

Abstract
Chronic pain conditions frequently co-occur, suggesting common risks and paths to prevention and treatment. Previous studies have reported genetic correlations among specific groups of pain conditions and reported genetic risk for within-individual multi-site pain counts (≤7). Here, we identified genetic risk for multiple distinct pain disorders across individuals using 24 chronic pain conditions and genomic structural equation modeling (Genomic SEM). First, we ran individual genome-wide association studies (GWASs) on all 24 conditions in the UK Biobank (N ≤ 436,000) and estimated their pairwise genetic correlations. Then we used these correlations to model their genetic factor structure in Genomic SEM, employing both hypothesis- and data-driven exploratory approaches. A complementary network analysis enabled us to visualize these genetic relationships in an unstructured manner. Genomic SEM analysis revealed a general factor explaining most of the shared genetic variance across all pain conditions and a second, more specific factor explaining genetic covariance across musculoskeletal pain conditions. Network analysis revealed a large cluster of conditions and identified arthropathic, back, and neck pain as potential hubs for cross-condition chronic pain. Additionally, we ran GWASs on both factors extracted in Genomic SEM and annotated them functionally. Annotation identified pathways associated with organogenesis, metabolism, transcription, and DNA repair, with overrepresentation of strongly associated genes exclusively in brain tissues. Cross-reference with previous GWASs showed genetic overlap with cognition, mood, and brain structure. These results identify common genetic risks and suggest neurobiological and psychosocial mechanisms that should be targeted to prevent and treat cross-condition chronic pain.
1. Introduction
Chronic pain is a well-documented burden on the patient [1,26,46,79,98,109] and the healthcare system [114]. The costs of pain management are driven by an incomplete understanding of pain chronification mechanisms, which impedes effective prevention and treatment. It is now widely understood that chronic pain is complex and involves changes in brain pathways as well as peripheral mechanisms [94, 102, 113]. This understanding has led to the introduction of chronic primary pain disease codes in version 11 of the International Classification of Diseases (ICD-11) [123]. Nonetheless, with few exceptions [134], pain conditions are still classified largely based on the body site affected and either treated in the primary care setting [110] or referred to specialists by body site (back pain to orthopedists, irritable bowel syndrome to gastroenterologists, etc.). Unfortunately, the processes that drive chronic pain across conditions remain insufficiently understood, and most current treatments do not work for most patients [31,132]. There is an urgent need for a fundamentally different approach.
Recent work in mental health epidemiology has revealed extensive co-occurrence across disorders, leading to identification of common factors underlying multiple conditions [14,54,89]. Co-occurrence across pain conditions with different pathologies (e.g., migraine with irritable bowel syndrome [IBS]) has also been documented [3,67,74,106,116]. Furthermore, chronic pain conditions are heritable (with estimates up to 45% [47]), and genetic risks for pain are shared across conditions [84]. Two recent genome-wide association studies (GWASs) in the UK Biobank (UKBB) identified genetic variants related to multi-site pain [57,60]. These important studies, however, focused on pain widespreadness (quantitative 0–7 [57] or binarized: 1 versus 2 or more affected body sites [60]), without tracking whether genetic risks were shared across conditions. Widespreadness may either arise from co-occurring pain conditions or itself be a pain condition that affects a number of musculoskeletal body sites [20]. Given that five of the seven body sites used in both studies are musculoskeletal, including the most prevalent site, back pain), the genetic associations they assessed are likely driven by the genetic risk for widespread musculoskeletal pain and may not identify genes that cut across distinct disorders.
Here, we examine shared genetic risks across 24 distinct pain conditions in over 400,000 individuals in the UK Biobank (UKBB) [13]. We use Genomic Structural Equation Modeling (SEM) [43] to identify common genetic factors across conditions. Genomic SEM applies the traditional techniques of SEM, a widely used method for latent factor modeling [61], to genetic correlations estimated from genome-wide association statistics. Then we annotate the factors with associated biological pathways.
Figure 1 provides a graphical overview of the study. The specific questions we address are: (1) Is there a general, condition-agnostic genetic risk factor for chronic pain? (2) Are there additional genetic factors underlying subsets of pain conditions? (3) Does the genetic structure correspond to organization of pain by symptom location or hypothesized etiology? (4) What biological pathways and tissues are associated with these genetic factors? Addressing these questions may shed light on trans-diagnostic causes of chronic pain and potential targets for prevention and treatment.
Figure 1.
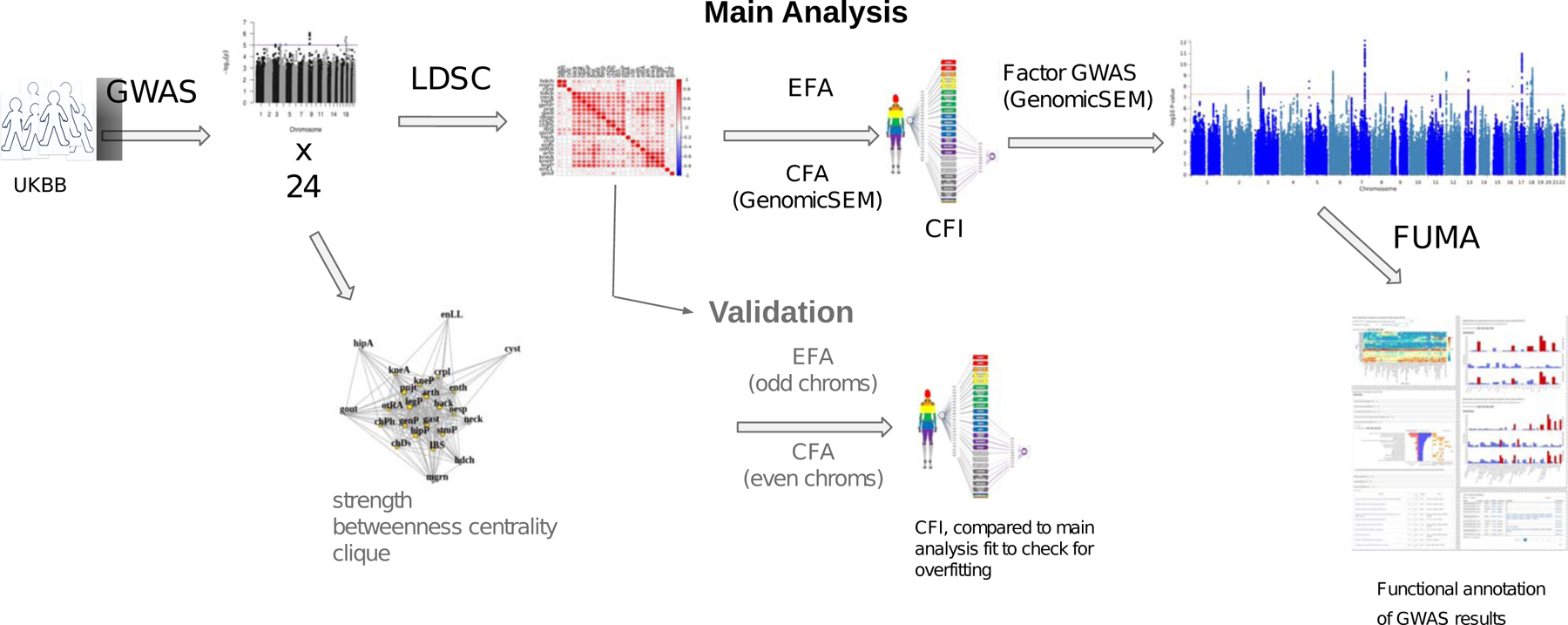
Scheme of study methods and analyses.
Abbreviations: GWAS, genome-wide association study; LDSC, linkage-disequilibrium score regression; EFA, exploratory factor analysis; CFA, confirmatory factor analysis; Genomic SEM, genomic structural equation modeling; CFI, comparative fit index.
2. Methods
The main steps in the analysis included: (1) GWAS, yielding association effect sizes for genetic variants (single-nucleotide polymorphisms, SNPs) with chronic pain conditions; (2) genetic correlations, which use the GWAS association effect sizes to estimate shared genetic risk across pain conditions; (3) Genomic SEM to identify common factors and compare factor models; (4) factor GWAS to identify SNPs associated with common factors; and (5) functional annotation of factor-associated SNPs in terms of likeliest implicated genes, pathways, and tissues.
2.1. Cohort
Participants in the UKBB were aged 40–69 and were recruited between 2006 and 2010 (UKBB data-request application 16651). The current standard in genetics is to limit analyses to samples of homogeneous ancestral background to avoid introducing confounds from population stratification [119]. We analyzed data from White Europeans, identified using UKBB-provided genomic principal components 1–4 [13], given that no other group had a sufficient sample size for our analyses (see Supplementary Table S2 for descriptive statistics of South Asians, the next highest sample size). Analyses in different ancestral groups will be a high priority when more data become available. Individuals who withdrew from the study by August 2020 were removed. Up to 435,971 people (54% female) were included in the analysis, with sample size varying by phenotype (Table 1).
Table 1:
Pain condition descriptive statistics
| Condition | Full name | Cases | Controls | Preva-lence | h2SNP(SE) | Reported h2SNP | Citations |
|---|---|---|---|---|---|---|---|
| *aCMC | Arthropathy of carpometacarpal joint | 1837 | 201439 | 0.009 | 0.09 (0.036) | NA | NA |
| arth | Arthropathies (non-specific, incl. osteoarthritis) |
80737 | 157458 | 0.339 | 0.09 (0.005) | NA | NA |
| back | Back pain | 119216 | 132641 | 0.473 | 0.09 (0.005) | 0.11/0.12/0.076 | [83, 35, 114] |
| chDs | Chest pain/discomfort | 72156 | 359415 | 0.167 | 0.08 (0.004) | ||
| chPh | Chest pain during physical activity | 2938 | 61044 | 0.046 | 0.13 (0.032) | ||
| *Crhn | Crohn’s Disease | 1826 | 201030 | 0.009 | 0.13 (0.038) | 0.47 | [121] |
| crpl | Carpal tunnel | 11912 | 424059 | 0.027 | 0.16 (0.011) | 0.02/0.01 | [135, 102] |
| CWP | Chronic widespread pain | 6021 | 427884 | 0.014 | 0.14 (0.014) | 0.10 | [56] |
| cyst | Cystitis | 15371 | 189253 | 0.075 | 0.03 (0.008) | ||
| *dbNr | Diabetic Neuropathy | 772 | 435199 | 0.002 | 0.13 (0.051) | 0.11 | [85] |
| enLL | Enthesopathies of lower limb | 7000 | 195713 | 0.035 | 0.06 (0.014) | ||
| enth | Enthesopathies | 28754 | 175077 | 0.141 | 0.06 (0.007) | ||
| *FM | Fibromyalgia | 2149 | 433822 | 0.005 | 0.10 (0.025) | 0.14 | [25] |
| gast | Gastritis | 41746 | 179970 | 0.188 | 0.07 (0.006) | ||
| gout | Gout | 15069 | 192253 | 0.073 | 0.20 (0.029) | ||
| hdch | Headache | 40222 | 345292 | 0.104 | 0.13 (0.008) | 0.21 | [81] |
| hipA | Hip arthrosis | 17676 | 193048 | 0.084 | 0.14 (0.012) | ||
| hipP | Hip pain | 41907 | 381055 | 0.099 | 0.08 (0.005) | 0.12 | [83] |
| IBS | Irritable bowel syndrome | 28419 | 182876 | 0.134 | 0.07 (0.008) | ||
| kneA | Knee arthrosis | 31267 | 184763 | 0.145 | 0.14 (0.009) | ||
| kneP | Knee pain | 78507 | 334812 | 0.190 | 0.10 (0.005) | 0.08 | [82] |
| legP | Leg pain | 41484 | 108241 | 0.277 | 0.10 (0.008) | ||
| mgrn | Migraine | 21586 | 189874 | 0.102 | 0.12 (0.009) | 0.15 | [39] |
| nksh | Neck/Shoulder pain | 72952 | 329192 | 0.181 | 0.08 (0.004) | 0.11 | [84] |
| oesp | Oesophagitis | 13003 | 195329 | 0.062 | 0.06 (0.010) | ||
| rhAt | Rheumatoid arthritis | 8685 | 198125 | 0.042 | 0.08 (0.014) | ||
| **plrh | Polymyalgia rheumatica | 2460 | 433511 | 0.006 | 0.09 (0.023) | ||
| pnjt | Pain in joint | 12016 | 423955 | 0.028 | 0.05 (0.008) | ||
| *prst | Prostatitis | 3604 | 199950 | 0.018 | 0.06 (0.020) | ||
| *seRA | Seropositive rheumatoid arthritis | 839 | 201957 | 0.004 | 0.15 (0.064) | ||
| stmP | Stomach pain | 21417 | 396116 | 0.051 | 0.08 (0.006) | 0.14 | [83] |
| **ulcC | Ulcerative colitis | 4211 | 199773 | 0.021 | 0.12 (0.022) | ||
| *urCl | Urinary colic | 4743 | 198679 | 0.023 | 0.06 (0.016) |
h2SNP is SNP heritability, variance in the phenotype explained by variance in genotypes (SNPs). S.E. is standard error. Reported h2SNP is provided where available.
phenotypes that did not have a significant h2SNP in either the odd or even autosome set.
phenotypes that did not load significantly onto either the common or specific factor in the EFA-informed CFA.
2.2. Phenotypes
The selected phenotypes were either chronic pain conditions, such as migraine or back pain lasting longer than three months, or conditions with persistent pain as a prevalent symptom, such as osteoarthritis. We drew an initial list of 91 phenotypes from five UKBB Categories: Medical conditions (100074), Health outcomes (713), Self-reported medical conditions (1003), Health and medical history (100036), and First occurrences (1712), downloaded in May 2021. We were able to greatly expand the number of pain conditions included in this study compared to previous studies [57,58,60,122], because of the First Occurrences dataset. This dataset gave researchers access to primary care and death register records to supplement self-reports and ICD-10 diagnoses that had been earlier available exclusively from hospital intake records. This updated UKKB dataset thus had, for the first time, more accurate case prevalences for a large number of conditions. We recoded these conditions into binary phenotypes (Supplementary Table S1) and pruned them to remove those that fell into one of the following categories: 1. heterogeneous disorders or groups of other conditions already included, such as ”Other diabetic polyneuropathies”; 2. branching traits (answers to questions dependent on endorsement of a previous question, with the exception of DF6159: ”Pain type(s) experienced in last month”, which was included as the branching question for pain experienced for more than three months); 3. disorders with case count < 500; 4. disorders that were not sufficiently related to genetics, with SNP heritability (see Section 2.2.1 below) less than or equal to 2 standard errors above zero (h2SNP - 2 ∗ SE <= 0), (see Supplementary Note). Genomic SEM models genetic covariances, and traits with low heritability cannot show significant genetic covariance with other traits; hence they were not included here. This pruning left 33 heritable chronic pain conditions (Table 1), which were further reduced to 24 during factor analysis (see Section 2.2.2 below).
2.3. GWAS
To prepare the data for GWAS, we used Plink [17], a commonly used, open-source toolset for genetic analyses (details in Supplementary Note). For the next step, GWAS, we estimated associations between each SNP and each chronic pain condition (phenotype) of interest using logistic regression, with the SNP variant as the predictor and the pain condition as the outcome. For these analyses, we used Regenie (details in Supplementary Note).
2.4. Factor analysis and structural equation model
2.4.1. Heritability and genetic correlations
We used linkage disequilibrium score regression (LDSC) software [12] to determine SNP heritability, h2SNP, and to estimate genetic correlations between every pair of pain conditions. For heritability, LDSC uses the pattern of SNP effects to estimate the variance in the phenotype that is attributable to all measured SNPs in aggregate (details in Supplementary Note). Table 1 reports the h2SNP for each condition, on a liability scale (Supplementary Note). For genetic correlations, LDSC determines the extent to which the pattern of genetic associations for one phenotype is correlated with the pattern of genetic associations for another phenotype. The resulting matrix of pairwise genetic correlations across pairs of chronic pain conditions is in Figure 2A. These genetic correlations provide the basis for factor analysis in Genomic SEM.
Figure 2.
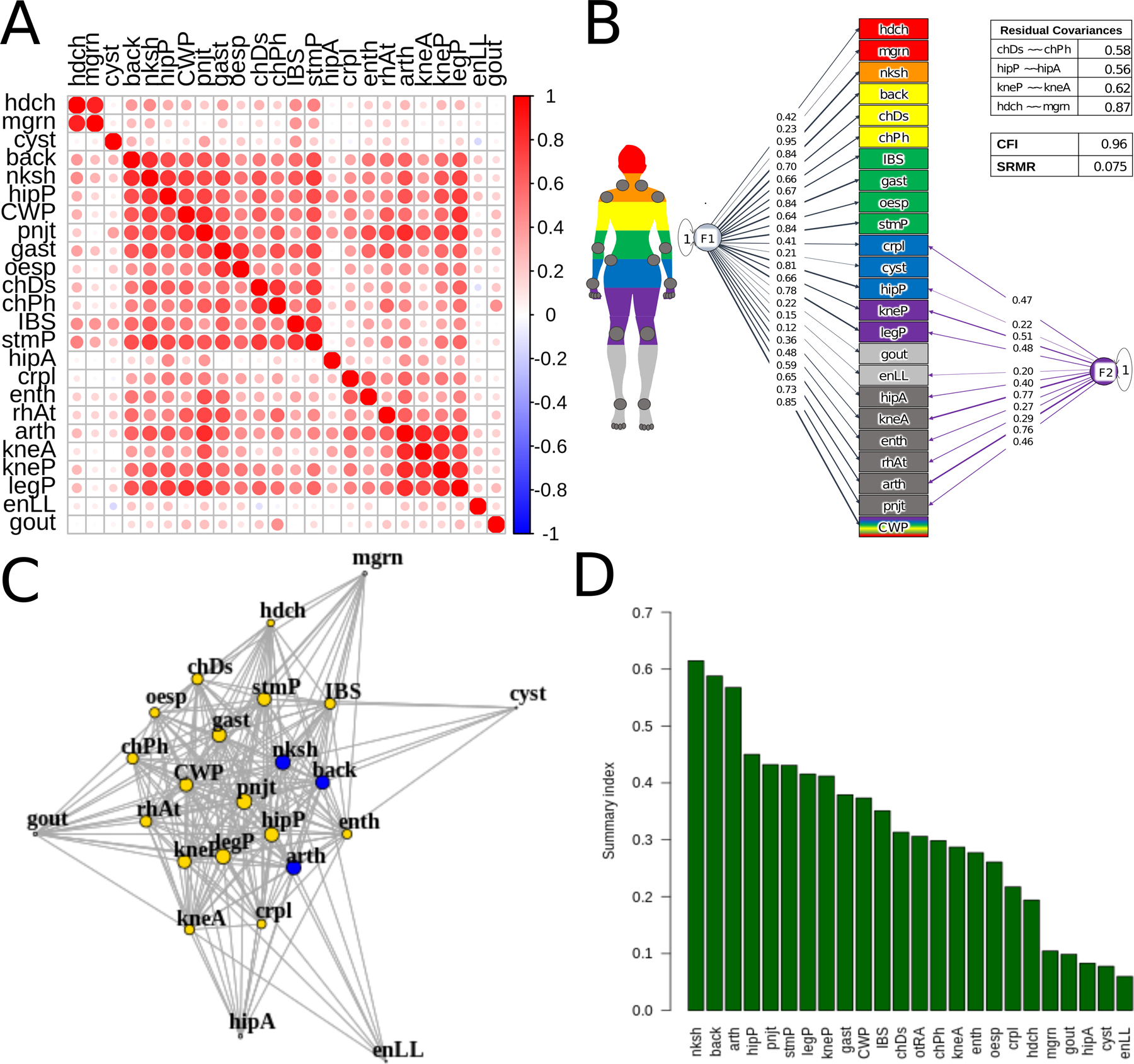
Genetic correlations, network, and Genomic SEM model.
Abbreviations: hdch, headache; mgrn, migraine; cyst, cystitis; back, back pain; nksh, neck/shoulder pain; hipP, hip pain; CWP, chronic widespread pain; pnjt, joint pain; gast, gastritis; oesp, oesphagitis; chDs, chest pain/discomfort; chPh, chest pain during physical activity; IBS, inflammatory bowel syndrome; stmP, stomach pain; hipA, hip arthrosis; crpl, carpal tunnel; enth, enthesopathies; rhAt, rheumatoid arthritis; arth, arthroses; kneA, knee arthrosis; kneP, knee pain; legP, leg pain; enLL, enthesopathies in lower limbs. (a) Genetic correlations for 24 pain conditions estimated using linkage disequilibrium score regression (LDSC) implemented in Genomic SEM. (b) Network of genetic correlations for 24 pain conditions, pruned for significance at FDR 0.01. The 19 conditions in yellow form a clique, complete subgraph. The 3 conditions in blue have the highest betweenness centrality, shortest path between 2 other nodes. Node size corresponds to strength, magnitude-weighted number of connections with other nodes. (c) EFA-CFA model for 24 pain conditions with residual covariances (~~) estimated for same body-site conditions (table in top left): hip arthrosis and pain; knee arthrosis and pain; headache and migraine; chest pain at baseline and during physical activity. F1 is the general factor with positive loadings from all conditions, and F2 is the musculoskeletal factor. CFI, comparative fit index; SRMR, standardized root mean squared residual. All loadings shown are significant at α=0.05. (d) Summary scores (overall measure of interconnectedness for each pain condition) obtained using F1 loadings from EFA-CFA and network strength and betweenness centrality, vector-normalized geometric means (y-axis). (More information on all conditions in Supplementary Table 1 (https://docs.google.com/spreadsheets/d/1S-vFvnwkD5iCP16La_iyjRDTxIqBoMRqAOV6SXpKhN8/edit#gid=0).
2.4.2. Factor analysis and Genomic SEM
We used Genomic SEM to test for evidence of shared genetic risk. SEM analyses test whether a hypothesized factor structure can adequately capture the observed correlations across a set of observed phenotypes (indicators). Genomic SEM applies the same method but to genetic correlations instead of phenotypic correlations; thus, the indicators are chronic pain conditions, but the data constitute correlations of genetic effects on phenotypes rather than phenotypic scores themselves. In confirmatory factor analysis (CFA), one specifies a series of hypothesis-driven models and compares their ability to accurately reproduce the pattern of correlations across conditions. Each model comprises a specification of which conditions load on which factors and whether the factors are themselves correlated. In exploratory factor analysis (EFA), one allows for the factor loadings to be determined in a data-driven fashion, identifying which groupings of conditions are best supported.
Our main analysis goal was to test for common factors without rigidly specifying groupings a priori. Thus, our main strategy combined EFA and CFA, using EFA to select the number of factors and their loadings and then evaluate the goodness of fit and compare with alternative models (see below for details). EFA-CFA, evaluated in [38] and recently used in [19,24,65], is a partially data-driven approach that captures observed groupings in the data while still permitting structure and inferences based on theory.
CFA can also be used to compare alternative theories about which types of common factors best explain observed correlations across chronic pain conditions. Here, we compared our EFA-guided model with two hypothesis-based CFA models: One based on shared genetic risks across conditions with similar body sites (‘anatomic’) and one based on shared risk for inflammatory conditions (‘etiologic’). The anatomic model included one general factor (on which all disorders load) and 6 specific factors that group conditions based on body site: Cranial, Gastrointestinal, Joint, Leg/Foot, Pelvic, and Torso. The etiologic model included the general factor and a specific factor for inflammatory conditions, which was the only putative etiology with a substantial number of representative conditions. We discussed a variety of other groupings, but as biological etiology is often unknown – a central problem in pain research – we did not reach clear consensus on additional etiological factors.
For all three models, the goal was to test a bifactor structure, which consisted of a general factor with loadings for all conditions and specific factors that are orthogonal to the general factor and have loadings for specific subsets of conditions. This type of model allowed us to test whether a common genetic factor underlay all tested pain conditions, while still allowing for shared variance for particular groups of conditions. Similar approaches have been used to model other multidimensional constructs, including personality [18] and psychopathology [9].
For the EFA portion of the EFA-CFA approach, we used the fa function in the ‘psych’ R package; a scree plot (Supplementary Figure S1) suggested three factors, allowing for correlations among factors using oblique rotation. The factor loadings were thresholded to define a CFA model for subsequent validation in Genomic SEM. The final model structure is shown in Figure 2C.
The use of EFA to guide model development requires validating the model’s fit on independent data, to avoid overfitting [33]. For this validation, we used a split-genome approach [34,42]. We developed a model using the EFA-CFA procedure described above in odd autosomes (1,3,...21), and assessed the fit of the final CFA in even autosomes (2,4,...22). This split created two independent sets, given that SNPs are not correlated across chromosomes (Mendel’s law of independent assortment).
Using an odd-even autosome split assumes that traits are polygenic (many genes contribute) and that relevant genes are distributed across autosomes, so that an estimate of genetic correlations from odd autosomes can be replicated in even ones. The polygenic nature of pain conditions, like other complex traits, was evident in this dataset (Supplementary Figure S2). However, to be conservative, we excluded conditions whose genetic associations were not heritable in both odd and even autosomes (Supplementary Note). This validation step led to a further exclusion of 7 conditions (arthropathy of carpometacarpal joint, diabetic neuropathy, Crohn’s disease, fibromyalgia, prostatitis, seropositive rheumatoid arthritis, and urinary colitis), whose heritability estimates were not significantly above 0 in at least one holdout set (Table 2). This final exclusion left 24 pain conditions for the validation step, which we also used in the main analysis and in the two hypothesis-driven approaches for consistency and comparability.
Table 2:
SNP heritability in whole genome, odd and even chromosomes
| Condition | Whole genome h2SNP (SE) | Odd chroms. h2SNP (SE) | Even chroms. h2SNP (SE) |
|---|---|---|---|
| *aCMC | 0.09 (0.036) | 0.09 (0.026) | 0.00 (0.023) |
| arth | 0.09 (0.005) | 0.05 (0.004) | 0.04 (0.004) |
| back | 0.09 (0.005) | 0.04 (0.003) | 0.05 (0.003) |
| chDs | 0.08 (0.004) | 0.04 (0.003) | 0.04 (0.002) |
| chPh | 0.13 (0.032) | 0.09 (0.025) | 0.05 (0.023) |
| *Crhn | 0.13 (0.038) | 0.08 (0.029) | 0.05 (0.025) |
| crpl | 0.16 (0.011) | 0.07 (0.008) | 0.08 (0.009) |
| cyst | 0.03 (0.008) | 0.01 (0.006) | 0.02 (0.006) |
| *dbNr | 0.13 (0.051) | 0.02 (0.033) | 0.11 (0.038) |
| enLL | 0.06 (0.014) | 0.03 (0.010) | 0.03 (0.009) |
| enth | 0.06 (0.007) | 0.03 (0.005) | 0.03 (0.005) |
| *FM | 0.10 (0.025) | 0.06 (0.017) | 0.04 (0.019) |
| gast | 0.07 (0.006) | 0.03 (0.004) | 0.04 (0.004) |
| CWP | 0.14 (0.014) | 0.07 (0.010) | 0.07 (0.010) |
| gout | 0.20 (0.029) | 0.08 (0.013) | 0.12 (0.024) |
| hdch | 0.13 (0.008) | 0.06 (0.004) | 0.07 (0.007) |
| hipA | 0.14 (0.012) | 0.07 (0.009) | 0.07 (0.008) |
| hipP | 0.08 (0.005) | 0.04 (0.003) | 0.04 (0.003) |
| IBS | 0.07 (0.008) | 0.04 (0.005) | 0.03 (0.004) |
| kneA | 0.14 (0.009) | 0.07 (0.005) | 0.08 (0.007) |
| kneP | 0.10 (0.005) | 0.05 (0.003) | 0.05 (0.003) |
| legP | 0.10 (0.008) | 0.05 (0.005) | 0.05 (0.005) |
| mgrn | 0.12 (0.009) | 0.06 (0.006) | 0.06 (0.007) |
| neck | 0.08 (0.004) | 0.04 (0.003) | 0.04 (0.003) |
| oesp | 0.06 (0.010) | 0.03 (0.007) | 0.03 (0.006) |
| rhAt | 0.08 (0.014) | 0.05 (0.010) | 0.04 (0.010) |
| **plrh | 0.09 (0.023) | 0.05 (0.016) | 0.03 (0.015) |
| pnjt | 0.05 (0.008) | 0.03 (0.006) | 0.02 (0.005) |
| *prst | 0.06 (0.020) | 0.03 (0.014) | 0.04 (0.013) |
| *seRA | 0.15 (0.064) | 0.17 (0.043) | −0.02 (0.047) |
| stmP | 0.08 (0.006) | 0.04 (0.004) | 0.04 (0.005) |
| **ulcC | 0.12 (0.022) | 0.07 (0.016) | 0.05 (0.015) |
| *urCl | 0.06 (0.016) | 0.02 (0.012) | 0.03 (0.012) |
h2SNP is SNP heritability, variance in the phenotype explained by variance in genotypes (SNPs). S.E. is standard error.
phenotypes that did not have a significant h2SNP in either the odd or even autosome set.
phenotypes that did not load significantly onto either the common or specific factor in the EFA-informed CFA. Condition definitions are in 1, and details are in Supplementary Table S1.
Models were evaluated using CFI (comparative fit index), which quantifies the extent to which the model fits better than a baseline model (one in which the variables are uncorrelated), and SRMR (standardized root mean residual), which quantifies the mean absolute difference between the observed correlations and the correlations predicted by the model [50]. A well-fitting model should generally have a CFI ≥ .95 and an SRMR ≤ .08 [50]. The models were additionally compared using AIC, (Akaike information criterion), which is a goodness-of-fit index favoring more parsimonious models (lower values indicate better fit) [61].
2.5. Factor GWAS
Common factors may have associations with genetic variants that are not detectable in analysis with individual conditions. To identify the SNPs with the largest associations with common chronic pain factors, we ran a factor GWAS in Genomic SEM (userGWAS function) (Supplementary Figure S3).
One issue to address when calculating GWAS on common factors is whether SNPs are associated primarily with the common factor or instead are more strongly associated with individual conditions that contribute to it. To assess each SNP for disproportionately strong or directionally opposing effects on a subset of conditions, we conducted a heterogeneity Q test [43,52] and discarded SNPs with heterogeneous effects as well as those correlated with them (Supplementary Note).
2.5.1. GWAS annotation
GWAS yields a list of SNPs significantly associated with a trait. Follow-up analyses are then needed to characterize those SNPs in terms of genes and pathways. Functional Mapping and Annotation (FUMA) of GWAS [137] is a platform developed to facilitate a number of standard GWAS follow-up analyses. To functionally characterize the genetic contributors to both individual phenotypes and the two factors, we submitted all GWAS results to FUMA for gene prioritization and functional annotation, using several integrated databases [137]. These analyses consisted of: 1. prioritizing SNPs based on their effect sizes and independence from each other; 2. mapping significant SNPs to genes as described below; 3. conducting a genome-wide gene-based association analysis using FUMA-implemented MAGMA (https://ctg.cncr.nl/software/magma) for gene analysis and gene property analyses; 4. gene set analysis for enrichment in known biological pathways; 5. gene property analysis, or testing for preferential expression of associated genes with 53 Gene-Tissue Expression repository (GTEX), version 8, tissues. We used FUMA’s default and standard significance thresholds and parameters, including p < 5×10−8 for lead SNPs (independent at r2 < 0.1); p < 0.05 for all other SNPs; r2 threshold for independent significant SNPs used for further annotations, including gene mapping: 0.6; reference panel population = UKB release 2b 10K European; minimum minor allele frequency = 0.01; maximum distance between LD blocks to merge into a locus = 250 kilobases. The r2 threshold represents a squared pairwise correlation for SNP variant alleles. The sample sizes for the two factors (general and musculoskeletal) identified in the final EFA-CFA model were 422,752 and 468,929, respectively, calculated using the method described in [76]. Variants from the reference panel that were in LD with GWAS lead SNPs were included to increase the chance of including causal variants.
Mappings of independent significant (as defined in FUMA, p < 5×10−8 and r2 < 0.6) SNPs onto genes was based on (1) positional distance (within 10 kilobases of gene start and stop coordinates); (2) statistical associations with transcription levels (expression quantitative trait locus, eQTL); and (3) chromatin interaction mapping, physical interactions with gene chromatin states (indicative of transcriptional accessibility). Only protein-coding genes were included, and the major histocompatibility (MHC) region was excluded from annotation. MAGMA analysis for gene-based (versus SNP-based) associations [23] was conducted with SNP assignment within windows of 10 kilobases of gene start and stop coordinates, and GTEx, version 8, [73] was used for gene expression analysis in 53 tissues. Given that the highest association statistic is not necessarily correlated with its relative importance (see this recent publication discussing negative selection as a mechanism for purging high-effect variants in critical gene loci [95]), our approach was to prioritize genes based on: 1. an a priori association p cut-off to ensure statistical rigor; and 2. convergent lines of evidence for functional importance, i.e. overlap in the three mapping approaches. In the resulting set, we interpret our findings in their entirety, without deference to the top association.
The gene-tissue expression analysis tested for association between highly expressed genes in 53 GTEx tissues and GWAS effect sizes for the same genes, which tests the relationship between the genes highly associated with the pain factors and highly expressed in different tissues (details in Supplementary Note). The parameters are summarized in Supplementary Table S3.
We used REVIGO [113] to assign, prune, and summarize biological pathways to the 25 genes with overlapping mappings (details in Supplementary Note).
2.6. Network analysis
While CFA has many strengths in permitting model comparison, some groups have emphasized that relationships among clinical conditions can have a complex causal structure that can be characterized in terms of networks of interacting variables [131]. We made no strong claims about the underlying causal structure and complemented the factor-analytic models with a network-based approach to characterize genetic relationships among conditions in terms of multiple local causes instead of a few latent causes. Network characterization and visualization was done in igraph in R [22]. Genetic correlations of the final 24 pain conditions were filtered for positive significant correlations, using a threshold of 0.01 false discovery rate (FDR)-corrected, calculated with fdrtool in R. We calculated two graph theoretic properties for each pain condition: (1) strength, calculated as the number of edges (genetic correlations with other pain conditions) weighted by their magnitude [7]; and (2) betweenness-centrality, the number of shortest paths between pairs of pain conditions that go through the pain condition in question) [11]. Strength identifies ‘hub’ conditions that are robustly genetically related to many other conditions and may thus be prominent indicators of multi-disorder susceptibility. Betweenness-centrality identifies ‘connector hubs’, conditions that are genetically related to multiple other conditions that are themselves less interrelated. ‘Connector hubs’ are thus key indicators of shared genetic vulnerability. These measures may themselves be correlated, and if so, combined into an overall index, as we did here (described below). At the network level, we estimated the largest clique, complete subgraph of intercorrelated pain conditions [29], which identifies a group of genetically interrelated conditions that may together serve as indicators of multi-disorder susceptibility.
2.7. Summary score
To summarize the evidence for which conditions are the most consistent key indicators of multi-disorder vulnerability, we combined results from Genomic SEM and network analysis, obtaining an overall measure of interconnectedness for each pain condition. Thus, we derived summary scores for all pain conditions using general factor loadings from EFA-CFA, network strength, and betweenness centrality, which are intercorrelated, r =.935 (general factor and strength), r =.614 (general factor and betweenness), r =.693 (strength and betweenness). We calculated a geometric mean of these 3 measures, after vector-normalizing them using the norm function in R.
3. Results
The work reported here is part of a project pre-registered on Open Science Foundation, OSF (Identifying and characterizing genetic susceptibility and its overlap with psychosocial traits, https://osf.io/4p5e3).
3.1. Univariate pain condition GWAS curation and annotation
We considered 91 potentially relevant pain phenotypes in the UK Biobank and selected 24 that (a) were indicative of chronic pain conditions, (b) had sufficient case counts (>500), and (c) were significantly heritable (see Methods; Table 1 and Supplementary Table S1). The sample size available for case assessment varied by condition and ranged from 63,982 (chest pain during physical activity) to 435,971 (several conditions). Prevalence ranged from 0.002 (772 cases, diabetic neuropathy) to 0.473 (119,216 cases, back pain). SNP heritability (variance in the phenotype explained by variance in the genotype) ranged from 0.03(SE, 0.008) for cystitis to 0.20(SE, 0.029) for gout.
Summaries of results from univariate GWAS are reported in Table 1 (SNP heritabilities), in Supplementary Figures S2 and S4 for Manhattan and quantile-quantile (QQ)-plots, and in Supplementary Table S14 for numbers of significant SNPs and genes.
3.2. Pain condition genetic correlations
Pairwise genetic correlations for the 24 pain conditions, Figure 2A, show a large cluster of interconnected vertices. This main cluster includes etiologically and anatomically diverse conditions, such as back pain, oesophagitis, irritable bowel syndrome (IBS), and carpal tunnel, suggesting shared genetic susceptibility among these disparate syndromes. Headache and migraine form a tight mini-cluster (top left), and cystitis, hip arthrosis, enthesopathies of the lower limb and gout show weaker correlations, suggesting more specific genetic risks for each of these four conditions.
A natural question is whether genetic correlations are potentially inflated when estimated in individuals with comorbid conditions. The answer is no. Conditions are primarily comorbid due to shared genetic risks or shared environmental risks. However, conditions with shared environmental risks would not correlate genetically, because alleles of different genes segregate independently from each other (Mendel’s law of independent assortment). Thus, given distinct genetic profiles for two conditions, the risk alleles for one condition would not correlate with the risk alleles for another. A similar genetic profile in many individuals with comorbid conditions indicates a true common predisposition.
3.3. Structural equation modeling
Using three approaches – hypothesis-driven anatomic (1) and etiologic (2), and largely data-driven exploratory-then-confirmatory (3) factor analyses (EFA-CFA) – we fit a bifactor model to test the loadings of all conditions onto a general factor, with differences in specific factor groupings in each approach. The anatomic model based on body site (Supplementary Figure S5, CFI= .875 and SRMR = .087) and the etiologic model, based on a grouping of inflammatory disorders (Supplementary Figure S6, CFI= .905, SRMR= .095) both had suboptimal fit (CFI≤ 0.95 and SRMR≥ .08), see Methods. The EFA-CFA model, shown in Figure 2C, produced an adequate overall fit (CFI= 0.956, SRMR = 0.075).
All pain conditions loaded positively and significantly onto the general factor. The specific factor had substantial positive loadings for arthropathies (which include osteoarthritis), carpal tunnel, enthesopathies of lower limb, other enthesopathies, hip arthrosis, hip pain, knee arthrosis, knee pain, leg pain, pain in joint, and rheumatoid arthritis. Given the pronounced musculoskeletal component among these indicators, we interpreted the specific factor as musculoskeletal. This factor is in line with the World Health Organization’s grouping of pain diseases of the musculoskeletal system, which groups conditions that affect joints, bones, muscles, the spine, and multiple body areas or systems [135]. The EFA-CFA was superior (AIC=4849.164) to both the anatomic (AIC=13184.43) and the etiologic (AIC=10024.93) models. In addition, the latter models had non-significant loadings on their specific factors (Leg/Foot, Pelvic, and Torso for the anatomic, Supplementary Figure S5, and Inflammatory for the etiologic, Supplementary Figure S6, suggesting that shared variance for those indicators was mainly explained by the general factor (details in the Supplementary Note). We validated this model by using the same approach, EFA on odd (CFI= .884 and SRMR= .123) and CFA testing on even (CFI= 0.903 and SRMR= .129) autosomes (details in the Supplementary Note). These comparable metrics in the odd/even and whole-genome datasets suggest that using EFA and CFA on the same dataset did not result in substantial overfitting.
3.4. Network analysis and central conditions
Network analysis provided additional evidence for substantial genetic overlap across pain conditions with a different theoretical model. Graph-theoretical properties of the network (Figure 2B) indicate shared genetic susceptibility, and node size corresponds to strength (magnitude-weighted number of connections). There is a complete subgraph of 19 interconnected conditions, highlighted in yellow: arthropathies, back pain, neck/shoulder pain, hip pain, knee pain, leg pain, chest pain (baseline and during physical activity), rheumatoid arthritis, knee arthrosis, joint pain, carpal tunnel, enthesopathies, widespread pain, gastritis, oesophagitis, stomach pain, headache, and IBS. Consistent with the CFA model, these conditions affect diverse body sites and span inflammatory and non-inflammatory as well as musculoskeletal and non-musculoskeletal forms of pain. Gout, hip arthrosis, enthesopathies of the lower limb, cystitis, and migraine lie outside the large cluster, but they still have more than 10 connections each. Overall, the network reveals a large core of pain syndromes with shared genetic vulnerability.
Some conditions were particularly central in the network, in several ways. Arthropathies, back, and neck/shoulder pain had the highest betweenness centrality (the highest number of shortest paths between node pairs that go through the index node), indicating that genetic associations between many conditions share genetic vulnerability with at least one of these three.
The summary score derived from the general factor, network node strength, and betweenness centrality, Figure 2D, reflects the highest degree of genetic overlap with other conditions. Once again, the top highest scorers were neck/shoulder pain, back pain, and arthropathies.
3.5. Factor GWAS and annotation
After running factor GWASs, we excluded QSNPs, which showed evidence of effects specific to certain pain conditions (not through the common factors), and conducted functional annotation of the GWAS output for each of these factors.
3.5.1. General Factor
The general factor GWAS yielded 33 genome-wide independent significant SNPs, Supplementary Table S4, Figure 3. FUMA mapped these to a total of 241 genes, using at least 1 of 3 methods (positional, eQTL, and chromatin interactions, see Methods), Supplementary Table S5: 26 by positional, 52 by eQTL, and 57 by chromatin interaction mappings. All 3 annotations were identified for 25 genes, highlighted in green in Supplementary Table S5.
Figure 3.
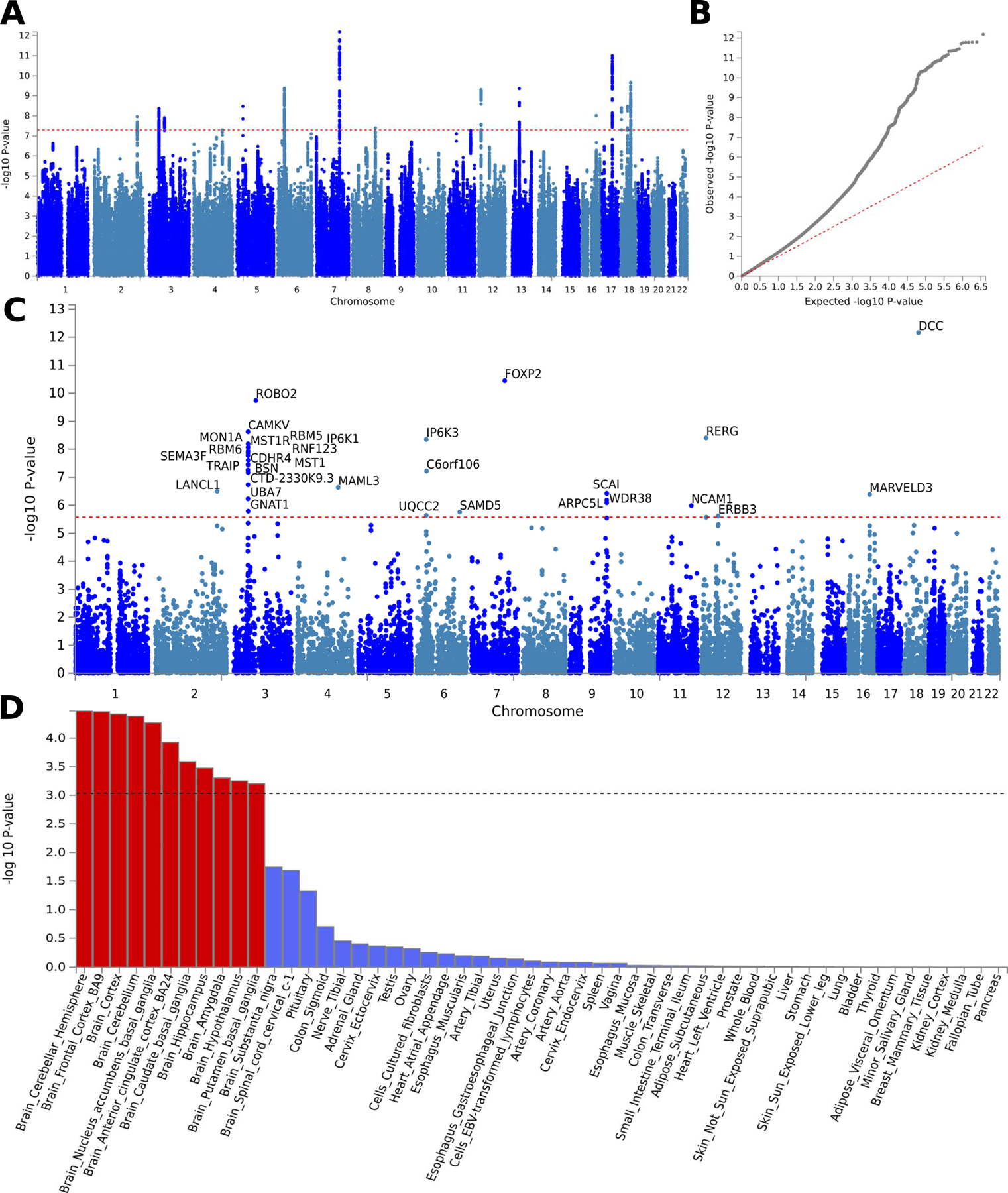
F1 factor GWAS output.
Genome-wide association study (GWAS) results for general pain factor (F1). SNP Manhattan (a) and quantile-quantile, QQ, (b) plots for F1 GWAS. (c) Gene-based genome-wide association Manhattan plot, with the top 31 associated genes labelled. Full gene names taken from the NCBI gene database, https://www.ncbi.nlm.nih.gov/gene/: ARPC5L, Actin-Related Protein 2/3 Complex Subunit 5-Like Protein; BSN, Bassoon Presynaptic Cytomatrix Protein; C6orf106, inflammation and lipid regulator with UBA-like and NBR1-like domains; CAMKV, CaM Kinase Like Vesicle Associated; CDHR4, Cadherin-Related Family Member 4; CTD-2330K9.3, Coats disease; DCC, Colorectal cancer suppressor; ERBB3, Erb-B2 Receptor Tyrosine Kinase 3; FOXP2, Forkhead Box P2; GNAT1, G Protein Subunit Alpha Transducin 1; IP6K1, Inositol hexakisphosphate kinase 1; IP6K3, Inositol hexakisphosphate kinase 3; LANCL1, LanC Like Glutathione S-Transferase 1; MAML3, Mastermind Like Transcriptional Coactivator 3; MARVELD3, MARVEL (Membrane-Associating) Domain Containing 3; MON1A, MON1 homolog A, secretory trafficking associated; MST1, macrophage stimulating 1; MST1R, macrophage stimulating 1 receptor; NCAM1, neural cell adhesion molecule 1; RBM5, RNA binding motif protein 5; RBM6, RNA binding motif protein 6; RERG, RAS like estrogen regulated growth inhibitor; RNF123, ring finger protein 123; ROBO2, roundabout guidance receptor 2; SAMD5, sterile alpha motif domain containing 5; SCAI, suppressor of cancer cell invasion; SEMA3F, semaphorin 3F; TRAIP, TRAF interacting protein; UBA7, ubiquitin like modifier activating enzyme 7; UQCC2, ubiquinol-cytochrome c reductase complex assembly factor 2; WDR38, WD repeat domain 38. (d) Gene property analysis for association between factor GWAS gene effects and gene expression levels in 53 specific tissues from GTEx, version 8.
REVIGO pathway analysis suggested that the pathways represented by these genes cover a broad range of biological processes, including organ development (gut, heart, muscle and brain), metabolism, catabolism, signaling, immunity, neuronal development, transcription, and DNA repair (Supplementary Table S6). FUMA gene set annotation showed a significant enrichment for a pathway involved in learned vocalization behavior or vocal learning (p = 8.93×10−07, Bonferroni-corrected p = .0138). Additionally, this analysis showed a trend towards significance in several other pathways involved in mechanosensory behavior, several neuronal development processes, and several biosynthesis and calcium channel regulation processes (Supplementary Table S7). Although these pathways did not reach corrected significance, we note them, because they are supported by previous findings [58, 60, 83, 117] and may be useful for hypothesis-generation purposes. We note, additionally, that biological pathways have roles in multiple functions, and our results do not imply a direct link between pain and the functions, such as vocal learning, associated with these pathways.
MAGMA-based tissue expression analysis, as implemented in FUMA, tested for association between highly expressed genes in 53 GTEx tissues and GWAS effect sizes for the same genes (Supplementary Note). Associations were significant only in brain tissues: cortical regions (the cerebral cortex, dorsomedial prefrontal cortex BA9, and anterior cingulate cortex BA24), nucleus accumbens, basal ganglia, amygdala, hippocampus, hypothalamus, and cerebellum, Figure 3D.
Additionally we used FUMA to cross-reference SNPs and genes with other GWAS reports. Of note is the overlap in SNPs (Supplementary Table S8), and significant enrichment for genes reported to be associated with chronic pain conditions (back pain, Crohn’s disease, IBS, and multi-site chronic pain), brain structural traits, anthropometric traits, cognition and intelligence-related phenotypes, sleep-related phenotypes, neuroticism, and mood phenotypes (Supplementary Figure S7). Genetic overlap with non-pain conditions is suggestive of the complexity of factors contributing to chronic pain and suggest potential pathways of susceptibility to pain chronification. Furthermore, DCC, the top gene associated with the general factor, is also the top gene reported in a recent study of chronic overlapping pain conditions, which used pain for more than 3 months in different body sites from the UKBB (head, face, neck/shoulder, back, stomach, hip, knee, all over the body) [60]. Of the 241 genes mapped to independent significant SNPs from the general factor GWAS, FKBP5 is the only one previously targeted in a candidate gene study (as opposed to GWAS) for posttraumatic musculoskeletal pain [10, 71, 148].
3.5.2. Musculoskeletal Factor
The musculoskeletal factor GWAS yielded 7 genome-wide significant lead SNPs (Supplementary Table S9 and Supplementary Figure S8). Positional mapping yielded 5 unique genes; eQTL mapping yielded 18 genes; and chromatin interaction mapping yielded 19 genes, with 5 genes mapped using all 3 methods, green: DPYD, MAPK6, GLIS3, COL27A1, and SLC44A2 (Supplementary Table S10).
REVIGO pathway analysis showed associations with genes involved in bone and neuronal development, cell cycle, transcription regulation and signal transduction, Supplementary Table S11. Gene set annotation showed a Bonferroni-corrected significant enrichment for regulation of RNA biosynthetic process and nominally significant (p < 0.05) enrichment for several other regulatory processes, chromatin organization, cell migration involved in heart development, and DNA damage response (Supplementary Table S12). MAGMA tissue expression analysis found no significant association between gene expression and GWAS effect sizes for 53 tissues (Supplementary Figure S8D).
Cross-referencing with other GWAS reports identified previously reported SNP associations with anthropometric traits (height, hip circumference, offspring birth weight), hip or knee osteoarthritis, sleep-related phenotypes, and type 2 diabetes (Supplementary Table S13), and significant overlap with genes reported to be associated with inflammatory skin disease, palmitic and stearic acid levels (Supplementary Figure S9). None of the genes previously targeted in candidate gene studies for pain [146] mapped to independent significant SNPs for the musculoskeletal factor.
4. Discussion
We ran Genomic SEM on 24 pain conditions in the UKBB to examine the structure of their shared genetic risk and characterize the genetic variants common to them. Our results identify a general factor that explains substantial genetic variance in pain conditions with different suspected etiologies and anatomic presentations and points to their shared systemic pathophysiology. Additionally, a second factor explains some of the shared genetic variance across musculoskeletal conditions. The two-factor model explains the pattern of genetic associations among disorders better than either the anatomic or etiologic grouping of known inflammatory disorders. The shared genetic burden is also apparent in our network analyses.
We identified 184 novel targets for cross-condition chronic pain (Supplementary Tables S5). Two lines of evidence suggest that genes associated with our general pain factor have an important role in the central nervous system (CNS). First, the genes most associated with the general factor for pain also have the highest expression in brain tissues. Second, the pathways of these genes include CNS development and maintenance. Our study further adds to existing evidence for the role of DCC [60, 104, 118], an axonal guidance mediator [144], in chronic pain. Beyond CNS, the pathways of genes associated with the general pain factor also implicate a broad range of other functions, such as gut development, locomotion, and protein secretion, suggesting that susceptibility to chronic pain may involve other systemic biological changes. The new molecular targets we identify can be cross-referenced with animal models in ‘reverse-translation’ approaches to better understand the pathophysiology of chronic pain and develop novel treatments.
We note overlap between the genetic variants associated with our general factor and those previously reported in GWAS for cognitive, structural, mood, sleep, and personality traits, regulation of inflammation and neuroplasticity, and psychiatric disorders. This overlap underscores the highly multifaceted nature of pain as a biopsychosocial condition, while elucidating the key genes and systems involved [15, 20, 21, 77, 128, 129]. This pleiotropy, or the association of genes with multiple conditions, together with the polygenic nature of the general factor we identified, exemplifies the frequently observed many-to-many mapping between genes and traits [136]. Identifying links between polygenic risk profiles of different disorders can provide important information on susceptibility and treatment.
As might be expected, the genes associated with the musculoskeletal factor are fewer, and their pathways are less diverse. They implicate skeletal development, choline transport, signalling, and transcription machinery. Notably, they do not implicate the nervous system. Overlap with previous GWAS results suggests involvement of variants affecting anthropometric traits and thereby body-structural mechanisms. Similar associations have been shown for musculoskeletal pain conditions before: genetic overlap in osteoarthritis with height and BMI [28], back pain [35] and multi-site musculoskeletal pain [124] with structural-anatomic genes.
Our work builds on earlier genetic analyses of combinations of pain conditions selected based on anatomic proximity or hypothesized etiology [45,75,99,133,140]. While most studies have been conducted in twins, several large-scale chronic pain GWAS, with strengths complementary to twin studies [36], have been published on pain in the past three years [35, 82, 83, 85, 86, 87, 117]. These reports, which used earlier releases of the UKBB prior to primary care data availability, include three on multi-site pain [57, 58, 60]. Most recently, another study reported a polygenic risk score for pain spreading that was associated with a phenotypic profile dominated by mood, sleep, and neuroticism [118].
Studies of multi-site pain differ from our study in a critical way. They may show genes associated with only one of the constituent conditions to be associated with the average count of pain sites (widespreadness), if that condition is present in several combinations of comorbid pain sites. Thus, given a genetic variant associated with hip pain and two three-site combinations -- hip, knee, back pain and hip, neck, stomach pain -- this variant will appear to be associated with a widespreadness score of 3 without having associations with any of the other constituent conditions in this example. Analyses of association with multi-site pain are thus not designed to identify the genetic variants shared among multiple distinct conditions. On the other hand, Genomic SEM is designed to identify genetic variants that are truly common to different conditions, and it enabled us to capture genetic risk for chronic pain, regardless of etiology or symptomatology.
The existence of cross-condition genetic risk factors challenges the current clinical practice of grouping and treating chronic pain conditions based on location of symptoms on the body or suspected etiology [30]. Evidence for central processes beyond local pathophysiology has been accumulating. Biopsychosocial factors [20, 21, 77, 128, 129], neuroinflammation and neuroplasticity [5, 6, 27, 37, 53, 88, 92, 97, 125, 147], and neuroimaging traits [2, 4, 16, 44, 47, 62, 68, 69, 78, 91, 96, 101, 107, 143], have all been reported to modulate pain experience and chronic pain risk. This work has culminated in a new classification system for chronic pain in ICD-11, which shifts pain category assignment to a hierarchical approach: etiology, then pathophysiology, then body site [122]. It also includes chronic primary pain as a diagnosis “agnostic with regard to etiology” [93]. These changes are important steps toward aligning diagnosis with pathophysiology, and genetics is an important piece of the puzzle. Our Genomic SEM model suggests that, in addition to condition-specific genetic susceptibility, there is a genetically encoded pathophysiology common to different chronic pain conditions and supports the view of chronic pain as a disorder involving systemic pathology [141]. This study further identifies genetic risk markers that are shared across distinct pain conditions contributing to a new, biologically grounded way of conceptualizing chronic pain conditions. The strong association between expression of chronic pain risk genes in the brain and cross-condition chronic pain provides additional evidence that the common genetic risk markers we identified may be associated with central sensitization and chronic primary pain, which have both been linked to alterations in the brain and spinal cord (135,141).
4.1. Limitations
There are several notable limitations of this study. First, although the annotated genetic associations for the general factor suggest a combination of systemic biological and psychological predispositions, the precise mechanisms underlying these predispositions need to be elucidated in future studies before they can be translated to clinical applications. In service of this goal, the pathways implicated by the SNPs and genes we have identified, can be targeted in follow-up studies to further elucidate the systemic mechanisms that lead to chronic pain.
The second limitation lies in the reliance of annotations obtained from FUMA on the information available in existing data repositories, which may be restricted by insufficient resolution or small sample sizes. Thus, although we did not find associations with inflammatory cytokines, evidence for their role in pain is abundant [41, 56, 112] and should be investigated further.
Third, we do not expect the genetic patterns we identified to be either selective or sufficient to explain cross-condition chronic pain. This is due to both pleiotropy (the effects of one gene on several different conditions) and environmental effects, which are certain to play a large part in the development of chronic pain.
Fourth, the genetic scores of our common factors should be validated for association with chronic pain using either association analysis in an independent sample. This validation would require obtaining a polygenic score and is the aim of a follow-up study.
Fifth, given sample size limitations in the UKBB for non-European individuals, we were not able to test our model for generalizability across ancestral populations, which we attempted to do in the next largest sample: South Asians (Supplementary Table 2).
By establishing genetic risk factors in a large sample, this study paves the way for more detailed assessments of pain prognosis and treatment response in targeted studies. For example, the ongoing Acute to Chronic Pain Signatures (A2CPS) study aims to establish risk factors for post-surgical pain from genetic, multi-omics, psychosocial, and neuroimaging measures in another large sample (2,800 patients; a2cps.org). Our factor scores could be tested alongside previously identified genetic patterns for multi-site pain and compared to them as prognostic risk factors for chronic post-surgical pain.
4.2. Conclusion
In summary, our findings confirm that there is a genetic susceptibility common to a broad range of diverse chronic pain conditions. The shared pathophysiology for the conditions examined here appears to lie partly in the CNS and partly scattered across many different systems and functional processes. Additionally, there is a body-wide, suggestively musculoskeletal system-specific genetic factor. Our study calls for new ways to diagnose and treat chronic pain, whereby a given chronic pain condition is not considered as only a symptom of a localized somatic disease by the clinician specializing in it, but is seen as a manifestation of an underlying shared pathology with concurrent risk for other pain conditions and previously unexplored centralized treatment targets. Future work will go beyond pain conditions and explore genetic links with psychological and physical traits to help identify patients who would benefit most from specific interventions.
Supplementary Material
5. Acknowledgments
This work is supported by National Institute of Drug Abuse grants DA046064 and DA017637. Special thanks to Amanda J. Neidermyer, MD, for her review and consultation. The authors declare no conflict of interest.
6. References
- [1].Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN and Chialvo DR [2004], ‘Chronic pain patients are impaired on an emotional decision-making task’, Pain 108(1–2), 129–136. [DOI] [PubMed] [Google Scholar]
- [2].Ayoub LJ, Seminowicz DA and Moayedi M [2018], ‘A meta-analytic study of experimental and chronic orofacial pain excluding headache disorders’, NeuroImage: Clinical 20, 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bair E, Gaynor S, Slade GD, Ohrbach R, Fillingim RB, Greenspan JD, Dubner R, Smith SB, Diatchenko L and Maixner W [2016], ‘Identification of clusters of individuals relevant to temporomandibular disorders and other chronic pain conditions: the OPPERA study’, Pain 157(6), 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baliki MN, Baria AT and Apkarian AV [2011], ‘The cortical rhythms of chronic back pain’, Journal of Neuroscience 31(39), 13981–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baliki MN, Mansour AR, Baria AT and Apkarian AV [2014], ‘Functional reorganization of the default mode network across chronic pain conditions’, PloS One 9(9), e106133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bannister K and Dickenson A [2017], ‘The plasticity of descending controls in pain: translational probing’, The Journal of physiology 595(13), 4159–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Barrat A, Barthelemy M, Pastor-Satorras R and Vespignani A [2004], ‘The architecture of complex weighted networks’, Proceedings of the national academy of sciences 101(11), 3747–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Becker S, Navratilova E, Nees F and Van Damme S [2018], ‘Emotional and motivational pain processing: Current state of knowledge and perspectives in translational research.’, Pain Research & Management . [DOI] [PMC free article] [PubMed]
- [9].Bornovalova MA, Choate AM, Fatimah H, Petersen KJ and Wiernik BM [2020], ‘Appropriate use of bifactor analysis in psychopathology research: Appreciating benefits and limitations’, Biological Psychiatry 88(1), 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bortsov AV, Smith JE, Diatchenko L, Soward AC, Ulirsch JC, Rossi C, Swor RA, Hauda WE, Peak DA, Jones JS et al. [2013], ‘Polymorphisms in the glucocorticoid receptor co-chaperone FKBP5 predict persistent musculoskeletal pain after traumatic stress exposure’, PAIN® 154(8), 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brandes U [2001], ‘A faster algorithm for betweenness centrality’, Journal of mathematical sociology 25(2), 163–177. [Google Scholar]
- [12].Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, Daly MJ, Price AL and Neale BM [2015], ‘LD score regression distinguishes confounding from polygenicity in genome-wide association studies’, Nature genetics 47(3), 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, OConnell J et al. [2018], ‘The UK Biobank resource with deep phenotyping and genomic data’, Nature 562(7726), 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Caspi A, Houts RM, Belsky DW, Goldman-Mellor SJ, Harrington H, Israel S, Meier MH, Ramrakha S, Shalev I, Poulton R et al. [2014], ‘The p factor: one general psychopathology factor in the structure of psychiatric disorders?’, Clinical psychological science 2(2), 119–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cay M, Gonzalez-Heydrich J, Teicher MH, van der Heijden H, Ongur D, Shinn AK and Upadhyay J [2022], ‘Childhood maltreatment and its role in the development of pain and psychopathology’, The Lancet Child & Adolescent Health [DOI] [PMC free article] [PubMed]
- [16].Čeko M, Shir Y, Ouellet JA, Ware MA, Stone LS and Seminowicz DA [2015], ‘Partial recovery of abnormal insula and dorsolateral prefrontal connectivity to cognitive networks in chronic low back pain after treatment’, Human brain mapping 36(6), 2075–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM and Lee JJ [2015], ‘Second-generation plink: rising to the challenge of larger and richer datasets’, Gigascience 4(1), s13742–015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen FF, Hayes A, Carver CS, Laurenceau J-P and Zhang Z [2012], ‘Modeling general and specific variance in multifaceted constructs: A comparison of the bifactor model to other approaches’, Journal of personality 80(1), 219–251. [DOI] [PubMed] [Google Scholar]
- [19].Chen P-Y, Yang C-M and Morin CM [2015], ‘Validating the cross-cultural factor structure and invariance property of the insomnia severity index: evidence based on ordinal EFA and CFA’, Sleep medicine 16(5), 598–603. [DOI] [PubMed] [Google Scholar]
- [20].Clauw DJ [2015], Fibromyalgia and related conditions, in ‘Mayo Clinic Proceedings’, Vol. 90, Elsevier, pp. 680–692. [DOI] [PubMed] [Google Scholar]
- [21].Clauw DJ, Essex MN, Pitman V and Jones KD [2019], ‘Reframing chronic pain as a disease, not a symptom: rationale and implications for pain management’, Postgraduate medicine 131(3), 185–198. [DOI] [PubMed] [Google Scholar]
- [22].Csardi G, Nepusz T et al. [2006], ‘The igraph software package for complex network research’, InterJournal, complex systems 1695(5), 1–9. [Google Scholar]
- [23].de Leeuw CA, Mooij JM, Heskes T and Posthuma D [2015], ‘Magma: generalized gene-set analysis of gwas data’, PLoS computational biology 11(4), e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Din HM and Minhatb HS [2021], ‘Psychometric properties of anxiety about aging scale among Malaysian youths’, Journal of Applied Structural Equation Modeling 5(1), 9–18. [Google Scholar]
- [25].Dutta D, Brummett CM, Moser SE, Fritsche LG, Tsodikov A, Lee S, Clauw DJ and Scott LJ [2020], ‘Heritability of the fibromyalgia phenotype varies by age’, Arthritis & Rheumatology 72(5), 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Eccleston C, Wastell S, Crombez G and Jordan A [2008], ‘Adolescent social development and chronic pain’, European Journal of Pain 12(6), 765–774. [DOI] [PubMed] [Google Scholar]
- [27].Edelmayer RM, Vanderah TW, Majuta L, Zhang E-T, Fioravanti B, De Felice M, Chichorro JG, Ossipov MH, King T, Lai J et al. [2009], ‘Medullary pain facilitating neurons mediate allodynia in headache-related pain’, Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 65(2), 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Elliott KS, Chapman K, Day-Williams A, Panoutsopoulou K, Southam L, Lindgren CM, Arden N, Aslam N, Birrell F, Carluke I et al. [2013], ‘Evaluation of the genetic overlap between osteoarthritis with body mass index and height using genome-wide association scan data’, Annals of the rheumatic diseases 72(6), 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eppstein D, Loffler M and Strash D [2010], Listing all maximal cliques in sparse graphs in near-optimal time, in ‘International Symposium on Algorithms and Computation’, Springer, pp. 403–414. [Google Scholar]
- [30].Fillingim RB, Loeser JD, Baron R and Edwards RR [2016], ‘Assessment of chronic pain: domains, methods, and mechanisms’, The journal of pain 17(9), T10–T20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS et al. [2015], ‘Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis’, The Lancet Neurology 14(2), 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Firth D [1993], ‘Bias reduction of maximum likelihood estimates’, Biometrika 80(1), 27–38. [Google Scholar]
- [33].Fokkema M and Greiff S [2017], ‘How performing pca and cfa on the same data equals trouble’.
- [34].Foote IF, Jacobs BM, Mathlin G, Watson CJ, Bothongo PL, Waters S, Dobson R, Noyce AJ, Bhui KS, Korszun A et al. [2021], ‘The genetic architecture of Alzheimer’s disease risk: A genomic structural equation modelling study’, medRxiv [DOI] [PubMed]
- [35].Freidin MB, Tsepilov YA, Palmer M, Karssen LC, Suri P, Aulchenko YS, Williams FM, Group CMW et al. [2019], ‘Insight into the genetic architecture of back pain and its risk factors from a study of 509,000 individuals’, Pain 160(6), 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Friedman NP, Banich MT and Keller MC [2021], ‘Twin studies to GWAS: there and back again’, Trends in Cognitive Sciences [DOI] [PMC free article] [PubMed]
- [37].Geha P and Waxman SG [2016], ‘Pain perception: multiple matrices or one?’, JAMA neurology 73(6), 628–630. [DOI] [PubMed] [Google Scholar]
- [38].Gerbing DW and Hamilton JG [1996], ‘Viability of exploratory factor analysis as a precursor to confirmatory factor analysis’, Structural Equation Modeling: A Multidisciplinary Journal 3(1), 62–72. [Google Scholar]
- [39].Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, Farh K-H, Cuenca-Leon E, Muona M, Furlotte NA et al. [2016], ‘Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine’, Nature genetics 48(8), 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Grace PM, Hutchinson MR, Maier SF and Watkins LR [2014], ‘Pathological pain and the neuroimmune interface’, Nature Reviews Immunology 14(4), 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gregory NS, Harris AL, Robinson CR, Dougherty PM, Fuchs PN and Sluka KA [2013], ‘An overview of animal models of pain: disease models and outcome measures’, The Journal of Pain 14(11), 1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Grotzinger AD, Mallard TT, Akingbuwa WA, Ip HF, Adams MJ, Lewis CM, McIntosh AM, Grove J, Dalsgaard S, Peter-Lesch K et al. [2020], ‘Genetic architecture of 11 major psychiatric disorders at biobehavioral, functional genomic, and molecular genetic levels of analysis’, medRxiv [DOI] [PMC free article] [PubMed]
- [43].Grotzinger AD, Rhemtulla M, de Vlaming R, Ritchie SJ, Mallard TT, Hill WD, Ip HF, Marioni RE, McIntosh AM, Deary IJ et al. [2019], ‘Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits’, Nature human behaviour 3(5), 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Han X, Ashar YK, Kragel P, Petre B, Schelkun V, Atlas LY, Chang LJ, Jepma M, Koban L, Losin EAR et al. [2022], ‘Effect sizes and test-retest reliability of the fMRI-based neurologic pain signature’, NeuroImage 247, 118844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hartvigsen J, Nielsen J, Kyvik KO, Fejer R, Vach W, Iachine I and Leboeuf-Yde C [2009], ‘Heritability of spinal pain and consequences of spinal pain: A comprehensive genetic epidemiologic analysis using a population-based sample of 15,328 twins ages 20–71 years’, Arthritis Care & Research 61(10), 1343–1351. [DOI] [PubMed] [Google Scholar]
- [46].Hengstebeck E, Roskos S, Breejen K, Arnetz B and Arnetz J [2017], ‘Chronic pain disrupts ability to work by interfering with social function: a cross-sectional study’, Scandinavian journal of pain 17(1), 397–402. [DOI] [PubMed] [Google Scholar]
- [47].Hocking L, Scotland G, Morris A, Dominiczak A, Porteous D and Smith B [2012], ‘Heritability of chronic pain in 2195 extended families’, European journal of pain 16(7), 1053–1063. [DOI] [PubMed] [Google Scholar]
- [48].Howard MA, Krause K, Khawaja N, Massat N, Zelaya F, Schumann G, Huggins JP, Vennart W, Williams SC and Renton TF [2011], ‘Beyond patient reported pain: perfusion magnetic resonance imaging demonstrates reproducible cerebral representation of ongoing postsurgical pain’, PloS one 6(2), e17096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hruschak V and Cochran G [2018], ‘Psychosocial predictors in the transition from acute to chronic pain: a systematic review’, Psychology, health & medicine 23(10), 1151–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hu L. t. and Bentler PM [1999], ‘Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives’, Structural equation modeling: a multidisciplinary journal 6(1), 1–55. [Google Scholar]
- [51].Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL, Danecek P, Malerba G, Trabetti E, Zheng H-F et al. [2015], ‘Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel’, Nature communications 6(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Huedo-Medina TB, Sanchez-Meca J, Marin-Martinez F and Botella J [2006], ‘Assessing heterogeneity in meta-analysis: Q statistic or I2 index?’, Psychological methods 11(2), 193. [DOI] [PubMed] [Google Scholar]
- [53].Jefferson T, Kelly CJ and Martina M [2021], ‘Differential rearrangement of excitatory inputs to the medial prefrontal cortex in chronic pain models’, Frontiers in Neural Circuits p. 159. [DOI] [PMC free article] [PubMed]
- [54].Jensen AR [2000], ‘The G factor: psychometrics and biology’, The nature of intelligence p. 37. [DOI] [PubMed]
- [55].Ji R-R, Nackley A, Huh Y, Terrando N and Maixner W [2018], ‘Neuroinflammation and central sensitization in chronic and widespread pain’, Anesthesiology 129(2), 343–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ji R-R, Xu Z-Z and Gao Y-J [2014], ‘Emerging targets in neuroinflammation-driven chronic pain’, Nature reviews Drug discovery 13(7), 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Johnston KJ, Adams MJ, Nicholl BI, Ward J, Strawbridge RJ, Ferguson A, McIntosh AM, Bailey ME and Smith DJ [2019], ‘Genome-wide association study of multisite chronic pain in UK Biobank’, PLoS genetics 15(6), e1008164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Johnston KJ, Ward J, Ray PR, Adams MJ, McIntosh AM, Smith BH, Strawbridge RJ, Price TJ, Smith DJ, Nicholl BI et al. [2021], ‘Sex-stratified genome-wide association study of multisite chronic pain in UK Biobank’, PLoS genetics 17(4), e1009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kambur O, Kaunisto MA, Winsvold BS, Wilsgaard T, Stubhaug A, Zwart JA, Kalso E and Nielsen CS [2018], ‘Genetic variation in P2RX7 and pain tolerance’, Pain 159(6), 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Khoury S, Parisien M, Thompson SJ, Vachon-Presseau E, Roy M, Martinsen AE, Winsvold BS, Pain HA-I, Mundal IP, Zwart J-A et al. [2021], ‘Genome-wide analysis identifies impaired axonogenesis in chronic overlapping pain conditions’, Brain [DOI] [PubMed]
- [61].Kline RB [2015], Principles and practice of structural equation modeling, Guilford publications. [Google Scholar]
- [62].Kong J, Spaeth B, Wey H-Y, Cheetham A, Cook AH, Jensen K, Tan Y, Liu H, Wang D, Loggia ML et al. [2013], ‘S1 is associated with chronic low back pain: a functional and structural MRI study’, Molecular pain 9, 1744–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kucyi A and Davis KD [2015], ‘The dynamic pain connectome’, Trends in neurosciences 38(2), 86–95. [DOI] [PubMed] [Google Scholar]
- [64].Kuner R and Flor H [2017], ‘Structural plasticity and reorganisation in chronic pain’, Nature Reviews Neuroscience 18(1), 20–30. [DOI] [PubMed] [Google Scholar]
- [65].Kyriazos TA et al. [2018], ‘Applied psychometrics: sample size and sample power considerations in factor analysis (EFA, CFA) and SEM in general’, Psychology 9(08), 2207. [Google Scholar]
- [66].Lacagnina MJ, Heijnen CJ, Watkins LR and Grace PM [2021], ‘Autoimmune regulation of chronic pain’, Pain reports 6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lau C-I, Lin C-C, Chen W-H, Wang H-C and Kao C-H [2014], ‘Association between migraine and irritable bowel syndrome: A population-based retrospective cohort study’, European Journal of Neurology 21(9), 1198–1204. [DOI] [PubMed] [Google Scholar]
- [68].Lee J-J, Kim HJ, Cěko M, Park B. y., Lee SA, Park H, Roy M, Kim S-G, Wager TD and Woo C-W [2021], ‘A neuroimaging biomarker for sustained experimental and clinical pain’, Nature medicine 27(1), 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee J, Protsenko E, Lazaridou A, Franceschelli O, Ellingsen D-M, Mawla I, Isenburg K, Berry MP, Galenkamp L, Loggia ML et al. [2018], ‘Encoding of self-referential pain catastrophizing in the posterior cingulate cortex in fibromyalgia’, Arthritis & rheumatology 70(8), 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lee SH, Wray NR, Goddard ME and Visscher PM [2011], ‘Estimating missing heritability for disease from genome-wide association studies’, The American Journal of Human Genetics 88(3), 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lobo JJ, Ayoub LJ, Moayedi M and Linnstaedt SD [2022], ‘Hippocampal volume, FKBP5 genetic risk alleles, and childhood trauma interact to increase vulnerability to chronic multisite musculoskeletal pain’, Scientific reports 12(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Loggia ML, Berna C, Kim J, Cahalan CM, Martel M-O, Gollub RL, Wasan AD, Napadow V and Edwards RR [2015], ‘The lateral prefrontal cortex mediates the hyperalgesic effects of negative cognitions in chronic pain patients’, The Journal of Pain 16(8), 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N et al. [2013], ‘The genotype-tissue expression (GTEx) project’, Nature genetics 45(6), 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Maixner W, Fillingim RB, Williams DA, Smith SB and Slade GD [2016], ‘Overlapping chronic pain conditions: implications for diagnosis and classification’, The Journal of Pain 17(9), T93– T107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Malkin I, Williams FM, LaChance G, Spector T, MacGregor AJ and Livshits G [2014], ‘Low back and common widespread pain share common genetic determinants’, Annals of human genetics 78(5), 357–366. [DOI] [PubMed] [Google Scholar]
- [76].Mallard TT, Linnér RK, Grotzinger AD, Sanchez-Roige S, Seidlitz J, Okbay A, de Vlaming R, Meddens SFW, Palmer AA, Davis LK et al. [2020], ‘Multivariate gwas of psychiatric disorders and their cardinal symptoms reveal two dimensions of cross-cutting genetic liabilities’, BioRxiv p. 603134. [DOI] [PMC free article] [PubMed]
- [77].Mansour A, Farmer M, Baliki M and Apkarian AV [2014], ‘Chronic pain: the role of learning and brain plasticity’, Restorative neurology and neuroscience 32(1), 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Martins D, Dipasquale O, Veronese M, Turkheimer F, Loggia ML, McMahon S, Howard MA and Williams SC [2021], ‘Transcriptional and cellular signatures of cortical morphometric remodelling in chronic pain’, Pain [DOI] [PMC free article] [PubMed]
- [79].Mayer S, Spickschen J, Stein KV, Crevenna R, Dorner TE and Simon J [2019], ‘The societal costs of chronic pain and its determinants: the case of austria’, PloS one 14(3), e0213889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mbatchou J, Barnard L, Backman J, Marcketta A, Kosmicki JA, Ziyatdinov A, Benner C, ODushlaine C, Barber M, Boutkov B et al. [2021], ‘Computationally efficient whole-genome regression for quantitative and binary traits’, Nature genetics 53(7), 1097–1103. [DOI] [PubMed] [Google Scholar]
- [81].Meints SM, Mawla I, Napadow V, Kong J, Gerber J, Chan S-T, Wasan AD, Kaptchuk TJ, McDonnell C, Carriere J et al. [2019], ‘The relationship between catastrophizing and altered pain sensitivity in patients with chronic low back pain’, Pain 160(4), 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Meng W, Adams MJ, Hebert HL, Deary IJ, McIntosh AM and Smith BH [2018], ‘A genome-wide association study finds genetic associations with broadly-defined headache in UK Biobank (n= 223,773)’, EBioMedicine 28, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Meng W, Adams MJ, Palmer CN, Shi J, Auton A, Ryan KA, Jordan JM, Mitchell BD, Jackson RD, Yau MS et al. [2019], ‘Genome-wide association study of knee pain identifies associations with GDF5 and COL27A1 in UK Biobank’, Communications biology 2(1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Meng W, Adams MJ, Reel P, Rajendrakumar A, Huang Y, Deary IJ, Palmer CN, McIntosh AM and Smith BH [2020], ‘Genetic correlations between pain phenotypes and depression and neuroticism’, European Journal of Human Genetics 28(3), 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Meng W, Chan BW, Harris C, Freidin MB, Hebert HL, Adams MJ, Campbell A, Hayward C, Zheng H, Zhang X et al. [2020], ‘A genome-wide association study finds genetic variants associated with neck or shoulder pain in UK Biobank’, Human molecular genetics 29(8), 1396–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Meng W, Deshmukh H, Van Zuydam N, Liu Y, Donnelly L, Zhou K, (WTCCC2), W. T. C. C. C. ., for Micro-and Macro-Vascular Hard Endpoints for Innovative Diabetes Tools (SUMMIT) Study Group, S. M., Morris A, Colhoun H et al. [2015], ‘A genome-wide association study suggests an association of Chr8p21. 3 (GFRA2) with diabetic neuropathic pain’, European Journal of Pain 19(3), 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Meng W, Reel P, Nangia C, Rajendrakumar A, Hebert H, Adams M, Zheng H, Lu Z, Ray D, Colvin L et al. [2021], ‘A meta-analysis of the genome-wide association studies on two genetically correlated phenotypes (self-reported headache and self-reported migraine) identifies four new risk loci for headaches (n= 397,385)’, medRxiv [DOI] [PMC free article] [PubMed]
- [88].Miller Neilan R, Majetic G, Gil-Silva M, Adke AP, Carrasquillo Y and Kolber BJ [2021], ‘Agent-based modeling of the central amygdala and pain using cell-type specific physiological parameters’, PLoS Computational Biology 17(6), e1009097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Miyake A, Friedman NP, Emerson MJ, Witzki AH, Howerter A and Wager TD [2000], ‘The unity and diversity of executive functions and their contributions to complex frontal lobe tasks: A latent variable analysis’, Cognitive psychology 41(1), 49–100. [DOI] [PubMed] [Google Scholar]
- [90].Morlion B, Coluzzi F, Aldington D, Kocot-Kepska M, Pergolizzi J, Mangas AC, Ahlbeck K and Kalso E [2018], ‘Pain chronification: what should a non-pain medicine specialist know?’, Current Medical Research and Opinion 34(7), 1169–1178. [DOI] [PubMed] [Google Scholar]
- [91].Napadow V and Harris RE [2014], ‘What has functional connectivity and chemical neuroimaging in fibromyalgia taught us about the mechanisms and management of centralized pain?’, Arthritis research & therapy 16(4), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Navratilova E and Porreca F [2014], ‘Reward and motivation in pain and pain relief’, Nature neuroscience 17(10), 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nicholas M, Vlaeyen JW, Rief W, Barke A, Aziz Q, Benoliel R, Cohen M, Evers S, Gi- amberardino MA, Goebel A et al. [2019], ‘The IASP classification of chronic pain for ICD-11: chronic primary pain’, Pain 160(1), 28–37. [DOI] [PubMed] [Google Scholar]
- [94].Niv D and Devor M [2004], ‘Chronic pain as a disease in its own right’. [DOI] [PubMed]
- [95].O’Connor LJ, Schoech AP, Hormozdiari F, Gazal S, Patterson N and Price AL [2019], ‘Extreme polygenicity of complex traits is explained by negative selection’, The American Journal of Human Genetics 105(3), 456–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Osborne NR, Anastakis DJ and Davis KD [2018], ‘Peripheral nerve injuries, pain, and neuro- plasticity’, Journal of Hand Therapy 31(2), 184–194. [DOI] [PubMed] [Google Scholar]
- [97].Ossipov MH, Morimura K and Porreca F [2014], ‘Descending pain modulation and chronification of pain’, Current opinion in supportive and palliative care 8(2), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Phillips CJ [2006], ‘Economic burden of chronic pain’, Expert review of pharmacoeconomics & outcomes research 6(5), 591–601. [DOI] [PubMed] [Google Scholar]
- [99].Plesh O, Noonan C, Buchwald DS, Goldberg J and Afari N [2012], ‘Temporomandibular disorder-type pain and migraine headache in women: a preliminary twin study’, Journal of orofacial pain 26(2), 91. [PubMed] [Google Scholar]
- [100].Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA and Reich D [2006], ‘Principal components analysis corrects for stratification in genome-wide association studies’, Nature genetics 38(8), 904–909. [DOI] [PubMed] [Google Scholar]
- [101].Quesada C, Kostenko A, Ho I, Leone C, Nochi Z, Stouffs A, Wittayer M, Caspani O, Finnerup NB, Mouraux A et al. [2021], ‘Human surrogate models of central sensitization: A critical review and practical guide’, European journal of pain (London, England) 25(7), 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Raffaeli W and Arnaudo E [2017], ‘Pain as a disease: an overview’, Journal of pain research 10, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, Narita A, Konuma T, Yamamoto K, Akiyama M et al. [2021], ‘A cross-population atlas of genetic associations for 220 human phenotypes’, Nature genetics 53(10), 1415–1424. [DOI] [PubMed] [Google Scholar]
- [104].Schubert A-L, Held M, Sommer C and Üçeyler N [2019], ‘Reduced gene expression of netrin family members in skin and sural nerve specimens of patients with painful peripheral neuropathies’, Journal of neurology 266(11), 2812–2820. [DOI] [PubMed] [Google Scholar]
- [105].Schug SA and Bruce J [2017], ‘Risk stratification for the development of chronic postsurgical pain’, Pain Reports 2(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Schur EA, Afari N, Furberg H, Olarte M, Goldberg J, Sullivan PF and Buchwald D [2007], ‘Feeling bad in more ways than one: comorbidity patterns of medically unexplained and psychiatric conditions’, Journal of general internal medicine 22(6), 818–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Seminowicz DA, Labus JS, Bueller JA, Tillisch K, Naliboff BD, Bushnell MC and Mayer EA [2010], ‘Regional gray matter density changes in brains of patients with irritable bowel syndrome’, Gastroenterology 139(1), 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Sibille KT, Steingrímsdottir ÓA, Fillingim RB, Stubhaug A, Schirmer H, Chen H, McEwen BS and Nielsen CS [2016], ‘Investigating the burden of chronic pain: an inflammatory and metabolic composite’, Pain Research and Management 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Simon LS [2012], ‘Relieving pain in america: A blueprint for transforming prevention, care, education, and research’, Journal of pain & palliative care pharmacotherapy 26(2), 197–198. [Google Scholar]
- [110].Smith BH and Torrance N [2011], ‘Management of chronic pain in primary care’, Current Opinion in Supportive and Palliative Care 5(2), 137–142. [DOI] [PubMed] [Google Scholar]
- [111].Smith SM, Dworkin RH, Turk DC, Baron R, Polydefkis M, Tracey I, Borsook D, Edwards RR, Harris RE, Wager TD et al. [2017], ‘The potential role of sensory testing, skin biopsy, and functional brain imaging as biomarkers in chronic pain clinical trials: Immpact considerations’, The Journal of Pain 18(7), 757–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Sommer C, Leinders M and Üçeyler N [2018], ‘Inflammation in the pathophysiology of neuropathic pain’, Pain 159(3), 595–602. [DOI] [PubMed] [Google Scholar]
- [113].Stanos S, Brodsky M, Argoff C, Clauw DJ, DArcy Y, Donevan S, Gebke KB, Jensen MP, Lewis&Clark E, McCarberg B et al. [2016], ‘Rethinking chronic pain in a primary care setting’, Postgraduate Medicine 128(5), 502–515. [DOI] [PubMed] [Google Scholar]
- [114].Steglitz J, Buscemi J and Ferguson MJ [2012], ‘The future of pain research, education, and treatment: a summary of the IOM report relieving pain in america: a blueprint for transforming prevention, care, education, and research’, Translational behavioral medicine 2(1), 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Supek F, Bošnjak M, Škunca N and Šmuc T [2011], ‘Revigo summarizes and visualizes long lists of gene ontology terms’, PloS one 6(7), e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Suri P, Morgenroth DC, Kwoh CK, Bean JF, Kalichman L and Hunter DJ [2010], ‘Low back pain and other musculoskeletal pain comorbidities in individuals with symptomatic osteoarthritis of the knee: data from the osteoarthritis initiative’, Arthritis care & research 62(12), 1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Suri P, Palmer MR, Tsepilov YA, Freidin MB, Boer CG, Yau MS, Evans DS, Gelemanovic A, Bartz TM, Nethander M et al. [2018], ‘Genome-wide meta-analysis of 158,000 individuals of european ancestry identifies three loci associated with chronic back pain’, PLoS genetics 14(9), e1007601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Tanguay-Sabourin C, Fillingim M, Parisien M, Guglietti GV, Zare A, Norman J, Da-ano R, Perez J, Thompson SJ, Martel MO et al. [2022], ‘A data-driven biopsychosocial framework determining the spreading of chronic pain’, medRxiv
- [119].Tian C, Gregersen PK and Seldin MF [2008], ‘Accounting for ancestry: population substructure and genome-wide association studies’, Human molecular genetics 17(R2), R143–R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Tracey I and Bushnell MC [2009], ‘How neuroimaging studies have challenged us to rethink: is chronic pain a disease?’, The journal of pain 10(11), 1113–1120. [DOI] [PubMed] [Google Scholar]
- [121].Treede R-D, Kenshalo DR, Gracely RH and Jones AK [1999], ‘The cortical representation of pain’, Pain 79(2–3), 105–111. [DOI] [PubMed] [Google Scholar]
- [122].Treede R-D, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB et al. [2015], ‘A classification of chronic pain for ICD-11’, Pain 156(6), 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Treede R-D, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB et al. [2019], ‘Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the international classification of diseases (ICD-11)’, Pain 160(1), 19–27. [DOI] [PubMed] [Google Scholar]
- [124].Tsepilov YA, Freidin MB, Shadrina AS, Sharapov SZ, Elgaeva EE, van Zundert J, Karssen L, Suri P, Williams FM and Aulchenko YS [2020], ‘Analysis of genetically independent phenotypes identifies shared genetic factors associated with chronic musculoskeletal pain conditions’, Communications biology 3(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Tylee DS, Sun J, Hess JL, Tahir MA, Sharma E, Malik R, Worrall BB, Levine AJ, Martinson JJ, Nejentsev S et al. [2018], ‘Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data’, American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 177(7), 641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Urien L and Wang J [2019], ‘Top-down cortical control of acute and chronic pain’, Psychosomatic medicine 81(9), 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Vachon-Presseau E, Berger SE, Abdullah TB, Griffith JW, Schnitzer TJ and Apkarian AV [2019], ‘Identification of traits and functional connectivity-based neurotraits of chronic pain’, PLoS biology 17(8), e3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Vachon-Presseau E, Centeno M, Ren W, Berger S, Tétreault P, Ghantous M, Baria A, Farmer M, Baliki M, Schnitzer T et al. [2016], ‘The emotional brain as a predictor and amplifier of chronic pain’, Journal of dental research 95(6), 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Vachon-Presseau E, Roy M, Martel M-O, Caron E, Marin M-F, Chen J, Albouy G, Plante I, Sullivan MJ, Lupien SJ et al. [2013], ‘The stress model of chronic pain: evidence from basal cortisol and hippocampal structure and function in humans’, Brain 136(3), 815–827. [DOI] [PubMed] [Google Scholar]
- [130].Vachon-Presseau E, Tétreault P, Petre B, Huang L, Berger SE, Torbey S, Baria AT, Mansour AR, Hashmi JA, Griffith JW et al. [2016], ‘Corticolimbic anatomical characteristics predetermine risk for chronic pain’, Brain 139(7), 1958–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].van Bork R, Epskamp S, Rhemtulla M, Borsboom D and van der Maas HL [2017], ‘What is the p-factor of psychopathology? some risks of general factor modeling’, Theory & Psychology 27(6), 759–773. [Google Scholar]
- [132].van der Gaag WH, Roelofs PD, Enthoven WT, van Tulder MW and Koes BW [2020], ‘Non- steroidal anti-inflammatory drugs for acute low back pain’, Cochrane Database of Systematic Reviews (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Vehof J, Zavos HM, Lachance G, Hammond CJ and Williams FM [2014], ‘Shared genetic factors underlie chronic pain syndromes’, PAIN® 155(8), 1562–1568. [DOI] [PubMed] [Google Scholar]
- [134].Walitt B, Ceko M, L Gracely J and H Gracely R [2016], ‘Neuroimaging of central sensitivity syndromes: key insights from the scientific literature’, Current rheumatology reviews 12(1), 55–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wang Z, Yuan M, Xiao J, Chen L, Guo X, Dou Y, Jiang F, Min W and Zhou B [2022], ‘Gray matter abnormalities in patients with chronic primary pain: A coordinate-based meta-analysis’, Pain physician 25(1), 1. [PubMed] [Google Scholar]
- [136].Watanabe K, Stringer S, Frei O, Umićević Mirkov M, de Leeuw C, Polderman TJ, van der Sluis S, Andreassen OA, Neale BM and Posthuma D [2019], ‘A global overview of pleiotropy and genetic architecture in complex traits’, Nature genetics 51(9), 1339–1348. [DOI] [PubMed] [Google Scholar]
- [137].Watanabe K, Taskesen E, Van Bochoven A and Posthuma D [2017], ‘Functional mapping and annotation of genetic associations with FUMA’, Nature communications 8(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].WHO [2019], ‘Musculoskeletal conditions’, https://www.who.int/news-room/fact-sheets/detail/musculoskeletal-conditions. Accessed: 2021–12-16.
- [139].Wiberg A, Ng M, Schmid AB, Smillie RW, Baskozos G, Holmes MV, Künnapuu K, Mägi R, Bennett DL and Furniss D [2019], ‘A genome-wide association analysis identifies 16 novel susceptibility loci for carpal tunnel syndrome’, Nature communications 10(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Williams FM, Spector TD and MacGregor AJ [2010], ‘Pain reporting at different body sites is explained by a single underlying genetic factor’, Rheumatology 49(9), 1753–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Woolf CJ [2011], ‘Central sensitization: implications for the diagnosis and treatment of pain’, pain 152(3), S2–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Wray NR, Wijmenga C, Sullivan PF, Yang J and Visscher PM [2018], ‘Common disease is more complex than implied by the core gene omnigenic model’, Cell 173(7), 1573–1580. [DOI] [PubMed] [Google Scholar]
- [143].Wrigley PJ, Press SR, Gustin SM, Macefield VG, Gandevia SC, Cousins MJ, Middleton JW, Henderson LA and Siddall P [2009], ‘Neuropathic pain and primary somatosensory cortex reorganization following spinal cord injury’, PAIN® 141(1–2), 52–59. [DOI] [PubMed] [Google Scholar]
- [144].Wu C-H, Yuan X-C, Gao F, Li H-P, Cao J, Liu Y-S, Yu W, Tian B, Meng X-F, Shi J et al. [2016], ‘Netrin-1 contributes to myelinated afferent fiber sprouting and neuropathic pain’, Molecular neurobiology 53(8), 5640–5651. [DOI] [PubMed] [Google Scholar]
- [145].Xiao Z, Martinez E, Kulkarni PM, Zhang Q, Hou Q, Rosenberg D, Talay R, Shalot L, Zhou H, Wang J et al. [2019], ‘Cortical pain processing in the rat anterior cingulate cortex and primary somatosensory cortex’, Frontiers in cellular neuroscience 13, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Zaitlen N and Kraft P [2012], ‘Heritability in the genome-wide association era’, Human genetics 131(10), 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Zhou H, Zhang Q, Martinez E, Dale J, Hu S, Zhang E, Liu K, Huang D, Yang G, Chen Z et al. [2018], ‘Ketamine reduces aversion in rodent pain models by suppressing hyperactivity of the anterior cingulate cortex’, Nature communications 9(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Zorina-Lichtenwalter K, Meloto CB, Khoury S and Diatchenko L [2016], ‘Genetic predictors of human chronic pain conditions’, Neuroscience 338, 36–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.