

Abstract
Objective:
We aimed to assess treatment response of infantile-onset epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) vs. other etiologies.
Methods:
We evaluated patients with ES from the CDKL5 Centers of Excellence and the National Infantile Spasms Consortium (NISC), with onset from 2 months to 2 years, treated with adrenocorticotropic hormone (ACTH), oral corticosteroids, vigabatrin, and/or ketogenic diet. We excluded children with Tuberous Sclerosis Complex, Trisomy 21, or unknown etiology with normal development because of known differential treatment responses. We compared the two cohorts for time to treatment and ES remission at 14 days and 3 months.
Results:
We evaluated 59 individuals with CDD (79% female, median ES onset 6 months) and 232 individuals from the NISC database (46% female, median onset 7 months). In the CDD cohort, seizures prior to ES were common (88%), and hypsarrhythmia and its variants was present at ES onset in 34%. Initial treatment with ACTH, oral corticosteroids, or vigabatrin started within one month of ES onset in 27/59 (46%) of the CDD cohort and 182/232 (78%) of the NISC cohort (p<0.0001). Fourteen-day clinical remission of ES was lower for the CDD group (26%, 7/27) than for the NISC cohort (58%, 106/182, p=0.0002). Sustained ES remission at 3 months occurred in 1/27 (4%) of CDD patients vs. 96/182 (53%) of the NISC cohort (p<0.0001). Comparable results were observed with longer lead time (>1 month) or prior treatment. Ketogenic diet, used within 3 months of ES onset, resulted in ES remission at 1 month, sustained at 3 months, in at least 2/13 (15%) individuals with CDD.
Significance:
Compared to the broad group of infants with ES, children with ES in the setting of CDD often experience longer lead time to treatment and respond poorly to standard treatments. Development of alternative treatments for ES in CDD is needed.
Keywords: CDD, National Infantile Spasms Consortium, hypsarrhythmia, adrenocorticotropic hormone, prednisolone, vigabatrin
Background
CDKL5 Deficiency Disorder (CDD) is an X-linked neurodevelopmental disorder in which epilepsy is a prominent feature. Epileptic spasms (ES), typically presenting in infancy, are the most common seizure type observed in individuals with CDD, affecting 82% of patients, 23% as the initial seizure type.1 Patients with CDD can present with typical clusters of ES, but more than a third report sequential seizures, including clusters of ES with other seizure types (hypermotor-tonic-spasms, hyperkinetic-spasms, tonic-clonic-spasms, and tonic-spasms-myoclonic).1–5
Epilepsy in individuals with CDD is highly refractory, and there are few reports concerning response to standard treatment regimens in individuals with CDD-related ES.1, 6 Clinical experience and initial data from three CDKL5 Centers of Excellence have suggested that response to first-line treatments for ES in individuals with CDD is poor.6 Despite the high rate of ES in CDD, the literature on treatment of seizures in CDD thus far is not specific to ES.7 Retrospective clinical studies and a family survey suggest limited or short-lived benefit from first-line treatments (ACTH, prednisolone, vigabatrin) in Infantile Epileptic Spasms Syndrome (IESS).8–11
Disease-specific information on treatment response in IESS is limited. Data from the National Infantile Spasms Consortium (NISC) database, consisting of individuals with ES with diverse etiologies, reported an overall response rate sustained at 3 months of 46% after first-line treatment (ACTH, oral corticosteroids, or vigabatrin), irrespective of etiology, and 44% at 60 days in an expanded cohort.12, 13 In the NISC study, receiving one of the above first-line therapies was the most important determinant of response to treatment of ES.12 The International Collaborative Infantile Spasms Study suggested better response in patients with no identified etiology compared to those with an identified etiology, genetic or otherwise, at presentation.14 Another study evaluating impact of etiology on treatment response reported that among patients with non-acquired etiology without recognizable dysmorphisms/syndromes, genetic etiology was associated with an unfavorable treatment outcome.15 None of these studies were powered to determine differential treatment responses for individual genetic epilepsies.12, 14, 15
In addition to standard therapies for IESS, clinical experience and the literature support use of the ketogenic diet for refractory epilepsy in infancy, including ES, particularly for some established genetic diagnoses, including CDD.8–11, 16–19
We aimed to evaluate response to first-line IESS treatment regimens in patients with CDD-related ES, compared to a cohort of patients with ES not related to pathogenic CDKL5 variants. Because treatment within 1 month of ES onset improves developmental outcomes and rate of electroclinical remission in larger studies of ES,12, 14 we separately evaluate initial treatment within 1 month compared to initial treatment with longer lead time to treatment or response to subsequent medications. In addition, we assessed response to ketogenic diet and to other pharmacological treatments in resolution of ES in both cohorts.
Methods
This retrospective observational study was approved by the Boston Children’s Hospital Institutional Review Board (IRB). De-identified data were collected with IRB approval at the CDKL5 Centers of Excellence (Boston Children’s, Children’s Hospital Colorado, Cleveland Clinic, Children’s Hospital of Philadelphia, Washington University, Children’s Hospital of Colorado, Baylor College of Medicine/Texas Children’s Hospital, and University of California Los Angeles)6 with participating site agreements and/or under the International CDKL5 Clinical Research Network protocol approved by the Children’s Hospital Colorado IRB.
Inclusion criteria for the CDD cohort included a pathogenic or likely pathogenic CDKL5 variant. The comparison cohort consisted of individuals without CDD from the NISC database, a subset of the multicenter Pediatric Epilepsy Research Consortium dataset.12, 13 We included patients with ES onset between 2012–2022 (CDD cohort) or 2012–2018 (NISC cohort), age of onset 2 months to 2 years, and adequate information to assess response to treatment. We excluded from the full NISC cohort (n = 644) individuals who did not receive treatment with ACTH, oral corticosteroids, vigabatrin or ketogenic diet; due to known differential responses of ES in these disorders, we excluded those with Tuberous Sclerosis Complex, Trisomy 21, or with unknown etiology and normal development at ES onset.13, 14, 20–23 We additionally excluded those in the NISC cohort with incomplete information or inconsistencies in the data on timing of response and those with ES onset at < 2 months or > 2 years of age.
Data collection and definitions:
Data were collected through standard data collection forms (Supplemental Table 1), review of medical records, and directly from families and treating neurologists. Age of seizure onset, age of ES onset, and age of treatment were collected in months. Lead time is defined from age of ES onset to age of first standard treatment. We evaluated response to the following standard treatments (1) hormonal therapy (ACTH or oral corticosteroids), (2) vigabatrin, or (3) combined hormonal/vigabatrin therapy, initiated together. We additionally collected information on response to ketogenic diet and other anti-seizure medications. Clinical remission was defined as resolution of observable ES. Electroclinical remission was defined as resolution of both ES and hypsarrhythmia or modified hypsarrhythmia on follow-up EEG (if present on the initial EEG). Clinical and electroclinical remission were evaluated at 14 days and 3 months from initiation of each treatment. Late remission was defined as clinical or electroclinical remission after 14 days from initiation of treatment but before 3 months. Partial response, available only in the CDD cohort, was defined as a reduction in the number of ES occurring per day (categorized when possible as ≥50% or <50% reduction), or a resolution of identifiable ES but with persistence of hypsarrhythmia.
For the CDD cohort, hypsarrhythmia and modified hypsarrhythmia were based on classification/interpretation in EEG reports, and, when available, re-review of original data. We additionally scored available EEG studies using the original 2015 BASED (Burden of AmplitudeS and Epileptiform Discharges) score (data collection pre-dated the newer version).24
Statistical analysis:
Data analysis and descriptive statistics were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA). Descriptive statistics were used to summarize patient demographic and clinical features. Counts and percentages were provided for categorical variables. Medians and interquartile ranges (IQRs) were provided for continuous variables. When performing two-group comparisons, Wilcoxon rank-sum test was performed for continuous variables, and Pearson chi-square test or Fisher’s exact test was used for categorical variables. Individuals with missing values were excluded for two-group comparisons.
For the primary analysis we included only those for whom the order of treatment and the clinical response were known. Specifically, we evaluated the proportion of individuals in the CDD vs. the NISC cohort with clinical remission within 14 days from initiation of treatment and whether remission was sustained at 3 months. We report this clinical remission at these time-points separately for 1) first medication, lead time within 1 month and 2) subsequent medication or lead time >1 month. This includes the standard medications as above, and separately describe response to ketogenic diet and other anti-seizure medications. When EEG data were available, we also evaluated for electroclinical remission.
Results
A cohort of 59 individuals with CDD (79.3% female) with ES met inclusion criteria and were compared to 232 individuals without CDD from the NISC database (45.7% female, Figure 1). Median age of ES onset was 6 months (IQR 3, 10 months) in the CDD cohort and 7 months (IQR 5, 9 months) in the NISC cohort; (p = 0.03) (Table 1).
Figure 1.
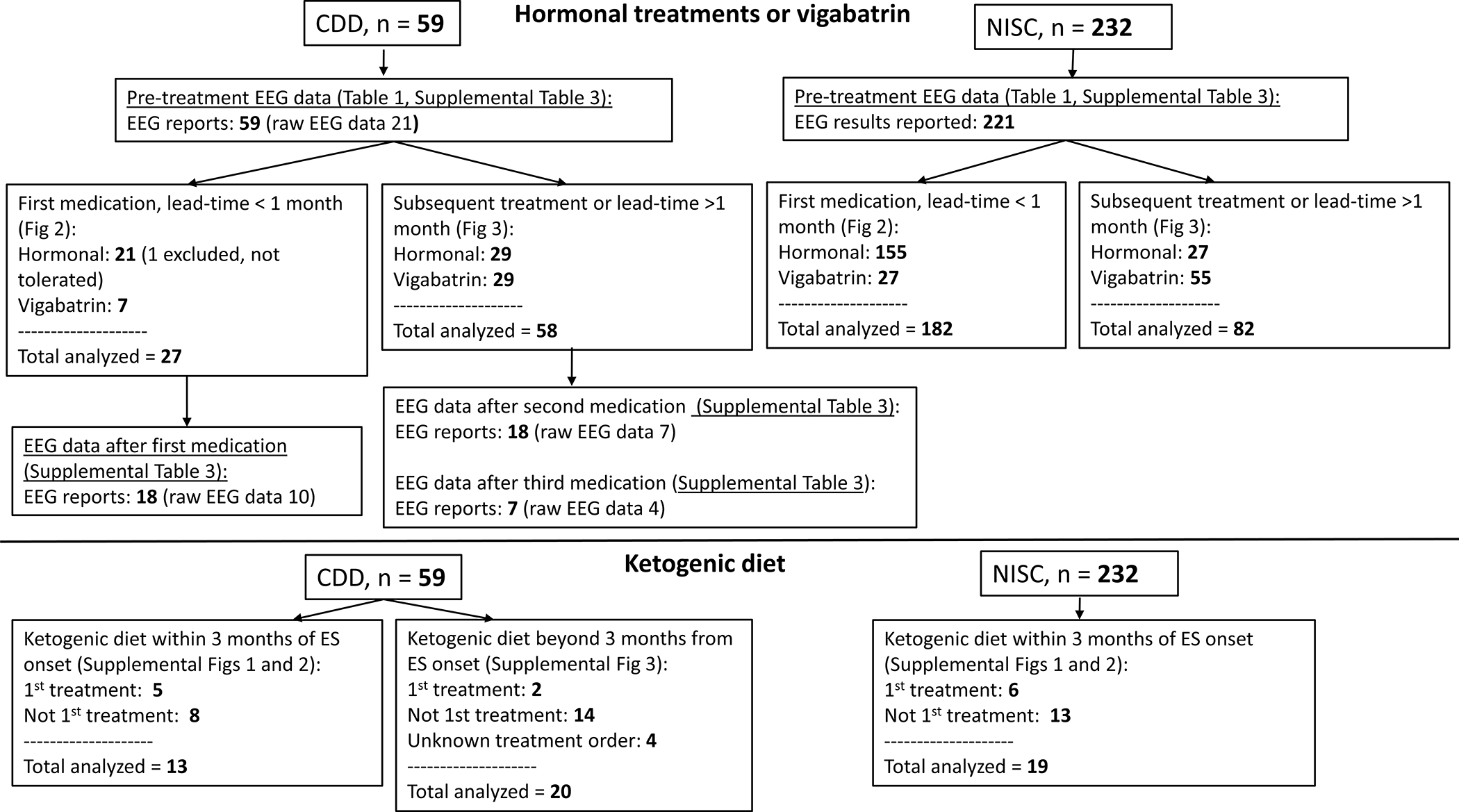
Flowchart of study cohorts and analysis.
Table 1.
Baseline characteristics of the study populations, including lead time from spasms onset for first-line treatment. The CDD cohort consisted of individuals seen at one of five CDKL5 Centers of Excellence in the United States with infantile spasms onset between 2012 and 2022. The National Infantile Spasms Consortium (NISC) cohort consisted of individual in the NISC database with infantile spasms of variable etiologies, enrolled from 2012–2019. This comparison cohort excluded individuals with an etiology known to have a differential response to first line IS treatments, specifically Tuberous Sclerosis Complex, Trisomy 21, or individuals with unknown etiology who presented with normal development at ES onset, and did not include known diagnosis of CDD.
| CDD cohort | NISC comparison cohort | P-value | |
|---|---|---|---|
|
| |||
| Total N (% Female) | 59 (79.7% Female) | 232 (45.7% Female) | <0.001 |
|
| |||
| Seizure onset (any type), weeks (median (IQR)) | 5 weeks (3, 8) | Not available | |
|
| |||
| Other seizure types prior to ES | 52 (88.1%) | 77 (33.9%), missing in 5 | <0.001 |
|
| |||
| ES onset, months (median (IQR)) | 6 months (3, 10) | 7 months (5, 9) | 0.03 |
|
| |||
| Hypsarrhythmia (full or modified) on EEG at ES onset prior to treatment? | 20 (33.9%) | 177 (80.1%) | <0.001 |
|
| |||
| Lead time (ES onset to first-line treatment) | |||
| Within 1 month of ES onset, n (%) | 36 (61.0%) | 196 (84.5%) | <0.001 |
| >1 month, n (%) | 18 (30.5%) | 14 (6.0%) | |
| Unknown, n (%) | 5 (8.5%) | 22 (9.5%) | |
|
| |||
| Duration of follow-up after ES, months (median, (IQR)) | 34.0 months (17.8, 53.2) | 2.9 months (2.0, 4.1) | <0.001 |
|
| |||
| Year of ES onset | 2012: 6 (10.2%) | 2012: 17 (7.3%) | <0.001 |
| 2013: 6 (10.2%) | 2013: 64 (27.6%) | ||
| 2014: 4 (6.8%) | 2014: 82 (35.3%) | ||
| 2015: 7 (11.9%) | 2015: 31 (13.4%) | ||
| 2016: 8 (13.6%) | 2016: 21 (9.0%) | ||
| 2017: 6 (10.2%) | 2017: 16 (6.9%) | ||
| 2018: 7 (11.9%) | 2018: 1 (0.4%) | ||
| 2019: 6 (10.2%) | |||
| 2020: 5 (5.1%) | |||
| 2021: 3 (5.1%) | |||
| 2022: 1 (1.7%) | |||
|
| |||
| Etiology | CDD 58 (100%) | Structural 88 (37.9%) | |
| Genetic 39 (16.8%) | |||
| Infectious 9 (3.9%) | |||
| Metabolic 7 (3.0%) | |||
| Other 11 (4.7%)# | |||
| Unknown 78 (33.6%) | |||
n=133, missing data for 57%
Other includes the following: atrophy, hypoglycemia, trauma, asymmetric ventriculomegaly, and tumor
Etiologies:
The 59 individuals with CDD included 17 with missense variants in the catalytic domain, 32 truncating variants (nonsense or frameshift), 8 partial gene deletions, and 2 intronic splice-affecting variants (Supplemental Table 2). NISC cohort etiologies included 88 (37.9%) structural (27 congenital, 61 acquired), 39 (16.8%) genetic, 9 (3.9%) infectious, 7 (3.0%) metabolic, 11 (4.7%) other, and 79 (33.6%) unknown (Table 1). The genetic category, when specified, included Trisomy 13, chromosomal copy number variants, adenylosuccinate lyase deficiency, neurofibromatosis, and STXBP1 and did not include any individuals with known CDD.
Epilepsy and associated phenotypes:
In the CDD cohort, 52 (88.1%) had had other seizure types prior to the onset of ES vs. 77 (33.9%) in the NISC cohort (Table 1). Median age of seizure onset was 5 weeks (IQR 3, 8 weeks) in the CDD cohort; data on age of onset were not collected for the NISC cohort.
Seizure types prior to ES in the CDD cohort included generalized motor (n=31, 63.3%), generalized non-motor (n=6, 13.3%), focal seizures (n=32, 66.7%), and unknown subtype (n=6, 13.9%). The most common specific seizure types prior to ES were focal motor (17), generalized tonic (n=13), GTC (n=7), and myoclonic (n=5). After ES onset, 44 (83.0%) of individuals had other seizure types documented as follows: generalized motor including tonic, myoclonic, GTC and atonic (n=41,95.3%), generalized non-motor (n=9, 22.5%), focal seizures (n=16, 38.1%), and unknown subtype (n=9, 25.7%).
Hypsarrhythmia or modified hypsarrhythmia was present at the onset of ES in 20/59 individuals (33.9%) in the CDD cohort compared to 177/ 221 individuals (80.1%) with available data in the NISC cohort (p <0.001, Supplemental Table 3). Six of 24 individuals in the CDD cohort for whom original EEG data were re-reviewed for classification had a BASED score of 4–5 (25.0%).
Developmental regression was documented in 22/52 individuals in the CDD cohort for whom data was available (42.3%), including 9 with regression at ES onset, 8 with regression not associated with ES, and 5 with timing of regression not specified.
Lead time, the time from ES onset to first-line treatment was within 1 month for 36 (61.0%) of the CDD cohort vs. 196 (84.5%) of the NISC cohort (Table 1); when lead time was >1 month, the median was 6 months (IQR 3, 10) for the CDD cohort vs. 2.2 months (IQR 1.6, 3.5) for the NISC cohort (p<0.001). Treatment responses are considered separately for those with lead time within 1 month compared to those with later treatment or subsequent medications.
Treatment regimens, first medication, lead time < 1 month:
Response to standard first-line medication for IESS, initiated within 1 month, was assessed in 27/59 (45.8%) individuals in the CDD cohort and 182/232 individuals (78.4%) in the NISC cohort (p <0.0001, Figure 1). One additional patient in the CDD cohort was started on prednisolone but discontinued treatment due to vomiting. In the CDD cohort, 7 (25.9%) were treated with ACTH, 13 (48.1%) with prednisolone, and 7 (25.9%) with vigabatrin. In the NISC cohort, 103 (56.5%) were treated with ACTH, 52 (28.6%) with oral corticosteroids, and 27 (14.8%) with vigabatrin. In addition to the 27 individuals in the CDD cohort treated with one of the first-line medications, one patient was treated with a combination of hormonal treatment and vigabatrin (partial response, no remission, not included in remission data below).
Clinical and electroclinical remission at 14 days, first medication, lead time < 1 month:
When considering only those treated with ACTH, oral corticosteroids, or vigabatrin, lead time less than 1 month, clinical ES remission by 14 days was reported in 25.9% (7/27) for all treatments combined in the CDD cohort vs. 58.2% (106/182) in the NISC cohort, with an additional 9.9% (18/182) with late clinical ES remission (>14 days) in the NISC cohort (Figure 2A, p = 0.0002). Of 7 CDD patients with clinical remission, electroclinical remission was confirmed by EEG in 3, no or partial response with EEG data in 2 and unknown in 2. In the CDD cohort, clinical ES remission by 14 days was reported in 3/7 with vigabatrin and 4/20 with hormonal treatment (p = 0.33). Partial early clinical response to treatment was documented in 7 individuals with CDD (2 with ACTH, 3 prednisolone, and 2 vigabatrin), 3 of whom had documentation of >50% reduction in ES.
Figure 2.
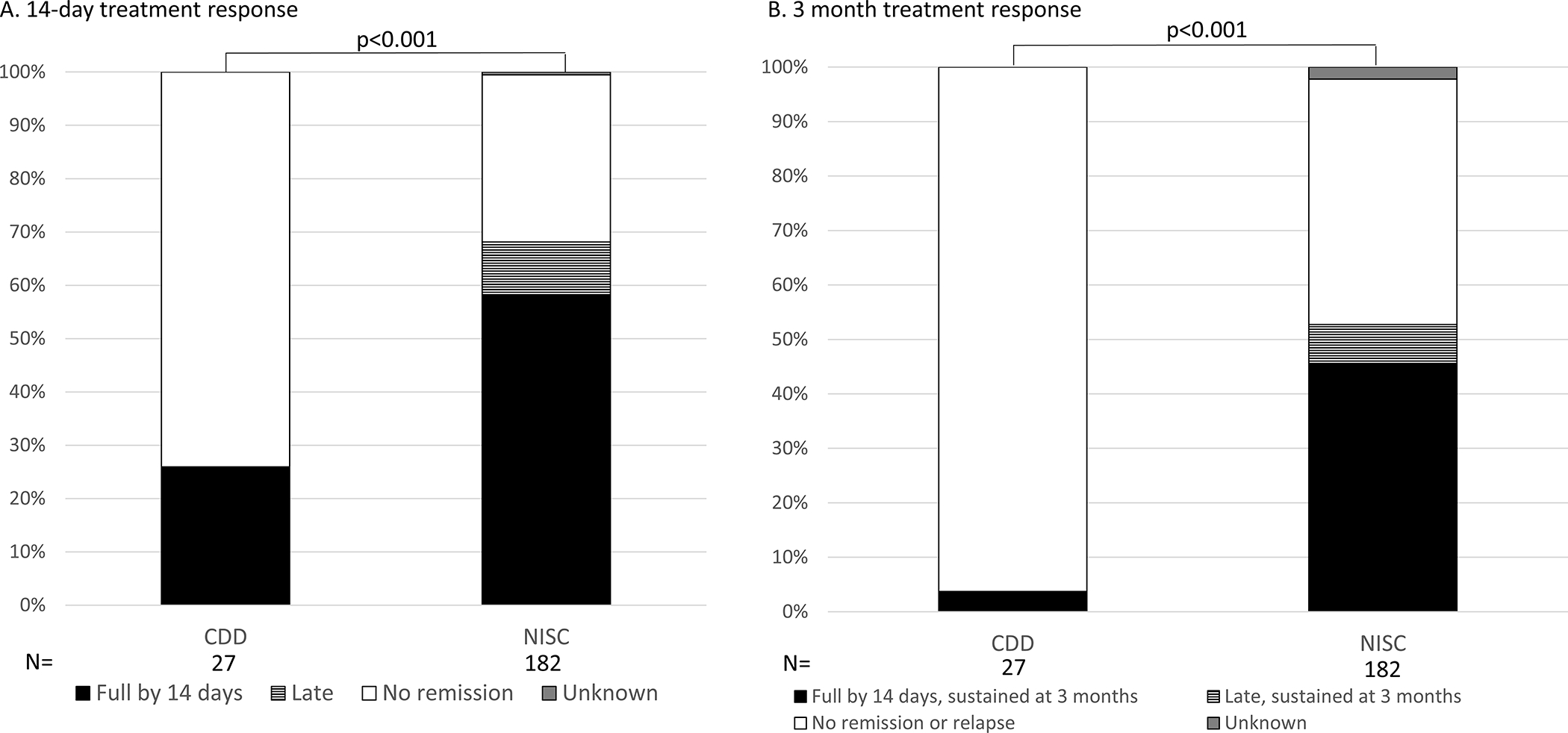
Clinical response to standard first-line treatment for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) when used as the first treatment, within one month of spasms onset. Standard treatments included hormonal treatments, adrenocorticotropic hormone (ACTH) and oral corticosteroids, and vigabatrin.
CDD cohort post-treatment EEGs, first medication, lead time < 1 month:
Hypsarrhythmia or modified hypsarrhythmia, if present pre-treatment, did not resolve with treatment including in 2 of 7 individuals with clinical remission of ES by 14 days (18 of 27 had both pre- and post- treatment EEG data available). Post-treatment EEGs showed hypsarrhythmia or modified hypsarrhythmia in 7 individuals (28.9%), normal in 2 (11.1%) and other patterns in 9 (50.0%) (Table S3). Post-treatment BASED scores in the CDD cohort were similar to pre-treatment (Table S3).
Clinical remission, sustained at 3 months, first medication, lead time < 1 month:
One of 27 individuals (3.7%) in the CDD cohort had a clinical (and confirmed electroclinical) ES remission by 14 days, sustained at 3 months (Fig 2B). This patient was treated with vigabatrin within 1 month of ES onset. In contrast, in the NISC cohort 83/106 individuals had ES remission by 14 days that was sustained at 3 months to any of the three treatments (77 with hormonal treatments, 6 vigabatrin), and 13/18 individuals with a late ES remission had a sustained remission at 3 months (10 with hormonal treatments, 3 with vigabatrin). ES remission was sustained at 3 months in the NISC cohort for 96 individuals (52.7%, Figure 2B).
Clinical and electroclinical remission at 14 days, subsequent medication or lead time >1 month:
When ACTH, prednisolone, or vigabatrin were used after another treatment standard or non-standard, e.g. vigabatrin after ACTH) or with a lead time of over 1 month, in the CDD cohort the overall clinical remission by 14 days was 22.4% (13/58) and 1 additional individual had a late clinical remission with vigabatrin with later relapse at 21 months (Figure 3A). These percentages remained lower than those of the NISC comparison cohort, in which 46/82 individuals (56.1%) had a clinical ES remission by 14 days and 4 individuals (4.9%) had a late ES remission, 1 with hormonal treatment and 4 with vigabatrin (p<0.0001). Of 29 individuals in the CDD cohort treated with vigabatrin after another treatment or with a lead time over 1 month, clinical remission was reported by 14 days in 8 and late remission in 1 (31.0% overall). Of 29 individuals in the CDD cohort treated with hormonal treatments, 5 (17.2%) had a clinical remission by 14 days (p = 0.12 comparing vigabatrin to hormonal treatments in the CDD cohort).
Figure 3.
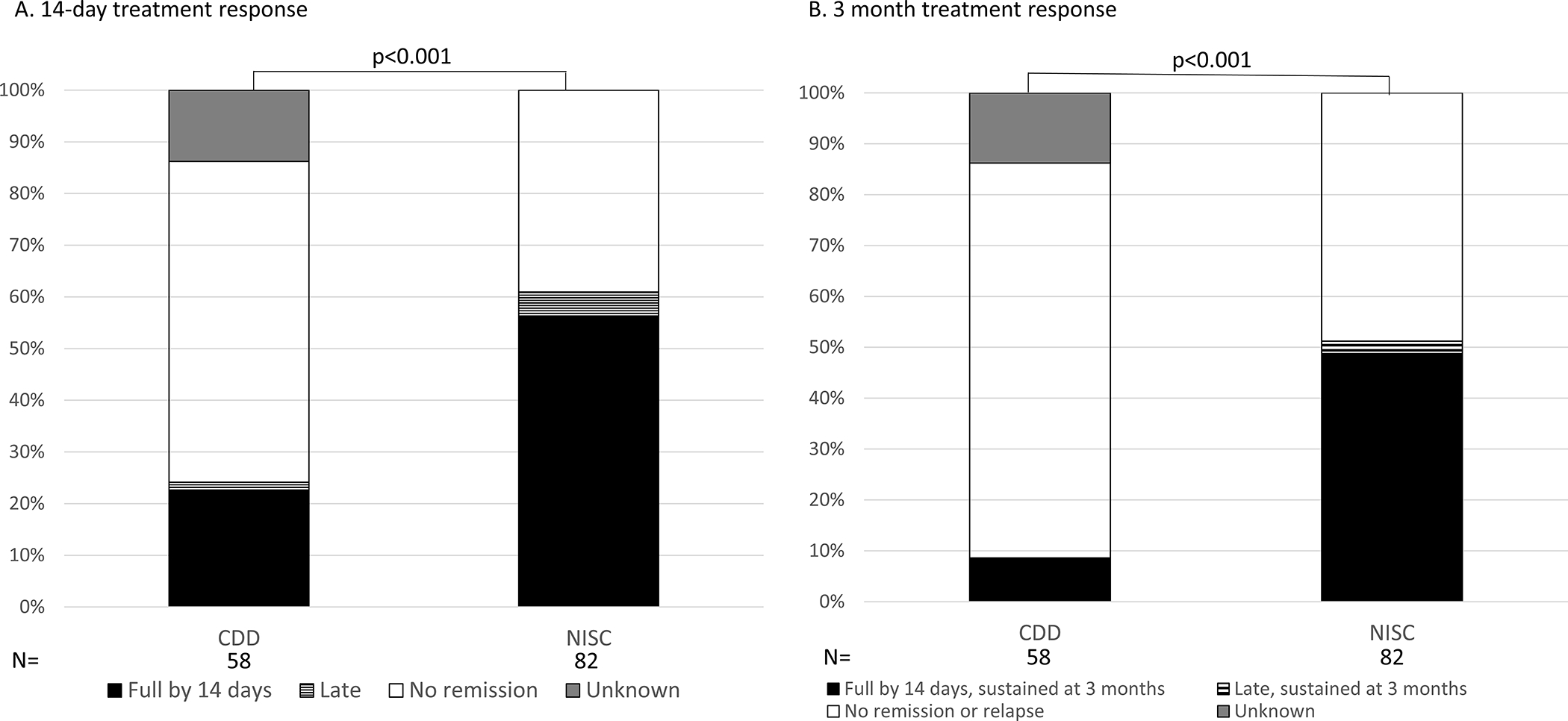
Clinical response to standard first-line treatment for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) when used as after another treatment and/or beyond one month from ES onset. Standard treatments included hormonal treatments, adrenocorticotropic hormone (ACTH) and oral corticosteroids, and vigabatrin.
Clinical remission, sustained at 3 months, subsequent medication or lead time >1 month:
Five of 58 individuals (8.6%) in the CDD cohort had a 14-day clinical ES remission sustained at 3 months, 4 on vigabatrin and one on ACTH. Forty of 82 individuals in the NISC cohort had a 14-day clinical ES remission sustained at 3 months and 2 had late response sustained at 3 months (p <0.0001 comparing the CDD to the NISC cohort in sustained 3-month response, Figure 3B). Thus, in the overall NISC cohort, 51.2% had a sustained ES remission at 3 months, including 16 treated with hormonal treatments and 26 treated with vigabatrin. EEG patterns after the second and third standard IESS treatments are included in Table S3.
ES remission with ketogenic diet:
Overall, ketogenic diet (initiated within 3 months of ES onset) resulted in clinical response at 1 month sustained at 3 months in at least 2/13 (15%) individuals in the CDD cohort and 1/19 (5%) in the NISC cohort. Detailed response data to ketogenic diet—divided into first treatment within 3 months of ES onset, treatment for refractory ES within 3 months of ES onset, and treatment beyond 3 months of ES onset—are included in Supplemental Figures 1–3.
Relationship of prior seizures with ES remission (ACTH, oral corticosteroids or vigabatrin only):
The clinical remission by 14 days did not vary when there were other seizure types prior to ES in the CDD cohort. When considering only the first medication treatment for ES regardless of lead time, 8/10 (80.0%) with clinical ES remission by 14 days and 24/28 (85.7%) without clinical ES remission by 14 days had other seizure types prior to ES (p = 0.64).
Discussion
In this retrospective study comparing treatment responses of ES in CDD to a comparison cohort from the NISC database, we report both longer lead time to treatment in the CDD cohort and poorer response to first-line therapy, even among those with a shorter lead time. While it has been shown that longer lead times are associated with poorer response to first line therapies,14 we found that in CDD patients, the response rates appeared to be similar between those with shorter vs longer lead times. We observed a high rate of prior seizures in the CDD cohort, 88% vs. 34% for NISC, but a relatively low rate of hypsarrhythmia or its variants (34% for CDD vs. 80% for NISC).12 This study is the first to evaluate ES treatment response in CDD, and we expand upon differential responses from larger studies of ES of diverse etiology, including NISC and the International Collaborative Infantile Spasms Study. Despite the limitations of retrospective studies, this approach was necessary and informative for this ultra-rare disorder with severe morbidity including related to ES.
The refractory nature of ES in CDD is consistent with prior literature demonstrating that seizures of all types are highly refractory to treatment in CDD.11 Prior literature has been limited by not differentiating treatment response to ACTH, oral corticosteroids, and vigabatrin in CDD by seizure type. One retrospective study reported greater than 50% reduction in seizure frequency (all types) at 3 months in 5/26 individuals (19%) with ACTH and in 12/23 individuals (52%) with vigabatrin.10 Vigabatrin is one of the most commonly used anti-seizure medications and also included among the most “effective” based on variable definitions in three retrospective studies and one caregiver survey.8–11 While the data from our study are also retrospective, they were collected using a standardized form from the CDKL5 Centers of Excellence with a focus specifically on response of ES to these first-line medications at appropriate time intervals as defined in prior studies of IESS.12, 14 In the CDD cohort, clinical remission by 14 days was slightly higher with vigabatrin than ACTH or prednisolone, but the number of CDD patients who were initially treated with vigabatrin (7) was small (not significant). Five individuals with CDD treated with vigabatrin at any point had a sustained clinical remission at 3 months.
The rare remission of ES to these first-line treatments in CDD raises the questions of whether benefit outweighs risks of side effects for the treatments, and whether other treatment approaches are warranted. Ketogenic diet was amongst the other treatments we evaluated; prior literature supported use of dietary therapy both for IESS of different etiologies and more broadly for epilepsy in CDD.8–11, 25–28 A 2018 review of randomized controlled trials and observational studies in patients with IESS noted that, within 6 months of follow-up, about one-third of patients were ES-free with the ketogenic diet and two-thirds had a greater than 50% reduction in ES.28 In the setting of established genetic diagnoses, such as pathogenic variants in CDKL5, KCNQ2, SCN2A, STXBP1, clinical experience and reports in the literature support use of the ketogenic diet for refractory epilepsy in infancy; 8, 11, 16, 17, 26, 29, 30 anecdotal reports from our Centers of Excellence suggest that this treatment may prevent or delay ES onset in infants with CDD. None of the data on ketogenic diet use in CDD is specific to ES, however, and initiation is often after 1 year of age.8–11, 16–19 In this retrospective cohort evaluating the treatment response of ES in CDD, firm conclusions regarding the efficacy of the ketogenic diet could not be made as the diet was initiated early after ES onset in only a few patients. Thus, future studies may be considered to assess efficacy of ketogenic diet in early infancy for the treatment or perhaps prevention of IESS in the CDD population.
In broader populations with ES of mixed etiologies, combinations of hormonal treatment plus vigabatrin have shown initial benefit but no difference in epilepsy or developmental outcomes at 18 months.14, 31, 32 Only one patient with CDD in this report received combination therapy with a partial response. While anti-seizure medications including valproic acid, clobazam, topiramate, rufinamide, felbamate, and others may be considered,33–36 we did not have adequate data to support any specific anti-seizure medication for treatment of ES.
Association of ES with hypsarrhythmia at onset has not been a predictor of treatment response in prior studies, and did not predict response in our CDD cohort.37 Hypsarrhythmia or its variants was present at ES onset in 34% of the CDD cohort in this study. Previous data reported from the CDKL5 Centers of Excellence, overlapping with the currently reported cohort, indicate that 47% of patients with spasms in CDD have hypsarrhythmia at some point in their disease course.1 We thus infer that some individuals with CDD develop hypsarrhythmia over time even if it is not present at the time of ES onset. Consistent with the poor rate of clinical remission with treatment for ES, the proportion of patients with hypsarrhythmia or modified hypsarrhythmia did not decline with treatment (Table S3). A prior study demonstrated treatment bias in that infants with hypsarrhythmia were more likely to be treated with standard treatments.37 Thus, lower rates of hypsarrhythmia at ES diagnosis may have contributed to the lower rates of use of first-line treatment in the CDD cohort.
Longer lead time from symptom onset to initiation of treatment has been negatively correlated with treatment response for IESS, not specific to etiology, in prior studies.12, 14 In this study, we observed longer lead time more often in the CDD cohort compared to the comparison cohort, but the rate of clinical remission was poor even in the subset with shorter lead time. Sequential seizures with multiple motor or non-motor phases occur frequently in CDD.2–5 While this study did not specifically evaluate timing of diagnosis or specific seizure patterns, we hypothesize that these seizure patterns with mixed motor phases may delay diagnosis and treatment of ES. It is also possible that ES presenting in individuals with prior seizures who are thus already on anti-seizure medications are inherently more refractory. Prior seizures or prior anti-seizure medications were associated with poorer response on univariate but not multivariate analysis in data from the NISC cohort.12, 13 In this study, a larger subset of individuals with CDD had prior seizures (87.9%) compared to the NISC cohort (33.9%), but prior seizures did not correlate with 14-day clinical response to treatment.
In CDD and related genetic epilepsies with high risk of IESS, early diagnosis and treatment may improve outcomes; thus, cautious clinical monitoring and anticipatory guidance about ES with or without EEG studies to serially screen for ES or hypsarrhythmia may be considered. Low rates of hypsarrhythmia and poor interrater reliability in assessing for hypsarrhythmia are limiting factors for EEG monitoring.38 Serial EEG and preventative treatment with vigabatrin at the onset of epileptiform activity in the EPISTOP study showed improved outcomes in Tuberous Sclerosis Complex, including delayed time of seizure onset, lower median proportion of days with seizures until age 2 years, and no patients in the preventative treatment arm developed ES.39
The retrospective nature of this work resulted in limitation in the precision and completeness of data. Data were extracted from medical records, including from outside centers, due to CDD being a rare disease.6 Individuals with CDD are often seen at the Centers of Excellence for quaternary care rather than for their primary neurology and epilepsy care. For this reason, and because of the long duration of the study from 2012–2021, primary EEG data were limited. The standard for IESS treatment response is electroclinical remission, but we took a pragmatic approach and evaluated clinical remission of ES because EEG data were not always available. We focused on data points that were most commonly available and on the subset of clinical treatment response for which we knew the timing and order of treatment as well as treatment responses. Additionally, data on specific dosing regimens were limited, so we are not able to comment on impact of dose. Prednisolone was more often used in the CDD cohort and ACTH in the NISC cohort, but response is comparable.40
In conclusion, we demonstrated that ES in CDD respond poorly to first-line treatment (ACTH, prednisolone, or vigabatrin). It is not clear how much of this effect is CDD-specific or the result of relatively late recognition of ES in CDD, which more often than with other etiologies, does not always present with hypsarrhythmia at onset of ES. Alternate therapies are not yet established, although ketogenic diet may be beneficial in select individuals in whom first-line treatments are ineffective. Future studies will need to evaluate, ideally prospectively, alternate or combination treatment approaches for ES in this population, and EEG screening for ES may be considered for more timely diagnosis and initiation of treatment. This study expands understanding of ES treatment response in specific genetic disorders. While it is difficult to adequately power disease-specific studies, it is possible that these observations in CDD may apply to other genetic developmental and epileptic encephalopathies. A prospective treatment study may better clarify patterns of treatment response by etiology and other factors over time.
Supplementary Material
Supplemental Figure 3. Clinical response to ketogenic diet for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) at 1 month (A) and sustained at 3 months (B) when initiated > 3 months from ES onset.
Supplemental Figure 1. Clinical response to ketogenic diet for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) at 1 month (A) and sustained at 3 months (B) when initiated as first-line treatment within 3 months of ES onset. No prior treatment with ACTH, oral corticosteroids or vigabatrin.
Supplemental Figure 2. Clinical response to ketogenic diet for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) at 1 month (A) and sustained at 3 months (B) when initiated for refractory ES within 3 months of ES onset.
Supplemental Table 1. RedCap form used for data collection from the CDKL5 Deficiency Disorder cohort
Supplemental Table 2. Table of genetic variants for the CDD cohort
Supplemental Table 3. EEG features pre-treatment of spasms in CDD and NISC cohorts.
Key points:
Lead time to treatment of epileptic spasms (ES) is often prolonged in CDKL5 Deficiency Disorder (CDD).
The majority (88% in this report) of patients with epileptic spasms in the setting of CDKL5 Deficiency Disorder have other seizure types preceding spasms.
The minority (34% in this report) of patients with epileptic spasms in the setting of CDKL5 Deficiency Disorder have hypsarrhythmia on EEG at spasms onset.
Response to first-line therapies for epileptic spasms is poor in patients with CDKL5 Deficiency Disorder.
Acknowledgments
The authors thank all of the individuals and families followed in our CDKL5 Centers of Excellence who have participated in our research studies. We thank our clinical colleagues for patient referrals and the Boston Children’s Hospital Epilepsy Genetics Program for thoughtful discussions. We thank Kelley Knupp, MD, for assistance with interpretation of the NISC data. We thank the Pediatric Epilepsy Research Consortium investigators who contributed to the NISC database. Funding for the CDKL5 Centers of Excellence was provided by the International Foundation for CDKL5 Research. Dr. Olson is supported by the National Institute of Neurologic Disorders and Stroke (NINDS K23 NS107646–05). Dr. Benke is funded by the Ponzio Family Chair in Neurology Research through Children’s Hospital Colorado Foundation.
Funding
International Foundation for CDKL5 Research, NINDS 1K23 NS107646–05, NINDS U01NS114312
Footnotes
Conflicts of Interest
Heather Olson – Dr. Olson received consulting fees from Takeda Pharmaceuticals and Zogenix regarding clinical trial design, Ovid Therapeutics regarding clinical trial results, Marinus Pharmaceuticals regarding CDKL5 Deficiency Disorder, and has done consulting for the FOXG1 Research Foundation.
Scott Demarest – Scott Demarest has consulted for Upsher-Smith, Biomarin, Neurogene, Marinus, Tysha, Zogenix and Ovid Therapeutics. He has funding from the NIH, International foundation for CDKL5 research, project 8P and Mila’s Miracle Foundation. He also serves on the advisory board for the non-profit foundations SLC6A1 Connect, Project 8P, Ring14 USA and FamilieSCN2A.
Elia Pestana-Knight – Elia Pestana-Knight has received consulting fees from Biomarin Pharmaceutical, Zogenix and Marinus Pharmaceuticals where she is a member of the Scientific Advisory Board (SAB).
Ahsan N. Moosa – No conflict of interest to disclose.
Xiaoming Zhang – XZ has no any conflict of interest to disclose.
José Renán Pérez-Perez – JP has no conflict of interest to disclose.
Judy Weisenberg – No conflict of interest to disclose.
Eric Marsh- Eric Marsh is a site PI and previous consultant with Stoke therapeutics. A site PI for Zogenix and Marinus Pharma. A site PI and consultant for Acadia Pharmaceuticals. A site PI and previous research support for GW Pharmaceuticals. He has research support from the NIH and from Eagles Autism Foundation, Penn Orphan Disease center- research support, RettSyndrome.org, RSRT, and International CDKL5 Research Foundation- Research Support/Clinical Center of Excellence Support
Rajsekar Rajaraman- Received honorarium from Zogenix Pharmaceuticals, consultation for UCB Pharmaceuticals, speaker for Marinus Pharmaceuticals.
Bernhard Suter – Consultancy for Taysha and Neurogene; and receives funding from International Rett Syndrome Foundation, International foundation for CDKL5 research, the NIH, the Blue Bird Circle foundation; Clinical Trials with Acadia, Avexis, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department.
Akshat Katyayan - No conflict of interest to disclose.
Isabel Haviland – No conflict of interest to disclose.
Carolyn Daniels – No conflict of interest to disclose.
Bo Zhang – No conflict of interest to disclose.
Caitlin Greene – No conflict of interest to disclose.
Michelle DeLeo - No conflict of interest to disclose.
Lindsay Swanson – Lindsay Swanson receives research support from the International Foundation for CDKL5 Research.
Jamie Love-Nichols – No conflict of interest to disclose.
Timothy Benke – Consultancy for AveXis, Ovid, GW Pharmaceuticals, International Rett Syndrome Foundation, Takeda, Taysha, CureGRIN, GRIN Therapeutics, Alcyone, Neurogene, and Marinus; Clinical Trials with Acadia, Ovid, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department
Chellamani Harini - No conflict of interest to disclose.
Annapurna Poduri – Annapurna Poduri is a member of the SAB for TevardBio, without person compensation.
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Demarest ST, Olson HE, Moss A, Pestana-Knight E, Zhang X, Parikh S, et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development Epilepsia. 2019. Aug;60:1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grosso S, Brogna A, Bazzotti S, Renieri A, Morgese G, Balestri P. Seizures and electroencephalographic findings in CDKL5 mutations: case report and review Brain Dev. 2007. May;29:239–242. [DOI] [PubMed] [Google Scholar]
- 3.Klein KM, Yendle SC, Harvey AS, Antony JH, Wallace G, Bienvenu T, et al. A distinctive seizure type in patients with CDKL5 mutations: Hypermotor-tonic-spasms sequence Neurology. 2011. Apr 19;76:1436–1438. [DOI] [PubMed] [Google Scholar]
- 4.Melani F, Mei D, Pisano T, Savasta S, Franzoni E, Ferrari AR, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life Dev Med Child Neurol. 2011. Apr;53:354–360. [DOI] [PubMed] [Google Scholar]
- 5.Melikishvili G, Epitashvili N, Tabatadze N, Chikvinidze G, Dulac O, Bienvenu T, et al. New insights in phenomenology and treatment of epilepsy in CDKL5 encephalopathy Epilepsy Behav. 2019. May;94:308–311. [DOI] [PubMed] [Google Scholar]
- 6.Olson HE, Demarest ST, Pestana-Knight EM, Swanson LC, Iqbal S, Lal D, et al. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review Pediatr Neurol. 2019. Aug;97:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong W, Haviland I, Pestana-Knight E, Weisenberg JL, Demarest S, Marsh ED, et al. CDKL5 Deficiency Disorder-Related Epilepsy: A Review of Current and Emerging Treatment CNS Drugs. 2022. Jun;36:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amin S, Majumdar A, Mallick AA, Patel J, Scatchard R, Partridge CA, et al. Caregiver’s perception of epilepsy treatment, quality of life and comorbidities in an international cohort of CDKL5 patients Hippokratia. 2017. Jul-Sep;21:130–135. [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi Y, Tohyama J, Takahashi Y, Goto T, Haginoya K, Inoue T, et al. Clinical manifestations and epilepsy treatment in Japanese patients with pathogenic CDKL5 variants Brain Dev. 2021. Apr;43:505–514. [DOI] [PubMed] [Google Scholar]
- 10.Muller A, Helbig I, Jansen C, Bast T, Guerrini R, Jahn J, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy Eur J Paediatr Neurol. 2016. Jan;20:147–151. [DOI] [PubMed] [Google Scholar]
- 11.Olson HE, Daniels CI, Haviland I, Swanson LC, Greene CA, Denny AMM, et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder J Neurodev Disord. 2021. Sep 16;13:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knupp KG, Coryell J, Nickels KC, Ryan N, Leister E, Loddenkemper T, et al. Response to treatment in a prospective national infantile spasms cohort Ann Neurol. 2016. Mar;79:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grinspan ZM, Knupp KG, Patel AD, Yozawitz EG, Wusthoff CJ, Wirrell E, et al. Comparative Effectiveness of Initial Treatment for Infantile Spasms in a Contemporary US Cohort Neurology. 2021. Jul 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Callaghan FJ, Edwards SW, Alber FD, Hancock E, Johnson AL, Kennedy CR, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial Lancet Neurol. 2017. Jan;16:33–42. [DOI] [PubMed] [Google Scholar]
- 15.Chourasia N, Yuskaitis CJ, Libenson MH, Bergin AM, Liu S, Zhang B, et al. Infantile spasms: Assessing the diagnostic yield of an institutional guideline and the impact of etiology on long-term treatment response Epilepsia. 2022. May;63:1164–1176. [DOI] [PubMed] [Google Scholar]
- 16.Ko A, Jung DE, Kim SH, Kang HC, Lee JS, Lee ST, et al. The Efficacy of Ketogenic Diet for Specific Genetic Mutation in Developmental and Epileptic Encephalopathy Front Neurol. 2018;9:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: Experience of >100 patients Epilepsia. 2017. Aug;58:1415–1422. [DOI] [PubMed] [Google Scholar]
- 18.Siri B, Varesio C, Freri E, Darra F, Gana S, Mei D, et al. CDKL5 deficiency disorder in males: Five new variants and review of the literature Eur J Paediatr Neurol. 2021. Jul;33:9–20. [DOI] [PubMed] [Google Scholar]
- 19.Ko A, Youn SE, Kim SH, Lee JS, Kim S, Choi JR, et al. Targeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy Epilepsy Res. 2018. Mar;141:48–55. [DOI] [PubMed] [Google Scholar]
- 20.van der Poest Clement E, Jansen FE, Braun KPJ, Peters JM. Update on Drug Management of Refractory Epilepsy in Tuberous Sclerosis Complex Paediatr Drugs. 2020. Feb;22:73–84. [DOI] [PubMed] [Google Scholar]
- 21.Daniels D, Knupp K, Benke T, Wolter-Warmerdam K, Moran M, Hickey F. Infantile Spasms in Children With Down Syndrome: Identification and Treatment Response Glob Pediatr Health. 2019;6:2333794X18821939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harvey S, Allen NM, King MD, Lynch B, Lynch SA, O’Regan M, et al. Response to treatment and outcomes of infantile spasms in Down syndrome Dev Med Child Neurol. 2022. Jun;64:780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elterman RD, Shields WD, Mansfield KA, Nakagawa J, Group USISVS. Randomized trial of vigabatrin in patients with infantile spasms Neurology. 2001. Oct 23;57:1416–1421. [DOI] [PubMed] [Google Scholar]
- 24.Mytinger JR, Hussain SA, Islam MP, Millichap JJ, Patel AD, Ryan NR, et al. Improving the inter-rater agreement of hypsarrhythmia using a simplified EEG grading scale for children with infantile spasms Epilepsy Res. 2015. Oct;116:93–98. [DOI] [PubMed] [Google Scholar]
- 25.Dressler A, Benninger F, Trimmel-Schwahofer P, Groppel G, Porsche B, Abraham K, et al. Efficacy and tolerability of the ketogenic diet versus high-dose adrenocorticotropic hormone for infantile spasms: A single-center parallel-cohort randomized controlled trial Epilepsia. 2019. Mar;60:441–451. [DOI] [PubMed] [Google Scholar]
- 26.Kim SH, Shaw A, Blackford R, Lowman W, Laux LC, Millichap JJ, et al. The ketogenic diet in children 3 years of age or younger: a 10-year single-center experience Sci Rep. 2019. Jun 19;9:8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nordli DR Jr., Kuroda MM, Carroll J, Koenigsberger DY, Hirsch LJ, Bruner HJ, et al. Experience with the ketogenic diet in infants Pediatrics. 2001. Jul;108:129–133. [DOI] [PubMed] [Google Scholar]
- 28.Prezioso G, Carlone G, Zaccara G, Verrotti A. Efficacy of ketogenic diet for infantile spasms: A systematic review Acta Neurol Scand. 2018. Jan;137:4–11. [DOI] [PubMed] [Google Scholar]
- 29.Su DJ, Lu JF, Lin LJ, Liang JS, Hung KL. SCN2A mutation in an infant presenting with migrating focal seizures and infantile spasm responsive to a ketogenic diet Brain Dev. 2018. Sep;40:724–727. [DOI] [PubMed] [Google Scholar]
- 30.Turkdogan D, Thomas G, Demirel B. Ketogenic diet as a successful early treatment modality for SCN2A mutation Brain Dev. 2019. Apr;41:389–391. [DOI] [PubMed] [Google Scholar]
- 31.Hahn J, Park G, Kang HC, Lee JS, Kim HD, Kim SH, et al. Optimized Treatment for Infantile Spasms: Vigabatrin versus Prednisolone versus Combination Therapy J Clin Med. 2019. Oct 2;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Callaghan FJK, Edwards SW, Alber FD, Cortina Borja M, Hancock E, Johnson AL, et al. Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial Lancet Child Adolesc Health. 2018. Oct;2:715–725. [DOI] [PubMed] [Google Scholar]
- 33.Dozieres-Puyravel B, Nasser H, Bellavoine V, Ilea A, Delanoe C, Auvin S. Felbamate for infantile spasms syndrome resistant to first-line treatments Dev Med Child Neurol. 2020. May;62:581–586. [DOI] [PubMed] [Google Scholar]
- 34.Hussain SA, Asilnejad B, Heesch J, Navarro M, Ji M, Shrey DW, et al. Felbamate in the treatment of refractory epileptic spasms Epilepsy Res. 2020. Mar;161:106284. [DOI] [PubMed] [Google Scholar]
- 35.Song JM, Hahn J, Kim SH, Chang MJ. Efficacy of Treatments for Infantile Spasms: A Systematic Review Clin Neuropharmacol. 2017. Mar/Apr;40:63–84. [DOI] [PubMed] [Google Scholar]
- 36.Olson HE, Loddenkemper T, Vendrame M, Poduri A, Takeoka M, Bergin AM, et al. Rufinamide for the treatment of epileptic spasms Epilepsy Behav. 2011. Feb;20:344–348. [DOI] [PubMed] [Google Scholar]
- 37.Demarest ST, Shellhaas RA, Gaillard WD, Keator C, Nickels KC, Hussain SA, et al. The impact of hypsarrhythmia on infantile spasms treatment response: Observational cohort study from the National Infantile Spasms Consortium Epilepsia. 2017. Dec;58:2098–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hussain SA, Kwong G, Millichap JJ, Mytinger JR, Ryan N, Matsumoto JH, et al. Hypsarrhythmia assessment exhibits poor interrater reliability: a threat to clinical trial validity Epilepsia. 2015. Jan;56:77–81. [DOI] [PubMed] [Google Scholar]
- 39.Kotulska K, Kwiatkowski DJ, Curatolo P, Weschke B, Riney K, Jansen F, et al. Prevention of Epilepsy in Infants with Tuberous Sclerosis Complex in the EPISTOP Trial Ann Neurol. 2021. Feb;89:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Zhong X, Hong S, Li T, Jiang L. Prednisolone/prednisone as adrenocorticotropic hormone alternative for infantile spasms: a meta-analysis of randomized controlled trials Dev Med Child Neurol. 2020. May;62:575–580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 3. Clinical response to ketogenic diet for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) at 1 month (A) and sustained at 3 months (B) when initiated > 3 months from ES onset.
Supplemental Figure 1. Clinical response to ketogenic diet for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) at 1 month (A) and sustained at 3 months (B) when initiated as first-line treatment within 3 months of ES onset. No prior treatment with ACTH, oral corticosteroids or vigabatrin.
Supplemental Figure 2. Clinical response to ketogenic diet for epileptic spasms (ES) in CDKL5 Deficiency Disorder (CDD) and a comparison cohort from the National Infantile Spasms Consortium (NISC) at 1 month (A) and sustained at 3 months (B) when initiated for refractory ES within 3 months of ES onset.
Supplemental Table 1. RedCap form used for data collection from the CDKL5 Deficiency Disorder cohort
Supplemental Table 2. Table of genetic variants for the CDD cohort
Supplemental Table 3. EEG features pre-treatment of spasms in CDD and NISC cohorts.