

SUMMARY
GPCRs mediate neuromodulation through activation of heterotrimeric G-proteins (Gαβγ). Classical models depict that G-protein activation leads to a one-to-one formation of Gα-GTP and Gβγ species. Each of these species propagates signaling by independently acting on effectors, but the mechanisms by which response fidelity is ensured by coordinating Gα and Gβγ responses remain unknown. Here, we reveal a paradigm of G-protein regulation whereby the neuronal protein GINIP biases inhibitory GPCR responses to favor Gβγ over Gα signaling. Tight binding of GINIP to Gαi-GTP precludes its association with effectors (adenylyl cyclase) and, simultaneously, with Regulator-of-G-protein-Signaling (RGS) proteins that accelerate deactivation. As a consequence, Gαi-GTP signaling is dampened whereas Gβγ signaling is enhanced. We show that this mechanism is essential to prevent imbalances of neurotransmission that underlie increased seizure susceptibility in mice. Our findings reveal an additional layer of regulation within a quintessential mechanism of signal transduction that sets the tone of neurotransmission.
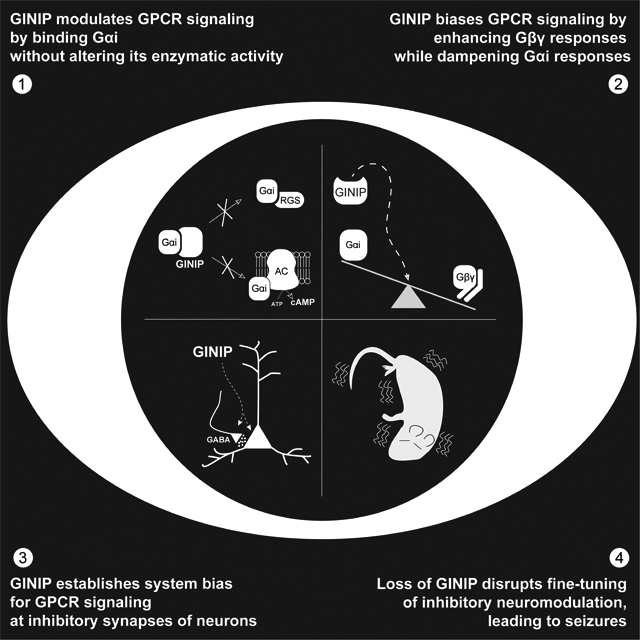
Graphical Abstract
Park, Luebbers et al. dissect a mechanism by which responses triggered by GPCRs, the largest family of cell surface receptors in humans, is finely tuned in neurons. By scaling different G-protein signaling branches immediately downstream of receptors, this mechanism ensures proper interpretation of signals that silence neurotransmission to prevent seizures.
INTRODUCTION
Understanding the molecular basis for how neurotransmission is orchestrated remains a central challenge in neuroscience. G protein-coupled receptors (GPCRs) are the largest family of metabotropic receptors, which collectively mediate responses to most neurotransmitters1–3. They can be found at pre-synaptic and post-synaptic structures, where they modulate synaptic transmission in a “slow” time scale of seconds to minutes due to the requirement of signaling intermediaries like heterotrimeric G-proteins (Gαβγ)3–5. GPCRs are also the largest class of druggable targets in the human genome, including numerous medications for neurological and neuropsychiatric diseases6–9. However, the molecular mechanisms that control GPCR-mediated neuromodulation are very complex and remain the subject of intense investigation.
Although some responses are mediated via β-arrestins, GPCRs primarily signal by promoting GTP loading on Gα subunits of heterotrimeric G-proteins (Gαβγ)2. Then, Gα-GTP and Gβγ dissociate, which allows their binding to and modulation of downstream effectors. Signaling is turned off upon GTP hydrolysis, leading to re-association of Gα with Gβγ. While this mechanism of G-protein regulation by GPCRs is well understood, much less is known about how it is influenced by a growing body of cytoplasmic proteins that act on G-proteins10–17. Most of these regulators have in common that they bind to Gα subunits and affect their enzymatic activity by modulating nucleotide binding or hydrolysis, thereby having a profound impact on the duration and amplitude of G-protein signaling. For example, Regulators of G-protein Signaling (RGS) are GTPase Activating Proteins (GAPs) that bind to Gα-GTP and accelerate the rate of nucleotide hydrolysis18,19. Other cytoplasmic regulators alter nucleotide exchange rates (i.e., GTP loading) by serving as Guanine-nucleotide Dissociation Inhibitors (GDIs)20–22 or as non-receptor Guanine-nucleotide Exchange Factors (GEFs)17,23–25.
Heterotrimeric G-proteins are broadly divided in four families based on sequence and functional similarity (Gs, Gi/o, Gq/11, G12/13)1. In the nervous system, Gi/o proteins mediate inhibitory neuromodulation triggered by neurotransmitters like GABA, dopamine, serotonin, or norepinephrine, among others. When activated, Gi/o proteins release free Gβγ subunits that modulate numerous targets, including inhibition of pre-synaptic voltage-gated Ca2+ channels (e.g., Cav2) or activation of post-synaptic G protein-gated inwardly rectifying K+ channels GIRK)26,27. These direct effects on ion activity suppress neurotransmission by preventing neurotransmitter release or reducing post-synaptic excitation, respectively. At the same time, Gi/o activation can also generate Gαi-GTP, which inhibits adenylyl cyclases28,29. This dampens intracellular cAMP levles, which has indirect effects on neurotransmission through the numerous targets affected by this ubiquitous second messenger30–34. Given the large complexity of signaling networks affected simultaneously by Gβγ and Gα-GTP, a central and unresolved question in GPCR signaling is: how is response specificity achieved? In particular, the fact that Gβγ and Gα-GTP are generated at equimolar amounts upon GPCR stimulation poses the conundrum of how different signaling cascades triggered by each one of the two species independently are coordinated to ensure the fidelity of the cellular response.
Here, we characterize a mechanism for biasing Gi responses in which Gαi-GTP signaling is inhibited while, simultaneously, Gβγ signaling is enhanced after receptor stimulation. We demonstrate that this mechanism of signaling coordination is required to ensure appropriate scaling of GPCR neuromodulatory influence over neuronal excitability. More specifically, our findings indicate that GINIP (aka KIAA1045), a protein previously identified as a binder of active Gαi35,36 and as a regulator of GABAB receptor (GABABR) signaling in dorsal root ganglia36,37, is a broad regulator of signaling via Gi-coupled GPCRs expressed across multiple brain regions in both inhibitory and excitatory neurons. At the molecular level, GINIP biases receptor responses to favor Gβγ signaling over Gαi-GTP signaling without affecting the enzymatic activity of Gα subunits directly. Overall, our findings reveal an additional layer of GPCR regulation through a previously unknown form of biased signaling that has a crucial role in modulating neurotransmission.
RESULTS
GINIP binds to active Gαi subunits with high affinity
GINIP is a protein well conserved in vertebrates (Fig. S1A) but does not have clear orthologues in invertebrates. A prior report showed that GINIP binds to GTPase-deficient Gαi mutants that mimic active G proteins36, but no rigorous biochemical characterization was performed. Using purified GINIP and Gαi3 proteins, we found that GINIP binds with high affinity (KD ~70 nM) to bona fide active Gαi3 (i.e., loaded with GTPγS, a non-hydrolysable GTP analog), as well as to the active transition state for GTP hydrolysis adopted upon binding of GDP-AlF4−38,39 (Fig. 1A). Binding to inactive, GDP-bound Gαi3 was undetectable (Fig. 1A). GINIP binds similarly to all three Gαi isoforms (Gαi1, Gαi2 and Gαi3), but not to other α-subunits of the Gi/o family like Gαo and Gαz, or to representative members of any of the other three G-protein families (Fig. 1B, Fig. S1B). These results indicate that GINIP is highly selective for Gαi isoforms, and that it binds with a marked G-protein state preference for active Gα.
Figure 1. GINIP binds to the effector binding region of Gαi without affecting its enzymatic activity.
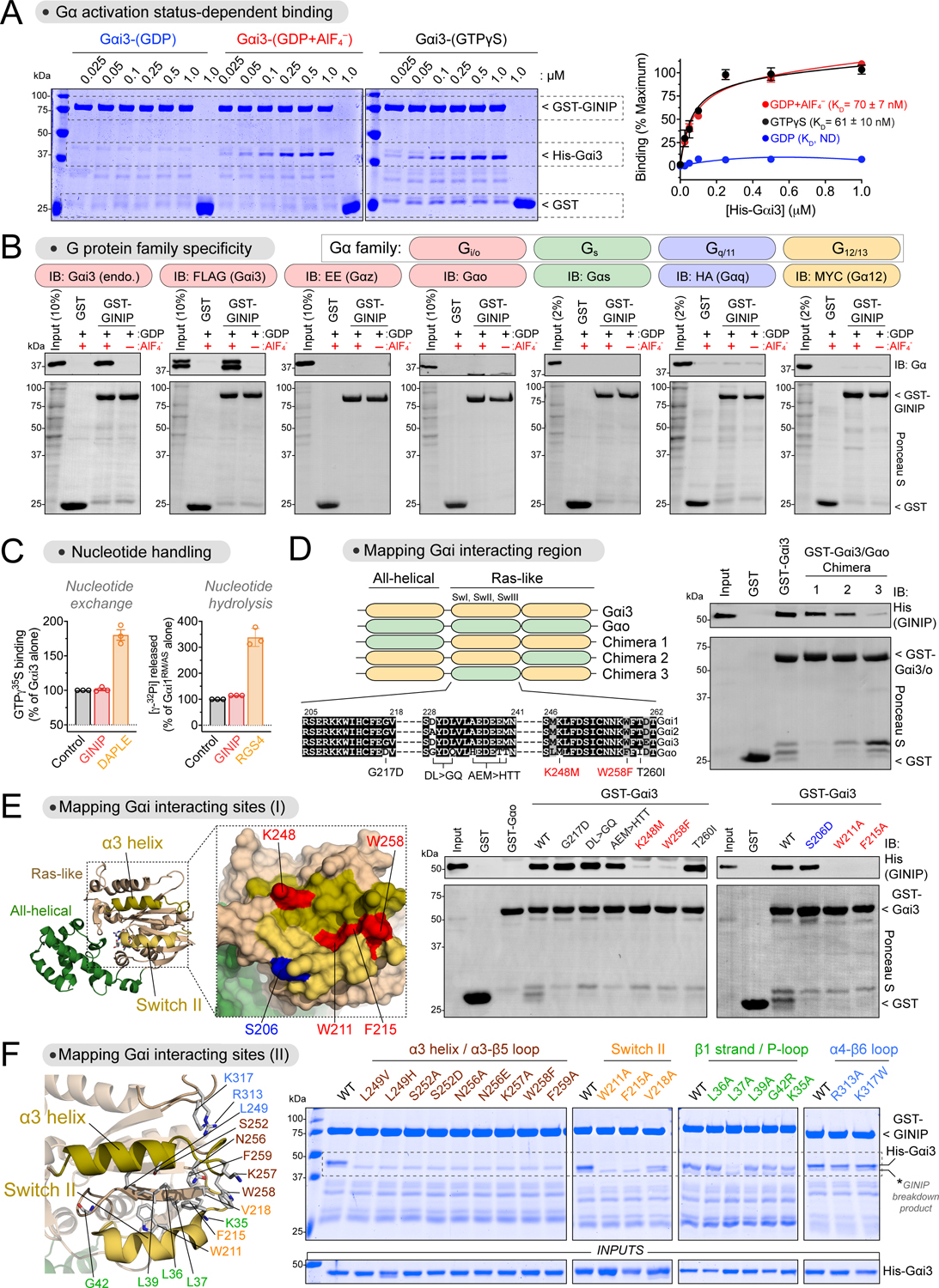
(A) GINIP binds to active but not inactive Gαi3. Left, Coomassie-stained gel showing binding of His-Gαi3, loaded as indicated (GDP, GDP·AlF4−, GTPγS), to immobilized GST-GINIP. Right, quantification of His-Gαi3 binding to GST-GINIP. Mean±S.E.M., n=3–4.
(B) GINIP binds to Gαi3, but not to other Gα’s of the same family (Gαo, Gαz), or other families (Gαs, Gαq, Gα12). Lysates of HEK293T cells expressing the indicated G-proteins were incubated with GST or GST-GINIP immobilized on glutathione-agarose beads in the presence of GDP or GDP·AlF4−. Bead-bound proteins were detected by Ponceau S staining or immunoblotting (IB).
(C) GINIP does not affect the enzymatic activity of Gαi. Nucleotide exchange on Gαi3 was determined by GTPγS binding, whereas nucleotide hydrolysis by Gαi1RM/AS was determined by the production of free phosphate (Pi) from GTP. GINIP, 2 μM, DAPLE, 1 μM, RGS4, 0.2 μM. Mean±S.E.M., n=3.
(D) Gαi3 region aa178–270 is required for GINIP binding. Left, diagram of Gαi3 (orange) /Gαo (green) chimeras. Sequence alignment of Gαi1, Gαi2, Gαi3, and Gαo, indicating mutations tested in panel (E). Right, binding of purified His-GINIP to the indicated G-proteins in the presence of GDP·AlF4−. Bead-bound proteins were detected by Ponceau S staining or immunoblotting (IB).
(E) Mutation of residues in the α3 helix and Switch II of Gαi ablate GINIP binding. Left, Structural model of Gαi1-(GDP·AlF4−) (PDB: 2G83). Red indicates residues in the α3/Switch II region that disrupt GINIP binding when mutated, whereas blue indicates an adjacent residue that does not affect GINIP binding when mutated. Center & Right, binding of purified His-GINIP to the indicated G-proteins in the presence of GDP·AlF4−. Bead-bound proteins were detected by Ponceau S staining or immunoblotting (IB).
(F) Mutation of residues within, but not adjacent to, the effector binding region (α3/Switch II groove) of Gαi impair GINIP binding. Left, Structural model of Gαi1-(GDP·AlF4−) (PDB: 2G83) displaying the residues investigated by site-directed mutagenesis. Right, binding of the indicated G-proteins loaded with GTPγS to GST-GINIP. Bead-bound proteins were detected Coomassie staining.
All protein electrophoresis results are representative of n ≥ 3 experiments. See also Figure S1.
GINIP does not affect the enzymatic activity of Gαi subunits
Since GINIP lacks any known catalytic domain that could justify its ability to serve as a G-protein effector, we investigated if GINIP affects nucleotide handling by Gαi. First, we found that nucleotide exchange by Gαi3, as determined by a GTPγS binding assay40,41, was not affected by GINIP, whereas the GEF DAPLE42 led to an increase in activity (Fig. 1C). Next, we determined if GINIP affects nucleotide hydrolysis using steady-state GTPase assays with a Gαi1 mutant (Gαi1RM/AS) for which nucleotide hydrolysis is the rate limiting step43. We found that GINIP did not affect nucleotide hydrolysis by Gαi1RM/AS, despite binding to it as strongly as to Gαi1 wild-type (Fig. S1B), whereas the GAP RGS419 led to a marked increase in activity (Fig. 1C). Taken together, these results show that GINIP does not affect the enzymatic activity of Gαi despite binding to it with high affinity.
GINIP binds to the groove formed by the α3 helix and the switch II region of Gαi
To begin elucidating the functional consequences of GINIP binding to Gαi, we carried out experiments to map its binding site on the G-protein. First, we leveraged the marked preference of GINIP for binding Gαi isoforms over the closely related protein Gαo (Fig. 1B, Fig.S1B) to design G-protein chimeras (Fig. 1D). We generated three Gαi/o chimeras by replacing, one at a time, three different regions of Gαi3 with the corresponding regions of Gαo and performed GINIP binding experiments with them. We found that the region between amino acid (aa) 178 and aa 270 of Gαi3 was required for GINIP binding (Fig. 1D). This segment is located in the Ras-like domain of Gα and encompasses the three “switch” regions that change conformation between GDP- and GTP-bound states39,44. Next, we systematically replaced Gαi3 residues in the aa178–270 region for their non-conserved counterparts in Gαo (Fig. 1D, E). Of these, K248M and W258F completely abolished GINIP binding to active Gαi3 (Fig. 1E). K248 is located in the α3 helix and W258 in the adjacent α3/β5 loop. Both residues are oriented towards a groove formed between the α3 helix and the switch II (SwII) region38,39 (Fig. 1E), which is a pocket utilized by Gα subunits to bind effectors45–50. Mutation of two SwII residues (W211 and F215) that contribute to this pocket also disrupted GINIP binding, whereas mutation of another SwII residue not oriented towards the α3/SwII groove (S206) did not (Fig. 1E).
We further pinpointed the structural determinants of Gαi3 required for GINIP binding by using a battery of mutants spanning regions within or adjacent to the α3/SwII groove (Fig. 1F). Of these, mutation of residues in the α3 helix (L249, S252, N256), α3/β5 loop (K257, W258, F259) and SwII (W211, F215, V218) oriented towards the α3/SwII groove resulted in loss of GINIP binding to active Gαi3 (Fig. 1F). Moreover, mutation of a residue of the β1 strand positioned at the bottom of the α3/SwII groove (L37) also resulted in loss of GINIP binding (Fig. 1F). In contrast, mutation of other residues adjacent to the elements forming the α3/SwII groove did not disrupt GINIP binding (K35, L36, L38, G42 in the β1 strand/P-loop, or R313, K317 in the α4/β6 loop, Fig. 1F). All mutants used have been shown to be functional based on their ability to adopt an active conformation upon GTP binding40,51. These results indicate that residues in the α3/SwII groove of Gαi are specifically involved in mediating GINIP binding. Consistent with this idea, we also found that GINIP competes for binding to Gαi3 with the effector-like peptide KB-1753, which binds on the α3/SwII groove46 (Fig. S1C). These results provide strong evidence that the α3/SwII groove of Gαi is a critical binding site for GINIP. Additional evidence from work in progress52 combining structure predictions, mass spectrometry, and cellular and biochemical assays support the conclusion that GINIP engages the effector binding site of Gαi.
GINIP dampens cAMP inhibition triggered by multiple Gi-coupled GPCRs
Since GINIP binds with high affinity to the effector binding site of Gαi-GTP, we hypothesized that GINIP would prevent the action of the G-protein on adenylyl cyclase. If so, GINIP should dampen cAMP inhibition upon Gi activation regardless of the GPCR involved. Consistent with a previous report36, we found that GINIP reduced the extent by which the GABABR inhibited forskolin-stimulated cAMP in HEK293T cells (Fig. 2A). Moreover, similar results were obtained with two other unrelated GPCRs that activate Gi, the α2A adrenergic receptor (α2A-AR) and the D2 dopamine receptor (D2R) (Fig. 2A). In contrast, GINIP expression did not affect forskolin-stimulated cAMP accumulation in the same assay format (0.66 ± 0.10 vs 0.60 ± 0.07 ΔBRET−1 in the presence or absence of GINIP, respectively, 10 min after stimulation with 1 μM forskolin, n=8, p>0.05, paired t-test), indicating that GINIP has no direct effect on adenylyl cyclase activity. These results support the idea that GINIP acts at the level of the G-protein rather than being receptor specific.
Figure 2. GINIP directly blocks Gαi-mediated regulation of adenylyl cyclase.
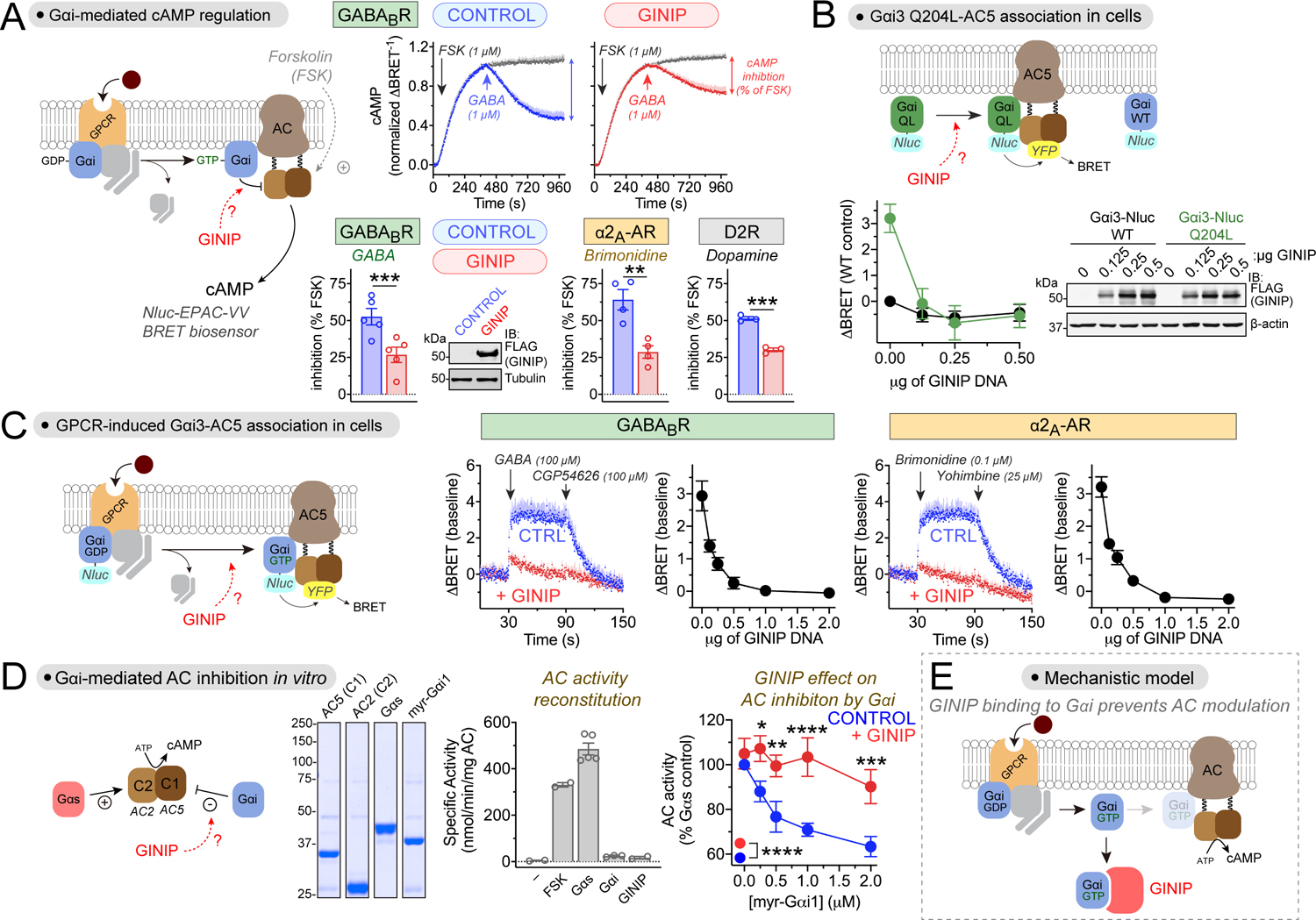
(A) GINIP prevents Gαi-mediated inhibition of adenylyl cyclase (AC) upon stimulation of 3 different GPCRs. Top row, kinetic traces of BRET measurement of cAMP in HEK293T cells expressing the GABABR in the presence or absence of GINIP treated with forskolin (FSK) and GABA as indicated. Bottom row, quantified inhibition of FSK-stimulated cAMP upon stimulation of GABABR, α2A-AR, or D2R with GABA (1 μM), Brimonidine (5 μM), or dopamine (0.2 μM). Immunoblot (IB) validates GINIP expression. Mean±S.E.M., n=3–5. **p<0.01, ***p<0.001, paired t-test.
(B) GINIP prevents the association of active Gαi3 with AC5 in cells. Left, changes in BRET (ΔBRET) were determined in HEK293T cells expressing Gαi3-Nluc WT or Gαi3-Nluc Q204L upon transfection of increasing amounts of GINIP. Mean±S.E.M., n=6. Right, validation of GINIP expression by immunoblotting (IB).
(C) GINIP prevents the association of Gαi3 with AC5 upon GPCR stimulation. BRET was measured in HEK293T cells expressing the GABABR or the α2A-AR upon transfection of different amounts of GINIP DNA. Kinetic traces correspond to cells expressing no GINIP (‘CTRL’ blue) or transfected with 2 μg of GINIP plasmid (red). Cells were treated with the indicated GPCR agonists/antagonists. Mean±S.E.M., n=3.
(D) GINIP blocks the regulation of AC by Gαi in vitro. Coomassie-stained gel shows the purified proteins used. Bar graph shows that FSK (5 μM) or Gαs-GTPγS (0.1 μM), but not myr-Gαi1 (2 μM) or GINIP (2 μM), promote the activation of reconstituted AC (AC5 (C1) + AC2 (C2)). Right, Gαs-stimulated AC activity in the presence of increasing concentrations of myr-Gαi1-GTPγS with (red) or withiot (blue) GINIP (2 μM). Mean±S.E.M., n=5. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA for presence/ absence of GINIP x myr-Gαi1 concentration, with multiple comparisons at each concentration using Fisher’s LSD test.
(E) Diagram summarizing the proposed mechanism action of GINIP on Gαi-mediated modulation of AC activity (competitive binding).
GINIP prevents the association of Gαi with adenylyl cyclase in cells
Next, we used a bioluminescence resonance energy transfer (BRET) assay to monitor the effect of GINIP on the interaction between Gαi3 and adenylyl cyclase 5 (AC5) in cells (Fig. 2B, C). For this, Gαi3 tagged with nanoluciferase (Nluc) was co-expressed with YFP-tagged AC5. First, we observed that a constitutively active mutant of Gαi3 (Gαi3 Q204L) that mimics Gα-GTP resulted in higher BRET levels than Gαi3 wild-type (WT) in the absence of GPCR stimulation (Fig. 2B), which is consistent with the expected preferential binding of AC5 with active G-proteins. Co-expression of increasing amounts of GINIP resulted in a decrease of BRET in cells expressing Gαi3 Q204L to levels equivalent to those observed for Gαi3 WT (Fig. 2B), indicating that GINIP prevents the association of active G-proteins with adenylyl cyclase. We also used the same assay to investigate the effect of GINIP on GPCR-stimulated Gαi3-AC5 association (Fig. 2C), and found that expression of increasing amounts of GINIP efficiently suppressed BRET responses induced by the stimulation of either GABABR or α2A-AR (Fig. 2C). These results not only indicate that GINIP prevents the association of Gαi with its effector adenylyl cyclase, but also that this effect is independent of the GPCR involved.
GINIP inhibits directly the regulation adenylyl cyclase by Gαi-GTP
To more definitely pinpoint that the mechanism of action of GINIP is direct blockade of Gαi-effector interaction, we used a reductionist approach with purified proteins. We reconstituted adenylyl cyclase activity in vitro with the purified C1 domain of AC5 and the purified C2 domain of AC2, as previously described53, which led to robust cAMP synthesis when incubated with forskolin or purified Gαs-GTPγS (Fig. 2D). As expected54, purified myristoylated Gαi1-GTPγS (myr-Gαi1) inhibited Gαs-stimulated adenylyl cyclase activity in a dose-dependent manner. This inhibitory effect of myr-Gαi1 was suppressed in the presence of GINIP (Fig. 2D), demonstrating that GINIP directly prevents the regulation of adenylyl cyclase by Gαi.
Taken together, our results support a mechanism in which GINIP regulates GPCR-dependent signaling by directly binding to Gαi-GTP and preventing its association with the effector adenylyl cyclase, therefore resulting in the suppression of Gα-GTP dependent signaling (Fig. 2E).
GINIP promotes free Gβγ signaling upon GPCR stimulation
After establishing how GINIP affects Gαi-GTP dependent signaling, we turned our attention to Gβγ, the other signaling species released upon G-protein activation by GPCRs. For this, we leveraged a BRET-based biosensor that directly detects “free” Gβγ competent for engaging effectors55,56 (Fig. 3A). We found that expression of GINIP in cells increased the amplitude of Gβγ responses elicited upon stimulation of GABABR with its agonist GABA at a submaximal concentration (1 μM) but not at a maximal concentration (100 μM) (Fig. 3A). However, when we determined the rate of Gβγ deactivation upon addition of an antagonist post-GABA stimulation, we found that GINIP delayed the rate of deactivation regardless of the concentration of agonist used (Fig. 3A). The magnitude of the change in deactivation rate was dependent on the amount of GINIP expressed (Fig. S2A, B). Similar effects of GINIP were observed with a second Gi-coupled GPCR, the α2A-AR (Fig. S2C). These results indicate that the overall effect of GINIP on Gβγ signaling is to enhance, rather than suppress as observed for Gαi-GTP signaling.
Figure 3. GINIP promotes Gβγ-mediated signaling by antagonizing the action of RGS GAPs on Gαi.
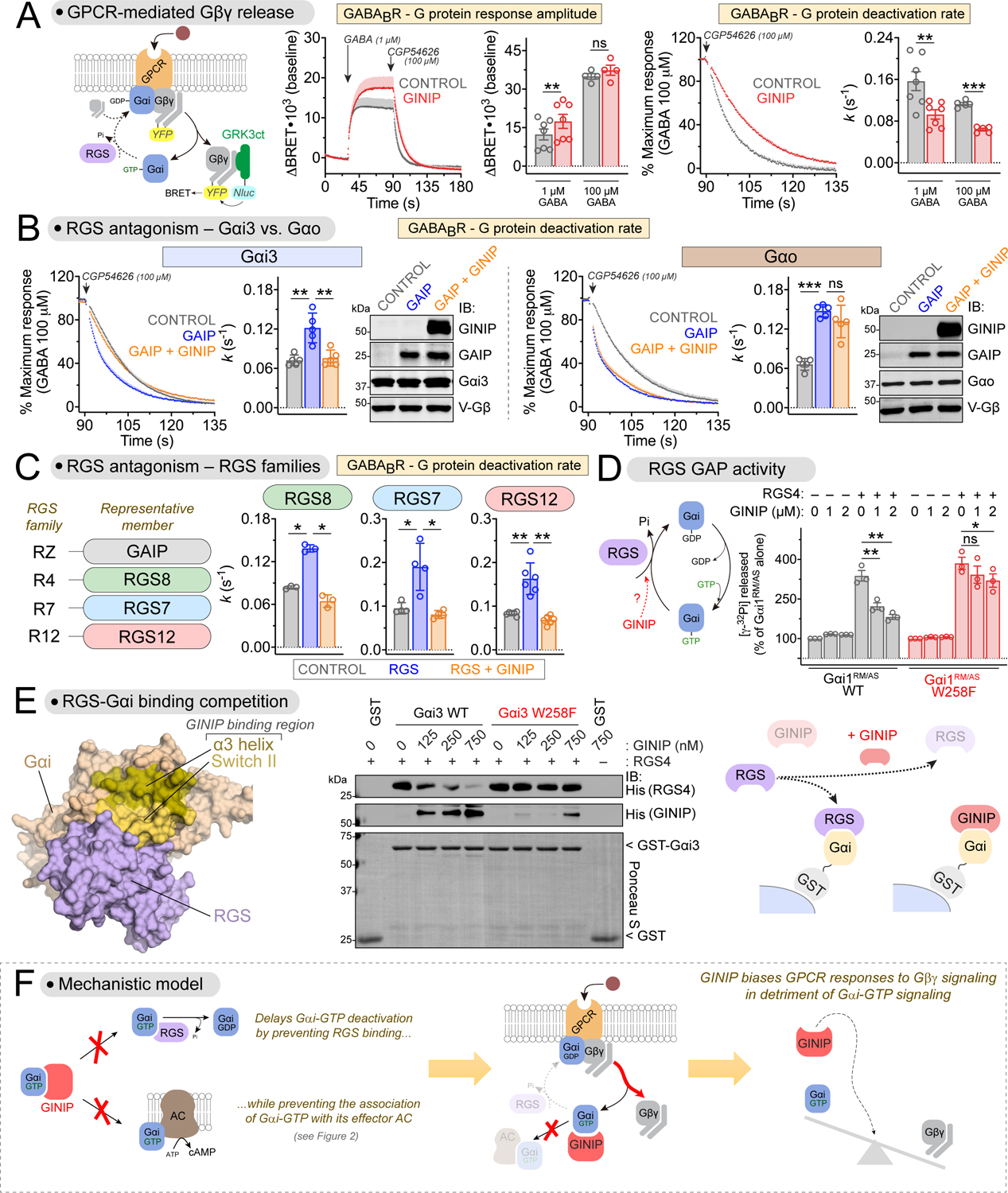
(A) GINIP enhances Gβγ-mediated signaling triggered by GABABR. Left, diagram of G-protein activation/deactivation cycle and BRET-based detection of free Gβγ. Center, BRET was measured in HEK293T cells expressing the GABABR in the absence (black) or presence (red) of GINIP. Cells were treated with GABA and CGP54626 as indicated. Right, G protein deactivation rates were determined by normalizing the BRET data and curve fitting to extract rate constant values (k). Mean±S.E.M., n=4–7. ns = not significant, **p<0.01, ***p<0.001, paired t-test.
(B) GINIP antagonizes GAIP-mediated acceleration of Gβγ deactivation for Gi but not Go proteins. BRET experiments were carried out and analyzed as in (A) with cells expressing Gαi3 or Gαo in the absence (grey) or presence of GAIP (blue) or GAIP plus GINIP (orange). GAIP and GINIP expression validated by immunoblotting (IB). Mean±S.E.M., n=5. ns = not significant, **p<0.01, ***p<0.001, one-way ANOVA corrected for multiple comparisons (Tukey).
(C) GINIP antagonizes the acceleration of Gβγ deactivation mediated by representative members of all RGS families. BRET experiments were carried out and analyzed as in (B), except that RGS8 (R4), RGS7 (R7), or RGS12 (R12) were used instead of GAIP (RZ). RGS7 was co-transfected with Gβ5 and R7BP. Mean±S.E.M., n=3–6. *p<0.05, **p<0.01, one-way ANOVA corrected for multiple comparisons (Tukey).
(D) GINIP antagonizes the GAP activity of RGS4 on Gαi in vitro. Nucleotide hydrolysis by Gαi1RM/AS (WT or W258F) was determined in the presence of RGS4 and/or GINIP. Mean±S.E.M., n=3. ns = not significant, *p<0.05, **p<0.01, one-way ANOVA corrected for multiple comparisons (Tukey).
(E) GINIP competes with RGS4 for binding to Gαi3. Left, Structural model of Gαi1-(GDP·AlF4−) bound to RGS4 (PDB: 1AGR). Right, increasing concentrations of purified His-GINIP and a fixed amount of His-RGS4 (20 nM) were incubated with GST or GST-Gαi3 (WT or W258F) immobilized on glutathione-agarose beads in the presence of GDP·AlF4−. Bead-bound proteins were detected by Ponceau S staining or by immunoblotting (IB). One representative result of three independent experiments is shown.
(F) Diagram summarizing the proposed mechanism by which GINIP biases G protein responses by favoring Gβγ-dependent signaling in detriment of Gαi-dependent signaling.
GINIP antagonizes RGS GAP-mediated deactivation of Gβγ signaling
The effects of GINIP on Gβγ signaling are reminiscent of those observed when the function of RGS GAPs is disabled57. When RGS GAP in function is disabled, Gβγ deactivation rates slow down and response amplitudes are potentiated57, much like what we found upon GINIP expression (Fig. 3A). Based on this, we hypothesized that GINIP might antagonize the action of RGS GAPs. Expression of the RGS protein GAIP (aka RGS19) accelerated the rate of Gβγ deactivation upon modulation of Gi3 protein activity by GABABR, as expected, and this effect was suppressed when GINIP was co-expressed with GAIP (Fig. 3B). Similar results were obtained with the α2A-AR (Fig. S2D), suggesting that GINIP antagonizes the inhibitory action of RGS proteins on Gi proteins independent of the receptor involved. In contrast, GINIP did not antagonize the acceleration of Gβγ deactivation by GAIP when the G-protein modulated by GABABR was Go instead of Gi3 (Fig. 3B). These results are consistent with the lack of binding of GINIP to Gαo (Fig. 1), and further confirm that GINIP exerts its effects on Gβγ signaling by acting at the level of the Gα subunit rather than any other component of the signaling system. The latter point is further substantiated by the observation that expression of GINIP does not affect the recruitment of arrestin-3 to two different GPCRs (Fig. S2E).
Next, we assessed if the effect of GINIP on GAIP-mediated regulation of Gβγ signaling was a phenomenon generalizable to other RGS proteins, with the expectation that it would be because all RGS proteins share a similar binding mode and mechanism of action on Gα38,58. In addition to the RZ family to which GAIP belongs, there are 3 other RGS proteins families: R4, R7 and R12. Our results confirmed that GINIP can suppress the acceleration of Gβγ deactivation exerted by representative members of each of these families (Fig. 3C). These findings show that GINIP antagonizes the regulation of Gβγ signaling by all four RGS protein families.
GINIP inhibits directly the regulation of Gαi by RGS proteins
Since GINIP binds directly to Gαi (Fig. 1), we tested if GINIP directly inhibits the GAP activity of RGS proteins by using purified proteins. We found that GINIP inhibited the GAP activity of RGS4 or GAIP on Gαi1RM/AS (Fig. 3D, Fig. S3A, B, D). In contrast, this effect of GINIP was not observed when the G-protein contained the W258F mutation (Fig. 3D, Fig. S3D) that precludes GINIP binding (Fig. 1) while preserving Gαi binding to RGS proteins (Fig. S3C and40). These results demonstrate that GINIP blocks the GAP activity of RGS proteins by binding to Gαi.
GINIP competes with RGS proteins for binding to Gαi
We reasoned that the mechanism by which GINIP blocks RGS GAP activity is competition for Gαi binding. This is not only because disrupting GINIP binding to Gαi prevents the inhibition of RGS GAP activity (Fig. 3D, Fig. S3D), but also because of the following two features of how GINIP binds Gαi: (1) GINIP has high affinity for the same conformation that is preferred by RGS proteins (Fig. 1A), i.e., the transition state mimicked by GDP-AlF4− loading38,59, and (2) it binds at a site (α3/SwII groove, Fig. 1) that is adjacent to the RGS binding region (Fig. 3E, left). To test this mechanism, we determined RGS4 binding to Gαi3-GDP-AlF4− in the absence or presence of GINIP using purified proteins (Fig. 3E). We found that binding of GINIP to Gαi3-GDP-AlF4− was accompanied by a marked reduction of RGS4 binding, which became barely detectable at the highest concentration of GINIP tested (Fig. 3E). In contrast, GINIP did not affect binding of RGS4 to the Gαi3 W258F mutant (Fig. 3E). These results demonstrate that GINIP antagonizes RGS GAP action by competing for binding to Gαi subunits.
Results presented so far from Fig.1 to Fig. 3 support an overarching model for the mechanism of action of GINIP on regulating GPCR signaling (Fig. 3F). Contrary to other Gα interacting proteins, GINIP behaves as a silent allosteric modulator that prevents the association of Gαi-GTP with downstream effectors (adenylyl cyclase) or negative regulators (RGS GAPs) while preserving intrinsic GTPase activity and nucleotide exchange. Preventing the association with RGS GAPs prolongs the lifetime of Gαi in the GTP-bound state and dissociated from Gβγ. While in this scenario the excess of Gβγ generated is competent for signaling to effectors, Gαi-GTP is not because GINIP prevents Gαi association with adenylyl cyclase. Consequently, G-protein signaling downstream of GPCR stimulation is biased towards Gβγ over Gαi-GTP signaling by the action of GINIP (Fig. 3F).
GINIP is highly expressed in brain
Next, we set out to investigate the physiological consequences of the signaling modulation mediated by GINIP. We found that GINIP expression was restricted to the nervous system across a battery of mouse tissues, being most abundant in brain even when compared to dorsal root ganglia (DRG) (Fig. 4A), where GINIP has been shown to modulate pain processing36. Motivated by this observation, we pursued the characterization of GINIP in brain. First, we found that brain-derived GINIP bound to active but not inactive Gαi3 (Fig. 4B), much like recombinant GINIP did (Fig.1A), suggesting that GINIP natively expressed in brain preserves the key function required to modulate GPCR signaling. Next, we characterized mice bearing a modified GINIP allele (i.e., GINIP 1a), which has an insertion of a LacZ-containing cassette in the GINIP genomic locus (Fig. 4C). This insertion is expected to result not only in a loss of GINIP protein (null allele), which was confirmed by western blotting (Fig. 4D), but also a reporter for GINIP expression. We leveraged the latter to assess the expression of GINIP in different brain regions, finding that GINIP expression is robust across the isocortex, hippocampus, striatum, amygdala, and, to a lesser extent, the thalamus (Fig. 4E). This expression pattern was confirmed by GINIP mRNA in situ hybridization in GINIP +/+ brains, for which GINIP 1a/1a served as a negative control demonstrating the specificity of the probe (Fig. 4F). These results suggested that GINIP may have a role in a wide range of neurons.
Figure 4. Loss of GINIP increases seizure susceptibility.
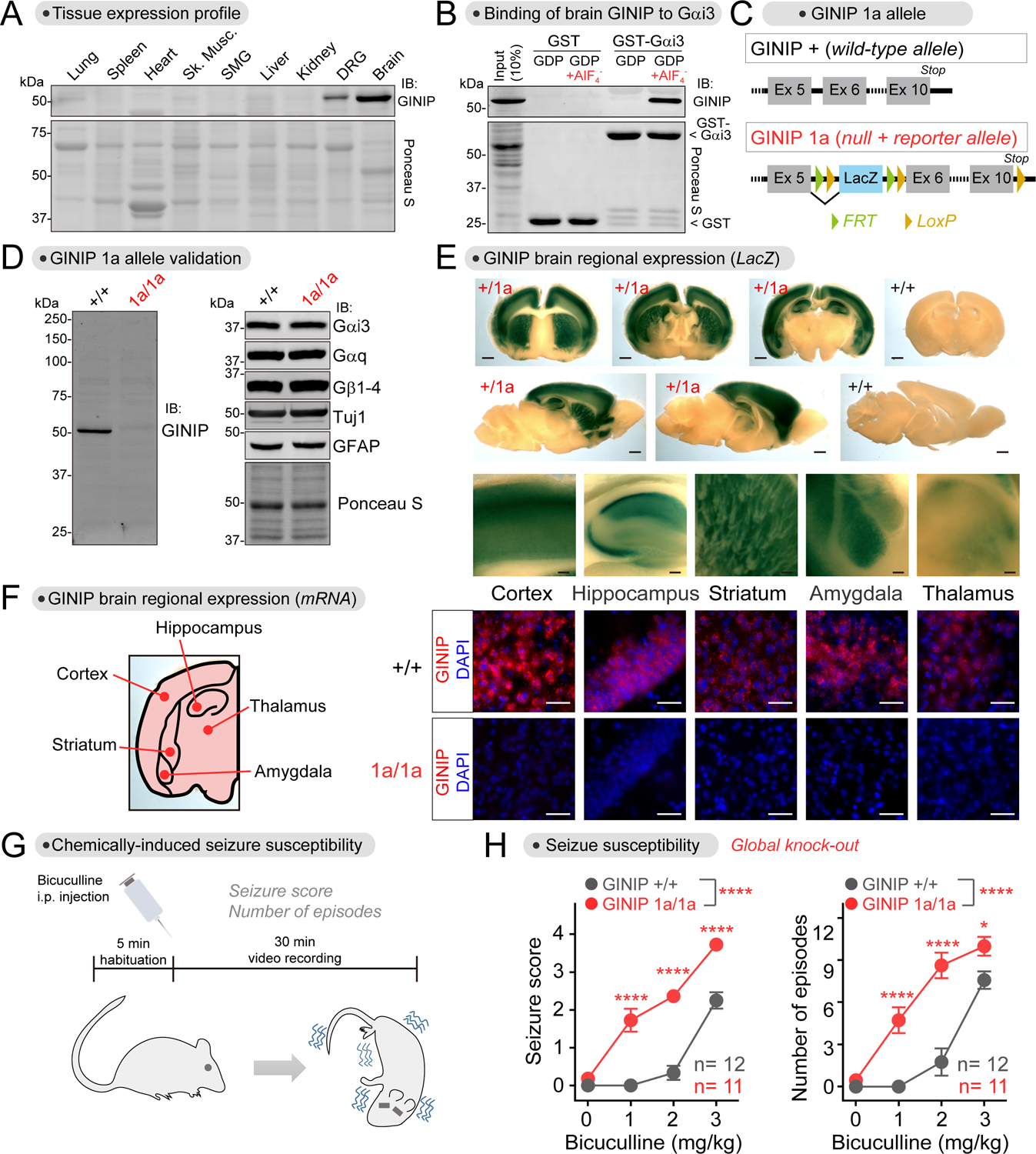
(A) GINIP expression is restricted to the nervous system and most abundant in brain. Proteins extracted from the indicated mouse tissues were analyzed by Ponceau S staining or by immunoblotting (IB). n=2.
(B) Active Gαi binds to brain-derived GINIP. GST or GST-Gαi3 immobilized on glutathione-agarose beads were incubated with mouse brain lysates in the presence of GDP or GDP·AlF4−. Bead-bound proteins were detected by Ponceau S staining or by immunoblotting (IB). n=3.
(C) Diagram depicting features of the GINIP 1a allele bearing a LacZ-containing cassette inserted between exon 5 and exon 6.
(D) GINIP expression is specifically ablated in GINIP 1a/1a mice. Mouse brain lysates were analyzed by immunoblotting (IB), n=3.
(E) GINIP is expressed in the cortex, hippocampus, striatum, amygdala and thalamus. β-galactosidase activity was detected by staining brain slices of GINIP +/1a or +/+ mice. Scale bars are 1 mm (whole sections) or 0.1 mm (enlarged areas). n=3.
(F) GINIP mRNA is expressed in the cortex, hippocampus, striatum, amygdala and thalamus. GINIP mRNA was detected in mouse brain coronal slices of GINIP +/+ or 1a/1a by fluorescence in situ hybridization. Scale bar = 50 μm. n=3.
(G, H) GINIP 1a/1a mice display increased susceptibility to bicuculline-induced seizures compared to GINIP +/+ mice. 11–12 mice (male and female) per genotype. See Fig. S4 for results stratified by sex. Mean±S.E.M. *p<0.05, ****p<0.0001, two-way ANOVA for genotype x concentration of bicuculline, with multiple comparisons at each concentration using Fisher’s LSD test.
See also Figure S4
Loss of GINIP increases seizure susceptibility
Motivated by an earlier report that identified GINIP as one of the candidate genes for spontaneous, generalized seizures in a strain of epileptic rats60, we explored the susceptibility of GINIP knock-out mice to epileptic seizures. Since GINIP is highly expressed in cortical brain regions typically involved in epileptogenesis61, we reasoned that seizure susceptibility would be a good proxy to assess the physiological relevance of brain-expressed GINIP. Although we did not observe spontaneous seizures in GINIP 1a/1a mice compared to GINIP +/+, there were marked differences in their susceptibility to chemically-induced seizures (Fig. 4G, H). To induce seizures, we treated mice with bicuculline, a GABAA receptor antagonist, to directly shift the balance of neurotransmission toward excitation62. We found that GINIP 1a/1a mice had higher sensitivity to bicuculline-induced seizures than GINIP +/+ littermates, displaying more severe seizures and more episodes at lower concentrations of bicuculline (Fig. 4H). Similar differences were observed when comparing groups with mixed males and females (Fig. 4H) or when analyzing males and females separately (Fig. S4A). These findings show that GINIP is required to prevent imbalances of neurotransmission that underlie increased seizure susceptibility.
GINIP biases GPCR-G protein signaling in neurons
We began exploring the role of GINIP in regulating neuromodulatory signaling via GPCRs in primary neurons. We cultured cortical neurons (with supportive glia) from neonatal mice and found that GINIP expression was as high as in brain tissue after 12–20 days in vitro (DIV) (Fig. 5A). GINIP was undetectable in cultured glia prepared in parallel from the same source (Fig. 5B), suggesting that GINIP is predominantly expressed in neurons. The latter was confirmed by co-staining GINIP with markers, finding that that almost all NeuN+ cells (neurons) expressed GINIP, whereas no GFAP+ cell (astrocytes) expressed GINIP (Fig. 5C). To investigate the role of GINIP in GPCR signaling in neurons, we generated mice bearing a floxed GINIP allele (GINIP flox) (Fig. S5A–C). Cortical neurons cultured from GINIP flox/flox mice were transduced with an adeno-associated virus (AAV) expressing Cre recombinase (AAV-Cre) to generate GINIP null cells that were compared to untransduced control cells (Fig. S5D–E). To monitor GPCR responses, we expressed a BRET biosensor named BERKY that is suitable for the detection of endogenous free Gβγ in response to endogenous neurotransmitter receptors in neurons63. We found that cells lacking GINIP had reduced Gβγ responses upon stimulation of two different Gi-coupled GPCRs, GABABR or α2-AR (Fig. 5D–E). In contrast, Gβγ responses elicited by stimulation of β-adrenergic receptors, which couple to Gs, were unaffected by the loss of GINIP (Fig. 5F). These results demonstrate that GINIP endogenously expressed in neurons promotes Gβγ signaling in response to GPCR stimulation, much like in cell lines expressing exogenous GINIP (Fig. 3). Next, we investigated if GINIP would also modulate cAMP changes in response to stimulation of a Gi-activating GPCR, like previously found in cell lines (Fig. 2). For this, we expressed a BRET-based biosensor for cAMP56 in neurons of GINIP flox/flox mice and compared responses with and without Cre-mediated ablation of GINIP as above. We found that the inhibition of forskolin-stimulated cAMP after stimulation of GABABR was enhanced upon loss of GINIP (Fig. 5G). These results suggest that, much like in cell lines, GINIP dampens Gαi-GTP-mediated inhibition of cAMP production by adenylyl cyclase. Collectively, these observations indicate that GINIP biases inhibitory GPCR responses in neurons by favoring Gβγ over Gαi signaling.
Figure 5. GINIP regulates Gi-coupled GPCR signaling in neurons.
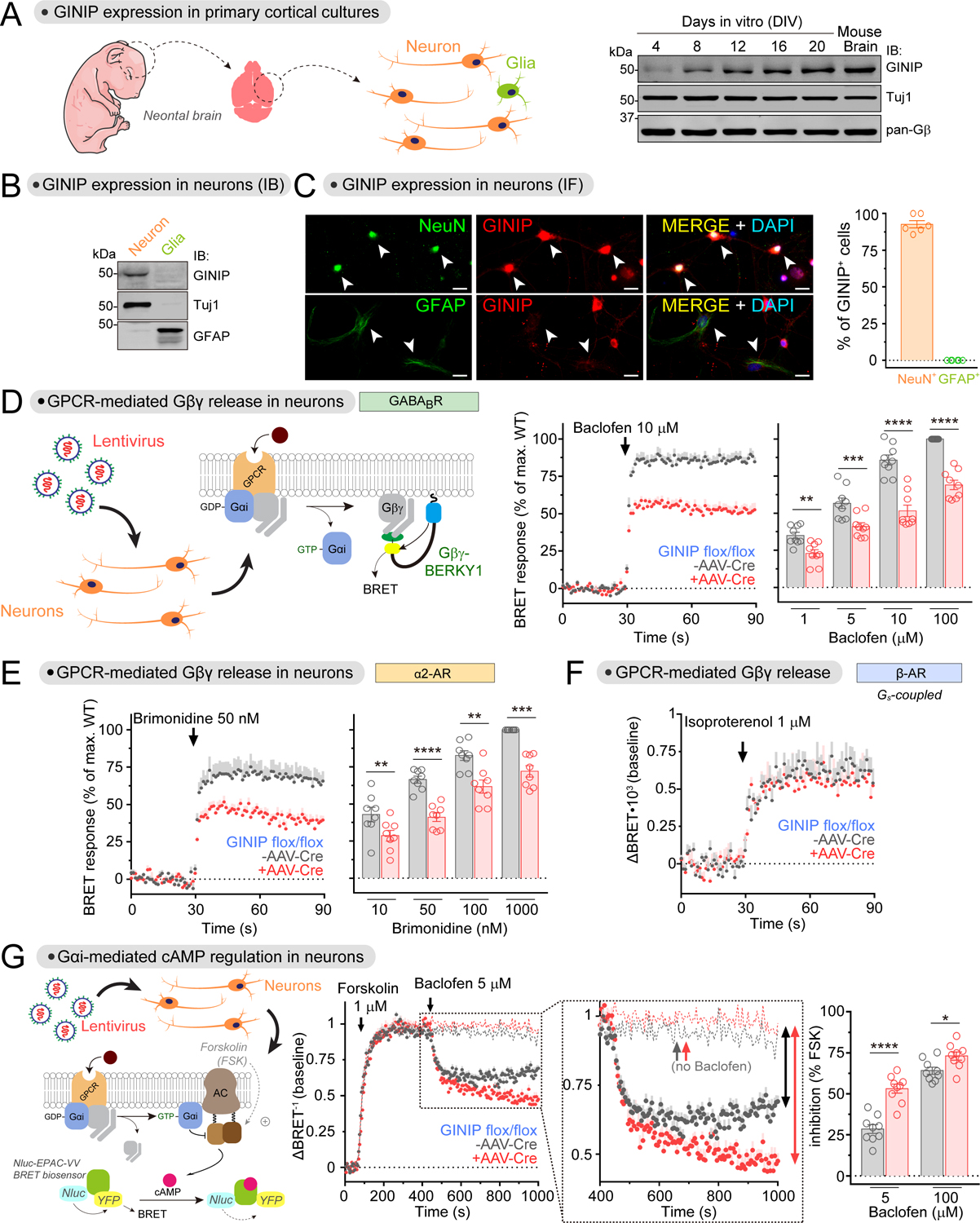
(A) GINIP is expressed in cortical neuron cultures. Neuron cultures established from the cortices of neonatal mouse brains were analyzed by immunoblotting (IB) on the indicated days in vitro (DIV). n=2.
(B) GINIP is not expressed in cortical glial cultures. Neuron and glial cultures established from the cortices of neonatal mouse brains were analyzed by immunoblotting (IB) on DIV12. n=3.
(C) GINIP is expressed in cortical neurons but not in glia. GINIP was co-stained with NeuN or GFAP in DIV12 cortical cultures. Arrows indicate NeuN+ or GFAP+ cells. The proportion of GINIP+ in NeuN+ or GFAP+ cells was quantified from three independent cultures (two image fields per experiment). Mean±S.E.M. Scale bar = 20 μm.
(D, E) Loss of GINIP decreases Gβγ responses triggered by GABABR (D) and α2-AR (E). BRET was measured in DIV12–14 cortical neurons from GINIP flox/flox mice that had been transduced (red) or not (black) with AAV-Cre. BRET responses were normalized to the maximum response of WT in each experiment. Mean±S.E.M., n=8–9. **p<0.01, ***p<0.001, ****p<0.0001, paired t-test.
(F) Loss of GINIP does not affect Gβγ responses triggered by β-AR. BRET was measured in DIV12–14 cortical neurons as in D and E. Mean±S.E.M. n=5
(G) Loss of GINIP enhances the inhibition of adenylyl cyclase upon stimulation of GABABR. cAMP was measured by BRET in DIV16–18 cortical neurons from GINIP flox/flox mice that had been transduced (red) or not (black) with AAV-Cre. Dotted lines in the kinetic traces indicate controls not stimulated with baclofen. Mean±S.E.M., n=9. *p<0.05, ****p<0.0001, paired t-test.
See also Figure S5
GINIP localizes to inhibitory but not excitatory synapses
Close inspection of cortical neurons in culture immunostained for GINIP revealed that the protein localized in puncta that partially overlapped with the synaptic marker synaptophysin (SYP) (Fig. 6A). The immunostaining was specific for GINIP because no signal was detected in neurons from GINIP 1a/1a mice (Fig. 6A). Further experiments co-staining with other markers revealed that GINIP is expressed in both inhibitory and excitatory neurons, identified by the expression of the GABAergic marker GAD65 or the glutamatergic marker vGlut1, respectively (Fig. 6B). However, GINIP colocalizes with markers of inhibitory but not excitatory synapses (Fig. 6C). For example, ~60–80% of GINIP+ puncta in dendrites colocalized with GAD65, a marker of inhibitory presynaptic terminals, and ~30% colocalized with gephyrin, a marker of inhibitory postsynaptic structures (Fig. 6C). In contrast, GINIP did not colocalize with vGlut1 or PSD95, which mark excitatory presynaptic and postsynaptic structures, respectively (Fig. 6C). Interestingly, the co-localization of SYP was bimodal— i.e., in dendrites of some neurons ~70% of GINIP+ puncta were positive for SYP, whereas co-localization was absent in dendrites of other neurons (Fig. 6C). In light of results obtained with other markers and the fact that SYP marks both inhibitory and excitatory presynaptic terminals, the most likely explanation for this is that GINIP co-localizes with SYP at inhibitory presynaptic terminals but not at excitatory presynapses. In summary, our results indicate that, even though GINIP is expressed in both excitatory and inhibitory cortical neurons, its subcellular distribution is restricted to inhibitory synapses, probably at both presynaptic and postsynaptic structures.
Figure 6. GINIP localizes to inhibitory but not excitatory synapses.
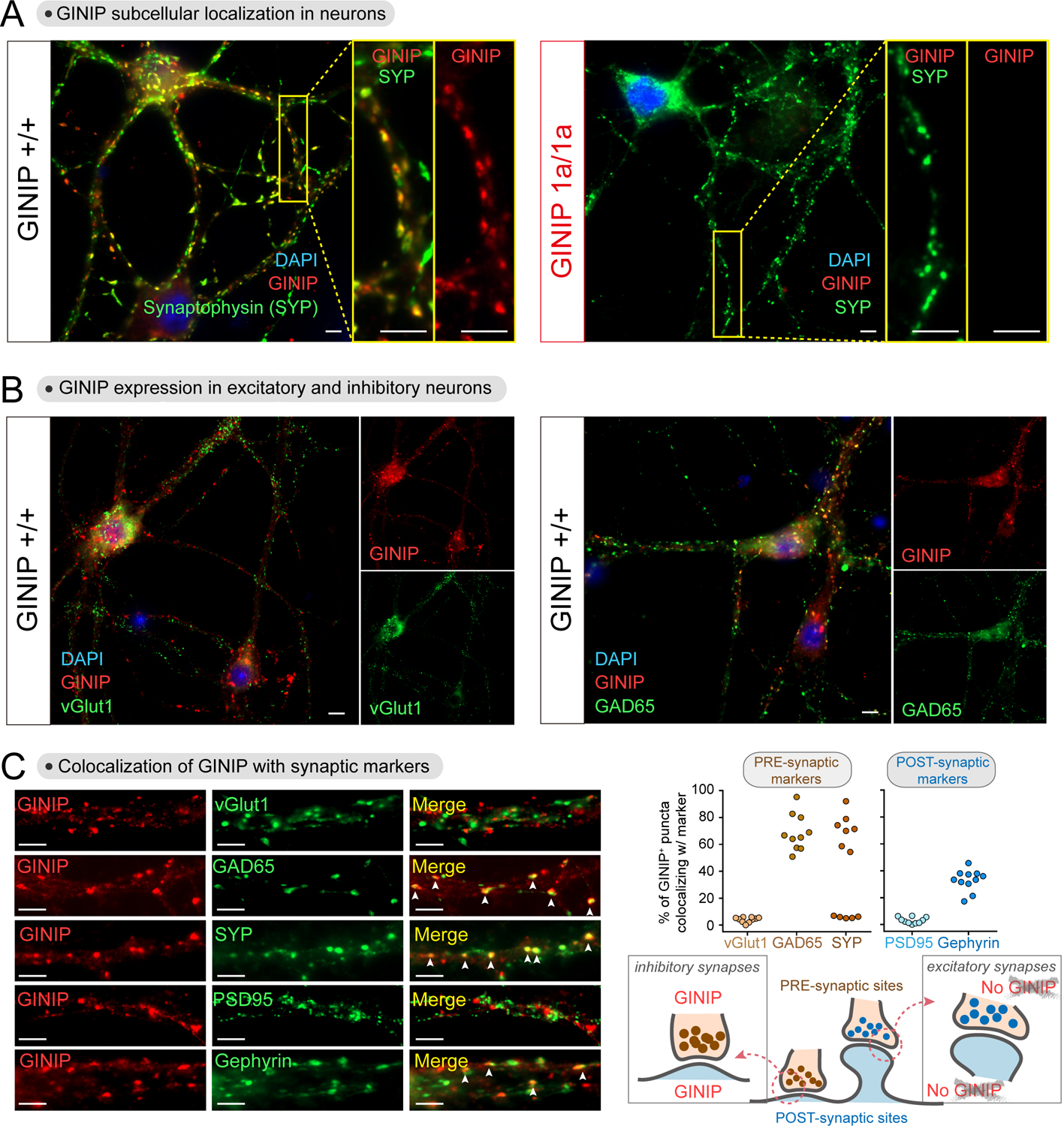
(A) GINIP protein localizes to dendritic puncta in cortical neurons. DIV21 cortical neurons from GINIP +/+ or 1a/1a mice were co-stained for GINIP and synaptophysin (SYP) before fluorescence imaging. Yellow boxes indicate areas enlarged on the right side of the main images.
(B) GINIP is expressed in both excitatory (vGlut1+) and inhibitory (GAD65+) neurons. DIV21 cortical neurons from GINIP +/+ mice were co-stained for GINIP and the indicated markers.
(C) GINIP co-localizes with markers of inhibitory but not excitatory synapses. Left, cortical neurons co-stained for GINIP and the indicated markers of different synaptic compartments were imaged by confocal fluorescence microscopy. Right, quantification of the colocalization of GINIP with synaptic markers. Scatter plot values are the percentage of GINIP positive puncta that were positive for each synaptic marker of one field (3–4 fields from 3 independent experiments).
All scale bars are 10 μm. All results are representative of n ≥ 3 experiments. See also Figure S6
GINIP affects GPCR-mediated neuromodulation in both excitatory and inhibitory neurons
Since GINIP is expressed in both excitatory and inhibitory cortical neurons in culture (Fig. 6), next we asked if GINIP regulated GPCR neuromodulation in excitatory or inhibitory neurons, or in both. For this, we carried out patch-clamp electrophysiological recordings in brain slices. We first confirmed that GINIP is expressed in both excitatory and inhibitory neurons of the cortex (Fig. 7A) by using mRNA in situ hybridization with brain slices. Next, we evaluated the impact of GINIP loss on neuromodulatory influence of GABABR using patch-clamp recordings of excitatory cortical pyramidal neurons in brain slices. Activation of postsynaptic GABABR in these neurons reduces excitability by activating potassium current thought to be largely mediated by G protein-gated inwardly rectifying K+ (GIRK) channels, a canonical Gβγ effector26,64. Indeed, application of GABABR agonist baclofen produced prominent K+ current (Fig. 7B). The amplitude of this current was reduced in GINIP 1a/1a compared to GINIP +/+ suggesting loss in the efficiency of postsynaptic GIRK channel activation by Gβγ in the absence of GINIP (Fig. 7B).
Figure 7. Loss of GINIP from either excitatory or inhibitory neurons affects inhibitory neuromodulation and increases seizure susceptibility.
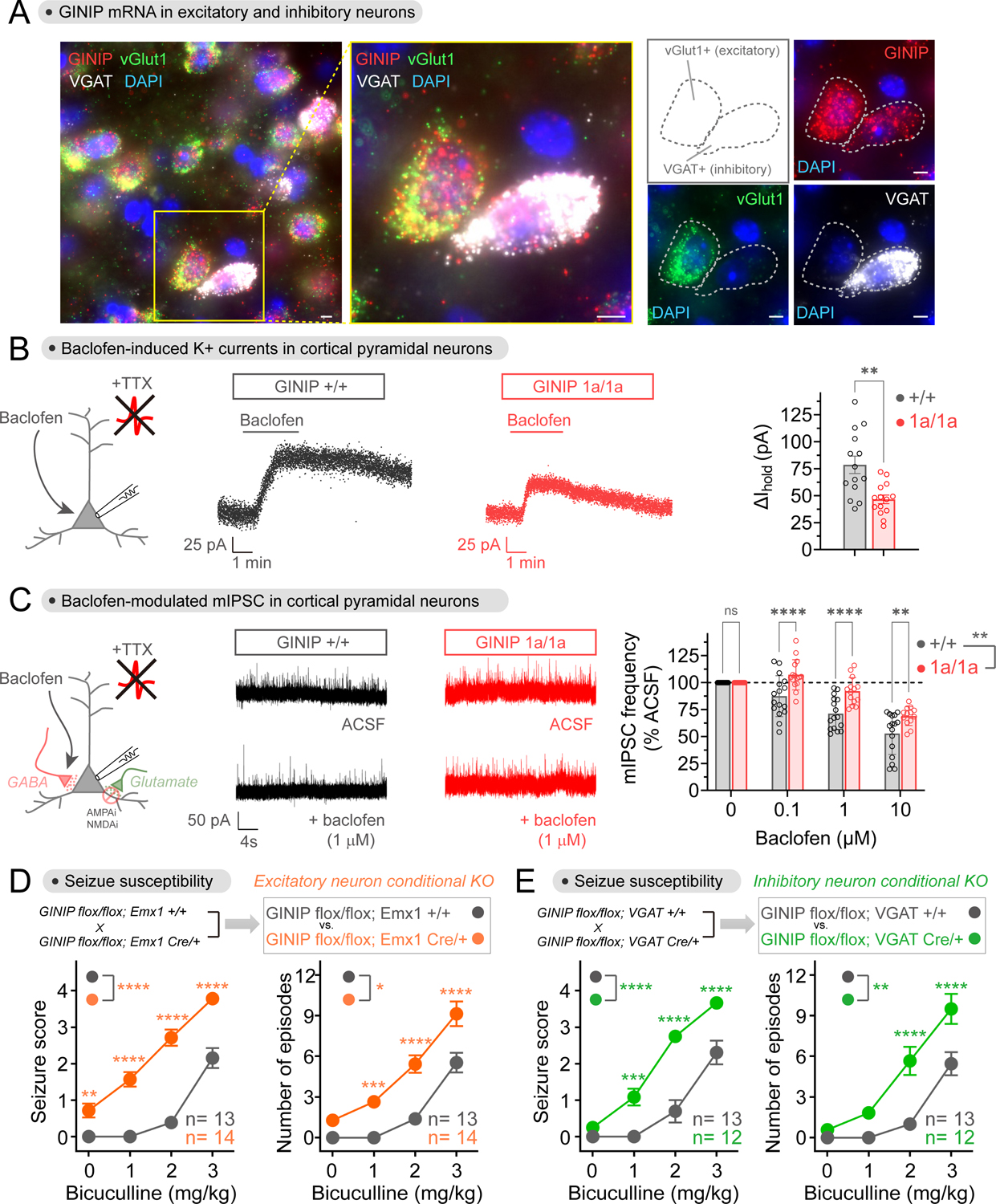
(A) GINIP mRNA is expressed in excitatory and inhibitory cortical neurons. GINIP, vGlut1 and VGAT mRNAs were simultaneously detected in mouse cortical slices. All scale bars are 10 μm. n ≥ 3 experiments.
(B) Loss of GINIP reduces GIRK currents in response to baclofen. Representative traces of baclofen-induced holding current change in cortical pyramidal neurons from GINIP +/+ (black) or GINIP 1a/1a (red) slices are shown in the left and middle, whereas quantification of peak amplitude across multiple cells is shown on the right. Mean±S.E.M. (n=14 per group), **p<0.01, unpaired t-test. Baclofen = 50 μM.
(C) Loss of GINIP dampens baclofen-induced reduction of mIPSC frequency. Representative traces of mIPSC recorded from GINIP +/+ (black) and GINIP 1a/1a (red) cortical pyramidal neuron before and after baclofen are shown in the left and middle, whereas quantification of mIPSC frequency for different concentrations of baclofen relative to controls is shown on the right. Mean±S.E.M. n=13–16 per group. **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA for GINIP genotype x baclofen concentration, with multiple comparisons at each concentration using Fisher’s LSD test.
(D, E) Loss of GINIP from Emx1+ (excitatory) neurons (B) or from VGAT+ (inhibitory) neurons (C) results in increased seizure susceptibility. 12–14 mice (male and female) per genotype. See Fig. S4 for results stratified by sex. Mean±S.E.M. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA for genotype x concentration of bicuculline, with multiple comparisons at each concentration using Fisher’s LSD test.
To evaluate the effect of GINIP in inhibitory neurons, we measured inhibition of GABA release mediated by activation of presynaptic GABABR autoreceptors. Here, Gβγ released upon activation of GABABR inhibits neurotransmission by inhibiting Ca2+ ion channels and by blocking SNARE-mediated vesicular fusion5,26. For this, we recorded miniature inhibitory postsynaptic currents (mIPSC) in pyramidal cortical neurons, reflecting neurotransmitter release from inhibitory, GABAergic neurons. In parallel, we also recorded spontaneous excitatory postsynaptic currents (sEPSC), which are also inhibited by presynaptic GABABR, but reflect neurotransmitter release from excitatory, glutamatergic neurons. We reasoned that the latter should not be affected by the loss of GINIP, since we could not detect GINIP in excitatory synapses (Fig. 6). As expected, application of baclofen to GINIP +/+ slices led to a reduction of the frequency of mIPSC (Fig. 7C) and of sEPSC (Fig. S6). While the effect of baclofen on sEPSC frequency was unaltered in GINIP 1a/1a compared to GINIP +/+ (Fig. S6), baclofen-induced reduction of mIPSC frequency in GINIP 1a/1a was lower than in GINIP +/+ (Fig. 7C), indicating a specific role of GINIP in inhibitory but not excitatory neurotransmission. In the absence of baclofen treatment, we found no differences by genotype in baseline mIPSC frequencies (4.72 Hz versus 4.75 Hz in GINIP 1a/1a and GINIP +/+, respectively, n=13–15 per group, p>0.05 unpaired t-test) or baseline sEPSC frequencies (2.22 Hz versus 2.20 Hz in GINIP 1a/1a and GINIP +/+, respectively, n=8–1 per group, p>0.05 unpaired t-test). Taken together, these results suggest that GINIP facilitates GPCR-mediated modulation of inhibitory, but not excitatory, neurotransmission in both excitatory and inhibitory neurons.
Loss of GINIP from either excitatory or inhibitory neurons increases seizure susceptibility
Based on the observed effects of GINIP on regulating neuromodulatory responses in both excitatory and inhibitory neurons, next we investigated if the phenotype of higher seizure susceptibility observed in global GINIP knock-out mice (Fig. 4G, H) was due to the function of GINIP in either excitatory or inhibitory neurons, or in both. For this, we crossed GINIP flox mice with different Cre driver lines to specifically ablate GINIP expression in excitatory or inhibitory neurons across the brain regions where GINIP is expressed. For the former, crosses were made with Emx1-Cre mice65, whereas VGAT-Cre line66 was used for the latter. We confirmed specific loss of GINIP from excitatory (vGlut1+) or inhibitory neurons (VGAT+) in GINIP flox/flox mice bearing the Emx1-Cre or VGAT-Cre allele, respectively, across all brain regions investigated (Fig. S7). We found that loss of GINIP from either excitatory or inhibitory neurons had a similar effect on bicuculine-induced seizure susceptibility, and that this effect was also similar to that observed with global GINIP knock-out (Fig. 4G, H)— i.e., it caused more severe seizures and more episodes at lower concentrations of bicuculline (Fig. 7E–F). As for the global GINIP knock-out, the increase in seizure susceptibility was observed when comparing groups with mixed males and females (Fig. 7E–F) or when analyzing males and females separately (Fig. S4B–C). These findings demonstrate that GINIP is required both in excitatory and inhibitory neurons to prevent imbalances of neurotransmission that underlie increased seizure susceptibility.
DISCUSSION
The main advance provided by this work is the identification of a unique mechanism of G-protein regulation that sets the tone of neurotransmission by fine tuning inhibitory neuromodulation triggered by GPCRs. This mechanism is mediated by the Gαi-binding protein GINIP, but fundamentally differs from other regulators that bind Gα subunits in that GINIP does not affect directly the enzymatic activity of the G-protein. Instead, GINIP functions as a silent allosteric modulator by binding tightly to a region of active Gαi comprising the α3 helix and the SwII to compete simultaneously with the binding of effectors and of RGS GAPs. This leads to a GPCR signaling outcome that is apparently paradoxical because some G-protein signals are enhanced while others are inhibited— i.e., Gi responses are biased to enhance the effects of one of its active signaling species (free Gβγ) in detriment of the effects of the other active signaling species (Gαi-GTP). This is explained by the prolonged lifetime of Gαi in its GTP-bound form upon RGS GAP displacement by GINIP, which is accompanied by the corresponding dissociation of Gβγ. However, Gαi-GTP in this scenario remains “sequestered” by GINIP in a complex that is incompetent for engagement with its effector, adenylyl cyclase. We show that this mechanism has a broad effect on GPCR signaling that is not limited to the previously described regulation GABABR signaling by GINIP in the peripheral nervous system36, but that potentially encompasses the regulation of any Gi-coupled GPCR co-expressed with GINIP across different brain regions. In this regard, based on its broad expression across different major types of neurons, like GABAergic and glutamatergic neurons, GINIP is poised to exert circuit level effects on neurotransmission in brain by controlling GPCR-mediated neuromodulation, an idea supported by our signaling, electrophysiological, and behavioral results.
The mechanism by which GINIP regulates GPCR signaling is different from those described for other regulators that bind to Gα subunits, like GDIs or GAPs. A first distinctive feature is that GINIP does not affect directly the ability of Gα subunits to bind and/ or hydrolyze nucleotides. Another distinctive feature is that GINIP has effects of opposite sign on signaling branches triggered by the same receptor depending on whether they are mediated by Gα or Gβγ subunits, whereas other regulators do not discriminate GPCR responses this way. For example, a previous report showed that the GDI LGN could enhance basal GIRK channel activation in neurons via Gβγ subunits released upon its binding to inactive, GDP-bound Gα in the absence of GPCR stimulation, but that GIRK responses were dampened by LGN upon GPCR stimulation67. Together, these observations suggest that GDIs like LGN inhibit all G-protein responses triggered by a GPCR by disrupting G-protein heterotrimers, which are the obligatory substrate for GPCRs. RGS GAPs also work as negative regulators of GPCR signaling regardless of whether the readout depends on Gα-GTP or Gβγ68.
Much attention has been paid in the recent years to the concept of ligand-induced bias in GPCR signaling69,70. The underlying idea is that the strength of different responses triggered by a GPCR can be modulated to different extents depending on the properties of the ligand that activates the receptor. One general hypothesis is that different ligands can facilitate different GPCR conformations, which in turn are better suited to engage preferentially some transducers over others. Most work has focused on differences between engagement of G-proteins vs β-arrestins, although there could also be ligand-induced bias for different types of G-proteins. Here, we describe a different type of signaling bias. Instead of regulating the transducer that engages the GPCR, GINIP determines functional outcome by biasing signaling mediated by different G-protein subunits. This is not necessarily determined by the nature of the ligand. Instead, the work presented here is a case of “system bias”— i.e., the relative sensitivity of pathways activated by the receptor that are hard-wired by the physiology of the system70,71. Although it is becoming recognized that system bias is a general impediment for the translation of in vitro pharmacology into useful therapeutics in vivo71, little is known about the mechanisms underlying this class of bias beyond attributing it to differences in receptor and transducer stoichiometry. Recent work has put forth the idea that receptor proximal events, like regulation of G-proteins by RGS GAPs, could also contribute to system bias by shaping the relative strength of GPCR signals mediated by different types of G-proteins in neurons or cardiac cells68,72. Our work presented here provides a detailed molecular understanding of a mechanism of system bias that operates specifically in neurons through the regulation of receptor proximal events to determine the sensitivity of different pathways under the control of not only the same receptor, but also the same G-protein type. A question that remains open is how the action of GINIP could be modulated to tune this bias. One obvious mechanism is through control of GINIP expression in specific neuron populations of different brain regions, which remains to be characterized in detail. However, even when expressed in a particular type of neuron, GINIP localization to different subcellular compartments (like synapses) will also determine the specific contexts in which GPCR signaling bias occurs.
The increased susceptibility to bicuculline-induced seizures observed upon loss of GINIP suggests an overall decrease in neuroinhibition, which is in agreement with other results from electrophysiological and G-protein signaling experiments in neurons. Our behavioral results are consistent with and expand on previous observations in male rats deficient for GINIP expression, which displayed increased spontaneous seizures60 or seizures induced using an experimental paradigm different from ours73. We not only observed similar effects in mice of both sexes, but also were able to leverage a conditional GINIP floxed allele to learn that loss of GINIP from either excitatory or inhibitory neurons increases similarly seizure susceptibility.
Gαo is the most abundant G-protein in brain, and GPCRs that activate Gi almost invariably also activate Go74. Yet, GINIP regulates responses by the former but not the latter. Why have a mechanism in place to regulate two closely related G-protein types that tend to be co-activated? While both Gαo and Gαi isoforms give rise to Gβγ subunits that might be functionally similar upon stimulation of same GPCRs, the key functional difference of Gαo with respect to Gαi is that it does not regulate adenylyl cyclase directly. It is tempting to speculate that GINIP comes into play to regulate GPCR responses by the G-protein subtype for which coordination between Gβγ and Gα-GTP is relevant— i.e., Gi. In this regard, it is also important to note that while GPCRs that activate Gi also co-activate Go, and that both G-proteins contribute to the formation of Gβγ, regulation of Gi by GINIP affects the overall levels of free Gβγ or Gβγ-dependent responses like post-synaptic GIRK channel regulation or pre-synaptic regulation of neurotransmitter release. This suggests that despite the abundance of Gαo in neurons, a significant fraction of Gβγ-dependent signaling is mediated through Gαi isoforms in brain. However, there is evidence that manipulation of the levels or activity of Gαo also affects inhibitory neurotransmission75,76, raising the question of how GINIP-dependent and GINIP-independent mechanism contribute to neuroinhibition. We speculate that GINIP might be spatially segregated within specific inhibitory synapses or molecular complexes therein where Gαi-type G proteins are the functional drivers of neuroinhibition.
In summary, this work provides detailed mechanistic insights into how GPCR-mediated neuromodulation is fine-tuned by coordinating the level of responses triggered by different G-protein subunits. By understanding the molecular basis of how receptor-mediated signaling is hard-wired in the native context of neurons, we might be able to envision better ways to pharmacologically target neurological and neuropsychiatric disorders.
LIMITATIONS OF THE STUDY
Our results indicate that GINIP expression in both inhibitory and excitatory neurons is important to prevent imbalances of neurotransmission that result in increased seizures. However, the mechanisms leading to a similar systems-level phenotype of decreased neuroinhibition are unknown. One possibility is that non-cell autonomous, circuit-level mechanisms lead to the similar overall phenotype. Similarly, another limitation in understanding how the function of GINIP operates at the systems level is that we do not know how cell-autonomous Gβγ vs Gαi-GTP responses regulated by GINIP are integrated differently depending on the type of neuron. Since both Gβγ and Gαi-GTP signaling tend to be neuroinhibitory but are regulated in opposite directions by GINIP, the net effects on regulating neurotransmission would depend on how they are integrated in a particular cell type.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to Lead Contact Mikel Garcia-Marcos (mgm1@bu.edu).
Materials availability
Plasmids generated in this study are available upon request or have been deposited to Addgene as indicated in the the Key resources table. There are restrictions to the availability of the C57BL/6N-Atm1Brd/aPhf24tm1a(EUCOMM)Hmgu/BcmMmucd mouse line because it was obtained from the MMRRC under an MTA that imposes certain conditions. The same restrictions apply to mice bearoing the GINIP flox allele derived from C57BL/6N-Atm1Brd/a Phf24tm1a(EUCOMM)Hmgu/BcmMmucd.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Gαi3 (C-10) | Santa Cruz Biotechnology | #sc-262 |
| FLAG M2 | Millipore Sigma | F1804 |
| Glu-Glu-epitope Tag | Millipore Sigma | AB3788 |
| Gαo (A2) | Santa Cruz Biotechnology | #sc-13532 |
| Gαs/olf (C-18) | Santa Cruz Biotechnology | #sc-383 |
| HA Tag | Roche | #11583816001 |
| Myc-Tag (9B11) Antibody | Cell Signaling Technology | #2276 |
| polyHistidine | Millipore Sigma | H1029 |
| α-Tubulin (DM1A) | Millipore Sigma | CP06 |
| β-Actin | LI-COR | #926-42212 |
| GAIP | 77 | N/A |
| Gβ (H-1) | Santa Cruz Biotechnology | #sc-166123 |
| GINIP (goat) | Santa Cruz Biotechnology | #sc-247284 |
| Gαq (E-17) | Santa Cruz Biotechnology | #sc-393 |
| β3 Tubulin (Tuj1) (2G10) | Santa Cruz Biotechnology | #sc-80005 |
| GFAP clone GA5 | Millipore Sigma | MAB360 |
| GINIP (rabbit) | Aviva | ARP70657_P050 |
| SYP/Synaptophysin (H-8) | Santa Cruz Biotechnology | #sc-55507 |
| vGlut1 (A-8) | Santa Cruz Biotechnology | #sc-377425 |
| GAD65 | Developmental Studies Hybridoma Bank | AB_528264 |
| PSD95 (7E3-1B8) | Abcam | #ab13552 |
| Gephyrin | Synaptic Systems | #147011 |
| Goat anti-rabbit Alexa Fluor 680 | Invitrogen | #A21077 |
| Goat anti-mouse Alexa Fluor 680 | Invitrogen | #A21058 |
| Goat anti-mouse IRDye 800 | LI-COR | #926-32210 |
| Goat anti-rabbit DyLight 800 | Thermo | #35571 |
| Donkey anti-goat IRDye 680RD | LI-COR | #926-68074 |
| Goat anti-mouse Alexa Fluor 488 | Invitrogen | #A11017 |
| Goat anti-rabbit Alexa Fluor 488 | Invitrogen | #A11070 |
| Goat anti-mouse Alexa Fluor 594 | Invitrogen | #A11020 |
| Goat anti-rabbit Alexa Fluor 594 | Invitrogen | #A11072 |
| Bacterial and Virus Strains | ||
| NEB 5-alpha competent E. coli | NEB | Cat# C2987I |
| BL21(DE3) Chemically Competent E. coli | Invitrogen | Cat# C600003 |
| AAV-hSyn-Cre | Addgene | Addgene Cat# 105553-AAV1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Forskolin | Tocris | Cat# 1099 |
| GABA | Tocris | Cat# 0344 |
| Brimonidine | Ark Pharm | Cat# AK-35795 |
| Dopamine HCl | Alfa Aesar | Cat# A11136 |
| CGP 54626 hydrochloride | Tocris | Cat# 1088 |
| Yohimbine | Alfa Aesar | Cat# J60185 |
| (+) - Bicuculine | TCI | Cat# B1890 |
| (R)-Baclofen | Tocris | Cat# 0796 |
| DL-isoproterenol hydrochloride | Alfa Aesar | Cat# J61788 |
| Tetrodotoxin citrate | Cayman | Cat# 18660-81-6 |
| CNQX disodium salt | Tocris | Cat# 1045 |
| DL-AP5 Na salt | Tocris | Cat# 3693 |
| Polyethylenimine (PEI) | Polysciences | Cat# 23966-1 |
| Q5 DNA polymerase | New England Biolabs | Cat# M0491L |
| SigmaFAST protease inhibitor cocktail | Sigma | Cat# S8830 |
| Critical Commercial Assays | ||
| NanoGlo Luciferase Assay System | Promega | Cat# N1120 |
| LANCE cAMP kit | PerkinElmer | Cat# AD0262 |
| RNAscope 2.0 Assay kit | Bio-techne | Cat# 320850 |
| Deposited Data | ||
| Original images from blotting and immunofluorescence staining | This paper | DOI: 10.17632/3ng65trfc7.1 |
| Experimental Models: Cell Lines | ||
| Human: 293T | ATCC | Cat# CRL-3216 |
| Human: Lenti-X 293T | Takara Bio | Cat# 632180 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River | Strain code: 027 |
| Mouse: C57BL/6N-Atm1Brd/a Phf24tm1a(EUCOMM)Hmgu/BcmMmucd | Mutant Mouse Resource & Research Centers (MMRRC) | #037754-UCD |
| Mouse: B6N.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/J | The Jackson Laboratory | #016226 |
| Mouse: B6.129S2-Emx1tm1(cre)Krj/J | The Jackson Laboratory | #005628 |
| Mouse: B6J.129S6(FVB)-Slc32a1tm2(cre)Lowl/MwarJ | The Jackson Laboratory | #028862 |
| Recombinant DNA | ||
| pLIC-His-GINIP | This paper | N/A |
| pLIC-His-RGS4 | This paper | N/A |
| pLIC-GST-GINIP | This paper | N/A |
| pET28b-Gαi3 (rat) | 78 | N/A |
| pGEX-4T-1-Gαi3 (rat) | 78 | N/A |
| pLIC-Gαi1(int.6xHis) | 35 | N/A |
| pET28b-Gαi2 | 79 | N/A |
| pET28b-Gαo | 79 | N/A |
| pET24a-Gαi3 (human) | 80 | N/A |
| pbb131-NMT | 81 | |
| pET28b-AC5 C1 | This paper | N/A |
| pET15b-hAC2 C2 | 82 | N/A |
| pGEX-GAIP | 83 | N/A |
| pGEX-KG-KB1753 | 83 | N/A |
| pHis6-Gαs | 84 | N/A |
| pGEX-4T-1-Gαi3/o chimera 1 (aka chimera 1 in this work) | 40 | N/A |
| pGEX-4T-1-Gai3/o chimera 3 (aka chimera 2 in this work) | 40 | N/A |
| pGEX-4T-1-Gai3/o chimera 4 (aka chimera 3 in this work) | 40 | N/A |
| pET28b-DAPLE CT (1650–2028) | 42 | N/A |
| p3xFLAG-CMV-14-GINIP | This paper | N/A |
| pcDNA3.1(+)-Gαz (EE-tagged) | cDNA Resource Center | GNA0Z0EI00 |
| pcDNA3-Gαq-HA | 85 | N/A |
| pcDNA3.1-Gα12-MYC | 86 | N/A |
| pcDNA3-YFP-hAC5 | 29 | N/A |
| pcDNA3.1(+)-FLAG-D2DR | 87 | N/A |
| pcDNA3-α2A-AR | 88 | N/A |
| pcDNA3.1(+)-GABABR1a | 89 | N/A |
| pcDNA3.1(+)-GABABR2 | 89 | N/A |
| pcDNA3.1-hRGS7 | 90 | N/A |
| pcDNA3.1-Gβ5 | 91 | N/A |
| pcDNA3.1-R7BP | 91 | N/A |
| pcDNA3.1-masGRK3ct-Nluc | 56 | N/A |
| pcDNA3.1-Nluc-EPAC-VV | 56 | N/A |
| pcDNA3.1-Venus(1–155)-Gγ2 (VN-Gγ2) | 55 | N/A |
| pcDNA3.1-Venus(155–239)-Gβ1 (VC-Gβ1) | 55 | N/A |
| pcDNA3.1(−)-3xHA-RGS8 | cDNA Resource Center | RGS080TN00 |
| pcDNA3-GAIP | 92 | N/A |
| psPAX2 | Addgene | Addgene cat# 12260 |
| pMD2.G | Addgene | Addgene cat# 12259 |
| pLenti-hSyn-Gβγ-BERKY1 | 63 | Addgene cat# 158429 |
| pLenti-hSynapsin-Nluc-EPAC-VV | This paper | N/A |
| P3VEA3–1-venus-arrestin-3 | 93 | N/A |
| α2A-AR-Rluc8 | 94 | N/A |
| D2R-Rluc8 | 94 | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Photoshop | Adobe | https://www.adobe.com/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/ |
| pClamp 11 | Molecular Devices | https://www.moleculardevices.com/ |
Data and code availability
All data is provided in the manuscript and original images of results presented in the figures have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the Key Resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
HEK293T (ATCC cat# CRL-3216) and Lenti-X 293T (Takara Bio Cat# 632180) cells were grown at 37°C, 5% CO2 in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2mM L-glutamine.
Mouse strains and breeding
All animal experiments were carried out in agreement with the institutional guidelines provided by the Institutional Animal Care and Use Committee at Boston University (protocol number PROTO 201800460), per applicable laws and regulations. Heterozygous mice bearing the reporter and null allele GINIP 1a (C57BL/6N-Atm1Brd/a Phf24tm1a(EUCOMM)Hmgu/BcmMmucd, Stock# 037754-UCD) were obtained from the Mutant Mouse Resource & Research Centers (MMRRC), and bred by intercrossing to generate cohorts of littermate animals for experiments and to maintain the line. The conditional GINIP flox allele was generated by crossing GINIP 1a/1a homozygous mice with Rosa26 Flpe homozygous mice (B6N.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/J, JAX stock# 016226). The resulting GINIP +/flox; Rosa26 +/Flpe animals were intercrossed to obtain animals bearing the GINIP flox allele but without the Rosa26 Flpe allele. GINIP flox mice were crossed with an Emx1 Cre driver line (B6.129S2-Emx1tm1(cre)Krj/J, JAX stock# 005628) or a VGAT Cre driver line (B6J.129S6(FVB)-Slc32a1tm2(cre)Lowl/MwarJ, JAX stock# 028862) to achieve specific GINIP knock-out (GINIP Δ null allele) in excitatory neurons of the telencephalon65 or in inhibitory neurons66, respectively. To obtain cohorts of littermates for experiments assessing the conditional ablation of GINIP in specific neuron populations, animals with the genotype GINIP flox/flox; Cre/+ were generated first and then crossed with GINIP flox/flox mice. Animal genotyping was carried out by PCR of genomic DNA extracted from tail clipping using a three-primer system. To distinguish between wild-type and 1a alleles, a forward primer targeting the intronic region between exons 5 and 6 (Fw.Intron5: TTAAAGTTGCACAACCCACTAGAAGC) and a forward primer targeting the gene trap cassette (Fw.1aTrap: GGGATCTCATGCTGGAGTTCTTCG) were used with a common reverse primer targeting the intronic region between exons 5 and 6 (Rv.Intron5: GAGCTGAGTGACTCTAGGGATGAACC) resulting in a 308 bp band for the wild-type allele and a 584 bp band for the 1a allele. The Fw.Intron5 and Rv.Intron5 primer set also allows for detection of the GINIP flox allele as 513 bp amplicon. Emx1 Cre95 and VGAT Cre96 lines were genotyped as previously described. To detect GINIP Δ null allele obtained after Cre-mediated recombination of the GINIP flox allele, the Fw.Intron5 primer described above was used with a reverse primer targeting downstream of exon 10 (Rv.5UTR: CCAGGCAGAAATCCACAAACTAGG) resulting in a 531 bp band. Both male and female mice were assessed and analyzed together or stratified by sex as indicated in the figures. The number of animals is indicated in the figure legends.
Mouse primary cortical cultures
Cortical neuron cultures were established from neonatal mouse brains (wild-type C57BL/6, Charles River, strain code 027, or the genotype(s) indicated for each experiment) as previously described97 with modifications. Newborn mouse pups (P0) were euthanized by decapitation, and brains rapidly placed in cold HBSS (Corning, 21–022-CV) after removal from the skull. The cerebrum was detached from other brain regions under a stereomicroscope by removal of the olfactory bulb and cerebellum, and the cortex dissected out with forceps. Tissue was minced into approximately 1–2 mm pieces using a sterile razor blade, and digested with 0.05% Trypsin in HBSS for 10 min at 37°C. Trypsinized tissue was washed three times with HBSS by cycles of gravity sedimentation of tissue and aspiration of buffer. Washed tissue was resuspended in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine (complete DMEM) before passing through a sterile 40 μm cell strainer (Fisherbrand, 22363547). Cells were counted and seeded on poly-L-lysine coated plates or coverslips. Coating was performed overnight at room temperature with 0.1 mg/mL poly-L-lysine hydrobromide (Sigma, P1955), followed by 3 washes with HBSS and addition of complete DMEM before seeding. Four hours after seeding, half of the medium was replaced by Neurobasal media (GIBCO, 21103049) with B-27 supplement (GIBCO, 17504001) and 1x Glutamax-I (GIBCO, 35050061) (complete neural medium). On day in vitro 3 (DIV3), one half of the media was replaced with complete neural media supplemented with 5 μM AraC to block glial cell proliferation. Beginning DIV5, half of the media was replaced by fresh complete neural medium every other day. For experiments comparing the expression of GINIP in neuron cultures and glial cell cultures by immunoblotting, 500,000 cells were seeded on 60 mm dishes and cultured as described above, except that for the glial cultures AraC was not added at DIV3. At DIV12, neuron or glial cells were washed twice by cold PBS and harvested by scraping followed by centrifugation at 10,000 × g at 4 °C. Cell pellets were lysed by resuspending with lysis buffer(20 mM HEPES, pH 7.2, 125 mM K(CH3COO), 0.4% (v/v) Triton X-100, 1 mM DTT, 10 mM β-glycerophosphate, and 0.5 mM Na3VO4 supplemented with a SigmaFAST protease inhibitor mixture) and cleared by centrifugation at 14,000 × g for 10 minutes at 4°C. Proteins concentration was quantified by Bradford and lysates boiled for 5 min in Laemmli sample buffer before “Protein Electrophoresis and Immunoblotting” (see below).
METHOD DETAILS
Plasmids
Plasmids for the bacterial expression of His-tagged human GINIP (pLIC-His-GINIP) or RGS4 (pLIC-His-RGS4) were generated using a previously described ligation-independent cloning (LIC) system98. Briefly, insert sequences containing LIC compatible flanking regions were amplified by PCR and inserted into the pLIC-His plasmid generously provided by D. Siderovski (UNT Health Science Center). Similarly, an LIC-compatible GINIP amplicon was inserted into the pLIC-GST plasmid kindly provided by J. Sondek (University of North Carolina at Chapel Hill)99 to generate pLIC-GST-GINIP. Plasmids for the bacterial expression of His-Gαi3 (rat), GST-Gαi3, His-Gαi1, His-Gαi2, and His-Gαo have been described previously78,79,100. The plasmid pET24a-Gαi3 used for the bacterial expression of human His-Gαi3 was a kind gift of I. Shimada51. A plasmid encoding rat Gαi1 containing a hexahistidine tag in the b/c loop (Gαi1-6xHis(int), required for producing myrisotoylated Gαi1) was kindly provided by C. Dessauer (UT Southwestern)54. Gαi1-6xHis(int) was subcloned without additional affinity tags between the NdeI and BglII sites of the pLIC-His vector (pLIC-Gαi1-6xHis(int)). The pbb131 plasmid encoding yeast N-myristoyltransferase (NMT)81 was a gift from Maurine Linder (Cornell University). A plasmid encoding the C1 domain (residues 444–751) of canine adenylyl cyclase 5 (AC5) was provided by C. Dessauer (UT Southwestern)54. AC5 C1 (with a C-terminal His tag) was subcloned between the NcoI and XhoI sites of the bacterial expression vector pET28b to generate pET28b-AC5 C1 without adding any other affinity tag. pET15b-hAC2 C2 encoding residues 871–1082 of human adenylyl cyclase 2 (AC2 C2) with an N-terminal His tag was provided by K. Shokat (University of Californa San Francisco)82. Plasmids for bacterial expression of GST-GAIP and GST-KB1753 have been described previously40,83. The plasmid for the expression of a His–tagged short isoform of bovine Gαs (pHis6-Gαs) in bacteria was kindly provided by N. Artemyev (University of Iowa). Plasmids for expression of GST-Gαi3/o chimeras have been described previously40. The plasmid for the expression of His-DAPLE CT, pET28b-DAPLE (1650–2028), was described previously42.
The plasmid for mammalian expression of C-terminally 3xFLAG-tagged GINIP (GINIP-FLAG) was generated for this paper by inserting the human GINIP sequence between EcoRI and BamHI sites of the p3xFLAG-CMV-14 vector and replacing the stop codon by a serine (p3xFLAG-CMV-14-GINIP). Plasmids encoding for C-terminally 3xFLAG-tagged Gαi3, untagged Gαi3, Gαo, and Gαs were described previously40,101–103, whereas the plasmid for the expression of EE-tagged Gαz was purchased from the cDNA Resource Center (GNA0Z0EI00) (Bloomsberg University, PA). The plasmid for mammalian expression of Gαq internally tagged with HA (pcDNA3-Gαq-HA) was kindly provided by P. Wedegaertner (Thomas Jefferson University)85, whereas the plasmid for mammalian expression of Gα12 internally tagged with MYC (pcDNA3.1-Gα12-MYC) was from T. Meigs (UNC Asheville)86. The plasmid for mammalian expression of YFP-tagged human adenylyl cyclase 5 (pcDNA3-YFP-hAC5) was a gift from C. Dessauer (UT Southwestern)29. The plasmid for mammalian expression of the long isoform of the human Dopamine 2 receptor (pcDNA3.1(+)-FLAG-D2DR) was provided by A. Kovoor (University of Rhode Island). The plasmid encoding α2A-AR (pcDNA3-α2A-AR) has been described previously104. pcDNA3.1(+)-GABABR1a and pcDNA3.1(+)-GABABR2 were a gift from Paul Slessinger, Mount Sinai NY. The following mammalian expression plasmids were described by us previously: pcDNA3.1-hRGS7, pcDNA3.1-Gβ5, pcDNA3.1-R7BP, pcDNA3.1-masGRK3ct-Nluc, pcDNA3.1-Nluc-EPAC-VV56. pcDNA3.1-Venus(1–155)-Gγ2 (VN-Gγ2), and pcDNA3.1-Venus(155–239)-Gβ1 (VC-Gβ1) were a gift from N. Lambert (Augusta University, GA)55. pcDNA3.1(−)-3xHA-RGS8 was acquired from the cDNA Resource Center (RGS080TN00) (Bloomsberg University, PA). pcDNA3-GAIP was a gift from M. Farquhar105. A plasmid encoding rat Gαi3 tagged with Nluc in the a/b loop, pcDNA3.1(−)-Gαi3-Nluc(a/b), was generated by inserting EcoRI and XhoI restriction sites between residues 91 and 92 of Gαi3, and then inserting Nluc by Gibson assembly at those sites, which were maintained in the final construct. The resulting construct contains a 5’ linker EF and 3’ linker SS flanking the Nluc sequence. The plasmids encoding venus-arrestin-3 (P3VEA3-1-venus-arrestin-3)93 and C-terminally Rluc8-fused GPCRs α2A-AR (α2A-AR-Rluc8) and D2R (D2R-Rluc8)94 were a gift from N. Lambert (Augusta University, GA).
Lentiviral packaging plasmids psPAX2 and pMD2.G, and the plasmid to produce a lentivirus for the expression of the Gβγ-BERKY1 biosensor in neurons (pLenti-hSyn-Gβγ-BERKY1) have been described previously63. The plasmid to produce lentiviral particles for the expression of the cAMP BRET biosensor Nluc-EPAC-VV in neurons was generated by amplifying the sequence from pcDNA3.1-Nluc-EPAC-VV and inserting it between the AgeI and EcoRI sites of pLenti-hSynapsin-Cre-WPRE (Addgene #86641;106) by Gibson assembly.
Protein expression and purification
His-tagged and GST-tagged proteins were expressed in BL21(DE3) E. coli transformed with the corresponding plasmids by overnight induction at 23 °C with 1 mM isopropyl β-D-1-thio-galactopyranoside (IPTG), with the exception of His-GINIP and GST-GINIP, which were induced for 5 hours at 23 °C. IPTG was added when the OD600 reached ~0.8. Unless otherwise indicated, protein purification was carried out following previously described protocols42,78. Briefly, bacteria pelleted from 1 liter of culture were resuspended at 4 °C in 25 ml of lysis buffer (50 mM NaH2PO4, pH 7.4, 300 mM NaCl, 10 mM imidazole, 1% (v/v) Triton X-100, supplemented with a protease inhibitor mixture of 1 μM leupeptin, 2.5 μM pepstatin, 0.2 μM aprotinin, and 1 mM phenylmethylsulfonyl fluoride). When purifying Gα subunits, this buffer was supplemented with 25 μM GDP and 5 mM MgCl2. After sonication (4 pulses of 30 s separated by 30 s intervals for cooling), the lysate was cleared by centrifugation at 12,000 × g for 30 min at 4 °C. The soluble fraction was used for affinity purification in batch on HisPur Cobalt (Thermo, 89964) or GSH-agarose resins (Thermo, 16100) by incubating lysate and beads with rotation for 2 hours at 4 °C. Resin was washed 3 times with lysis buffer and then eluted with lysis buffer supplemented with 250 mM imidazole or with 50 mM Tris-HCl, pH 8, 100 mM NaCl, 30 mM reduced GSH, respectively. Proteins were buffer exchanged to PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4) by overnight dialysis (10,000 Da cut-off) at 4 °C, except for Gα proteins, which were buffer exchanged to 20 mM Tris-HCl, pH 7.4, 20 mM NaCl, 1 mM MgCl2, 1 mM DTT, 10 μM GDP, 5% (v/v) glycerol using a HiTrap desalting column (Cytiva, 29048684) connected to an AKTA FPLC. All protein samples were aliquoted and stored at −80 °C. Gαi1RM/AS was stored as single use aliquots for experiments to avoid freeze/thaw cycles.
Myristoylated Gαi1 (myr-Gαi1) was purified as described above from BL21(DE3) E. coli bacteria co-expressing the plasmid encoding Gαi1-6xHis(int) with a plasmid encoding N-myristoyl transferase (NMT), except that after the cobalt affinity purification step the eluate was subjected to ion-exchange chromatography in a HiTrapQ HP column (Cytiva, 17115401). AC5 C1 was induced and purified as described above for other His-tagged proteins except that 1 mM β-mercaptoethanol was present in all purification buffers and after the cobalt affinity purification step the eluate was subjected to ion-exchange chromatography in a HiTrapQ HP column. AC2 C2 was expressed by overnight induction at 23 °C with 40 μM IPTG. Protein was purified as described above for other His-tagged proteins except that after the cobalt affinity purification step the eluate was subjected to ion-exchange chromatography in a HiTrapQ HP column. AC5 C1 and AC2 C2 were then buffer exchanged to 20 mM HEPES pH 8.0, 1 mM EDTA, 20 mM NaCl, 2 mM DTT, and 5% (v/v) glycerol in a HiTrap desalting column connected to an AKTA FPLC and aliquoted before storage at −80 °C.
Purification of His-Gαs was carried out using a previously described protocol107 that differs from the one described above for other His-tagged proteins. Briefly, His-Gαs was expressed in BL21(DE3) E. coli transformed with the corresponding plasmid by overnight induction at 23 °C with 0.1 mM IPTG when OD600 reached ~0.5. Bacteria were pelleted and resuspended at 4 °C in lysis buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5 mM MgCl2, 50 μM GDP, and 5 mM β-mercaptoethanol, supplemented with a protease inhibitor mixture of 1 μM leupeptin, 2.5 μM pepstatin, 0.2 μM aprotinin, and 1 mM PMSF). After sonication, the lysate was cleared by centrifugation at 12,000 × g for 30 min at 4 °C. The supernatant was adjusted to 500 mM NaCl and 20 mM imidazole before affinity purification by incubation with nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen, 30210) for 90 min at 4 °C. Resin was washed four times with lysis buffer, and protein was eluted with lysis buffer supplemented with 100 mM imidazole. The eluted fraction was adjusted to 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5 mM MgCl2 using a protein concentrator with a 10-kDa cutoff (Millipore, UFC801024 before loading onto a HiTrap Q HP column connected to an ÄKTA FPLC. Proteins were eluted by applying a 50–500 mM NaCl gradient, and fractions containing His-Gαs were pooled and supplemented with 10 μM GDP and 5 mM β-mercaptoethanol before concentration in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM MgCl2, 5% (w/v) glycerol, 10 μM GDP, and 5 mM β-mercaptoethanol, and storage at −80 °C.
Pulldown assays
The following GST-fused proteins were immobilized on GSH-agarose beads (Thermo, 16100) for 90 min at room temperature in PBS (range of protein amounts used in different experiments is in parenthesis): GST (2–20 μg), GST-GINIP (4–12 μg), GST-Gαi3 (1–8 μg), GST-KB1753 (15 μg), GST-GAIP (3 μg). Beads were washed twice with PBS and resuspended in 300–400 μl of binding buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.4% (v/v) Nonidet P-40, 5 mM EDTA, 2 mM DTT) supplemented with the following additives depending on the conditions indicated in figures and legends: 30 μM GDP (GDP condition), or 30 μM GDP, 30 μM AlCl3, and 10 mM NaF (GDP·AlF4− condition), or 30 μM GTPγS (GTPγS condition).
For experiments using purified proteins as source of soluble binding ligands, the following His-tagged proteins were used (range of protein amounts used in different experiments is in parenthesis): rat His-Gαi3 (0.3–14 μg), human His-Gαi3 (8 μg), His-GINIP (1–12 μg), His-Gαi1 (1 μg), His-Gαi2 (1 μg), His-Gαo (1 μg), His-RGS4 (0.2 μg). Aliquots of protein stored at −80 °C were quickly thawed and cleared by centrifugation at 14,000 × g for 2 minutes before addition to tubes containing the GST-fused proteins immobilized on GSH-agarose beads in a final volume of 400 μl. Tubes were incubated for 4 h at 4 °C with constant rotation. Beads were washed three times with 1 ml of wash buffer (4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4, 137 mM NaCl, 2.7 mM KCl, 0.1% (v/v) Tween 20, 10 mM MgCl2, 5 mM EDTA, 1 mM DTT) supplemented with GDP, GDP·AlF4− or GTPγS as indicated above, and resin-bound proteins were eluted with Laemmli sample buffer by incubation at 65 °C for 10 min. Proteins were separated by SDS-PAGE and immunoblotted with antibodies as indicated under “Protein Electrophoresis and Immunoblotting.”
For experiments using lysates of cultured cells as a source of soluble binding ligands, HEK293T cells (ATCC CRL-3216) were grown at 37 °C, 5% CO2 in DMEM (Gibco, 11965-092) supplemented with 10% fetal bovine serum (Hyclone, SH30072.03), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine (Corning, 30-009-CI). Approximately two million HEK293T cells were seeded on 10-cm dishes and transfected the day after using the calcium phosphate method with plasmids encoding the following constructs (DNA amounts in parentheses): Gαi3-FLAG (6 μg), Gαz-EE (6 μg), Gαo (6 μg), Gαs (6 μg), Gαq-HA (6 μg), Gα12-MYC (6 μg). Cell medium was changed 6 h after transfection, and approximately 24 h later, cells were lysed at 4 °C with 700 μl of lysis buffer (20 mM HEPES, pH 7.2, 125 mM K(CH3COO), 0.4% (v/v) Triton X-100, 1 mM DTT, 10 mM β-glycerophosphate, and 0.5 mM Na3VO4 supplemented with a SigmaFAST protease inhibitor mixture (Sigma, cat# S8830)). Cell lysates were cleared by centrifugation at 14,000 × g for 10 minutes, and supplemented with GDP or GDP·AlF4− as indicated above. One hundred microliters (~400 μg of total protein) of these cell lysates were added to the GST-fused proteins immobilized on GSH-agarose beads in a final volume of 400 μl. Tubes were incubated for 4 h at 4 °C with constant rotation, then washed and eluted according to the protocol described above for purified soluble ligands.
For experiments using brain tissue as a source of endogenous GINIP as binding ligand, the lysates were prepared as follows. Whole brains of adult mice were isolated and lysed in one ml (per brain) of modified RIPA buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% (v/v) Nonidet P-40, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 0.1% (v/v) Triton X100 supplemented with SigmaFAST protease inhibitor mixture) by grinding using tissue homogenizer (Fisherbrand, 15340167) on ice. Lysates were cleared by centrifugation at 14,000 × g for 10 minutes at 4 °C twice, aliquoted and stored at −80 °C as single use aliquots for experiments to avoid freeze/thaw cycles. After thawing, lysates were cleared by centrifugation at 14,000 × g for 10 minutes at 4 °C. Twenty-five μl of brain lysate (~500 μg of total protein) were added to GST-fused proteins immobilized on GSH-agarose beads in volume of 300 μl of binding buffer supplemented with GDP or GDP·AlF4− as indicated above. Tubes were incubated for 4 h at 4 °C with constant rotation, then washed according to the protocol described above for purified soluble ligands and eluted with Laemmli sample buffer by incubation at 37 °C for 10 min.
Protein electrophoresis and immunoblotting
Protein samples were prepared in Laemmli sample buffer as described in previous sections. For the analysis of GINIP expression in mouse tissues, samples were prepared using the same buffer and procedure as for the preparation of brain lysates as described in “Pulldown assays.” Proteins were separated by SDS-PAGE and transferred to PVDF membranes, which were blocked with 5% (w/v) nonfat dry milk and sequentially incubated with primary and secondary antibodies. For protein-protein-binding experiments with GST-fused proteins, PVDF membranes were stained with Ponceau S and scanned before blocking. The primary antibodies used were the following (dilution in parenthesis): rabbit Gαi3, SCBT #sc-262 (1:1000); mouse FLAG, Sigma #F1804 (1:1000); rabbit EE (Glu-Glu), Millipore #AB3788 (1:1000); mouse Gαo, SCBT #sc-13532 (1:1000); rabbit Gαs/olf, SCBT #sc-383 (1:1000); mouse HA, Roche #11583816001 (1:1000); mouse MYC, Cell Signaling #2276 (1:1000); mouse His, Sigma #H1029 (1:2500); mouse α-tubulin, Sigma #DM1A (1:2000); rabbit β-actin, LI-COR #926–42212 (1:1000); rabbit GAIP, serum gifted by M. Farquhar77,108(1:2000); rabbit pan-Gβ, SCBT #sc-166123 (1:250); goat GINIP, SCBT #sc-247284 (1:1000); mouse Gαq, SCBT #sc-393 (1:1000); mouse Tuj1, SCBT #sc80005 (1:2000); mouse GFAP, Merck #MAB360 (1:2000). The secondary antibodies were (dilution in parenthesis): goat anti-rabbit Alexa Fluor 680, Invitrogen #A21077 (1:10,000); goat anti-mouse Alexa Fluor 680, Invitrogen #A21058 (1:10,000); goat anti-mouse IRDye 800, LI-COR #926–32210 (1:10,000); goat anti-rabbit DyLight 800, Thermo #35571 (1:10,000); donkey anti-goat IRDye 680RD, LI-COR #926–68074. Infrared imaging of immunoblots was performed using an Odyssey CLx Infrared Imaging System (LI-COR). Images were processed using ImageJ software (National Institutes of Health) or Image Studio software (LI-COR), and assembled for presentation using Photoshop and Illustrator software (Adobe).
GTPγS-binding assays
GTPγS-binding assays were performed as described previously (40,107. Purified His-Gαi3 (100 nM) was diluted in assay buffer (20 mM Na-HEPES, pH 8, 100 mM NaCl, 1 mM EDTA, 25 mM MgCl2, 1 mM DTT, 0.05% (w/v) C12E10) and preincubated with purified proteins (as indicated in the figures) for 15 min at 30 °C. Reactions were initiated by adding an equal volume of assay buffer containing 1 μM [35S]GTPγS (~50 cpm/fmol, Perkin Elmer) at 30 °C. Duplicate aliquots (25 μl) were removed after 15 minutes and binding of radioactive nucleotide was stopped by addition of 3 ml of ice-cold wash buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 25 mM MgCl2). The quenched reactions were rapidly passed through BA-85 nitrocellulose filters (GE Healthcare, 10402506) and washed with 4 ml of cold wash buffer. Filters were dried and subjected to liquid scintillation counting. Background [35S]GTPγS detected in the absence of G protein was subtracted from each reaction. Data are expressed as percentage of [35S]GTPγS binding relative to a control reaction containing the G protein alone (% of control).
GTPase assays
Assays to measure GTPase activity under steady-state conditions were performed as described previously40,107. These experiments were carried out with Gαi1 R178M/A326S (His-Gαi1RM/AS), a previously described43 double mutant that simultaneously increases the rate of nucleotide exchange and decreases GTP hydrolysis. The rate limiting step of GTPase reactions for Gαi1RM/AS is nucleotide hydrolysis43 as opposed to the limiting rate of nucleotide exchange with the wild-type protein14,40, which allows to measure changes in nucleotide hydrolysis such as those caused by GAPs under steady-state conditions. His-Gαi1RM/AS (100 nM) was diluted in assay buffer (20 mm Na-HEPES, pH 8, 100 mm NaCl, 1 mm EDTA, 2 mm MgCl2, 1 mm DTT, 0.05% (w/v) C12E10) with purified proteins (as indicated in the figures) at 4 °C. Reactions were initiated by adding an equal volume of assay buffer containing 1 μm [γ−32P]GTP (~50 cpm/fmol, Perkin Elmer) at 30 °C. Duplicate aliquots (25 μl) were removed after 30 minutes, and the reaction was stopped by the addition of 975 μl of ice-cold 5% (w/v) activated charcoal in 20 mM H3PO4, pH 3. Samples were centrifuged for 10 min at 10,000 × g, and 500 μl of the resultant supernatants were subjected to liquid scintillation counting to quantify the amount of [32P]Pi released. Background [32P]Pi detected in the absence of G protein was subtracted from each reaction. Data are expressed as percentage of [γ-32P]Pi released relative to a control reaction containing the G protein alone (% of control).
cAMP measurements in HEK239T cells by BRET
HEK293T cells (ATCC CRL-3216) were grown at 37 °C, 5% CO2 in DMEM medium (Gibco, 11965-092) supplemented with 10% fetal bovine serum (Hyclone, SH30072.03), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine (Corning, 30-009-CI). Approximately 400,000 cells/well were seeded on 6-well plates coated with 0.1% gelatin and transfected ~24 hr later using the calcium phosphate method with plasmids encoding the following constructs (DNA amounts in parentheses): Nluc-EPAC-VV (0.05 μg), GABABR1a (0.2 μg), GABABR2 (0.2 μg), α2A-AR (0.2 μg), FLAG-D2R (0.2 μg), Gαi3 WT (0.5 μg), and GINIP-FLAG (2 μg). Total DNA amount per well was equalized by supplementing with empty pcDNA3.1 as needed. Cell medium was changed 6 h after transfection, and approximately 16–24 h after transfection, cells were washed and gently scraped in room temperature PBS, centrifuged (5 min at 550 × g), and resuspended in BRET buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 0.37 mM NaH2PO4, 20 mM HEPES and 0.1% glucose, pH 7.4) at a concentration of ~106 cells/ml. ~25,000 cells/well were added to a white opaque 96-well plate (Opti-Plate, PerkinElmer Life Sciences, 6005290) and mixed with the nanoluciferase substrate Nano-Glo (Promega, N1120, final dilution 1:200) before measuring luminescence. Luminescence signals at 460 ± 80 and 535 ± 35 nm were measured at 28 °C every 2 s in a BMG Labtech POLARStar Omega plate reader and BRET was calculated as the ratio between the emission intensity at 535 nm divided by the emission intensity at 460 nm. Since the Nluc-EPAC-VV construct reports cAMP binding as a decrease in BRET, results were processed as the inverse of the BRET ratio (BRET−1) to make it more intuitive. After subtraction of a basal signal measured for 30 s before stimulation with forskolin (ΔBRET−1), results were normalized to the maximum response of forskolin detected prior to the addition of GPCR agonists (cAMP (normalized ΔBRET−1). For calculation of the agonist-mediated inhibition of cAMP induced by forskolin “inhibition (%FSK),” the average of the last six time points of ΔBRET−1 curves were used. Baseline ΔBRET−1 values from cells not treated with forskolin were subtracted from values obtained from forskolin and agonist treated or forskolin treated conditions. Inhibition (%FSK) was represented as the ratio of forskolin and agonist treated divided by forskolin treated.
Measurement of Gαi3-AC5 association in HEK293T cells by BRET
HEK293T cells were cultured, transfected, and harvested as described in “cAMP measurements in HEK239T cells by BRET.” Cells were transfected with plasmids encoding the following constructs (DNA amounts in parentheses): GABABR1a (0.2 μg), GABABR2 (0.2 μg), α2A-AR (0.2 μg), YFP-hAC5 (0.5 μg), Gαi3-Nluc(a/b) WT or Q204L (0.1 μg), GINIP-FLAG (0.1–2 μg). Total DNA amount per well was equalized by supplementing with empty pcDNA3.1 as needed. Luminescence measurements were carried out as in “cAMP measurements in HEK239T cells by BRET,” except that signals were recorded every 0.24 s for kinetic measurements of Gαi3-AC5 association upon modulation of GPCR activation. For endpoint measurements comparing the association of Gαi3 WT or Gαi3 Q204L (QL) under steady-state conditions, signals were recorded as an average of 3 measurements 30 s apart. BRET was calculated as the ratio between the emission intensity at 535 nm divided by the emission intensity at 460 nm for both kinetic and endpoint measurements. Results for the kinetic measurements were presented as increase in BRET after subtraction of the basal signal measured for 30 s before GPCR stimulation (ΔBRET (baseline)), whereas for endpoint measurements they were presented as difference in BRET compared with the BRET signal in cells expressing Gαi3-Nluc WT and YFP-hAC5 in the absence of GINIP (ΔBRET (WT control)).
Free Gβγ measurements in HEK293T cells by BRET
HEK293T cells were cultured, transfected, and harvested as described in “cAMP measurements in HEK239T cells by BRET.” Cells were transfected with plasmids encoding the following constructs (DNA amounts in parentheses): VN-Gγ2 (0.2 μg), VC-Gβ1 (0.2 μg), masGRK3ct-Nluc (0.2 μg), Gαi3 WT (1 μg), Gαo WT (1 μg), GABABR1a (0.2 μg), GABABR2 (0.2 μg), α2A-AR (0.2 μg), GAIP (0.5 μg), RGS7 (1 μg), Gβ5 (1 μg), R7BP (1 μg), RGS8 (2 μg), RGS12 (0.2 μg), GINIP-FLAG (2 μg). Luminescence measurements and BRET calculations were carried out as in “cAMP measurements in HEK239T cells by BRET,” except signals were recorded every 0.24 s. Results were presented as increase in BRET after subtraction of the basal signal measured for 30 s before any stimulation (ΔBRET (baseline)). For the calculation of response amplitudes, the difference between the raw BRET ratio before and 60 s after agonist stimulation was calculated. For the presentation and calculation of G protein deactivation rates, the minimum BRET value reached after the addition of antagonist (plateau signal) was subtracted from the raw BRET ratio of each timepoint (recovery corrected ΔBRET), and each resulting value was scaled to as the percentage of the maximal BRET value right before the addition of antagonist (“% Maximum response”). G protein deactivation rate constants (k) were determined by fitting the recovery-corrected ΔBRET values after the addition of antagonist to a one-phase decay equation (Y=(Y0 − Plateau) × e(−kX) + Plateau) in Prism 9 (Graphpad), where Y0 is the starting value right before the addition of antagonist (constrained to 100) and Plateau is the near-zero minimum estimated by the fit.
Measurement of arrestin-3 recruitment to GPCRs by BRET in HEK293T cells
HEK293T cells were cultured, transfected, and harvested as described in “cAMP measurements in HEK239T cells by BRET,” with modifications. The morning after seeding, cells were transfected using the calcium phosphate method with plasmids encoding the following constructs (DNA amounts in parentheses): venus-arrestin-3 (1 μg) and α2A-AR-Rluc8 (0.01 μg) or D2R-Rluc8 (0.025 μg). Cell medium was changed 6 h after transfection, followed immediately by transfection with 2 μg of plasmid DNA encoding GINIP-FLAG or the same amount of an empty plasmid (pcDNA3.1(+)) using polyethylenimine (PEI) (Longo et al., 2013) at a 2:1 PEI:DNA ratio. Cell medium was changed the following morning. Approximately 18–24 h after PEI transfection luminescence measurements and BRET calculations were carried out as in “cAMP measurements in HEK239T cells by BRET,” except that 20 μM coelenterezine-h (Promega, S2011) was used as luciferase substrate and luminescence was recorded every 2 s. Results were presented as increase in BRET after subtraction of the basal signal measured for 60 s before any stimulation (ΔBRET (baseline)).
Adenylyl cyclase activity in vitro
Regulation of adenylyl cyclase (AC) activity by purified G proteins was determined by measuring the production of cAMP upon reconstitution of the enzyme activity using the purified AC5 C1 and AC2 C2 domains as previously described54 with modifications. Before AC activity measurements myr-Gαi1 and His-Gαs were loaded with GTPγS by incubating them at 30 °C with 150 μM GTPγS in buffer (20 mM Tris-HCl, 20 mM NaCl, 1 mM MgCl2, 1 mM DTT, 5 % glycerol (v:v)) for 3 hours or 45 minutes, respectively. GTPγS-loaded G proteins were aliquoted and stored at −80 °C. AC reactions were carried out in technical duplicates in a final volume of 40 μl, and all reactants were diluted in assay buffer (50 mM HEPES, 2 mM MgCl2, 1 mM EDTA, 0.5 mg/mL BSA, 1 mM DTT, pH 8.0) unless otherwise indicated. 8 μl of 250 nM AC5 C1, 8 μl of 5X concentrated stocks of myr-Gαi1-GTPγS (or the same volume of assay buffer without Gαi1), and 8 μl of 10 μM GST-GINIP (or the same volume of PBS for conditions without GINIP) were mixed and incubated on ice for 15 minutes. Simultaneously, a stock of 1.66 μM AC2 C2 and 0.33 μM Gαs-GTPγS was mixed and incubated on ice for 15 minutes. 12 μl of AC2 C2/ Gαs-GTPγS was added to the tubes containing 24 μl AC5 C1, myr-Gαi1-GTPγS, and GST-GINIP. Samples were then incubated for 15 minutes at room temperature before starting the reactions by addition of 4 μl of ATP and MgCl2 solution (10 mM ATP, 50 mM MgCl2) and rapidly transferring the tubes to a heat block at 30 °C. The final concentrations of reactants were: AC5 C1, 50 nM; myr-Gαi1-GTPγS, 0.25–2 μM; GST-GINIP, 2 μM; AC2 C2, 0.5 μM; Gαs-GTPγS, 0.1 μM; ATP, 1 mM; MgCl2, 5 mM. Reactions were stopped after 10 minutes by rapidly transferring the tubes to a heat block at 95 °C for 5 minutes. Samples were centrifuged at 5,000 × g for 2 min, and an aliquot from the supernatant was collected to quantify cAMP using the LANCE cAMP kit (Perkin Elmer, cat#AD0262) according to the manufacturer protocol. Time-resolved fluorescence measurements to quantify cAMP were done on a TECAN Infinite M1000 plate reader in white 384-well ProxiPlates (Perkin Elmer, 6008280). Total cAMP was determined based on a standard curve, and each technical duplicate was measured individually before averaging cAMP values. Specific activity was calculated as nmol cAMP / min / mg AC; mg AC was based on the concentration of AC5 C1, the limiting reactant in the C1/C2 complex. AC activity was background corrected by subtracting the signal obtained for AC5 C1 and AC2 C2 in the absence of Gαs-GTPγS. AC activity was then presented as a percentage of Gαs-GTPγS stimulated AC activity.
β-galactosidase staining
GINIP +/1a and GINIP +/+ littermates (3 month-old) were anesthetized and transcardially perfused with cold PBS. Brains were rapidly removed from the skull, and placed in an acrylic matrix (Electron Microscopy Sciences, 69080-C or 69080-S) to make either coronal or sagittal slices (1 mm thickness). Slices were rinsed twice with PBS and incubated in staining solution (0.1 M phosphate buffer, pH 7.3, 20 mM Tris HCl, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 1 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside)) at room temperature with gentle rocking for 1 hour. Staining was stopped by removal of the staining solution, and samples were washed with PBS 3 times, followed by fixation in 4% (w/v) paraformaldehyde in PBS at room temperature for 30 min. Slices were moved to the stage of an Olympus SZX16 stereo microscope equipped with a digital camera (U-TV0.63XC) and with a SDF PLAPO 0.5XPF objective lens and imaged by bright-field microscopy at room temperature using QImaging (Teledyne) software. Individual images were assembled for presentation in Photoshop and Illustrator software (Adobe).
Fluorescence mRNA in situ hybridization (RNAScope®)
Mice (4–5 month-old, genotype-matched littermates) were anesthetized and transcardially perfused with cold PBS. Brains were rapidly removed from the skull, submerged in cryo-embedding medium (OCT) (Fisherbrand, 23730571), and frozen in dry-ice prior to storage of the OCT tissue blocks at −80 °C. Blocks were equilibrated to −20 °C and sectioned in a HM 550 VP cryostat microtome (MICROM GmbH, 956444). Twenty μm sections were mounted onto Superfrost Plus slides (Fisherbrand, 22037246) and stored at −80 °C in air-tight, light-proof containers until use. Frozen slides were placed in a Tissue Tek Slide Rack (StatLab, LWS2124), and immediately immersed in pre-chilled fixative solution (10% neutral buffered formalin, Fisherbrand, 305510) for 15 min at 4 °C. After fixation, sections were washed at room temperature with PBS twice by dipping the rack in the solution container 3–5 times during a period of ~3 min for each wash. Sections were dehydrated by sequential 5-minute incubations at room temperature in 50%, 75% and 100% (v/v) ethanol. Slides were air-dried by incubation at room temperature for approximately 5 min before drawing a hydrophobic barrier around each section with an Immedge™ hydrophobic barrier pen (Vector, H-4000). Subsequent steps were carried out using the RNAscope 2.0 Assay kit following the manufacturer’s instructions (Bio-techne, 320850). Briefly, ~5 drops of Protease IV solution (Bio-techne, 322340) were added to each section, followed by incubation at room temperature for 30 min. Slides were washed with PBS twice by dipping the slide rack in the solution container 3–5 times during a period of ~3 min for each wash. After removing the excess of liquid by flicking, the Amp 1-FL reagent containing the hybridization probe(s) for GINIP (Mm-Phf24-O1), vGlut1 (Mm-Slc17a7-C2) and/or VGAT (Mm-Slc32a1-C3), all from Bio-techne, was added to cover the slides, followed by a 30 min incubation at 40 °C in a HybEZ™ II hybridization oven (Bio-techne, 240200ACD). After two washes with 1X Wash buffer (Bio-techne, 310091) slides were sequentially incubated in 100 μl of the following reagent solutions containing the signal amplifiers (Amp 2-FL and Amp 3-FL) and the fluorescent tracers (Amp 4-FL) for each mRNA probe at 40 °C in a HybEZ™ II hybridization oven for 30 min, 15 min, and 30 min, respectively. Samples were washed twice with 1X Wash buffer after each incubation. The slides were stained with DAPI for 1 minute (Bio-techne, 320858), and mounted in ProLong Diamond Antifade (Invitrogen, P36965). Mounted slides were cured overnight at room temperature prior to imaging. Stained sections were imaged by wide-field microscopy at room temperature using a Zeiss Axio Observer Z1 microscope equipped with a digital camera (C10600/ORCA-R2 Hamamatsu Photonics). Images were taken with a 63x oil-immersion objective (NA = 1.4; working distance = 0.19 mm) using ZEN software. Individual images were assembled for presentation in Photoshop and Illustrator software (Adobe). The custom-made probe for GINIP mRNA (Mm-Phf24-O1, Bio-techne) recognizes the region from nucleotide 857 to 1491 of the mouse GINIP mRNA sequence NM_001346526.1. This region is present in mRNA from the GINIP wild-type and GINIP flox alleles, but not in the GINIP 1a allele or the GINIP flox allele after Cre-mediated recombination (GINIP Δ null allele).
Immunofluorescence
Cortical neuron cultures were established from neonatal mouse brains as described in “Mouse primary cortical cultures.” 50,000 cells were plated on poly-L-lysine coated 12 mm coverslips in 24 well plates. DIV12 cells were used for experiments evaluating neuronal versus glial marker expression, whereas DIV21 cells were used for experiments evaluating the presence of different synaptic markers in dendrites. Cells were washed with PBS twice for 2 minutes at room temperature and fixed with 4% (w/v) PFA in PBS for 15 minutes at room temperature. After 2 additional washes with PBS, cells were permeabilized and blocked at room temperature for 1 hour in blocking buffer (5% (w/v) Bovine Serum Albumin (BSA), 5% (v/v) goat serum, 0.1% (v/v) Triton X-100 in PBS). Coverslips were placed upside down over 25 μl of buffer (1% (w/v) BSA, 1% (v/v) goat serum, 0.1% (v/v) Triton X-100 in PBS) spotted on parafilm containing primary antibodies in the following dilutions: rabbit GINIP, Aviva ARP70657_P050 (1:100); mouse Tuj1, SCBT sc-80005 (1:200); mouse GFAP Merck MAB360 (1:200); mouse synaptophysin, SCBT sc-55507 (1:200); mouse vGlut1, SCBT sc-377425 (1:50); mouse GAD65, DSHB AB_528264 (1:50); mouse PSD95, ABcam ab13552 (1:200); mouse Gephyrin, Synaptic Systems 147011 (1:200). After overnight incubation at 4°C in a humid chamber, coverslips were washed with PBS 3 times for 5 minutes at room temperature, placed upside down over 30 μl of buffer (1% (w/v) BSA, 1% (v/v) goat serum, 0.1% (v/v) Triton X-100 in PBS) spotted on parafilm containing secondary antibodies in the following dilutions: goat anti-mouse Alexa Fluor 488, Life Technologies A11017 (1:400); goat anti-rabbit Alexa Fluor 488, Life Technologies A11070 (1:400); goat anti-mouse Alexa Fluor 594, Life Technologies A11020 (1:400), and goat anti-rabbit Alexa Fluor 594, Life Technologies A11072 (1:400). After incubation for 1 hour at room temperature in a humid chamber, coverslips were washed with PBS 3 times for 10 minutes at room temperature, followed by staining with DAPI (1:10,000) for 5 minutes at room temperature. After one wash with PBS, coverslips were mounted with ProLong Diamond Antifade (Invitrogen, P36970) and cured overnight at room temperature prior to imaging.
Wide-field microscopy imaging was performed at room temperature with a Zeiss Axio Observer Z1 microscope equipped with a digital camera (C10600/ORCA-R2 Hamamatsu Photonics). Images were taken with a 63x oil-immersion objective (NA = 1.4; working distance = 0.19 mm) using ZEN software. Confocal microscopy imaging was carried out at room temperature with a Zeiss LSM 700. Single sections of confocal images of 0.321 μm thickness along the z axis were taken with a 63x oil-immersion objective (NA 1.4, working distance 0.19 mm) using ZEN software.
For experiments to determine the expression of GINIP in neuron versus glia (Fig. 5), the number of NeuN (neuronal marker) or GFAP (astrocyte marker) positive cells that were also positive for GINIP was quantified as a percentage from 6 randomly chosen fields of three independent cultures at DIV12 (2 fields/ experiment). For experiments to determine the subcellular localization of GINIP (Fig. 6), the number of GINIP positive puncta (0.25–0.75 μm2 area) on dendrites that were also positive for each synaptic marker was quantified as a percentage from 3–4 fields per experiment. Three independent experiments corresponding to separate cultures at DIV21 were quantified. Individual images were assembled for presentation in Photoshop and Illustrator software (Adobe).
Chemically induced seizures
Animals (n = 11 – 14 per group; approximately 5 – 6 months old; males and females) were assessed prior to start of experiment for any health issues and were individually habituated to sterile empty housing cages prior to start of study. During the study, all subjects were injected with bicuculline (i.p. Bio-Techne Co., Minneapolis, MN USA) at increasing doses (0, 1, 2 and 3 mg/kg), immediately placed into sterile empty housing cage, and tracked (Logitech C920 HD Pro; Logitech International S.A., Newark, CA USA. Canon VIXIA HF MF80 HD; Canon USA, Melville, NY USA) for 30 minutes. Behavior was scored by intensity of 0 to 4 (0 = normal behavior, 1 = wild running, 2 = tonic seizure, 3 = clonic seizure, 4 = cardiac arrest/death) and by number of seizure episodes. The genotypes of all animals were blinded prior to start of study.
Lentivirus packaging and transduction of primary cortical neurons
Lentiviruses used for the transduction of cultured neurons were concentrated after large scale packaging as described next. HEK293T cells (Lenti-X 293T, Cat# 632180, Takara Bio) were plated on 150 mm diameter dishes (~2.5 million cells / dish) and cultured at 37°C, 5% CO2 in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. After 16–24 h, cells were co-transfected with plasmids encoding Gβγ-BERKY1 (pLenti-hSyn-Gβγ-BERKY1) (27 μg / dish), or Nluc-EPAC-VV (pLenti-hSyn-Nluc-EPAC-VV) (27 μg / dish) along with the packaging plasmid psPAX2 (18 μg / dish) and the envelope plasmid pMD2.G (11.25 μg / dish) using the polyethylenimine (PEI) method109 at a 2:1 PEI:DNA ratio. Approximately 16 h after transfection, media was replaced with serum-free media. Lentivirus containing medium was collected 24 h and 48 h after the initial media change (a total of ~70 ml per dish and 4 dishes for each construct). Media was centrifuged for 5 min at 900 × g and filtered through a 0.45 μm sterile PES filter (Fisherbrand, cat# FB12566505). Filtered media was centrifuged for overnight (~16 h) at 17,200 × g at 4 °C (Sorvall RC6+, ThermoScientific F12–6×500 LEX rotor) to sediment lentiviral particles. Pellets were washed and gently resuspended in 1 ml of PBS and centrifuged at 50,000 × g for 1 h at 4°C (Beckman Optima MAX-E, TLA-55 rotor). The resulting pellets were resuspended in 500 μl of PBS to obtain concentrated lentiviral stocks that were stored at −80°C in aliquots. Each aliquot was thawed only once for subsequent experiments.
Measurement of free Gβγ in primary cortical neurons by BRET
Cortical neuron cultures were established from neonatal brains of GINIP flox/flox mice as described in “Mouse primary cortical cultures.” For experiments aimed at measuring the release of free Gβγ in neurons, 100,000 cells were seeded on poly-L-lysine coated 5 mm coverslips in 96 well plates. On DIV6, primary cortical neurons were transduced with AAV encoding Cre recombinase (AAV-hSyn-Cre, Addgene 105553-AAV1) by replacing half of the volume of media in the well with media containing the AAV (final virus dilution 1:500 in 200 μl per well). Two hours later, 100 μl of media were replaced by fresh complete neural medium. Controls were subjected to the same media changes but omitting the AVV. At DIV8, all conditions were transduced with lentiviruses encoding Gβγ-BERKY1 by replacing one half of the media with complete neural media containing lentivirus (final lentivirus dilution 1:200 in 200 μl per well). Two hours after addition of the lentivirus, one half of the medium was replaced with fresh complete neural media and culturing carried on as described in “Mouse primary cortical cultures”. BRET measurements were carried out between DIV12 and DIV16. For this, coverslips were transferred to white opaque 96-well plate (Opti-Plate, PerkinElmer Life Sciences) containing 100 μl of BRET buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 0.37 mM NaH2PO4, 24 mM NaHCO3, 10 mM HEPES and 0.1% glucose, pH 7.4) with nanoluciferase substrate Nano-Glo (Promega, final dilution 1:200). Luminescence signals at 460 ± 80 and 535 ± 35 nm were measured at 28 °C every 0.96 s in a BMG Labtech POLARStar Omega plate reader, and BRET was calculated as the ratio between the emission intensity at 535 nm divided by the emission intensity at 460 nm. Results were calculated as increase in BRET after subtraction of the basal signal measured for 30 s before any stimulation, followed by normalization to the maximum increase in BRET observed in control neurons (from GINIP flox/flox mice without Cre recombination) observed upon stimulation with saturating concentration of the agonist for the GPCR under investigation (insert here how this is called in the Y axis of the corresponding result).
Measurement of cAMP in primary cortical neurons by BRET
Cortical neuron cultures were established from neonatal brains of GINIP flox/flox mice as described in “Mouse primary cortical cultures.” For experiments aimed at measuring intracellular cAMP levels cells were processed as described above in “Measurement of free Gβγ in primary cortical neurons by BRET”, except that the lentiviral transductions were carried out with particles for the expression of the BRET-based cAMP biosensor Nluc-EPAC-VV. BRET measurements were carried out as described in “Measurement of free Gβγ in primary cortical neurons by BRET.” Luminescence signals at 460 ± 80 and 535 ± 35 nm were measured at 28 °C every 5 seconds in a BMG Labtech POLARStar Omega plate reader and BRET was calculated as the ratio between the emission intensity at 535 nm divided by the emission intensity at 460 nm. Since the Nluc-EPAC-VV construct reports cAMP binding as a decrease in BRET, results were processed as the inverse of the BRET ratio (BRET−1) to make it more intuitive. Results are presented as the change in BRET (ΔBRET−1) relative to the BRET baseline measured for 30 s before stimulation with forskolin.
Brain Slice Electrophysiology
Brains from adult (2–5 month-old) GINIP +/+ and GINIP 1a/1a littermate mice were sectioned fresh to generate coronal slices (220 μm thickness) using a Leica VT1000S vibratome (Leica Microsystems). Mice were anesthetized, perfused and decapitated in accordance with national and institutional guidelines. Perfusion was carried out with modified artificial cerebral spinal fluid (mACSF) containing (in mM): 92 NMDG, 20 HEPES, 25 glucose, 30 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 5 L-ascorbic acid, 3 sodium pyruvate, 2 thiourea, 10 MgSO4, 0.5 CaCl2, 12 N-acetyl-l-cysteine, 300−310 mOsm, pH 7.3−7.4 adjusted with 4N HCl. Brain slices were sectioned in cold mACSF, recovered in the same buffer at 32 °C for 10 min, and further transferred to a holding ACSF solution containing (in mM): 92 NaCl, 20 HEPES, 25 glucose, 30 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 5 L-ascorbic acid, 3 sodium pyruvate, 2 thiourea, 1 MgSO4, 2 CaCl2, 12 N-acetyl-l-cysteine, 300−310 mOsm, at pH 7.3−7.4 at room temperature to recover for 1h before electrophysiological recordings. All solutions were continuously saturated with 95% O2 and 5% CO2. Slices were continuously perfused (2 ml/min) during electrophysiological recordings with carbogen saturated ACSF containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 26 NaHCO3, 11 glucose, 2.4 CaCl2, 300− 310 mOsm, at pH 7.3−7.4 at 32°C. Pyramidal neurons from the somatosensory cortex (layer III/IV) were visualized with infrared differential interference contrast video microscopy (FV3000, Olympus) under 40x water-immersion lens and recorded in the whole-cell configuration using a MultiClamp 700B amplifier (2 kHz low-pass Bessel filter and 10 kHz digitization) with pClamp 11 software (Molecular Devices). Patch pipettes were pulled from OD/ID 1.5/0.86 mm capillary glass in a horizontal pipette puller (Model P-1000, Sutter Instrument). Series resistance and whole cell capacitance were monitored during the experiments and recordings with access resistance changes >50% were discarded.
Potassium currents were recorded in the presence of 1 μM tetrodotoxin (TTX) in the bath solution and with the borosilicate glass electrodes (3−5 MΩ) filled with an internal solution containing (in mM): 120 mM potassium gluconate, 20 KCl, 0.05 EGTA, 10 HEPES, 1.5 MgCl2, 2.18 Na2-ATP, 0.38 Na-GTP, 10.19 sodium phosphocreatine, 275−285 mOsm, and pH 7.3−7.4. Holding current was recorded in voltage clamp mode at a corresponding resting membrane potential of −55–65 mV. Started 10 min after stable whole cell access was obtained, K+ currents were induced by 50 μM baclofen followed by wash with ACSF. Change (Δ) in holding current (pA) after baclofen addition was presented as mean ± S.E.M. from 14 cells per group (representing at least 4–5 animals per group).
Miniature inhibitory postsynaptic currents (mIPSCs) were recorded by adding 1 μM tetrodotoxin (TTX), 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and 10 μM d-2-amino-5 phosphonopentanoic acid (d-AP5) in the bath solution, and inhibiting K+ channels by filling the electrodes with internal solution containing (in mM): 140 mM cesium gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 0.1 EGTA, 2 Na2-ATP, 0.4 Na-GTP, 275−285 mOsm, at pH 7.3−7.4. The holding potential was set to +10 mV and current was recorded in voltage clamp mode. Ten min of stable baseline was confirmed after obtaining whole cell access. Different concentrations of baclofen (0.1, 1 and 10 μM) were perfused for 5 min. The pharmacological cocktail described above to isolate mIPSCs was maintained throughput the recordings. Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded as described for mIPSCs except that 50 μM picrotoxin was present in the ACSF instead of the cocktail of TTX, CNQX, and d-AP5. Positive-going outward IPSCs or negative-going outward EPSCs were separately detected at the same baseline using Clampfit analysis software and a template was constructed by averaging >50 mIPSCs or sEPSCs for automatic event frequency detection with amplitude above threshold of 5 pA. Between 8 and 14 cells from 4–5 different animals were recorded per group. Data were reported as mean ± SEM.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, experiments were independently repeated a minimum of three times. The value of n is representative of an independent experiment; for experiments using animals, the value of n is representative of number of animals. For experiments displaying pooled data, individual data points and/or mean ± S.E.M (Standard error of the mean) is depicted. Statistical details including statistical tests, value of n, and defined confidence intervals based on p-values for all experiments are indicated in the figure legend of each panel. For other experiments, like immunoblot images, one representative result is presented. All statistical comparisons were calculated in GraphPad Prism 9. A Student’s t-test was used in cases where two conditions were compared. One-way ANOVA with correction for multiple comparisons using Tukey post-hoc analysis was used in cases where 3 or more conditions were compared. In the case where differences in quantitative variables for two independent variables were compared a two-way ANOVA was used without correction for multiple comparisons using Fisher’s LSD test.
Supplementary Material
HIGHLIGHTS.
GINIP modulates GPCR signaling by binding Gαi without altering its enzymatic activity
GINIP biases GPCR signaling by enhancing Gβγ responses while dampening Gαi responses
GINIP establishes system bias for GPCR signaling at inhibitory synapses of neurons
Loss of GINIP disrupts fine-tuning of inhibitory neuromodulation, leading to seizures
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01NS117101 (to MG-M) and F31NS115318 (to AL). We thank Vickery Trinkaus-Randall for access to confocal microscopes. We thank the following investigators for providing DNA plasmids: N. Lambert, J. Blumer, J. Sondek, and M. Linder. We also thank C. Dessauer for providing plasmids and protocols for the in vitro adenylyl cyclase experiments. We thank members of the Garcia-Marcos laboratory for reading a draft of this manuscript and making comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no conflict of interests.
REFERENCES
- 1.Gilman AG (1987). G proteins: transducers of receptor-generated signals. Annual review of biochemistry 56, 615–649. 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 2.Weis WI, and Kobilka BK (2018). The Molecular Basis of G Protein-Coupled Receptor Activation. Annual review of biochemistry 87, 897–919. 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth BL (2019). Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nature structural & molecular biology 26, 535–544. 10.1038/s41594-019-0252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greengard P (2001). The neurobiology of slow synaptic transmission. Science 294, 1024–1030. 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- 5.Zurawski Z, Yim YY, Alford S, and Hamm HE (2019). The expanding roles and mechanisms of G protein-mediated presynaptic inhibition. The Journal of biological chemistry 294, 1661–1670. 10.1074/jbc.TM118.004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nature reviews. Drug discovery 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hopkins AL, and Groom CR (2002). The druggable genome. Nat Rev Drug Discov 1, 727–730. [DOI] [PubMed] [Google Scholar]
- 8.Sriram K, and Insel PA (2018). GPCRs as targets for approved drugs: How many targets and how many drugs? Mol Pharmacol, Jan 3. pii: mol.117.111062. doi: 1110 10.111124/mol.111117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, and Overington JP (2017). A comprehensive map of molecular drug targets. Nature reviews. Drug discovery 16, 19–34. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato M, Blumer JB, Simon V, and Lanier SM (2006). Accessory proteins for G proteins: partners in signaling. Annual review of pharmacology and toxicology 46, 151–187. 10.1146/annurev.pharmtox.46.120604.141115. [DOI] [PubMed] [Google Scholar]
- 11.Siderovski DP, and Willard FS (2005). The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. International journal of biological sciences 1, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cismowski MJ, Takesono A, Ma C, Lizano JS, Xie X, Fuernkranz H, Lanier SM, and Duzic E (1999). Genetic screens in yeast to identify mammalian nonreceptor modulators of G-protein signaling. Nature biotechnology 17, 878–883. 10.1038/12867. [DOI] [PubMed] [Google Scholar]
- 13.De Vries L, Zheng B, Fischer T, Elenko E, and Farquhar MG (2000). The regulator of G protein signaling family. Annual review of pharmacology and toxicology 40, 235–271. [DOI] [PubMed] [Google Scholar]
- 14.Ross EM, and Wilkie TM (2000). GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annual review of biochemistry 69, 795–827. [DOI] [PubMed] [Google Scholar]
- 15.De Vries L, Mousli M, Wurmser A, and Farquhar MG (1995). GAIP, a protein that specifically interacts with the trimeric G protein G alpha i3, is a member of a protein family with a highly conserved core domain. Proceedings of the National Academy of Sciences of the United States of America 92, 11916–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dohlman HG, and Thorner J (1997). RGS proteins and signaling by heterotrimeric G proteins. The Journal of biological chemistry 272, 3871–3874. 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- 17.DiGiacomo V, Marivin A, and Garcia-Marcos M (2018). When Heterotrimeric G Proteins Are Not Activated by G Protein-Coupled Receptors: Structural Insights and Evolutionary Conservation. Biochemistry 57, 255–257. 10.1021/acs.biochem.7b00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson N, Linder ME, Druey KM, Kehrl JH, and Blumer KJ (1996). RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature 383, 172–175. 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 19.Berman DM, Wilkie TM, and Gilman AG (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86, 445–452. 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 20.Peterson YK, Bernard ML, Ma H, Hazard S 3rd, Graber SG, and Lanier SM (2000). Stabilization of the GDP-bound conformation of Gialpha by a peptide derived from the G-protein regulatory motif of AGS3. The Journal of biological chemistry 275, 33193–33196. [DOI] [PubMed] [Google Scholar]
- 21.De Vries L, Fischer T, Tronchere H, Brothers GM, Strockbine B, Siderovski DP, and Farquhar MG (2000). Activator of G protein signaling 3 is a guanine dissociation inhibitor for Galpha i subunits. Proceedings of the National Academy of Sciences of the United States of America 97, 14364–14369. 10.1073/pnas.97.26.14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimple RJ, De Vries L, Tronchere H, Behe CI, Morris RA, Gist Farquhar M, and Siderovski DP (2001). RGS12 and RGS14 GoLoco motifs are G alpha(i) interaction sites with guanine nucleotide dissociation inhibitor Activity. The Journal of biological chemistry 276, 29275–29281. 10.1074/jbc.M103208200. [DOI] [PubMed] [Google Scholar]
- 23.Cismowski MJ, Ma C, Ribas C, Xie X, Spruyt M, Lizano JS, Lanier SM, and Duzic E (2000). Activation of heterotrimeric G-protein signaling by a ras-related protein. Implications for signal integration. The Journal of biological chemistry 275, 23421–23424. 10.1074/jbc.C000322200. [DOI] [PubMed] [Google Scholar]
- 24.Tall GG, Krumins AM, and Gilman AG (2003). Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. The Journal of biological chemistry 278, 8356–8362. 10.1074/jbc.M211862200. [DOI] [PubMed] [Google Scholar]
- 25.Lee MJ, and Dohlman HG (2008). Coactivation of G protein signaling by cell-surface receptors and an intracellular exchange factor. Curr Biol 18, 211–215. 10.1016/j.cub.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betke KM, Wells CA, and Hamm HE (2012). GPCR mediated regulation of synaptic transmission. Progress in neurobiology 96, 304–321. 10.1016/j.pneurobio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Csanady L (2017). A new target for G protein signaling. eLife 6. 10.7554/eLife.31106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostrom KF, LaVigne JE, Brust TF, Seifert R, Dessauer CW, Watts VJ, and Ostrom RS (2022). Physiological roles of mammalian transmembrane adenylyl cyclase isoforms. Physiological reviews 102, 815–857. 10.1152/physrev.00013.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadana R, and Dessauer CW (2009). Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neuro-Signals 17, 5–22. 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosaka M, Hammer RE, and Sudhof TC (1999). A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24, 377–387. 10.1016/s0896-6273(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 31.Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, and Linden DJ (2003). Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115, 49–60. 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- 32.Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, and Sorensen JB (2004). Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41, 417–429. 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 33.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, and Scott JD (1999). Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 285, 93–96. 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 34.Kandel ER (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Molecular brain 5, 14. 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maziarz M, Broselid S, DiGiacomo V, Park JC, Luebbers A, Garcia-Navarrete L, Blanco-Canosa JB, Baillie GS, and Garcia-Marcos M (2018). A biochemical and genetic discovery pipeline identifies PLCdelta4b as a nonreceptor activator of heterotrimeric G-proteins. The Journal of biological chemistry 293, 16964–16983. 10.1074/jbc.RA118.003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaillard S, Lo Re L, Mantilleri A, Hepp R, Urien L, Malapert P, Alonso S, Deage M, Kambrun C, Landry M, et al. (2014). GINIP, a Galphai-interacting protein, functions as a key modulator of peripheral GABAB receptor-mediated analgesia. Neuron 84, 123–136. 10.1016/j.neuron.2014.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Wang F, Fischer G, Hogan QH, and Yu H (2016). Peripheral nerve injury induces loss of nociceptive neuron-specific Galphai-interacting protein in neuropathic pain rat. Molecular pain 12. 10.1177/1744806916646380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tesmer JJ, Berman DM, Gilman AG, and Sprang SR (1997). Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell 89, 251–261. 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 39.Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, and Sprang SR (1994). Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412. 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Marcos M, Ghosh P, Ear J, and Farquhar MG (2010). A structural determinant that renders G alpha(i) sensitive to activation by GIV/girdin is required to promote cell migration. The Journal of biological chemistry 285, 12765–12777. 10.1074/jbc.M109.045161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leyme A, Marivin A, Casler J, Nguyen LT, and Garcia-Marcos M (2014). Different biochemical properties explain why two equivalent Galpha subunit mutants cause unrelated diseases. The Journal of biological chemistry 289, 21818–21827. 10.1074/jbc.M114.549790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aznar N, Midde KK, Dunkel Y, Lopez-Sanchez I, Pavlova Y, Marivin A, Barbazan J, Murray F, Nitsche U, Janssen KP, et al. (2015). Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. eLife 4, e07091. 10.7554/eLife.07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zielinski T, Kimple AJ, Hutsell SQ, Koeff MD, Siderovski DP, and Lowery RG (2009). Two Galpha(i1) rate-modifying mutations act in concert to allow receptor-independent, steady-state measurements of RGS protein activity. Journal of biomolecular screening 14, 1195–1206. 10.1177/1087057109347473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, and Sprang SR (1995). Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis. Science 270, 954–960. 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- 45.Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, and Harden TK (2010). Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 330, 974–980. 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnston CA, Lobanova ES, Shavkunov AS, Low J, Ramer JK, Blaesius R, Fredericks Z, Willard FS, Kuhlman B, Arshavsky VY, and Siderovski DP (2006). Minimal determinants for binding activated G alpha from the structure of a G alpha(i1)-peptide dimer. Biochemistry 45, 11390–11400. 10.1021/bi0613832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, and Tesmer JJ (2005). Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science 310, 1686–1690. 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Singer WD, Sternweis PC, and Sprang SR (2005). Structure of the p115RhoGEF rgRGS domain-Galpha13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nature structural & molecular biology 12, 191–197. 10.1038/nsmb888. [DOI] [PubMed] [Google Scholar]
- 49.Tesmer JJ, Sunahara RK, Gilman AG, and Sprang SR (1997). Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 278, 1907–1916. 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 50.Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, and Sigler PB (2001). Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature 409, 1071–1077. 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 51.de Opakua AI, Parag-Sharma K, DiGiacomo V, Merino N, Leyme A, Marivin A, Villate M, Nguyen LT, de la Cruz-Morcillo MA, Blanco-Canosa JB, et al. (2017). Molecular mechanism of Galphai activation by non-GPCR proteins with a Galpha-Binding and Activating motif. Nature communications 8, 15163. 10.1038/ncomms15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luebbers A, Zhou M, Eyles S, and Garcia-Marcos M (2023). Dissecting the molecular basis for the modulation of neurotransmitter GPCR signaling by GINIP. bioRxiv : the preprint server for biology 10.1101/2023.04.20.537566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sunahara RK, Dessauer CW, Whisnant RE, Kleuss C, and Gilman AG (1997). Interaction of Gsalpha with the cytosolic domains of mammalian adenylyl cyclase. The Journal of biological chemistry 272, 22265–22271. 10.1074/jbc.272.35.22265. [DOI] [PubMed] [Google Scholar]
- 54.Dessauer CW, Tesmer JJ, Sprang SR, and Gilman AG (1998). Identification of a Gialpha binding site on type V adenylyl cyclase. The Journal of biological chemistry 273, 25831–25839. 10.1074/jbc.273.40.25831. [DOI] [PubMed] [Google Scholar]
- 55.Hollins B, Kuravi S, Digby GJ, and Lambert NA (2009). The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cellular signalling 21, 1015–1021. 10.1016/j.cellsig.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, and Martemyanov KA (2015). Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Science signaling 8, ra123. 10.1126/scisignal.aab4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lambert NA, Johnston CA, Cappell SD, Kuravi S, Kimple AJ, Willard FS, and Siderovski DP (2010). Regulators of G-protein signaling accelerate GPCR signaling kinetics and govern sensitivity solely by accelerating GTPase activity. Proceedings of the National Academy of Sciences of the United States of America 107, 7066–7071. 10.1073/pnas.0912934107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, Gileadi C, Fedorov OY, Dowler EF, Higman VA, et al. (2008). Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proceedings of the National Academy of Sciences of the United States of America 105, 6457–6462. 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berman DM, Kozasa T, and Gilman AG (1996). The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. The Journal of biological chemistry 271, 27209–27212. 10.1074/jbc.271.44.27209. [DOI] [PubMed] [Google Scholar]
- 60.Kuramoto T, Voigt B, Nakanishi S, Kitada K, Nakamura T, Wakamatsu K, Yoshihara M, Suyama M, Uemura R, Tanaka M, et al. (2017). Identification of Candidate Genes for Generalized Tonic-Clonic Seizures in Noda Epileptic Rat. Behavior genetics 47, 609–619. 10.1007/s10519-017-9870-2. [DOI] [PubMed] [Google Scholar]
- 61.Goldberg EM, and Coulter DA (2013). Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nature reviews. Neuroscience 14, 337–349. 10.1038/nrn3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, and Toth M (2001). Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–1050. [DOI] [PubMed] [Google Scholar]
- 63.Maziarz M, Park JC, Leyme A, Marivin A, Garcia-Lopez A, Patel PP, and Garcia-Marcos M (2020). Revealing the Activity of Trimeric G-proteins in Live Cells with a Versatile Biosensor Design. Cell 182, 770–785 e716. 10.1016/j.cell.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luscher C, and Slesinger PA (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nature reviews. Neuroscience 11, 301–315. 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 6309–6314. 20026564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vong L, Ye C, Yang Z, Choi B, Chua S Jr., and Lowell BB (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiser O, Qian X, Ehlers M, Ja WW, Roberts RW, Reuveny E, Jan YN, and Jan LY (2006). Modulation of basal and receptor-induced GIRK potassium channel activity and neuronal excitability by the mammalian PINS homolog LGN. Neuron 50, 561–573. 10.1016/j.neuron.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 68.Masuho I, Balaji S, Muntean BS, Skamangas NK, Chavali S, Tesmer JJG, Babu MM, and Martemyanov KA (2020). A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell 183, 503–521 e519. 10.1016/j.cell.2020.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kolb P, Kenakin T, Alexander SPH, Bermudez M, Bohn LM, Breinholt CS, Bouvier M, Hill SJ, Kostenis E, Martemyanov KA, et al. (2022). Community guidelines for GPCR ligand bias: IUPHAR review 32. British journal of pharmacology 179, 3651–3674. 10.1111/bph.15811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kenakin T (2019). Biased Receptor Signaling in Drug Discovery. Pharmacological reviews 71, 267–315. 10.1124/pr.118.016790. [DOI] [PubMed] [Google Scholar]
- 71.Kenakin T (2018). Is the Quest for Signaling Bias Worth the Effort? Molecular pharmacology 93, 266–269. 10.1124/mol.117.111187. [DOI] [PubMed] [Google Scholar]
- 72.Anderson A, Masuho I, Marron Fernandez de Velasco E, Nakano A, Birnbaumer L, Martemyanov KA, and Wickman K (2020). GPCR-dependent biasing of GIRK channel signaling dynamics by RGS6 in mouse sinoatrial nodal cells. Proceedings of the National Academy of Sciences of the United States of America 117, 14522–14531. 10.1073/pnas.2001270117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Serikawa T, Kunisawa N, Shimizu S, Kato M, Alves Iha H, Kinboshi M, Nishikawa H, Shirakawa Y, Voigt B, Nakanishi S, et al. (2019). Increased seizure sensitivity, emotional defects and cognitive impairment in PHD finger protein 24 (Phf24)-null rats. Behavioural brain research 369, 111922. 10.1016/j.bbr.2019.111922. [DOI] [PubMed] [Google Scholar]
- 74.Hauser AS, Avet C, Normand C, Mancini A, Inoue A, Bouvier M, and Gloriam DE (2022). Common coupling map advances GPCR-G protein selectivity. eLife 11. 10.7554/eLife.74107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng H, Yuan Y, Williams MR, Roy AJ, Leipprandt JR, and Neubig RR (2022). Mice with monoallelic GNAO1 loss exhibit reduced inhibitory synaptic input to cerebellar Purkinje cells. Journal of neurophysiology 127, 607–622. 10.1152/jn.00720.2020. [DOI] [PubMed] [Google Scholar]
- 76.Bouchet CA, McPherson KB, Li MH, Traynor JR, and Ingram SL (2021). Mice Expressing Regulators of G protein Signaling-insensitive Galphao Define Roles of mu Opioid Receptor Galphao and Galphai Subunit Coupling in Inhibition of Presynaptic GABA Release. Molecular pharmacology 100, 217–223. 10.1124/molpharm.121.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vries LD, Elenko E, McCaffery JM, Fischer T, Hubler L, McQuistan T, Watson N, and Farquhar MG (1998). RGS-GAIP, a GTPase-activating Protein for GαiHeterotrimeric G Proteins, Is Located on Clathrin-coated Vesicles. 9, 1123–1134. 10.1091/mbc.9.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garcia-Marcos M, Ghosh P, and Farquhar MG (2009). GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proceedings of the National Academy of Sciences of the United States of America 106, 3178–3183. 10.1073/pnas.0900294106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia-Marcos M, Parag-Sharma K, Marivin A, Maziarz M, Luebbers A, and Nguyen LT (2020). Optogenetic activation of heterotrimeric G-proteins by LOV2GIVe, a rationally engineered modular protein. eLife 9. 10.7554/eLife.60155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mase Y, Yokogawa M, Osawa M, and Shimada I (2012). Backbone resonance assignments for G protein alpha(i3) subunit in the GTP-bound state. Biomolecular NMR assignments 6, 217–220. 10.1007/s12104-012-9361-6. [DOI] [PubMed] [Google Scholar]
- 81.Mumby SM, and Linder ME (1994). Myristoylation of G-protein alpha subunits. Methods in enzymology 237, 254–268. 10.1016/s0076-6879(94)37067-2. [DOI] [PubMed] [Google Scholar]
- 82.Hu Q, and Shokat KM (2018). Disease-Causing Mutations in the G Protein Galphas Subvert the Roles of GDP and GTP. Cell 173, 1254–1264 e1211. 10.1016/j.cell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leyme A, Marivin A, Maziarz M, DiGiacomo V, Papakonstantinou MP, Patel PP, Blanco-Canosa JB, Walawalkar IA, Rodriguez-Davila G, Dominguez I, and Garcia-Marcos M (2017). Specific inhibition of GPCR-independent G protein signaling by a rationally engineered protein. Proceedings of the National Academy of Sciences of the United States of America 114, E10319–E10328. 10.1073/pnas.1707992114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Natochin M, and Artemyev NO (1998). A single mutation Asp229 --> Ser confers upon Gs alpha the ability to interact with regulators of G protein signaling. Biochemistry 37, 13776–13780. 10.1021/bi981155a. [DOI] [PubMed] [Google Scholar]
- 85.Wedegaertner PB, Chu DH, Wilson PT, Levis MJ, and Bourne HR (1993). Palmitoylation is required for signaling functions and membrane attachment of Gq alpha and Gs alpha. The Journal of biological chemistry 268, 25001–25008. [PubMed] [Google Scholar]
- 86.Meigs TE, Juneja J, DeMarco CT, Stemmle LN, Kaplan DD, and Casey PJ (2005). Selective uncoupling of G alpha 12 from Rho-mediated signaling. The Journal of biological chemistry 280, 18049–18055. 10.1074/jbc.M500445200. [DOI] [PubMed] [Google Scholar]
- 87.Celver JP, Lowe J, Kovoor A, Gurevich VV, and Chavkin C (2001). Threonine 180 is required for G-protein-coupled receptor kinase 3- and beta-arrestin 2-mediated desensitization of the mu-opioid receptor in Xenopus oocytes. The Journal of biological chemistry 276, 4894–4900. 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- 88.Oner SS, An N, Vural A, Breton B, Bouvier M, Blumer JB, and Lanier SM (2010). Regulation of the AGS3.Galphai signaling complex by a seven-transmembrane span receptor. The Journal of biological chemistry 285, 33949–33958. 10.1074/jbc.M110.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boyer SB, Clancy SM, Terunuma M, Revilla-Sanchez R, Thomas SM, Moss SJ, and Slesinger PA (2009). Direct interaction of GABAB receptors with M2 muscarinic receptors enhances muscarinic signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 15796–15809. 10.1523/JNEUROSCI.4103-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Masuho I, Xie K, and Martemyanov KA (2013). Macromolecular composition dictates receptor and G protein selectivity of regulator of G protein signaling (RGS) 7 and 9–2 protein complexes in living cells. The Journal of biological chemistry 288, 25129–25142. 10.1074/jbc.M113.462283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martemyanov KA, Yoo PJ, Skiba NP, and Arshavsky VY (2005). R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. The Journal of biological chemistry 280, 5133–5136. 10.1074/jbc.C400596200. [DOI] [PubMed] [Google Scholar]
- 92.De Vries DD, Went LN, Bruyn GW, Scholte HR, Hofstra RM, Bolhuis PA, and van Oost BA (1996). Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia. American journal of human genetics 58, 703–711. [PMC free article] [PubMed] [Google Scholar]
- 93.Zhan X, Gimenez LE, Gurevich VV, and Spiller BW (2011). Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. Journal of molecular biology 406, 467–478. 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuravi S, Lan TH, Barik A, and Lambert NA (2010). Third-party bioluminescence resonance energy transfer indicates constitutive association of membrane proteins: application to class a g-protein-coupled receptors and g-proteins. Biophysical journal 98, 2391–2399. 10.1016/j.bpj.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoo T, Cho H, Park H, Lee J, and Kim E (2019). Shank3 Exons 14–16 Deletion in Glutamatergic Neurons Leads to Social and Repetitive Behavioral Deficits Associated With Increased Cortical Layer 2/3 Neuronal Excitability. Frontiers in cellular neuroscience 13, 458. 10.3389/fncel.2019.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weaver DR, van der Vinne V, Giannaris EL, Vajtay TJ, Holloway KL, and Anaclet C (2018). Functionally Complete Excision of Conditional Alleles in the Mouse Suprachiasmatic Nucleus by Vgat-ires-Cre. Journal of biological rhythms 33, 179–191. 10.1177/0748730418757006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaech S, and Banker G (2006). Culturing hippocampal neurons. Nature protocols 1, 2406–2415. 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 98.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, and Donnelly MI (2002). A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein expression and purification 25, 8–15. 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 99.Cabrita LD, Dai W, and Bottomley SP (2006). A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC biotechnology 6, 12. 10.1186/1472-6750-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Marivin A, Maziarz M, Zhao J, DiGiacomo V, Olmos Calvo I, Mann EA, Ear J, Blanco-Canosa JB, Ross EM, Ghosh P, and Garcia-Marcos M (2020). DAPLE protein inhibits nucleotide exchange on Gα(s) and Gα(q) via the same motif that activates Gαi. The Journal of biological chemistry 295, 2270–2284. 10.1074/jbc.RA119.011648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garcia-Marcos M, Jung BH, Ear J, Cabrera B, Carethers JM, and Ghosh P (2011). Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 25, 590–599. 10.1096/fj.10-167304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ghosh P, Garcia-Marcos M, Bornheimer SJ, and Farquhar MG (2008). Activation of Galphai3 triggers cell migration via regulation of GIV. The Journal of cell biology 182, 381–393. 10.1083/jcb.200712066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beas AO, Taupin V, Teodorof C, Nguyen LT, Garcia-Marcos M, and Farquhar MG (2012). Gαs promotes EEA1 endosome maturation and shuts down proliferative signaling through interaction with GIV (Girdin). Molecular biology of the cell 23, 4623–4634. 10.1091/mbc.E12-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oner SS, An N, Vural A, Breton B, Bouvier M, Blumer JB, and Lanier SM (2010). Regulation of the AGS3·G{alpha}i signaling complex by a seven-transmembrane span receptor. The Journal of biological chemistry 285, 33949–33958. 10.1074/jbc.M110.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Vries L, Elenko E, Hubler L, Jones TL, and Farquhar MG (1996). GAIP is membrane-anchored by palmitoylation and interacts with the activated (GTP-bound) form of G alpha i subunits. Proceedings of the National Academy of Sciences of the United States of America 93, 15203–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sakurai K, Zhao S, Takatoh J, Rodriguez E, Lu J, Leavitt AD, Fu M, Han BX, and Wang F (2016). Capturing and Manipulating Activated Neuronal Ensembles with CANE Delineates a Hypothalamic Social-Fear Circuit. Neuron 92, 739–753. 10.1016/j.neuron.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leyme A, Marivin A, Casler J, Nguyen LT, and Garcia-Marcos M (2014). Different biochemical properties explain why two equivalent Gα subunit mutants cause unrelated diseases. The Journal of biological chemistry 289, 21818–21827. 10.1074/jbc.M114.549790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Garcia-Marcos M, Ear J, Farquhar MG, and Ghosh P (2011). A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. 22, 673–686. 10.1091/mbc.e10-08-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Longo PA, Kavran JM, Kim MS, and Leahy DJ (2013). Transient mammalian cell transfection with polyethylenimine (PEI). Methods in enzymology 529, 227–240. 10.1016/B978-0-12-418687-3.00018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is provided in the manuscript and original images of results presented in the figures have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the Key Resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.