

Abstract
Objective:
To determine macrophage-specific alterations in epigenetic enzyme function contributing to development of abdominal aortic aneurysms (AAAs).
Summary Background Data:
AAA are a life-threatening disease, characterized by pathological vascular remodeling driven by an imbalance of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). Identifying mechanisms regulating macrophage-mediated extracellular matrix degradation is of critical importance to developing novel therapies.
Methods:
The role of SETDB2 in AAA formation was examined in human aortic tissue samples by single-cell RNA sequencing and in a myeloid-specific SETDB2 deficient murine model induced by challenging mice with a combination of a high-fat diet and angiotensin II.
Results:
Single-cell RNA sequencing of human AAA tissues, identified SETDB2 was upregulated in aortic monocyte/macrophages and murine AAA models compared to controls. Mechanistically, interferon-β regulates Setdb2 expression via JAK/STAT signaling which trimethylates histone 3 lysine 9 (H3K9) on the TIMP1–3 gene promoters thereby suppressing Timp1–3 transcription and leading to unregulated MMP activity. Macrophage-specific knockout of SETDB2 (Setdb2f/fLyz2Cre+) protected mice from AAA formation with suppression of vascular inflammation, macrophage infiltration, and elastin fragmentation. Genetic depletion of SETDB2 prevented AAA development due to removal of the repressive H3K9-trimethylation mark on the Timp1–3 gene promoter resulting in increased TIMP expression, decreased protease activity, and preserved aortic architecture. Lastly, inhibition of the JAK/STAT pathway with an FDA approved inhibitor, Tofacitinib, limited Setdb2 expression in aortic macrophages.
Conclusions:
These findings identify SETDB2 as a critical regulator of macrophage-mediated protease activity in AAAs and identify SETDB2 as a mechanistic target for the management of AAAs.
Keywords: Inflammation, Aneurysm, Vascular Biology
MINI ABSTRACT
Using single-cell RNA sequencing and murine models we characterize the role of the epigenetic enzyme SETDB2 in abdominal aortic aneurysm development. SETDB2 trimethylates the TIMP1–3 promoters suppressing transcription and leading to unregulated MMP activity. Genetic depletion of SETDB2 prevents AAA development and identifies SETDB2 as a therapeutic target for AAAs.
INTRODUCTION
Abdominal aortic aneurysms (AAA) are a common vascular disease that can progress to aortic rupture which has a mortality of over 80% 1. Recent studies have suggested that AAAs have a reported incidence of 1.5 to 2 per 1,000 people per year in the United States 2. Despite advances in the surgical and endovascular management of AAAs, to date there are no proven pharmacological interventions that slow AAA growth or prevent rupture 3. Cumulative efforts to understand mechanisms that contribute to AAA dilation have consistently demonstrated inflammation and extracellular matrix degradation, two processes primarily controlled by aortic macrophages, as critical drivers of aneurysmal development 4–7. Extracellular matrix (ECM) degradation of aortic tissue is primarily mediated by the matrix metalloproteinase (MMP) family of proteolytic enzymes. Under inflammatory conditions, infiltrating macrophages secrete pro-MMPs; where cleavage of the pro-MMP subunit activates the MMPs, causing ECM degradation. MMP activity may be naturally suppressed by tissue inhibitors of MMPs (TIMPs), which comprise a family of 4 protease inhibitors: TIMP-1, TIMP-2, TIMP-3, and TIMP-4 8. An improper balance between MMPs and TIMPs shifts the equilibrium towards matrix degradation during AAA development. Studies have shown that genetic and pharmacological approaches to mitigate MMP proteolytic activity can protect against AAA development in several murine models 9–11. Despite the critical importance of MMPs in AAA development and expansion, the molecular mechanisms that program and sustain macrophage TIMP production and MMP activity in AAA disease have not been identified.
Macrophage functional plasticity and gene expression is tightly regulated by transcriptional reprogramming, which is achieved by modifications of chromatin accessibility dictated by the epigenetic landscape. Accumulating evidence suggests that epigenetic regulation of gene expression, via mechanisms such as histone modification, is a major influence on innate immune cell phenotypes in both normal and pathologic conditions 12–14. Histone methylation is important as it maintains active or suppressed gene expression, depending on the methylation site, and thereby controls downstream protein expression patterns. Prior work by our lab and others has identified that epigenetic modifications regulate immune-mediator expression in macrophages both in vitro and in vivo 14,15. The chromatin modifying enzyme SET Domain Bifurcated Histone Lysine Methyltransferase 2 (SETDB2), a histone methyltransferase acts to methylate lysine residues on histones in a sequence-specific fashion. SETDB2 specifically trimethylates lysine 9 (K9) on histone 3 (H3) (H3K9me3) and keeps chromatin in a conformation where the promoter is not accessible for transcription factor binding, effectively silencing gene transcription 16. SETDB2 has been previously reported to be involved in innate and adaptive immunity, proinflammatory responses, and T cell differentiation through modulation of the expression of NF-κB target genes and type I IFN responses 15,16. From a cardiovascular standpoint, SETDB2 has been shown to be an important regulator of macrophage inflammation and atherosclerosis progression 17,18. Independently, we have shown that SETDB2 is instrumental to macrophage polarization and gene expression in diabetic wound healing 17. Despite the importance of histone modification on macrophage function, there remains a paucity of data on epigenetic-based mechanisms that regulate AAA formation 19,20. Additionally, little is known about the upstream mechanisms that regulate SETDB2 in in vivo aortic macrophages.
Herein, we provide experimental evidence that SETDB2 directs macrophage-mediated TIMP expression and aortic aneurysm formation in human tissue samples and two well-established murine AAA models (elastase-induced AAAs and angiotensin (Ang) II-induced AAAs). Mechanistically during AAA development, macrophage-specific SETDB2 reduces TIMP expression by increasing the repressive histone methylation mark H3K9me3 on TIMP gene promoters. Loss of TIMP expression leads to unregulated MMP activity, aortic wall degradation, and aneurysmal formation. Mechanistically, SETDB2 expression is regulated by interferon (IFN) β via the janus kinase (JAK) / signal transducer and activator of transcription 3 (STAT3) pathway as genetic depletion of the interferon receptor reduced SETDB2 expression and prevented AAA development. Further targeted inhibition of SETDB2 via myeloid-specific genetic depletion of SETDB2 (Setdb2f/fLyz2Cre+) augmented TIMP expression as well as decreased MMP activity and prevented AAA expansion. Overall, these findings identify SETDB2 as a critical regulator of monocyte/macrophage-mediated TIMP expression and MMP activity during aneurysmal progression and demonstrate translational implications as cell-targeted manipulation of this pathway may reduce AAA development.
MATERIALS AND METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Frank Davis (davisfr@umich.edu) and Katherine Gallagher (kgallag@med.umich.edu).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were maintained in the University of Michigan pathogen-free animal facility, and all protocols were approved by and in accordance with the guidelines established by the Institutional Animal Care and Use Committee (UCUCA). Male mice were used for AAA experiments as detailed in the American Heart Association Council statement.21 Only male mice were used for these studies as female mice do not adequately develop AAAs 21. Mouse strains include: C57BL/6J mice maintained on a normal diet (ND) (13.5% kcal fat; LabDiet 5001) were purchased at 8–10 weeks from The Jackson Laboratory (Bar Harbor, ME). Setdb2f/f were created as previously published by our lab 15. Setdb2f/f mice were then bred with B6.129P2-Lyz2tm1(Cre)Ifo/J (Lyz2Cre) mice from the Jackson Laboratory to obtain mice deficient in Setdb2 in monocytes, macrophages and granulocytes 22. Lyz2cre was chosen for the cell specific line as this affects myelomonocytic cells. Currently, there is no Cre-transgenic line that is perfectly specific for macrophages 23. Interferon-α/β receptor−/− (Ifnar−/−), Stat3f/fLyz2Cre-, and Stat3f/fLyz2Cre+ were obtained from Christiane Wobus, Ph.D. (University of Michigan, Ann Arbor, MI) and maintained in breeding pairs at the Unit of laboratory animal medicine facilities. Animals were housed in a barrier facility on a light:dark cycle of 14 : 10 hours (ambient temperature of 22°C) with free access to water, food (Lab diet 5001), and bedding (Andersons Lab Bedding Bed o’Cobs combo). Animals underwent all procedures at 8–10 weeks of age.
Production and Injection of Adeno-associated Viral (AAV) Vectors
Adeno-associated virus (AAV) vectors (serotype 8) were produced by the Viral Vector Core at the University of Pennsylvania (https://gtp.med.upenn.edu/core-laboratories-public/vector-core). These AAV vectors contained inserts expressing mouse PCSK9D377Y mutation (equivalent to human PCSK9D374Y gain-of-function mutation). Empty AAV vector (null AAV) was used as control. AAV vectors were diluted in sterile PBS (200 μl per mouse) and injected intraperitoneally as reported previously 24,25. Mice inclusion and randomization were conducted as previously specified. Further, mice had a predefined exclusion from the data analysis if plasma total cholesterol concentrations were < 250 mg/dl 3 weeks and < 500 mg/dl 6 weeks after PCSK9D377Y.AAV injection.24 Briefly, mice received injections of AAVs containing either a null insert or a mouse PCKS9 insert expressing D377Y mutation. Immediately after AAV injections, normolipidemic mice were fed a diet containing saturated fat (milk fat 21% wt/wt; Diet #TD.88137, Envigo) for 2 weeks at which time they underwent implantation of mini osmotic pumps as described below. Body weights were determined prior to experimentation.
Osmotic Mini Pump Implantation and AngII Infusion
To induce AAAs, 8–10 week male C57BL/6J mice were injected with AAV and started on a saturated fat diet as detailed above. Following two weeks of saturated fat diet feeding, mice were randomized to receive mini osmotic pumps (Model 2004; Alzet) containing AngII (1,000 ng/min per kilogram, Cat# H-1706; Bachem) or saline were implanted subcutaneously in the neck region of anesthetized mice following a protocol described previously 26. Briefly, mice were anesthetized in a closed chamber with isoflurane (3%) in oxygen for 2 to 5 minutes until immobile. Each mouse was then removed and taped on a heated (35 to 37°C) procedure board with isoflurane (1.0% to 1.5%) administered via nosecone during minor surgery. Pumps were implanted subcutaneously on the right flank of each mouse which provided AngII or saline infusion for 28 days. Incisions were closed with surgical staples and post-operative analgesia (buprenophine, 0.05 mg/kg/12 h, intraperitoneal) was administered. All AngII groups experienced equivalent elevations of blood pressure. Animal experiments were conducted following the NIH guidelines and were approved by the IACUC of the University of Michigan.
Elastase treatment model of aneurysm formation
A murine elastase treatment model of AAA formation was used as described by Laser et al. 27. In brief, the infrarenal aorta was treated topically with 30 μl elastase reconstituted with normal saline (5 U/mg protein) or 30 μl heat-inactivated elastase (at 90°C for 30 min) as a control group. The topical application was accomplished by dropping the elastase on the anterior aorta from a 2 cm height for 5 min. Video micrometric measurements of aortic diameters were made in situ before perfusion, after perfusion, and before harvesting the aorta on day 14. Maximum infrarenal aortic diameter and ratio of treated vs. untreated section of the aorta was calculated.
Quantification of Aortic Pathologies
For in vivo imaging of the abdominal aorta in mice, 2-dimensional (B-mode) ultrasound images were obtained 27 days after the implantation of osmotic pumps using a VisualSonics Vevo2100 imaging system with a mechanical transducer (MS400) from the University of Michigan Frankel Center for Physiology. The maximum diameter observer in each mouse in the abdominal aortic region was documented. Two independent investigators measured aortic diameters at systole with no significant interobserver or intraobserver variability.At the completion of each murine aneurysm experiment (day 28), mice were deep anesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg). At termination after blood collection, right atrium was cut open, and saline was perfused through the left ventricle to remove blood in aortas. Subsequently, aortas were dissected and placed in either RNA later or 10% neutrally buffered formalin overnight at room temperature. After fixation, periaortic adventitia were removed thoroughly. Maximal outer diameters of suprarenal aortas were measured ex vivo as a parameter for abdominal aortic aneurysm (AAA) quantification using ImageJ software (National Institutes of Health, Bethesda, MD).
Necropsy was performed for mice that died during AngII infusion. Aortic rupture was defined as observation of blood clots in either the thoracic cavity (thoracic aortic rupture) or retroperitoneal cavity (abdominal aortic rupture). There was no difference in early rupture between treatment groups and genotypes.
Histology/Immunofluorescence
For aortic histology, aortas were excised as above and fixed in formalin (10%) overnight before embedding in paraffin. Sections (5 μm) were stained with Verhoeff Van Gieson stain for elastin (Sigma HT25A). Images were captured using Olympus BX43 microscope and Olympus cell Sens Dimension software. Elastin fragmentation was defined as the presence of free ends in what seems to be an otherwise continuous elastin fiber. Elastin fragmentation was quantified by determining the number of elastin breaks in the whole aortic section at 40X magnification and then averaging the number of breaks across three serial sections of the aorta. Two independent observers blinded to mouse treatment group were used.
For immunohistochemistry, formalin-fixed, paraffin-embedded tissue slides obtained from patients with AAA and aortic healthy control were heated for 30 min at 60°C, deparaffinized, and rehydrated. Slides were placed in Ph9 antigen retrieval buffer and heated at 95C for 20 minutes in a hot water bath. After cooling, slides were treated with 3% H2O2 (5 minutes) and blocked using 10% goat serum (30 minutes). Overnight incubation (4C) was then performed using first antibody at a working concentration. Slides were then washed, treated with secondary antibody, peroxidase (30 minutes) and diaminobenzidine substrate. Antibody used was human anti-SETDB2 (Applied Biosystems, RRID:AB_10982748). Antibody specicifty was verified in our publication.28 Images were quantified ImageScope software and Image J at 20 X magnification.
Peripheral Macrophage Isolation and Magnetic-Activated Cell Sorting
Peripheral circulating macrophages were isolated using magnetic activated cell sorting. Briefly, single cell suspensions were incubated with fluorescein isothiocyanate–labeled anti-CD3, anti-CD19, anti-NK1.1, and anti-Ly6G (BioLegend) followed by EasySep™ Mouse Streptavidin RapidSpheres (Stem Cell Technologies 19860). Flow-through was then incubated with EasySep™ Mouse CD11b Positive Selection Kit (Stem Cell Technologies18970) to isolate the non-neutrophil, non-natural killer (NK) cells, non-lymphocyte, CD11b+ cells. Of note we did not negatively select for CD11c+ dendritic cells. When indicated, peripheral macrophages were stimulated with/without IFNβ (100U/ml) (PBL Assay Science, catalog 12400–01). Cells were saved in Trizol (Invitrogen) for quantitative RT-PCR analyses or were processed for chromatin immunoprecipitation as described below.
Cell Culture
Bone marrow derived macrophages (BMDMs) were collected by flushing mouse femurs and tibias at day 28 following angiotensin II or saline infusion with RPMI. BMDMs were cultured as previously detailed 29. On day 6, the cells were replated, and after resting for 24 h, they were incubated with or without IFNβ (10U/ml) (PBL Assay Science, catalog 12400–01). For JAK1, 3 inhibition, cells were treated with 50nM tofacitinib (Cayman Chemicals) at the time of stimulation with IFNβ. After appropriate time for stimulation, cells were fixed in paraformaldehyde for ChIP analysis or placed in Trizol (Invitrogen) for RNA analysis.
ChIP Assay
Chromatin immunoprecipitation (ChIP) assay was performed as described previously.30 Briefly, cells fixed in paraformaldehyde were lysed and sonicated to generate 100–300bp fragments. To immunoprecipitated samples were incubated in anti-H3K27trimethyl antibody (Active Motif 39155) or isotype control (rabbit polyclonal IgG) (Millipore) in parallel samples overnight followed by addition of protein A Sepharose beads (Thermo-Fisher). Bound DNA was eluted and purified using phenol:chloroform:isoamyl alcohol extraction and ethanol precipitation. Primers were designed using the Ensembl genome browser to search the Timp1, Timp2, and Timp3 promoter for NFκB within the promoter region and then NCBI Primer-BLAST was used to design primers that flank this site. Data are representative of 2–3 independent experiments. Primer sequences are available in Supplemental Table I.
RNA Analysis
Total RNA extraction was performed using Trizol (Invitrogen) or Trizol LS (for human samples) according to manufacturer’s instructions. RNA was then reversed transcribed to cDNA using iScript (Biorad). PCR was performed with 2X Taqman PCR mix using the 7500 Real-Time PCR System. Primers for Timp1 (Mm0134136_m1), Timp2 (Mm00441825_m1), Timp3 (Mm00441826_m1), murine Setdb2 (Mm01318752_m1), and human SETDB2 (HS01126262_m1)18S was used as the internal control. Data were then analyzed relative to 18s ribosomal RNA using the 2-ΔΔCT methods with values standardized to the control cohort. All samples were assayed in triplicate. The threshold cycle values were used to plot a standard curve. Data are representative of 2–3 independent experiments were compiled in Microsoft Excel and presented using Prism software (GraphPad).
MMP Activity Detection
Total MMP activity was determined in protein extracts from suprarenal aortas with an MMP Assay Kit (catalog No. 112146; Abcam). Protein samples were incubated with 4-aminophenylmercuric acetate for 3 hours at 37°C. Then, the MMP Green substrate was added, and the fluorescence signal was measured in a fluorescent microplate reader (Gemini XPS Spectrofluorometer; Molecular Devices, Sunnyvale, CA).
Gelatin zymography
Gelatin zymography was performed as described previously with slight modification. Whole aortic tissue was homogenized and dissolved in RIPA buffer. 4.5 μg of aortic tissue homogenate was then dissolved in zymogram sample buffer (BIO-RED, # 161–0764) and loaded onto zymography gel (EC617S) to run on SDS running buffer for 90 minutes. Gels were then renatured in Zymogram Renaturing Buffer (Invitrogen, # LC2670) for 30 minutes at room temperature and developed in Zymogram Developing Buffer (Invitrogen, # LC2671) for 30 min at 37C incubator. Fresh developing buffer was then added for 24 hours. The gel was then washed with ddH20 and stained with Simply-Blue Stain (Invitrogen, # LC6060) for 20 minutes following which it was imaged.
In situ zymography
In situ zymography was performed as described previously 31. Briefly, 8–10 micron sections of frozen tissue samples were cut in optimal cutting temperature compound (OCT). Sections were incubated overnight in the dark at room temperature with a 20 µg/mL solution of DQ-gelatin (D-12054 Molecular Probes) diluted in zymogram development buffer (161–0766 BioRad), in the presence or absence of 20 mM EDTA. After 24 hours, slides were washed in PBS and mounted in Vectashield Hardset Mounting Medium with DAPI (VECTASHIELD). Gelatinase activity was visualized as green fluorescence.
Human Tissue
Full-thickness aortic wall tissue specimens were collected from the infrarenal abdominal aorta from patients undergoing open aortic aneurysm repair (n=19) or open aorto-bifemoral bypass (n=6). The aneurysmal samples were taken from the midportion of the aneurysmal sac. For control samples, aortic tissue was isolated from patients with atherosclerotic occlusive disease but no history of aneurysmal disease at the time of open aorto-bifemoral bypass. Patient medical comorbidities are represented in Supplemental Table II and III. All aortic samples were processed for both histology and protein/RNA analyses. For histology, human aortas were placed in formalin (10%) for 24 hrs prior to paraffin embedding. For protein/RNA analysis, specimens were stored at −80oC for future protein and RNA analyses. For single cell RNA-sequencing (scRNA-seq) a second cohort of samples were retrieved from the infrarenal abdominal aorta of patients undergoing open aortic aneurysm repair (n=4) or open aorto-bifemoral bypass (n=2). Patient medical comorbidities can be seen in Supplemental Table II. These samples were immediately processed as described below. This study was approved by the University of Michigan Institutional Review Board HUM00098915.
Single Cell RNA-Seq and Bioinformatics Analysis
Generation of single cell suspensions for scRNA-seq was performed as follows: Aortic tissue from aneurysmal and non-aneurysmal controls was harvested at the time of surgical intervention. Briefly samples were minced, digested in 0.2% Collagenase II (Life Technologies) and 0.2% Collagenase V (Sigma) in plain medium for 1 hour at 37°C, and strained through a 70μM mesh. The scRNA-seq samples were analyzed by the University of Michigan Advanced Genomics Core on the 10X Chromium system. Libraries were sequencing on the Illumina NovaSeq 6000 sequencer to generate 151-bp paired end reads. Data processing including quality control, read alignment and gene quantification were conducted using the 10X Cell Ranger software. Clustered cells were mapped to corresponding cell types by matching cell cluster gene signatures with putative cell-type specific markers. Data Availability Statement: The single cell RNA-sequencing data for this paper is available at GEO accession number of GSE166676 as previously described 32.
Statistical Analysis
Data were analyzed using GraphPad Prism software version 6. Data are represented as means ±SEM. Shapiro-Wilk test was used to determine normality of data, Brown-Forsythe or F test to determine variances, ROUT test was used to identify outliers in given datasets, and then a parametric or nonparametric test was performed accordingly. Parametric statistical analysis was performed using unpaired Student’s t test (2 tailed) between 2 groups and 1-way ANOVA followed by post hoc analysis for analysis of differences between >2 groups. Nonparametric statistical analysis was performed using Mann-Whitney U test. P values <0.05 were considered significant.
RESULTS
TIMP Expression in AAA Development is Regulated by SETDB2
Since extracellular matrix degradation due to an imbalance of MMP activity is a pathologic hallmark of aortic dilation 5,33, we examined the expression of endogenous MMP inhibitors, TIMPs, in murine macrophages using an established angiotensin (Ang) II-induced murine model 24. Specifically, male C57BL/6J mice were injected intraperitoneally with a single dose of an AAV vector expressing the mouse D377Y gain-of-function proprotein convertase subtilisin/kexin type 9 (PCSK9), which resulted in sustained hypercholesterolemia as described previously 24. Following this, mice were fed a saturated fat enrich diet for 6 weeks and received either saline or AngII infusion (1000 ng/min/kg) for the last 4 weeks. The use of a saturated fat diet and AngII infusion to induce AAAs recapitulates important clinical risk factors including dyslipidemia and hypertension. We then isolated in vivo infiltrating monocyte/macrophages (CD11b+[CD3−CD19−Nk1.1−Ly6G−]) by cell sorting on day 28 from mice infused with AngII or saline given that the predominance of inflammatory macrophages witin the aortic wall during AAA develop arise from infiltrating monocyte/macrophages rather than resident tissue macrophages that populate the aorta during development.34 Timp1, Timp2, and Timp3 expression were significantly reduced in in vivo macrophages in AngII-induced AAAs in comparison to controls (Figure 1A and Supplemental Figure 1A, B). This was also associated with an increase in extracellular matrix degradation and elastin fragmentation in AngII-induced AAAs (Figure 1B).
Figure 1. SETDB2 is Increased Murine AAA Macrophages and Increases the Repressive H3K9 Trimethylation on Timp Gene Promoters.
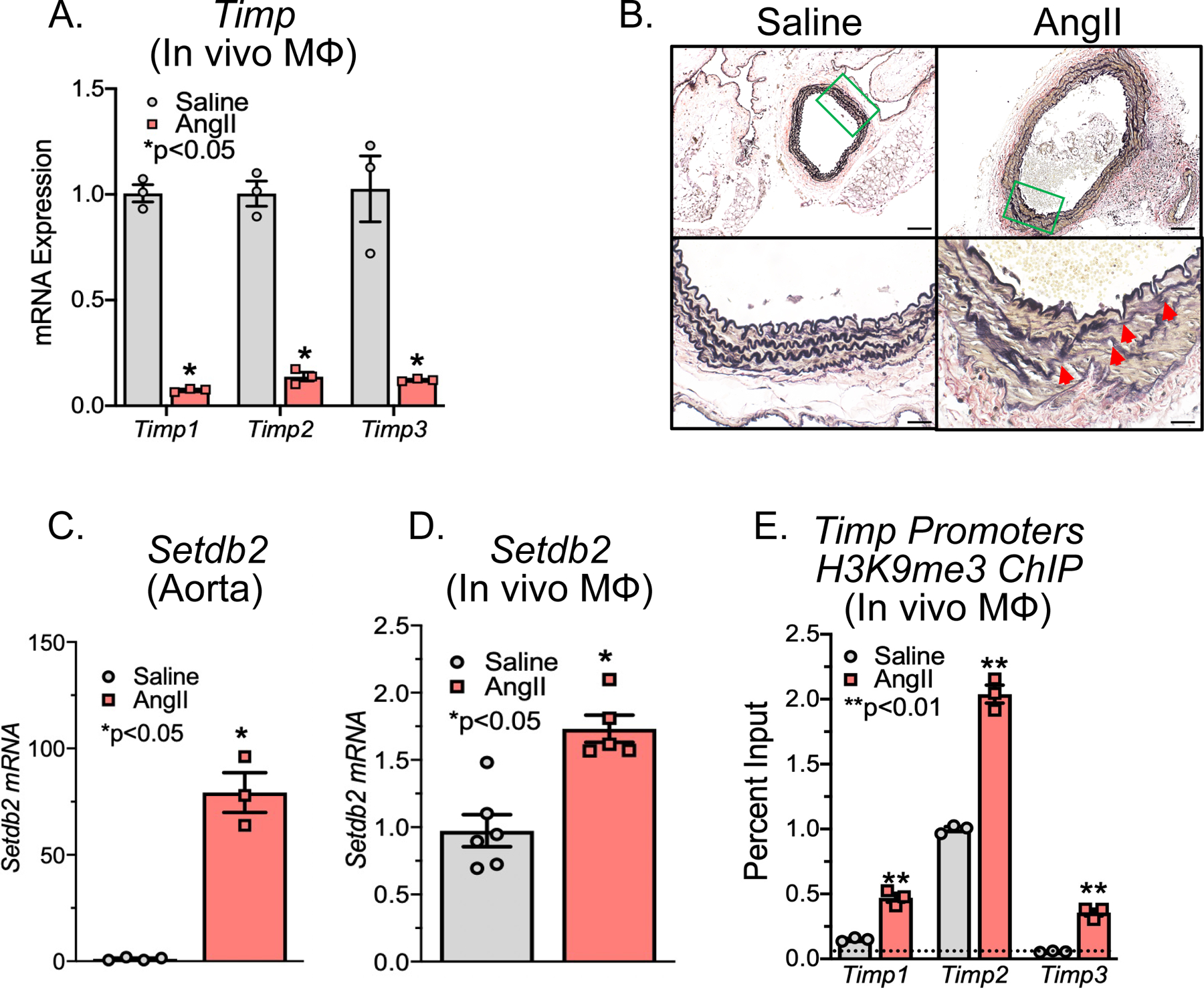
A. Male C57BL/6J mice were injected intraperitoneally with an AAV containing mouse PCSK9D377Y and fed saturated fat diet for 6 wk. Mice were infused with saline or AngII (1,000 ng/min/kg) for 4 weeks. Quantitative PCR analysis of Timp1, Timp2, and Timp3 isolated from in vivo macrophages (MΦ) (CD11b+[CD3−CD19−Nk1.1−Ly6G−]) of mice exposed to saline or AngII for 28 days (n =3/group run in triplicate). *p<0.05, **p<0.01 for Welch’s t-test. B. Representative Verhoeff–van Gieson elastin staining of abdominal aortic sections at 10X and 40X showing disrupted aortic structure in AngII mice compared with saline control mice; scale bar is 50 μm or 10 μm in Verhoeff–van Gieson stain; arrows represent elastin fragmentation. C, D. Quantitative PCR analysis of Setdb2 isolated from aortas or MΦs (CD11b+[CD3−CD19−Nk1.1−Ly6G−]) in mice infused with either saline or Ang II for 28 days (n = 3–4/group run in triplicate). *p<0.05 for Mann-Whitney U test. E. ChIP analysis for H3K9me3 at Timp1, Timp2, and Timp3 promoter was performed (n=3/group run in triplicate). For all ChIP experiments, isotype-matched IgG was run in parallel. Dotted line represents isotype-matched control. **p<0.01 for Mann-Whitney U test.
Given the decreased gene expression of multiple Timps in murine aneurysmal aortic tissue, we examined expression of Setdb2, a methyltransferase associated with gene repression, in both the murine AngII-induced and elastase-induced AAA model. This demonstrated Setdb2 expression was increased in both aneurysmal tissue and in vivo macrophages in the AngII-induced and elastase-induced AAA murine model compared to nonaneurysmal controls (Figure 1C, D and Supplemental Figure 1C). SETDB2 has been shown to act via H3K9me3-mediated mechanism on gene promoters. As a methytransferase, increased SETDB2 methylates the H3K9 site which renders gene promoters inaccessible to transcription factor binding, resulting in gene suppression. To examine the transcriptional effects of SETDB2 in aortic tissue, chromatin immunoprecipitation (ChIP) was performed on aortic tissue from AngII-induced AAAs compared to those from saline-infused controls. Utilizing primers for the NFκB binding sites on the Timp1, Timp2, and Timp3 promoters, we identified that H3K9me3 was increased at the NFκB binding sites in the AngII-induced AAA aortic tissue (Figure 1E). Collectively, these results suggested that upregulation of SETDB2 may play a causative role for aberrant Timp expression and protease activity in murine AAA development.
Human Aortic Single Cell Transcription Profiling Reveals Elevated SETDB2 and Inflammatory Pathway Expression in Myeloid Cells
In order to translate our murine findings to human disease and to further characterize the structural and infiltrating cells within the aortic wall, we examined the presence of SETDB2 in human aortic tissue samples isolated from infrarenal AAAs and atherosclerotic aorto-iliac occlusive controls. We found expression of SETDB2 was markedly increased in AAA tissue samples compared with atherosclerotic control aortic tissues (Figure 2A, Supplemental Table II). Immunostaining showed that SETDB2 was highly expressed in the aortic media and adventitia of diseased aortas (Figure 2B). Next, to further characterize the impact of SETDB2 expression in structural and infiltrating cells within the aortic wall we analyzed single-cell RNA sequencing (scRNA-seq) analysis on a cohort of patients undergoing abdominal aortic surgery (Supplemental Table III) 32. Cluster analysis using the uniform manifold approximation and projection (UMAP) technique identified 21 different cell clusters present in the aortic tissue. We attributed clusters to their putative identities and hierarchical similarities by differentially expressed gene signatures (Figure 2C). Given our murine findings, we sought to determine if SETDB2 contributes to the gene expression and pathway alterations in AAA monocyte/macrophage. As such, we screened for alterations in known epigenetic enzymes in AAA macrophages/monocytes and found that SETDB2 expression was markedly elevated in the macrophage and monocyte clusters in human AAA tissue in comparison to control samples (Figure 2D) with minimal expression in smooth muscle cells and T- and B-lymphocyte sub-clusters, confirming our findings from the murine model. On analysis of SETDB2 + macrophages/monocytes compared to SETDB2− macrophages/monocytes from human AAA samples, gene ontology analysis and differential gene expression analysis demonstrated that multiple chromatin and transcription regulatory complexes were elevated in SETDB2+ monocytes in comparison to SETBD2− monocytes. Additionally, pathways instrumental to cell-cell adhesion and the extracellular space as well as gene expression of TIMP1 and TIMP2 were substantially altered in SETDB2+ monocytes further confirming the associated between SETDB2, histone regulation and extracellular matrix organization (Figure 3E and Supplemental Table IV). Taken together, these results suggest human AAAs are characterized by a significant upregulation of the histone methytransferase SETDB2 in monocytes that likely leads to alterations in transcriptional regulation and extracellular protease activity.
Figure 2. Human Aortic Single Cell Transcription Profiling Reveals Elevated SETDB2 and Extracellular Matrix Organization Pathways in Infiltrating Monocyte/Macrophages.
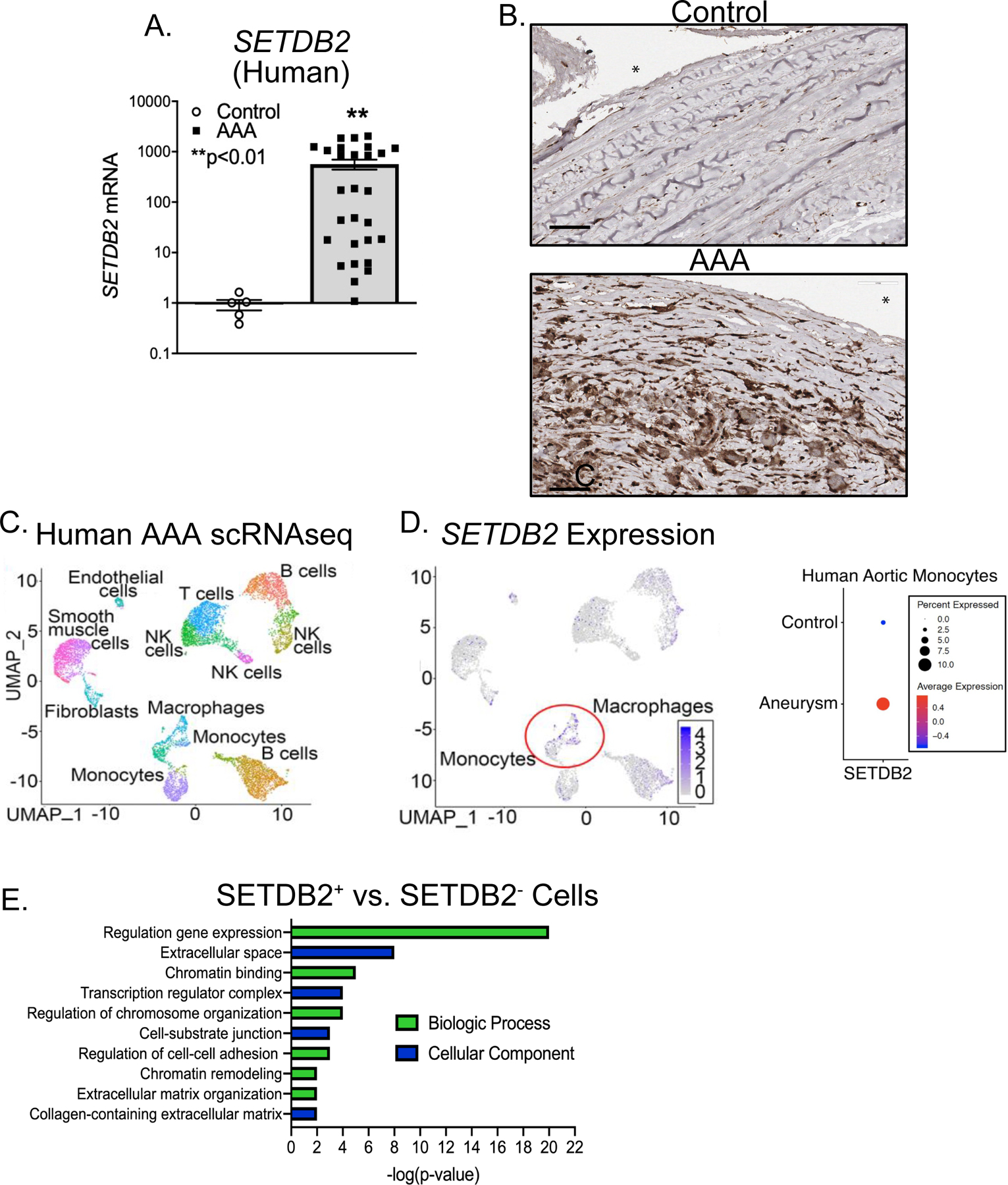
A. Aortic tissue from patients with AAA (n=19) and atherosclerotic controls (n=6) were collected. No statistical differences were found between groups with respect to sex, age, or comorbid conditions. SETDB2 gene expression was measured by qPCR with log scale. *p<0.05 by Welch’s t test replicated twice. B. Immunohistochemistry was performed for SETDB2 in human control and AAA samples. Representative slides are shown at 40X and scale bar is 60 μm. C. Cluster analysis using the uniform manifold approximation and projection (UMAP) technique of single cell sequencing from human AAA (n=4) and nonaneurysmal (n=2) samples revealed 21 distinct cell clusters (representative). D. Feature plots displaying the single cell gene expression of SETDB2 across cell clusters. E. Gene Ontology biological process or cellular component enrichment analysis of differentially expressed genes SETDB2+ vs. SETDB2− cells. The combined score metric corresponds to the P value (two-tailed Fisher’s exact test) multiplied by the Z-score of the deviation from the expected rank, and q values determined by Benjamini–Hochberg correction.
Figure 3. AAA Formation is Inhibited in Macrophage-specific SETDB2-deficient Mice.
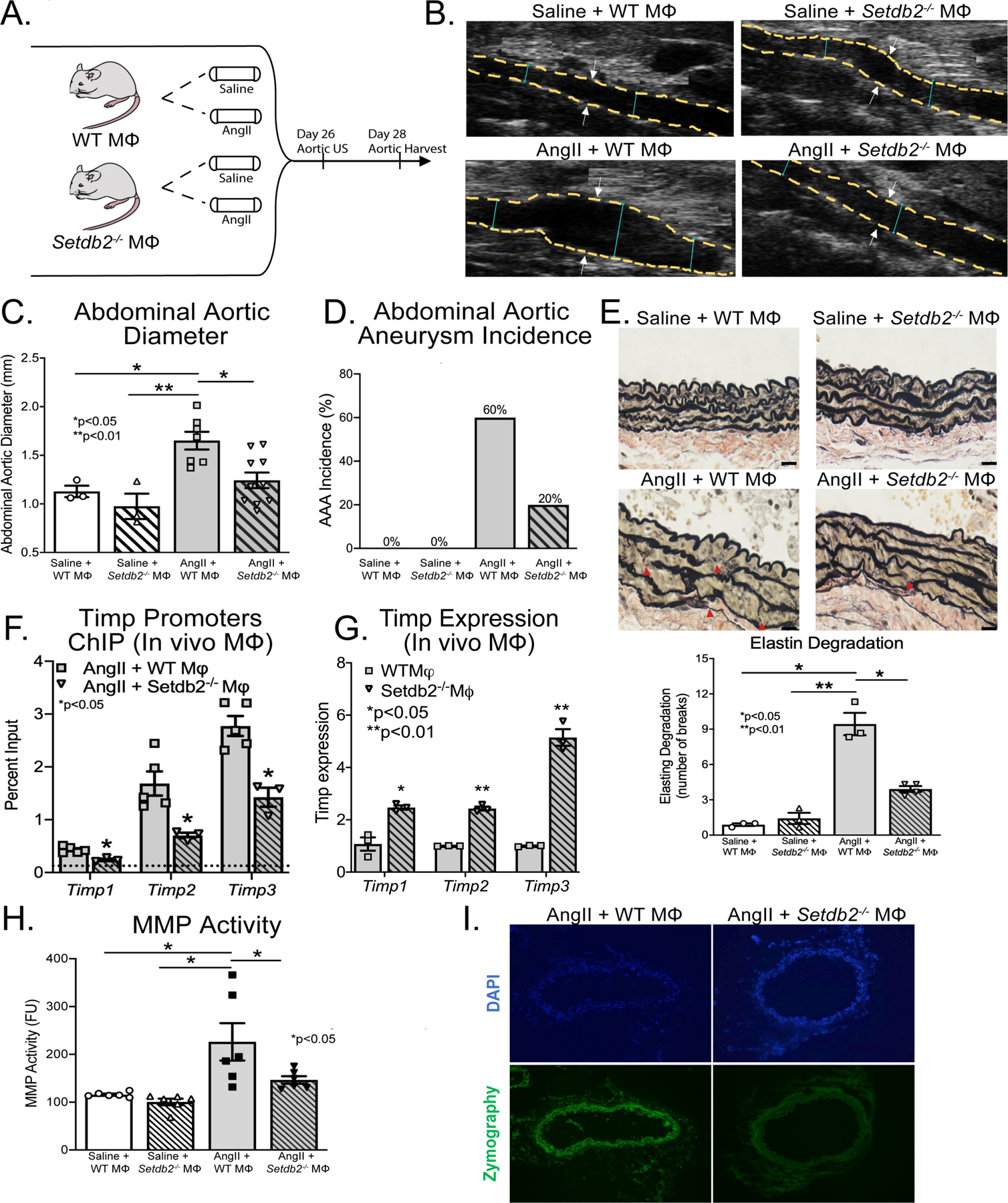
A. Experimental design of macrophage-specific SETDB2-deficiency in murine AAA model. Wild-type (WT M𝛷) mice and mice with macrophage-specific SETDB2 deficiency (Setdb2−/− M𝛷) were fed high fat diet for 6 weeks and infused with saline or AngII infusion (1,000 ng/min/kg) for 4 weeks. B. Representative ultrasound images of the abdominal aorta at day 28 in WT M𝛷 or Setdb2−/− M𝛷 following either saline or AngII infusion. Dotted line represents aortic contour and arrows represent aortic wall diameter. C, D. Maximal abdominal aortic diameter and aneurysm incidence as determined by ultrasound measured by two observers in WT M𝛷 or Setdb2−/− M𝛷 infused with either saline or AngII (n= 6 in saline infused cohorts and 20 in AngII infused cohorts). *p<0.05; **p<0.001 by ANOVA with Newman-Keuls Multiple Comparison test. Data are presented as the mean±SEM. E. Representative Verhoeff–van Gieson elastin staining of abdominal aortic sections showing preserved aortic structure in Setdb2−/− M𝛷 + AngII compared with WT M𝛷 + AngII mice; scale bar is 200 μm in Verhoeff–van Gieson stain; arrows represent elastin fragmentation. Average number of elastin fragmentation per high power field. F. ChIP analysis for H3K9me3 at Timp1, Timp2, and Timp3 promoter was performed on macrophages (CD11b+[CD3−CD19−Nk1.1−Ly6G−]) isolated from AngII + WT M𝛷 and Setdb2−/− M𝛷 at day 28 (n=5 mice/group pooled and run in triplicate). For all ChIP experiments, isotype-matched IgG was run in parallel. Dotted line represents isotype-matched control. **p<0.01 for Mann-Whitney U test. G. Timp1, Timp2, and Timp3 expression were measured by quantitative PCR in in vivo M𝛷s isolated from WT M𝛷 or Setdb2−/− M𝛷 following AngII infused mice on day 28 (n=3/group run in triplicate). *p<0.05, **p<0.01 by Mann-Whitney U Test. H. MMP activity was measured in protein abdominal aorta extracts by fluorometry and measured in fluorometric units. Data represent the mean±SEM from n=6 animals per group. Statistical analysis was performed by 1-way ANOVA (Newman-Keuls post hoc test). *p<0.05. I. MMP activity in abdominal aortic sections by in situ zymography. Representative images are shown with green signal corresponding to active MMPs; blue signal corresponding to DAPI staining for cell nuclei.
Myeloid-specific Deletion of SETDB2 Augments TIMP Expression and Prevents Aortic Wall Degeneration
SETDB2-mediated epigenetic modifications in monocytes/macrophages altered histone regulatory complexes and gene expression, hence, we investigated the translational potential of SETDB2 inhibition in regulating AAA formation and protease activity. As shown in the schematic in Figure 3A, we examined the effects of macrophage-specific genetic deletion of SETDB2 on AAA development in a murine model. To define the cell-specific function of SETDB2 in macrophages during AAA development, we uniquely generated a myeloid-specific Setdb2-deficient mouse. The Setdb2flox/floxLys2Cre+ (Setbd2−/−MΦ) and Setdb2flox/floxLys2Cre−/− (WTMΦ) control mice showed no phenotypic defects at baseline. Setdb2−/−MΦ and WTMΦ mice underwent AngII-induced AAA induction with temporal monitoring of aortic dilation. Setdb2−/−MΦ mice were less susceptible to AngII-induced AAA formation with significantly decreased maximum abdominal aortic diameter and AAA incidence (Figure 3B–D). This was accompanied by a preservation of aortic architecture in the Setbd2−/−MΦ mice who displayed decreased elastin fragmentation compared to WTMΦ following AngII infusion (Figure 3E). There was no difference in aortic rupture or ascending aortic diameter (Supplemental Figure 2).
To determine the transcriptional effects of macrophage-specific SETDB2 deficiency on Timp gene expression in vivo macrophages were sorted from our Setdb2−/−MΦ and WTMΦ mice after 28-days of saline or AngII infusion. We found a significant reduction in H3K9me3 levels on Timp1, Timp2, and Timp3 gene promoters in Setdb2−/−MΦ mice following AngII infusion in comparison to WTMΦ (Figure 3F). This was associated with a substantial increase in Timp1–3 gene expression in vivo macrophages from Setdb2−/−MΦ mice (Figure 3G). To further determine the functional impact of altered Timp1–3 expression we analyzed MMP activity within the aortic wall and observed decreased MMP activity in Setdb2−/−MΦ mice following AngII infusion in comparison to WTMΦ mice that underwent AngII-induced AAA induction (Figure 3H, I and Supplemental Figure 3A). These data identify that SETDB2 is important in myeloid cells for AAA development and that in vivo macrophage Timp gene expression and protease activity is controlled, at least partly, by SETDB2.
IFNβ/JAK/STAT3 Regulate Setdb2 Expression Resulting in Increased H3K9me3 at NFκB Binding Sites on TIMP Gene Promoters in Monocytes/Macrophages
Type 1 interferons (IFN-I) serve as cell signaling molecules that bind to cell surface receptors and ultimately phosphorylate tyrosine kinases inducing specific transcription factors to activate inflammatory genes 35. Although IFN-I has been well-studied in viral disorders and autoimmune diseases, very little is known about the role of IFN-I in both normal and pathologic vascular remodeling 36–38. Prior investigations have demonstrated that IFN-Is are increased in both human tissue samples and murine models of AAAs. Further, downstream IFN-I signaling pathways, janus kinase 1 (JAK1) / signal transducer and activator of transcription 1 (STAT3) pathway, have also been shown to be highly upregulated in human AAA tissue samples 38 and administration of an anti-IFN receptor 1 antibody attenuated AAA development in a murine model36,39. It has recently been suggested in diabetic cutaneous macrophages that transcriptional regulation of SETDB2 by IFNβ and STAT signaling may drive inflammatory responses 28. IFNβ has been recently showed to be correlated with advanced age and aortic aneurysmal disease progression40, however, the mechanisms by which IFNβ and JAK/STAT signaling contribute to SETDB2 regulation in aneurysmal development remain undefined.
Our results support altered levels of IFNβ in the AngII-induced and elastase induced AAAs (Supplemental Figure 1D). To determine if IFNβ stimulation regulated Setdb2 expression in macrophages, we sorted macrophages stimulated them ex vivo with IFNβ (100U) for 6 hours and examined Setdb2 gene expression. In macrophages, Setdb2 was significantly increased following IFNβ stimulation (Figure 4A). To confirm that SETDB2 regulated Timp expression, ChIP analysis was conducted for H3K9me3 on the NFκB binding sites on Timp1–3 gene promoters demonstrating significant increase in repressive H3K9me3 following IFNβ stimulation (Figure 4B). This also corresponded with a significant decrease in Timp1–3 expression (Figure 4C). To confirm that loss of IFNβ signaling decreased macrophage Setdb2 expression, we isolated macrophages from Ifnar−/− mice and Ifnar+/+ controls. Following six-hour IFNꞵ stimulation, Setbd2 was significantly reduced in the Ifnar−/− macrophages compared to matched controls suggesting that IFNβ signaling alters SETDB2 expression in macrophages (Figure 4D). IFN-I signals through the JAK/STAT pathway to promote gene transcription during viral infection, however, the role of IFN-I/JAK/STAT in AAA pathogenesis remains undefined. We thereby isolated macrophages and stimulated them with IFNβ (100U) with or without the JAK1 inhibitor, tofacitinib (50nM). Setdb2 was significantly decreased in macrophages treated with the JAK1 inhibitor following stimulation with IFNβ (Figure 4E). To further confirm that tofacitinib impacted AAA development in vivo, mice underwent AngII-induced AAA induction with and without tofacitinib treatment (20mg/kg intraperitoneal injection three times weekly) and temporal monitoring of aortic dilation was conducted. Tofacitinib therapy significantly decreased AAA development (Supplemental Figure 3B). Given the prior findings implicating STAT in AAA tissue, we further investigated JAK/STAT3 downstream signaling, we analyzed Setdb2 expression in macrophages from Stat3−/− mice and matched controls (Stat3+/+). Stat3−/− mice demonstrated decreased Setdb2 expression in response to IFNβ stimulation (Figure 4F). Taken together, these results suggest that the increased SETDB2 responsible for the TIMP regulation during AAA development may be altered through upstream therapeutic manipulation of the IFNβ/JAK1/STAT3 pathway.
Figure 4. IFNβ/JAK/STAT3 pathway induced Setdb2/H3K9me3 on Timp Promoters in Macrophages.
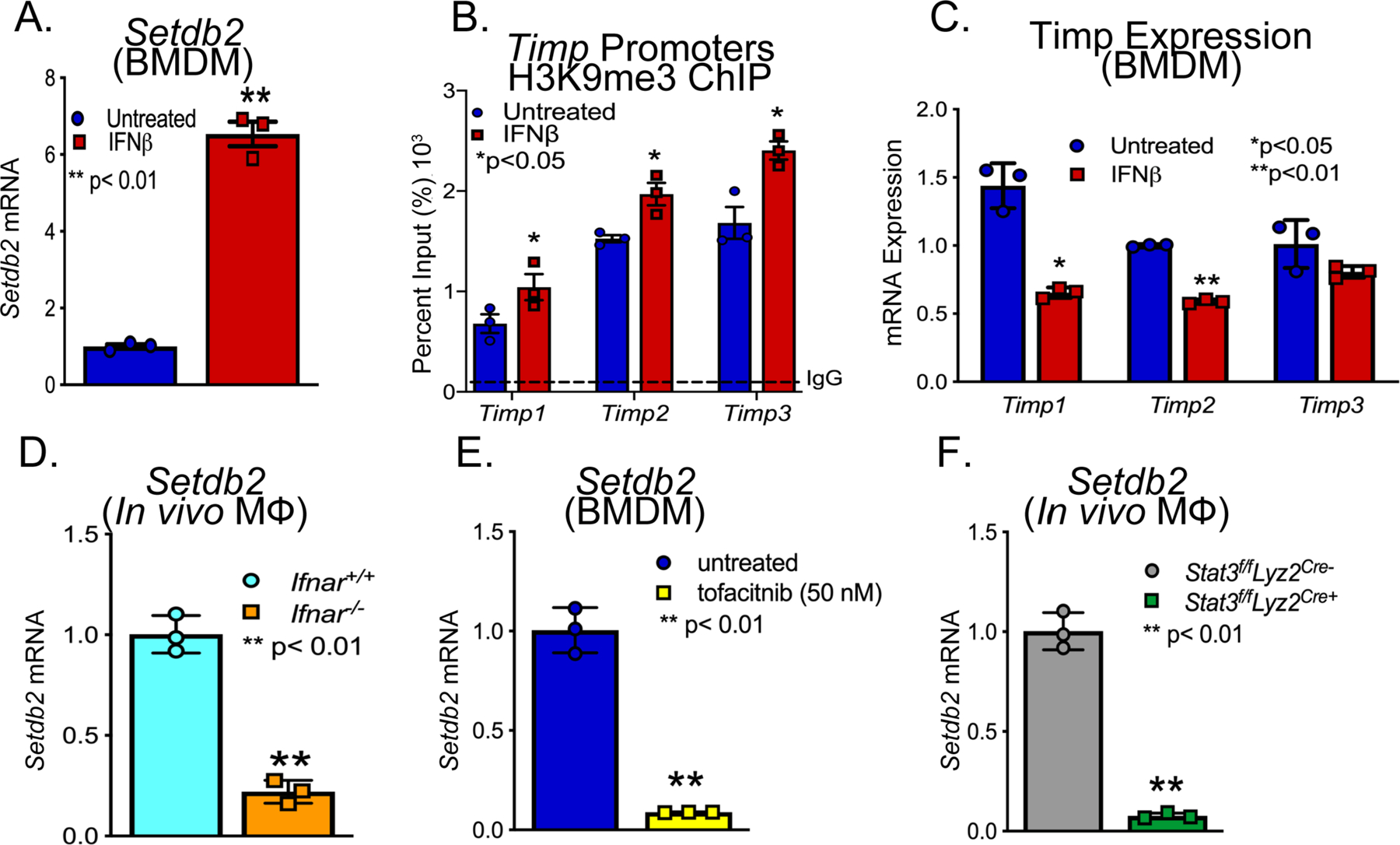
A. Setdb2 expression in BMDM treated ex vivo with IFNβ for 8 hrs (n = 6 mice per group). B. ChIP analysis of BMDMs from controls treated ex vivo with IFNβ (100U) for 6 hrs and analyzed for H3K9me3 at the NF-κB binding site on the Timp1, Timp2, and Timp3 promoter (n = 6 mice per group). C. Timp1, Timp2, and Timp3 expression in BMDM treated ex vivo with IFNβ for 8 hrs (n = 6 mice per group). *p<0.05 D. Quantitative PCR analysis of Setdb2 was conducted in macrophages (MΦ) (CD11b+[CD3−CD19−Nk1.1−Ly6G−]) isolated from Ifnar−/− and littermate controls (Ifnar+/+) (n = 3 mice/group run in triplciated). **p<0.01 for Mann-Whitney U test. E. Setdb2 expression in BMDMs treated with IFNβ +/− tofacitinib (JAK inhibitor; 50 nM) (n = 3 mice per group). **p<0.01 for Mann-Whitney U test. F. Quantitative PCR analysis of Setdb2 was conducted in macrophages (MΦ) (CD11b+[CD3−CD19−Nk1.1−Ly6G−]) isolated from Stat3f/fLyz2Cre- and littermate controls (Stat3f/fLyz2Cre+) (n = 3 mice/group run in triplciated). **p<0.01 for Mann-Whitney U test.
DISCUSSION
Our results have uncovered dynamic mechanisms that bridge the imbalance of protease mediated extracellular matrix degradation and natural inhibitors of proteases during AAA expansion. Further, we have identified the epigenetic chromatin modifying enzyme, SETDB2, as an instrumental signal in perpetuating the aortic wall degradation (Figure 5). Herein, using human AAA tissue samples and two murine models (AngII-induced AAAs and elastase AAA model), we identified that the epigenetic enzyme SETDB2 is a crucial regulator of Timp expression and subsequent MMP activity. Mechanistically, IFNβ signals through the JAK/STAT3 pathway to increase SETDB2 which co-localizes to NFκB binding sites on TIMP1–3 gene promoters, where it leads to inciting the repressive histone methylation mark H3K9me3, thereby inhibiting Timp gene expression. Additionally, single cell transcriptomics demonstrated human monocytes and tissue macrophages have increased SETDB2 expression resulting in the activation of multiple chromatin regulatory and extracellular matrix pathways. Ultimately, manipulation of this pathway, using a macrophage-specific genetic model (Setdb2flox/floxLys2Cre+) increased Timp expression, limited extracellular matrix protease activity, and decreased AAA formation.
Figure 5.
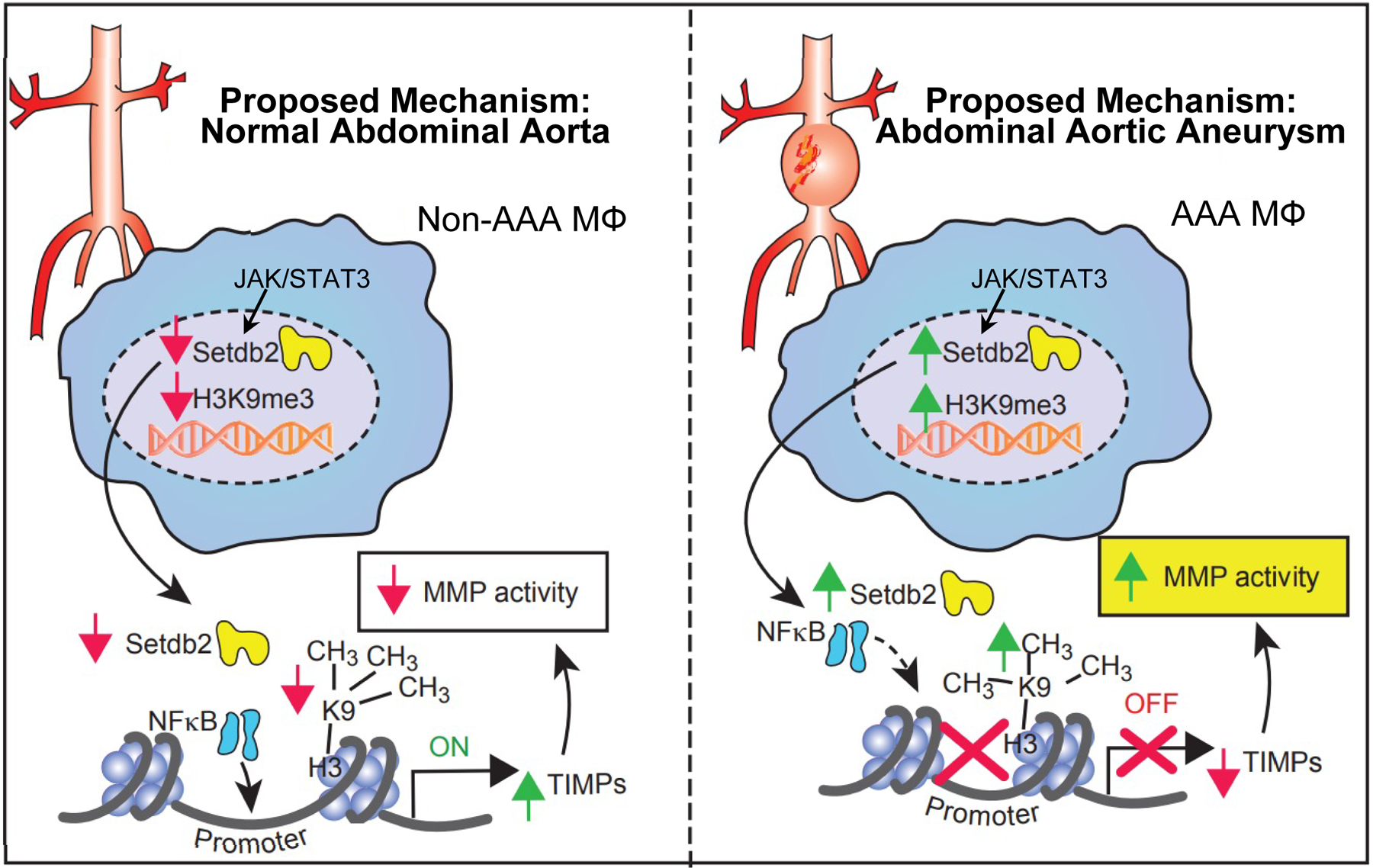
Schematic of SETDB2-mediated regulation of macrophage Timp expression and MMP during AAA development.
Macrophages present in aortic tissue arise from either hematopoietic progenitor cell proliferation/differentiation or mobilization of splenic monocytes 41,42. These recruited macrophages serve to promote AAA formation, in part, through increased proteolytic enzyme production that weakens the aortic wall. The main class of proteolytic enzymes associated with impaired aortic tissue integrity are the matrix metalloproteinases. These enzymes degrade elastin, collagen, and the extracellular matrix allowing for negative remodeling and loss of vascular smooth muscle cells. Although some papers have reported how altered MMP activity are critical for the regulation of vascular remodeling during development, within AAA formation MMP activity has consistently been demonstrated to be pathologic 43,44. MMPs are regulated at the level of mRNA expression, pro-enzyme activation and inhibitory action of tissue inhibitors of metalloproteinases (TIMPs). These endogenous naturally occurring MMP inhibitors have a protective effect against the development of aneurysms as they inhibit the proteolytic activity of MMPs by forming protein complexes with their specific pairs45,46. The molecular mechanisms that program and sustain macrophage gene expression profiles in AAA disease and allow dysregulation of MMP/TIMPs have not previously been explored. Hence elucidating the precise mechanisms responsible for how macrophages regulate proteolytic enzyme production will allow for development of more precise and targeted therapeutics.
Prior investigations by our group and others have begun to explore the role of epigenetics on macrophage-mediated inflammation in human disease20,47–49, however few studies have looked at epigenetic regulation of macrophage function in the setting of aortic pathologies. Initial investigations found histone deacetylases were increased in AAA tissues from humans and AngII-infused mice. Further, inhibition of class I or class IIa histone deacetylases improved survival and decreased AAA formation in mice however the cell-specific mechanisms were not fully investigated and the genes these deacetylases regulated were unknown 50–52. More recent studies have attempted to provide cell specific information by analyzing immune cell subsets within AAA disease. Specially, isolation of regulatory T lymphocytes (FOXP3+ CD4+ CD25+) from human AAA tissue had reduced acetylation on histone 3 and increased histone deacetylase enzymes compared to healthy controls but failed to correlate changes in histone acetylation to regulatory T lymphocyte function 53,54. In contrast to histone acetylation, the role of histone methylation has not been extensively investigated in AAA disease. Specific to this, our group recently published on the role of an epigenetic enzyme histone demethylase, JMJD3, on macrophage mediated inflammation in AAAs, however this work did not examine the role of other epigenetic enzymes or macrophage regulation of proteases 20. In the current investigation, our human single cell sequencing identified SETDB2 to be significantly increased in macrophages from AAA tissue and that SETDB2 adds a repressive H3K9me3 mark onto the TIMP promoters in monocyte/macrophages resulting in decreased TIMP expression and unregulated MMP activity. Further, mice with macrophage-specific deletion of SETDB2 demonstrated decreased AAA formation and improved TIMP/MMP ratios, consistent with alterations in the protease levels and decreased elastin degradation. SETDB2 has recently been shown to be important to the regulation of macrophage inflammation in a variety of disease states including in atherosclerosis18,47; however, no prior publication has examined the role of SETDB2 in AAA macrophages or in any disease state with respect to protease function. Our findings suggest the SETDB2-mediated regulation of TIMP/MMP imbalance in the aortic wall that degrades the structural integrity of the aorta may be a more influential driver of aortic aneurysmal dilation than SETDB2-mediated regulation of inflammation. Additionally, from a translational standpoint, it is well established clinically that diabetic patients have a reduced prevalence of AAAs and a slower AAA growth rate of established aneurysms55–57. However, the mechanism behind the negative association between AAAs and diabetes remains unknown. We have previously demonstrated that SETDB2 is profoundly reduced in macrophages from diabetic patients and multiple diabetic animal models resulting in altered macrophage function during wound healing47. As such a potential driver behind the reduction in AAA incidence seen in diabetic patients is a decrease in SETDB2 expression and an alteration of TIMP/MMP balance in the aortic wall in favor of maintenance of the aortic wall architecture. However, additional preclinical and clinical investigations focusing only on diabetic patients are warranted to further establish this mechanistic link.
Presently there is no pharmacological inhibitors of SETDB2, however we examined the upstream signaling pathway whereby JAK/STAT signaling increases SETDB2 in aortic macrophages. Multiple clinical trails have attempted to investigate medical therapy for AAA disease with the recent N-TA3CT trial investigating the utility of doxycycline, a broad MMP inhibitor, on the reduction of aortic dilation. Unfortunately within the N-TA3CT, doxycycline failed to prevent AAA progression58. This lack of clinical impact of doxycycline on AAA disease despite multiple promising preclinical investigations was likely related to the inability of doxycycline to inhibit MMP activity in comparison to placebo controls in the serum of N-TA3CT participants. In contrast to doxycycline therapy that broadly targets MMPs, within this manuscript we chose to target cell specific SETDB2 epigenetic modifications within macrophages in order to prevent AAA dilation. As macrophages exhibit different functional phenotypes during tissue repair 59, the ability to modulate macrophage phenotype at a particular time after injury is an attractive therapeutic strategy. Indeed, epigenetic therapies have been shown to be effective in the treatment of cancer with drugs resulting in dose dependent inhibition of cell proliferation, invasion, and cell migration 60–62. More recently, epigenetic therapies have are beginning to be examined in cardiovascular disease 14,63. For example, epigenetic therapies for atherosclerosis are currently undergoing phase III clinical trials, suggesting that methods to alter epigenetic enzymes show promise in controlling cardiovascular disease 64. Despite these preclinical and ongoing trials for cardiovascular disease, the role of JAK/STAT3 signaling and SETDB2 inhibition in AAA development has not been vigorously investigated. Prior preclinical studies on focusing on STAT3 pharmacological and genetic inhibition have yielded conflicting results demonstrating both inhibition and progression of aneurysmal disease as well as limited translational ability due to low oral bioavailability of pharmacological STAT3 inhibitors.65 Our results demonstrate SETDB2 and potential inhibition with tofacitinib may be an attractive therapeutic target, either in peripheral monocytes or aortic tissue macrophages, due to its capacity to decrease vascular remodeling.
Although this study provides mechanism(s) behind important advances related to dysregulated macrophage function in AAA development, there are several limitations to this study. Regarding the human samples, these are taken from patients with AAA that meets repair criteria, and thus represent the end-stages of disease. It is possible that SETDB2 is less relevant during the earlier stages where AAAs are developing and the elastin is less fragmented. Second, within our myeloid-specific SETDB2 murine model we utilized the Lyz2Cre system which we have previously demonstrates efficiently depletes SETDB2 in myeloid cell population.28 We acknowledge that gene expression profiles between monocytes/macrophages, neutrophils and dendritic cells overlap due to their close lineage relationship. As such there is no Cre-transgenic line that is perfectly specific for macrophages 23. However, the contributions of neutrophils and dendritic cells for AAA development have been shown to be minor in comparison to macrophage pathophysiology 33,66,67. Lastly, we recognize that other epigenetic enzymes or vascular cell types may regulate aberrant macrophage function during AAA development as well 68.
In conclusion, our study provides important insight where JAK/STAT signaling regulates SETDB2 in aortic macrophages that results in increased protease activity that ultimately increases AAA development. Increased SETDB2 in macrophages increases the repressive histone methylation mark H3K9me3 on the TIMP gene promoters which alters the MMP/TIMP imbalance and leads to adverse vascular remodeling and aortic dilation. Targeting the JAK/STAT/SETDB2 pathway in a cell specific manner will allow us to modulate macrophage-mediated protease activity in aortic tissue and may lead to decreased AAA formation and rupture.
Supplementary Material
Figure S1. SETDB2 is Increased in Murine Elastase AAA Model. A-D. Quantitative PCR analysis of Timp1, Timp2, Timp3,Setdb2, Mmp2, Mmp9, and Ifnb in aortic tissues from control or elastase-induced AAA mice (n=3 mice/group). *p<0.05; Mann-Whitney Test.
Figure S2. No Difference in Ascending Aortic Diameter in WT M𝛷 or Setdb2−/− M𝛷. A. Wild-type (WT M𝛷) mice and mice with macrophage-specific SETDB2 deficiency (Setdb2−/− M𝛷) were fed high fat diet for 6 weeks and infused with saline or AngII infusion (1,000 ng/min/kg) for 4 weeks. Maximum ascending aortic diameter was quantified with in vivo ultrasound on day 28 (n= 6 in saline infused cohorts and 20 in AngII infused cohorts). Data are presented as the mean±SEM.
Figure S3. Tofacitinib reduces AngII-induced AAA Development. A. Gelatin gel zymography to detect MMP2 and 9 activity in aortic tissue extracts from different groups of mice. B. Representative ultrasound images of the abdominal aorta at day 28 in wild type mice following either saline or AngII infusion with or without Tofacitinib injection (20mg/kg IP injection three times per week). Dotted line represents aortic contour and arrows represent aortic wall diameter. Maximal abdominal aortic diameter as determined by ultrasound measured by two observers in wild type mice following either saline or AngII infusion with or without Tofacitinib injection (20mg/kg IP injection three times per week). n= 3–5. *p<0.05; **p<0.001 by ANOVA with Newman-Keuls Multiple Comparison test. Data are presented as the mean±SEM.
Acknowledgements:
We thank Robin Kunkel for her assistance with the graphical illustrations.
Funding Sources:
This work is supported in part by National Institutes of Health grants NIH 1R01HL156274 (KG and FD), Society of University Surgeons Junior Faculty Research Award (FD), Vascular and Endovascular Surgery Society Early Career Investigator Award (FD), P30 AR075043 (JG, LT), R01-AR069071 (JG), and the Doris Duke Foundation (KG).
Abbreviations
- AAA
Abdominal aortic aneurysm
- AngII
Angiotensin II
- H3
Histone 3
- H3K9me3
Histone 3 lysine 9 trimethylation
- IFN-I
Type 1 interferons
- Interferon β
IFN β
- Janus kinase
JAK
- SETDB2
SET Domain Bifurcated Histone Lysine Methyltransferase 2
- PCSK9
Proprotein convertase subtilisin/kexin type 9
- scRNA-seq
single cell RNA sequencing
- STAT3
Signal transducer and activator of transcription 3
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
REFERENCES
- 1.Nordon IM, Hinchliffe RJ, Loftus IM, et al. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nature Reviews Cardiology 2011;8:92–102. [DOI] [PubMed] [Google Scholar]
- 2.Folsom AR, Yao L, Alonso A, et al. Circulating Biomarkers and Abdominal Aortic Aneurysm Incidence: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation 2015;132:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation 2008;117:1883–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kent KC. Clinical practice. Abdominal aortic aneurysms. N Engl J Med 2014;371:2101–8. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Wang W, Li L, et al. MMPs and ADAMs/ADAMTS inhibition therapy of abdominal aortic aneurysm. Life Sciences;253 . Epub ahead of print July 15, 2020. DOI: 10.1016/j.lfs.2020.117659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis FM, Rateri DL, Daugherty A. Mechanisms of aortic aneurysm formation: Translating preclinical studies into clinical therapies. Heart 2014;100:1498–1505. [DOI] [PubMed] [Google Scholar]
- 7.Hadi T, Boytard L, Silvestro M, et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat Commun;9 . Epub ahead of print December 1, 2018. DOI: 10.1038/s41467-018-07495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nosoudi N, Nahar-Gohad P, Sinha A, et al. Prevention of Abdominal Aortic Aneurysm Progression by Targeted Inhibition of Matrix Metalloproteinase Activity with Batimastat-Loaded Nanoparticles. Circ Res 2015;117:e80–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Longo GM, Xiong W, Greiner TC, et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest 2002;110:625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadoglou NP, Liapis CD. Matrix metalloproteinases: Contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Current Medical Research and Opinion 2004;20:419–432. [DOI] [PubMed] [Google Scholar]
- 11.Prescott MF, Sawyer WK, Von Linden-Reed J, et al. Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpressing MMP-3, MMP-12, and MMP-13 and on restenosis in rats after balloon injury. In: Annals of the New York Academy of Sciences New York Academy of Sciences:179–190. [DOI] [PubMed] [Google Scholar]
- 12.Obata Y, Furusawa Y, Hase K. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol 2015;93:226–232. [DOI] [PubMed] [Google Scholar]
- 13.Hoeksema MA, De Winther MPJ. Epigenetic Regulation of Monocyte and Macrophage Function. Antioxidants and Redox Signaling 2016;25:758–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuznetsova T, Prange KHM, Glass CK, et al. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nature Reviews Cardiology 2020;17:216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroetz DN, Allen RM, Schaller MA, et al. Type I Interferon Induced Epigenetic Regulation of Macrophages Suppresses Innate and Adaptive Immunity in Acute Respiratory Viral Infection. PLOS Pathog 2015;11:e1005338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schliehe C, Flynn EK, Vilagos B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol 2015;16:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimball AS, Davis FM, denDekker A, et al. The Histone Methyltransferase Setdb2 Modulates Macrophage Phenotype and Uric Acid Production in Diabetic Wound Repair. Immunity;51 . Epub ahead of print 2019. DOI: 10.1016/j.immuni.2019.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Sun J, Canfrán-Duque A, et al. Deficiency of histone lysine methyltransferase SETDB2 in hematopoietic cells promotes vascular inflammation and accelerates atherosclerosis. JCI Insight;6 . Epub ahead of print June 22, 2021. DOI: 10.1172/jci.insight.147984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis FM, Gallagher KA. Epigenetic Mechanisms in Monocytes/Macrophages Regulate Inflammation in Cardiometabolic and Vascular Disease. Arteriosclerosis, thrombosis, and vascular biology 2019;39:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis FM, Tsoi LC, Melvin WJ, et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J Exp Med;218 . Epub ahead of print June 7, 2021. DOI: 10.1084/jem.20201839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinet P, Milewicz DM, Cassis LA, et al. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arterioscler Thromb Vasc Biol 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clausen BE, Burkhardt C, Reith W, et al. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999;8:265–77. [DOI] [PubMed] [Google Scholar]
- 23.Shi J, Hua L, Harmer D, et al. Cre driver mice targeting macrophages. In: Methods in Molecular Biology Humana Press Inc.:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu H, Howatt DA, Balakrishnan A, et al. Hypercholesterolemia induced by a PCSK9 gain-of-function mutation augments angiotensin II-induced abdominal aortic aneurysms in C57BL/6 mice-brief report. Arterioscler Thromb Vasc Biol 2016;36:1753–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu C, Xu Y, Lu H, et al. Cys18-Cys137 disulfide bond in mouse angiotensinogen does not affect AngII-dependent functions in vivo. Hypertension 2015;65:800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu H, Howatt DA, Balakrishnan A, et al. Subcutaneous Angiotensin II Infusion using Osmotic Pumps Induces Aortic Aneurysms in Mice. J Vis Exp . Epub ahead of print September 28, 2015. DOI: 10.3791/53191. [DOI] [PMC free article] [PubMed]
- 27.Laser A, Lu G, Ghosh A, et al. Differential gender- and species-specific formation of aneurysms using a novel method of inducing abdominal aortic aneurysms. J Surg Res 2012;178:1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimball AS, Davis FM, denDekker A, et al. The Histone Methyltransferase Setdb2 Modulates Macrophage Phenotype and Uric Acid Production in Diabetic Wound Repair. Immunity 2019;51:258–271.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishii M, Wen H, Corsa CAS, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009;114:3244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii M, Wen H, Corsa CAS, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009;114:3244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George SJ, Johnson JL. In situ zymography. Methods Mol Biol 2010;622:271–277. [DOI] [PubMed] [Google Scholar]
- 32.Davis FM, Tsoi LC, Ma F, et al. Single-cell Transcriptomics Reveals Dynamic Role of Smooth Muscle Cells and Enrichment of Immune Cell Subsets in Human Abdominal Aortic Aneurysms. Ann Surg 2022;276:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis FM, Rateri DL, Daugherty A. Mechanisms of aortic aneurysm formation: Translating preclinical studies into clinical therapies. Heart 2014;100:1498–1505. [DOI] [PubMed] [Google Scholar]
- 34.Raffort J, Lareyre F, Clément M, et al. Monocytes and macrophages in abdominal aortic aneurysm. Nature Reviews Cardiology 2017;14:457–471. [DOI] [PubMed] [Google Scholar]
- 35.Rayamajhi M, Humann J, Kearney S, et al. Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence;1 . Epub ahead of print September 27, 2010. DOI: 10.4161/viru.1.5.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohno T, Aoki H, Ohno S, et al. Cytokine Profile of Human Abdominal Aortic Aneurysm: Involvement of JAK/STAT Pathway. Ann Vasc Dis 2018;11:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lv Y-C, Tang Y-Y, Zhang P, et al. Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. PLoS One 2016;11:e0157265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao M, Xu J, Clair AJ, et al. Local and systemic alterations in signal transducers and activators of transcription (STAT) associated with human abdominal aortic aneurysms. J Surg Res 2012;176:321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shoji T, Guo J, Ge Y, et al. Type I Interferon Receptor Subunit 1 Deletion Attenuates Experimental Abdominal Aortic Aneurysm Formation. Biomolecules;12 . Epub ahead of print October 1, 2022. DOI: 10.3390/biom12101541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyrrell DJ, Chen J, Li BY, et al. Aging Alters the Aortic Proteome in Health and Thoracic Aortic Aneurysm. Arterioscler Thromb Vasc Biol 2022;42:1060–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peshkova IO, Aghayev T, Fatkhullina AR, et al. IL-27 receptor-regulated stress myelopoiesis drives abdominal aortic aneurysm development. Nat Commun 2019;10:5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mellak S, Ait-Oufella H, Esposito B, et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe−/− mice. Arterioscler Thromb Vasc Biol 2015;35:378–88. [DOI] [PubMed] [Google Scholar]
- 43.Massaro M, Scoditti E, Carluccio MA, et al. Oxidative stress and vascular stiffness in hypertension: A renewed interest for antioxidant therapies? Vascular Pharmacology 2019;116:45–50. [DOI] [PubMed] [Google Scholar]
- 44.Simões G, Pereira T, Caseiro A. Matrix metaloproteinases in vascular pathology. Microvascular Research;143 . Epub ahead of print September 1, 2022. DOI: 10.1016/j.mvr.2022.104398. [DOI] [PubMed]
- 45.Cui N, Hu M, Khalil RA. Biochemical and Biological Attributes of Matrix Metalloproteinases. In: Progress in Molecular Biology and Translational Science Elsevier B.V.:1–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Zhang H, Cao L, et al. The Role of Macrophages in Aortic Dissection. Frontiers in Physiology;11 . Epub ahead of print February 5, 2020. DOI: 10.3389/fphys.2020.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimball AS, Davis FM, denDekker A, et al. The Histone Methyltransferase Setdb2 Modulates Macrophage Phenotype and Uric Acid Production in Diabetic Wound Repair. Immunity 2019;51:258–271.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis FM, Kimball A, DenDekker A, et al. Histone methylation directs myeloid TLR4 expression and regulates wound healing following cutaneous tissue injury. J Immunol;202 . Epub ahead of print 2019. DOI: 10.4049/jimmunol.1801258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis FM, Tsoi LC, Wasikowski R, et al. Epigenetic regulation of the PGE2 pathway modulates macrophage phenotype in normal and pathologic wound repair. JCI Insight;5 . Epub ahead of print September 3, 2020. DOI: 10.1172/jci.insight.138443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Y, Tanios F, Reeps C, et al. Histone acetylation and histone acetyltransferases show significant alterations in human abdominal aortic aneurysm. Clin Epigenetics 2016;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galán M, Varona S, Orriols M, et al. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: Therapeutic potential of HDAC inhibitors. DMM Dis Model Mech 2016;9:541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vinh A, Gaspari TA, Liu H Bin, et al. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J Vasc Res 2008;45:143–52. [DOI] [PubMed] [Google Scholar]
- 53.Xia Q, Zhang J, Han Y, et al. Epigenetic regulation of regulatory T cells in patients with abdominal aortic aneurysm. FEBS Open Bio 2019;9:1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang H, Xin S, Yan Y, et al. Abnormal acetylation of FOXP3 regulated by SIRT-1 induces Treg functional deficiency in patients with abdominal aortic aneurysms. Atherosclerosis 2018;271:182–192. [DOI] [PubMed] [Google Scholar]
- 55.Lederle FA, Johnson GR, Wilson SE, et al. Prevalence and associations of abdominal aortic aneurysm detected through screening. Ann Intern Med 1997;126:441–449. [DOI] [PubMed] [Google Scholar]
- 56.Bhak RH, Wininger M, Johnson GR, et al. Factors associated with small abdominal aortic aneurysm expansion rate. JAMA Surg 2015;150:44–50. [DOI] [PubMed] [Google Scholar]
- 57.Chun KC, Teng KY, Chavez LA, et al. Risk factors associated with the diagnosis of abdominal aortic aneurysm in patients screened at a regional veterans affairs health care system. In: Annals of Vascular Surgery Ann Vasc Surg:87–92. [DOI] [PubMed] [Google Scholar]
- 58.Baxter BT, Matsumura J, Curci JA, et al. Effect of Doxycycline on Aneurysm Growth among Patients with Small Infrarenal Abdominal Aortic Aneurysms: A Randomized Clinical Trial. JAMA - J Am Med Assoc 2020;323:2029–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watanabe S, Alexander M, Misharin AV., et al. The role of macrophages in the resolution of inflammation. Journal of Clinical Investigation 2019;129:2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verma SK, Tian X, Lafrance LV., et al. Experimental Methods ( Chemistry ). ACS Med Chem Lett 2012;3:1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mottamal M, Zheng S, Huang TL, et al. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015;20:3898–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shukeir N, Stefanska B, Parashar S, et al. Pharmacological methyl group donors block skeletal metastasis in vitro and in vivo. Br J Pharmacol 2015;172:2769–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luque-Martin R, Van Den Bossche J, Furze RC, et al. Targeting histone deacetylases in myeloid cells inhibits their maturation and inflammatory function with limited effects on atherosclerosis. Front Pharmacol;10 . Epub ahead of print 2019. DOI: 10.3389/fphar.2019.01242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ray KK, Nicholls SJ, Ginsberg HD, et al. Effect of selective BET protein inhibitor apabetalone on cardiovascular outcomes in patients with acute coronary syndrome and diabetes: Rationale, design, and baseline characteristics of the BETonMACE trial. Am Heart J 2019;217:72–83. [DOI] [PubMed] [Google Scholar]
- 65.Aoki H, Majima R, Hashimoto Y, et al. Ying and Yang of Stat3 in pathogenesis of aortic dissection. Journal of Cardiology 2021;77:471–474. [DOI] [PubMed] [Google Scholar]
- 66.Raffort J, Lareyre F, Clément M, et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol 2017;14:457–471. [DOI] [PubMed] [Google Scholar]
- 67.Davis FM, Rateri DL, Daugherty A. Abdominal aortic aneurysm: Novel mechanisms and therapies. Curr Opin Cardiol;30 . Epub ahead of print 2015. DOI: 10.1097/HCO.0000000000000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kruidenier L, Chung CW, Cheng Z, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012;488:404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SETDB2 is Increased in Murine Elastase AAA Model. A-D. Quantitative PCR analysis of Timp1, Timp2, Timp3,Setdb2, Mmp2, Mmp9, and Ifnb in aortic tissues from control or elastase-induced AAA mice (n=3 mice/group). *p<0.05; Mann-Whitney Test.
Figure S2. No Difference in Ascending Aortic Diameter in WT M𝛷 or Setdb2−/− M𝛷. A. Wild-type (WT M𝛷) mice and mice with macrophage-specific SETDB2 deficiency (Setdb2−/− M𝛷) were fed high fat diet for 6 weeks and infused with saline or AngII infusion (1,000 ng/min/kg) for 4 weeks. Maximum ascending aortic diameter was quantified with in vivo ultrasound on day 28 (n= 6 in saline infused cohorts and 20 in AngII infused cohorts). Data are presented as the mean±SEM.
Figure S3. Tofacitinib reduces AngII-induced AAA Development. A. Gelatin gel zymography to detect MMP2 and 9 activity in aortic tissue extracts from different groups of mice. B. Representative ultrasound images of the abdominal aorta at day 28 in wild type mice following either saline or AngII infusion with or without Tofacitinib injection (20mg/kg IP injection three times per week). Dotted line represents aortic contour and arrows represent aortic wall diameter. Maximal abdominal aortic diameter as determined by ultrasound measured by two observers in wild type mice following either saline or AngII infusion with or without Tofacitinib injection (20mg/kg IP injection three times per week). n= 3–5. *p<0.05; **p<0.001 by ANOVA with Newman-Keuls Multiple Comparison test. Data are presented as the mean±SEM.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.