) that correlate with prognosis, including ductular reaction (DR). This has greatly limited AH therapeutic development even though many targets and pathways have shown promise in preclinical models. In fact, several studies have shown that mechanistic target of rapamycin complex 1 (mTORC1), a master regulator of cell growth, energy metabolism, and autophagy, is activated in AH and inhibition of mTORC1 can reverse ethanol-induced liver injury(2). However, the effect of mTORC1 on alcohol-associated DR and its associated liver injury remains unknown.
Some of the questions surrounding mTORC1 in ALD are answered in this issue of Hepatology by Chao et al.(3). Chao et al detected increased mTORC1 activation in chronic-plus-binge ethanol fed mouse and human AH liver samples, and such elevated mTORC1 activation is associated with decreased protein levels of tuberous sclerosis complex 1 (TSC1), a negative regulator of mTORC1. These findings led the authors to characterize ethanol-induced liver injury in liver specific Tsc1 knockout (L-Tsc1 KO) mice that exhibit constitutive mTORC1 activation. ALT levels were not different between wild-type (WT) and L-Tsc1 KO mice subjected to the control diet, indicating that baseline liver injury may not be different. However, L-Tsc1 KO mice on the control diet exhibited increased liver size, DR, fibrosis, and immune cell infiltration compared to their WT counterparts. Ethanol feeding exacerbated many of these characteristics in the L-Tsc1 KO mice compared to WT, which resembled the human AH phenotype, indicating that the L-Tsc1 KO mice may be predisposed to increased liver injury due to ethanol. The most striking finding was that the DR in the ethanol-fed L-Tsc1 KO mice reproduced the DR found in human AH patients. Since the chronic-binge model of ethanol feeding induces very little DR in WT mice, the DR phenotype in ethanol-fed L-Tsc1 KO mice was an exciting finding that the authors decided to follow.
To further characterize the phenotype of the L-Tsc1 KO mice, the authors performed RNA-seq and looked at cell-type specific markers. They found that L-Tsc1 KO mice exhibited decreased gene signatures of hepatocytes, and increased gene signatures of cholangiocytes, hepatic stellate cells (HSCs), and macrophages, and these changes were more pronounced with ethanol feeding, similar to gene signatures in human AH samples. Accordingly, hepatocyte marker hepatocyte nuclear factor 4 alpha isoform p1 (HNF4α-P1) was decreased in L-Tsc1 KO mice indicating hepatocyte degeneration, while yes associated protein (YAP) and sry-box transcription factor 9 (SOX9) were increased, indicating increases in liver progenitor cells (LPCs) and cholangiocytes. Interestingly, proliferating cell nuclear antigen (PCNA) levels were elevated in both control and ethanol-fed L-Tsc1 KO mice, and the cells positive for PCNA seemed to be non-parenchymal (NPCs) or LPCs with few hepatocytes. Together, these experiments indicate that that liver mTORC1 activation may bias liver cell repopulation after injury towards cell types other than hepatocytes, and the combination of mTORC1 inhibitors and hepatocyte regeneration promoting agents may induce liver repopulation with hepatocytes over NPCs and LPCs. Future studies should evaluate therapies that have shown promise for AH, such as IL-22(4), in ethanol-treated L-Tsc1 KO mice to characterize how these therapies interact with constitutive liver mTORC1 activation seen in human AH and determine whether combination treatment with a mTORC1 inhibitor is effective in treating AH.
Chao et al. then investigated the fibrotic, inflammatory, and cholestatic phenotype of the L-Tsc1 KO mice. In accordance with the RNA-seq data, the L-Tsc1 KO mice on the control diet exhibited increases in fibrosis, neutrophil infiltration, and Kupffer cells/infiltrated macrophages that were all further increased after ethanol feeding. Interestingly, most macrophages are adjacent to SOX9+ DR cells in the livers of L-Tsc1 KO mice. Several studies have suggested that there is a codependent relationship between inflammatory cells and SOX9+ cholangiocytes in multiple types of liver disease. Liver injury leads to immune cell infiltration near SOX9+ cholangiocytes, and immune cells promote the inflammatory phenotype to exacerbate DR and cholestasis, becoming a vicious cycle(5). Since recent studies have shown that inflammatory cells can increase cholangiocyte proliferation and impair bile acid secretion(6), it will be important to determine whether the TSC1 deletion on its own is sufficient to drive DR in macrophage-depleted animals. The L-Tsc1 KO mice also exhibit a cholestatic phenotype, with increases in total serum bile acids similar to AH patients. However, the mechanism behind this impaired bile acid homeostasis needs further study, as changes in the genes involved in bile acid disposition did not point to a specific pathway.
Since the L-Tsc1 KO mice have Tsc1 deletion in both hepatocytes and cholangiocytes, the authors sought to separate the effects of mTORC1 activation in these two cell types by utilizing AAV8-TBG-Cre and Ad-CK19-CreERt2 in the Tsc1 flox/flox mice to specifically delete Tsc1 in hepatocytes and cholangiocytes, respectively. They found that Tsc1 deletion in cholangiocytes leads to significant increases in DR and fibrosis and shows trends of higher ALT and immune cell infiltration compared to hepatocyte-specific Tsc1 KO. In combination with their data showing that Ki67/EPCAM double positive cells are increased in L-Tsc1 KO livers and that Tsc1 deletion increases cell proliferation in an immortalized cholangiocyte cell line, the authors make a compelling case that cholangiocyte TSC1 deficiency drives the DR, inflammatory, and fibrotic responses seen in the L-Tsc1 KO mice and potentially in human AH.
While the overall data points towards cholangiocyte TSC1 deficiency mediating the liver injury phenotype seen in L-Tsc1 KO mice, the cell-type specific approaches need further study. Cre-based approaches to knock-out genes have enabled unprecedented insight into cell-specific protein functions, but also have inherent disadvantages, including the expression of cre-driver genes outside of the cells of interest. The Krt19 gene (CK19) is also expressed in pancreatic ductular cells in developed animals, and therefore, studies with the Ad-CK19-CreERt2 in the future should evaluate whether pancreatic ductal TSC1 deficiency has any effect on the phenotypes in the Ad-CK19-CreERt2 animals. In addition, the AAV-null- and Ad-null-mediated liver injury and phenotype data were not presented, making it hard to evaluate the effect of the viruses themselves in this model. Keeping these caveats in mind, there are several interesting questions raised by these experiments. Previous studies show decreases in TFEB after ethanol-feeding in mice and in human AH samples, which were recapitulated in ethanol-fed cholangiocyte Tsc1 KO mice. Counterintuitively, ethanol-treated hepatocyte Tsc1 KO mice exhibited a trend of higher liver TFEB expression compared to controls, indicating that hepatocyte mTORC1 activation may affect liver autophagy differently than cholangiocyte mTORC1. These cell-specific Tsc1 KO mice have hinted that mTORC1 activation in different cell types may have drastically different effects, and these models should be further investigated to see whether crosstalk between mTORC1, YAP, and SOX9 in the different cell types contribute to the phenotype of the L-Tsc1 KO mice.
The authors also showed that administration of the mTORC1 inhibitor Torin1 on Day 6, 8, and 10 of the chronic plus binge ethanol feeding model in L-Tsc1 KO mice ameliorated DR, inflammatory cell infiltration, and fibrosis, demonstrating the potential therapeutic effect of mTORC1 inhibition in AH. However, it is important to keep in mind that chronic mTORC1 inhibition has been shown to increase inflammation in mice and humans, and exacerbate the development of hepatocellular carcinoma (HCC) in mice(7), so treating AH with mTORC1 inhibitors should be designed as a short-term intervention. In addition, the apparent cholangiocyte-specificity of deleterious mTORC1 activation brings both opportunities and challenges for therapeutic targeting. Both nanoparticles and antibody drug conjugates (ADCs) have shown promise in cell-specific targeting by using surface receptors on the cell type of interest to induce internalization and release of the drug into the cell(8). Of these approaches, ADCs may provide a better therapeutic approach due to the mechanism of drug delivery. In general, when ADCs with non-cleavable linkers bind to cell surface receptors, they are internalized into an early endosome which eventually becomes a lysosome, leading to degradation of the antibody and drug release into the cell. As the mTORC1 complex is located in the lysosome, this delivery strategy may enable lower drug doses in addition to increased cell specificity. In this case, the identification of a cell surface marker that is highly expressed in AH cholangiocytes but has low expression in other tissues will be the key to successful ADC development. As other ductular cells in the body express similar cell surface markers, it will be important to evaluate off-target effects of mTORC1 inhibition, especially in pancreatic ductular cells. Regardless, the high mortality rate and limited treatment options for AH means that even untargeted mTORC1 inhibition should be further characterized as a therapeutic strategy in AH patients.
Overall, these studies have provided a solid mechanistic basis for the formation of DR in AH via mTORC1 hyperactivation in cholangiocytes (Fig. 1). Based on these findings, further investigations of the mTORC1-YAP-SOX9 interaction and cell-type specific mTORC1 activation-mediated crosstalk with other liver cell types are needed. Future studies should investigate whether this mechanism is common to the different histopathological phenotypes of AH that have been identified(9), since a common target between different phenotypes would be ideal for therapeutic development.
Figure 1. mTORC1 activation in cholangiocytes, but not hepatocytes, exacerbates ethanol-induced ductular reaction.
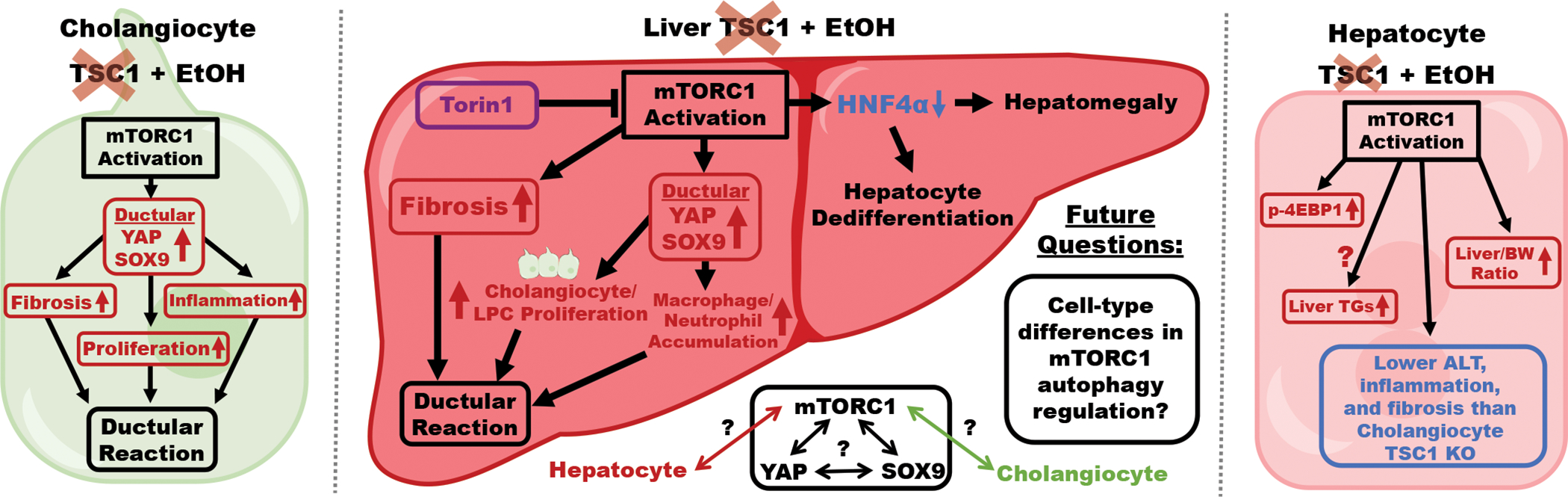
Ethanol-fed L-Tsc1 KO mice with constitutive mTORC1 activation exhibit increases in liver YAP and SOX9, which drives cholangiocyte/LPC proliferation and immune cell infiltration, and fibrosis, all of which contribute to the exacerbated ductular reaction compared to WT mice. mTORC1 activation in L-Tsc1 KO mice also downregulates HNF4α, leading to liver enlargement and hepatocyte dedifferentiation. The ductular reaction phenotype in L-Tsc1 KO mice is recapitulated in ethanol-fed cholangiocyte-specific Tsc1 KO mice but not hepatocyte-specific Tsc1 KO mice. Outstanding questions surrounding the mTORC1-YAP-SOX9 axis, hepatocyte-cholangiocyte mTORC1 crosstalk, and cell-type differences in autophagy regulation by mTORC1 should be addressed in future studies.
Acknowledgments:
The authors’ laboratory was supported by the intramural program of NIAAA, NIH.
Footnotes
COI Statement: The authors declare that no conflict of interest exists.
References
- 1.Devarbhavi H, Asrani SK, Arab JP, Nartey YA, Pose E, Kamath PS. Global burden of Liver Disease: 2023 Update. Journal of Hepatology 2023. [DOI] [PubMed] [Google Scholar]
- 2.Chao X, Williams SN, Ding W-X. Role of mechanistic target of rapamycin in autophagy and alcohol-associated liver disease. American Journal of Physiology-Cell Physiology 2022;323:C1100–C1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chao X, Wang S, Ma X, Zhang C, Qian H, Williams SN, Sun Z, et al. Persistent mTORC1 activation due to loss of liver tuberous sclerosis complex 1 promotes liver injury in alcoholic hepatitis. Hepatology 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arab JP, Sehrawat TS, Simonetto DA, Verma VK, Feng D, Tang T, Dreyer K, et al. An Open-Label, Dose-Escalation Study to Assess the Safety and Efficacy of IL-22 Agonist F-652 in Patients With Alcohol-associated Hepatitis. Hepatology 2020;72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guillot A, Winkler M, Silva Afonso M, Aggarwal A, Lopez D, Berger H, Kohlhepp MS, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology 2023. [DOI] [PubMed] [Google Scholar]
- 6.Aguilar-Bravo B, Rodrigo-Torres D, Ariño S, Coll M, Pose E, Blaya D, Graupera I, et al. Ductular Reaction Cells Display an Inflammatory Profile and Recruit Neutrophils in Alcoholic Hepatitis. Hepatology 2019;69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umemura A, Park Eek J, Taniguchi K, Lee Jun H, Shalapour S, Valasek Mark A, Aghajan M, et al. Liver Damage, Inflammation, and Enhanced Tumorigenesis after Persistent mTORC1 Inhibition. Cell Metabolism 2014;20:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuentes-Antrás J, Genta S, Vijenthira A, Siu LL. Antibody–drug conjugates: in search of partners of choice. Trends in Cancer 2023;9:339–354. [DOI] [PubMed] [Google Scholar]
- 9.Ma J, Guillot A, Yang Z, Mackowiak B, Hwang S, Park O, Peiffer BJ, et al. Distinct histopathological phenotypes of severe alcoholic hepatitis suggest different mechanisms driving liver injury and failure. The Journal of Clinical Investigation 2022;132. [DOI] [PMC free article] [PubMed] [Google Scholar]