

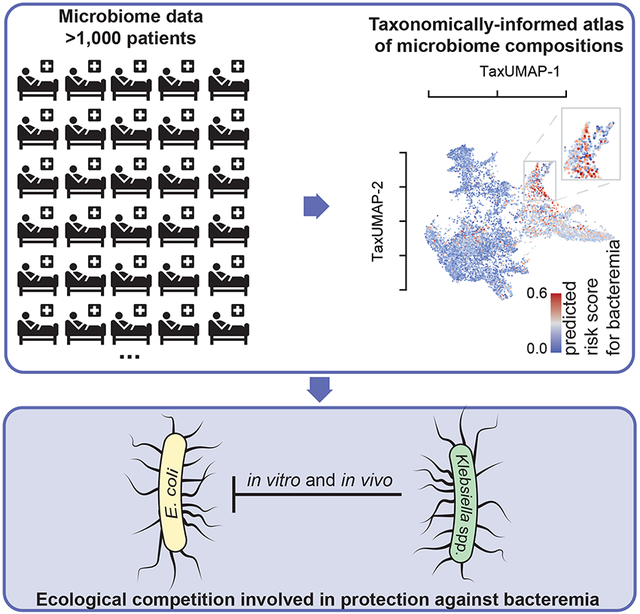
SUMMARY
Longitudinal microbiome data provide valuable insight into disease states and clinical responses but are challenging to mine and view collectively. To address these limitations, we present TaxUMAP, a taxonomically-informed visualization for displaying microbiome states in large clinical microbiome datasets. We used TaxUMAP to chart a microbiome atlas of 1,870 cancer patients during therapy-induced perturbations. Bacterial density and diversity were positively associated, but the trend was reversed in liquid stool. Low diversity states (dominations) remained stable after antibiotic treatment, and diverse communities had a broader range of antimicrobial resistance genes than dominations. When examining microbiome states associated with risk for bacteremia, TaxUMAP revealed that certain Klebsiella species associated with lower risk for bacteremia localize in a region of the atlas that is depleted in high-risk enterobacteria. This indicated a competitive interaction that was validated experimentally. Thus, TaxUMAP can chart comprehensive longitudinal microbiome datasets, enabling insights into microbiome effects on human health.
Graphical Abstract
eTOC
Schluter, Djukovic, Taylor et al. present TaxUMAP—a taxonomically-informed visualization tool for microbiome datasets. TaxUMAP charts an atlas of patient microbiome states, facilitating the exploration of microbiome effects on health. The authors observed competitive interactions between certain low bacteremia risk-associated Klebsiella spp. and other high-risk enterobacteria, which they validated experimentally.
INTRODUCTION
The gut microbiome contains the most numerous and diverse bacterial community in the human body. Some species of gut bacteria have been associated with human health 1,2, for example by reducing the risk of gut colonization by incoming pathogens3,4 . Antibiotics perturb the microbiota composition5, reduce the density of strict anaerobes associated with resistance to pathogen colonization6,7 and lead to dominations by antibiotic-resistant bacteria that increase the risk of bacteremia in immune-compromised patients8,9.
The dominations of the gut microbiome that happen after antibiotics are strongly associated with higher risks of bacteremia in cancer patients receiving systemic chemotherapy8,9,14. But establishing causal effects for even such strong clinical associations remains difficult. Finding the mechanisms underlying other patient outcomes, such as cancer progression and response to therapy where any association with microbiome composition is perhaps indirect, may be even harder15. Experimental tests of cause-and-effect relationships between the microbiome and host phenotypes are easier in animal models; however, due to different ecological and host environments, among other factors, the experimental findings only sometimes translate to humans16. Large longitudinal datasets of patient microbiomes provide an alternative: the longitudinal data collected from patients—as they experience microbiome compositional changes in response to treatments such as antibiotics—could be mined to infer relationships of cause and effect17. This is conceptually similar to how “natural experiments”, events or policy changes in real life, can enable economists to study the impacts of these phenomena in human societies18.
Here we introduce a technique to study large-scale microbiome data in the context of clinical and host metadata. The method, called TaxUMAP, extends the Uniform Manifold Approximation and Projection (UMAP)19,20 to include hierarchical taxonomic relations between the bacterial sequencing variants identified in a sample to cluster microbiome community samples with similar global ecosystem features while retaining fine-grained differences. We use TaxUMAP to chart an atlas of microbiome compositions using >10,000 samples from 1,870 patients hospitalized at Memorial Sloan Kettering to receive allogeneic hematopoietic stem cell transplantation (allo-HCT)21,22. We use the atlas to explore microbiome states associated with a high risk of bloodstream infections. We find a region of the atlas enriched for certain species of Klebsiella that seem to exclude other enterobacteria associated with a high risk of infection. This observation was intriguing: independent studies have shown that the same types of Klebsiella are important for resistance to colonization by pathogenic Enterobacteriaceae in mice23,24. We then isolated strains from corresponding patient samples and experimentally validated their ability to outcompete pathogenic isolates in vitro and in vivo. Our results illustrate how TaxUMAP facilitates the identification of ecological mechanisms in the gut microbiome with implications for host health.
RESULTS
TaxUMAP charts an atlas from longitudinal clinical microbiota data.
We built TaxUMAP to visualize a large longitudinal microbiome data set obtained from hospitalized cancer patients21 and extract biological insights. Our work has previously shown that the microbiome perturbations experienced by these immune-compromised patients can exacerbate certain bacterial populations’ health effects and make them more visible25. HCT is a complex procedure with three phases: pre-transplant chemotherapeutic conditioning (phase I), a prolonged period of low immune cell counts (neutropenia, phase II), and immune reconstitution (phase III, Figure 1A). The duration of each phase varies among patients. We split samples into early, mid, and late-phase, providing a comparable pseudo time point (Figure 1B-D). Anti-infective drugs are administered to patients prophylactically during phase I and II and as needed during any phase for patients with suspected infections. Antibiotics cause collateral damage to the gut ecosystem5,7,11. The microbiota α-diversity tends to drop from phase I to phase II, and to stay low in phase III (Figure 1C, D)10.
Figure 1. The TaxUMAP algorithm effectively visualizes different microbiome states.

A) Three distinct clinical phases of a HCT therapy. B) Gut microbiome bacterial compositions over time from one patient; bars represent the relative taxon abundances measured in a stool sample by 16S rRNA gene sequencing. C) Compositions in 12,546 samples; clinical phases at collection indicated. D) Bacterial α-diversity (inverse Simpson index) over pseudo-time by assigning each sample to an early, mid, or late phase of the respective clinical phase (shading as per colors in A) during which a sample was obtained; mean (black solid) and median (black dashed) diversity (n=12,546, shaded: 95%C.I. of the mean). E) ASV level principal component, and principal coordinate plots of all samples (unweighted- and weighted UniFrac distances). F) UMAP embedding at the ASV level. G) Comparison between UMAP and TaxUMAP on a simulated data set; colors indicate the hypothetical genus, indicated on the simulated taxonomy tree, with the highest abundance in a sample. H) Euclidean distances between in silico generated samples with the same dominant family in UMAP vs. TaxUMAP embedding (n=1,000, ***:p<10−4, Wilcoxon rank sum test). I) TaxUMAP embedding of patient samples. E, F, I) color by most abundant taxon.
A fecal microbiome can contain hundreds or even thousands of different bacterial taxa. An ideal way to map the microbiome states effectively should reduce the data dimensionality without losing crucial detail. Common ordination methods such as principal component analysis (PCA), or principal coordinate analyses using UniFrac distances (PCoA) can be applied to allo-HCT patient data, but they only resolve the most frequently observed, low diversity, single-taxon enriched microbiota states (Figure 1E) at the expense of many other distinct states of microbiota composition that become less visible. The recently developed nonlinear dimensionality reduction method UMAP20 is especially useful for working with large sample numbers and high data dimensionality26 and reveals microbiota states left unclear in other methods (Figure 1F). However, it needs crucial biological similarity information in the form of taxonomic relationships between ASVs. To demonstrate this, we simulated 1,000 bacterial compositions from 600 AVSs belonging to 30 hypothetical species, ten families, and 2 phyla (Figure 1G). While the UMAP places samples with similar species composition together, it neglects similarities between samples based on higher taxonomic levels (Figure 1F, G). When applying UMAP to our patient samples at the ASV level, for example, samples dominated by different ASVs from the genus Akkermansia (in shades of purple, highlighted by circles, Figure 1F), or the family Enterobacteriaceae (in shades of red, highlighted by rectangles, Figure 1F) end up far from each other.
Our open-source TaxUMAP algorithm combines ecological distance matrices computed at different taxonomy levels. Depending on the research question, each taxonomic level can be emphasized or disregarded by the user. The resulting taxonomically aware pairwise sample distances matrix is the input for UMAP. When applying this technique to our in silico data (Figure 1G), formerly separate clusters of samples with different species but similar families are now embedded significantly closer together (Figure 1H). In our patient data, the highlighted clusters that were artefactually far apart (Figure 1F) are now moved together (Figure 1I), facilitating a biologically interpretable visualization.
Atlas annotation with clinical data reveals collateral damage caused by antibiotics.
The TaxUMAP atlas can be annotated by charting clinical metadata onto microbiome states. For example, we can see that while the dataset has slightly more men than women, the atlas reveals that sex does not play an important role since patients of both sexes visit the same regions (Figure 2A). Nor does the underlying disease (Figure 2B). Every patient, independent of sex or disease, received antibiotic prophylaxis. Phase I samples fall in the bottom left corner of the map (Figure 2C), with the earliest samples (Figure 2D) preceding anti-infective treatment (Figure 2E). Antibiotics precede changes in the gut microbiome composition, particularly a loss of diversity (Figure 2F) and reduction in the relative abundances of obligate anaerobes (Figure 2G, S1A). Alongside these high-level shifts in the microbiome, the consistency of stool also changes from well-formed to semi-formed and liquid (Figure 2H). The total bacterial density (16S rRNA gene copies per gram of stool) varies widely, from ~1010 copies per gram of stool, to almost undetectable in some samples (Figure 2I, S1B, limit of detection: 2.84 copies/g stool). Bacterial density correlates positively with community diversity in non-liquid samples but negatively in liquid samples (Figure S1C). Microbiome compositional data alone suffers from a lack of total population density information. Comparing microbiota composition (Figure 1I) with bacterial loads (Figure 2I) reveals that states enriched in Streptococacceae or Staphylococcaceae tend to have the lowest densities. Similarly, low-diverse states populated predominantly by Enterobacteriaceae or Enterococcaceae also exhibit low population densities in many samples; however, they also include samples with high bacterial loads, indicating that these bacteria can expand in numbers in the gut during periods where they dominate the community (Figure 1I, 2I).
Figure 2. The atlas of the HCT bacterial gut microbiota using TaxUMAP.

Each scatter plot is accompanied by histograms (grey) of the displayed metadata distribution. A) Samples from female and male donors, with B) different diseases. C) Samples from different clinical phases; phase I samples are concentrated in a distinct region corresponding to D) samples taken early during therapy. In the same region, samples are concentrated where no anti-infective administrations were recorded in the 7 days prior to sample collection (E). F) Bacterial α-diversity (measured by the inverse Simpson index). G) Relative abundances of obligate anaerobe commensal taxa. See also Figure S1A. H) Stool composition (liquid, semi-formed, formed stool). I) Total bacterial abundance estimated by total 16S gene copy numbers per gram of stool. See also Figure S1B-C. J) Volatility of the bacterial community (most volatile in red: volatility >0.9; least volatile in blue: volatility<0.1). See also Figure S1D-G. K) Unique antimicrobial resistance phenotypes detected per sample. See also Figure S1H-K. L) Vancomycin-resistance conveying vanA gene detected in rectal swab. See also Figure S1K-M.
The high temporal frequency of the longitudinal data allows us to analyze consecutive time points from the same patient and assess compositional volatility (Figure 2J). We could find volatile switches, defined as a compositional turnover of 90% or more between two consecutive days, throughout the entire atlas (Figure S1D), indicating that any microbiome state in allo-HCT patients is prone to a sudden change. Interestingly, the endpoints of volatile switches, which tended to be localized in low-diversity states, also harbored some of the most stable states, i.e. where volatility scores were the lowest (Figure 2J, blue). This suggests that the microbiota, once injured, may remain depleted, possibly due to continued selection pressure, for example by antibiotics or because, according to ecological theory, less diverse microbiome ecosystems tend to be more stable27,28.
To test if microbiome domination states persist due to continuous antibiotic pressure, we analyzed antibiotic treatment records and correlated them to microbiome composition and diversity (Figure S1E). The data showed that several low-diversity intestinal domination states could persist beyond the antibiotic administration window. Biodiversity recovered slowly after the last day of antibiotic treatment, and the diversity loss had a typical half-life of 9 days (Figure S1F). The half-life for eight of the most frequent domination states post-antibiotics showed that Enterococcus (the most frequent) has a half-life of 6.4 days after antibiotics, while Streptococcus (the second most frequent) has 15.5 days half-life (Figure S1G). While antibiotics may persist in the gut lumen even after administration was stopped, these results suggest that the persistence of dominations might at least partly be due to the nature of the dominating species or the interactions between the rest of the microbiome, including the host, with the dominating species.
To understand how antibiotics drive these shifts in microbiome composition, we analyzed shotgun metagenomic sequences from 395 samples from 49 unique patients. As done before29, we counted each sample’s unique, qualitative antibiotic resistance genes (ARG). Plotting the number of unique ARGs identified in each sample on the TaxUMAP suggested a negative association with bacterial α-diversity (Figure 2F, K): there were more diverse sets of antimicrobial resistance genes in ecologically diverse microbial samples than in samples dominated by a single taxon (Figure 2K, S1H). This was an interesting observation because domination states tend to occur after antibiotics, and they are assumed to be caused by selection pressures benefiting antibiotic resistant taxa. Our results suggest, however, that antibiotics, which deplete ecological diversity, act consistent with a model where they purge antimicrobial resistance genes from the ecological community that do not convey resistance to the applied antibiotic, and thereby these antibiotics may also decrease the diversity of the antimicrobial resistance gene pool.
We performed a statistical analysis of the diversity-depleting effects of different antibiotics to show how antibiotics can increase the abundance of a specific antimicrobial-resistance gene. Similar to previous results7, we identified significant associations between piperacillin/tazobactam, metronidazole, meropenem and vancomycin with microbiome diversity loss (see Figure S1I for a full list). Then, we investigated which ARGs correlated with administering those four antibiotics. As expected, the number of genes statistically significantly correlated with the antibiotics was only a fraction of the total number of the ARGs (Figure S1J). ARGs significantly associated with resistance to these four antibiotics include blaZ (encoding a beta-lactamase) and mecA (an alternative penicillin-binding protein) of S. aureus a taxon that in our data increased in abundance with the administration of the beta-lactam antibiotic piperacillin/tazobactam, as well as several genes of the vanA operon associated with vancomycin resistance in E. faecium, a taxon that increased in abundance with the administration of oral vancomycin. We detected vanA in 54.5% of our shotgun-sequenced samples, all of which showed a sequence identity of >98% with the matched vanA gene of E. faecium (Figure S1K). Vancomycin is among the most frequently administered antibiotics in these patients (24.6% of all administrations). We detected the presence of vanA in 7,589 samples using a PCR test (Figure S1L). We saw that those PCR-positive samples were localized across the TaxUMAP, but more frequently in the low-diversity samples dominated by Enterococcus (Figure 2L). This agrees with the notion that strong selection for vancomycin resistance, facilitated by the antibiotic-induced loss of obligate anaerobe bacteria, which might otherwise hinder its expansion4,30,31, drives enterococcal domination. In support of this idea, the patients who were vanA positive already in phase I, i.e. before vancomycin prophylaxis, tended to lose more diversity in phases II and III (Figure S1M) and become dominated by Enterococcus (Figure S1M).
Risk of bloodstream infection changes with microbiota ecology.
To investigate how strong selection and gut microbiota ecology affect patients’ health, we turned to the most immediate impact of altered microbiome community in these patients: the translocation of gut bacteria into the blood leading to life-threatening bacteremia32. Previous studies have linked gut dominations by Enterococcus and gram negative proteobacteria to bacteremia8,9. For our analyses, we, therefore, selected the last stool sample collected ⩽7 days before a patient developed an enterococcal or gamma proteobacterial bacteremia (Figure 3A). This showed—consistent with previous studies—that most enterococcal infections followed Enterococcaceae domination and most gamma proteobacterial infections followed gamma proteobacteria domination. To identify specific ASVs associated with infection, we compiled a list of 10 candidate ASVs that were either the most abundant or had the largest increase before infections (see methods). BLAST analysis revealed that their 16S rRNA gene sequences matched the bloodstream isolate; each of these 10 ASVs are therefore a potential risk factor for gut-to-blood translocation, especially if they were enriched in patients who later had a BSI relative to similar patients who did not have a BSI. To test this statistically, we conducted a multivariate Bayesian logistic regression comparing BSI-cases to matched samples from uninfected patients (methods, Figure 3B); the model takes as input the abundances of all 10 candidate ASVs in a stool sample and predicts the infection risk (AUC: 0.73, Figure 3C-E). The posterior distributions confirmed that 3 out of those 10 ASVs—one belonging to the Enterococcaceae and two to the gamma proteobacterial Enterobacteriaceae family—were positively associated with a higher risk of infection (Figure 3C). We displayed the calculated risk score predicted by the model on the atlas to chart regions of highest infection risk (Figure 3B). While high-risk samples (Figure 3B) localize in both the Enterococcaceae- and Enterobacteriaceae-dominated regions (Figure 3D), the enterobacterial-enriched region (Figure 3B inset) had the highest risk. This region is enriched in ASV-3, classified as a group of Escherichia-Shigella and further characterized by metagenomic sequencing (Figure 3E, Figure 4).
Figure 3. BSI risk scores across the TaxUMAP suggests reduced risk-associated ASV-36-Klebsiella may exclude high risk-associated Escherichia.

A) TaxUMAP visualization of samples followed by a BSI within the next week, no infection (grey), BSI by gamma proteobacteria (dark red), or enterococcal species (green); histogram shows relative frequencies of blood isolate taxa. B) TaxUMAP of the mean a posteriori predicted BSI risk per sample from a Bayesian logistic regression comparing stool compositions between BSI cases and uninfected patients using the log10-relative ASV abundances of 10 predictor ASVs, the histogram shows the distribution of posterior risk predictions across all samples; inset: magnified region with the highest predicted risk samples. C) Posterior coefficient distributions (circle: mean, box: HDI50, whiskers HDI95). D) High risk samples are located in the Enterobacteriaceae dominated TaxUMAP region (top: TaxUMAP with samples labeled by the most prevalent taxon as in Figure 1I); ASV-3 and ASV-568 (increased risk) and ASV-36 (reduced risk) relative abundances across the TaxUMAP and in the magnified Enterobacteriaceae dominated region. E) Receiver-operator characteristic of the Bayesian model. F) Relative abundances of ASV-3 and ASV-36 across all samples where Enterobacteriaceae represented the most abundant family (red), or not (black). Blood isolate organism abbreviations) EFVR: E. faecium vancomycin resistant, Esch: Escherichia, KP: K. pneumoniae, Ebct: Enterobacter, EF: E. faecium, P: Pseudomonas, SM: Stenotrophomonas maltophilia, EVR: Enterococcus vancomycin resistant, EFlis: E. faecalis, K: Klebsiella, C: Citrobacter; abbreviations in ROC, E) TPR: true positive rate, FPR: false positive rate.
Figure 4. Ecological interference by ASV-36-Klebsiella isolates prevents expansion of BSI-associated ASV-3-Escherichia isolates in in vitro competitive exclusion assays.

A) Phylogenetic tree representing 17 ASV-36-Klebsiella isolates and five ASV-3-E. coli isolates; colors indicate isolates coming from the same patient. See also Figure S2 and Table S1. B) Results of ecological invasion assays. Bars represent mean CFU counts of ASV-3-E. coli isolates after 12h of incubation. Rows correspond to each of the ASV-3-E. coli isolates: red and orange bars represent isolates grown in absence of a resident ASV-36-Klebsiella community; other bars correspond to ASV-3-E. coli CFUs grown in presence of different ASV-36-Klebsiella isolates. The colors of the bars and their order indicate which of the 17 ASV-36-Klebsiella strains represented on the tree in A) was used as a competitor in the assay. Error bars: standard deviation, symbols indicate three independent experiments. CFU: colony forming unit.
Interestingly, the model revealed that ASV-36 was negatively associated with infection (Figure 3C). Visual inspection of the TaxUMAP confirmed that ASV-36 is enriched in a low-risk sub-region of the Enterobacteriaceae-enriched region (Figure 3D). ASV-3 and ASV-568, two Enterobacteriaceae taxa positively associated with infection, were highly abundant in directly adjacent regions. This suggested that high risk associated ASV-3 may not co-occur with low risk associated ASV-36; this exclusion was supported by comparing the relative abundances of ASV-3 and ASV-36 across all samples (Figure 3F). ASV-36 had made it to the list of 10 candidates because it was highly abundant preceding three BSI cases where the bacteria isolated from the patient’s blood were annotated as “Klebsiella sp.”. The 16S rRNA gene sequence of ASV-36 precisely matches several species of the Klebsiella genus, including K. michiganensis and K. oxytoca (Figure S2), but not K. pneumoniae.
Experiments validate ASV-36-Klebsiella ability to exclude bacteria associated with high infection risk.
Our analysis so far shows that ASV-36-Klebsiella contribute negatively to the infection risk model, and the TaxUMAP suggested this ASV may exclude other bacteria associated with higher risk of infection like ASV-3-E. coli. This finding was especially intriguing because two recent studies showed in mice that similar strains of Klebsiella could protect against pathogenic Enterobacteriaceae by mechanisms of nutrient competition23,24. Our results suggest ASV-36-Klebsiella could be an ecological competitor of other bacteria associated with high risk of infection in humans.
We then sought to characterize ASV-36-Klebsiella beyond the resolution of 16S amplicon sequencing. We first analyzed the subset of 16 shotgun-sequenced samples from 4 unique patients containing ASV-36-Klebsiella and identified eight metagenomically-assembled genomes (MAGs, Figure S2). Two of those MAGs came from one patient and matched K. michiganensis; six other MAGs came from another patient and matched K. grimontii. We then obtained 17 strain isolates by plating patients’ stool samples on selective MacConkey agar (Table S1). Whole genome sequencing allowed the construction of a phylogenetic tree of the 17 ASV-36 isolates, which also comprised type strains and the murine K. michiganensis ARO112 strain23, as well as the high-risk associated ASV-3 isolates (Figure 4A). The tree shows that isolates from the same patient are clonal, indicating a larger inter- than intra-individual variability among strains subsumed by ASV-36-Klebsiella. A larger phylogenetic tree constructed from our isolates alongside all published genomes containing the ASV-36-Klebsiella 16S rRNA gene sequence shows that isolates from three patients colocalized in the branch defined mostly by K. michiganensis genomes, while isolates obtained from one patient positioned on the branch dominated by K. oxytoca genomes (Figure S2). These results indicate that ASV-36-Klebsiella represents several related species of Klebsiella, including K. oxytoca and K. michiganensis, but not K. pneumoniae.
The TaxUMAP atlas (Figure 3D) suggested a competitive exclusion between ASV-36-Klebsiella and other, potentially more virulent Enterobacteriaceae, such as ASV-3. If this was the case, therefore, an established community of ASV-36-Klebsiella would interfere with ASV-3 colonization. To validate this mechanism experimentally we isolated 5 different ASV-3 strains (Table S1), and confirmed they classify as E. coli. To test our first hypothesis—that an established ASV-36-Klebsiella community could prevent ASV-3-E. coli colonization, as suggested by the TaxUMAP—we performed a simple experiment to test the minimum requirement for such competition. We first established communities of each of the 17 isolated strains in vitro by growing them in LB media to stationary phase. We then introduced ASV-3-E. coli and measured their growth compared to their growth in a sterile LB flask. If ASV-3-E. coli and ASV-36-Klebsiella shared an ecological niche, including similar or identical nutrient requirements, we would expect a growth reduction of ASV-3-E. coli. Indeed, after 12 more hours of incubation, ASV-3-E. coli reached densities of up to 108 CFUs when unimpeded by an established ASV-36-Klebsiella (Figure 4B). But the counts dropped precipitously if their niche was previously colonized by ASV-36-Klebsiella (Figure 4B). Out of the 17 ASV-36-Klebsiella isolates, 11 entirely prevented expansion of all five tested ASV-3-E. coli isolates (limit of detection: 1x106 CFUs/100μl, see Methods). The 6 remaining ASV-36-Klebsiella isolates also strongly inhibited expansion of ASV-3-E. coli. In summary, every one of the tested ASV-36-Klebsiella isolates prevented expansion of ASV-3-E. coli. This, together with the findings from ARO112 in mice23, suggests that the phenotype of ecological exclusion is conserved across a taxonomic group that includes K. oxytoca and K. michiganensis, potentially explaining the pattern seen on the TaxUMAP which suggests mutual exclusivity (Figure 3D).
In vitro competition reveals carbon sources that provide an advantage to ASV-36-Klebsiella over ASV-3-E. coli.
In order to determine the conditions that would favor ASV-36-Klebsiella versus ASV-3-E. coli, we quantified their fitness ratio through direct competition in BIOLOG Phenotype MicroArray- PM1 and PM2a plates containing 190 different carbon sources. The post-competition ratio measured after 24h (Figure 5) showed that the majority of the tested carbon sources favored ASV-36 in aerobiosis: when we analyzed carbon sources where the ASV-36 to ASV-3 ratio was significantly different from the one in the inoculum we saw that ASV-36 outcompeted ASV-3 on 76 carbon sources (61%, p<0.05 after multiple hypothesis correction from linear mixed-effects model; experiment run in triplicate; Table S2). In anaerobic conditions, ASV-36 outcompeted ASV-3 on 59 carbon sources (49%, p<0.05 after multiple hypothesis correction, linear mixed-effects model; experiment run in triplicate; Table S3). Forty-three of the tested carbon sources favored ASV-36 regardless of the presence of oxygen (Figure 5, upper right quadrant). This includes metabolites such as the sugars sucrose and maltose, known to be available to gut bacteria and important for their fitness33,34. From this observation, we hypothesized the existence of simple common chemical characteristics across carbon sources that favor ASV-36; but a correlation of the competition index (ASV-36 to ASV-3 ratio) with the molecular weights, as well as with the number of carbons in each carbon source showed no significant trend (Figure S3). Of note, however, the in vitro experiments revealed that the outcome of the competition in certain carbon sources might change with oxygen levels: 10 of the carbon sources that favored ASV-3 in anaerobic conditions changed to favoring ASV-36 in aerobic conditions (Figure 5, lower right quadrant), while one carbon source worked in the opposite direction, i.e. favoring ASV-36 in anaerobic conditions but ASV-3 in aerobic conditions (D-Lactic Acid Methyl Ester, Figure 5, upper left quadrant). This indicates that a switch from anaerobic to aerobic environments provides an almost exclusive advantage to ASV-36. All together, these results indicate that perturbations which compromise the anaerobiosis of the gut lumen would tend to favor ASV-36-Klebsiella.
Figure 5. ASV-36-Klebsiella and ASV-3-E. coli in vitro competition results depend on the available nutrients and oxygen concentration.

On the left is the scheme representing the setup of in vitro competition experiments. On the right is a scatter plot representing log10 ratio of ASV-36 to ASV-3 after 24 hours of growth in 190 tested carbon sources in aerobic and anaerobic conditions. ASV-36-Klebsiella outcompetes ASV-3-E. coli in 59 different carbon sources in the absence of oxygen, (upper half of the scatter plot; p < 0.05 after multiple hypothesis testing; linear mixed-effects model; N=3 independent experiments). In aerobiosis the number of carbon sources that favor ASV-36 rises to 76 (right half of the plot; p < 0.05 after multiple hypothesis testing; linear mixed-effects model; N=3 independent experiments). In blue are marked carbon sources that favor ASV-36, in red those that favor ASV-3 and in gray those in which ASV-36 to ASV-3 ratio differences were non-significant with respect to the inoculum. The numbers of carbon sources favoring ASV-36 (blue) and ASV-3 (red) are displayed. See also Figure S3, Table S2 and Table S3.
AS-36-Klebsiella are more competitive in vivo.
Our in vitro assays showed that environmental conditions could alter the outcome of competition between ASV-36 and ASV-3. The implications of this context-dependent fitness for patients’ microbiomes are difficult to extrapolate: nutrients available in the gut depend on several factors including diet35,36 and the microbiome composition of other gut bacteria that will also consume and release metabolites37,38. Several factors can impact anaerobiosis, including the host’s intestinal inflammation an epithelial damage39-41, which is frequent in allo-HCT patients, and could raise the concentration of oxygen in the lumen.
To evaluate competition between ASV-3 and ASV-36 in vivo, we conducted two types of experiments, measuring competition either during simultaneous inoculation (Figure 6A, “race scenario”), and in the form of successive invasion into an established community (Figure 6B, “invasion scenario”). In both scenarios, we first treated mice with a cocktail of antibiotics (ampicillin, vancomycin and neomycin: AVN).
Figure 6. ASV-36-Klebsiella and ASV-3-E. coli competition in the gut of antibiotic and DSS treated animals favors ASV-36.

A) ASV-36-Klebsiella outcompetes ASV-3-E. coli in vivo in a mouse model of antibiotic-induced co-colonization and in the model of epithelial damage through antibiotic treatment in combination with dextran sodium sulfate (DSS) when both strains are inoculated simultaneously (“race scenario”). CFU counts collected over time show a significantly faster decline of ASV-3 in mix with ASV-36 (p<10−3, linear mixed effects model, N=5 per group). B) ASV-36-Klebsiella outcompetes ASV-3-E. coli in vivo in a mouse model of antibiotic-induced co-colonization when strains are orally gavaged in a staggered manner (“invasion scenario”). ASV-3 levels decrease faster in the group of animals pre-colonized with ASV-36 (p=0.065, linear mixed effects model; N=5 animals per group). See also Table S4.
In the first experiment, we mono-colonized mice with ASV-3-E.coli or ASV-36-Klebsiella and colonized one group with a mixed culture in 1:1 ratio. Both ASV-3 and ASV-36 could colonize the gut after AVN treatment (Figure 6A), and, as expected23, their abundances decreased over time during microbiome recovery from antibiotics. A joint analysis with a mixed effects model of the CFU time series data indicated that in mono-colonized mice, ASV-3 CFU counts decreased 73% faster than ASV-36 counts (coefficient ASV-36: −0.045, and ASV-3: −0.078, p<10−3 and p=0.017, respectively) and that this ASV-3 decrease was further accelerated in the mix with ASV-36 (p=0.06), whereas the ASV-36 decrease was significantly slowed in presence of ASV-3 (p=0.003). This suggested that in co-colonized animals and over time ASV-36 could outcompete ASV-3 (Figure 6A, Table S4). Next, to determine if host-induced environmental perturbations impacted the advantage of ASV-36 mixed with ASV-3, we conducted an additional experiment (Figure 6A) where we treated mice with dextran sodium sulfate (DSS) in addition to AVN. DSS is a model for inflammatory bowel disease (IBD) that leads to increased oxygenation of the gut, perturbs the intestinal barrier function and increases intestinal permeability more so than what is typically observed in IBD patients42; DSS treatment thus constitutes a major perturbation to epithelial homeostasis that is also expected in chemotherapeutically treated cancer patients. In AVN+DSS treated mice inoculated with both ASV-3 and ASV-36, ASV-3 decreased 240% faster than ASV-36 (coefficient ASV-36: −0.029, and ASV-3: −0.099, p<10−3 and p=0.017, respectively), a stronger effect than in mice not treated with DSS (Figure 6A). Taken together, these results show that while in mono-colonized mice, both ASVs’ CFU counts decrease over time, ASV-36-Klebsiella outcompete ASV-3-E. coli in vivo when inoculated simultaneously, and that this competitive advantage is robust to extreme environmental perturbations.
Next, we asked if previous colonization by ASV-36 would impair ASV-3 colonization when invading the gut at a later time, or vice versa, similar to our in vitro assay (Figure 4). To test this, we first colonized mice with ASV-36 or with ASV-3, and one week later, we orally gavaged the other strain (with corresponding mono-colonized controls Figure 6B). Consistent with our “race” scenario experiments, we observed an accelerated decline of ASV-3 in mice previously colonized by ASV-36 (Figure 6B, p=0.065). In summary, the data obtained from our in vivo experiments support a model where ASV-36 colonizes the mouse gut more robustly and may benefit from ASV-3 presence, while ASV-3 clearance is accelerated in the presence of ASV-36.
DISCUSSION
Here we present TaxUMAP, a taxonomy-aware tool to display large longitudinal clinical microbiome data. Across all our samples, antibiotic-exposed and not exposed, the most diverse gut microbiota samples—which tended to be those collected prior to antibiotic treatment—also harbored the most diverse repertoire of antimicrobial resistance genes. This may result from fierce competition between commensal microbes that wield antibiotics to kill other species and have counteracting resistance mechanisms as part of the microbial warfare arsenal43,44. Antibiotic exposure was associated with reduced microbiota diversity and the number of antimicrobial resistance genes. This correlation between the number of resistance genes and microbiota diversity may result from selection for resistance to specific antibiotics. This finding has implications for interpreting metagenomic counting of antimicrobial resistance genes: antibiotics select for specific genes rather than expanding the antibiotic resistance gene repertoire overall. Therefore, a large resistance repertoire could be part of a diverse microbiome flora. We also saw that microbiota α-diversity correlates with the total bacterial load when we corrected for stool consistency. This observation confirms that stool consistency is an important—though often neglected—variable45.
Related bacteria can form nutrient-exchanging food webs with potentially mutually beneficial effects on their growth37; however, most microbiome species and bacterial taxa generally compete with one another28,47-49. Such facilitation or interference could reveal tangible microbiome engineering principles if the direction of interactions could be identified49. We investigated facilitation or interference between strains with a plausible causal health effect on hospitalized patients. The TaxUMAP atlas suggested that ASV-36-Klebsiella, associated with a reduced risk for BSIs, exclude high BSI-risk associated ASV-3-E. coli. By testing if high-risk associated ASV-3 isolates would establish themselves in an existing community of ASV-36-Klebsiella we confirmed experimentally the exclusion suspected from the TaxUMAP visualization.
Such an exclusion effect might be expected when cell densities in an established culture are already high. Many nutrients might have been consumed such that newly arriving stains fail to grow on this spent medium, but only when the competing strains share nutrient requirements and can utilize the same available nutrients rather than facilitate each other by cross feeding37. When we tested the growth capacity of ASV-36-Klebsiella and ASV-3-E.coli in different nutrient sources we found a range of carbon sources in which both strains can grow. Among carbon sources that boosted ASV-36 growth in both aerobic and anaerobic conditions are beta-Methyl-D-Glucoside, D-cellobiose, sucrose, inositol, that were previously described to provide growth advantage to protective K. oxytoca strains over multidrug-resistant K. pneumoniae24 Interestingly, aerobiosis favored ASV-36 in more carbon sources than what was observed in anaerobic conditions. This might be relevant for patients receiving antibiotics and suffering from gut inflammation, since both factors may increase oxygen in the gut50,51. Thus, our in vitro results demonstrate that ASV-36 and ASV-3 might compete for a niche and that this competition might be tilted in favor of ASV-36 in a more oxygenated environment.
Our experiments in vivo indicate that ASV-36 outcompetes ASV-3, and we provide evidence that ASV-36 may interfere with ASV-3. However, since ASV-3 clearance from the gut of mice appeared to occur generally more rapidly, the impact of this accelerated clearance via interference from ASV-36 in patients is unclear. Nevertheless, when we tested how staggered colonization would impact ASV-3 colonization levels, we confirmed that ASV-3 remained the poorer colonizer when ASV-36 was first to colonize the gut, as well as when ASV-3 was given the advantage of the first colonizer.
Our experiments demonstrate that ASV-36 outcompetes ASV-3 both in antibiotic-treated animals, with or without an epithelial barrier damaged by DSS, both in simultaneous and staggered co-colonization experiments. These results agree with recent mouse studies where related strains of Klebsiella competitively excluded pathogenic Enterobacteriaceae23,24. Our study indicates that the same mechanism occurs in patients: a low-risk gram-negative enterobacterium could exclude the subsequent establishment of pathobiont enterobacteria in a perturbed microbiome, thereby reducing the risk of infection. In addition to the ongoing attempts to utilize complex consortia for therapeutic approaches, low-risk relatives of health-risk species may be mined for potential interventions.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Joao B. Xavier (xavierj@mskcc.org).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
Sequencing files generated by whole genome sequencing of bacterial isolates have been submitted to NCBI and are publicly available (Table S1 and key resources table). Genomes used for the construction of phylogenetic trees are publicly available (key resources table). This paper analyzes existing, publicly available patients’ data. The accession numbers for the datasets are listed in the key resources table.
The TaxUMAP algorithm is available on GitHub (https://github.com/jsevo/taxumap). DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AD9 | This paper | SRR14131380 |
| AD10 | This paper | SRR14131379 |
| AD11 | This paper | SRR14131378 |
| AD12 | This paper | SRR14131376 |
| AD24 | This paper | SRR14131383 |
| AD25 | This paper | SRR14131382 |
| AD26 | This paper | SRR14131381 |
| AD30 | This paper | SRR14131374 |
| AD31 | This paper | SRR14131373 |
| AD32 | This paper | SRR14131372 |
| AD33 | This paper | SRR14131389 |
| AD34 | This paper | SRR14131387 |
| AD35 | This paper | SRR14131386 |
| AD36 | This paper | SRR14131385 |
| AD37 | This paper | SRR14131384 |
| AD38 | This paper | SRR14131400 |
| AD39 | This paper | SRR14131399 |
| AD40 | This paper | SRR14131388 |
| AD41 | This paper | SRR14131377 |
| AD42 | This paper | SRR14131375 |
| AD48 | This paper | SRR16077561 |
| AD49 | This paper | SRR16077560 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin | Nova Plus | Cat#0781-9408-80 |
| Vancomycin hydrochloride | Fisher Scientific | Cat#J62790.06 |
| Neomycin sulfate hydrate | Fisher Scientific | Cat#J61499.14 |
| Dextran Sulfate Sodium Salt | Fisher Scientific | Cat#AAJ6278722 |
| LB broth, Miller | Fisher Scientific | Cat#BP1426-500 |
| Critical commercial assays | ||
| MacConkey agar II | Fisher Scientific | Cat#B21172X |
| BIOLOG PM1 plates | Fisher Scientific | Cat#NC9040419 |
| BIOLOG PM2A plates | Fisher Scientific | Cat#NC0058821 |
| QIAamp Fast DNA Stool Kit | Qiagen | Cat#51604 |
| Quant-iT dsDNA Assay Kit, High Sensitivity | Invitrogen | Cat#Q33120 |
| Nextera XT DNA Library preparation kit | Illumina | Cat#FC-131-1096 |
| MiSeq Reagent Kit v3 | Illumina | Cat#15043895 |
| Deposited data | ||
| Liao et al. (2021); Yan et al. (2022) | SRA | PRJNA394877, PRJNA607574, PRJNA606262, PRJNA548153, PRJNA545312 |
| Genomes of the isolates reported in this paper | This paper | PRJNA545312; PRJNA606262; PRJNA607574 |
| Genome of Klebsiella sp. Kd70 TUC-EEAOC | SRA | PRJNA285494 |
| Genome of Klebsiella michiganensis strain ARO112 | SRA | PRJNA590204 |
| Genome of Klebsiella oxytoca strain CAV1374 | SRA | PRJNA246471 |
| Genome of Klebsiella oxytoca KCTC 1686 | SRA | PRJNA65523 |
| Genome of K. pneumoniae subsp. pneumoniae HS11286 | SRA | PRJNA84387 |
| Genome of K. michiganensis DSM25444 | SRA | PRJNA388837 |
| Genome of K. grimontii 06D021 | SRA | PRJEB21765 |
| Genome of K. oxytoca DSM5175 | SRA | PRJNA543274 |
| Genome of E. coli ATCC11775 | SRA | PRJNA472652 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6/J | The Jackson Laboratory | JAX Cat#000664; RRID: IMSR_JAX:000664; Room #EM03 |
| Oligonucleotides | ||
| PCR for Sanger sequencing: 16S Forward 27F 5’-AGAGTTTGATCMTGGCTCAG-3’ | IDT | N/A |
| PCR for Sanger sequencing: 16S Reverse 803R 5’-CTACCRGGGTATCTAATCC-3’ | IDT | N/A |
| Software and algorithms | ||
| TaxUMAP | This paper | https://github.com/jsjse/taxumap; DOI: 10.5281/zenodo.7958500 |
| Python v3.7.9 | Python | python.org |
| Matlab v2020a | Matlab | https://www.mathworks.com/products/matlab.html |
| Bhatt lab workflow for the genomes’ assembly | Siranosian et al., 2022 | github.com/bhattlab/bhattlab_workflows |
| PATRIC Comprehensive Genome Analysis Tool | Wattam et al., 2017 | https://www.bvbrc.org/app/ComprehensiveGenomeAnalysis |
| Prokka | Seemann, 2014 | https://github.com/tseemann/prokka |
| Roary | Page et al., 2015 | https://github.com/sanger-pathogens/Roary |
| FastTree2 | Price et al., 2010 | http://www.microbesonline.org/fasttree/ |
| LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB 2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB 477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al.1 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al.2) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children's Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: Cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard3 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al.4 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al.5 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al.6 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al.7 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg8 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al.9 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.10 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
| PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185-5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 μm, 90 mm | Scientificfilters.com/TischScientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365-0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al.1 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.2 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvacphi.com/en/products/xps/phi-quantera-ii/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Bacterial strains
All bacterial strains were routinely grown on MacConkey agar plates (BD MacConkey II agar) in both aerobic and anaerobic conditions at 37°C for 24-48h. Anaerobic conditions inside the anaerobic chamber were maintained with a gas mix containing 5% carbon dioxide, 7.5% hydrogen and nitrogen as a balance. Hydrogen levels were maintained above 3%.
Mouse model
Mice used in this study were C57BL/6J specific pathogen-free mice purchased from The Jackson Laboratories. Animals were 6-8 weeks old females. During the study, mice were single-housed in autoclaved cages with ad libitum access to autoclaved and acidified reverse osmosis water (pH, 2.5 to 2.8) and irradiated feed (LabDiet 5053, PMI, St Louis, MO). The animal holding room is maintained at 72 ± 2 °F (21.5 ± 1 °C), relative humidity between 30% and 70%, and a 12:12 hour light:dark photoperiod. Animal use is approved by Memorial Sloan Kettering Cancer Center's IACUC. The institution’s animal care and use program is AAALAC-accredited and operates in accordance with the recommendations provided in the Guide.
Human participants
Data obtained from human participants analyzed in this study has been previously published and is publicly available. The accession numbers for the datasets are listed in the key resources table.
METHOD DETAILS
Isolation of ASV-36 and ASV-3 strains
For isolation, patients’ samples with high relative abundance (>90%) of desired ASVs were plated on MacConkey agar plates (BD MacConkey II Agar). Plates were incubated in aerobic conditions at 37°C for 24-48h. Individual colonies were picked, and Sanger sequenced to confirm their identity based on 16S rRNA gene. For whole genome sequencing (WGS) a single colony was grown overnight in LB liquid media. Next day 1ml of the culture was centrifuged at 15 000rpm for 5 minutes and pellet was subjected to DNA extraction with Qiagen Fast DNA Stool Mini Kit. Nextera XT DNA Library Preparation Kit was used for library generation according to the manufacturer's instructions. Libraries were quantified with Quant-iT™ dsDNA Assay Kit, normalized and sequenced using MiSeq Reagent Kit V3. Comprehensive Genome Analysis tool in PATRIC (Wattam et al., 2017) was used for sequence processing: genomes were assembled using SPAdes (Bankevich et al., 2012) and annotated using RAST Tool Kit (Brettin et al., 2015). Phylogenetic Tree tool in PATRIC was used for codon tree generation from 1,000 randomly selected single-copy genes by using the RAxML (Stamatakis, 2006). Genome sequences are available at NCBI (Table S1).
Phylogenetic tree of ASV-36-Klebsiella
The Klebsiella genomes that contain the same 16S gene sequence as ASV-36 were obtained from NCBI. The previously studied strains K. oxytoca CAV1374, K. oxytoca KCTC1686, K. michiganensis strain ARO112, and K. sp. Kd70 TUC-EEAOC were also included and they belong to ASV-36 as well. All Klebsiella genomes including the metagenome-assembled genomes (MAGs) and our isolated ASV-36-Klebsiella strains were annotated using prokka (Seemann, 2014) and the core genome alignment was generated by roary (Page et al., 2015) to further construct the phylogenetic tree using FastTree2 (Price et al., 2010). The K. pneumoniae reference genome K. pneumoniae subsp. pneumoniae HS11286 that does not belong to ASV-36 was used to root the tree. The species information was taken from the NCBI genome annotation.
Assembly of Klebsiella genomes
The draft genomes were assembled from 16 shotgun samples (1042X, FMT.0009X, FMT.0009Y, FMT.0103Y, 668DD, 668T, 668EE, 668Y, 668CC, 668FF, 668X, 668GG, 668Z, 668W, 668MM, 668BB; accessible from Bioproject PRJNA545312) which contain at least 1% of ASV-36-Klebsiella based on 16S amplicon sequencing. The assembly pipeline was downloaded from the Bhatt lab github repository (https://github.com/bhattlab/bhattlab_workflows) and installed locally. Briefly, the computational workflows used Megahit (Li et al., 2015) for genome assembly and both Metabat2 (Kang et al., 2019) and CONCOCT (Alneberg et al., 2014) for metagenomic binning.
Ecological invasion experiments
Competitive exclusion of ASV-3-E. coli isolate invasion by resident ASV-36-Klebsiella was tested by challenging established ASV-36-Klebsiella communities with ASV-3-E. coli. To generate an established ASV-36-Klebsiella community to be invaded by ASV-3-E. coli, ASV-36 strains were grown in liquid LB medium from fresh overnight culture until reaching stationary phase. To do this ASV-36-Klebsiella strains AD9, AD10, AD11, AD12, AD30, AD31, AD32, AD33, AD34, AD35, AD36, AD37 were grown for 12h and strains AD38, AD39, AD40, AD41, AD42 were grown for 20h. The ASV-36-Klebsiella incubation times were selected based on the different growth rates among our isolates that resulted in some of the isolates reaching the stationary phase earlier than the others. Once the ASV-36-Klebsiella isolates reached the stationary phase they were inoculated (1:50) with ASV-3-E. coli isolates in the exponential phase of growth. As a control, LB medium not containing ASV-36-Klebsiella culture was also inoculated with ASV-3-E. coli. After 12h of incubation in aerobic conditions at 37°C and with shaking cultures were plated on MacConkey agar and colony forming units (CFUs) of ASV-3-E. coli in mixed cultures were compared to those observed when ASV-3-E. coli were grown alone. All ASV-36-Klebsiella isolates were tested against all ASV-3-E. coli isolates making a total of 85 tested combinations. The limit of detection was 1x106 CFUs/100μl. This limit was due to the fact that detection of both competing strains was done on the same agar plate. Overgrowth of ASV-36-Klebsiella on more concentrated plates prevented a higher resolution that could allow a lower limit of detection of ASV-3-E. coli.
Competition in different nutrients
To test in which conditions ASV-36-Klebsiella might outcompete ASV-3-E.coli we setup in vitro competition assay that tested their growth in different carbon sources in BIOLOG Phenotype MicroArray (PM) 1 and 2a in presence or absence of oxygen. A single colony of ASV-36 (AD9 strain) and ASV-3 (AD24 strain) was used to inoculate 5ml of LB, after which cultures were grown overnight at 37°C and with shaking. Next day, both cultures were centrifuged at maximum speed for 5 minutes and re-suspended in M9 minimal media lacking carbon source. This washing step was performed twice. After the third and final centrifugation step, strains were re-suspended in M9 minimal media until reaching the same OD600 absorbance of 0.05. ASV-36 and ASV-3 were mixed in 1:1 ratio and 100ul was aliquoted in each well of the BIOLOG PM1 and PM2a plates. Each BIOLOG plate contains 95 different carbon sources and allows for assessing the growth capacity of tested strains on each one of them, meaning that we tested 190 carbon sources in total. Plates were incubated inside of plate readers at 37°C and with shaking both inside the anaerobic chamber (anaerobiosis) and on the bench top (aerobiosis). After 24h aliquots from each well were serially diluted and plated on MacConkey agar plates. ASV-36 and ASV-3 colonies were counted and ASV-36 to ASV-3 ratio was determined for each BIOLOG well. The initial inoculum was plated the same way to determine the starting ASV-36/ASV-3 ratio. Experiment was done in triplicate. The limit of detection for aerobic growth was 1x106 CFUs/100μl, and for anaerobic growth 1x105 CFUs/100μl.
In vivo competition assays
In order to test the outcome of ASV-36-Klebsiella and ASV-3-E. coli competition in vivo the following experiment was performed. We treated 6-8 weeks old C57BL/6J female mice with a cocktail of ampicillin (0.5g/l), vancomycin (0.5g/l) and neomycin (1g/l) for one week in drinking water. Antibiotics were changed once during the course of the treatment. Animals were single housed in autoclaved cages. Autoclaved water supplemented with antibiotics and 5053 irradiated food was provided ad libitum. The day after antibiotic cessation animals were orally gavaged with ~107 CFUs of ASV-36-Klebsiella, ASV-3-E. coli or 1:1 mix of both strains in PBS. To test whether profound epithelial damage could alter the result of competition, one group of animals was treated with the same antibiotic cocktail and 3% dextran sodium sulfate (DSS) in the drinking water for a week. The water with antibiotics and DSS was changed once, as previously mentioned. Animals were orally gavaged with ~107 CFUs of 1:1 mix of ASV-36 and ASV-3 the day after treatment withdrawal. To test how invasion by ASV-3 and ASV-36 strains would affect competition in mice that were already colonized with opposite strains, mice were treated with the antibiotic cocktail as in the previous experiment. One day after removal of the antibiotics two groups of mice were orally gavaged with ~108 CFUs of ASV-36 and ASV-3 respectively. After one week the same animals were re-challenged with opposite strains (a group that was initially gavaged with ASV-36 was now gavaged with ~108 CFUs of ASV-3 and vice versa). At the same time, 2 other groups were orally gavaged with ~108 CFUs of ASV-36 alone and ~108 CFUs of ASV-3 strain alone as a control. In all of the experiments the levels of colonization were monitored by collecting fecal pellets and plating them in MacConkey agar plates.
vanA PCR from rectal swabs
Data obtained from clinical records; rectal swabs are routinely collected and analyzed for presence of the vanA gene via PCR on the same day a corresponding stool sample was taken.
α-diversity calculation
We calculate the inverse Simpson index () in sample based upon the relative ASV abundances () using the following equation:
Pseudo time visualization of clinical samples
While HCT is a strictly planned therapy, per patient variability arises. This leads to different clinical and medication exposures per patient per day, and for visualization purposes of trends over treatment regimens, we harmonize this by introducing a pseudo time that splits each patient’s treatment timeline into 9 segments corresponding to 3 points per each of the 3 clinical phases. We provide an antibiotic exposure summary table (Table S5) for each of the three phases, and visualize antibiotic exposure per treatment day as well as per pseudo timepoints (Figure S4).
The TaxUMAP algorithm
The TaxUMAP algorithm is publicly available as a package for the Python programming language and as a command line application. We provide an extensive online tutorial on https://github.com/jsevo/taxumap, including data used in the manuscript, as well as smaller published data sets for exploration. The algorithm starts by calculating the sample-by-sample distances on ASV relative abundances. The algorithm allows users to choose from a range of different distance metrics; here, we used the cityblock distance on relative abundances throughout, defined as between samples and represented by the composition vectors and , i.e. the element-wise absolute difference in each taxon out of all taxa. Then, the algorithm calculates additional sample-by-sample distance metrics based on user-chosen taxonomic aggregations of the relative abundances of bacterial taxa. For example, the sample by sample distances at the genus level are calculated by first summing the relative abundances of all ASVs belonging to the same genus and then calculating the L1-norm (or user defined distance metric) across all samples. The ranges of the sample-by-sample cityblock distances are the same for each taxonomic aggregation, but their magnitudes are on average equal or smaller at higher taxonomic aggregations. The TaxUMAP algorithm allows users to scale different taxonomic aggregations by assigning a weight to each aggregation; here we chose to weight distances at ASV, family, and phylum level equally, discarding distances at other levels. We also set this as the default implementation and explain in the documentation and tutorials how to change these settings (Figure S5). For example, a researcher may be less interested in phylum-level differences between samples but choose to weigh more heavily how dissimilar samples are with respect to their genus composition. The choice of the default implementation was inspired by the traditional importance of phylum level difference in microbiome samples (Ley et al., 2006), the reported conservation of important traits at the family level (Goldford et al., 2018), and the ASV level as the highest resolution data available to us. Before applying the UMAP algorithm (McInnes et al., 2018b), the weighted distance matrices are added. The UMAP algorithm is then applied with a minimum distance of , where is the weight at taxon level of taxonomy levels, 120 neighbors, 103 epochs. Each of these parameters can be altered by the user. We provide recommendations on the minimum distances and neighborhoods in the online documentation.
Analysis of TaxUMAP performance
We simulated 1,000 microbiome compositions (simSamples) of 600 simulated ASVs (simASVs), with taxonomic relationships assigned randomly such that simulated genera will have the same number of simASVs on average, the simulated families the same number of genera on average, etc. We simulated 2 phyla, with a total of 10 families, with a total of 30 genera for the 600 simASVs. The simASV abundances in a simSample were drawn from the distribution of ASV relative abundances observed in our patient dataset, and normalized to ensure they sum to 1 in a simSample. We assigned color schemes to the simulated taxonomy that visually separates the two simulated phyla (red-orange colors, and blue-green colors, respectively), with a unique color for each simulated genus. We then computed UMAP and TaxUMAP projections on the L1 sample by sample distances of the simulated dataset. For TaxUMAP, we used the same weights per taxonomic level as in the main analyses (i.e. phylum=family=ASV, and zero weights for other taxonomic levels). While non-linear dimensionality reduction techniques such as TaxUMAP prevent a simple interpretation of local and global distances between embedded data points, here we compute the 2-dimensional Euclidean distance between simSamples with the same dominant simulated genus in the UMAP or, respectively, TaxUMAP embedding, to test the improved visual appearance of TaxUMAP over naïve UMAP. We repeated this process for 10 simulated datasets. This enabled a statistical analysis of clustering of samples that are simulated to be biologically more similar, i.e. have the same dominant genus even if driven by different simASVs.
Identification of risk-associated ASVs
To analyze the associations between the microbiome bacterial composition and identified bacteria of a BSI, we sought to associate relative ASV abundances with a risk score. For this we took the following steps: 1) match BSI organism to its bacterial family, 2) identify a list of candidate translocating ASVs in BSI-preceding stool samples, 3) analyze the risk of the ASVs to cause bacteremia by comparing their abundances in BSI cases vs. control samples.
Step 2) Identification of candidate taxa
We chose ASV candidates from among ASVs belonging to the same family as the BSI causing bacterium. We selected ASVs that were most abundant and/or increased the most in the seven days prior to a BSI event. The resulting list includes specific candidate translocator ASVs. We appended a single ASV, ASV_6997, to this list corresponding to a singleton Citrobacter BSI which could not be analyzed as the other, repeatedly observed BSIs. Manual inspection showed that the corresponding stool sample prior to the Citrobacter infection (sample 727C) was enriched for an Enterobacteriaceae ASV, ASV_6997, the sequence of which was mapped to Citrobacter sedlakii via BLAST.
Step 3) Matching case and control samples from uninfected patients
We created a matched case-control data set: case samples were the stool samples closest in time prior to a BSI event; controls were samples from patients without recorded BSI event chosen such that the sex of the patient and the clinical phase of the HCT therapy during which a sample was collected matched; we chose 4 controls for every case sample.
Step 3) Bayesian logistic regression
We performed a Bayesian logistic regression using the matched cases-control data with a constrained intercept, a, of −1.38, i.e. inverse logit(a) = 0.2, such that our model assumes the defined case-control probability a priori, with standard Normal predictor coefficient prior distributions. We implemented the model using the pymc3 package for the Python programming language and sampled 4 independent posterior chains using the No-U-turn sampling method (Homan and Gelman, 2014). For posterior predictions, we used the relative abundances of predictor taxa in a given sample and calculated the mean predicted probability of BSI from 10,000 posterior samples with an intercept correction , where is the fraction of cases, the posterior intercept estimate, and the population incidence rate of BSIs (King et al., 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of the patients’ data
In all the analysis of the patients’ data significance was defined as a p-value lower than 0.05. For each of the plots the statistical test that was applied is stated in the legend of the figure, together with a p-value and sample size. Where applicable, confidence intervals are displayed.
Competition in different nutrients
Generalized linear mixed-effects model implemented in Matlab 2020a was used to determine which carbon sources had ASV-36 to ASV-3 ratio significantly different than the one detected in corresponding starting inoculum. Briefly, the model determined how different are ASV-36 CFU counts in each of the tested carbon sources with respect to the inoculum and for each of the biological replicates:
where “ASV36CFUs” represents ASV-36 CFUs in a metabolite of interest, “metaboliteCategorical” represents 190 different carbon sources and inoculum that is treated as the reference category. The distribution used for the model was binomial. The model yielded p-values for each of the carbon sources that were then subjected to false discovery rate (FDR). Values of FDR that were lower than 0.05 were considered significant. Estimates, standard errors (SE), p-values and FDR predicted by the model are displayed in Table S2 for aerobic conditions and Table S3 for anaerobic. The model was implemented in the MATLAB programming language.
In vivo competition assays
A linear mixed-effects model was used to determine the effects of ASV-36 presence on ASV-3 CFU counts in colonized mice over time. In all of the experiments the levels of colonization were monitored by collecting fecal pellets and plating them in MacConkey agar plates. We analyzed ASV-3 or ASV-36 counts over time in mono-colonized mice using the model formula:
where Time is a continuous variable representing the days of the experiment; we accounted for repeated measurements from the same mouse by including a random intercept term. To analyze the effect of co-existence in co-colonized mice, we jointly inferred interaction coefficients between time, mixed inoculation, and DSS addition, pooling information across all CFU measurements
where Mix is a boolean variable that represents co-inoculation and is zero in the mono-colonized control. The model included interaction terms between Mix and Time, as well as the interactions between these combined terms and the ASV measured. To analyze the effect of DSS perturbation, we performed a similar analysis and modeled the CFU counts observed with the model formula:
We analyzed the invasion experiment in a similar way. To model the effect of presence of a resident ASV on the dynamics of an invader, we modeled the CFU counts observed with the formula:
where Resident is a boolean variable that represents the presence of a previously inoculated resident, and is zero in the mono-colonized control. Interference results and p-values are provided in Table S4.
Supplementary Material
Table S2. Total number of ASV-36 and ASV-3 colonies counted in each well of BIOLOG PM1 and PM2A plates after 24 h incubation in aerobiosis and model summary results. Related to Figure 5.
Table S3. Total number of ASV-36 and ASV-3 colonies counted in each well of BIOLOG PM1 and PM2A plates after 24 h incubation inside of anaerobic chamber and model summary results. Related to Figure 5.
HIGHLIGHTS.
TaxUMAP charts a patient microbiome atlas with ecological and clinical context
Biodiversity and bacterial density are generally positively correlated
Antibiotics deplete biodiversity and reduce the antimicrobial resistance genes’ numbers
Certain Klebsiella spp. are associated with a lower bacteremia risk by gut pathogens
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (NIH) grants DP2 AI164318-01 to J.S.; U01 AI124275 to E.P. and J.B.X.; R01 AI137269 to J.B.X. and Y.T.; NHLBI NIH Award K08HL143189, the MSKCC Cancer Center Core Grant NCI P30 CA008748, the Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center to J.U.P.; NIH R01 AI14862303, NIH R01 AI143757, CDC BAA 75D30118C02921, Sloan Foundation Fellowship, V Foundation Fellowship, Stand Up 2 Cancer award 6206 to A.S.B.; National Cancer Institute award numbers R01-CA228358, R01-CA228308, P30 CA008748 MSK Cancer Center Support Grant/Core Grant and P01-CA023766, National Heart, Lung, and Blood Institute (NHLBI) award number R01-HL123340 and R01-HL147584, National Institute of Aging award number P01-AG052359, Starr Cancer Consortium, Tri Institutional Stem Cell Initiative, The Lymphoma Foundation, The Susan and Peter Solomon Divisional Genomics Program, Cycle for Survival, and the Parker Institute for Cancer Immunotherapy to M.R.M.v.d.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.S. is cofounder of Postbiotics Plus Research LLC. J.U.P. reports research funding, intellectual property fees, and travel reimbursement from Seres Therapeutics, and consulting fees from DaVolterra, CSL Behring, and from MaaT Pharma, serves on an Advisory board of and holds equity in Postbiotics Plus Research LLC, has filed intellectual property applications related to the microbiome (reference numbers #62/843,849, #62/977,908, and #15/756,845). M.-A.P. received honoraria from AbbVie, Bellicum, Celgene, Bristol Myers Squibb, Incyte, Merck, Novartis, Nektar Therapeutics, Omeros, and Takeda, served on data safety monitoring boards for Cidara Therapeutics, Servier, and Medigene and scientific advisory boards for MolMed and NexImmune, has received research support for clinical trials from Incyte, Kite/Gilead, and Miltenyi Biotec, has served as a volunteer for and as a member of the Board of Directors of American Society for Transplantation and Cellular Therapy and Be The Match (National Marrow Donor Program), and on the Center for International Blood and Marrow Transplant Research Cellular Immunotherapy Data Resource Committee. A.S.B. is on the advisory board of Caribou Biosciences, and ArcBio, and has served as a paid consultant for BiomX and Guardant Health. M.R.M.v.d.B. has received research support and stock options from Seres Therapeutics and stock options from Notch Therapeutics and Pluto Therapeutics; has received royalties from Wolters Kluwer; has consulted, received honorarium from or participated in advisory boards for Seres Therapeutics, WindMIL Therapeutics, Rheos Medicines, Merck & Co, Inc., Magenta Therapeutics, Frazier Healthcare Partners, Nektar Therapeutics, Notch Therapeutics, Forty Seven Inc., Ceramedix, Lygenesis, Pluto Therapeutics, GlaskoSmithKline, Da Volterra, Novartis (Spouse), Synthekine (Spouse), and Beigene (Spouse), has IP Licensing with Seres Therapeutics and Juno Therapeutics, he holds a fiduciary role on the Foundation Board of DKMS (a nonprofit organization). E.G.P. has received speaker honoraria from Bristol Myers Squibb, Celgene, Seres Therapeutics, MedImmune, Novartis, and Ferring Pharmaceuticals and is an inventor on patent application #WPO2015179437A1 (entitled “Methods and compositions for reducing Clostridium difficile infection”) and #WO2017091753A1 (entitled “Methods and compositions for reducing vancomycin-resistant enterococci infection or colonization”) and holds patents that receive royalties from Seres Therapeutics. All other authors declare no competing interests.
References
- 1.Manzo VE, and Bhatt AS (2015). The human microbiome in hematopoiesis and hematologic disorders. Blood 126, 311–318. 10.1182/blood-2015-04-574392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, and Brodin P (2018). Stereotypic Immune System Development in Newborn Children. Cell 174, 1277–+. 10.1016/j.cell.2018.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buffie CG, and Pamer EG (2013). Microbiota-mediated colonization resistance against intestinal pathogens. Nature Reviews Immunology 13, 790–801. 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caballero S, Kim S, Carter RA, Leiner IM, Susac B, Miller L, Kim GJ, Ling LL, and Pamer EG (2017). Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enterococcus faecium. Cell Host & Microbe 21, 592–+. 10.1016/j.chom.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences of the United States of America 108, 4554–4561. 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling LL, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–U207. 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morjaria S, Schluter J, Taylor BP, Littmann ER, Carter RA, Fontana E, Peled JU, van den Brink MRM, Xavier JB, and Taur Y (2019). Antibiotic-Induced Shifts in Fecal Microbiota Density and Composition during Hematopoietic Stem Cell Transplantation. Infection and Immunity 87, e00206–19. 10.1128/iai.00206-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoma I, Littmann ER, Peled JU, Giralt S, van den Brink MRM, Pamer EG, and Taur Y (2021). Compositional Flux Within the Intestinal Microbiota and Risk for Bloodstream Infection With Gram-negative Bacteria. Clinical Infectious Diseases 73, e4627. 10.1093/cid/ciaa068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, Lee YJ, Dubin KA, Socci ND, Viale A, et al. (2012). Intestinal Domination and the Risk of Bacteremia in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Clinical Infectious Diseases 55, 905–914. 10.1093/cid/cis580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, Weber D, Hashimoto D, Slingerland AE, Slingerland JB, et al. (2020). Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. New England Journal of Medicine 382, 822–834. 10.1056/NEJMoa1900623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djukovic A, Garzón MJ, Canlet C, Cabral V, Lalaoui R, García-Garcerá M, Rechenberger J, Tremblay-Franco M, Peñaranda I, and Puchades-Carrasco L (2022). Lactobacillus supports Clostridiales to restrict gut colonization by multidrug-resistant Enterobacteriaceae. Nature Communications 13, 5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Djukovic A, González-Barberá EM, Sanz J, Artacho A, Peñaranda I, Herrera B, Garzón MJ, Salavert M, López-Hontangas JL, and Xavier KB (2020). High heterogeneity of multidrug-resistant Enterobacteriaceae fecal levels in hospitalized patients is partially driven by intravenous β-lactams. Antimicrobial agents and chemotherapy 64, e01415–01419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernard-Raichon L, Venzon M, Klein J, Axelrad JE, Zhang CZ, Sullivan AP, Hussey GA, Casanovas-Massana A, Noval MG, Valero-Jimenez AM, et al. (2022). Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia. Nature Communications 13, 5926. 10.1038/s41467-022-33395-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamburini FB, Andermann TM, Tkachenko E, Senchyna F, Banaei N, and Bhatt AS (2018). Precision identification of diverse bloodstream pathogens in the gut microbiome. Nature Medicine 24, 1809–+. 10.1038/s41591-018-0202-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xavier JB, Young VB, Skufca J, Ginty F, Testerman T, Pearson AT, Macklin P, Mitchell A, Shmulevich I, Xie L, et al. (2020). The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends in Cancer 6, 192–204. 10.1016/j.trecan.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter J, Armet AM, Finlay BB, and Shanahan F (2020). Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 180, 221–232. 10.1016/j.cell.2019.12.025. [DOI] [PubMed] [Google Scholar]
- 17.Gerber GK (2014). The dynamic microbiome. Febs Letters 588, 4131–4139. 10.1016/j.febslet.2014.02.037. [DOI] [PubMed] [Google Scholar]
- 18.Card D, and Krueger AB (1993). Minimum wages and employment: A case study of the fast food industry in New Jersey and Pennsylvania. National Bureau of Economic Research Cambridge, Mass., USA. [Google Scholar]
- 19.Kobak D, and Linderman GC (2021). Initialization is critical for preserving global data structure in both t-SNE and UMAP. Nature Biotechnology 39. 10.1038/s41587-020-00809-z. [DOI] [PubMed] [Google Scholar]
- 20.McInnes L, Healy J, and Melville J (2018). Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426. [Google Scholar]
- 21.Liao C, Taylor BP, Ceccarani C, Fontana E, Amoretti LA, Wright RJ, Gomes ALC, Peled JU, Perales MA, van den Brink MRM, et al. (2021). Compilation of longitudinal microbiota data and hospitalome from hematopoietic cell transplantation patients. Scientific Data 8, 71. 10.1038/s41597-021-00860-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan JY, Liao C, Taylor BP, Fontana E, Amoretti LA, Wright RJ, Littmann ER, Dai AQ, Waters N, Peled JU, et al. (2022). A compilation of fecal microbiome shotgun metagenomics from hematopoietic cell transplantation patients. Scientific Data 9, 219. 10.1038/s41597-022-01302-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliveira RA, Ng KM, Correia MB, Cabral V, Shi HD, Sonnenburg JL, Huang KC, and Xavier KB (2020). Klebsiella michiganensis transmission enhances resistance to Enterobacteriaceae gut invasion by nutrition competition. Nature Microbiology 5, 630–+. 10.1038/s41564-019-0658-4. [DOI] [PubMed] [Google Scholar]
- 24.Osbelt L, Wende M, Almasi E, Derksen E, Muthukumarasamy U, Lesker TR, Galvez EJC, Pils MC, Schalk E, Chhatwal P, et al. (2021). Klebsiella oxytoca causes colonization resistance against multidrug-resistant K. pneumoniae in the gut via cooperative carbohydrate competition. Cell Host & Microbe 29, 1663–+. 10.1016/j.chom.2021.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Schluter J, Peled JU, Taylor BP, Markey KA, Smith M, Taur Y, Niehus R, Staffas A, Dai AQ, Fontana E, et al. (2020). The gut microbiota is associated with immune cell dynamics in humans. Nature 588. 10.1038/s41586-020-2971-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armstrong G, Martino C, Rahman G, Gonzalez A, Vazquez-Baeza Y, Mishne G, and Knight R (2021). Uniform Manifold Approximation and Projection (UMAP) Reveals Composite Patterns and Resolves Visualization Artifacts in Microbiome Data. Msystems 6, e00691–21. 10.1128/mSystems.00691-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.May RM (1972). Will a large complex system be stable? Nature 238, 413–414. [DOI] [PubMed] [Google Scholar]
- 28.Coyte KZ, Schluter J, and Foster KR (2015). The ecology of the microbiome: Networks, competition, and stability. Science 350, 663–666. 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- 29.Montassier E, Valdes-Mas R, Batard E, Zmora N, Dori-Bachash M, Suez J, and Elinav E (2021). Probiotics impact the antibiotic resistance gene reservoir along the human GI tract in a person-specific and antibiotic-dependent manner. Nature Microbiology 6, 1043–+. 10.1038/s41564-021-00920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ubeda C, Bucci V, Caballero S, Djukovic A, Toussaint NC, Equinda M, Lipuma L, Ling LL, Gobourne A, No D, et al. (2013). Intestinal Microbiota Containing Barnesiella Species Cures Vancomycin-Resistant Enterococcus faecium Colonization. Infection and Immunity 81, 965–973. 10.1128/iai.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MRM, Kamboj M, and Pamer EG (2010). Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. Journal of Clinical Investigation 120, 4332–4341. 10.1172/jci43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whangbo J, Ritz J, and Bhatt A (2017). Antibiotic-mediated modification of the intestinal microbiome in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplantation 52, 183–190. 10.1038/bmt.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones SA, Jorgensen M, Chowdhury FZ, Rodgers R, Hartline J, Leatham MP, Struve C, Krogfelt KA, Cohen PS, and Conway T (2008). Glycogen and maltose utilization by Escherichia coli O157 : H7 in the mouse intestine. Infection and Immunity 76, 2531–2540. 10.1128/iai.00096-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Townsend GE, Han WW, Schwalm ND, Raghavan V, Barry NA, Goodman AL, and Groisman EA (2019). Dietary sugar silences a colonization factor in a mammalian gut symbiont. Proceedings of the National Academy of Sciences of the United States of America 116, 233–238. 10.1073/pnas.1813780115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–+. 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson AJ, Vangay P, Al-Ghalith GA, Hillmann BM, Ward TL, Shields-Cutler RR, Kim AD, Shmagel AK, Syed AN, Walter J, et al. (2019). Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans. Cell Host & Microbe 25, 789–+. 10.1016/j.chom.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 37.Rakoff-Nahoum S, Foster KR, and Comstock LE (2016). The evolution of cooperation within the gut microbiota. Nature 533, 255–+. 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ze XL, Duncan SH, Louis P, and Flint HJ (2012). Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. Isme Journal 6, 1535–1543. 10.1038/ismej.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chanin RB, Winter MG, Spiga L, Hughes ER, Zhu WH, Taylor SJ, Arenales A, Gillis CC, Buttner L, Jimenez AG, et al. (2020). Epithelial-Derived Reactive Oxygen Species Enable AppBCX-Mediated Aerobic Respiration of Escherichia coli during Intestinal Inflammation. Cell Host & Microbe 28, 780–+. 10.1016/j.chom.2020.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Litvak Y, Byndloss MX, and Baumler AJ (2018). Colonocyte metabolism shapes the gut microbiota. Science 362, 1017–+, eaat9076. 10.1126/science.aat9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rigottier-Gois L (2013). Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. Isme Journal 7, 1256–1261. 10.1038/ismej.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cochran KE, Lamson NG, and Whitehead KA (2020). Expanding the utility of the dextran sulfate sodium (DSS) mouse model to induce a clinically relevant loss of intestinal barrier function. Peerj 8, e8681. 10.7717/peerj.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donia MS, Cimermancic P, Schulze CJ, Brown LCW, Martin J, Mitreva M, Clardy J, Linington RG, and Fischbach MA (2014). A Systematic Analysis of Biosynthetic Gene Clusters in the Human Microbiome Reveals a Common Family of Antibiotics. Cell 158, 1402–1414. 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mavridou DAI, Gonzalez D, Kim W, West SA, and Foster KR (2018). Bacteria Use Collective Behavior to Generate Diverse Combat Strategies. Current Biology 28, 345–+. 10.1016/j.cub.2017.12.030. [DOI] [PubMed] [Google Scholar]
- 45.Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, and Raes J (2016). Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65, 57–62. 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhai B, Ola M, Rolling T, Tosini NL, Joshowitz S, Littmann ER, Amoretti LA, Fontana E, Wright RJ, Miranda E, et al. (2020). High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nature Medicine 26, 59–+. 10.1038/s41591-019-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foster KR, and Bell T (2012). Competition, Not Cooperation, Dominates Interactions among Culturable Microbial Species. Current Biology 22, 1845–1850. 10.1016/j.cub.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 48.Foster KR, Chluter JS, Oyte KZC, and Rakoff-Nahoum S (2017). The evolution of the host microbiome as an ecosystem on a leash. Nature 548, 43–51. 10.1038/nature23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmer JD, and Foster KR (2022). Bacterial species rarely work together. Science 376, 581–582. 10.1126/science.abn5093. [DOI] [PubMed] [Google Scholar]
- 50.Cevallos SA, Lee JY, Velazquez EM, Foegeding NJ, Shelton CD, Tiffany CR, Parry BH, Stull-Lane AR, Olsan EE, Savage HP, et al. (2021). 5-Aminosalicylic Acid Ameliorates Colitis and Checks Dysbiotic Escherichia coli Expansion by Activating PPAR-gamma Signaling in the Intestinal Epithelium. Mbio 12, e03227–20. 10.1128/mBio.03227-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu GG, Velazquez EM, Lebrilla CB, Winter SE, and Baumler AJ (2016). Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host & Microbe 19, 443–454. 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Total number of ASV-36 and ASV-3 colonies counted in each well of BIOLOG PM1 and PM2A plates after 24 h incubation in aerobiosis and model summary results. Related to Figure 5.
Table S3. Total number of ASV-36 and ASV-3 colonies counted in each well of BIOLOG PM1 and PM2A plates after 24 h incubation inside of anaerobic chamber and model summary results. Related to Figure 5.
Data Availability Statement
Sequencing files generated by whole genome sequencing of bacterial isolates have been submitted to NCBI and are publicly available (Table S1 and key resources table). Genomes used for the construction of phylogenetic trees are publicly available (key resources table). This paper analyzes existing, publicly available patients’ data. The accession numbers for the datasets are listed in the key resources table.
The TaxUMAP algorithm is available on GitHub (https://github.com/jsevo/taxumap). DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AD9 | This paper | SRR14131380 |
| AD10 | This paper | SRR14131379 |
| AD11 | This paper | SRR14131378 |
| AD12 | This paper | SRR14131376 |
| AD24 | This paper | SRR14131383 |
| AD25 | This paper | SRR14131382 |
| AD26 | This paper | SRR14131381 |
| AD30 | This paper | SRR14131374 |
| AD31 | This paper | SRR14131373 |
| AD32 | This paper | SRR14131372 |
| AD33 | This paper | SRR14131389 |
| AD34 | This paper | SRR14131387 |
| AD35 | This paper | SRR14131386 |
| AD36 | This paper | SRR14131385 |
| AD37 | This paper | SRR14131384 |
| AD38 | This paper | SRR14131400 |
| AD39 | This paper | SRR14131399 |
| AD40 | This paper | SRR14131388 |
| AD41 | This paper | SRR14131377 |
| AD42 | This paper | SRR14131375 |
| AD48 | This paper | SRR16077561 |
| AD49 | This paper | SRR16077560 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin | Nova Plus | Cat#0781-9408-80 |
| Vancomycin hydrochloride | Fisher Scientific | Cat#J62790.06 |
| Neomycin sulfate hydrate | Fisher Scientific | Cat#J61499.14 |
| Dextran Sulfate Sodium Salt | Fisher Scientific | Cat#AAJ6278722 |
| LB broth, Miller | Fisher Scientific | Cat#BP1426-500 |
| Critical commercial assays | ||
| MacConkey agar II | Fisher Scientific | Cat#B21172X |
| BIOLOG PM1 plates | Fisher Scientific | Cat#NC9040419 |
| BIOLOG PM2A plates | Fisher Scientific | Cat#NC0058821 |
| QIAamp Fast DNA Stool Kit | Qiagen | Cat#51604 |
| Quant-iT dsDNA Assay Kit, High Sensitivity | Invitrogen | Cat#Q33120 |
| Nextera XT DNA Library preparation kit | Illumina | Cat#FC-131-1096 |
| MiSeq Reagent Kit v3 | Illumina | Cat#15043895 |
| Deposited data | ||
| Liao et al. (2021); Yan et al. (2022) | SRA | PRJNA394877, PRJNA607574, PRJNA606262, PRJNA548153, PRJNA545312 |
| Genomes of the isolates reported in this paper | This paper | PRJNA545312; PRJNA606262; PRJNA607574 |
| Genome of Klebsiella sp. Kd70 TUC-EEAOC | SRA | PRJNA285494 |
| Genome of Klebsiella michiganensis strain ARO112 | SRA | PRJNA590204 |
| Genome of Klebsiella oxytoca strain CAV1374 | SRA | PRJNA246471 |
| Genome of Klebsiella oxytoca KCTC 1686 | SRA | PRJNA65523 |
| Genome of K. pneumoniae subsp. pneumoniae HS11286 | SRA | PRJNA84387 |
| Genome of K. michiganensis DSM25444 | SRA | PRJNA388837 |
| Genome of K. grimontii 06D021 | SRA | PRJEB21765 |
| Genome of K. oxytoca DSM5175 | SRA | PRJNA543274 |
| Genome of E. coli ATCC11775 | SRA | PRJNA472652 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6/J | The Jackson Laboratory | JAX Cat#000664; RRID: IMSR_JAX:000664; Room #EM03 |
| Oligonucleotides | ||
| PCR for Sanger sequencing: 16S Forward 27F 5’-AGAGTTTGATCMTGGCTCAG-3’ | IDT | N/A |
| PCR for Sanger sequencing: 16S Reverse 803R 5’-CTACCRGGGTATCTAATCC-3’ | IDT | N/A |
| Software and algorithms | ||
| TaxUMAP | This paper | https://github.com/jsjse/taxumap; DOI: 10.5281/zenodo.7958500 |
| Python v3.7.9 | Python | python.org |
| Matlab v2020a | Matlab | https://www.mathworks.com/products/matlab.html |
| Bhatt lab workflow for the genomes’ assembly | Siranosian et al., 2022 | github.com/bhattlab/bhattlab_workflows |
| PATRIC Comprehensive Genome Analysis Tool | Wattam et al., 2017 | https://www.bvbrc.org/app/ComprehensiveGenomeAnalysis |
| Prokka | Seemann, 2014 | https://github.com/tseemann/prokka |
| Roary | Page et al., 2015 | https://github.com/sanger-pathogens/Roary |
| FastTree2 | Price et al., 2010 | http://www.microbesonline.org/fasttree/ |
| LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB 2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB 477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al.1 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al.2) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children's Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: Cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard3 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al.4 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al.5 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al.6 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al.7 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg8 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al.9 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.10 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
| PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185-5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 μm, 90 mm | Scientificfilters.com/TischScientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365-0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al.1 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.2 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvacphi.com/en/products/xps/phi-quantera-ii/ |