

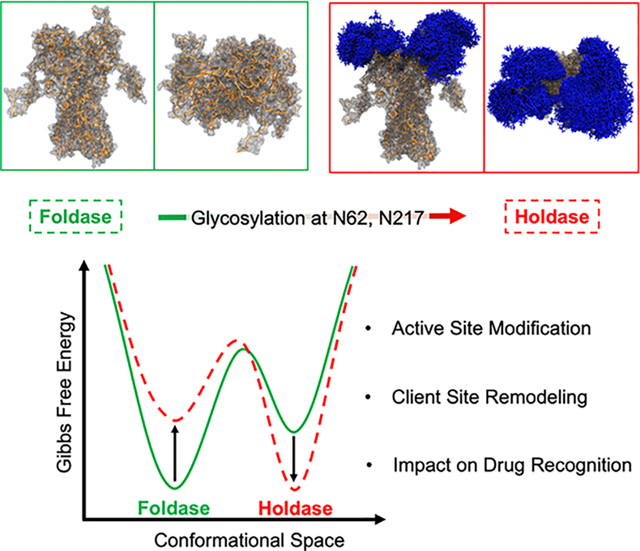
Summary
Protein assembly defects due to an enrichment of aberrant conformational protein variants are emerging as a new frontier in therapeutics design. Understanding the structural elements that rewire the conformational dynamics of proteins and pathologically perturb functionally-oriented ensembles, is important for inhibitor development. Chaperones are hub proteins for the assembly of multiprotein complexes and an enrichment of aberrant conformers can affect the cellular proteome, and in turn, on phenotypes. Here, we integrate computational and experimental tools to investigate how N-glycosylation of specific residues in glucose-regulated protein 94 (GRP94) modulates internal dynamics and alters the conformational fitness of regions fundamental for the interaction with ATP and synthetic ligands, and impacts substructures important for the recognition of interacting proteins. N-glycosylation plays an active role in modulating the energy landscape of GRP94, and we provide support for leveraging the knowledge on distinct glycosylation variants to design molecules targeting GRP94 disease-associated conformational states and assemblies.
eTOC BLURB
Castelli et al. describe at atomic-resolution how the glycosylation of the chaperone GRP94, an important protein for cell life control, impacts on its functional spectrum remodeling interaction networks and affects disease phenotypes. The results are actionable in the selection of drugs that specifically target disease-related forms of GRP94.
Graphical Abstract
Introduction
Protein function, and in turn malfunction, is determined by the extent conformational ensembles populate distinct states, each endowed with specific activities, and by the mechanisms of dynamic transition from a conformational state to another1–7. Changes in state distributions are governed by both non-covalent events, such as allosteric ligand binding, and covalent events such as post-translational modifications (PTMs)8,9. The importance of PTMs cannot be overstated, and recent investigations have started to shed light on their significance in regulating both physiological and disease conditions10–12. In the context of a dynamic model of conformational regulation, PTMs work by shifting populations of protein ensembles and thus modulate the abundance of specific forms. This may have a dramatic impact on cellular networks, as different conformers may engage in distinct interactions, remodeling protein-protein interactions (PPI) and the assembly of the multimolecular assemblies required to regulate biomolecular pathways.
Among PTMs, glycosylation plays a host of important roles13–15. This fact is vividly illustrated by recent studies on the SARS-CoV-2 Spike protein, whereby glycosylation modulates mechanisms at the basis of viral entry16,17. Glycosylation has also emerged as a mechanism in cancer and inflammation where cellular localization as well as protein assemblies could be pathologically remodeled by this PTM10,15,18. For example, we recently reported on a glycosylation-dependent disease-specific variant of the chaperone GRP9415. GRP94 is a protein of the Hsp90 family, which consists of several paralogs, namely HSP90 in the cytosol, GRP94 in the endoplasmic reticulum (ER), and TRAP1 in the mitochondria. Proteins of this family act as chaperones helping the correct folding and activation of a wide array of proteins named “clients”. GRP9419, like other Hsp90 proteins, is a homodimer with each individual chain consisting of three globular domains, the N-terminal (NTD), Middle and C-terminal (CTD) (Figure 1). Mechanistically, conformational changes in the chaperone are linked to the folding of the client, which is recruited to the chaperone in a partially unfolded form, in a sequence governed by ATP: ATP binding at the NTD20 shifts the chaperone to a partially closed, and then into an asymmetric closed conformation that is significantly strained at the Middle:CTD interface, the region involved in protein client binding21. Upon ATP hydrolysis, rearrangement in the client binding-site residues takes place and this is subsequently mechanically translated into conformational changes of the bound client.
Figure 1. Structural and domain organization of GRP94.
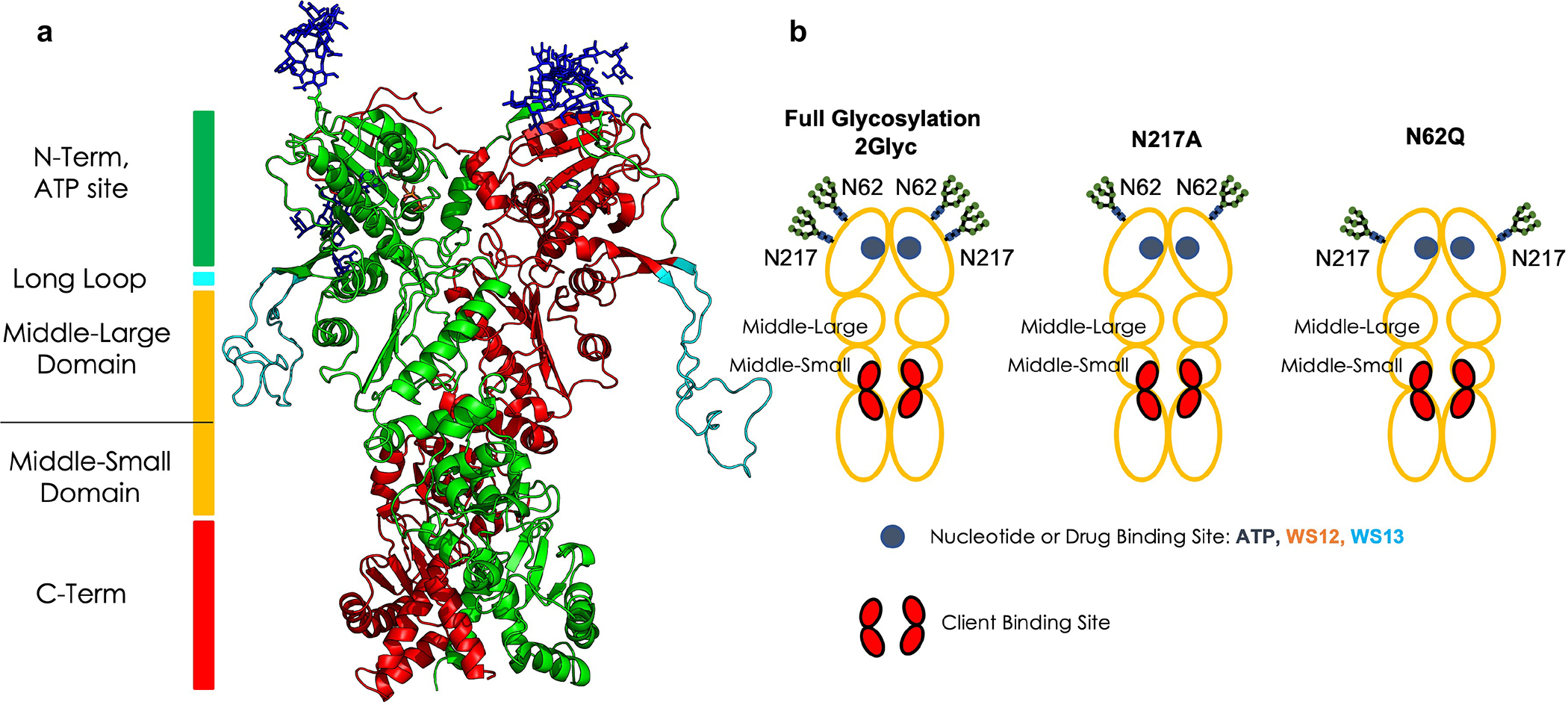
(a) The 3D structure of 2Glyc Grp94. The two protomers are colored in green and red respectively, while the glycosyl PTMs are shown in sticks.
(b) Schematic of the domains, binding sites, glycosylation sites, and glycosylation states, as they are referred to throughout the paper. The red areas indicate the client binding site regions.
In normal physiological conditions, GRP94 is glycosylated at N217, is confined to the ER and folds client proteins through transient interactions 22,23. Aberrant glycosylation on N62 promotes a conformational state in GRP94 that allows for plasma membrane (PM) localization and stable interactions with PM proteins15. Through this complexation stabilization, the functions of PM proteins are enhanced, and downstream cellular protein pathways are aberrantly remodeled. N-glycosylation thus transforms GRP94 from a folding to a scaffolding protein that in turn remodels protein assembly and connectivity, with an end result of proteome-wide dysfunction24,25.
Aggregating these findings, aberrantly glycosylated protein variants and their altered assemblies have emerged as promising pharmacological targets in infectious diseases, inflammation and cancer15,24–28. Despite its clear disease relevance, an atomistic view on how glycosylation may influence proteins is largely missing. Specifically, the molecular mechanisms underlying the modulation of conformational dynamics and of molecular recognition properties with regards to substrate and drug-like inhibitors, as well as their impact on functions remain poorly understood. Without this knowledge, the development of inhibitors and clinical candidates targeting specific disease-associated, glycosylation-dependent, conformational states and assemblies is hampered.
Here, we set out to unveil at atomistic resolution the mechanism through which N-glycans at different sites modulate the conformational dynamics of GRP94, affecting both its interaction mode with clients and nucleotide processing. The degree of glycosylation and the specific positioning of the glycosyls are reconnected to their impact on function. On this basis, we rationalize the effects of distinct ligands on different forms of GRP94. In this context, we document the reach and implications of our approach through chemical biology tests on the activity of different designed molecules. We use long-timescale Molecular Dynamics (MD) simulations (~ 40 micro-seconds in total) to expose the links between protein dynamics and the establishment of specific interaction patterns in different protein forms, and leverage this knowledge to build a simple model of PTM-induced functional perturbation mechanisms. Finally, we use these results to rationalize productive and nonproductive engagement of conformational states by active vs. inactive inhibitors, thus providing key insights into the design of ligands targeting the disease-specific, aberrantly glycosylated GRP94 variant.
Our work reveals an essential structural role of N-glycans in modulating the internal dynamics of the protein linked to the activation of functional motions: the results show that PTMs favor distinct states of the protein, which will ultimately be presented to different pools of interactors for the assembly of complexes determining cell phenotypes. The insights we provide here lay the foundation for the design of ligands that act on GRP94 forms in a context-dependent way, which could be exploited to develop drugs and chemical tools to untangle the intricate roles of post-translationally modified chaperones in disease. While focused on GRP94, our approaches and models are entirely transferable to other systems, setting the stage for a generalized understanding of the interplay between protein covalent modification, the modulation of dynamics, and their impact on the motions associated to functions and interactions in cells.
Results
Ligand preference for GRP94 and disease variants: experimental investigations
GRP94 is a protein widely explored for the discovery of small molecule binders, and several GRP94 ligands were reported over the past few years. These include ligands based on the purine-scaffold (eg. N-ethylcarboximinoadenosine, cytosine-containing adenosine analogs, PU-WS13 and others) 29–31, resorcinol type compounds (eg. BnIm and KUNG94) and benzamide scaffold derivatives 32–34. These compounds, while tight binders of recombinant GRP94 protein, elicit non-overlapping biological profiles when tested in a variety of disease models. For example, some but not all are toxic to cancer cells characterized and driven by Receptor Tyrosine Kinase (RTK)-overexpression (eg. HER2- and EGFR-overexpressing breast cancer cells)32,35,36. In these cells, the N62 glycosylated GRP94 variant (referred to further as Glyc62GRP94) is enriched at the plasma membrane, where it acts to reduce RTK internalization and to maintain the RTK in a state that is competent for constitutively enhanced downstream signaling15. Thus RTK-overexpressing cancer cells require Glyc62GRP94 to promote survival, and its inhibition should be toxic in such context, consistent with previous findings for GRP94 knockdown15,29,37.
The lack of biological activity observed for some GRP94 ligands but not others in RTK-overexpressing breast cancer cells has thus been puzzling. It was proposed, but never formally tested, that distinct ligands might favor and/or engage different context-specific GRP94 conformers and assemblies, each endowed with distinct biological function35. To test this hypothesis, and thus provide molecular insights for atomistic studies through MD simulations, we selected a sub-library of closely-related GRP94 ligands of the purine-scaffold series (i.e. PU-series) (Figure 2a). We tested each ligand in a battery of assays designed to distinguish active ligands from those inactive on Glyc62GRP94-related biological functions (see Figure 2a–e and SI Figure S1a–c). We contrasted these ligands and their activity to BnIm, a ligand reported to productively engage GRP94 in cells, and modulate GRP94- but not Glyc62GRP94-related functions15,32,35,36.
Figure 2. Biochemical and functional evaluation of GRP94 ligands for biological activity via Glyc62GRP94.
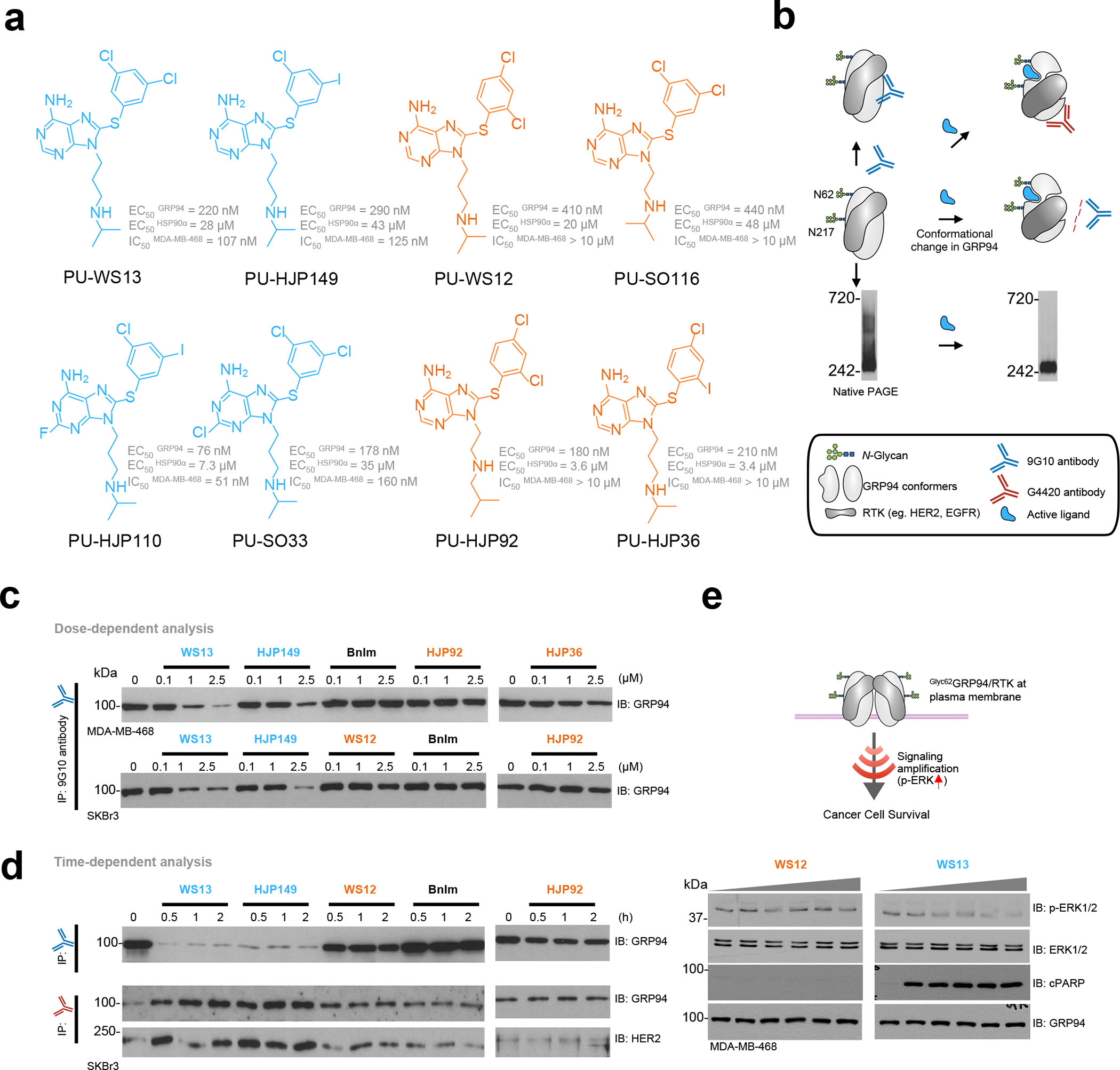
(a) Chemical structure of GRP94 small molecule ligands. EC50GRP94 and EC50HSP90, relative binding affinity constant to recombinant GRP94 and HSP90α, respectively. IC50MDA-MB-468, half maximal inhibitory concentration determined in the MDA-MB-468 EGFR-overexpressing and Glyc62GRP94-expressing breast cancer cells (ATP quantitation assay). Values, mean of three independent experiments.
(b) Schematic showing the conformational changes induced in GRP94 expected from small molecule ligands that act on Glyc62GRP94.
(c,d) Immuno-capture as in (b) of SKBr3 HER2-overexpressing or MDA-MB-468 EGFR-overexpressing (both Glyc62GRP94-expressing) breast cancer cells treated with the indicated ligands in a dose- (for 4 h) and time-dependent (at 2.5 μM) manner. Experiments were repeated trice with similar results.
(e) Western blot analysis for inhibition of Glyc62GRP94 function in MDA-MB-468 cancer cells. Downstream signaling (p-ERK/ERK) and induction of apoptosis (cleaved PARP, cPARP) were analyzed for MDA-MB-468 cancer cells treated for 24 h with 0, 0.5, 1, 2.5, 5 and 10 μM of the indicated inhibitors. GRP94, loading control. Gels are representative of three independent results. See also Figure S1.
We found most ligands exhibit a comparable binding affinity for recombinant GRP94 under conditions of equilibrium binding (Figure 2a, see EC50 rGRP94, 120–450 nM for the PU-series and 1.2 μM for BnIm, as reported 15,32,35,36). Remarkably, even in the structurally closely related PU-type ligands, some but not all, were active in cancer cells dependent on Glyc62GRP94 for survival, even at the highest tested concentration of 10 μM (Figure 2a).
To provide mechanistic insights, we implemented a testing pipeline that probes biochemically and functionally productive engagement of Glyc62GRP94. It is based on the observations that preferential and productive binding to Glyc62GRP94 is dependent on a GRP94 conformation being stabilized by N62-glycosylation15. This conformation is sensitive to detection by GRP94 antibodies, such as 9G10, which recognizes the charged linker region (residues 290–350), and G4420, which recognizes amino acids 733–750 in the CTD of GRP94 (Figure 2b). We treated RTK-overexpressing breast cancer cells with the ligands prior to immunocapture with the two GRP94 antibodies (Figure 2c,d). Active ligands, but none of those inactive, dramatically reduced the amount of GRP94 captured by the 9G10 antibody, indicating that active inhibitors change GRP94 to a conformation that is no longer recognized by the antibody. Active ligands also increased the amount of both GRP94 captured by the G4420 antibody and HER2 pulled down by the immunocapture of ligand-bound GRP94, indicating that active ligands promote or stabilize a conformation that favors HER2 association. Cell permeability or efflux pump mechanisms were not accountable for the activity difference (see activity in cell homogenates vs intact cells, SI Figure S1a).
Native PAGE analyses confirmed the immunoprecipitation results (SI Figure S1b). Under non-denaturing gel conditions, cells expressing Glyc62GRP94 exhibit a number of distinct and indistinct high-molecular-weight GRP94 species above the main 242-kDa band (Figure 2b), a biochemical signature of Glyc62GRP94 incorporation into epichaperomes, high molecular weight pathologic scaffolding structures formed of chaperones, co-chaperones and other factors15,38. The signal observed in native PAGE reflects both GRP94 complexation and retained native conformation of the protein, which is recognized by the 9G10 antibody. We observed that compounds active in cells via Glyc62GRP94, and not those GRP94-active but Glyc62GRP94-inactive, induced a change in the epichaperome species detected on Native PAGE (SI Figure S1b–d).
Glyc62GRP94 is required to reduce RTK internalization and maintain the RTK in a state that is competent for constitutively enhanced downstream signaling15. Compounds active in cells via Glyc62GRP94, and not those GRP94-active but Glyc62GRP94-inactive, inhibited RTK downstream signaling as measured by western blot analysis (see p-ERK, Figure 2e).
Overall, active ligands remodeled glycosylated GRP94’s conformation upon binding, as measured by antibody binding, HER2 capture, and native gel mobility, and inhibited Glyc62GRP94 function in cells as measured by cell death induction and RTK function inhibition in Glyc62GRP94-expressing RTK+ cancer cells (eg. SKBr3 and MDA-MB-468). Unlike active ligands, inactive ligands produced no or partial decrease in the conformation detected by 9G10 and G4420, no change in glyc62GRP94-incorporating epichaperomes, and exhibited no cellular activity via Glyc62GRP94. These findings are important in the context of our MD studies because they clearly show not every GRP94 ligand can optimally engage the Glyc62GRP94 variant, but more importantly they indicate that we can study, and elucidate, the structure of Glyc62GRP94 by investigating its dynamics and conformation in relationship to active and inactive ligands.
MD simulations: design and implications for GRP94 functional investigations
The energy landscape model can help rationalize the mechanisms of how PTMs work. In this theoretical framework, biomolecules are viewed as dynamical entities that are continuously interconverting between a variety of structures: distinct structures may be endowed with different functions depending on the conditions. In this model, a PTM’s effect is both local and distal, as it modifies a residue and its immediate interaction networks, and importantly, it may propagate its effect throughout the structure, even at distant regions, eventually determining a protein population shift2,3,8,9,39. As a consequence, a PTM-modified protein may be devoid of enzymatic reactivity or show different mechanisms and affinities for ligand binding, while removal of the PTMs may shift the protein back to the catalytically competent structural ensemble, restoring enzymatic activity, ligand recognition mechanisms, and PPI interaction profiles40–43.
To understand how PTMs and inhibitor binding modulate the dynamics of GRP94, we set out to analyze and compare fully solvated atomistic simulations of different models of the protein. Specifically, we analyzed the protein with glycosylation at both N62 and N217 (simulations with double glycosylation, labeled 2Glyc), bound to the endogenous activator ATP (simulation labeled 2GlycATP) or to two different inhibitors, PU-WS13 (a prototype active inhibitor, simulation labeled 2GlycPU-WS13) and PU-WS12 (ligand that binds but is functionally inactive on the tumor Glyc62GRP94 variant, i.e. prototype of inactive inhibitor, labeled 2GlycPU-WS12) (Figure 1a,b).
To understand the roles of the sugars in modulating functional dynamics, we generated two experimentally inspired mutants, namely N62Q (no glycosylation at position 62 while retaining the glycosylation at 217; simulations labeled N62Q) and N217A (no glycosylation at position 217 while retaining glycosylation at 62; simulations labeled N217A, mimicking experimentally studied Glyc62GRP94) (Figure 1a,b). Each mutant was simulated in the presence of ATP, PU-WS12, or PU-WS13 in the active site: the labeling for these states follows the logic described above.
Overall, we considered and cross-compared 9 systems. Each was simulated for 3 microseconds in total (3 independent replicates of 1 microsecond length each), for a total of 27 microseconds. Throughout the paper, we use the fully glycosylated ATP state (2GlycATP) as the reference state. Additionally, as controls, we simulated three completely unglycosylated systems (3 microseconds each), labeled Wild Type (WT): these models are intended to report on the dynamics of GRP94 in the absence of PTMs. In the three control simulations, GRP94 is in complex with ATP (labeled NoGlycATP), PU-WS13 (labeled NoGlycPU-WS13), PU-WS12 (labeled NoGlycPU-WS12). All simulations, their conditions, and labels are reported in Table 1.
Table 1.
Simulated systems, ligand states and labels for each of the simulation described in the text.
| GRP94 Properties | Ligand State | Simulation Length | Simulation Label |
|---|---|---|---|
| GRP94, Full Glycosylation at N62 and N217 | ATP | 3 microseconds | 2GlycATP |
| GRP94, Full Glycosylation at N62 and N217 | PU-WS12 | 3 microseconds | 2GlycPU-WS12 |
| GRP94, Full Glycosylation at N62 and N217 | PU-WS13 | 3 microseconds | 2GlycPU-WS13 |
| GRP94 N62Q mutant, Glycosylation at N217 | ATP | 3 microseconds | N62Q-ATP |
| GRP94 N62Q mutant, Glycosylation at N217 | PU-WS12 | 3 microseconds | N62Q-PU-WS12 |
| GRP94 N62Q mutant, Glycosylation at N217 | PU-WS13 | 3 microseconds | N62Q-PU-WS13 |
| GRP94 N217A mutant, Glycosylation at N62 | ATP | 3 microseconds | N217-ATP |
| GRP94 N217A mutant, Glycosylation at N62 | PU-WS12 | 3 microseconds | N217A-PU-WS12 |
| GRP94 N217A mutant, Glycosylation at N62 | PU-WS13 | 3 microseconds | N217A-PU-WS13 |
| GRP94 No Glycosylation | ATP | 3 microseconds | NoGlycATP |
| GRP94 No Glycosylation | PU-WS12 | 3 microseconds | NoGlycPU-WS12 |
| GRP94 No Glycosylation | PU-WS13 | 3 microseconds | NoGlycPU-WS13 |
Glycosylation modulates the global and local dynamics of GRP94
To quantify the effect of glycosyl moieties on GRP94 dynamic states, we first visually analyzed the ensembles of equally interspersed conformations of each glycan sampled along each 3 μs metatrajectory17. This representation returns a view of the mechanisms by which sugars modulate access to GRP94 interactors (Figure 3a–c).
Figure 3. Glycan dynamics on the surfaces of GRP94.
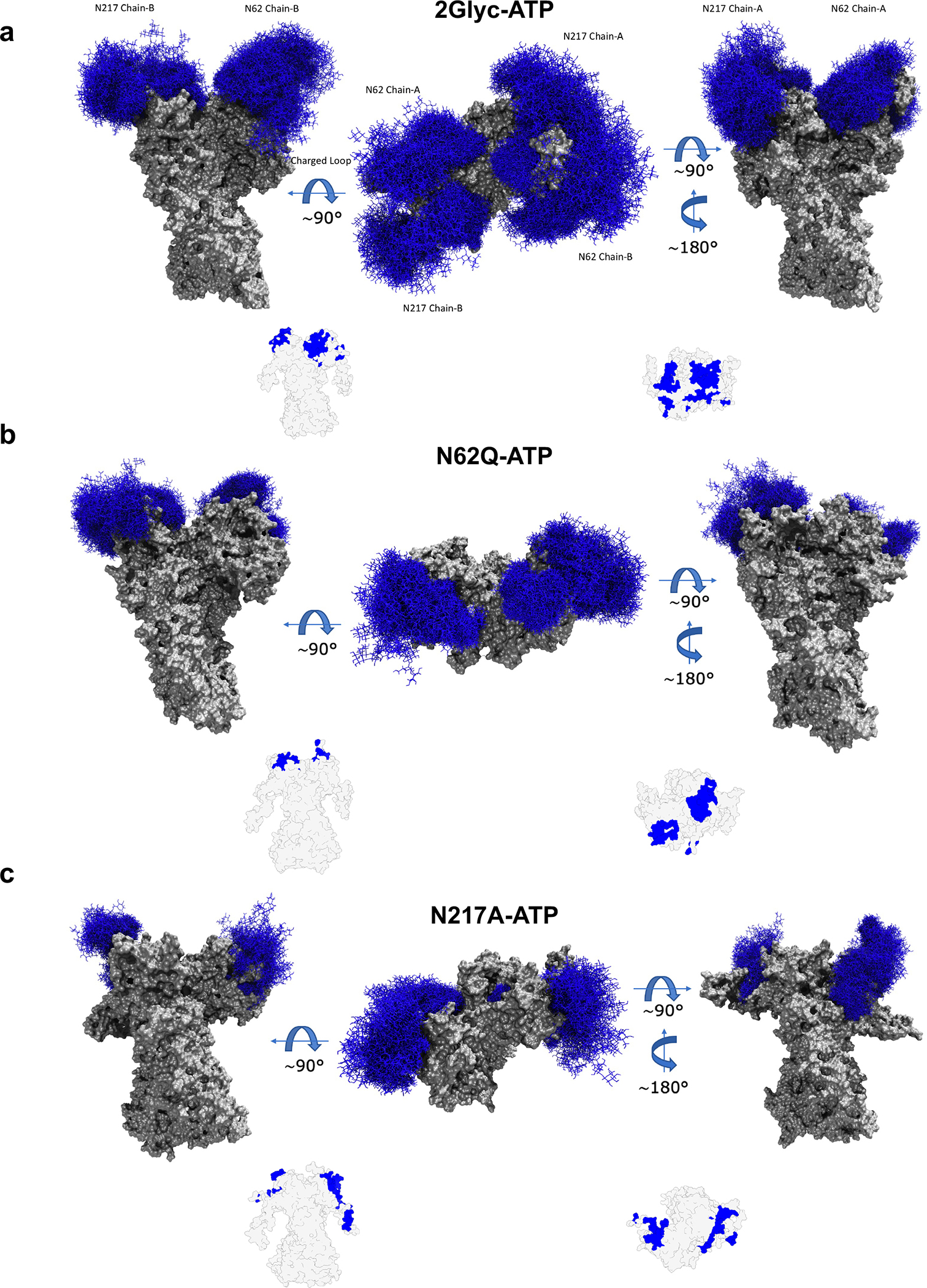
(a) Molecular representation of the 2GlycATP system. Glycans at several frames (namely, 300 frames, one every 30 ns from one replica) are represented with blue lines, while the protein is shown as a gray surface.
(b) Same as a), for the N62Q mutant in complex with ATP; c) Same as a), for the N217A mutant in complex with ATP.
Insets: Analysis of accessible area for more than 70% of the simulation time; gray shading indicates the areas that are accessible for more than 70% of the simulation time; in blue the ones that are shielded by carbohydrates.
In the ATP state of the fully glycosylated protein (2GlycATP), the sugars completely cover the NTD surface, with N-glycans at N62 extending out to form interactions with the charged loop that connects the NTD (where ATP binds) to the M-domain hosting the client-binding site (Figure 3a) 41–44. Interestingly, removal of this glycan in the N62Q mutant removes the shielding from a large part of the NTD and releases the long N-M connecting loop (Figure 3b) (vide infra). In contrast, removal of N-glycans from N217 (N217A mutant, mimic of Glyc62GRP94) with conservation of glycosylation on N62 further exposes the tendency for the latter to interact with the long loop (Figure 3c). These data are further integrated by calculating the surface area of the protein that is shielded by the glycans in different variants (SI Figure S2): in the presence of both glycans, as expected, a large portion of the surface is inaccessible to solvent. Mutations N271A and N62Q have slightly different effects, with the former (corresponding to pathologic Glyc62GRP94) causing a larger shielding than the latter. Significant shielding by the sugars can disfavor binding of ATP to the NTD by rendering the nucleotide binding site less accessible, thus decreasing the efficiency of GRP94 in the folding cycle.
We next set out to characterize the residues that are shielded by carbohydrates for more than 70 percent of the simulation time to find different roles for the two carbohydrate chains: importantly, the chain at N62 is seen to make extensive stable contacts with the charged loop (See insets in Figure 3). Supplementary Information reports the list of residues in the NTD and M-domain that remain accessible. These data are complemented by the calculation of the frequency with which a contact between the sugar chain and the protein is formed (SI Figure S3–S5; full data provided electronically Data_Glyc_Contacts.zip, see Key Resource Table).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-GRP94 | Enzo Life Sciences | Cat# ADI-SPA-850, RRID:AB_11179746 |
| Rabbit polyclonal anti-GRP94 | Sigma-Aldrich | Cat# G4420, RRID:AB_477017 |
| Rabbit polyclonal anti-HER2 | Invitrogen | Cat# 28-0004, RRID: AB_86903 |
| Rabbit monoclonal anti-EGFR | Cell Signaling Technology | Cat# 4267, RRID:AB_2246311 |
| Goat anti-rat IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2006, RRID:AB_1125219 |
| Goat anti-Rabbit IgG-HRP | SouthernBiotech | Cat# 4010-05, RRID:AB_2632593 |
| Rabbit polyclonal anti-cleaved parp | Cell Signaling Technology | Cat# 5625, RRID: AB_10699459 |
| Rabbit polyclonal anti-phospho-ERK (T202/Y204) | Cell Signaling Technology | Cat# 4377, RRID:AB_331775 |
| Rabbit polyclonal anti-ERK | Cell Signaling Technology | Cat# 4695, RRID:AB_390779 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PU-WS13 | Chiosis Lab | N/A |
| PU-SO-33 | Chiosis Lab | N/A |
| PU-HJP-149 | Chiosis Lab | N/A |
| PU-HJP-110 | Chiosis Lab | N/A |
| BnIm | Clevenger and Blagg et al. 2004; Duerfeldt et al. 2012. | N/A |
| PU-WS12 | Chiosis Lab | N/A |
| PU-SO-116 | Chiosis Lab | N/A |
| PU-HJP-92 | Chiosis Lab | N/A |
| PU-HJP-36 | Chiosis Lab | N/A |
| GRP94 (canine) recombinant protein | Enzo Life Sciences | Cat# ADI-SPP-766 |
| HSP90α (human) recombinant protein | Enzo Life Sciences | Cat# ADI-SPP-776 |
| Critical Commercial Assays | ||
| ProteoExtract® Subcellular Proteome Extraction Kit | Millipore Sigma | Cat# 539790 |
| Bio-Safe coomassie stain | Biorad | Cat# 1610786 |
| CellTiter-Glo luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Deposited Data | ||
| Data on Glycosyl Contacts | This paper | https://doi.org/10.5281/zenodo.7912736 |
| Data on Ligand/Loop Contacts | This paper | https://doi.org/10.5281/zenodo.7912736 |
| Docking results data | This paper | https://doi.org/10.5281/zenodo.7912736 |
| Experimental Models: Cell Lines | ||
| Human: MDA-MB-468 | ATCC | Cat# HTB-132, RRID:CVCL_0419 |
| Human: SKBr3 | ATCC | Cat# HTB-30, RRID:CVCL_0033 |
| Software and Algorithms | ||
| Prism V6 | GraphPad software | https://www.graphpad.com |
| SOFTmax Pro 4.3.1 | Molecular Devices | https://www.moleculardevices.com |
| MAESTRO - GLIDE | Schrodinger Inc | https://www.schrodinger.com |
| GLYCAM | Woods Lab | https://www.glycam.org |
| AMBER 18 | Amber | https://ambermd.org |
| Xmgrace | Wis Plasma Lab | https://plasma-gate.weizmann.ac.il/Grace/ |
| INPUT FILES for SIMULATIONS | This paper | https://doi.org/10.5281/zenodo.7912686 |
| Code for Analysis of Simulations | This paper | https://github.com/colombolab |
| Other | ||
| Protein A Agarose | Roche | Cat# 11134515001 |
| Protein G Agarose | Roche | Cat# 11243233001 |
| Pierce™ ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32109 |
| McCoy’s 5A with 1% Penicillin/Streptomycin | MSKCC Media Core | N/A |
| DMEM with 1% Penicillin/Streptomycin | MSKCC Media Core | N/A |
| FBS | VWR | Cat# 97068-085 |
The N-M charged loop plays an important role in regulating the conformational cycle of the chaperone upon ATP hydrolysis, connecting nucleotide processing in the NTD with structural reorganizations of the M-domain. In our model the N-glycan chain at N62, the defining characteristic of Glyc62GRP94, modulates the motions of the loop forming stable interactions. The motions around the hinge between the N and M-domains, or the population of the structural ensemble endowed with client-folding activities, can thus be perturbed with blockage of the foldase activity of the protein. Our data also imply that the role of N-glycans at N217 is less relevant for functional regulation.
To further characterize the impact of carbohydrate PTMs, we ran Principal Component Analysis (PCA) on the various metatrajectories. To shed light on the impact of glycosylation on the charged linker, we started by aligning all the structures visited during the trajectories to the reference frame (the first structure), except for the sequence of the linker, to allow observing the maximum possible diversity in its conformations. Then the covariance matrices of the linker in the various states were calculated. Our results indicate that indeed the presence of the sugars cause the loop to explore different parts of the conformational space (SI Figure S6). Consistent with contact analysis (vide infra), in the ATP bound states the protein with a single glycosylation at N62 in most cases spans an area of the landscape that is not shared with N62Q. PU-WS13 significantly limits the conformational exploration compared to ATP, particularly in the fully and N62-glycosylated states. We then ran PCA on the whole protein, starting from simulation 2GlycATP. We projected the whole trajectory on the plane defined by the resulting first two principal eigenvectors. The 2Glyc-ATP system is seen to span a large portion of the plane (SI Figure S7). In contrast, projection on the same plane of the trajectories of the monoglycosylated versions of GRP94 shows different global dynamics for these systems. Specifically, the presence of glycosylation on N62 (in the N217A mutant) appears to substantially limit the span of the trajectory (SI Figure S7).
Overall, these results support a mechanistic model in which glycosyl modifications play an active role in modifying the conformational landscape of GRP94, switching its functions from a foldase protein to a protein-assembly platform: specifically, glycosyls shield the ATP-binding site in the NTD lowering the efficiency in binding and processing ATP, and perturb the dynamics of the charged linker connecting the NTD and the M-Domain, relaying the impact of PTM modifications in the NTD to the domain in which client remodeling takes place.
Glycans differentially modulate GRP94 internal dynamics, response to ligands, and client binding.
In the energy landscape framework, the effects of glycosylation can diffuse throughout the structure and be sensed by groups of distal residues in functionally relevant regions of the protein 45–47. To investigate these aspects, we next calculated the coordination patterns between distal residues, using Distance Fluctuation (DF) analysis. This analysis, applied to every residue-pair in the protein, evaluates the dynamic coordination between any two residues as a function of the mean square fluctuation of their distance. Small DF values define (groups of) mechanically connected aminoacids that move in a cooperative manner giving the highest contribution in modulating functional motions. The DF profile may change in the presence/absence of a PTM and/or ligand allowing to link internal dynamics with biological effects45,46. Differences between the DF profiles of the 2GlycATP reference state and differentially glycosylated as well as inhibitor-bound states can shed light on the regions that are dynamically responsive to the modifications. Changes on short time-scales in the structural dynamics of GRP94, captured by DFs, can cooperate in modulating the motions of regions that determine biological functions: this is achieved through the local disruption and reassembly of interactions, which eventually result in specific structural deformations and modifications on longer time scales2,7,48–51. Our working hypothesis is that distinct collective GRP94 fluctuation patterns that emerge in a particular ligand/glycosylation state may underpin key functional perturbations.
The representative pairwise mean-square distance fluctuations DF matrix for the reference state is reported in a color-coded form in Figure 4a. For the sake of brevity and simplicity, the fully detailed matrices are reported in SI Figures S8–S9. In Figure 4, we report a simplified view of the GRP94 domains whose internal dynamics is mostly perturbed compared to the native state according to the DF metric. This representation reflects the difference DF matrices reported in SI Figures S8–S9.
Figure 4. Residue-Pair Distance Fluctuations and Domain Cross-Talk for GRP94 in Different Glycosylation and Ligand Conditions.
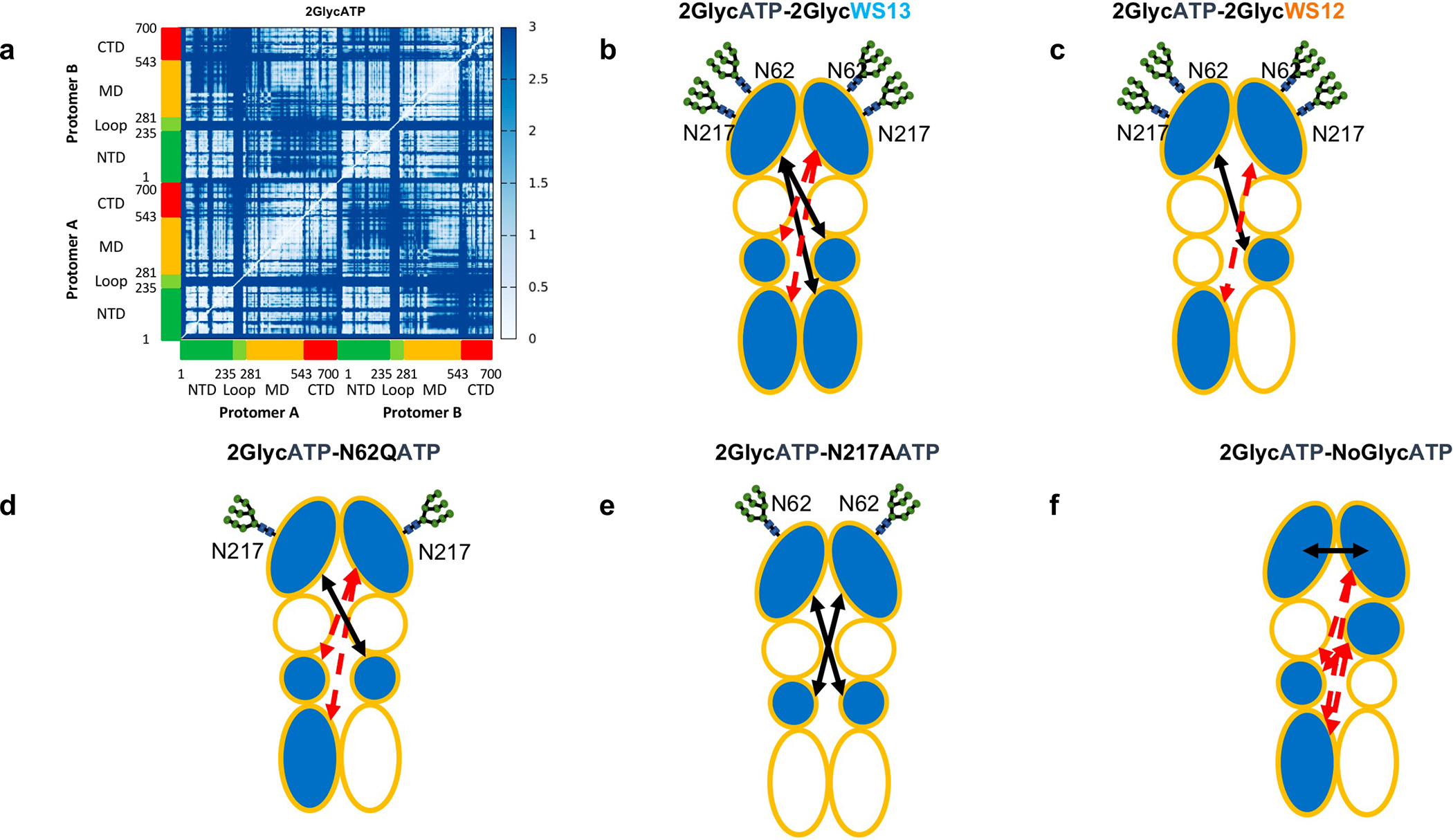
The internal dynamics of GRP94 characterized from the various trajectories.
(a) The original Distance Fluctuation matrix is reported for the fully-glycosylated ATP-bound state. The axes report the residue numbering and domain organisation, as depicted also in Figure 2. In this view, lighter pixels correspond to highly coordinated residue pairs, while darker ones report on low coordination pairs. The single original matrices for all 9 conditions, together with the difference matrices obtained by subtracting the DF matrix of a certain state from the reference one pertaining to 2GlycATP are reported in Supplementary Information.
(b-f) Domain based representations of the modulation of internal flexibility as a function of ligand-state and/or glycosylation state. This representation is a simplified graphical translation of the difference matrices reported in the Supplementary Information (Figures S8–S9). A certain domain is colored in light-blue if its coordination with other domains changes as a function of the condition. The variation in internal dynamics is evaluated as a difference for the DF of the system under exam from that of the fully glycosylated ATP-bound state, 2GlycATP. A black arrow indicates increased coordination (rigidity) between two domains with respect to the fully glycosylated ATP-bound state, 2GlycATP. A red, broken arrow indicates decreased coordination (flexibility) between two domains with respect to the fully glycosylated ATP-bound state, 2GlycATP. The various ligand states are reported in each subfigure.
From a general point of view, all DF matrices exhibit a common block character, which reflects the alternation of regions with small and large pair fluctuations consistent with the tripartite domain organization of GRP94 protomers. Differences emerge in analyzing the finer details of the matrices which point to glycosylation-dependent modulations of the internal dynamics in different complexes. Relevant differences are also seen in the comparison between the active and inactive ligands. From the detailed analysis, we find that the fully glycosylated ATP (2GlycATP) state is characterized by extensive regions of coordination between the NTD, M-domain, and CTD within each protomer, between the NTD of one protomer and the NTD and M-domain of the other, and between M-Domains (Figure 4a, SI Figure S8). In the PU-WS13-fully glycosylated complex (2GlycPU-WS13), an increase in the fluctuations of pairwise distances can be noticed (large red areas in the difference matrix, SI Figure S8). This entails in particular the relative fluctuations between the NTD of protomer 2 (chain B) and the Middle-Small/CTD substructure (which hosts the client binding site) of protomer 1 (chain A) compared to the ATP one (Figure 4b). The reported effects on the internal dynamics of the fully glycosylated version of the protein can be correlated with the observed efficacy of PU-WS13 in targeting this form in tumor models expressing it: the ligand in fact significantly perturbs the internal dynamics of the functional regions of protein, in particular the one dedicated to engage clients. The population of different dynamic states induced by the inhibitor can thus remodel the conformational fitness of GRP94 to interact with the set of proteins (such as RTKs) characterized in tumor cells where the connectivity of the fully glycosylated chaperone is a determinant of pathogenic dysfunction15. Importantly, these effects are significantly less pronounced with PU-WS12 (Simulations 2GlycPU-WS12, Figure 4c)(SI Figure S8).
Removal of the N-glycan in N62Q is seen to cause a decrease in the degree of coordination between the NTD of protomer 2 and the M-small/CTD region of protomer 1, showing DF patterns for N62Q-ATP state that are similar to the ones observed for the inhibitor-bound fully glycosylated complex (Figure 4d)(SI Figure S9a). In general, the presence of synthetic ligands (in particular of the active PU-WS13) is characterized by a more marked effect on the internal dynamics and coordination patterns. Removal of the glycan on N217 (N217A mutant) in the presence of ATP, appears to have a much less pronounced effect than N62Q (SI Figure S9a). Indeed, the effect of maintaining N62, in terms of internal coordination, keeps the protein in a state qualitatively similar to the reference state (Figure 4e, SI Figure S9a).
Importantly, these data are qualitatively consistent with experiments showing that N62Q mimicks the effects of full GRP94 Knock Out or PU-WS13 inhibition. Similar patterns are observed as the two synthetic ligands are bound to N62Q or N217A (Figure 5a–f)(SI Figure S9b,c): the small molecules in both mutants modify mainly the internal coordination of the NTDs. Interestingly, PU-WS13 perturbs the intraprotomer dynamics (i.e. the coordination between the NTD with the M-Domain and the CTD) more than PU-WS12.
Figure 5. Residue-pair Distance Fluctuations and Domain Cross-Talk for GRP94 in Different Glycosylation and Ligand Conditions.
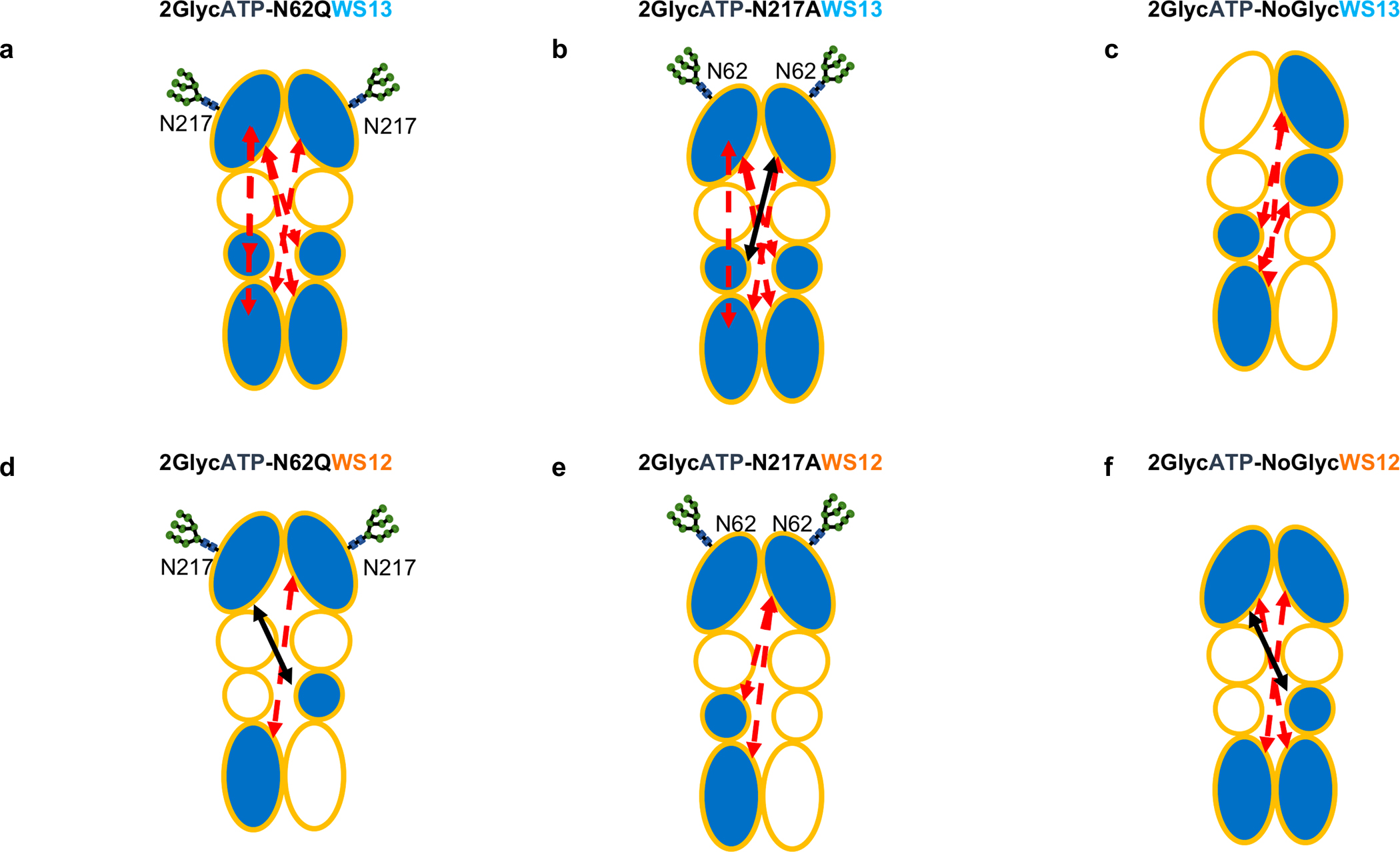
Same as Figure 4(b–f), for systems with different degrees of glycosylation.
Interestingly, the fully unglycosylated WT system in the ATP-state (NoGlycATP) shows the highest degree of coordination between the two NTDs, required to form the catalytically competent state 46,52. Significant flexibility is observed at the level of the Middle/CTD substructures (Figure 4f)(SI Figure S9a). These distinct dynamic traits of the NoGlycATP state can be reconnected to the foldase role of this GRP94 form. First, the closed-catalytic state is required for the proper organization of the ATPase enzymatic machinery; moreover, in a sequential model of ATP hydrolysis, the high coordination between the two NTDs favors sensing of the first ATP hydrolysis by the domain containing the second ATP molecule; finally, the flexibility of the client binding region supports the conformational changes that are required to adapt to and carry out the remodeling of client proteins52.
In this framework, the interactions that the glycosyls (at N62 in particular) establish with the protein, specifically at the level of the N-M charged loop, aptly perturb the structures of the client binding site required for client-folding53–56. Representative structures in Figure 6a–d, show how PTMs differentially remodel the client binding pocket: a closure of the space between the two protomers at the level of the client-binding site can indeed be noticed. In contrast, in the NoGlycATP state, the intraprotomer space in the lumen is substantially larger. In the glycosylated form, the reduction of the intraprotomer space hinders recognition, binding, and the threading of the client through the lumen. In the NoGlycATP state, the lower steric hindrance of the binding pocket, combined to its increased flexibility, can support adaptation to the client. Interestingly, the N217A mutant (aberrant Glyc62GRP94), while displaying a slight opening of the pocket, never reaches the states observed for unglycosylated WT.
Figure 6. Conformational dynamics of GRP94 Client-Binding Site.
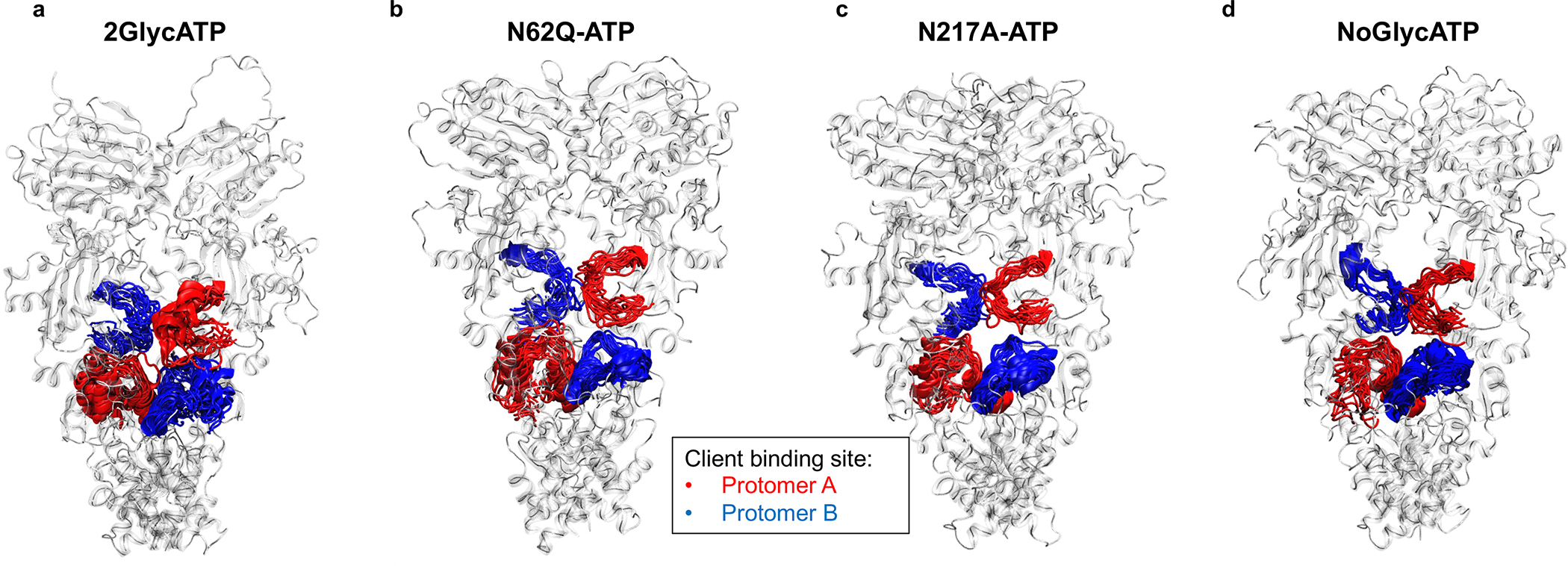
The client binding site is depicted as red (Protomer A) or blue (protomer B) ribbons, whereas the rest of the protein is depicted as transparent cartoons.
(a) 2GlycATP system.
(b) Same as A, for the N62Q mutant in complex with ATP.
(c) Same as A, for the N217A mutant in complex with ATP.
(d) Same as A, for the completely unglycosylated NoGlyc state in complex with ATP.
Overall, these data show that PTMs remodel the connectivity and coordination between distal substructures of the protein required for proper functioning in normal conditions. The impact of PTMs on the internal dynamics of GRP94 is reflected in the remodeling of the structure of the client binding site, which tends to close-up in the glycosylated form of the protein, thus potentially hindering the threading of the client through the chaperone lumen. Interestingly, PU-WS13 has a strong effect on the internal dynamics of protein variants with glycosylation at N62, a trait that can help explain the activity of this drug on highly glycosylated GRP94. In contrast, the impact of PU-WS12 is much less pronounced, which correlates with the apparent inactivity of this ligand.
Ligands and PTMs cooperate to modulate the conformations of the active site lid and of the nucleotide binding site
As a final step, we characterized the fine details of the interactions of glycosyls and ligands on the conformational profiles of the active site. The active site region is known to undergo large conformational variations, which entail open to close state transitions and modifications of the folded state of Helix 4 (H4), the active site lid. Figure 7 reports three different lid conformations via a specific color code for distinct states56–58.
Figure 7. Simplified representation of the structural ensembles of GRP94 ATP-lid.
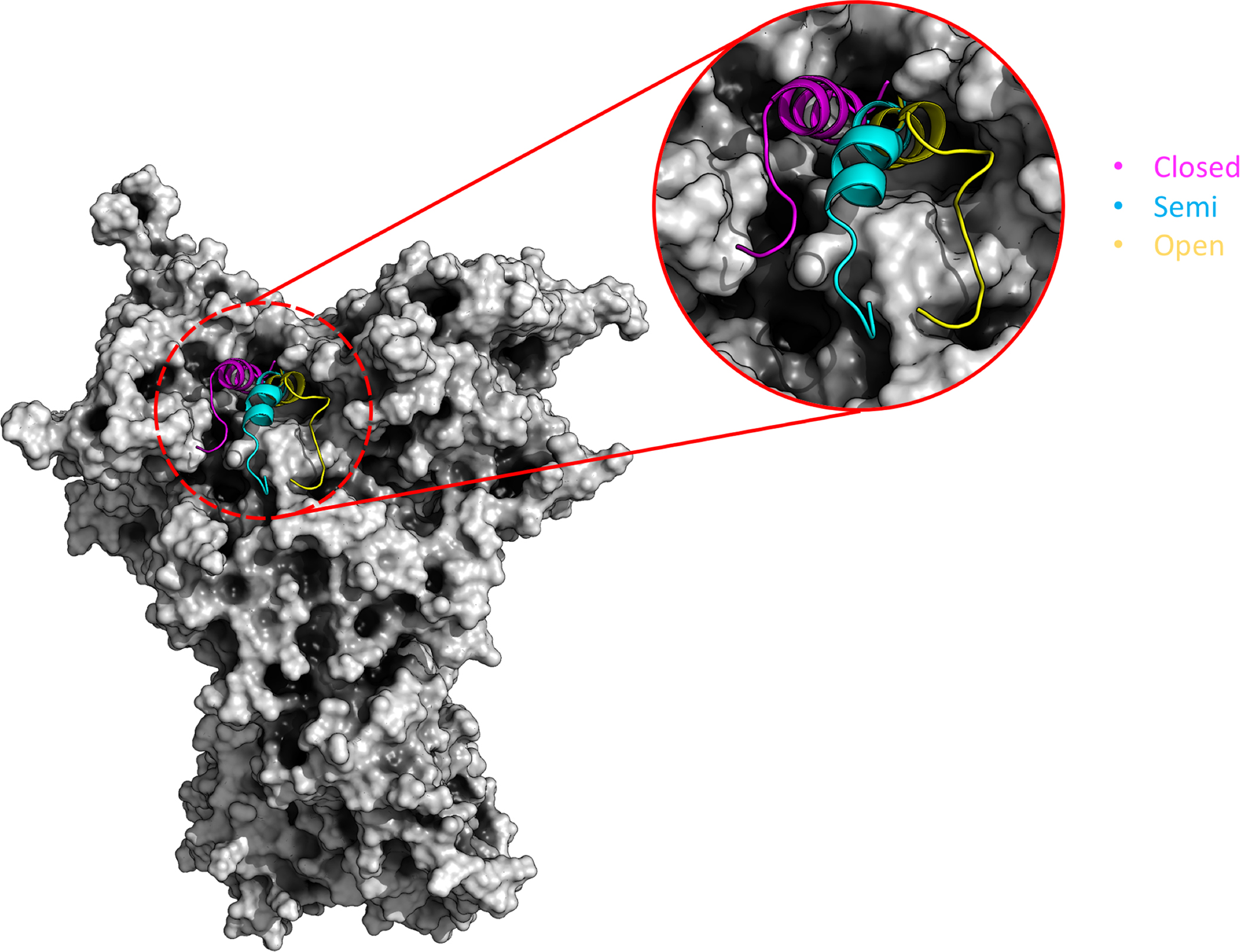
The cartoon shows the whole GRP94 represented as a surface. The zoom on the lid structure shows the three main conformational families of the lid covering the active site of Grp94 in the closed (magenta), semi-open (cyan), open (yellow).
Simulations start with the lid in the closed state over the binding site. In 2GlycATP, the lid passes through closed and open states (Figure 8a). The figure shows in red the interspersed conformations of the lid sampled along the 3 μs metatrajectory and in blue sticks the conformations of the sugars interacting with it. As the interaction is lost (N62Q mutant bound to ATP, Figure 8b), the distribution of lid conformations for protomer A shifts to the preferentially closed state. Here, the carbohydrate moiety bound to N217 folds on H4 stabilizing the closed conformation. Deletion of N217 (Figure 8c) favors an intermediate state for both protomers, with the ATP-lid of protomer B still preferentially closed.
Figure 8. Conformational dynamics of the ATP-lid covering the active site of Grp94 and its interactions with sugar chains in the open or closed forms.
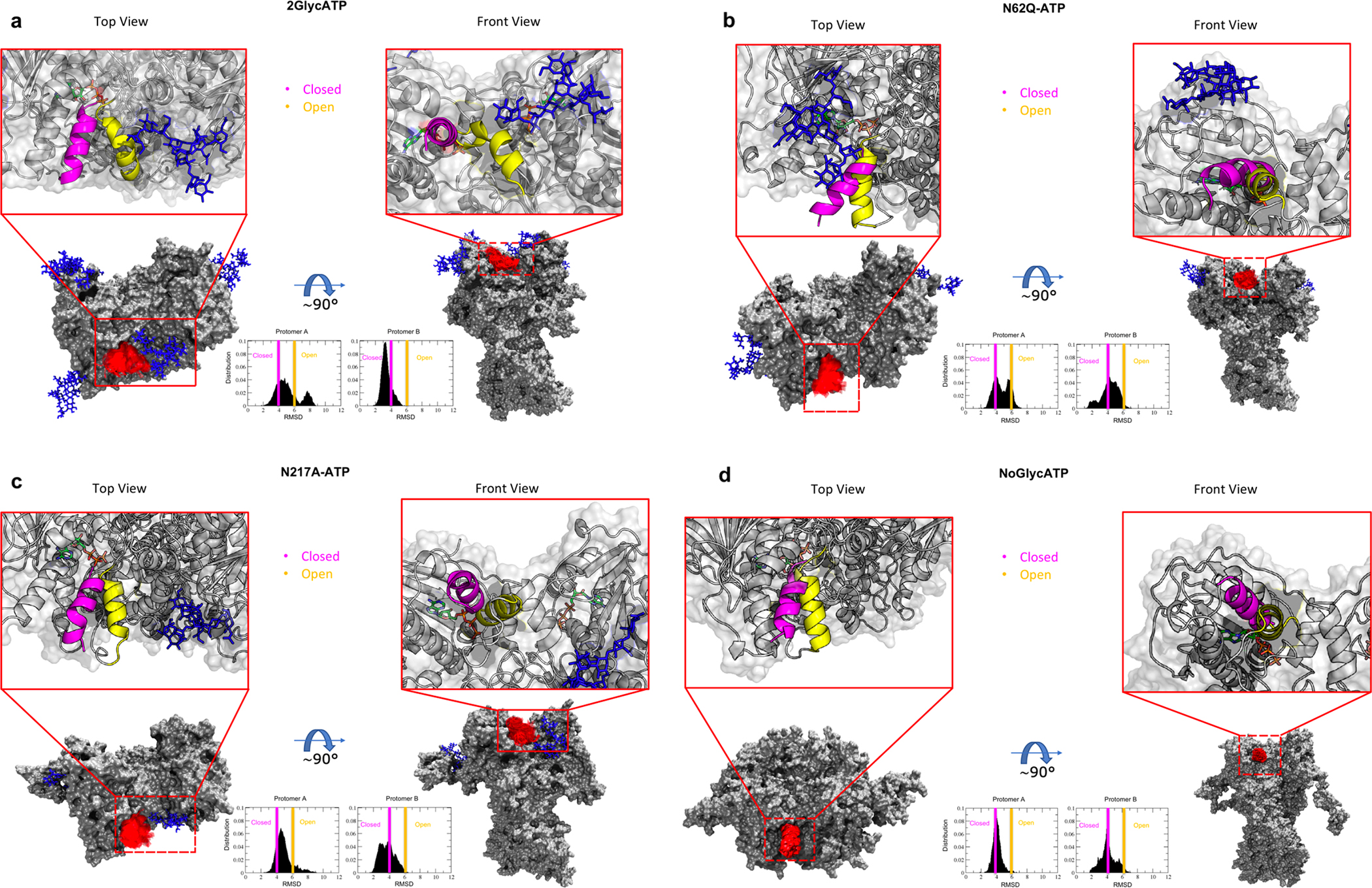
Two orientations are depicted. ATP-lid and glycans at several frames (300 frames) are represented with red and blue lines, respectively. The insets show the distributions of RMSD values from the conformation presented in crystal structure PDB 5ULS.pdb. The blue and yellow vertical lines show the RMSD threshold for the closed and open states, respectively.
(a) 2GlycATP.
(b) N62Q mutant in complex with ATP.
(c) N217A mutant in complex with ATP.
(d) NoGlyc system in complex with ATP.
Overall, in the presence of ATP, the dynamics of the lid are regulated mainly by the glycan on N62, which stabilizes the open structure. As the closed lid-state is induced in the presence of ATP, the glycan on N217 may play an accessory role in stabilizing this conformation. Indeed, N217A mutation does not completely abolish the population of the open state. In contrast, as N62 is mutated to Q, the percentage of open lid conformations, in particular in protomer A, is significantly decreased.
To confirm the role of the glycans, we characterized the structural behavior of this ATP-lid in the unglycosylated form (Figure 8d). The difference in the selection of conformational ensembles compared to the cases reported above is striking, showing a clear preference for the closed state.
To shed light on the impact of the synthetic GRP94 ligand with high affinity for the fully glycosylated form, we analyzed the conformational dynamics of the lid in simulations with PU-WS13 bound to the active site (Figure 9a). A clear partial unfolding of lid Helix H4 immediately emerges. In parallel, the long disordered loop in the lid is remodeled into a helical conformation, a variation that is not observed in the simulation with ATP. In these three replicates, the opening and closing of the lid is observed more frequently than in the simulations with ATP. The N62 glycan stabilizes the open lid, as observed previously in the ATP simulations. The Pre-N-terminal (PN) strap also participates in this interaction. In the presence of PU-WS13, the N217-glycan can act as a stabilizer of the closed form of the H4 of the lid.
Figure 9. The effect of ligands on the conformational dynamics of the ATP-lid covering the active site of GRP94 and its interactions with sugar chains in the open or closed forms.
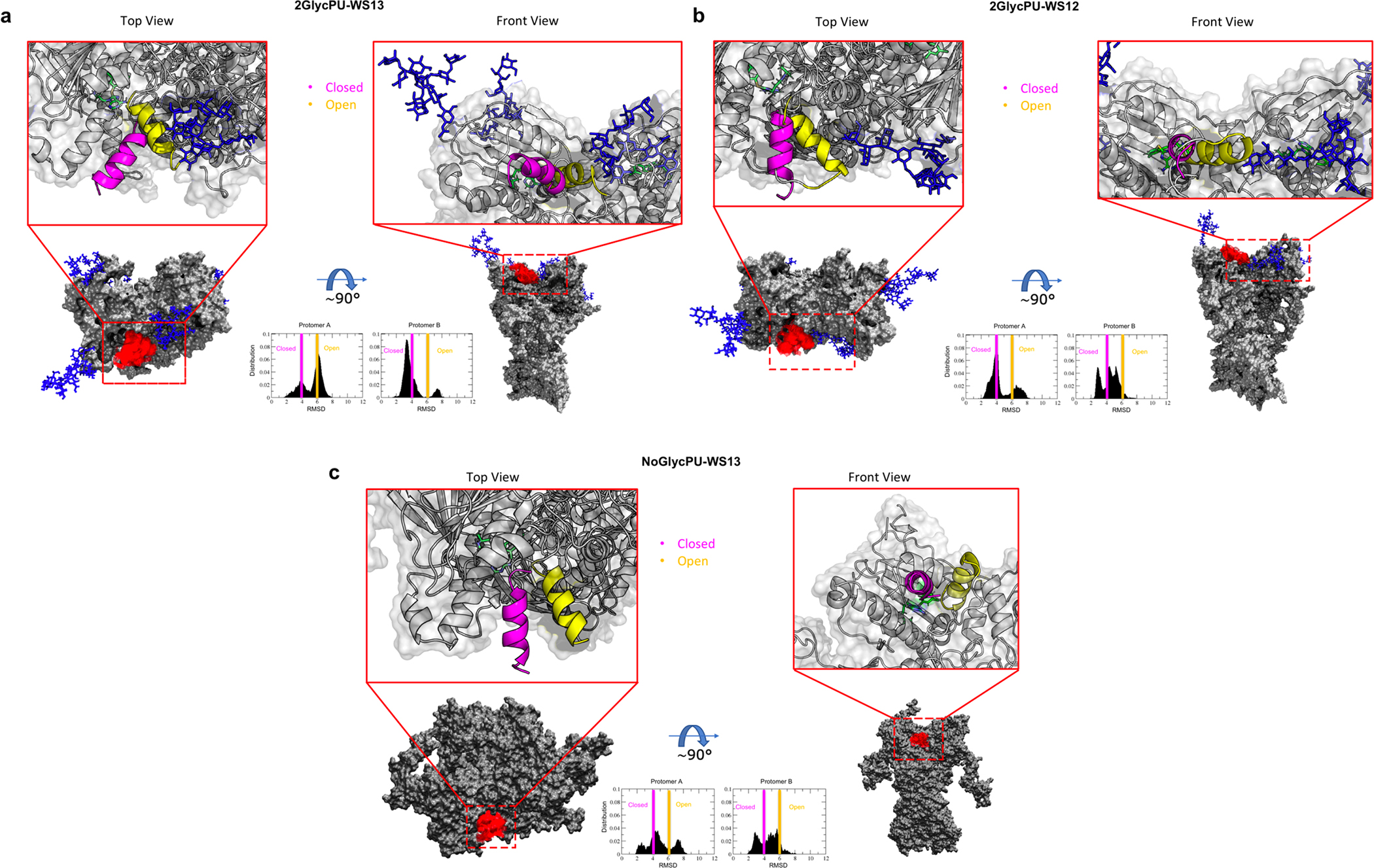
Same as Figure 8.
(a) 2Glyc system in complex with PU-WS13.
(b) 2Glyc system in complex with PU-WS12.
(c) NoGlyc system in complex with PU-WS13.
In summary, PU-WS13 reconfigures the lid, making it more dynamic and prone to spontaneously open. The effect of the glycans appears to be less pronounced than in the ATP-bound case, although the interactions that they establish are substantially the same as observed above.
In N62Q (Simulation N62Q-PU-WS13, SI Figure S10a), PU-WS13 induces a significant remodeling of the lid, which shows an increased tendency to populate an open conformation. No interactions with the lid for the N217 glycan are observed, a factor that would further favor an open configuration of the binding site. On this basis, it may be hypothesized that the residence time of PU-WS13 at the binding site would be significantly lower than that observed for the fully glycosylated form: a consistently open site can aptly facilitate the escape of the ligand from the protein. This model is consistent with the observed insensitivity to PU-WS13 of N62Q mutant cells15. Importantly, the dynamics of the lid in the complex with PU-WS13 and the N217A mutant of GRP94 are qualitatively similar to the fully glycosylated GRP94/PU-WS13 complex (SI Figure S10b). This observation, in turn, is consistent with the fact that there is no effect on the binding of this ligand in the N217A mutant.
Finally, when PU-WS12 is bound, the lid appears to be partially unfolded as in the simulations with PU-WS13. The lid undergoes opening/closing transitions with and without interactions with the glycans. Also in this case, the presence of the ligand confers more mobility to the lid than is observed with ATP (Figure 9b). The glycan on N62 stabilizes the open form and the closed one can in some cases be stabilized by the glycan on N217 (Figure 9b)(See SI Figure S11a,b).
In the NoGlyc form both ligands stabilize the open state of the lid, with PU-WS13 strongly favoring the open form: the impact of PU-WS13 in preventing the protein to stabilize the closed state required for catalysis and to enter the chaperone cycle is one of the factors that determine the efficacy of this molecule in tumor treatment (Figure 9c) (for NoGlyc-PU-WS12 See SI Figure S11c)
Focusing on the ligand-binding site, we next calculated the frequency with which a contact is formed between the ligand and the surrounding residues of the protein: We describe the results in SI and in a pictorial manner in the SI Figure S12 (See also Key Resource Table). The low-activity ligand PU-WS12, in particular, appears to populate different ensembles of conformations in the complex, establishing in general mostly transient contacts with the binding site. These data also support a model whereby PU-WS13 features a longer residence time on the glycosylated target, determining a favorable kinetic profile.
These observations are further corroborated by analyzing the distributions of RMSD values of the residues making up the active site compared to their respective positions in the ATP-bound crystal structure of GRP94 (see SI Figure S13). In the fully glycosylated protein, the active site in the presence of ATP preferentially visits one ensemble of conformations, for both protomers. The same is noticeable for PU-WS13, which clearly locks the active site in a restricted portion of configurational space. PU-WS12, in contrast, explores a larger ensemble of configurations (larger distributions, with more than one peak). Importantly, removal of the glycosyl moieties in the WT system, determines markedly different profiles: a configuration similar to the one observed in the crystallographic closed structure is predominantly populated only for the ATP state in protomer A, while the structure of the binding site in protomer B appears to be endowed with larger conformational freedom. The sequential and deterministic model for ATP hydrolysis20,52,59 provides a rationale for this observation. ATP hydrolysis occurs with different rates in the two WT protomers: while protomer A is ready to favor the reaction of the nucleotide, the other protomer needs to be flexible and explore different possible rearrangements for the second hydrolysis. Both PU-WS13 and PU-WS12 determine a variation in the RMSD distributions: interestingly, PU-WS13 is the ligand that forces the WT system to differentiate more substantially from the ATP-bound state.
In summary, the comparative analysis of the lid and active site dynamics defines a model in which the specific propensity of the GRP94 ATP-lid to populate open conformations is further amplified by the interactions established in particular by the glycosyl moiety at N62. Favoring the open lid contrasts the population of the ATPase competent state. The results are consistent with the experimental characterization of N62-glycosylation as a key factor for the switch of GRP94 from a folding ER chaperone to a templating protein. Interestingly, the active ligand PU-WS13 further favors lid opening and unfolding of H4 in the N62 glycosylated versions of the protein. At the same time PU-WS13 populates one dominant configuration in the active site, establishing stable interactions with it. Qualitatively speaking, this can be expected to determine a longer persistence time of this ligand compared to PU-WS12, providing further mechanistic support to the observed activity of PU-WS13 in Glyc62GRP94 cancer cells.
Binding of different scaffolds: Glycan-induced GRP94 conformational preferences underlie the selection of active ligands
Several GRP94 ligands with different structures and scaffolds were reported over the past few years. These include for instance resorcinol type compounds such as BnIm, which are GRP94 active but Glyc62GRP94 inactive (see Figure 1 and SI Figure 1) 33,34. Here, we tested whether these types of compounds could preferentially bind to protein conformations representative of different ensembles. This test is also intended to assess if the conformations obtained from the simulations are actionable in the selection of ligands for distinct protein forms. To this end we docked BnIm33 into the active site of the central structures of the most populated conformational clusters obtained from the trajectories of the fully glycosylated and the WT forms bound to ATP, respectively 2GlycATP and NoGlycATP (SI Figure S14)(Full data are provided electronically, see Key Resource Table). Interestingly, while in NoGlycATP the best docking pose is observed for Bnlm binding to the most visited conformation, in the fully glycosylated 2GlycATP form, the best docking poses are observed for the states that are visited less frequently. This result is consistent with the fact that BnIm33 was experimentally shown to be unable to inhibit glycosylated GRP94, as evidenced biochemically (see the effect on GRP94 conformation, Figure 2c,d and SI Figure 1c,d) and functionally (no cytotoxicity and no effect on HER2 signaling in SKBr3 cells at the highest tested concentration of 20 μM, 1-log higher than its binding affinity for GRP94)35.
Summarizing, our data propose a model whereby PTMs and ligands cooperate in defining different GRP94 dynamic states. In particular, the observed modulation of the active site provides a structural framework to rationalize the distinct effects of the ligands on the various PTM-forms of GRP94, and, as a consequence, their impact on the biology of cells where these forms are highly expressed.
Discussion
Our study unveils the mechanisms through which N-glycans at different protein sites modulate the conformational dynamics of GRP94, affecting both its nucleotide processing activity and interaction modes with clients, at atomistic resolution. Specifically, we build a structural and dynamic model in which the degree of glycosylation and the specific positioning of the glycosyl moieties are correlated with the observed switch of function for PTM-modified GRP94 from a folding to a scaffolding protein. In our model, stress-induced glycosylation, in particular at position N62, favors dynamic states of the chaperone with peculiar traits of internal coordination, distinct structures of active and client-binding sites, and specific responses to nucleotide and inhibitor binding.
Our data support an active role for glycosyl modifications: consistent with experiments15, in the presence of glycosylation at N62, the combined perturbation of the communication between the NTD and the M-Domain, the expected lower efficiency in binding and processing ATP due to sugar-shielding, and the closure of the lumen can block remodeling of the client15. In this context, glycosylation affects the conformational modifications of the chaperone and its ability to relay the signal encoded by ATP to the client binding site25,60.
The carbohydrate moiety at N62 specifically engages the charged linker connecting the NTD and M-Domain. Here, the perturbative role of glycosyls on GRP94 functions can originate from the modulation of the conformational ensembles of the charged linker and in the modification of the statistical weights of protein subpopulations61 that are poised to efficiently translate the nucleotide-encoded signal into folding functions. In this framework, activity is determined by the weights of the different conformational ensembles of the species present at specific timepoints. It is the fraction of molecules that have domains in a defined active/inactive conformation, and the interaction networks associated with these structures (in the ATP and/or client binding sites) that ultimately define observed biological activities. While non-covalent interactions between single monosaccharide moieties and proteins are generally weak in nature, the presence of long carbohydrate chains and the diverse conformations available to the long charged linker expand (and favor) the spectrum of possible binding modes between the two motifs via interactions that are multivalent in nature62. While this picture may be different from the interaction mechanisms found in ordered portions of proteins where a dominant binding structure is selected, this dynamic model of interaction between dynamic parts of the protein63,64 can help explain the functional effects brought about by PTMs.
The overall outcome is the malfunction of the glycosylated protein: in this picture, PTMs perturb the population of GRP94 structural ensembles that are presented to the other partners necessary to assemble an efficient folding machinery, and consequently of the kinetics/timing of the chaperone cycle40–43,65,66. The net result is a perturbation of the foldase function. In terms of PPI networks, this translates into a remodeling of protein connectivity and, given the centrality of GRP94, the final effect is amplified in proteome-wide dysfunction.
As a caveat, it is important to note here that in the crowded environment of the cell, a number of other factors may play a role. One of these is the abundance of Ca2+ ions in the ER, ions that are known to bind the charged loop and may modify GRP94 functions in transport processes. Given the high concentration of Ca2+ required to properly model the physiological environment and the limitations of classical forcefields in treating highly charged systems, we opted for a minimal and manageable model. While this approach may have limitations, it has been shown to provide important, experimentally validated, biochemical insight in previous studies of Hsp90 systems67–70.
Our extensive atomistic simulations also shed light on the determinants of GRP94 response to ligands. Interestingly, PU-WS13 dramatically perturbs the dynamics of the protein variants with glycosylation at N62, while forming a stable complex with the protein, traits that can help explain the activity of this drug in cells with high levels of Glyc62GRP94. In contrast, the effects of PU-WS12 are much less pronounced with the ligand unable to populate a stable configuration for the bound state, consistent with its apparent inactivity.
A key aspect of our work is that the comparative analysis of the dynamics of different glycosylated variants of GRP94 supports an active key role for glycosylation in regulating the conformational dynamics of the active site, where ATP and ATP-competitive ligands bind. The ATP-lid of GRP94 has a distinctive propensity to adopt the open structure, thus presenting an energetic barrier to closure of the dimer into its active conformation15,56,58,71. While lid closure is required for ATP hydrolysis and for the chaperone cycle to proceed, N62-glycans stabilize the lid in open states, determine additional barriers for the lid to reach the closed conformation, and disfavor the population of the foldase state of the chaperone15. It is worth noting here that the conformational dynamics of the lid is substantially asymmetric between the two protomers, a theme already observed in the regulation of the functions of various members of the Hsp90 family20,44,46,52,54,55,72–75. Asymmetric dynamics has in fact been observed as a convenient mechanism for the expansion of the conformational space of chaperones and fine-tuning of their functions52.
In the case studied here, the observed sugar-induced asymmetry provides further potential for the expansion of the ensembles of GRP94 conformations that can interact with different partners, supporting the role of the aberrantly glycosylated chaperone as the hub for proteome-wide interaction perturbations. We suggest a picture whereby PTMs modulate the dynamics of the ATP-state as well as the efficiency of the catalytic hydrolysis cycle of the nucleotide, potentially providing a convenient mechanism of GRP94 adaptation to different cell conditions, in particular to chronic cellular stress states76–78, inducing extensive protein covalent modifications10,69,79.
The potential for expansion of chaperone states may be even greater if one considers asymmetric PTMs. In the crowded cell environment and under stress conditions, protein modifying enzymes can be expected to work differently on either protomers, potentially providing a diversity of chaperone-species with a wide spectrum of (detrimental) functional properties. These PTM-modified chaperones define a chaperone-code that needs to be decrypted via the combination of experiments and computational approaches 80. Importantly, the approach we describe here is completely general and can immediately be translated to the study of other PTM-modified systems.
Finally, our data provide a structure-based rationale for the observed (in)sensitivity of Glyc62GRP94 to some GRP94 inhibitors. These results have important reach and implications in chemical biology and drug design as they suggest the possibility to leverage the structural and dynamic models generated here as templates to guide the selection and development of small molecules able to selectively act on cells where GRP94 aberrant glycosylation is a factor in determining pathologic states. More specifically, these small molecules could function by specifically engaging PTM-induced dynamic states, representing an important expansion of the molecular diversity space of chaperone targeting drugs 25,60,81. An additional advantage is the opportunity to selectively interfere with the disease-associated forms of the protein, leaving unmodified forms of normal cells untouched. Together with the development of other isoform specific ligands, this could decisively enrich our arsenal of chemical tools to clinically target and study the complex mechanisms of chaperones in their endogenous context.
Significance
Aberrant N-glycosylation remodels GRP94 conformational dynamics, and, in turn, its selection of, and interaction with, protein partners. The final result is a remodeling of the connectivity within the PPI networks of which GRP94 is a central hub. The outcome at the cell level is that modified protein interaction pathways determine aberrant disease phenotypes. From these results, we propose a general model that can establish a link between the fundamental physicochemical basis of protein conformational behavior and the cellular functions it is involved in. The impact of protein modifications reflects the extent of the conformational dynamic perturbation and population shift between the functional and the altered forms of the protein. When considered in large systems, with multiple complexes that need to form and/or disrupt in a highly regulated, orderly manner with numerous players involved, these modifications can have significantly amplified effects on physiological signaling and deregulation in disease. Protein modifications and the variation of protein assembly mechanisms can thus reverberate in wide-spread proteome malfunctions determined by altered PPIs, which ultimately causes disease states.
The overall approach we present here may represent an effective means of exploiting the functional dynamics of different forms of GRP94, modulated by covalent modifications, in the selection and design of drugs that are specific and selective for a given form of the protein. Such drugs would intervene by specifically addressing PTM-induced dynamic states and by selectively interfering with the disease-associated form of the protein, leaving the unmodified forms of normal cells untouched. Together with the development of isoform specific ligands, this could decisively enrich our arsenal of chemical tools to clinically target and study the complex mechanisms of chaperones in their endogenous context. While based on the case of GRP94, our models and considerations are fully general, and readily transferable to other targets and contexts for cancers and other diseases.
STAR+METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for materials and resource should be directed to Dr. Giorgio Colombo (g.colombo@unipv.it).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
1. The input files, topologies and restart files are provided in a Zenodo link reported in the Key Resource Table. 2. Code used in this work is available from https://github.com/colombolab. 3. Any additional information required to reanalyze the data reported in this work is available at the link in the Key resource table.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human Cell Lines
The breast cancer cell lines MDA-MB-468 (HTB-132) and SKBr3 (HTB-30) were obtained from the American Type Culture Collection (ATCC) and were cultured in DMEM and McCoy’s 5A mediums supplemented with 10% FBS, 1% Glutamax and 1% penicillin and streptomycin (Pen/Strep), respectively. Both cell lines were incubated in the humidified cell incubators with 5% CO2 at 37 °C. Cells were authenticated using short tandem repeat profiling and tested for mycoplasma before and after use.
METHOD DETAILS
Reagents
Glyc62GRP94 active ligands (PU-WS13, PU-SO33, PU-HJP149, PU-HJP110) and inactive ligands (BnIm, PU-WS12, PU-SO116, PU-HJP92, PU-HJP36) were synthesized using previously reported protocols 15,29,35. Briefly, for PU-WS13, a CuI-catalyzed coupling of 8-mercaptoadenine with 3,5-dichloroiodobenzene at 110°C resulted in 8-(3,5-dichloro-phenylsulfanyl)adenine in 72% yield, which was heated with 3-(tertbutoxycarbonyl-isopropyl-amino)-propyl tosylate in DMF at 80°C under nitrogen protection for 30 min. The boc-deprotection of resulting tert-butyl(3-(6-amino-8-((3,5-dichlorophenyl)thio)-9H-purin-9-yl)propyl)(isopropyl) carbamate from the previous step with TFA gave crude material which upon purification by preparatory TLC with CH2Cl2:MeOH-NH3 (7N) at 20:1 gave PU-WS13 in 45% yield. 1H NMR (600 MHz, CDCl3/CD3OD): δ 8.21 (s, 1H), 7.26 (t, J = 1.7 Hz, 1H), 7.24 (d, J = 1.9 Hz, 2H), 4.23 (t, J = 6.9 Hz, 2H), 2.63 (septet, J = 6.2 Hz, 1H), 2.46 (t, J = 6.4 Hz, 2H), 1.89 (quintet, J = 6.9 Hz, 2H), 0.97 (d, J = 6.3 Hz, 6H); 13C NMR (150 MHz, CDCl3): δ 154.7, 153.1, 151.3, 144.1, 135.9, 133.7, 128.8, 128.7, 119.7, 48.6, 43.4, 41.6, 29.8, 22.4; HRMS (ESI): m/z [M+H]+ calcd for C17H21Cl2N6S 411.0925, found 411.0917. For PU-WS12, CuI-catalyzed coupling of 8-mercaptoadenine with 1-iodo-2,4-dichlorobenzene at 110°C resulted in 8-((2,4-dichlorophenyl)thio)-9H-purin-6-amine in 87% yield, which was heated with 3-(tertbutoxycarbonyl-isopropyl-amino)-propyl tosylate in DMF at 80°C under nitrogen protection for 30 min. The boc-deprotection of resulting tert-butyl (3-(6-amino-8-((2,4-dichlorophenyl)thio)-9H-purin-9-yl)propyl)(isopropyl)carbamate from the previous step with TFA gave crude material which upon purification by preparatory TLC with CH2Cl2:MeOH-NH3 (7N) at 20:1 yielded PU-WS12 (35% yield). 1H NMR (600 MHz, CDCl3/CD3OD): δ 8.23 (s, 1H), 7.55 (s, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 4.34 (t, J = 6.7 Hz, 2H), 2.92 (septet, J = 5.8 Hz, 1H), 2.69 (t, J = 6.4 Hz, 2H), 2.13 (quintet, J = 6.5 Hz, 2H), 1.16 (d, J = 5.7 Hz, 6H); HRMS (ESI): m/z [M+H]+ calcd for C17H21Cl2N6S 411.0925, found 411.0907. Synthesis of PU-SO33 includes cyclocondensation of 2,4,5,6-tetraaminopyrimidine sulfate with CS2 in ethanol to obtain 2,6-diamino-9H-purine-8-thiol in quantitative yield. Next, copper-catalyzed coupling of 2,6-diamino-9H-purine-8-thiol with 3,5-dichloroiodobenzene resulted in 65% yield of 8-((3,5-dichlorophenyl)thio)-9H-purine-2,6-diamine. Transformation of C2-amino group of 8-((3,5-dichlorophenyl)thio)-9H-purine-2,6-diamine to Cl was achieved with SbCl3 and t-BuONO in 10:1 mixture of DCE and DMSO at 80°C resulting in 2-chloro-8-((3,5-dichlorophenyl)thio)-9H-purin-6-amine. The sequential reaction of 2-chloro-8-((3,5-dichlorophenyl)thio)-9H-purin-6-amine with 1,3-dibromopropane, followed by treating the bromo derivative from the previous step with excess isopropylamine resulted in crude PU-SO33. The crude residue was purified by preparatory TLC (CH2Cl2-MeOH-NH3 (7N), 20:1) to give 60% yield of PU-SO33. 1H NMR (500 MHz, CDCl3) δ 7.33 – 7.31 (m, 1H), 7.30 – 7.28 (m, 2H), 5.75 (br s, 2H), 4.29 (t, J = 6.9 Hz, 2H), 2.78 – 2.70 (m, 1H), 2.56 (t, J = 6.6 Hz, 2H), 1.99 – 1.91 (m, 2H), 1.05 (d, J = 6.2 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 155.0, 154.5, 152.7, 144.6, 135.9, 134.0, 128.7, 128.5, 118.9, 48.9, 43.6, 42.0, 30.1, 22.7; MS m/z 444.92 [M+H]+. For methyl 2-(2-(1-benzyl-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (BnIm)36,82, the reaction of methyl 2,4-dihydroxy-6-methylbenzoate with tert-butyldimethylsilyl chloride in the presence of NaH provided TBS-protected methyl benzoate derivative, which was treated with calcium hypochlorite in 10% AcOH/H2O to achieve chlorination. The resulting compound from the previous step reacted with ally bromide and lithium diisopropylamide at −78°C in THF to obtain methyl 2-(but-3-en-1-yl)-4,6-bis((tert-butyldimethylsilyl)oxy)-3-chlorobenzoate in 45% yield. Next, the oxidation of 2-(but-3-en-1-yl)-4,6-bis((tert-butyldimethylsilyl)oxy)-3-chlorobenzoate with ozone and the reaction of resulting aldehyde compound with benzylamine and glyoxal, followed by TBS-deprotection with TBAF provided crude BnIm compound. The purification was achieved using column chromatography (CH2Cl2-MeOH, 20:1) which provided 22% yield of BnIm. 1H NMR (600 MHz, CDCl3) δ 11.35 (s, 2H), 7.40 – 7.36 (m, 2H), 7.35 – 7.32 (m, 1H), 7.10 – 7.07 (m, 3H), 6.90 (d, J = 1.3 Hz, 1H), 6.52 (s, 1H), 5.14 (s, 2H), 3.86 (s, 3H), 3.58 – 3.54 (m, 2H), 3.01 – 2.97 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 170.8, 162.8, 158.1, 147.6, 141.5, 136.1, 129.0, 128.1, 126.6, 120.1, 114.9, 105.8, 102.7, 52.5, 49.5, 30.8, 26.0; HRMS (ESI): m/z [M+H]+ calcd for C20H20ClN2O4 387.1112, found 387.1116. For the synthesis and characterization of PU-HJP149, PU-HJP110, PU-SO116 and PU-HJP92 and PU-HJP36 refer to Methods, Chemical synthesis and information.
Fluorescence polarization (FP) measurements
The FP competition assays were performed on an Analyst GT instrument (Molecular Devices, Sunnyvale, CA) and carried out in black 96-well micro-plates (Corning, no. 3650) in a total volume of 100 μL in each well. A stock of 10 μM cy3B-GM 83 was prepared in DMSO and diluted with Felts buffer (20 mM Hepes (K), pH 7.3, 50 mM KCl, 2 mM DTT, 5 mM MgCl2, 20 mM Na2MoO4 and 0.01% NP40 with 0.1 mg/mL BGG). To each well was added 6 nM cy3B-GM, 10 nM recombinant GRP94 or HSP90α protein and tested inhibitor (initial stock in DMSO) in a final volume of 100 μL Felts buffer. Compounds were added in triplicate wells. For each assay, background wells (buffer only), tracer controls (free, cy3B-GM only) and bound controls (cy3B-GM in the presence of protein) were included on each assay plate. The assay plate was incubated on a shaker at 4 °C for 24 h, and the FP values (in mP) were measured. The fraction of cy3B-GM bound GRP94 was correlated to the mP value and plotted against values of competitor concentrations. The inhibitor concentration at which 50% of bound cy3B-GM was displaced was obtained by fitting the data. An excitation filter at 530 nm and an emission filter at 580 nm were used with a dichroic mirror of 561 nm. All of the experimental data were analyzed using SOFTmax Pro 4.3.1 and plotted using Prism 6.0 (GraphPad Software Inc., San Diego, CA). Binding affinity values were given as relative binding affinity values (EC50, concentration at which 50% of fluorescent ligand was competed off by compound).
SDS-PAGE and Immunoblotting
Cells were either treated with DMSO (vehicle) or indicated compounds and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% sodium deoxycholate and 0.5% NP40) supplemented with cocktail protease inhibitors (Roche) to produce whole-cell lysates (WCLs). Membrane fraction was prepared by ProteoExtract Subcellular Proteome Extraction Kit (Millipore, 539790) according to the manufacturer’s instructions. Protein concentrations were determined using the BCA kit (Pierce). Ten to fifty micrograms of total protein were examined by immunoblotting with indicated antibodies. GRP94 antibody (SPA-850, 1:3,000) from Enzo, HER2 antibody (28–0004, 1:2,000) from Invitrogen, EGFR (#4267, 1:1,000), p-ERK1/2 (#4377, 1:3,000) and total ERK1/2 (#4695, 1:5,000) antibodies from Cell Signaling Technology were used. The blots were washed with TBS/0.1% Tween20 and incubated with appropriate HRP-conjugated secondary antibodies (1:3,000). Chemiluminescent signal was detected with Enhanced Chemiluminescence Detection System (GE Healthcare) according to the manufacturer’s instructions.
Western blot analysis of signaling and viability
MDA-MB-468 cells were plated in 6-well plates at 3 × 106 cells per plate and treated next day with the indicated concentrations of PU-WS12, PU-WS13 or vehicle. The cells were collected after 24 h of treatment and protein extracts were prepared in RIPA buffer. Twenty micrograms of total protein were examined by immunoblotting with cleaved PARP (5625, 1:1,000), phospho-ERK (T202/Y204) (4377, 1:3,000) and ERK (4695, 1:5,000) antibodies purchased from Cell Signaling Technology. The blots were processed as described in the immunoblotting section.
GRP94 conformation detection by immunoprecipitation (IP)
In cell IP: MDA-MB-468 cells or SkBr3 cells were plated in 10 cm plates at 3 × 106 cells per plate and treated next day with the indicated compounds at indicated concentrations. At the end of the treatment cells were washed with cold PBS and harvested. Protein extracts were prepared in the RIPA buffer. 100 μg whole cell lysates were incubated with 2 μg either 9G10 (SPA-850, Enzo) or G4420 (Sigma) antibody for 3 hr at 4°C, followed by incubation with Protein A/G agarose beads (Roche) for another 2 hr at 4°C. The beads were washed with cold Felts buffer three times, eluted by SDS loading buffer and subjected to immunoblotting as described above. In lysate IP: MDA-MB-468 cells were washed with cold PBS, harvested by centrifugation and lysed in RIPA buffer. 200 μg cell lysates were diluted to 100 μL with Felts buffer and incubated with 1 μM PU-WS13 or PU-WS12 for 2 hr at 4°C. 2 μg 9G10 antibody (SPA-850, Enzo) was added to the samples and incubated at 4°C overnight, followed by incubation with Protein G agarose beads for another 2 hr at 4°C. The beads were washed with cold Felts buffer three times, eluted by SDS loading buffer and subjected to immunoblotting as described above.
Native Gel Electrophoresis and GRP94 epichaperome detection
Cells were lysed in the RIPA buffers (WCL) and diluted with Felts buffer (20 mM Hepes pH 7.3, 50 mM KCl, 2 mM DTT, 5 mM MgCl2, 20 mM Na2MoO4 and 0.01 % NP40 with 0.1 mgmL−1 BGG). Twenty-five to one hundred μg of protein were loaded onto 5% native gel and resolved at 4°C. The gels were stained with Bio-Safe Coomassie stain (Biorad, 1610786), or soaked in Tris-Glycine-SDS running buffer for 15 min prior to gel transfer for 75 min at 100V and immunoblotting for GRP94 using the 9G10 antibody as described above.
ATP based cell viability assessment.
Cell viability was assessed using CellTiter-Glo luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. The method determines the number of viable cells in culture based on quantification of ATP amount, which signals the presence of metabolically active cells, and was performed as previously reported (Yan 2020 cell reports). Briefly, MDA-MB-468 cells were plated in clear bottom black 96-well plates (Corning) at 6,000 cells per well and treated with serial dilutions of the indicated compounds for 72 h. After incubation, 100 μL of CellTiter-Glo reagent was added to each well. Plates were incubated with the reagent for 10 min at room temperature and the luminescence signal was measured using Analyst GT microplate reader (Molecular Devices). IC50 values for each compound were determined using Prism GraphPad.
Chemical synthesis and information
Chemistry: General experimental
Chemistry general considerations.
All commercial chemicals and solvents were reagent grade and used without further purification. The identity and purity of each product was characterized by MS, HPLC, TLC, and NMR. 1H/13C NMR spectra were recorded on either a Bruker 400, 500 or 600 MHz instrument. Chemical shifts are reported in δ values in ppm downfield from TMS as the internal standard. 1H data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constant (Hz), integration. 13C chemical shifts are reported in δ values in ppm downfield from TMS as the internal standard. Low resolution mass spectra were obtained on Waters Acquity Ultra Performance LC with electrospray ionization and SQ detector. Purity of target compounds has been determined to be >95% by LC/MS on a Waters Autopurification system with PDA, MicroMass ZQ and ELSD detector and a reversed phase column (Waters X-Bridge C18, 4.6 × 150 mm, 5 μm) eluted with water/acetonitrile gradients, containing 0.1% TFA. Column chromatography was performed using 230–400 mesh silica gel. Analytical thin layer chromatography was performed on 250 μM silica gel F254 plates. Preparative thin layer chromatography was performed on 1000 μM silica gel F254 plates. Flash chromatography was performed using CombiFlash®Rf instrument. Compounds 1, 7, 13, and 16 were synthesized as reported previously 15,35,84,85.
Synthesis of PU-HJP36
For synthetic scheme see SI Scheme S1.
Step 1: Preparation of 8-((4-Chloro-2-nitrophenyl)thio)-9H-purin-6-amine (2).
8-Mercaptoadenine (1, 1.6 g, 8.8 mmol), neocuproine hydrate (540 mg, 2.6 mmol), Cul (330 mg, 1.73 mmol), NaOtBu (1.7 g, 17.38 mmol), 4-chloro-1-iodo-2-nitrobenzene (3.2 g, 11.2 mmol), and anhydrous DMF (25 mL) were taken in a round bottom flask flushed with nitrogen. The flask was sealed with Teflon tape, heated at 120 °C, and magnetically stirred for 20 h under nitrogen. Solvent was removed under reduced pressure and the resulting residue was purified using flash chromatography (CH2Cl2:MeOH, 10:0 to 10:1) to provide 2 in 95 % yield (2.7 g). MS (ESI): m/z 322.8 [M + H]+.
Step 2 and 3: Preparation of 9-(3-Bromopropyl)-8-((4-chloro-2-nitrophenyl)thio)-9H-purin-6-amine (3) and 8-((4-chloro-2-nitrophenyl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine (4).
Compound 2 (2 g, 6.211 mmol) was dissolved in DMF (20 mL) and Cs2CO3 (2.43 g, 7.45 mmol) and 1,3-dibromopropane (3.15 mL, 31.05 mmol) were added and the mixture was stirred under nitrogen at room temperature for 2 h. After completion of reaction, Cs2CO3 was filtered off and the residue was washed with DMF (3 mL). The filtrate containing crude compound 3 was taken into an RBF and directly reacted with isopropylamine (0.31 mmol) overnight. Following solvent removal, the crude material was purified by column chromatography (CH2Cl2:CH3OH-NH3 (7N), 100:1 to 20:1) to afford desired product 4 in 23% yield (600 mg). 1H NMR (600 MHz, CDCl3/CD3OD) δ 8.35 (s, 1H), 8.30 (d, J = 2.2 Hz, 1H), 7.43 (dd, J = 8.7, 2.2 Hz, 1H), 6.81 (d, J = 8.7 Hz, 1H), 4.29 (t, J = 6.9 Hz, 2H), 2.67–2.73 (m, 1H), 2.52 (t, J = 6.8 Hz, 2H), 1.93–1.98 (m, 2H), 1.03 (d, J = 6.3 Hz, 6H); 13C NMR (150 MHz, CDCl3/CD3OD) δ 155.3, 153.9, 151.3, 145.8, 142.0, 134.5, 133.3, 131.9, 129.7, 126.2, 120.4, 48.7, 43.3, 41.9, 30.1, 22.3; HRMS (ESI) m/z [M+H]+ calcd. for C17H21ClN7OS, 422.1166; found 422.1170.
Step 4: Preparation of 8-((2-Amino-4-chlorophenyl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine (5).
A mixture of 4 (300 mg, 0.711 mmol) and iron powder (150 mg, 2.488 mmol) in acetic acid (5 mL) was stirred at room temperature for 4 hrs. On completion, the reaction was neutralized by adding solid Na2CO3 at 0 °C and washed with EtOAc (75 ml X 3). Following drying the organic layer over MgSO4 and solvent removal, the crude material was purified by column chromatography (CH2Cl2:CH3OH-NH3 (7N), 50:1 to 15:1) to afford 5 in 93% yield (260 mg). 1H NMR (600 MHz, CDCl3/CD3OD) δ 8.16 (s, 1H), 7.39 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 2.2 Hz, 1H), 6.73 (dd, J = 8.3, 2.2 Hz, 1H), 4.36 (t, J = 6.7 Hz, 2H), 3.40–3.41 (m, 1H), 2.83 (t, J = 6.8 Hz, 2H), 2.26–2.32 (m, 2H), 1.34 (d, J = 6.5 Hz, 6H); 13C NMR (150 MHz, CDCl3/CD3OD) δ 154.2, 151.9, 151.4, 150.5, 147.3, 138.3, 138.1, 118.9, 115.7, 107.0, 74.4, 50.6, 42.1, 40.4, 31.1, 19.8; HRMS (ESI) m/z [M+H]+ calcd. for C17H23ClN7S, 392.1424; found 392.1419.
Step 5: 8-((4-Chloro-2-iodophenyl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine (6, PU-HJP36).
A mixture of 5 (260 mg, 0.665 mmol), NaNO2 (57 mg, 0.831 mmol) and potassium iodide (221 mg, 1.3 mmol) in acetic acid (5 mL) was stirred at 0°C for 15 min. On completion, water (10 mL) was added, and the reaction was neutralized by adding solid Na2CO3 at 0°C and further washed with EtOAc (75 ml X 3). Following drying over MgSO4 and solvent removal, the crude material was purified by column chromatography (CH2Cl2:CH3OH-NH3 (7N), 80:1 to 20:1) to afford PU-HJP36 (6) in 79% yield (260 mg). 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 7.87 (d, J = 2.2 Hz, 1H), 7.23 (dd, J = 8.5, 2.2 Hz, 1H), 7.07 (d, J = 2.2 Hz, 1H), 5.87 (br s, 2H), 4.30 (t, J = 6.9 Hz, 2H), 2.68–2.72 (m, 1H), 2.56 (t, J = 6.8 Hz, 2H), 1.95–1.99 (m, 2H), 1.03 (d, J = 6.2 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 154.7, 153.3, 151.6, 144.8, 139.3, 135.9, 134.1, 131.1, 129.5, 120.4, 99.8, 48.7, 43.8, 41.9, 30.3, 22.9; HRMS (ESI) m/z [M+H]+ calcd. for C17H21IClN6S, 503.0282; found 503.0277.
Synthesis of PU-SO116
For synthetic scheme see SI Scheme S2.
Step 1: 9-(2-Bromoethyl)-8-((3,5-dichlorophenyl)thio)-9H-purin-6-amine (8).
To a solution of 7 (200 mg, 0.640 mmol) in DMF (5 mL) was added Cs2CO3 (335 mg, 1.025 mmol) and 1,2-dibromoethane (277 μL, 3.2 mmol) and the mixture was stirred under nitrogen at rt for 4 h. Solvent was removed under reduced pressure and the resulting residue was chromatographed (CH2Cl2:MeOH:AcOH, 20:1:0.5) to afford 8 in 38 % yield (102 mg). NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.28 (s, 2H), 7.25 (s, 1H), 6.54 (s, 2H), 4.66 (t, J = 6.5 Hz, 2H), 3.74 (t, J = 6.5 Hz, 2H); MS (ESI): m/z 420.2 [M+H]+.
Step 2: 8-((3,5-Dichlorophenyl)thio)-9-(2-(isopropylamino)ethyl)-9H-purin-6-amine (9, PU-SO116).
To 8 (20 mg, 0.047 mmol) in dry DMF (2 mL) was added isopropylamine (121 μL, 1.41 mmol) and the reaction mixture was stirred at rt for 72 hours. Solvent was removed under reduced pressure and the residue was purified by preparatory TLC (CH2Cl2:MeOH-NH3 (7N), 20: 1) to afford 12.6 mg (68%) of PU-SO116. 1H NMR (600 MHz, CDCl3): δ 8.36 (s, 1H), 7.26–7.28 (m, 3H), 5.82 (br s, 2H), 4.34 (t, J = 6.5 Hz, 2H), 2.97 (t, J = 6.5 Hz, 2H), 2.74–2.76 (m, 1H), 0.96 (d, J = 6.2 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 154.8, 153.5, 151.5, 144.0, 135.7, 135.3, 128.1, 127.8, 120.4, 48.5, 46.1, 44.5, 22.8. HRMS (ESI) m/z [M+H]+ calcd. for C16H19N6SCl2, 397.0769; found 397.0765.
Synthesis of PU-HJP149
For synthetic scheme see SI Scheme S3.
Step 1: 8-((3-chloro-5-iodophenyl)thio)-9H-purin-6-amine (10).
8-Mercaptoadenine (1, 1 g, 5.988 mmol), neocuproine hydrate (0.25 g, 1.1976 mmol), Cul (0.23 g, 1.1976 mmol), NaOtBu (1.45 g, 11.976 mmol), 1-chloro-3,5-diiodobenzene (3.3 g, 8.982 mmol), and anhydrous DMF (25 mL) were taken in a round bottom flask flushed with nitrogen. The flask was sealed with Teflon tape, heated at 120 °C, and magnetically stirred for 20 h under nitrogen. Solvent was removed under reduced pressure and the resulting residue was chromatographed (CH2Cl2:MeOH: 10:1) to provide 10 in 67 % yield (1.6 g). 1H NMR (500 MHz, DMSO-d6) δ 13.47 (br s, 1H), 8.13 (s, 1H), 7.81 (s, 1H), 7.72 (s, 1H), 7.50 (s, 1H), 7.37 (br s, 2H); MS (ESI): m/z 403.7 [M + H]+.
Step 2 and 3: 9-(3-Bromopropyl)-8-((3-chloro-5-iodophenyl)thio)-9H-purin-6-amine (11) and 8-((3-chloro-5-iodophenyl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine (12, PU-HJP149).
To a solution of 10 (500 mg, 1.24 mmol) in DMF (15 mL) was added Cs2CO3 (485 mg, 1.48 mmol) and 1,3-dibromopropane (630 μL, 6.2 mmol). The reaction mixture was stirred under nitrogen at room temperature for 2 h. On completion, the solid was filtered off and washed with DMF (4 mL). The filtrate containing crude compound 11 was taken into an RBF and directly reacted with isopropylamine (750 μL) at room temperature overnight. Solvent was removed under reduced pressure and the resulting residue was purified by preparative TLC (CH2Cl2:CH3OH-NH3 (7N), 20: 1 or 60: 1) to afford PU-HJP149 (12) in 20% yield (125 mg). 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 1H), 7.62 (dt, J = 7.4 Hz, 1.4 Hz, 2H), 7.34 (t, J = 1.7 Hz, 1H), 6.13 (br s, 2H), 4.32 (t, J = 7.0 Hz, 2H), 2.67–2.73 (m, 1H), 2.55 (t, J = 6.8 Hz, 2H), 1.92– 1.98 (m, 2H), 1.03 (d, J = 6.2 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 154.8, 153.4, 151.6, 143.6, 136.7, 136.6, 135.7, 134.7, 129.3, 120.2, 94.4, 48.7, 43.8, 41.8, 30.3, 22.9; HRMS (ESI) m/z [M+H]+ calcd. for C17H21IClN6S, 503.0282; found 503.0260.
Synthesis of PU-HJP92
For synthetic scheme see SI Scheme S4.
Step 1: 9-(2-bromoethyl)-8-((2,4-dichlorophenyl)thio)-9H-purin-6-amine (14).
A mixture of 13 (400 mg, 1.28 mmol), Cs2CO3 (630 mg, 1.92 mmol), and 1,2-dibromopropane (1.21 g, 0.55 mL, 6.43 mmol) in DMF (10 mL) under nitrogen protection was stirred at rt for 3 h. Following solvent removal, the crude material was purified by preparatory TLC (CH2Cl2:CH3OH:AcOH, 20:1:0.1) to afford 14 in 36% yield (190 mg). 1H NMR (500 MHz, CDCl3/CD3OD) δ 8.27 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.26 (dd, J = 8.4, 2.2 Hz, 1H), 4.68 (t, J = 6.5 Hz, 2H), 3.77 (t, J = 6.5 Hz, 2H); 13C NMR (125 MHz, CDCl3/CD3OD) δ 154.6, 153.1, 150.9, 145.0, 136.3, 135.7, 133.9, 130.3, 128.4, 128.2, 119.8, 45.0, 28.5; MS (ESI): m/z 417.9 [M + H]+.
Step 2: 8-((2,4-dichlorophenyl)thio)-9-(2-(isobutylamino)ethyl)-9H-purin-6-amine (15, PU-HJP92).
A mixture of 14 (9 mg, 0.022 mmol) and isobutylamine (53.5 μL, 0.54 mmol) in DMF (1 mL) under nitrogen protection was stirred at room temperature overnight. Following solvent removal, the crude material was purified by preparative TLC (CH2Cl2:CH3OH-NH3 (7N), 20:1) to afford PU-HJP92 in 81% yield (7.3 mg). 1H NMR (600 MHz, CDCl3/CD3OD) δ 8.21 (s, 1H), 7.43 (d, J = 2.0 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.16 (dd, J = 8.5 Hz, 2.0 Hz, 1H), 4.31 (t, J = 5.9 Hz, 2H), 2.94 (t, J = 5.7 Hz, 2H), 2.34–2.37 (m, 2H), 1.62–1.66 (m, 1H), 0.80 (d, J = 6.5 Hz, 6H); 13C NMR (150 MHz, CDCl3/CD3OD) δ 154.5, 152.9, 151.2, 145.2, 136.1, 135.4, 133.7, 130.3, 130.1, 128.9, 128.2, 119.8, 57.3, 48.6, 43.7, 28.0, 20.4; HRMS (ESI) m/z [M+H]+ calcd. for C17H21Cl2N6S, 411.0925; found 411.0917.
Synthetic scheme of PU-HJP110
For synthetic scheme see SI Scheme S5.
Step 1: 8-((3-chloro-5-iodophenyl)thio)-9H-purine-2,6-diamine (17).
Compound 16 (2 g, 10.989 mmol), 1-chloro-3,5-diiodobenzene (5 g, 13.736 mmol), neocuprine hydrate (0.46 g, 2.1978 mmol), Cul (0.42 g, 2.1978 mmol), NaOtBu (2.11 g, 21.978 mmol) and DMF (35 mL) were charged in a nitrogen protected dry vessel. The reaction vessel was sealed and placed in an oil bath (120°C) and stirred for 20 h. The reaction mixture was then cooled to room temperature and DMF was removed in vacuo. The crude material was purified by silica gel column chromatography (CH2Cl2:CH3OH, 10:0 to 10:1) to afford 3.3 g (71.8 %) of 17. MS (ESI): m/z 418.6 [M + H]+.
Step 2: 8-((3-chloro-5-iodophenyl)thio)-2-fluoro-9H-purin-6-amine (18).
To a cooled solution (0 °C) of 17 (1 g, 2.392 mmol) in HF/pyridine (1.5 mL) was slowly added NaNO2 (185 mg, 3.11 mmol). The resulted mixture was stirred at 0°C for 10 min and then at room temperature for 1.5 h. The reaction mixture was quenched by stirring with saturated CaCO3 solution (5 mL) for 1 h. The crude material was taken up in EtOAc, washed with water, and dried over anhydrous Na2SO4. Following solvent removal, the residue was purified using flash chromatography (CH2Cl2:CH3OH, 100:0 to 10:1) to afford 18 (100 mg, 11% yield). MS (ESI): m/z 421.8 [M + H]+.
Step 3: 9-(3-bromopropyl)-8-((3-chloro-5-iodophenyl)thio)-2-fluoro-9H-purin-6-amine (19).
A mixture of 18 (40 mg, 0.0951 mmol), Cs2CO3 (40.51 mg, 0.1236 mmol), and 1,3-dibromopropane (48.3 μL, 0.476 mmol) in DMF (2 mL) under nitrogen protection was stirred at room temperature for 2 h. Following solvent removal, the crude material was purified by preparatory TLC (CH2Cl2:CH3OH:AcOH, 40:1:0.1) to afford 19 in 45% yield (23 mg). 1H NMR (500 MHz, CDCl3) δ 7.64 (dt, J = 4.1, 1.5 Hz, 2H), 7.36 (t, J = 1.7 Hz, 1H), 6.42 (br s, 2H), 4.34 – 4.29 (m, 2H), 3.39 (t, J = 6.3 Hz, 2H), 2.36 – 2.29 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 159.38 (d, J = 212.5 Hz), 156.61 (d, J = 20.2 Hz), 152.94 (d, J = 19.4 Hz), 143.71 (d, J = 2.8 Hz), 137.10, 136.90, 135.89, 134.19, 129.58, 118.46 (d, J = 3.8 Hz), 94.58, 42.82, 32.03, 29.18; MS m/z 541.8 [M + H]+.
Step 4: 8-((3-chloro-5-iodophenyl)thio)-2-fluoro-9-(3-(isopropylamino)propyl)-9H-purin-6-amine (20, HJP-VI-110).
To a solution of 19 (23 mg, 0.00423 mmol) in dry DMF (2 ml) was added isopropylamine (173 μL, 2.1194 mmol) and the reaction mixture was stirred at rt for 8 h. Then, the solvent was removed under reduced pressure and the crude product was purified by prep TLC (CH2Cl2:MeOH-NH3 (7N), 20:1) to afford 15.2 mg (69%) of 20 (HJP-V-110). 1H NMR (600 MHz, CDCl3) δ 7.63 (t, J = 1.6 Hz, 1H), 7.61 (t, J = 1.5 Hz, 1H), 7.33 (t, J = 1.7 Hz, 1H), 6.34 (br s, 2H), 4.25 (t, J = 7.1 Hz, 2H), 2.75 – 2.68 (m, 1H), 2.56 (t, J = 6.8 Hz, 2H), 1.94 (p, J = 6.8 Hz, 2H), 1.04 (d, J = 6.3 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 159.3 (d, J = 212.0 Hz), 156.5 (d, J = 20.3 Hz), 152.9 (d, J = 19.2 Hz), 143.6 (d, J = 2.7 Hz), 136.9, 136.5, 135.8, 134.6, 129.3, 118.4 (d, J = 3.7 Hz), 94.5, 48.8, 43.8, 42.1, 30.2, 22.8; MS m/z 520.9 [M + H]+.
COMPUTATIONAL METHODS
System preparation
The starting structure for all simulations of the glycosylated models of full-length GRP94, either with ATP or ligands bound at the active site, is crystal structure 5ULS.pdb56; both protomers were reconstructed without gaps by modeling the missing fragments with Modeller 86. As the original structure contains AMPPNP, to reconstruct ATP, the Nβ atom of the adenylyl-imidodiphosphate molecule that is present in each protomer’s active site was replaced by oxygen. To predict ligand binding modes and concomitant structural changes in the receptor for the designed inhibitors, the adenylyl-imidodiphosphate molecule was removed and ligands PU-WS12 and PU-WS13 were docked into the binding site using the Glide docking program (Glide, version 6.9, Schrödinger, LLC, New York, NY, 2021) using the Induced Fit Docking (IFD) protocol, based on Glide and the Refinement module in Prime. Docking calculations were performed in standard precision mode (SP) with the OPLS_2005 force field. No modifications were applied to the default settings. The best poses according to the docking score function were selected as a starting point for MD simulations. All hydrogens were added (or replaced post-docking) using AmberTools’ tleap utility (version 19) 87, with residues calculated to be in their standard protonation states at physiological pH, (as predicted by PROPKA, v. 3.1. 88). Glycans were connected to residues N62 and N217 using the glycoprotein construction tool http://www.glycam.org: The following sequence was used to build the sugar chain: DManp α1,2 DManp α1,6 [DManp α1,2 DManp α1,3] DManp α1,6 [DManp α1,2 DManp α1,2 DManp α1,3] DManp ß1,4 DGlcpNAc ß1,4 DGlcpNAc - Asn. This type of glycosylation motif, while simplified, was chosen based on the consideration that ER glycosylation motifs are mainly mannosyl-based and on the basis of data reported by Yan et al.15 The exact structure and composition of the N62 and N217 glycans are unknown. However, limited sugar removal that retains the N-acetylglucosamine attached to Asn (such as by EndoH) partially reduces PU-WS13 binding (Yan et al. cell Reports 2020). Conversely, complete deglycosylation (such as by PNGase F), abolishes GRP94’s binding to PU-WS13 (Yan et al. cell Reports 2020), indicating the residue being modified, rather than the exact sugar composition, are of most importance in drug binding. Also, both Glyc62GRP94 and Glyc217GRP94 are sensitive to deglycosylation by EndoH (see deglycosylation studies using the N217A and N62Q mutans, ref. (Yan et al. cell Reports 2020)) indicating a high mannose glycan characteristic. High mannose glycans are involved in initial steps in N-glycan processing and control of protein folding in the ER, which are subsequently trimmed back in the process of generating complex type glycans through Golgi-localized processing. High mannose glycans are not commonly observed on the cell membranes of mammalian cells but are prevalent on various tumor cells. Thus, high mannose cell surface GRP94, as in the aberrantly glycosylated GRP94, is an incompletely processed pathologic GRP94, of high mannose character, and we chose the Man9 glycan to model such feature.
Forcefield parameters for molecular dynamics simulations and parametrization of PU-WS12 and PU-WS13
During Molecular Dynamics simulations (MD), protein residues are modeled using ff14SB forcefield 89 parameters, except glycans and glycosylated asparagines N62 and N217 which were modeled using the GLYCAM_06j force field 90. ATP is treated using ad hoc parameters by Meagher and coworkers91. Compounds PU-WS12 and PU-WS13 parametrization were carried out with the aid of AmberTools’ antechamber and parmchk2 87. Derivation of point charges was carried out using the Gaussian16 program (www.gaussian.com) in conjunction with antechamber: first, compounds were structurally optimized at the B3LYP/6-31G(d) level of density functional theory (DFT); subsequently, ESP charges 92 around each atom are calculated based on the electrostatic potential, calculated at the Hartree-Fock/6-31G(d) level, and sampled over 10 shells per atom at a density of 17 grid points per square Bohr; final atomic point charges are assigned after RESP 93 fitting performed by antechamber. The chosen water model is TIP3P 94 which is compatible with parameters by Joung and Cheatham 95 chosen to treat Na+ cations used to neutralize the charge of the system.
Molecular dynamics simulations
MD simulations were carried out using the Amber suite (version 18) 87 with the Sander MD engine employed in the early preproduction stages (minimization, heating and equilibration) of each MD run, and the GPU-accelerated pmemd.cuda utility 96 thereafter. Three independent MD replicas (atomic velocities assigned from different random seeds) are carried out for each system, comprising minimization, heating, equilibration, and production. First, the prepared solute was energy-minimized in vacuo using 200 steps of steepest descent and an additional 200 steps using conjugate gradients. A cutoff of 10 Å was used for Coulomb and van der Waals interactions. The minimized system was then solvated using the TIP3P water model in a truncated octahedral box and rendered electroneutral by adding sodium counterions. To remove any bad contacts between solute and solvent, the system was minimized with a two-stage approach. In the first stage we restrained the solute coordinates using a 500 kcal mol−1 Å−2 harmonic constant force and minimized the positions of water and ions with 500 steps of steepest descent followed by 500 steps of conjugate gradient, a. Then, in the second stage, the entire system was minimized with 1000 steps of steepest descent and 1500 conjugate gradient steps without restraints. The temperature of the system was then increased from 0 to 300 K in the NVT ensemble, running a rapid heating step of 20 ps of MD with weak positional restraints on the solute (force constant of 10 kcal kcal mol−1 Å−2) and the Langevin thermostat97 (collision frequency of 1.0 ps-1) to avoid any large fluctuations. All bonds involving hydrogen were constrained using the SHAKE98 algorithm. The systems were then equilibrated with no restraints for 500 ps with a 2 fs time step, with the SHAKE 98 algorithm and periodic boundary conditions in the NPT ensemble. Initial velocities for each replicate were obtained from a Maxwellian distribution at the initial temperature of 300 K. The temperature was kept constant with the Langevin thermostat (collision frequency of 1.0 ps−1) 97, and a constant pressure of 1 bar was introduced via Berendsen’s barostat (2ps relaxation time)99. The electrostatic interactions were treated using the particle mesh Ewald method100 with a cutoff of 10 Å. The same cutoff was used even for short-range Lennard-Jones interactions. Each production run (3 for each system) was extended to 1 μs with an identical setup to the final equilibration condition. Postprocessing and analysis of MD trajectories were conducted with the CPPTRAJ tool101 whereas the VMD platform was used for visual inspection102.
Analysis of MD trajectories
Distance fluctuations (DF) analysis.
The replicates for each system were combined into 1 comprehensive metatrajectory (one metatrajectory for each system reported in Table 1). The matrix of the fluctuations of pairwise amino acid distances has been obtained by calculating the DF parameter for any pair of residues 45. This parameter is defined as , where is the (time-dependent) distance of the Cα atoms of amino acids i and j, and the brackets indicate the time average over the trajectory. The DF matrix was calculated for the each of the meta-trajectory of all the simulated systems.
Analysis of contacts.
Analysis of contacts between the glycans (or the ligands) and the protein over the trajectories have been performed using the nativecontacts command implemented in the CPPTRAJ program distributed within the AmberTools suite (version 19) 87. To count as a contact, the distance between residues i and j in a structure should be lower than 7 Å. The frequency of each contact has been determined with in-house python script. Calculations were carried out independently for each sugar chain (or for each ligand).
Root-mean-square-deviation (RMSD).
RMSD of protein backbone atoms was computed using the rmsd command implemented in the CPPTRAJ program distributed within the AmberTools suite (version 19) 87. Alignment of each snapshot of the analyzed trajectory using the coordinates of the corresponding backbone atoms of the crystal structure (PDB code 5ULS, 56 as a reference was done before RMSD calculations. Calculations was carried out independently for each protomer.
Cluster analysis.
Cluster analysis was carried out using hierarchical agglomerative algorithm from cluster command in CPPTRAJ distributed within the AmberTools suite (version 19) 87 Clustering was carried out in the following way: first, all snapshots from the metatrajectories of each simulated system are aligned using the coordinate of backbone atoms of the entire protein of the starting conformation, then the coordinated of the backbone atoms of the lid in each snapshot are used to define the different conformational families, using as distance metric option the RMSD with the nofit option. For each simulated system, 10 representative conformations were collected, with the complete-linkage method, which uses the maximum distance between members of two clusters.
Docking calculations.
Compound Bnlm was docked with the GLIDE docking program (Glide, version 9.6, Schrödinger, LLC, New York, NY, 2022) at the ATP binding site of the central structures of the most populated conformational clusters obtained from the trajectories of the fully glycosylated and the WT forms bound to ATP, respectively 2GlycATP and NoGlycATP. Rigid receptor and flexible ligand docking calculations were performed in standard precision mode (SP) with the OPLS_2005 force field, nonplanar conformations of amide bonds were penalized, van der Waals radii were scaled by 0.80, and the partial charge cut-off was fixed to 0.15. No further modifications were applied to the default settings.
QUANTIFICATION AND STATISTICAL ANALYSIS
Any quantification and statistical analysis that were applied in the experiments are described in Method details, as well as in the main and supplemental figure legends.
Supplementary Material
N-glycosylation and ligands actively modify functional dynamics of the chaperone GRP94
Dynamic mechanisms favor GRP94 transition from a folder to a scaffolding protein
Dynamic GRP94 mechanisms rationalize the biological activity of small molecules
Pathologically N-glycosylated GRP94 variants are selectively actionable
ACKNOWLEDGMENTS
This work is supported in part by the US National Institutes of Health (NIH) (R56 AG061869, P01 CA186866, P30 CA08748) for Gabriela Chiosis. Giorgio Colombo acknowledges funding from Fondazione AIRC (Associazione Italiana Ricerca Sul Cancro) grant IG 20019 and from PRIN (grant 20209KYCH9).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
G.Chiosis, A.R., C.S.D. and P.Y. are inventors on patents covering PU-WS13 and associated composition of matter. G.Chiosis is a founder of Samus Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 14 figures and can be found with this article online at the journal website
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nussinov R, Ma B, and Tsai CJ (2014). Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys. Chem. 186, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei G, Xi W, Nussinov R, and Ma B (2016). Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 116, 6516–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nussinov R, Tsai C-J, and Jang H (2019). Protein ensembles link genotype to phenotype. PLoS computational biology 15, e1006648–e1006648. 10.1371/journal.pcbi.1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreiro DU, Hegler JA, Komives EA, and Wolynes PG (2011). On the role of frustration in the energy landscapes of allosteric proteins. Proc. Natl. Acad. Sci. U. S. A. 108, 3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gfeller D, Michielin O, and Zoete V (2013). Shaping the interaction landscape of bioactive molecules. Bioinformatics 29, 3073–3079. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Wiewiora RP, Meng F, Babault N, Ma A, Yu W, Qian K, Hu H, Zou H, Wang J, et al. (2019). The dynamic conformational landscape of the protein methyltransferase SETD8. eLife 8, e45403. 10.7554/eLife.45403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pontiggia F, Pachov D, Clarkson M, Villali J, Hagan M, Pande V, and Kern D (2015). Free energy landscape of activation in a signalling protein at atomic resolution. Nat. Commun. 6, 7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai C-J, and Nussinov R (2014). A Unified View of “How Allostery Works”. PLoS computational biology 10, e1003394. 10.1371/journal.pcbi.1003394.s004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wodak SJ, Paci E, Dokholyan NV, Berezovsky IN, Horovitz A, Li J, Hilser VJ, Bahar I, Karanicolas J, Stock G, et al. (2019). Allostery in Its Many Disguises: From Theory to Applications. Structure (London, England : 1993) 27, 566–578. 10.1016/j.str.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Backe SJ, Sager RA, Woodford MR, Makedon AM, and Mollapour M (2020). Post-translational modifications of Hsp90 and translating the chaperone code. Journal of Biological Chemistry 295, 11099–11117. 10.1074/jbc.REV120.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramazi S, and Zahiri J (2021). Post-translational modifications in proteins: resources, tools and prediction methods. Database 2021, baab012. 10.1093/database/baab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santos AL, and Lindner AB (2017). Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Medicine and Cellular Longevity 2017, 5716409. 10.1155/2017/5716409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schjoldager KT, Narimatsu Y, Joshi HJ, and Clausen H (2020). Global view of human protein glycosylation pathways and functions. Nature Reviews Molecular Cell Biology 21, 729–749. 10.1038/s41580-020-00294-x. [DOI] [PubMed] [Google Scholar]
- 14.Mereiter S, Balmaña M, Campos D, Gomes J, and Reis CA (2019). Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 36, 6–16. [DOI] [PubMed] [Google Scholar]
- 15.Yan P, Patel HJ, Sharma S, Corben A, Wang T, Panchal P, Yang C, Sun W, Araujo TL, Rodina A, et al. (2020). Molecular Stressors Engender Protein Connectivity Dysfunction through Aberrant N-Glycosylation of a Chaperone. Cell Reports 31, 107840. 10.1016/j.celrep.2020.107840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harbison AM, Fogarty CA, Phung TK, Satheesan A, Schulz BL, and Fadda E (2022). Fine-tuning the spike: role of the nature and topology of the glycan shield in the structure and dynamics of the SARS-CoV-2 S. Chemical Science 13, 386–395. 10.1039/D1SC04832E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casalino L, Gaieb Z, Goldsmith JA, Hjorth CK, Dommer AC, Harbison AM, Fogarty CA, Barros EP, Taylor BC, McLellan JS, et al. (2020). Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS central science 6, 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mollapour M, and Neckers L (2012). Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 1823, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marzec M, Eletto D, and Argon Y (2012). GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta. 1823, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, and Agard DA (2014). Structural asimmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol. Cell. 53, 330–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genest O, Reidy M, Street TO, Hoskins JR, Camberg JL, Agard DA, Masison DC, and Wickner S (2013). Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol Cell 49, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu BX, Hong F, Zhang Y, Ansa-Addo E, and Li Z (2016). GRP94/gp96 in Cancer: Biology, Structure, Immunology, and Drug Development. Adv. Cancer Res. 129, 165–190. [DOI] [PubMed] [Google Scholar]
- 23.Lee AS (2014). Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nature Reviews Cancer 14, 263–276. 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillarsetty N, Jhaveri K, Taldone T, Caldas-Lopes E, Punzalan B, Joshi S, Bolaender A, Uddin MM, Rodina A, Yan P, et al. (2019). Paradigms for Precision Medicine in Epichaperome Cancer Therapy. Cancer Cell 36, 559–573.e557. 10.1016/j.ccell.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan P, Wang T, Guzman ML, Peter RI, and Chiosis G (2020). Chaperome Networks – Redundancy and Implications for Cancer Treatment. Advances in Experimental Medicine and Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi S, Gomes EDG, Wang T, Corben A, Taldone T, Gandu S, Xu C, Sharma S, Buddaseth S, Yan P, et al. (2021). Pharmacologically controlling protein-protein interactions through epichaperomes for therapeutic vulnerability in cancer. Communications Biology 4, 1333. 10.1038/s42003-021-02842-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sumitomo T, Nakata M, Nagase S, Takahara Y, Honda-Ogawa M, Mori Y, Akamatsu Y, Yamaguchi M, Okamoto S, and Kawabata S (2021). GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection. mBio 12, e03269–03220. 10.1128/mBio.03269-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaumonnot K, Masson S, Sikner H, Bouchard A, Baverel V, Bellaye P-S, Collin B, Garrido C, and Kohli E (2021). The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death & Disease 12, 114. 10.1038/s41419-020-03288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel PD, ...., and Chiosis G. (2013). Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol 9, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huck JD, Que NLS, Immormino RM, Shrestha L, Taldone T, Chiosis G, and Gewirth DT (2019). NECA derivatives exploit the paralog-specific properties of the site 3 side pocket of Grp94, the endoplasmic reticulum Hsp90. Journal of Biological Chemistry 294, 16010–16019. 10.1074/jbc.RA119.009960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pechalrieu D, Assemat F, Halby L, Marcellin M, Yan P, Chaoui K, Sharma S, Chiosis G, Burlet-Schiltz O, Arimondo PB, and Lopez M (2020). Bisubstrate-Type Chemical Probes Identify GRP94 as a Potential Target of Cytosine-Containing Adenosine Analogs. ACS Chemical Biology 15, 952–961. 10.1021/acschembio.9b00965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tosh DK, Brackett CM, Jung Y-H, Gao Z-G, Banerjee M, Blagg BSJ, and Jacobson KA (2021). Biological Evaluation of 5′-(N-Ethylcarboxamido)adenosine Analogues as Grp94-Selective Inhibitors. ACS Medicinal Chemistry Letters 12, 373–379. 10.1021/acsmedchemlett.0c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crowley VM, Khandelwal A, Mishra S, Stothert AR, Huard DJE, Zhao J, Muth A, Duerfeldt AS, Kizziah JL, Lieberman RL, et al. (2016). Development of Glucose Regulated Protein 94-Selective Inhibitors Based on the BnIm and Radamide Scaffold. Journal of Medicinal Chemistry 59, 3471–3488. 10.1021/acs.jmedchem.6b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khandelwal A, Crowley VM, and Blagg BSJ (2017). Resorcinol-Based Grp94-Selective Inhibitors. ACS Medicinal Chemistry Letters 8, 1013–1018. 10.1021/acsmedchemlett.7b00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel HJ, Patel PD, Ochiana SO, Yan P, Sun W, Patel MR, Shah SK, Tramentozzi E, Brooks J, Bolaender A, et al. (2015). Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem 58, 3922–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, and Blagg BSJ (2012). Development of a Grp94 inhibitor. Journal of the American Chemical Society 134, 9796–9804. 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Sun L, Hou J, Gui M, Ying J, Zhao H, Lv N, and Meng S (2015). Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. International Journal of Cancer 137, 512–524. [DOI] [PubMed] [Google Scholar]
- 38.Rodina A, Wang T, Yan PR, Gomes ED, Dunphy MPS, Pillarsetty N, Koren J, Gerecitano JF, Aldone TT, Zong HL, et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–+. 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nussinov R, Tsai C-J, and Li J (2014). Principles of Allosteric Interactions in Cell Signaling. J. Am. Chem. Soc. 136, 17692–17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zierer BK, Rubbelke M, Tippel F, Madl T, Schopf FH, Rutz DA, Richter K, Sattler M, and Buchner J (2016). Importance of cycle timing for the function of the molecular chaperone Hsp90. Nat Struct Mol Biol 23, 1020–1028. 10.1038/nsmb.3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmid S, Götz M, and Hugel T (2018). Effects of Inhibitors on Hsp90′s Conformational Dynamics, Cochaperone and Client Interactions. ChemPhysChem 0. 10.1002/cphc.201800342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmid S, and Hugel T (2020). Controlling protein function by fine-tuning conformational flexibility. eLife, e57180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolf S, Sohmen B, Hellenkamp B, Thurn J, Stock G, and Hugel T (2020). Allosteric action of nucleotides on Hsp90 across several time- and length scales. bioRxiv, 2020.2002.2015.950725. 10.1101/2020.02.15.950725. [DOI] [Google Scholar]
- 44.Schopf FH, Biebl MM, and Buchner J (2017). The HSP90 chaperone machinery. Nature Reviews Molecular Cell Biology 18, 345–360. 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 45.Morra G, Potestio R, Micheletti C, and Colombo G (2012). Corresponding Functional Dynamics across the Hsp90 Chaperone Family: Insights from a Multiscale Analysis of MD Simulations. Plos Comput. Biol. 8, e1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moroni E, Agard DA, and Colombo G (2018). The Structural Asymmetry of Mitochondrial Hsp90 (Trap1) Determines Fine Tuning of Functional Dynamics. J. Chem. Theory Comput. 14, 1033–1044. [DOI] [PubMed] [Google Scholar]
- 47.Ferraro M, D’Annessa I, Moroni E, Morra G, Paladino A, Rinaldi S, Compostella F, and Colombo G (2019). Allosteric modulators of Hsp90 and Hsp70: Dynamics meets Function through Structure-Based Drug Design. Journal of Medicinal Chemistry 62, 60–87. 10.1021/acs.jmedchem.8b00825. [DOI] [PubMed] [Google Scholar]
- 48.Wolynes PG (2005). Energy landscapes and solved protein-folding problems. Philos. Trans. R. Soc., A 363, 453. [DOI] [PubMed] [Google Scholar]
- 49.Okazaki K, Koga N, Takada S, Onuchic JN, and Wolynes PG (2006). Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 103, 11844–11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, and Kern D (2007). A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–918. [DOI] [PubMed] [Google Scholar]
- 51.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, et al. (2007). Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838–844. [DOI] [PubMed] [Google Scholar]
- 52.Elnatan D, Betegon M, Liu Y, Ramelot T, Kennedy MA, and Agard DA (2017). Symmetry broken and rebroken during the ATP hydrolysis cycle of the mitochondrial Hsp90 TRAP1. eLife 6, e25235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verba KA, Wang RY, Arakawa A, Liu Y, Shirouzu M, Yokoyama S, and Agard DA (2016). Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noddings CM, Wang RY-R, Johnson JL, and Agard DA (2022). Structure of Hsp90–p23–GR reveals the Hsp90 client-remodelling mechanism. Nature 601, 465–469. 10.1038/s41586-021-04236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang RY-R, Noddings CM, Kirschke E, Myasnikov AG, Johnson JL, and Agard DA (2022). Structure of Hsp90–Hsp70–Hop–GR reveals the Hsp90 client-loading mechanism. Nature 601, 460–464. 10.1038/s41586-021-04252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huck JD, Que NS, Hong F, Li Z, and Gewirth DT (2017). Structural and functional analysis of GRP94 in the closed state reveals an essential role for the pre-N domain and a potential client-binding site. Cell reports 20, 2800–2809. 10.1016/j.celrep.2017.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dollins DE, Warren JJ, Immormino RM, and Gewirth DT (2007). Structures of GRP94-Nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell. 28, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ephraim AA-A, Jessica T, Feng H, Bill XW, Yongliang Z, Caroline WF, Alessandra M, Brian R, Katelyn W, Daniel TG, et al. (2016). Clients and Oncogenic Roles of Molecular Chaperone gp96/grp94. Current Topics in Medicinal Chemistry 16, 2765–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verba KA, and Agard DA (2017). How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends in biochemical sciences 42, 799–811. 10.1016/j.tibs.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bolaender A, Zatorska D, He H, Joshi S, Sharma S, Digwal CS, Patel HJ, Sun W, Imber BS, Ochiana SO, et al. (2021). Chemical tools for epichaperome-mediated interactome dysfunctions of the central nervous system. Nature Communications 12, 4669. 10.1038/s41467-021-24821-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hilser VJ (2010). An Ensemble View of Allostery. Science 327, 653–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mammen M, Choi S-K, and Whitesides GM (1998). Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem Int Ed 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- 63.Jin F, Yu C, Lai L, and Liu Z (2013). Ligand Clouds around Protein Clouds: A Scenario of Ligand Binding with Intrinsically Disordered Proteins. PLoS Comput Biol 9 e1003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, and Nussinov R (2016). The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chemical Reviews 116, 6391–6423. 10.1021/acs.chemrev.5b00623. [DOI] [PubMed] [Google Scholar]
- 65.Luo Q, Boczek EE, Wang Q, Buchner J, and Kaila VRI (2017). Hsp90 dependence of a kinase is determined by its conformational landscape. Scientific Reports 7, 11, 43996. 10.1038/srep43996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mickler M, Hessling M, Ratzke C, Buchner J, and Hugel T (2009). The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 16, 281–286. [DOI] [PubMed] [Google Scholar]
- 67.Morra G, Neves MAC, Plescia CJ, Tsustsumi S, Neckers L, Verkhivker G, Altieri DC, and Colombo G (2010). Dynamics-Based Discovery of Allosteric Inhibitors: Selection of New Ligands for the C-terminal Domain of Hsp90. Journal of Chemical Theory and Computation 6, 2978–2989. 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sattin S, Tao J, Vettoretti G, Moroni E, Pennati M, Lopergolo A, Morelli L, Bugatti A, Zuehlke A, Moses M, et al. (2015). Activation of Hsp90 Enzymatic Activity and Conformational Dynamics through Rationally Designed Allosteric Ligands. Chemistry 21, 13598–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rehn A, Moroni E, Zierer BK, Tippel F, Morra G, John C, Richter K, Colombo G, and Buchner J (2016). Allosteric Regulation Points Control the Conformational Dynamics of the Molecular Chaperone Hsp90. J. Mol. Biol. 428, 4559–4571. [DOI] [PubMed] [Google Scholar]
- 70.Sanchez-Martin C, Moroni E, Ferraro M, Laquatra C, Cannino G, Masgras I, Negro A, Quadrelli P, Rasola A, and Colombo G (2020). Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone TRAP1. Cell Reports 31, 107531. [DOI] [PubMed] [Google Scholar]
- 71.Wang T, Rodina A, Dunphy MP, Corben A, Modi S, Guzman ML, Gewirth DT, and Chiosis G (2019). Chaperome heterogeneity and its implications for cancer study and treatment. Journal of Biological Chemistry 294, 2162–2179. 10.1074/jbc.REV118.002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Retzlaff M, Hagn F, Mitschke L, Hessling M, Gugel F, Kessler H, Richter K, and Buchner J (2010). Asymmetric activation of the hsp90 dimer by its cochaperone Aha1. Mol. Cell. 37, 344–354. [DOI] [PubMed] [Google Scholar]
- 73.Li J, and Buchner J (2013). Structure, function and regulation of the Hsp90 machinery. Biomed. J. 36, 106–117. [DOI] [PubMed] [Google Scholar]
- 74.Mader SL, Lopez A, Lawatscheck J, Luo Q, Rutz DA, Gamiz-Hernandez AP, Sattler M, Buchner J, and Kaila VRI (2020). Conformational dynamics modulate the catalytic activity of the molecular chaperone Hsp90. Nature Communications 11, 1410. 10.1038/s41467-020-15050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y, Elnatan D, Sun M, Myasnikov AG, and Agard DA (2020). Cryo-EM reveals the dynamic interplay between mitochondrial Hsp90 and SdhB folding intermediates. bioRxiv, 2020.2010.2006.327627. 10.1101/2020.10.06.327627. [DOI] [Google Scholar]
- 76.Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL, and Chiosis G (2018). Adapting to stress — chaperome networks in cancer. Nature Reviews Cancer 18, 562–575. 10.1038/s41568-018-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ratna A, Lim A, Li Z, Argemi J, Bataller R, Chiosis G, and Mandrekar P (2021). Myeloid Endoplasmic Reticulum Resident Chaperone GP96 Facilitates Inflammation and Steatosis in Alcohol-Associated Liver Disease. Hepatology Communications 5, 1165–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ginsberg SD, Joshi S, Sharma S, Guzman G, Wang T, Arancio O, and Chiosis G (2021). The penalty of stress - Epichaperomes negatively reshaping the brain in neurodegenerative disorders. Journal of Neurochemistry 159, 958–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, Taldone T, Pullen L, Zierer BK, Lee MJ, et al. (2013). Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. . Oncotarget 4, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Truman AW, Bourboulia D, and Mollapour M (2021). Decrypting the chaperone code. Journal of Biological Chemistry 296. 10.1016/j.jbc.2021.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pugh KW, Alnaed M, Brackett CM, and Blagg BSJ (2022). The biology and inhibition of glucose-regulated protein 94/gp96. Medicinal Research Reviews 42, 2007–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clevenger RC, and Blagg BSJ (2004). Design, Synthesis, and Evaluation of a Radicicol and Geldanamycin Chimera, Radamide. Organic Letters 6, 4459–4462. 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 83.Moulick K, Clement CC, Aguirre J, Kim J, Kang Y, Felts S, and Chiosis G (2006). Synthesis of a red-shifted fluorescence polarization probe for Hsp90. Bioorg Med Chem Lett 16, 4515–4518. [DOI] [PubMed] [Google Scholar]
- 84.Chang L, Lee S-Y, Leonczak P, Rozenski J, De Jonghe S, Hanck T, Müller CE, and Herdewijn P (2014). Imidazopyridine- and Purine-Thioacetamide Derivatives: Potent Inhibitors of Nucleotide Pyrophosphatase/Phosphodiesterase 1 (NPP1). Journal of Medicinal Chemistry 57, 10080–10100. 10.1021/jm501434y. [DOI] [PubMed] [Google Scholar]
- 85.He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, Wu N, Immormino RM, Gewirth DT, and Chiosis G (2006). Identification of Potent Water Soluble Purine-Scaffold Inhibitors of the Heat Shock Protein 90. Journal of Medicinal Chemistry 49, 381–390. 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 86.Sali A, and Blundell TL (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779. [DOI] [PubMed] [Google Scholar]
- 87.Case DA, Cerutti DS, Cheatham TEI, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, et al. (2018). AMBER 2018. University of California, San Francisco. [Google Scholar]
- 88.Søndergaard CR, Olsson MHM, Rostkowski M, and Jensen JH (2011). Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. Journal of Chemical Theory and Computation 7, 2284–2295. 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 89.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, and Woods RJ (2008). GLYCAM06: A generalizable biomolecular force field. Carbohydrates. Journal of Computational Chemistry 29, 622–655. 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meagher KL, Redman LT, and Carlson HA (2003). Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry 24, 1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 92.Lee C, Yang W, and Parr RG (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 93.Bayly CI, Cieplak P, Cornell W, and Kollman PA (1993). A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. The Journal of Physical Chemistry 97, 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- 94.Jorgensen WL, Chandrasekhar J, Madura J, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- 95.Joung IS, and Cheatham TE (2008). Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. The Journal of Physical Chemistry B 112, 9020–9041. 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, and Walker RC (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. Journal of Chemical Theory and Computation 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 97.Loncharich RJ, Brooks BR, and Pastor RW (1992). Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 32, 523–535. [DOI] [PubMed] [Google Scholar]
- 98.Miyamoto S, and Kollman PA (1992). SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comp. Chem. 13, 952–962. [Google Scholar]
- 99.Berendsen HJC, Postma JPM, van Gunsteren WF, Di Nola A, and Haak JR (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- 100.Darden T, York D, and Pedersen L (1993). Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 98. [Google Scholar]
- 101.Roe DR, and Cheatham III TE (2018). Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. Journal of Computational Chemistry 39, 2110–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Humphrey W, Dalke A, and Schulten K, , . (1996). VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
1. The input files, topologies and restart files are provided in a Zenodo link reported in the Key Resource Table. 2. Code used in this work is available from https://github.com/colombolab. 3. Any additional information required to reanalyze the data reported in this work is available at the link in the Key resource table.