

Abstract
Background:
The isolation of cell-free DNA (cfDNA) from the bloodstream can be used to detect and analyze somatic alterations in circulating tumor DNA (ctDNA) and multiple cfDNA targeted sequencing panels are now commercially available for FDA-approved biomarker indications to guide treatment. More recently, cfDNA fragmentation patterns have emerged as a tool to infer epigenomic and transcriptomic information. However, most of these analyses used whole-genome sequencing, which is insufficient to identify FDA-approved biomarker indications in a cost-effective manner.
Patients and methods:
We used machine-learning models of fragmentation patterns at the first coding exon in standard targeted cancer gene cfDNA sequencing panels to distinguish between cancer vs. non-cancer patients, as well as the specific tumor type and subtype. We assessed this approach in two independent cohorts: a published cohort from GRAIL (breast, lung, and prostate cancers, non-cancer, N=198) and an institutional cohort from the University of Wisconsin (UW; breast, lung, prostate, bladder cancers, N=320). Each cohort was split 70/30% into training and validation sets.
Results:
In the UW cohort, training cross validated accuracy was 82.1%, and accuracy in the independent validation cohort was 86.6% despite a median ctDNA fraction of only 0.06. In the GRAIL cohort, to assess how this approach performs in very low ctDNA fractions, training and independent validation were split based on ctDNA fraction. Training cross validated accuracy was 80.6%, and accuracy in the independent validation cohort was 76.3%. In the validation cohort where the ctDNA fractions were all <0.05 and as low as 0.0003, the cancer vs. non-cancer AUC was 0.99.
Conclusion:
To our knowledge, this is the first study to demonstrate that sequencing from targeted cfDNA panels can be utilized to analyze fragmentation patterns to classify cancer types, dramatically expanding the potential capabilities of existing clinically used panels at minimal additional cost.
Keywords: cell-free DNA, cancer, fragmentomics
INTRODUCTION
Profiling of genomic driver alterations in cancer has become increasingly important, not only for studying the biological underpinnings of cancer, but also in identifying clinically actionable alterations for targeted therapies in clinical trials and practice. Historically, tumor samples have been required, but obtaining tissue specimens for molecular profiling is not always feasible and can be especially challenging in the metastatic setting. Cell-free DNA (cfDNA) from cancer patients provides a minimally invasive approach for assessing molecular events in the tumor by detecting alterations in the tumor-derived cfDNA, also called circulating tumor DNA (ctDNA)1. This is a mature technology, with multiple commercially available next-generation sequencing (NGS) ctDNA panels2. These panels primarily profile the coding regions of select oncogenes and tumor suppressors, as they are designed for the identification of targetable DNA alterations for FDA-approved biomarker indications to guide treatment selection.
The stability of cfDNA in circulation is dependent on its association with proteins and protein complexes which offer protection against DNAses found in the blood3, 4. The nucleosome complex is the most common protector of cfDNA which is reflected in the size distribution of cfDNA fragments showing a mode fragment size of 167 bp corresponding to the wrapping of DNA around a single nucleosome, with a smaller proportion of fragments at 334 bp corresponding to a di-nucleosome complex5–8. Other studies have also described smaller peaks at a periodicity of approximately 10 bp at lower fragment sizes representing the accessibility of DNA minor grooves to endonuclease cleavage as it wraps around the histone complex, as well as the binding of transcription factors or other small DNA binding proteins7–11. Distinct fragmentation patterns around the transcription start site (TSS) have been shown to reflect binding of transcriptional machinery, and these fragmentation patterns have been demonstrated to correlate with gene expression9, 12–16. Additionally, coverage depth of ctDNA at both the TSS and the first exon-intron junction have been shown to mirror gene expression, with weaker but still significant correlations extending into the gene body and transcription termination sites15. The study of cfDNA fragmentation patterns has been referred to as “fragmentomics” and cancer patients display unique fragmentomic patterns that have been used to non-invasively investigate the biology of the tumor17–19. The use of fragmentomics for differentiating between cancer and heathy patients with machine learning models has recently been reported through its use to infer nucleosome binding17, 20, copy number alterations21, gene expression14, transcription factor binding profiles22, 23, and DNAse processing motifs24. These models have also been used to differentiate between specific cancer subtypes. For example, the use of fragmentation patters as a surrogate for gene expression was able to differentiate between lung adenocarcinoma and squamous cell carcinoma with an AUC of 0.9014. Similarly, this study was also able to differentiate between molecular subtypes of diffuse large B cell lymphoma (DLBCL) with a strong correlation between the predicted subtype and the predictions of standard assays for DLBCL subtype classification14. Recently, the analysis of cfDNA fragmentation patterns around known DNA accessibility sites and transcription factor binding sites was able to differentiate between ER+ and ER-breast cancer subtypes with an overall AUC of 0.9623. Additionally, in silico fragment size filtering has been applied to increase the sensitivity of mutation detection, as tumor-derived cfDNA tends to form smaller fragments than cfDNA from typical immune cell sources21, 25, 26. However, to our knowledge, none have reported successful identification of multiple cancer types.
Almost all clinical fragmentomic studies to date have utilized whole-genome sequencing (WGS) to assess fragmentation patterns across the genome in an unbiased manner27. While WGS has the advantage of breadth of coverage, there is generally low sequencing depth making it unsuitable for cfDNA somatic alteration detection as it has poor sensitivity, especially at low ctDNA fractions28. Conversely, cfDNA targeted panels allow for deeper sequencing at areas of interest, which are typically coding regions of important cancer genes. Previous cfDNA fragmentomics analyses have generally focused on WGS which affords probing of fragmentation patterns at all genomic regions in an unbiased manner, as the investigated biological phenomena are typically not unique to regions profiled by target panels (e.g. exonic regions). For example, many analyses of fragmentation patterns have focused on the assessment of histone binding, which requires relatively uniform read support across large areas of the genome7, 9, 17, 20, 23. This type of read support is not provided by targeted panel sequencing.
While previous studies have focused on fragmentation patterns across the whole genome, we hypothesized that cfDNA fragmentation patterns in the coding regions of important oncogenes and tumor suppressors could provide important insights for distinguishing between tumor and normal samples, as well as between different tumor types and subtypes. Given its known association with gene expression9, 14, 15, we specifically focused on fragmentation patterns overlapping the first coding exon of targeted genes. To evaluate this, we examined the fragmentomic patterns in both a publicly available multi-cancer cfDNA dataset profiled using the GRAIL cfDNA assay29, as well as an institutional multi-cancer cohort profiled using a custom cfDNA panel. We found that analysis of the fragmentation patterns of first coding exons could distinguish between cancer types as well as between cancer vs. normal. The use of fragmentation patterns from targeted cfDNA panels would allow for the advantages of both variant calling and fragmentomics in a single assay which could be leveraged on any existing panels that are already commercially available.
METHODS
UW patient cohort
Peripheral blood samples were collected from patients with metastatic cancer enrolled in an IRB-approved liquid biopsy collection protocol at the University of Wisconsin-Madison (2014–1214), as well as from two ongoing clinical trials (NCT03090165, NCT03725761).
UW cfDNA sample collection, preparation, and sequencing
Blood was collected in 10 mL K2 EDTA (BD Vacutainer) or CellSave™ preservative blood collection tubes (Menarini Silicon Biosystems). Whole blood was processed within 4 hours (EDTA) or 36 hours (CellSave) from time of collection and was centrifuged at 300xg for 10 minutes. Plasma (3–6 mL) was harvested and centrifuged at 1500xg for 10 minutes, then stored at -80°C. cfDNA was isolated from 2–6mL plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen). Germline DNA (gDNA) was isolated from matched peripheral blood mononuclear cells using the DNeasy blood and tissue kit (Qiagen) and fragmented using the NEBNext Ultra II FS DNA module (New England Biolabs). The Agilent Bioanalyzer high sensitivity DNA chip was used to quantify and assess cfDNA and fragmented gDNA quality. 50ng cfDNA or 50ng fragmented gDNA were subjected to library preparation with unique molecular indexes using the xGen Prism DNA library preparation kit (Integrated DNA technologies). For samples with less than 50ng available cfDNA, 1, 10, or 25ng DNA input was used. 8–12 libraries were pooled at 500ng per library followed by hybridization and capture with a custom 822-gene panel using the xGen hybridization capture of DNA libraries kit (Integrated DNA technologies). Paired end sequencing (2x150bp) was performed on a NovaSeq 6000 at the University of Wisconsin sequencing core, with a target depth of 20 million reads per germline sample and 50 million reads per cfDNA sample.
Sequencing data processing
UW sequencing was aligned to the hg38 genome using BWA-mem30 (v0.7.17) followed by deduplication of the aligned BAM files with Connor v0.6.1 (https://github.com/umich-brcf-bioinf/Connor) which uses both start-stop position and UMIs along with filtering of low quality reads. A minimum family size threshold of 1 (-s 1) was used to keep all unique reads. BAM files were filtered for properly paired reads (samtools flags -f3 -F2308), sorted by read name, then converted to BEDPE files using bedtools31 (v2.30.0) bamtobed using the -bedpe flag. The start and stop positions of each read were extracted from the BEDPE file to yield a BED file of the sequencing reads to use for subsequent overlaps. GRAIL cfDNA sequencing data and metadata29 were accessed and downloaded through the European Genome Archive (Dataset ID EGAD00001005302). As raw FASTQ files were not available, the hg19-prealigned BAM files were deduplicated using start-stop position and UMI followed by BAM to BED conversion as described above for the UW samples.
Fragmentomics
For each sample, a global fragmentation distribution was calculated from the BED file by extracting the read insert size from the mapped end of the template and the mapped start of the template (stop – start) and then counting the number of reads at each size. The number of reads at each size was divided by the total number of reads in the sample to return the proportion of reads at each fragment size. Individual fragment distributions were plotted using the proportion of reads at each fragment size.
Shannon Entropy for first coding exon
Canonical exon coordinates were downloaded as BED files from the UCSC Genome Browser using the Table Browser tool for both hg38 and hg19 (https://genome.ucsc.edu/cgi-bin/hgTables). The BED file of each cfDNA sample was then overlapped with the respective exon file (hg38 for UW data, hg19 for GRAIL data) using bedtools intersect (v2.30.0) to yield reads overlapping with canonical exons. A minimum of 1 bp overlap was required for a read to be considered overlapped with an exon of interest. Reads overlapping the first coding exon of each gene were extracted, and a fragment size distribution was calculated for each gene using only the reads overlapping exon 1. Throughout the manuscript, references to “exon 1” or “E1SE” refer to the first coding exon of the respective gene or genes. Shannon entropy was calculated with the entropy function from the “entropy” package (v1.3.1) in R (v4.0.4) using the count of read fragments at each fragment size. This returned a single Shannon entropy value for reads overlapping the first exon of each gene in each sample. Given the association between the number of fragments analyzed and Shannon entropy (Figure 2F), with low fragment count leading to a less accurate estimation of Shannon entropy, we required a minimum of 500 reads to overlap an exon across all samples to be included in the final dataset.
Figure 2: cfDNA fragmentation patterns from targeted panels.
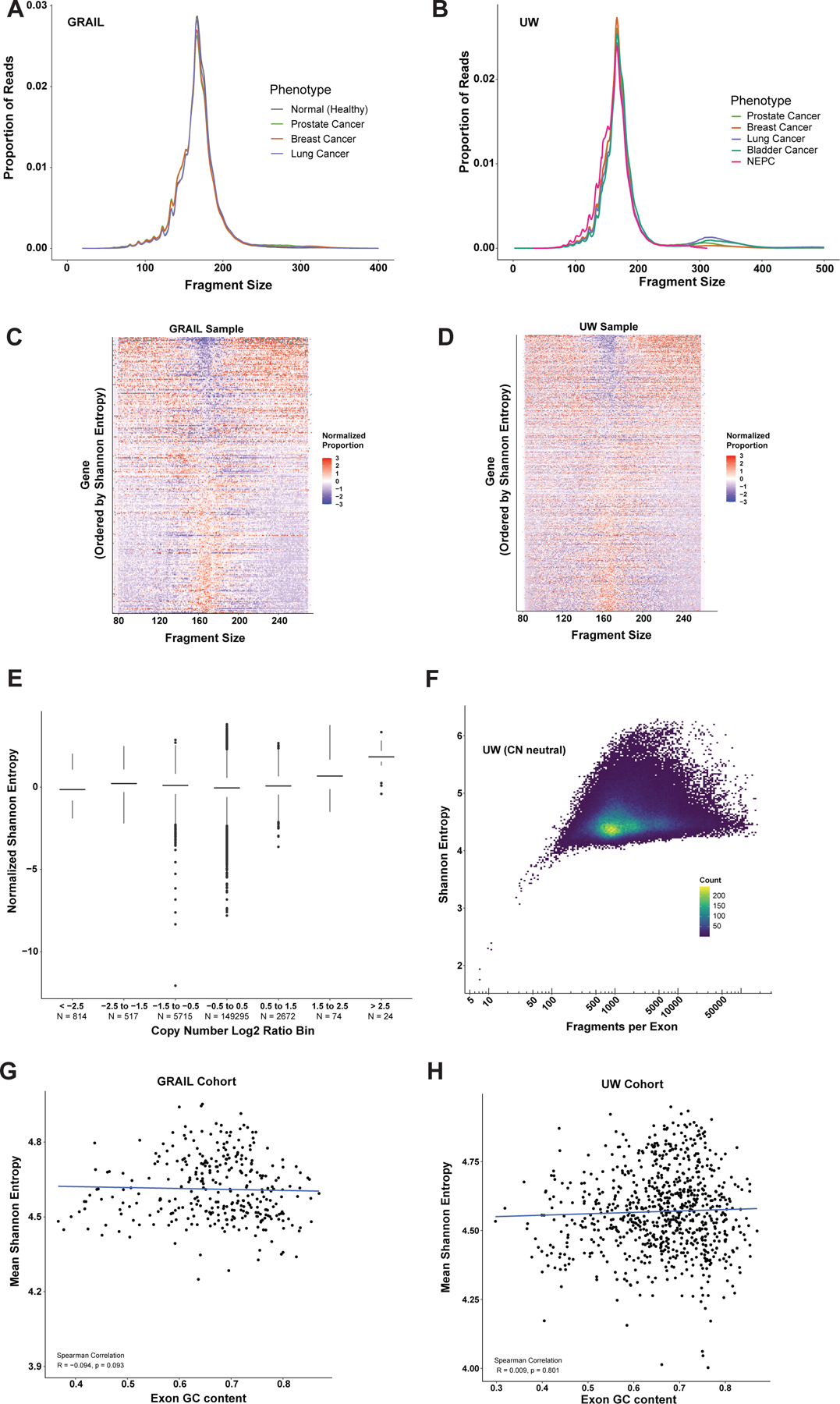
Average total fragment distribution across tumor types in the (A) GRAIL and (B) UW datasets respectively. Heatmap of the fragment size distributions at exon 1 across all genes from the GRAIL targeted panel (C) and UW targeted panel (D) in a single representative sample from each cohort. Genes are ordered by exon 1 Shannon entropy (E1SE) with high E1SE genes at the top and low E1SE genes at the bottom. Fragment size proportions are normalized within each fragment size across all genes analyzed. Plot demonstrates that genes with high E1SE are depleted for fragments near the mono-nucleosome peak (167bp) and enriched for fragments at lower (<120 bp) and higher (> 200 bp) sizes, while genes with low E1SE display the opposite pattern. (E) Copy number calls from the UW cohort compared to Shannon entropy. Copy number was calculated for each gene for each patient. Each point represents a single gene-patient pair. Copy number data was binned as shown, and Shannon entropy distributions are shown for each bin. E1SE was normalized by centering and scaling on a per-gene basis before plotting. This transforms the E1SE distribution for each gene such that the mean is zero and the standard deviation is one, eliminating inter-gene variability. Data from all genes and patients are plotted. Only the UW cohort was used because the exact panel design was required to accurately determine CN, but this was not available for the GRAIL cohort (F) Shannon entropy as a function of fragments per exon in the UW cohort at copy number neutral regions (Log2 ratio between -0.5 and 0.5). Correlation between GC content and mean Shannon entropy at each exon analyzed in the (G) GRAIL cohort and (H) UW cohort.
GRAIL training, cross validation, and independent validation
Using the E1SE values for each gene in the GRAIL panel as features, multinomial regression using a generalized linear model with elastic net penalty (GLMNET) was used to predict cancer types. Samples were split into 70% training and 30% validation with low ctDNA fraction samples placed in the validation cohort. For all model training, a range of α and λ values were selected using latin hypercube sampling, and the best AUC on 10-fold cross validation was used select the final parameters. To estimate performance in the training cohort, 10-fold cross validation was performed, and training and parameter fitting (using 10-fold cross validation nested within the training set of each fold) was performed within each fold separately to avoid any information leakage. Predictions from the hold-out test sets for each fold were combined to calculate accuracy and ROC curves. A final model was then trained using the full training cohort. The independent validation cohort was then entered into the model to yield prediction scores, again with no information leakage between training and validation. These prediction scores were used to calculate accuracy and ROC curves.
UW training, cross validation, and independent validation
A similar approach was used for the UW cohort, which was also split into 70% training and 30% training. However, due to more missing ctDNA fraction data and imbalanced tumor types, the split was random while stratifying by tumor type, such that the relative proportions were similar across training and validation. Otherwise, training, cross validation, and independent validation were all performed the same as in GRAIL.
Identification of somatic mutations in the UW cohort
Somatic variant identification was performed using VarDictJava v1.8.332 in paired sample mode using standard filter settings. Somatic mutations were required to have a minimum of 10 supporting reads, a minimum of 20 total reads covering the position, and up to 2 mismatches in the cfDNA samples, and a minimum of 20 total reads in the matched gDNA samples. For SNVs, the average mapping quality of mutation supporting reads was required to be at least 50 and the average distance of the mutant allele from the nearest read end was required to be at least 15 bases. We then conservatively removed germline mutations and somatic mutations related to clonal hematopoiesis of indeterminate potential (CHIP) by removing mutations to have more than 1 supporting read in any gDNA sample and removing any of 4,938 CHIP related mutations compiled by Bick et al.33. Lastly, mutations in the low-complexity genomic regions and shared common mutations in dbSNP (dbSNP_G5) were discarded.
Copy number analysis in the UW cohort
Deduplicated BAM files were further filtered for uniquely mapped reads with high mapping quality using sambamba v0.8.2 (-F “mapping quality >= 30 and not ([XA] != null or [SA] != null)”. Using the deduplicated, filtered, sorted, and indexed bam files as input, we ran CNVkit v0.9.934 to call somatic copy number alterations. CNVkit is a read-depth approach and utilizes both targeted and non-targeted regions to infer copy number more evenly across the genome. An accessibility bed file was created (cnvkit.py access -s 10000) to remove unmappable regions (i.e. large stretches of “N” characters) from the reference genome. CNVkit was run in batch mode for all cfDNA samples with a flat reference, which assumes equal coverage in all bins. Bin-level read depth was corrected for GC content, sequence repeats, and target density, and individually compared with the flat reference to calculate read depth ratio (log2). Genes with copy number gain or loss were identified using the genemetrics command with minimum absolute log2 copy ratio threshold (log2) of 0.5. Genes with less than three bins (probes) and read depth (depth) less than 1000 in each sample were discarded. CN was only used to compare against E1SE in our analysis. As ctDNA fraction impacts both fragmentomic patterns and copy number, copy number was therefore not corrected for tumor content.
Estimation of ctDNA fraction in the UW cohort
The proportion of tumor-derived cfDNA (ctDNA fraction) was estimated based on VAF of autosomal somatic mutations. VAF in autosomes is elevated if a mutant allele is accompanied by deletion of the other allele (i.e., loss of heterozygosity, LOH). Assuming a diploid tumor model and that the mutation with the highest VAF displays LOH, ctDNA fraction and the highest VAF can be related as . To account for stochastic variation, we modeled the mutant allele read count with a binomial distribution as suggested by Vandekerkhove et al.35 and calculated what the true VAF would be if the observed mutant allele read count was a 95% quantile outlier. After calculating ctDNA fraction for each somatic mutation in a given sample, the highest estimate of ctDNA fraction was used for the given sample as the mutation with the highest VAF is the most likely to be clonal. While the classification of LOH for the highest VAF is an assumption, many other reports utilize this method when analyzing targeted cfDNA sequencing15, 35–41. Data for ctDNA fraction for samples from the GRAIL cohort were obtained from their previously published report29 in the supplemental data (Source Data Fig. 2; tab “Fig_2f”)
Data Availability
Raw sequencing data from the GRAIL dataset is available at the European Genome Archive (Dataset ID EGAD00001005302). Our institutional protocol did not allow unrestricted public access to the raw sequencing data. Therefore, data sharing requests must be submitted to the University of Wisconsin-Madison for approval. For samples from the two clinical trials (NCT03090165, NCT03725761), these trials are still ongoing, and data sharing requests must be submitted to the trial organizers.
RESULTS
Overview of two independent targeted ctDNA panels and cohorts
We examined two cohorts of cfDNA profiled using targeted cancer gene exon panels. The first was a previously published multi-cancer cohort of 198 cfDNA samples assessed using the commercial assay from GRAIL, covering 508 genes (~2MB) at a sequencing depth of >60,000X across breast, lung, and prostate cancer patients along with healthy donors29. The second cohort was an institutional multi-cancer cohort from the University of Wisconsin (UW) with 320 samples across breast, lung, bladder, prostate, and neuroendocrine prostate cancers. Profiling was performed using a custom panel broadly covering the exons of 822 cancer genes, covering ~2.4MB of the genome at an average sequencing depth of 3,042X. Details of differences between GRAIL and UW cohorts can be found in Supplemental Methods. Previous reports have shown that nucleosome positioning and other DNA-binding factors can affect cfDNA fragmentation patters at transcription start sites (TSS) which can inform gene expression patterns7, 14. This correlation was shown to additionally extend to exon 1 of genes14 which we hypothesized could be used to inform tumor of origin using cfDNA sequencing from targeted panels which cover these regions in greater depth. To quantify the cfDNA fragmentation patterns at each exon 1 analyzed, the exon 1 Shannon entropy (E1SE) of the distribution was calculated which summarizes the diversity of fragments in the region. We then use these E1SEs to train models to predict tumor type. Both the UW and GRAIL cohorts were split into 70% training in which cross-validation was used to assess performance, and 30% independent validation In the GRAIL cohort, training was specifically performed on the 70% samples with the highest ctDNA fraction, and validation was performed on the lowest 30% by ctDNA fraction (Figure 1).
Figure 1: Schematic of fragmentomics experimental setup.
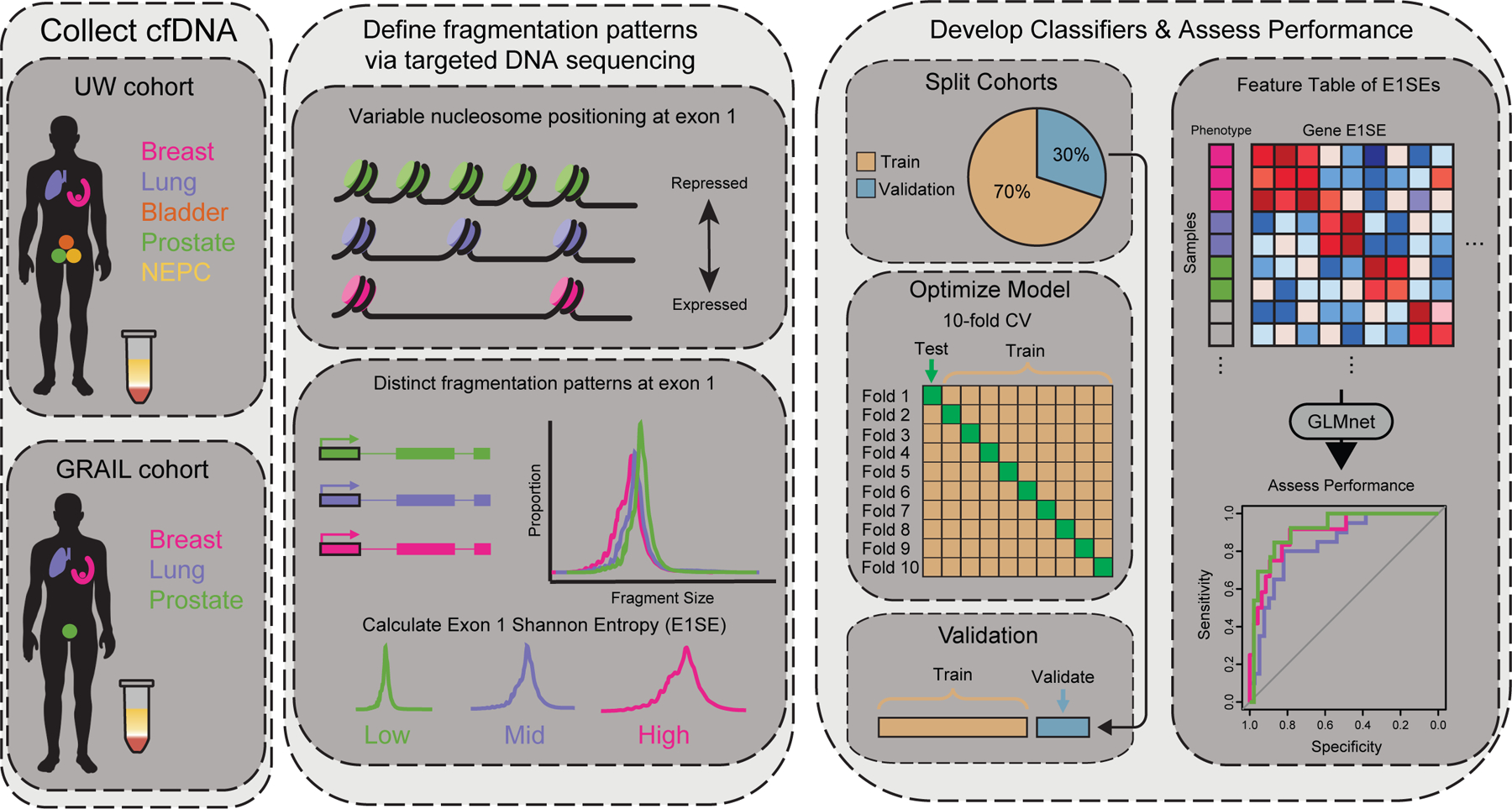
Liquid biopsies from patients from two independent cohorts with various cancer types are collected and cfDNA is isolated using targeted exon panels. Unique histone distributions across cancer types lead to variable fragmentation patterns at targeted exons. Exon 1 shows particular variability due to its proximity to promoter regions and is correlated with gene expression. The diversity of fragmentation distributions at each coding exon 1 are measured via Shannon entropy for each sample. Machine learning models are built to predict tumor type for each cohort, with training performed on 70% of the data and 30% withheld for validation. Ten-fold cross validation performed on the training data. In the UW cohort, samples are randomly selected for training and validation, while the GRAIL cohort is trained on high ctDNA samples and validated on low ctDNA samples.
Fragment distributions in targeted panels
The narrow breadth of genomic coverage in targeted panels compared to WGS may bias fragmentomic patterns. When we assessed the total distribution of fragment sizes from each targeted panel, the average global fragment distributions within each phenotype across both cohorts and assays were similar. In both, we observed a main peak at 167bp corresponding to a single nucleosome, as well as a smaller peak at 334bp corresponding to two nucleosomes. In addition, we observed subnucleosomal peaks at smaller fragment sizes with roughly 10 bp periodicity which likely corresponding to the accessibility of DNA minor grooves to endonuclease digestion as the DNA wraps around the histone core, as well as the binding of transcription factors and other DNA-binding proteins7, 8 (Figure 2A, 2B). The fragment distribution from these targeted panels was similar to previously published cfDNA fragment patterns which use WGS8, 12, 14, 17, 21, 26, 42, suggesting that fragmentomics might be successfully applied to targeted exon panels (Figure S1A, S1B).
Fragmentomic patterns from WGS around the transcription start site (TSS), have been shown to infer binding of transcriptional complexes, and thus gene expression9, 12–16. Previous reports measuring the diversity of fragments in these TSS regions using Shannon entropy has found strong correlations between this metric and gene expression14. Repressed genes contain high nucleosome occupancy at their TSS, leading to a more uniform distribution of fragment reads at 167 bp14, 16, 43–46. In contrast, actively expressed genes have more open chromatin at their TSS, allowing the cfDNA originating from this region to be cleaved in a more random manner, leading to a more diverse distribution of DNA fragment sizes14, 16, 43–46. These changes can be detected out to 2000 bp from the TSS, which overlaps most first coding exons7, 14, 47. Additionally, when we compared the fragment coverage around the TSS and first coding exon in highly expressed vs. lowly expressed genes from deep WGS in a separate cohort48, we found that the lower coverage observed at the TSS of highly expressed genes extended well into the first coding exon, indicating that fragmentation profiles in the first coding exon are linked to gene expression (Figure S2). This is important because the majority of standard targeted cancer gene panels, including the GRAIL and UW panels, do not include the TSS in most cases and instead start at the first coding exon of targeted genes. Epigenetics of the first coding exon can influence transcription49–51, and correlation between gene expression and fragmentomic patterns at the first exon of genes14, 16 and at the first exon-intron junction15, 16 have been described.
To assess the diversity of fragment sizes at the first coding exon of each gene, Shannon entropies were calculated for each individual gene in the respective sequencing panels for each patient using the distribution of fragment sizes overlapping the first coding exon. We defined this metric as Exon 1 Shannon Entropy (E1SE). To visualize the relationship between E1SE and fragment size distribution, we plotted the fragment distributions of all analyzed genes from highest to lowest E1SE within individual samples from each cohort, and noted that as expected, high E1SE genes were depleted in fragments around the mode of 167 bp with an increased proportion of fragments at lower (<120 bp) and higher (>200 bp) sizes (Figure 2C, 2D; individual representative sample shown for each cohort). Conversely, low E1SE genes displayed a higher proportion of fragments at the mono-nucleosome peak (167 bp) suggesting a more closed chromatin structure at exon 1 of those genes. These observations are consistent with previously reports which assessed the diversity of cfDNA fragments at gene TSSs14. We additionally noted that the E1SE of the androgen receptor gene (AR) was significantly higher in prostate cancer samples compared to all other cancer types and normal samples in both the GRAIL and UW cohorts (Figure S3A, S3B). Further, AR E1SE was observed to be higher in high ctDNA fraction prostate cancer samples, but not lung cancer or breast cancer samples, suggesting that the high AR E1SE originates from tumor-derived cfDNA (Figure S4). This example highlights how differences in E1SE levels could help distinguish between tumor types and subtypes.
Copy number alterations are common in cancer and can affect the number of reads mapping to each gene which could potentially bias the measurement of fragment size diversity via E1SE. However, we did not observe a clear relationship between copy number and E1SE (Figure 2E). E1SE did start to trend up at very high copy numbers, though this should be interpreted with caution as there were only a small number of high copy number genes across our samples. Another possible influence on E1SE is the total number of observations used in its calculation, which corresponds in our application to the number of fragments analyzed per exon. Variation in depth of sequencing at each exon can occur through variations in targeted probe pull-down efficiency and other technical factors. To isolate this effect from copy number, we analyzed the effect of the number of fragments per exon on E1SE only in copy number neutral regions. The total number of reads mapped to an exon did not affect E1SE above a count of ~100 (Figure 2F). GC content has also been shown to potentially bias cfDNA sequencing and various studies have corrected for this bias when performing fragmentomics analyses through shallow whole genome sequencing17, 20, 23. However, we did not find a significant correlation between exon 1 GC content and E1SE in either cohort (Figure 2G, 2H), possibly because these panels target a much smaller proportion of the genome and are comprised primarily of coding DNA. Thus, we sought to assess the potential utility of E1SE in classifying and subtyping tumors using targeted panel fragmentomics, while simultaneously allowing for standard ctDNA somatic alteration identification.
E1SE fragmentomics distinguishes tumor subtypes
First, we examined if the E1SE fragmentation patterns could be used to reliably classify different cancer types in our institutional cohort and panel. The UW cohort contained 320 samples from patients with metastatic disease from six different tumor types: breast cancer (N = 100), bladder cancer (N = 22), lung cancer (N = 39), and prostate cancer (N = 144). In addition, we had samples from patients with metastatic neuroendocrine prostate cancer (N = 15, NEPC), a molecularly and clinically distinct subtype of prostate cancer.
Fragmentomic differences are subtle, and many studies use machine learning approaches to assess fragmentomic biomarkers. We used elastic-net regression to train a multi-class classifier to distinguish the different tumor types in the UW cohort, which was split into 70% training and 30% independent validation. In the training cohort, we utilized 10-fold cross validation to assess performance and compared this to the independent validation. We found that in the training cohort, the E1SE model was able to distinguish the different tumor types with an overall accuracy of 82.1% on cross-validation. The performance was similar in the independent validation cohort, with an overall accuracy of 86.6% (Figure 3A). We additionally tested the performance of the model using the middle and last coding exon of each gene and found that accuracy was highest when using the first coding exon (Figure S5). When we examined the ROC curves for each tumor type, the AUCs for all tumor types were ≥0.89 (bladder cancer = 0.98, breast cancer = 0.98, lung cancer = 0.89, prostate cancer = 0.99, NEPC = 1.00, Figure 3B) indicating that E1SE is able to distinguish between tumor types and subtypes. These results were achieved despite a median ctDNA fraction of only 0.06 (Table S1). Prediction accuracy remained high across ctDNA fractions, though numbers are small in some subgroups (Figure 3C). We additionally analyzed the prediction scores for each sample within each cancer type to determine if incorrect predictions within a cancer type were biased toward a certain cancer. In all cancer types, the majority of samples had prediction scores matching the diagnosed cancer type for that patient (Figure 3D).
Figure 3: Predicting tumor type in the UW panel and cohort.
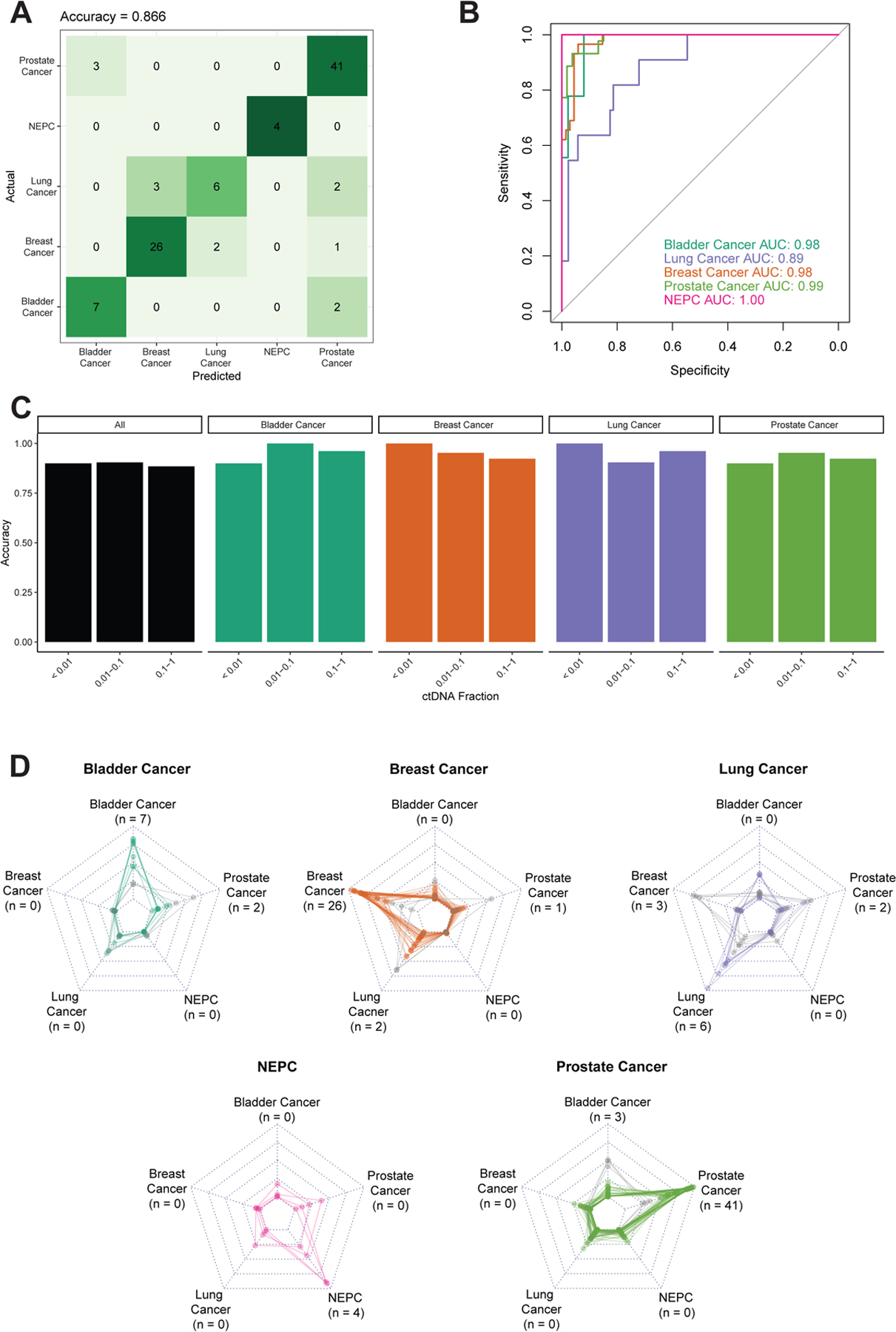
The UW data was split into 70% training and 30% independent validation, the latter of which is shown. Performance was assessed by (A) confusion matrix of classifier accuracy in CV data comparing predicted vs. actual phenotypes and (B) ROC curves of classifier AUCs in CV data. (C) Accuracy as a function of ctDNA fraction in CV data. ctDNA fractions ranged from 0.003–0.771. NEPC samples are not shown due to the lack of germline sequencing for this cohort which are required for ctDNA fraction estimation. Only samples with available germline sequencing, and thus ctDNA fraction estimation, are shown. The number of samples in each ctDNA fraction bin are: <0.01: n = 10; 0.01–0.1: n = 21; 0.1–1.0: n = 26. (D) Radar plots depicting the prediction score, where each plot represents one pathologic diagnosis (noted in bold above the plot), and each line in the plot represents model prediction for a single patient. The vertices of each graph represent the continuous prediction scores from the E1SE models for each of the predicted phenotypes, with the outer ring denoting a prediction score of 1 and the inner ring a prediction score of 0. For each patient, the final model prediction is the highest-scoring predicted phenotype which is correct in the majority of cases. The number of predictions for each tumor type are noted next to the label of each vertex (matching panel A). Correctly predicted patients are represented by colored lines, whereas incorrectly predicted patients are represented by light gray lines.
E1SE fragmentomics distinguishes tumor types and tumor vs. normal in low ctDNA fraction samples
Given the multiplicity of targeted cfDNA sequencing platforms currently in clinical and research use that can differ quite substantially in targeted genes and depth of sequencing, we sought to test whether our approach was reproducible, robust, and independent of the specific targeted sequencing panel used. Due to differences in panel construction, an independent model would be needed for each platform of interest. We therefore performed a similar approach in the GRAIL panel and cohort, which contained 198 samples from patients with lung cancer (N=49), breast cancer (N=48), prostate cancer (N=54), as well as patients without cancer (N=47)29. Approximately 347 of the genes overlap between the GRAIL and UW targeted sequencing panels. Because of the different panel designs, model training was performed again using the GRAIL cohort and panel. The median ctDNA fraction in the GRAIL cohort was 0.076 and the depth of sequencing was much higher than in our institutional cohort allowing an order of magnitude greater resolution of very low ctDNA fraction samples. Therefore, we sought to investigate the sensitivity of E1SE in distinguishing tumor types and normal samples at low ctDNA fractions. To assess this, we split the GRAIL cohort into 70% training and 30% validation based on ctDNA fractions, where the validation cohort consisted of the samples with the lowest ctDNA fractions, all <0.0481, and the training cohort contains all remaining samples.
We found that in the training cohort, the E1SE model was able to distinguish the different tumor types with an overall accuracy of 80.6% on cross-validation. Remarkably, in the independent validation, even at these low ctDNA fractions, the E1SE model had an overall accuracy of 76.3% (Figure 4A). As with the UW cohort, we additionally tested model performance using the middle and last coding exon of each gene and found that accuracy was highest when using the first coding exon. (Figure S5). When we examined the ROC curves for each tumor type, the AUCs were all ≥0.83 (breast cancer = 0.90, lung cancer = 0.83, prostate cancer = 0.91, tumor vs. normal = 0.99, Figure 4B). Prediction accuracy was high in ctDNA fractions down to 0.001, with an accuracy of 85.7% in samples with ctDNA fractions from 0.001 to 0.01 (Figure 4C). Unsurprisingly, accuracy was 0% in predicting tumor type in ctDNA fractions <0.001, thus identifying the lower limit of distinguishing different tumor types with this approach. Notably, when considering the three tumor types grouped together into a single “cancer” category, the accuracy of distinguishing cancer samples from normal samples was 100% in samples with ctDNA fraction <0.001, with the lowest ctDNA fraction being 0.0003. When we analyzed the prediction scores for each cancer type, as with the UW cohort, the majority of samples were correctly predicted as their true cancer type (Figure 4D).
Figure 4: Predicting tumor type in the GRAIL panel and cohort.
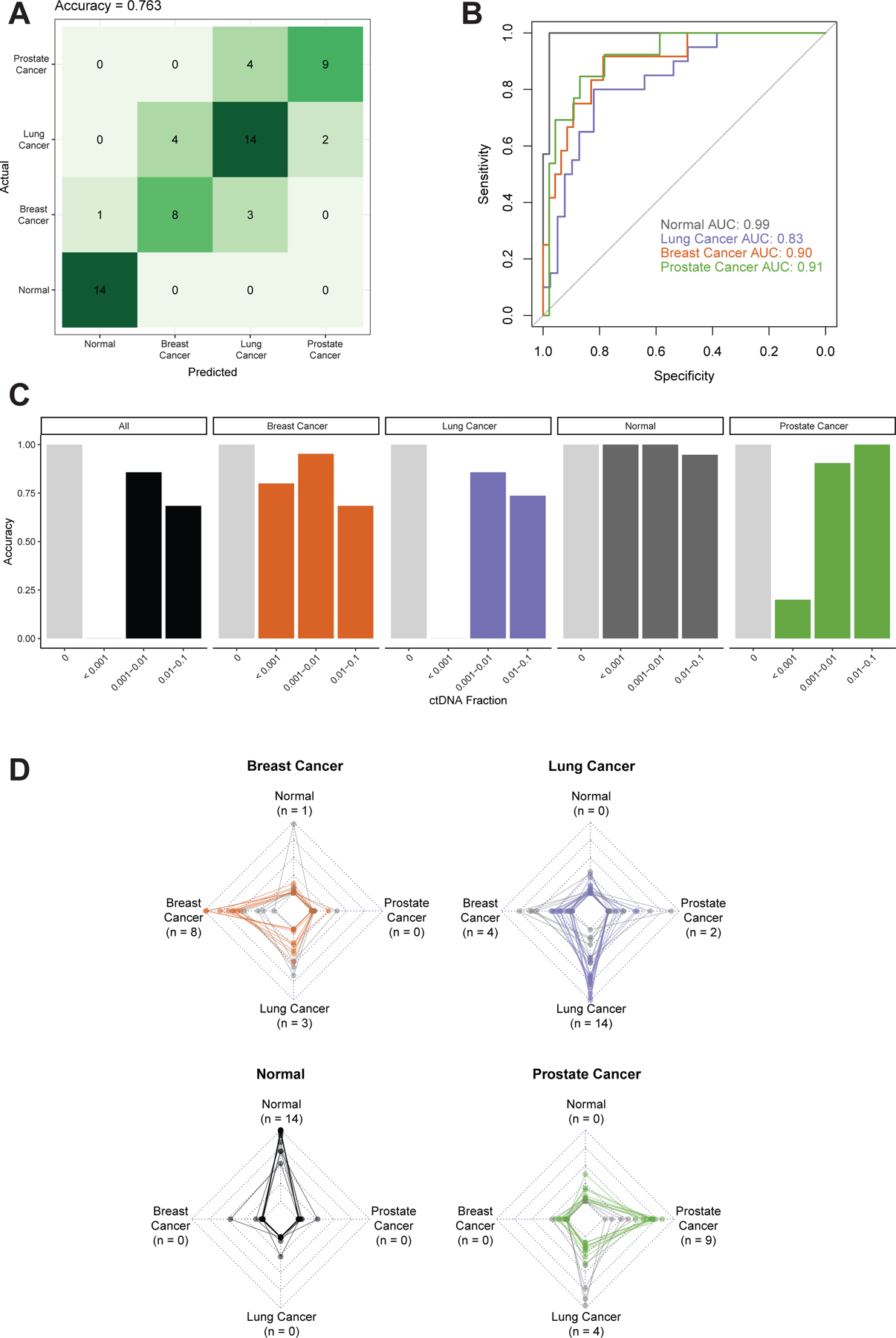
The GRAIL data was split into 70% training and 30% independent validation, the latter of which is shown. The validation data contained the lowest ctDNA fraction samples, all <0.05. Performance was assessed by (A) confusion matrix of classifier accuracy in validation data and (B) ROC curves of classifier AUCs in validation data. (C) Accuracy as a function of ctDNA fraction in validation data. ctDNA fractions ranged from 0.0003–0.925 for cancer samples. Light grey bars represent normal samples with a ctDNA fraction of 0. The number of samples in each ctDNA fraction bin are: 0 (Normal): n = 33; <0.25: n = 28; 0.25–1.0: n = 32. (D) Radar plots depicting the prediction score, where each plot represents one specific pathologic diagnosis (noted in bold above the plot), and each line in the plot represents the model prediction for a single patient. The vertices of each graph represent the continuous prediction scores from the E1SE models for each of the predicted phenotypes, with the outer ring denoting a prediction score of 1 and the inner ring a prediction score of 0. For each patient, the final model prediction is the highest-scoring predicted phenotype which is correct in the majority of cases. The number of predictions for each tumor type are noted next to the label of each vertex (matching panel A). Correctly predicted patients are represented by colored lines, whereas incorrectly predicted patients are represented by light gray lines.
Assessing performance as a function of sequencing depth
Since the cost of NGS is not trivial, we wanted to evaluate how performance of the E1SE fragmentomics model varied as a function of depth of sequencing. To do this, we performed down-sampling of GRAIL cohort after the de-duplication step as this assessed the effect of unique read depth on model performance. Due to the increased depth of sequencing from the GRAIL data, we were able to down-sample all samples to 100, 50, 25, 10, 5, and 1 million de-duplicated reads which correspond to sequencing depths of roughly 15000X, 7500X, 3750X, 1500X, 750X, and 150X respectively for a 2Mb panel. After down-sampling, E1SE were calculated as described above. This down-sampling process was repeated ten times at each level to account for variability, and the resulting E1SE tables were used for model training, with assessment being performed in the independent validation cohort as above. Interestingly, we found that reduced sequencing had only a modest impact on model performance, with AUCs between 100 million and 10 million reads remaining stable for breast (0.841 vs 0.888), prostate (0.929 vs 0.942), lung (0.814 vs. 0.781), and tumor vs. normal (1.00 vs 0.996) (Figure 5A). Predicting tumor vs. normal is particularly robust, with the mean AUC remaining close to 1 when down-sampled to 1M reads (AUC = 0.996). Similarly, down-sampling was found to have limited effect on the accuracy of the model, both overall and within cancer types down to 1 million reads (Figure 5B). These results indicate that high levels of depth are not required for tumor type prediction using fragmentomics approaches within targeted panels and allows for its application to sequencing depths used in standard variant calling.
Figure 5: Effect of downsampling on model performance in the GRAIL cohort.
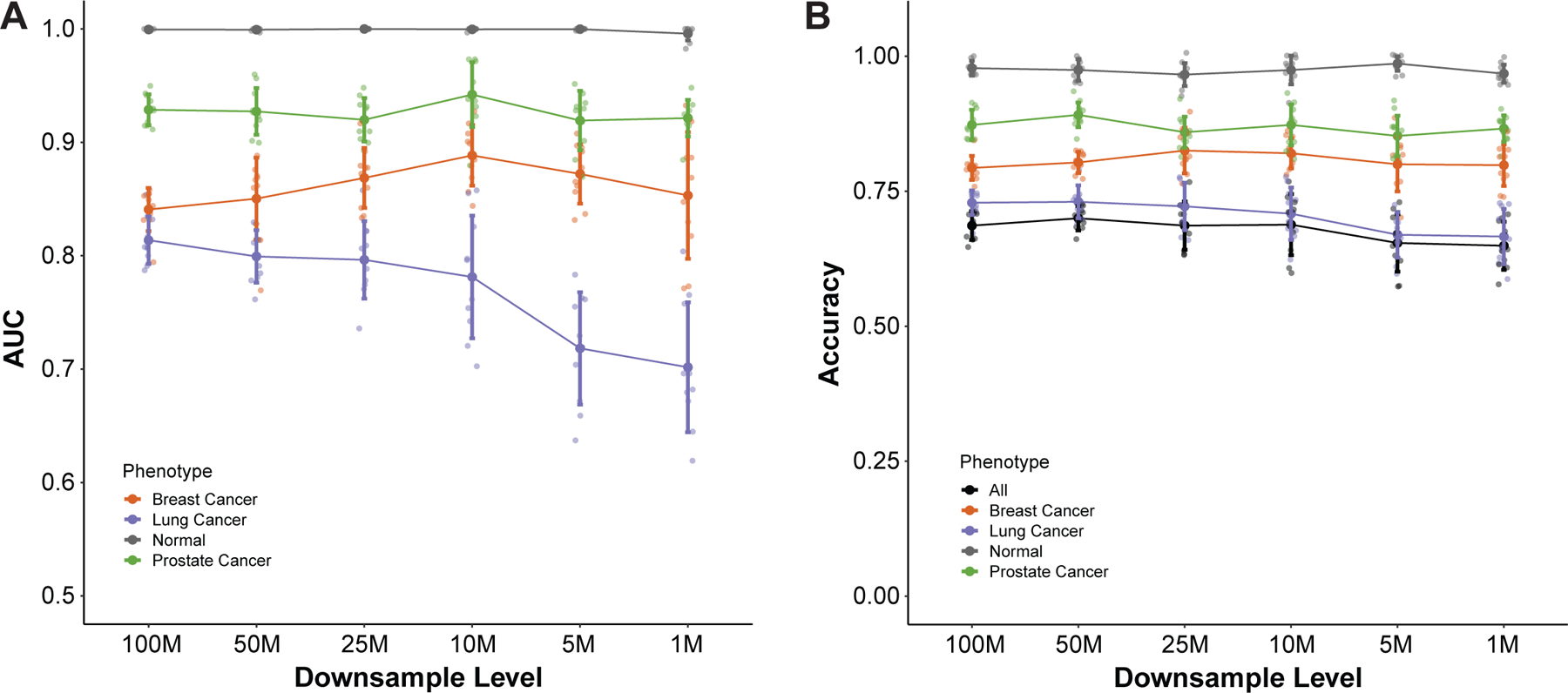
Downsampling of the GRAIL cohort was performed to levels ranging from 100M to 1M reads 10 times for each downsampling level. For each replicate and downsampling level, Shannon entropies were calculated for the fragment distributions at the first exon of each gene in the panel as described previously. Training and validation using the new downsampled feature tables was performed and results for (A) ROC AUC and (B) accuracy are shown for each phenotype in the cohort. Small points represent individual values, large solid points represent mean values, and error bars represent +/- 1 standard deviation.
DISCUSSION
Fragmentomic patterns of cfDNA are non-uniform and may reflect transcriptional and epigenetic changes from their cell of origin, thus providing complementary information to the ctDNA somatic alteration information currently used in clinical practice. However, a major challenge with current fragmentomic approaches is the requirement for WGS, which cannot be cost-effectively used to identify somatic alterations and thus is not the current standard for clinical assays. Herein, we describe the first fragmentomic approach that can use existing targeted cancer gene cfDNA panels to accurately classify tumor vs. normal as well as tumor types and subtypes, which performs in the same range as commercial WGS fragmentomics approaches17, 18. This approach remains accurate at distinguishing different tumor types and subtypes down to a ctDNA fraction of 0.001. At this ctDNA fraction, the GRAIL assay only has a sensitivity for detecting variants of 65–75%28. The ability to distinguish prostate cancer adenocarcinoma from NEPC suggests that fragmentomics on targeted panels may also be useful in identifying clinically relevant biological subtypes for other cancers, though additional samples are needed to develop such signatures. Remarkably, this approach is nearly perfect at distinguishing tumor vs. normal samples even in samples with ctDNA fractions ranging from 0.001 to 0.0003. Sensitivity at such low ctDNA fractions suggests potential clinical applications such as multi-cancer early detection (MCED) and minimal residual disease (MRD) detection.
The applicability of fragmentomics to standard targeted ctDNA panels represents a tremendous practical advancement to the field. Most of the existing clinical ctDNA data are from this type of assay and will continue to be, barring a precipitous decrease in sequencing costs. Fragmentomics represents an essentially “free” orthogonal information stream that can complement the somatic alteration detection for which these assays are currently being used. A single assay therefore could provide multiple layers of information depending on ctDNA fraction. Tumor type from fragmentomics can be identified reliably down to 0.1% ctDNA with high depth of sequencing, lower than many assays can even reliably detect somatic alterations28, 52, 53. Below that, tumor vs. normal can still be identified using fragmentomic approaches. Since ctDNA fraction is unknown prior to sequencing, a single unified assay provides the maximum data regardless, and is also cost effective. In addition, a single targeted panel cfDNA sequencing assay allows for maximal use of a plasma sample, as splitting a sample for multiple assays can decrease the sensitivity of each, especially at very low ctDNA quantities. Of note, while ctDNA fraction is a useful metric for these analyses, it is not always possible to obtain due to the lack of germline sequencing, which is required for accurate ctDNA fraction estimation. An advantage of our fragmentomics approach is that it does not require germline sequencing and could be applied to standard commercial target ctDNA sequencing panels which commonly omit germline sequencing.
In conclusion, fragmentomics of standard targeted ctDNA panels is not only feasible, but can accurately distinguish tumor site of origin, tumor subtypes, and tumor vs. normal even in low ctDNA samples. A single assay combining fragmentomics and somatic alteration detection provides tremendous performance, logistical, and cost benefits compared to separate assays for each. This approach merits incorporation into all existing and future targeted ctDNA studies considering its implementation can be performed without any additional sample or additional sequencing cost. The institutional assay described herein is currently being tested in multiple clinical trials across cancer types.
Supplementary Material
HIGHLIGHTS.
cfDNA fragmentomics in targeted cancer gene panels, not just WGS, can infer key phenotypic features of cancer
Fragmentomics models of standard targeted cfDNA panels can distinguish between cancer types and subtypes
Fragmentomics models of standard targeted cfDNA panels can distinguish cancer vs. normal
ACKNOWLEDGEMENTS
We would like to thank all the patients who participated in this study. We would also like to acknowledge the assistance of Katie Kovacich, Laura Ruelle, Hannah Ranous, Lyndsey Deverman, the UWCCC Biospecimen collection team, the Circulating Biomarker Core, and the Big Ten Cancer Research Consortium.
FUNDING
We would also like to acknowledge funding from the National Institutes of Health (DP2 OD030734 to SGZ, 1UH2CA260389 to SGZ, AJA, JML, R01CA247479 to JML, DJB), Department of Defense (PC190039 to SGZ, PC200334 to SGZ, JML, PC180469 to JML), Prostate Cancer Foundation (Movember Foundation – PCF Challenge Award to JML, 2022 Janssen – PCF Special Challenge Award to DAQ, 2022 Point Biopharma Young VAlor Investigator Award to MNS, 2021 Michael and Patricia Berns-PCF Young Investigator Award to MS), the University of Wisconsin Office of the Vice Chancellor for Research and Graduate Education (PICI award to SGZ), and the Doris Duke Charitable Foundation (Physician Scientist Fellowship #2021088 to MNS). Shared research services at the UWCCC are supported by Cancer Center Support Grant P30 CA014520.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
KTH has a family member who is an employee of Epic Systems. MLB has a family member who is an employee of Luminex. SGZ reports unrelated patents licensed to Veracyte, and that a family member is an employee of Artera and holds stock in Exact Sciences. KTH, SGZ, and the University of Wisconsin have filed a provisional patent on the work herein. All remaining authors have declared no conflicts of interest. S.M. Dehm reports consulting relationships with BMS, Oncternal therapeutics, Janssen R&D/J&J and a grant from Pfizer/Astellas/Medivation (the grant was submitted to Medivation, ultimately funded by Astellas and then moved to Pfizer). F.Y. Feng reports personal fees from Janssen Oncology, Bayer, PFS Genomics, Myovant Sciences, Roivant Sciences, Astellas Pharma, Foundation Medicine, Varian, Bristol Myers Squibb (BMS), Exact Sciences, BlueStar Genomics, Novartis, and Tempus; other support from Serimmune and Artera outside the submitted work. IDT assisted in a pilot project to assess the performance characteristics the UW panel prior to purchase, but played no role in this study.
REFERENCES
- 1.Diaz LA Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014; 32 (6): 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen M, Zhao H. Next-generation sequencing in liquid biopsy: cancer screening and early detection. Hum Genomics 2019; 13 (1): 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao W, Mei C, Nan X et al. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: A qualitative study. Gene 2016; 590 (1): 142–148. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Takada S, Mizuta R. Cell-free DNA in blood circulation is generated by DNase1L3 and caspase-activated DNase. Biochem Biophys Res Commun 2019; 516 (3): 790–795. [DOI] [PubMed] [Google Scholar]
- 5.Fan HC, Blumenfeld YJ, Chitkara U et al. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A 2008; 105 (42): 16266–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo YM, Chan KC, Sun H et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010; 2 (61): 61ra91. [DOI] [PubMed] [Google Scholar]
- 7.Snyder MW, Kircher M, Hill AJ et al. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016; 164 (1–2): 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez C, Roch B, Mazard T et al. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021; 6 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulz P, Thallinger GG, Auer M et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet 2016; 48 (10): 1273–1278. [DOI] [PubMed] [Google Scholar]
- 10.Rao S, Han AL, Zukowski A et al. Transcription factor-nucleosome dynamics from plasma cfDNA identifies ER-driven states in breast cancer. Sci Adv 2022; 8 (34): eabm4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luger K, Mäder AW, Richmond RK et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389 (6648): 251–260. [DOI] [PubMed] [Google Scholar]
- 12.Ivanov M, Baranova A, Butler T et al. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 2015; 16 Suppl 13 (Suppl 13): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramachandran S, Ahmad K, Henikoff S. Transcription and Remodeling Produce Asymmetrically Unwrapped Nucleosomal Intermediates. Mol Cell 2017; 68 (6): 1038–1053.e1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esfahani MS, Hamilton EG, Mehrmohamadi M et al. Inferring gene expression from cell-free DNA fragmentation profiles. Nat Biotechnol 2022; 40 (4): 585–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herberts C, Annala M, Sipola J et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature 2022; 608 (7921): 199–208. [DOI] [PubMed] [Google Scholar]
- 16.Zhu G, Guo YA, Ho D et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat Commun 2021; 12 (1): 2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cristiano S, Leal A, Phallen J et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019; 570 (7761): 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathios D, Johansen JS, Cristiano S et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun 2021; 12 (1): 5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun K, Jiang P, Cheng SH et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res 2019; 29 (3): 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peneder P, Stütz AM, Surdez D et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat Commun 2021; 12 (1): 3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mouliere F, Chandrananda D, Piskorz AM et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018; 10 (466). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulz P, Perakis S, Zhou Q et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 2019; 10 (1): 4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doebley AL, Ko M, Liao H et al. A framework for clinical cancer subtyping from nucleosome profiling of cell-free DNA. Nat Commun 2022; 13 (1): 7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang P, Sun K, Peng W et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov 2020; 10 (5): 664–673. [DOI] [PubMed] [Google Scholar]
- 25.Underhill HR, Kitzman JO, Hellwig S et al. Fragment Length of Circulating Tumor DNA. PLoS Genet 2016; 12 (7): e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang P, Chan CW, Chan KC et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A 2015; 112 (11): E1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y At the dawn: cell-free DNA fragmentomics and gene regulation. Br J Cancer 2022; 126 (3): 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herberts C, Wyatt AW. Technical and biological constraints on ctDNA-based genotyping. Trends Cancer 2021; 7 (11): 995–1009. [DOI] [PubMed] [Google Scholar]
- 29.Razavi P, Li BT, Brown DN et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 2019; 25 (12): 1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25 (14): 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26 (6): 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai Z, Markovets A, Ahdesmaki M et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 2016; 44 (11): e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bick AG, Weinstock JS, Nandakumar SK et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020; 586 (7831): 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talevich E, Shain AH, Botton T et al. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 2016; 12 (4): e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandekerkhove G, Lavoie JM, Annala M et al. Plasma ctDNA is a tumor tissue surrogate and enables clinical-genomic stratification of metastatic bladder cancer. Nat Commun 2021; 12 (1): 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wyatt AW, Annala M, Aggarwal R et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J Natl Cancer Inst 2017; 109 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Annala M, Taavitsainen S, Khalaf DJ et al. Evolution of Castration-Resistant Prostate Cancer in ctDNA during Sequential Androgen Receptor Pathway Inhibition. Clin Cancer Res 2021; 27 (16): 4610–4623. [DOI] [PubMed] [Google Scholar]
- 38.Annala M, Vandekerkhove G, Khalaf D et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov 2018; 8 (4): 444–457. [DOI] [PubMed] [Google Scholar]
- 39.Vandekerkhove G, Struss WJ, Annala M et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur Urol 2019; 75 (4): 667–675. [DOI] [PubMed] [Google Scholar]
- 40.Vandekerkhove G, Todenhöfer T, Annala M et al. Circulating Tumor DNA Reveals Clinically Actionable Somatic Genome of Metastatic Bladder Cancer. Clin Cancer Res 2017; 23 (21): 6487–6497. [DOI] [PubMed] [Google Scholar]
- 41.Mizuno K, Sumiyoshi T, Okegawa T et al. Clinical Impact of Detecting Low-Frequency Variants in Cell-Free DNA on Treatment of Castration-Resistant Prostate Cancer. Clin Cancer Res 2021; 27 (22): 6164–6173. [DOI] [PubMed] [Google Scholar]
- 42.Markus H, Chandrananda D, Moore E et al. Refined characterization of circulating tumor DNA through biological feature integration. Sci Rep 2022; 12 (1): 1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hesson LB, Sloane MA, Wong JW et al. Altered promoter nucleosome positioning is an early event in gene silencing. Epigenetics 2014; 9 (10): 1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 2009; 10 (3): 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee CK, Shibata Y, Rao B et al. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 2004; 36 (8): 900–905. [DOI] [PubMed] [Google Scholar]
- 46.Lai WKM, Pugh BF. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 2017; 18 (9): 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davuluri RV, Grosse I, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nat Genet 2001; 29 (4): 412–417. [DOI] [PubMed] [Google Scholar]
- 48.Herberts C, Annala M, Sipola J et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature 2022; 608 (7921): 199–208. [DOI] [PubMed] [Google Scholar]
- 49.Bieberstein NI, Carrillo Oesterreich F, Straube K et al. First exon length controls active chromatin signatures and transcription. Cell Rep 2012; 2 (1): 62–68. [DOI] [PubMed] [Google Scholar]
- 50.Brenet F, Moh M, Funk P et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 2011; 6 (1): e14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiszbein A, Krick KS, Begg BE et al. Exon-Mediated Activation of Transcription Starts. Cell 2019; 179 (7): 1551–1565.e1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller L, Belloum Y, Wikman H et al. Clinical relevance of blood-based ctDNA analysis: mutation detection and beyond. Br J Cancer 2021; 124 (2): 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang DK, Park BH. Circulating tumor DNA: current challenges for clinical utility. J Clin Invest 2022; 132 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data from the GRAIL dataset is available at the European Genome Archive (Dataset ID EGAD00001005302). Our institutional protocol did not allow unrestricted public access to the raw sequencing data. Therefore, data sharing requests must be submitted to the University of Wisconsin-Madison for approval. For samples from the two clinical trials (NCT03090165, NCT03725761), these trials are still ongoing, and data sharing requests must be submitted to the trial organizers.