

Abstract
Proteins are expressed from genes via sequential biological processes of transcription, mRNA processing, export and translation, and play their roles in maintaining cellular functions via interactions with proteins, DNAs or RNAs. Thus, it is important to study the protein interactions during biological processes in living cells towards understanding their mechanisms-of-action in real time. Methodologies have been developed over the years to study protein interactions in vivo. One state-of-the-art approach is formaldehyde crosslinking-based immuno- or chemi-precipitation to analyze selective as well as genome/proteome-wide interactions in living cells. It is a popular and widely used methodology for cellular analysis of the protein-protein and protein-nucleic acid interactions. Here, we describe this approach to analyze protein-protein/nucleic acid interactions in vivo.
Keywords: Formaldehyde-based crosslinking, chromatin immunoprecipitation, RNA immunoprecipitation, immunoprecipitation, and chromatin fractionation
A. Introduction:
Genes are expressed to proteins that function in various cellular processes (1). Proteins do not generally function alone, but rather deliver their roles at the cellular/organismal levels via interactions with DNAs, RNAs or proteins (2, 3). Therefore, to decipher functional mechanisms of proteins, it is important to analyze protein interactions. Further, such interaction analysis could help to understand disease pathogenesis and/or decipher therapeutic targets, since altered protein interactions could lead to cellular/organismal dysfunctions (and hence cellular pathologies/diseases). There are a variety of methodologies available to study protein interactions in vitro (e.g., 4–16). However, protein interaction analysis in vitro may not be functionally relevant in vivo, as these ex vivo studies are generally performed in the purified systems (or in isolation) in the absence of many other factors independently of the biological processes, but rather under artificial conditions. Further, the in vitro interaction results could vary from study to study, depending on the experimental variables such as salt and detergent concentrations. Therefore, the in vitro results need to be validated in vivo with associated functions. Thus, methodologies have been developed over the years to study protein interactions in the living cells (e.g., 17–34). One such procedure is the formaldehyde-based in vivo crosslinking during the biological processes in the growing cells in real time, followed by immuno- or chemi-precipitation of DNAs, RNAs or proteins interacting with the protein of interest. It is a state-of-the-art methodology to study selective as well as global (genome/proteome-wide) protein interactions during the biological processes in living cells. We describe below this formaldehyde crosslinking-based methodology to analyze protein interactions in vivo.
B. Formaldehyde crosslinking-based analysis of protein-DNA interactions in vivo:
We have described below formaldehyde-based in vivo crosslinking and ChIP (Chromatin immunoprecipitation) methodology to analyze protein-DNA interactions. In this methodology, the growing cells are treated with ~1% formaldehyde for 15 min. Being a small molecule, formaldehyde easily penetrates into the cell, and produces covalent bonds between the interacting proteins and nucleic acids, as schematically shown in Figure 1. The chemically reactive groups for formaldehyde-mediated crosslinking are the primary and secondary amino groups (−NH2 and >NH, respectively) of the proteins and nucleic acids (35–37). Thus, bases/nucleotides of the nucleic acids, and amino acids such as lysine, histidine, tryptophan, asparagine, glutamine, arginine of the proteins are reactive to formaldehyde-mediated crosslinking (35–37; Figure 1). Further, the thiol (−SH) group of cysteine contributes to the formaldehyde-mediated crosslinking (35–37). Furthermore, tyrosine can also participate in the formaldehyde-mediated crosslinking (35–37). However, the reactivities of these amino acids depend on the concentration and duration of formaldehyde used. Predominant formaldehyde-mediated crosslinking occurs between lysine and dG (Deoxy guanosine) via the formation of aminal linkage, while the second most prominent crosslinking takes place between cysteine and dG through the formation of hemiaminal thioether linkage (36). Formaldehyde being a small molecule crosslinker, the reactive groups of the interacting proteins and nucleic acids need to be in close proximity (2.3–2.7 A°) for crosslinking (35).
Figure 1:
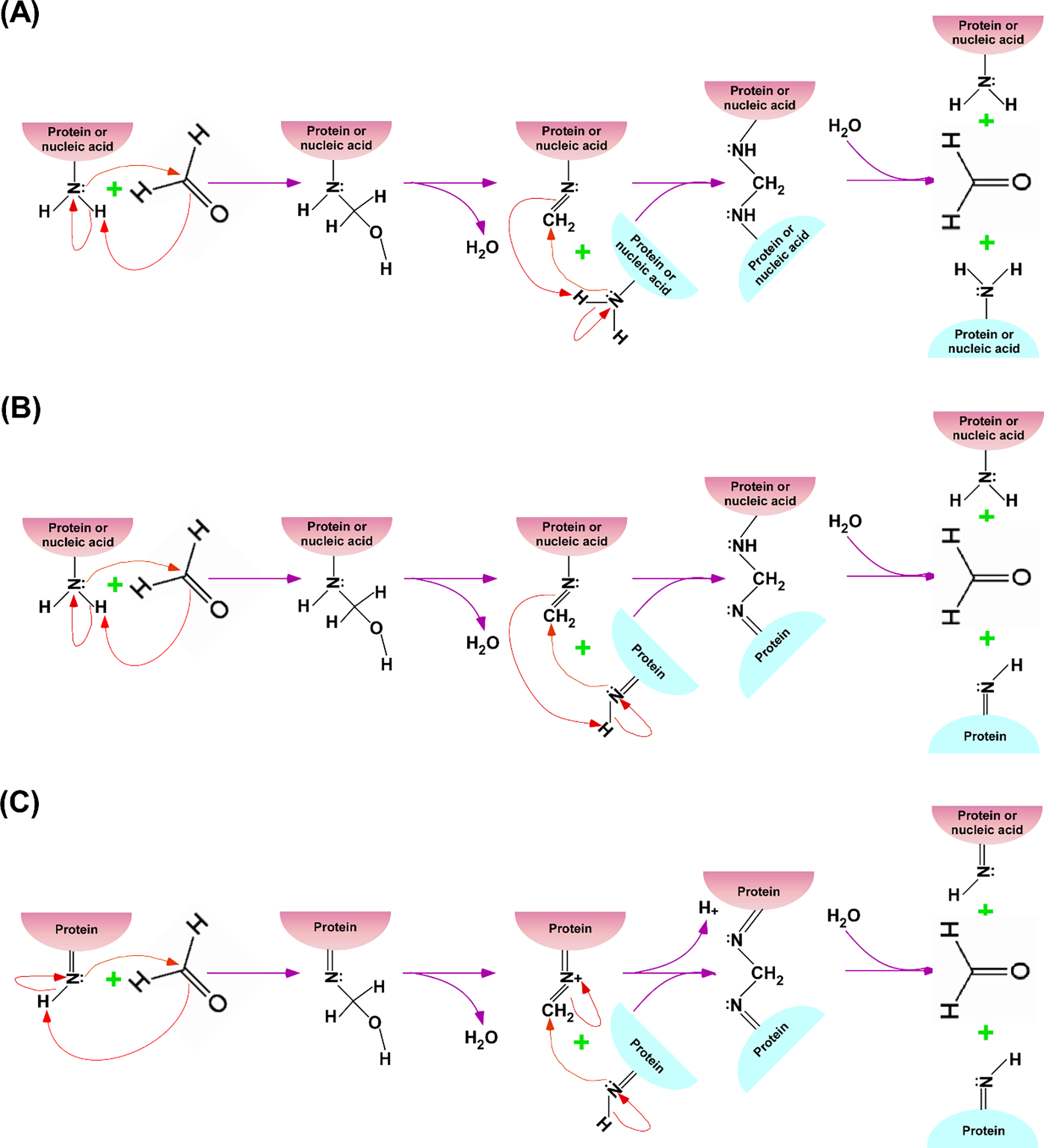
Schematic representations of the proposed mechanisms for formaldehyde (HCHO)-mediated crosslinking among primary and secondary amino groups (-NH2 and >NH, respectively) of the interacting proteins and nucleic acids (35–37). (A) Formaldehyde-based crosslinking between primary amino groups of the interacting proteins or between protein and nucleic acid. The primary amino group of protein/nucleic acid reacts with formaldehyde to form methylol intermediate which loses water (H2O) to produce Schiff base that reacts with nearby primary amino group of the interacting protein/nucleic acid to covalently crosslink protein-protein or protein-nucleic acid interaction. Formaldehyde-mediated covalent crosslinking can be reversed by hydrolysis (which can be enhanced at high temperature), as shown in the last step. (B) Formaldehyde-based crosslinking between primary and secondary amino groups of the interacting proteins and nucleic acids. The primary amino group of protein/nucleic acid reacts with formaldehyde to form methylol intermediate which loses water to produce Schiff base that reacts with nearby secondary amino group of the interacting protein to crosslink protein-protein or protein-nucleic acid interaction. Formaldehyde-mediated crosslinking can be reversed by hydrolysis, as shown in the last step. (C) Formaldehyde-based crosslinking between secondary amino groups of the interacting proteins. The secondary amino group of a protein reacts with formaldehyde to form methylol intermediate which loses water to produce Schiff base that reacts with nearby secondary amino group of the interacting protein to crosslink protein-protein interaction. Formaldehyde-mediated crosslinking can be reversed by hydrolysis, as shown in the last step.
When a protein interacts with nucleic acid or other protein during a biological process, the addition of formaldehyde in the growing cells can fix/freeze these interactions in living cells via crosslinking, provided the formaldehyde-reactive groups of the interacting protein and nucleic acid are present in the close proximity at the interaction surface (Figure 1). Importantly, numerous protein interactions have been successfully fixed by formaldehyde in living cells, supporting its general applicability for protein interaction analysis in vivo. These fixed protein interactions in vivo are detected ex vivo by preparing sonicated WCE (Whole cell extract) for subsequent immunoprecipitation/purification for selective as well as genome/proteome-wide interaction analyses, as schematically shown in Figure 2. We described below formaldehyde crosslinking-based analysis of protein-DNA interactions in vivo.
Figure 2:
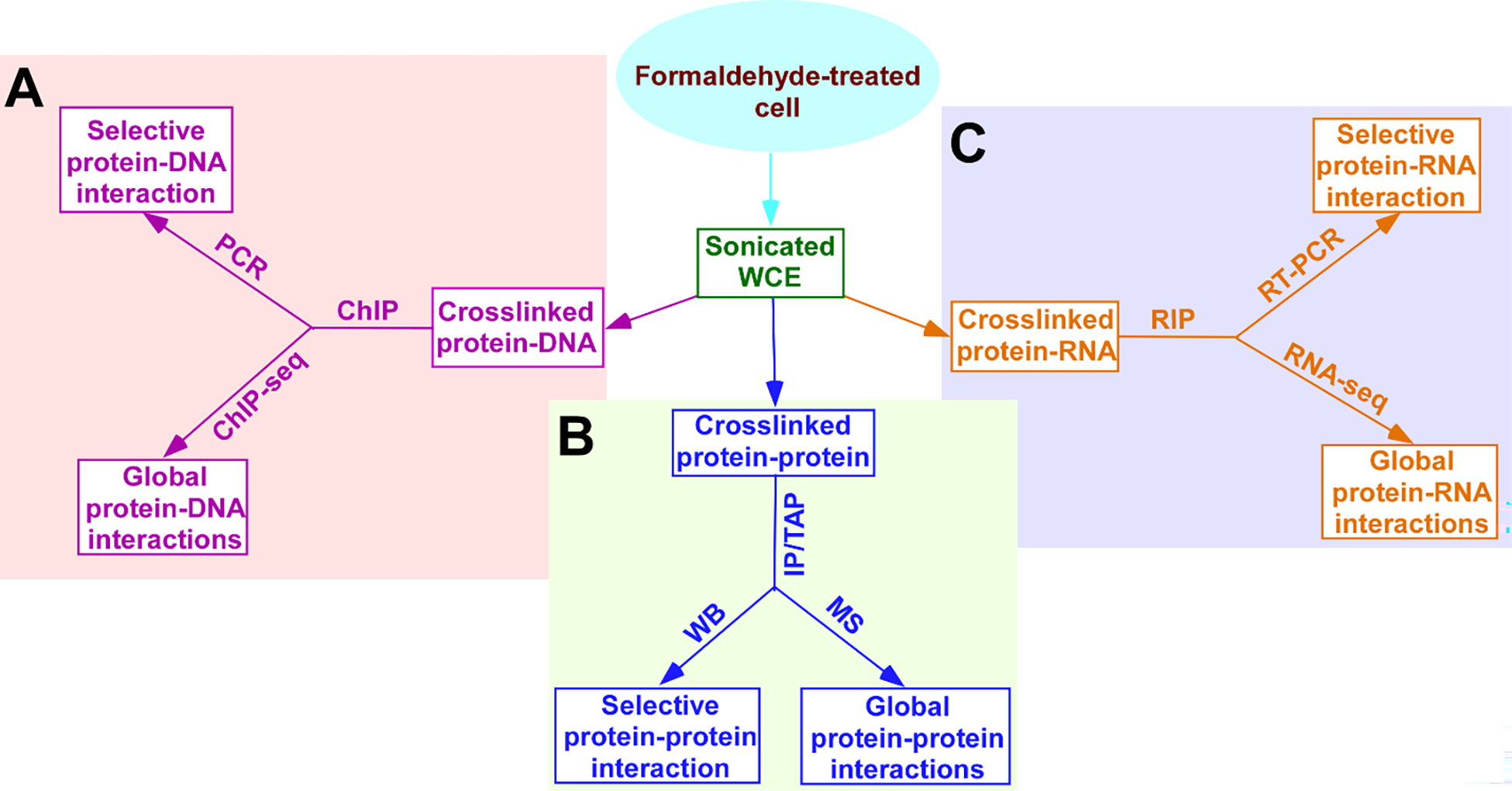
Schematic diagrams showing the strategies/approaches for formaldehyde crosslinking-mediated selective as well as global protein-protein, protein-DNA and protein-RNA interactions in living cells. Growing cells are treated with formaldehyde to crosslink proteins with interacting nucleic acids (DNAs and RNAs) and proteins during biological processes in living cells in real time, and subsequently, cells are harvested, lysed and sonicated for analysis of the protein-protein/nucleic acid interactions. The sonicated WCE (Whole cell extract) contains in vivo crosslinked protein-DNA, protein-protein and protein-RNA interactions, which can be analyzed by ChIP (Chromatin immunoprecipitation), IP/TAP (Immunoprecipitation/Tandem affinity purification) and RIP (RNA immunoprecipitation), respectively (A, B and C). (A) Protein-DNA interaction in vivo. Following ChIP, isolated DNAs can be sequenced (ChIP-seq, ChIP-sequencing) to analyze genome-wide interactions/locations of a protein of interest. The isolated DNAs can also be analyzed for selective protein-DNA interaction by PCR (Polymerase chain reaction) using the specific primer pair targeted to a gene/DNA of interest. (B) Protein-protein interaction in vivo. Following IP/TAP, isolated proteins can be analyzed by MS (Mass spectrometry) to decipher proteome-wide interactions of a protein of interest. The isolated proteins can also be analyzed for selective protein-protein interaction by WB (Western blotting) using a specific antibody against a protein of interest. (C) Protein-RNA interaction in vivo. Following RIP, isolated RNAs can be sequenced (RNA-seq) to analyze global RNA interactions of a protein of interest. The isolated RNAs can also be analyzed for selective protein-RNA interaction by RT-PCR (Reverse transcriptase - PCR).
B.1. Standard ChIP analysis for protein-DNA interactions in vivo.
The efficiency of protein-DNA crosslinking (and hence ChIP signal) depends on the strengths (or association constant/on-rate) of the protein-DNA interactions as well as the close proximity of the chemical moieties of the interacting proteins and DNAs at the interaction surfaces for formaldehyde-mediated crosslinking. If a protein interacts with DNA directly and strongly with chemical moieties located at the close vicinity for formaldehyde-mediated crosslinking, it would be crosslinked well with DNA in the growing cells with formaldehyde treatment, which can be detected ex vivo following sonicated WCE preparation, immunoprecipitation of DNA using the antibody against the interacting protein of interest, and subsequent PCR (Polymerase chain reaction) analysis. We describe below the detailed protocol for detection of the protein-DNA interactions in living yeast cells using the standard ChIP assay. Similar approach can be adopted in mammalian cells.
B.1.1. Cell growth, crosslinking and harvesting.
Inoculate yeast strain (from freshly streaked yeast strain on solid growth medium in the petri dish) in 55 ml of YPD (Yeast extract, peptone plus 2% dextrose), and let it grow overnight at 30 °C in an incubator shaker with a speed of 190 rpm (Revolutions per minute) up to an OD600 (Optical density at 600 nm) of 1.
Add 1.25 ml of 37% formaldehyde solution (v/v; Fisher) to the growing yeast culture (50 ml), and put on the working bench for 15 min with occasional shaking by hand (in every 2–3 min or so for about 30 s).
After 15 min treatment with formaldehyde, add 2.5 ml of 2.5 M glycine to the yeast cell culture and keep on the working bench for 5 min with occasional shaking by hand (in every 2 min or so for about 30 s).
Harvest yeast cells by centrifugation in Sorvall LegandRT for 5 min at 4 °C with a speed of 3500 rpm.
Discard supernatant and suspend the cells in 2 ml ice-cold TBS [10 mM Tris-HCl buffer (pH 8.0) containing 1 mM EDTA and 150 mM NaCl].
Transfer yeast cell suspension to two 1.5 ml Eppendorf tubes with equal volume in each.
Spin down yeast cells in the microcentrifuge machine (Eppendorf centrifuge 5415C) with a speed of 13000 rpm for 2 min at 4 °C.
Discard supernatant and resuspend cell pellet in 2 ml ice-cold TBS.
Spin down yeast cells in the microcentrifuge machine with a speed of 13000 rpm for 2 min at 4 °C.
Discard supernatant and store the cell pellet in the −80 °C freezer or continue to prepare WCE in the next step.
B.1.2. WCE preparation and sonication.
Take out cell pellet from the −80 °C freezer and put in ice for thawing (if stored in the −80 °C freezer in the above step 10 of B.1.1. Otherwise, proceed to the next step).
After thawing, suspend the cells in 200 μl ice-cold FA lysis buffer containing protease inhibitors. FA lysis buffer: 50 mM HEPES, pH 7.5 with 140 mM NaCl, 1 mM EDTA, 1% Triton X-100 and 0.1% Na-deoxycholate. Protease inhibitors: 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, 25 μg/ml tosyl lysyl chloromethylketone (TLCK), 50 μg/ml tosyl phenylalanyl chloromethylketone (TPCK), 10 μg/ml aprotinin, 20 μg/ml antipain, 1 μg/ml leupeptin and 1 μg/ml pepstatin. PMSF is added just before vortexing in the step 4 below.
Add 200 μl volume-equivalent of spherical-shaped acid-washed glass beads (425–600 μm, Sigma).
Vortex the cell suspension with glass beads at a maximum speed for 30 min using the TOMY micro tube mixer MT-360 (Peninsula Laboratories, Inc.) at 4 °C.
Punch a hole at the bottom of the Eppendorf tube containing cell lysate with a 21G11⧸2 needle in an inverted position and put onto a new Eppendorf tube to collect the lysate by a brief centrifugation (~20s at 6000 rpm) in the microcentrifuge machine at room temperature.
Combine the lysates of the two Eppendorf tubes to get the larger volume of cell suspension (>400 μl) needed for sonication.
Sonicate WCE using a microtip probe (with an amplitude of 40) five times for 10 s each in a Misonix sonicator (XL4000; Misonix Inc.). To avoid heating the WCE during sonication, dip the lower half of the WCE-containing Eppendorf tube quickly in the ethanol-dry ice mixture before sonication, and put the WCE in ice for 2 min between pulses. For smaller DNA fragments and better resolution of the protein-DNA interactions, sonication cycle can be increased to 7.
Centrifuge the sonicated lysate in the microcentrifuge machine with a speed of 13000 rpm for 5 min at 4° C.
Collect supernatant in a new Eppendorf tube, which will be used for immunoprecipitation in the next step. Keep 5 μl lysate in the −80 °C freezer for the input DNA control to be used later in the following step 3 in B.1.5.
B.1.3. Immunoprecipitation.
Put 100 μl lysate in a new Eppendorf tube and add 300 μl FA lysis buffer with protease inhibitors.
Add the antibody against the protein of interest to the above lysate, and incubate with rotation in the rotisserie rotator (Barnstead/Thermolyne Labquake) for 3 h at 4 °C.
Add 50 μl of protein A (Invitrogen) or protein A/G plus agarose (Santa Cruz Biotechnology, Inc.) beads to the above lysate-antibody mixture, and then incubate for 1 h with rotation in the rotisserie rotator at 4 °C. Other beads can also be used depending on the nature of the primary antibody.
Centrifuge above lysate with antibody and beads at room temperature in the microcentrifuge machine with a speed of 3000 rpm for 5 min.
Remove supernatant carefully including last traces of supernatant (using a 1-ml syringe affixed with a thin needle), and keep the beads bound to protein-DNA complex for washing in the next step.
B.1.4. Washing beads after immunoprecipitation.
Resuspend the beads after immunoprecipitation with 1 ml of FA lysis buffer (without protease inhibitors) and put in a rotisserie rotator for 5 min at the room temperature.
Centrifuge above suspension at room temperature in the microcentrifuge machine with a speed of 3000 rpm for 5 min.
Remove the supernatant carefully (Wash 1: Step 1–3).
Suspend the beads with 1 ml of FA lysis buffer containing 1 M NaCl and put in a rotisserie rotator for 5 min at the room temperature.
Centrifuge above suspension at the room temperature in the microcentrifuge machine with a speed of 3000 rpm for 5 min.
Remove the supernatant carefully (Wash 2: Step 4–6).
Repeat step 4–6 (Wash 3).
Suspend the beads with 1 ml of FA-W3 buffer (10 mM Tris–HCl, pH 8.0 with 0.25 M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate and 1 mM EDTA) and put in a rotisserie rotator for 5 min at the room temperature.
Centrifuge above suspension at the room temperature in the microcentrifuge machine with a speed of 3000 rpm for 5 min.
Remove the supernatant carefully (Wash 4: Step 8–10).
Suspend the beads with 1 ml of TE (10 mM Tris-HCl and 1 mM EDTA, pH 8.0) and put in a rotisserie rotator for 5 min at the room temperature.
Centrifuge above suspension at the room temperature in the microcentrifuge machine with a speed of 3000 rpm for 5 min.
Remove the supernatant carefully including last traces of TE (using a 1-ml syringe affixed with a thin needle) (Wash 5: Step 11–13).
B.1.5. Proteinase K treatment, reverse crosslinking and DNA precipitation.
Suspend washed beads in 200 μl proteinase K buffer (100 mM Tris–HCl, pH 7.5 with 12.5 mM EDTA, 150 mM NaCl and 1% SDS).
Transfer suspended beads to a new Eppendorf tube that contains 3 μl of 10 mg/ml proteinase K (Sigma), and then incubate in the 37 °C water bath for 5 h.
Prepare input DNA by adding 5 μl of the sonicated WCE from the step 9 of B.1.2 to 180 μl proteinase K buffer and 10 μl of 10 mg/ml proteinase K, and subsequently incubating at 55 °C for 5 h.
Reverse formaldehyde-mediated crosslinking by incubating these samples of above steps 2 and 3 (i.e., input and immunoprecipitated DNA samples) at 65 °C for 6 h.
Vortex the samples from the above step 4 and spin down the beads in the microcentrifuge with a speed of 13000 rpm for 2 min at the room temperature.
Carefully collect the supernatant of the immunoprecipitated DNA samples to the new Eppendorf tubes, and leave the input DNA samples in the same Eppendorf tubes.
Add 200 μl phenol:chloroform:isoamyl alcohol (25:24:1; Fisher Scientific) to the above supernatants and input DNA samples of step 6, and vortex for 30 s at a maximum speed in the vortexer (Fisher Scientific).
Centrifuge above mixtures at the room temperature in the microcentrifuge machine with a speed of 13000 rpm for 10 min.
Collect the upper aqueous phase of each sample in the new Eppendorf tube.
To optimize DNA extraction, add 100 μl of 50 mM Tris–HCl (pH 8.0) containing 0.3 M sodium acetate to the lower phenol phase of the step 9 (after collecting aqueous phase), and vortex for 30 s at a maximum speed in the vortexer at the room temperature. Centrifuge as in step 8. Collect the upper aqueous phase carefully, and combine with the collected aqueous phase of the step 9.
Add 10 μg glycogen (Molecular biology grade; Thermo Scientific) and ~700 μl (2.5 volume equivalent of aqueous phase) ice-cold absolute ethanol (Molecular Biology grade; Fisher) to the above collected aqueous phase, and put in the −80 °C freezer for 1 h.
Spin down precipitate in the microcentrifuge machine with a speed of 13000 rpm for 15 min at 4 °C.
Discard supernatant carefully. Add 1 ml of 70% ethanol at the room temperature to the precipitate and rotate in the rotisserie rotator for 5 min at the room temperature.
Spin down precipitate in the microcentrifuge machine with a speed of 13000 rpm for 5 min at the room temperature.
Remove supernatant carefully by leaving about 50 μl of 70% ethanol.
Dry the precipitate at 65 °C in the Eppendorf vacufuge™.
Dissolve the immunoprecipitated and input DNA samples in 20 and 100 μl TE (pH 8.0), respectively, for about 10 min at the room temperature, vortex at a maximum speed, and spin down in the microcentrifuge machine at 13000 rpm for 1 min at the room temperature. Repeat this step one more time, and then store these samples in the −20 °C/−80 °C freezer.
B.1.6. PCR analysis.
Above immunoprecipitated and input DNAs following proteinase K treatment and reverse crosslinking can be analyzed at the known target DNA/gene in comparison to the non-specific DNA and/or antibody by radioactive PCR using the primer pairs for the specific and non-specific DNA sequences or genes, and subsequent gel electrophoresis followed by autoradiography, as described below. As an alternative to the radioactive PCR and gel electrophoresis, real time PCR or qPCR (Quantitative PCR) can also be used for analysis of the association of the protein of interest with the target DNA/gene.
Use 1 μl each of the immunoprecipitated and input DNA samples for PCR analysis in a total reaction volume of 25 μl that contains 25 pmol primer (each), 0.2 mM each of dCTP, dGTP, dATP and dTTP, and 0.25 μl [α32-P]dATP (10 mCi/ml). PCR is generally set in a strip of 8 PCR tubes. Input and immunoprecipitated DNAs are serially diluted to access the linear range of the PCR amplifications. 1 μl input DNA generates PCR signal within the linear range (and thus below PCR saturation), using the PCR program, as described below in the step 2. Immunoprecipitated DNA samples are generally diluted to match with the intensity of the PCR signal of the input DNA sample to avoid PCR saturation of the immunoprecipitated DNA sample.
-
Perform PCR amplifications under following conditions.
94 °C for 2 min.
-
23 cycles of the following three steps
Melting: 94 °C for 30 s.
Annealing: 50 °C (typically 5 °C below the primer melting temperature) for 30 s.
Synthesis: 72 °C for 1 min.
72 °C for 7 min.
Hold at 10 °C.
To analyze the PCR products by native DNA gel electrophoresis (6% polyacrylamide) using a mixture of 8 μl of the PCR reaction products and 4 μl DNA-loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol and 20% glycerol).
After electrophoresis, transfer the gel onto the Whatman paper and cover by saran wrap. Cut and remove the lower part (below the dye front) of the gel.
Dry the gel in the gel dryer (Fisher Scientific). Put two layers of Whatman paper below the gel in gel dryer.
Put the dried gel between two amplified screens in the cassette, and keep in the −80 °C/−20 °C freezer prior to the development using the X-ray film developer. Phosphorimager can also be used as an alternative to this step.
Quantitate the immunoprecipitated and input DNA signals using NIH image 1.62 (or by Phosphorimager analysis, if phosphorimager is used in the above step). For NIH image analysis, use lighter exposure of the X-ray films to avoid signal saturation. Results are expressed as the ratio of the immunoprecipitated DNA to the input DNA signals.
B.1.7. ChIP-seq (Sequencing) analysis.
Above target specific immunoprecipitated DNA analysis in B.1.6 does not provide information regarding the protein-DNA interactions genome-wide in vivo. The ChIP-seq methodology has been developed to analyze immunoprecipitated DNA fragments genome-wide by sequencing the immunoprecipitated and input DNAs and subsequently overlapping/aligning with the genome to decipher genome-wide location of the protein of interest (or genome-wide protein-DNA interactions) (38), as described below.
Measure the concentrations of above immunoprecipitated and input DNA samples, using nanodrop (Thermo Scientific).
Prepare libraries using 7 to 14 ng of immunoprecipitated and input DNA samples.
Sequence DNA libraries using a NovaSeq S4 (LL) 300 cycle sequencing run with 2 × 150 reads, targeting 20 million reads per ChIP-seq library.
Trim the sequences to remove adapters using Cutadapt (parameter -m 20).
Align to the Saccharomyces cerevisiae genome (SacCer3) using Bowtie2 (parameters -X 1000–very-sensitive –no-mix –no-unal). Use SAMtools to sort, index, and remove PCR duplicates from the bam files. The number of paired-end reads for ChIP-seq becomes ~49.0 million after alignment. Analyze the ChIP-seq bam files, using bamR (https://github.com/rchereji/bamR).
B.2. ChIP analysis of weak and/or indirect protein-DNA interactions in vivo.
When a protein interacts weakly with DNA (i.e., high dissociation constant/off-rate or less residence time) and/or has a relatively a smaller number of formaldehyde-reactive amino acids at the close proximity to the DNA bases for crosslinking, the ChIP signal following the above protocol in B.1 may not be observed. Further, a protein of interest may contact DNA via other proteins. Like protein-DNA crosslinking, protein-protein interactions also get crosslinked by formaldehyde when the protein interaction surface contain formaldehyde-reactive groups at the close proximity for crosslinking, as described above (Figure 1). The efficiency of the crosslinking of a protein of interest with DNA decreases with the increase in the number of the bridging proteins between them. Under these situations, one can detect the ChIP signal by increasing the crosslinking time from 15 min to 25 min (that would increase crosslinking) and/or increasing the volume of sonicated WCE from 100 μl to 400 μl (which would increase the amount of precipitate) with appropriate controls such as non-specific DNA (an inactive region within chromosome V), non-specific antibody (e.g., an anti-HA antibody when anti-Myc antibody is used as a positive/experimental antibody) and non-specific gene (or gene location) target, as done in our studies (39–46). In such analysis, all the steps would remain the same as in B.1 except the cell volume, crosslinking time and/or volume of the sonicated WCE to be used for immunoprecipitation, as described below. Concentration for formaldehyde could also be increased with appropriate controls. Moreover, one can also use protein-protein crosslinker [e.g., Dimethyl adipimidate (DMA), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), Disuccinimidyl glutarate (DSG), and Dimethyl pimelimidate (DMP)] in addition to formaldehyde to enhance the ChIP signal, as described previously (47, 48).
Inoculate yeast strain in 105 ml of YPD, and let it grow at 30 °C in an incubator shaker with a speed of 190 rpm up to an OD600 of 1.
Add 2.5 ml of 37% formaldehyde solution to the 100 ml growing yeast culture for 15 min (or 25 min if less or no ChIP signal is observed with 15-min formaldehyde treatment).
After formaldehyde treatment, add 5 ml of 2.5 M glycine to the yeast cell culture and keep on the working bench for 5 min with occasional shaking by hand. Subsequently, harvest cells by centrifugation in Sorvall LegandRT for 5 min at 4 °C with a speed of 3500 rpm.
Discard supernatant and suspend the cells in 4 ml ice-cold TBS. Transfer yeast cell suspension to four 1.5 ml Eppendorf tubes with equal volume in each.
Spin down cells in the microcentrifuge machine with a speed of 13000 rpm for 2 min at 4 °C.
Discard supernatant and resuspend cell pellet in 1 ml ice-cold TBS in each Eppendorf tube.
Spin down cells in the microcentrifuge machine with a speed of 13000 rpm for 2 min at 4 °C.
Discard supernatant and cell pellet can be stored in the −80 °C freezer or continued to the next step.
Resuspend cells in 200 μl ice-cold FA lysis buffer containing protease inhibitors (Add PMSF just before vortexing in the next step). Add 200 μl volume-equivalent of spherical-shaped acid-washed glass beads.
Vortex the cell suspension with glass beads at a maximum speed for 30 min in the TOMY micro tube mixer MT-360 at 4 °C. At the end, punch a hole at the bottom of the Eppendorf tube containing cell lysate with a 21G11⧸2 needle in an inverted position and collect the lysate in the new Eppendorf tube by centrifugation.
Combine the lysates of the two Eppendorf tubes to get the larger volume of cell suspension (>400 μl) needed for sonication. Sonicate WCE using a microtip probe (with an amplitude of 40) for seven times for 10 s each in a Misonix sonicator.
Centrifuge the sonicated lysate in the microcentrifuge machine with a speed of 13000 rpm for 5 min at 4° C. Carefully collect sonicated WCE for immunoprecipitation. Keep 5 μl lysate in the −80 °C freezer for the input DNA control to be used later in the proteinase K treatment step.
Put 400 μl lysate in each of the two new Eppendorf tubes and add 10 μl (2 μg) anti-Myc antibody (Santa Cruz Biotechnology, Inc.) against Myc-tagged protein of interest to the lysate in the Eppendorf tube. Add 10 μl (2 μg) anti-HA antibody (Santa Cruz Biotechnology, Inc.) to the lysate in other Eppendorf tube as a non-specific antibody control. Incubate these Eppendorf tubes with rotation in the rotisserie rotator for 3 h at 4 °C.
Add 100 μl of protein A/G plus agarose beads to the above lysate-antibody mixture, and then incubate for 1 h with rotation in the rotisserie rotator at 4 °C.
Centrifuge above lysate with antibody and beads at room temperature in the microcentrifuge machine with a speed of 3000 rpm for 5 min.
Remove the supernatant carefully, and wash the beads bound to protein-DNA complex following the steps as described above in B.1.4. Subsequently, treat the washed beads with proteinase K followed by reverse crosslinking and DNA precipitation steps, as described above in B.1.5.
Dissolve the immunoprecipitated and input DNA samples in 10 and 100 μl TE (pH 8.0), respectively.
Use 1 μl each of input and immunoprecipitated DNA samples for radioactive PCR and gel electrophoresis.
Use ~10 ng input and immunoprecipitated DNA samples for ChIP-seq analysis.
B.3. Chemi-precipitation analysis for protein-DNA interactions in vivo
It is possible that protein is crosslinked well with DNA by formaldehyde, but DNA could not be detected by immunoprecipitation, using the antibody against the protein of interest. This could be due to the fact that antibody in the ChIP assay might not be able to recognize/access the epitope of the target protein of interest. To address this, one can perform above described assay in B.1 and B.2 by isolating protein-DNA complex under protein denaturation condition (i.e., in 6M Guanidinium chloride), using Ni2+-NTA (Nitrilotriacetic acid) agarose beads and hexahistidine-tagged protein of interest (Our unpublished data). One could also use different epitope-specific antibodies of the protein of interest or switch the epitope tag from carboxy to amino terminal. Further, crosslinked protein-DNA could be isolated using biotin/streptavidin system (49). For the Ni2+-NTA-based chemi-precipitation analysis of protein bound to DNA, all steps in B.1 are the same except the steps in B.1.1 and B.1.3. In this approach, the use of antibody is replaced by Ni2+-NTA agarose beads and the protein of interest is tagged by hexahistidine (that forms the coordinate bonds with Ni2+ of the Ni2+-NTA beads; Figure 3; 50). Ni2+-NTA-based protein-DNA interaction analysis can also be performed under native conditions (Our unpublished data). We briefly describe below the steps for chemi-precipitation for analysis of the protein-DNA interactions in vivo, using Ni2+-NTA agarose beads.
Figure 3:
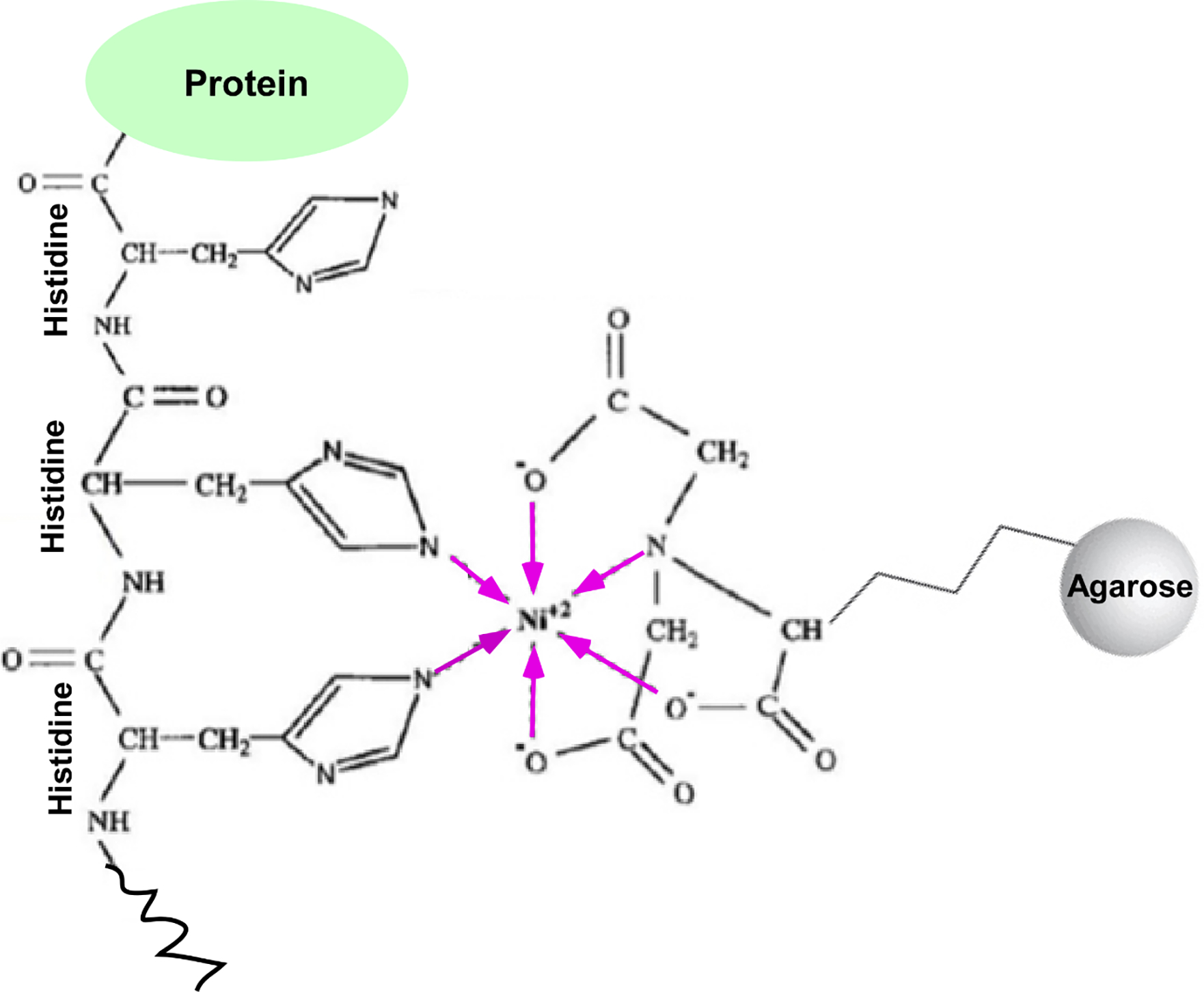
Schematic diagram showing the formation of two coordinate bonds (marked by pink colored arrows) by the histidine-tagged protein with Ni2+ of Ni2+-NTA (Nitrilotriacetic acid)-agarose beads. NTA forms four coordinate bonds with Ni2+. Ni2+ being a hexadentate, Ni2+ of Ni2+NTA forms four coordinate bonds (marked by pink colored arrows) with NTA and two coordinate bonds with H2O in the absence of histidine-tagged protein. In the presence of histidine-tagged protein, two H2O molecule are displaced by two histidines via formation of the coordinate bond with Ni2+. Such formation of coordinate bonds with histidine helps to isolate hexahistidine-tagged proteins and associated factors.
1. Inoculate cells expressing hexahistidine-tagged protein of interest in 100 ml medium, grow up to an OD600 of 1, prepare WCE and sonicate as described in B.2. Hexahistidine-tagged protein can be expressed from plasmid in the cell. It can also be expressed from its endogenous promoter following the experimental strategies, as described previously (31–33, 51).
2. Take 400 μl sonicated lysate in a new Eppendorf tube and add 100 μl pre-equilibrated Ni2+ NTA agarose (Qiagen) beads (100 μl sonicated lysate can also be used with 50 μl pre-equilibrated Ni2+ NTA agarose beads). Keep at the rotisserie rotator at 4 °C for 1 h followed by 45 min incubation with rotation in the rotisserie rotator at the room temperature. Keep 5 μl of sonicated WCE in the −80 °C freezer to be used later in the proteinase K treatment step.
3. After 1 h 45 min incubation, spin down the beads in the microcentrifuge machine at 3000 rpm for 4 min at the room temperature.
4. Carefully remove supernatant, and wash the beads as described in B.1.4.
5. Treat the beads as well as input DNA with proteinase K followed by reverse crosslinking and DNA precipitation as in B.1.5.
6. Dissolve the chemi-precipitated and input DNA samples in 20 and 100 μl TE (pH 8.0), respectively.
16. Use 1 μl each of input and chemi-precipitated DNA samples for radioactive PCR and gel electrophoresis.
17. Use ~10 ng input and chemi-precipitated DNA samples for ChIP-seq analysis.
B.4. Analysis of chromatin-bound proteins.
If the protein-DNA interaction surface does not have formaldehyde-reactive proximal groups for crosslinking (or at the best very minimal), the ChIP (or chemi-precipitation) signal would not be observed following the ChIP (or chemi-precipitation) analysis, as described above in B.1, B.2 and B.3. For example, Mediator is required for transcription, but it is not detected at the active gene by the ChIP assay (52). Likewise, there could be other proteins which need to be associated with genes for transcription or other DNA transacting processes, but cannot be detected by the ChIP assay. Under such situation, one can analyze chromatin association of a protein of interest by chromatin fractionation (53), as described below.
Inoculate yeast strain in 30 ml of YPD, and let it grow in an incubator shaker with a speed of 190 rpm at 30 °C up to an OD600 of 1.
Harvest 25 ml of cell culture by centrifugation in Sorvall Legend™ machine with a speed of 3000 rpm at 4 °C for 5 minutes.
Discard supernatant and resuspend cell pellet in 1 ml ice cold STOP solution (150mM NaCl, 50 mM NAF, 10 mM EDTA, 1 mM NaN3, pH 8 .0), and transfer the cell suspension to a 1.5 ml Eppendorf tube.
Spin down cell suspension for 5 minutes at 4 °C in the microcentrifuge machine (Eppendorf 5425) with a speed of 400 g.
Discard supernatant and resuspend cell pellet in 1 ml ice cold STOP solution. Spin down cell suspension for 5 minutes at 4 °C in the microcentrifuge machine (Eppendorf 5425) with a speed of 400 g.
Discard supernatant and the cell pellet can be stored in the −80 °C freezer or continue to next step.
Resuspend slowly the cell pellet in 500 μl of PEMS buffer (100 mM PIPES [piperazine-N,-N′bis(2-ethanesulfonic acid)], 50 mM EDTA, 10 mM MgSO4, 1.2 M Sorbitol, pH 6.9), and add 20 μl of lyticase (stock= 10 mg/ml; Sigma).
Tap the Eppendorf tube at the bottom to ensure proper mixing of lyticase.
Incubate cell suspension at 37 °C with rotation in a rotisserie rotator for 30 minutes. Measure OD600 immediately and 25 min after adding lyticase to monitor the progress of cell wall digestion. When a minimum of 80% cell wall digestion occurs (i.e., when the OD600 is reduced by 80%), proceed to the next step. Keep 50 μl aside to be analyzed later (in the gel electrophoresis) as the WCE in the −80 °C freezer.
After cell wall digestion, spin down spheroplast (cell with membrane) suspension at 400 g for 5 min at 4 °C in the microcentrifuge machine (Eppendorf 5425).
Carefully remove supernatant, and wash spheroplast pellet twice by resuspending in 1 ml of 1.2 M sorbitol to avoid bursting of spheroplast, followed by centrifugation at 400g for 5 minutes at 4 °C.
Resuspend the spheroplast in 428 μl of lysis buffer (HBS buffer [25 mM MOPS {morpholinepropanesulfonic acid}, 60 mM beta-glycerophosphate, 15 mM MgCl2, 15 mM EGTA, 15 mM p-nitrophenylphosphate, 0.2 mM Na3VO4, pH 7.2], 0.4 M sorbitol, 1 mM dithiothreitol [DTT] and 1 complete minitablet of protease inhibitors [Roche]). Add PMSF separately in this cell suspension just before the next step.
Incubate it in ice for 5 minutes. Subsequently, add Triton X-100 (20%) to a final concentration of 1%. Tap gently or invert it and then rotate in the rotisserie rotator for 15 min at 4 °C.
After rotation, vortex it mildly for 15 s on the VWR minivortexer with a low speed of 5. Spin down the lysate at 22000g for 15 minutes at 4 °C in the microcentrifuge machine (Eppendorf 5425).
Collect supernatant as a chromatin unbound fraction.
Suspend pellet in 1 ml of lysis buffer followed by centrifugation at 22000g for 5 min at 4 °C in the microcentrifuge machine (Eppendorf 5425) to wash pellet. Discard supernatant.
Repeat above step 15 (Second wash).
Resuspend the pellet in 450 μl of lysis buffer with protease inhibitors, and save it as a chromatin bound fraction.
Add SDS-PAGE (Sodium dodecyl sulfate – polyacrylamide gel electrophoresis) loading dye to the chromatin bound and unbound fractions for SDS-PAGE. Heat the samples at 95 °C for 20 min in the heat block.
After heating, vortex the samples and spin it down at 13000 rpm for 2 minutes at the room temperature in the microcentrifuge machine. Load the samples in the gel and perform SDS-PAGE.
After gel electrophoresis, transfer the protein from gel to the PVDF (Polyvinylidene difluoride) membrane for western blot analysis (following the standard protocol), using the primary antibody against the protein of interest. As controls, use antibodies against known chromatin bound and unbound proteins in the western blot analysis.
C. Formaldehyde crosslinking-based analysis of protein-protein interactions in vivo.
As described above, formaldehyde crosslinks interacting proteins (Figure 1), provided the reactive groups are present in the close vicinity at the interaction surface. Thus, following formaldehyde-based crosslinking in the growing cells, one can study protein-protein interactions in the living cells (Figure 2). However, such analysis may not distinguish direct vs indirect protein-protein interactions, as formaldehyde can crosslink multiple interacting proteins. Thus, formaldehyde crosslinking-mediated protein-protein interaction in vivo needs to be further pursued to dissect direct vs indirect by other approaches such as FRET (Fluorescence resonance energy transfer; 32–34), genetic and biochemical methodologies. Nonetheless, formaldehyde crosslinking-based protein-protein interaction analysis would provide valuable information for selective studies using different approaches. In this approach, all the steps up to bead washing (i.e., B.1.4) in B.1 are the same, and subsequent steps are described below.
C.1. SDS-PAGE and western blotting.
Add 100 μl 1.5X SDS-PAGE loading dye in the washed beads after immunoprecipitation (i.e., B.1.4).
Heat the suspended beads in the SDS-PAGE loading dye at 95 °C heating block for 20 min.
After heating, vortex the samples at room temperature at a maximum speed, and then spin down the beads at 13000 rpm for 2 minutes at the room temperature.
Carefully collect the supernatant, and run SDS-PAGE.
After gel electrophoresis, perform western blot following the standard protocol using the antibody against the protein of interest.
C.2. MS (Mass spectrometry) analysis.
One can analyze protein-protein interactions globally in living cells following formaldehyde-crosslinking and TAP (Tandem affinity purification)-mediated isolation of the TAP-tagged protein of interest and its crosslinked protein interactors with subsequent MS analysis, as described previously (38, 54, 55). Briefly, grow TAP-tagged protein of interest in large volume with crosslinking by formaldehyde, prepare WCE, sonicate and perform TAP-mediated isolation of the interacting proteins, following the protocol, as described previously (38, 54, 55). MS analysis of the isolated proteins would identify the interactors of the protein of interest during biological processes in the growing cells.
D. Formaldehyde crosslinking-based analysis of protein-RNA interactions in vivo:
In addition to crosslinking proteins with proteins and DNAs, formaldehyde also crosslinks proteins with interacting RNAs following similar chemical reaction mechanisms, as described above in Figure 1. Thus, one can study protein-RNA interactions by immunoprecipitating RNA using the antibody against the protein of interest to be tested for interaction with RNA (Figure 2), following similar experimental strategies as described above in B.1 with the addition of RNAase free DNAase in the sonicated WCE and use of the DEPC (Diethylpyrocarbonate)-treated water throughout, as described previously (56, 57). Further, RNA-protein interactions can be captured chemically using Ni2+-NTA agarose beads and hexahistidine-tagged protein of interest. Isolated RNA can be detected selectively by RT-PCR (Reverse transcriptase-PCR) or globally by RNA-seq. Thus, formaldehyde-based in vivo crosslinking approach can be adopted for protein-RNA interaction analysis in living cells.
E. Concluding Remarks.
We have described above the protocols used for analysis of the protein-protein/nucleic acid interactions in living yeast cells. These protocols can be generally used for any protein of interest to study protein-protein and/or protein-nucleic acid interactions for the selective as well as global (genome-wide and/or proteome-wide) studies (Figure 2). Similar approach can also be applicable to the mammalian cells. Formaldehyde crosslinks the interacting proteins and nucleic acids in living cells (Figure 1). While both short- and long-range protein interaction networks could be crosslinked by formaldehyde, the efficiency of long-range crosslinking (and hence the population of the long-range crosslinked protein interaction networks) would be relatively less as compared to the short-range protein interaction networks. This could be due to the fact that the probability of formaldehyde crosslinking of the interacting proteins and nucleic acids with many proteins present between them is low in the same population (or in a particular long-range interaction network) with treatment of ~1% formaldehyde for 15 min or so, thereby leading to the less population of crosslinked long-range protein interaction network in vivo. Therefore, long-range interaction networks may not be detected or could generate weak signal (using the standard ChIP assay conditions), which could be further pursued with increased crosslinking time, WCE volume or other conditions, as described above in B.2. Since formaldehyde crosslinks the interacting proteins and nucleic acids, the identified interactions using the above protocols could be direct or indirect. Therefore, the identified interactions from above protocols need to be further elucidated/validated via mutational and other analyses (e.g., FRET, genetic and biochemical) to resolve direct vs. indirect interactions. Further, the identified interactions may not be functionally relevant, which need to be followed up by analyzing the biological functions of the identified interactors. Nonetheless, the formaldehyde crosslinking-based approach for protein interaction analysis is widely used to obtain selective as well as large scale genome/proteome-wide data, which provide valuable resources towards understanding the biological processes with impact on future research directions.
Highlights:
Analysis of the protein-nucleic acid interactions in vivo.
Analysis of the protein-protein interactions in vivo.
Acknowledgements:
Work in the Bhaumik laboratory was supported by the grants from the National Institutes of Health (2R15GM088798–03), American Heart Association (15GRNT25700298), Simmons Cancer Institute (Team Science Grant) and SIU School of Medicine (Research Seed and Near-miss grants). AK was supported by a predoctoral fellowship from American Heart Association (19PRE34380855).
Footnotes
Declaration of Interest Statement:
Authors declare no conflict of interest.
CRediT authorship contribution statement:
Priyanka Barman: Conceptualization, Writing–original draft, Writing–review and editing. Amala Kaja: Conceptualization, Writing–review and editing. Pritam Chakraborty: Writing–review and editing. Sukesh R. Bhaumik: Conceptualization, Funding acquisition, Supervision, Writing – review and editing.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Braun P and Gingras AC (2012) History of protein-protein interactions: from egg-white to complex networks. Proteomics, 12: 1478–1498. [DOI] [PubMed] [Google Scholar]
- 2.Yanagida M (2002) Functional proteomics; current achievements. Journal of Chromatography B, 771: 89–106. [DOI] [PubMed] [Google Scholar]
- 3.Berggård T, Linse S, and James P (2007) Methods for the detection and analysis of protein-protein interactions. Proteomics, 7: 2833–2842. [DOI] [PubMed] [Google Scholar]
- 4.Pedamallu CS, and Posfai J (2010) Open source tool for prediction of genome wide protein-protein interaction network based on ortholog information. Source Code for Biology and Medicine, 5: article 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srinivasa Rao V, Srinivas K, Sujini GN, Sunand Kumar GN (2014) Protein-Protein Interaction Detection: Methods and Analysis. International Journal of Proteomics, 2014: 147648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, and Seraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology, 17: 1030–1032. [DOI] [PubMed] [Google Scholar]
- 7.Gavin A-C, Bösche M, Krause R et al. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature, 415: 141–147. [DOI] [PubMed] [Google Scholar]
- 8.Pitre S, Alamgir M, Green JR, Dumontier M, Dehne F, and Golshani A (2008) Computational methods for predicting protein-protein interactions. Advances in Biochemical Engineering/Biotechnology, 110: 247–267. [DOI] [PubMed] [Google Scholar]
- 9.MacBeath G and Schreiber SL (2000) Printing proteins as microarrays for high-throughput function determination. Science, 289: 1760–1763. [DOI] [PubMed] [Google Scholar]
- 10.Michnick SW, Ear PH, Landry C, Malleshaiah MK, and Messier V (2011) Protein-fragment complementation assays for large-scale analysis, functional dissection and dynamic studies of protein-protein interactions in living cells. Methods in Molecular Biology, 756: 395–425. [DOI] [PubMed] [Google Scholar]
- 11.Moresco JJ, Carvalho PC, and Yates III JR (2010) Identifying components of protein complexes in C. elegans using co-immunoprecipitation and mass spectrometry. Journal of Proteomics, 73: 2198–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.SR Bhaumik HM Sonawat (1999) Glucose Dehydrogenase from Halobacterium salinarum: Fluorescence Quenching and Binding to NADPH. Physiological Chemistry and Physics and Medical NMR, 31: 85–92. [Google Scholar]
- 13.Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science, 228: 1315–1317. [DOI] [PubMed] [Google Scholar]
- 14.Tong AHY, Drees B, Nardelli G et al. (2002) A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science, 295: 321–324. [DOI] [PubMed] [Google Scholar]
- 15.O’Connell MR, Gamsjaeger R, and Mackay JP (2009) The structural analysis of protein-protein interactions by NMR spectroscopy. Proteomics, 9: 5224–5232. [DOI] [PubMed] [Google Scholar]
- 16.Gao G, Williams JG, and Campbell SL (2004) Protein-protein interaction analysis by nuclear magnetic resonance spectroscopy. Methods in Molecular Biology, 261: 79–92. [DOI] [PubMed] [Google Scholar]
- 17.Uetz P, Glot L, Cagney G et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature, 403: 623–627. [DOI] [PubMed] [Google Scholar]
- 18.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, and Sakaki Y (2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Sciences of the United States of America, 98: 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llères D, Swift S, and Lamond AI (2007) Detecting protein-protein interactions in vivo with FRET using multiphoton fluorescence lifetime imaging microscopy (FLIM). Current Protocols in Cytometry, 12: Unit12.10. [DOI] [PubMed] [Google Scholar]
- 20.Tong AHY, Evangelista M, Parsons AB et al. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science, 294: 2364–2368. [DOI] [PubMed] [Google Scholar]
- 21.Rutherford SL (2000) From genotype to phenotype: buffering mechanisms and the storage of genetic information. BioEssays, 22: 1095–1105. [DOI] [PubMed] [Google Scholar]
- 22.Hartman IV JL, Garvik B, and Hartwell L (2001) Cell biology: principles for the buffering of genetic variation. Science, 291: 1001–1004. [DOI] [PubMed] [Google Scholar]
- 23.Bender A and Pringle JR (1991) Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Molecular and Cellular Biology, 11: 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ooi SL, Pan X, Peyser BD et al. (2006) Global synthetic-lethality analysis and yeast functional profiling. Trends in Genetics, 22: 56–63. [DOI] [PubMed] [Google Scholar]
- 25.Brown JA, Sherlock G, Myers CL et al. (2006) Global analysis of gene function in yeast by quantitative phenotypic profiling. Molecular Systems Biology, 2: Article ID 2006.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall DB, Struhl K (2002) The VP16 activation domain interacts with multiple transcriptional components as determined by protein-protein cross-linking in vivo. J Biol Chem, 277: 46043–46050. [DOI] [PubMed] [Google Scholar]
- 27.Lahudkar S, Shukla A, Bajwa P, Durairaj G, Stanojevic N, Bhaumik SR (2011) The mRNA cap-binding complex stimulates the formation of pre-initiation complex at the promoter via its interaction with Mot1p in vivo. Nucleic Acids Res, 39: 2188–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malik S, Chaurasia P, Lahudkar S, Durairaj G, Shukla A, Bhaumik SR (2010) Rad26p, a transcription-coupled repair factor, is recruited to the site of DNA lesion in an elongating RNA polymerase II-dependent manner in vivo. Nucleic Acids Res, 38: 1461–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sen R, Ferdoush J, Kaja A, Bhaumik SR. 2016. Fine-tuning of FACT by the ubiquitin proteasome system in regulation of transcriptional elongation. Mol Cell Biol 36:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen R, Kaja A, Ferdoush J, Lahudkar S, Barman P, Bhaumik SR (2017) An mRNA capping enzyme targets FACT to the active gene to enhance the engagement of RNA polymerase II into transcriptional elongation. Mol Cell Biol, 37: e00029–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhaumik SR, Green MR (2001) SAGA is an essential in vivo target of the yeast acidic activator Gal4p. Genes Dev. 15: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhaumik SR, Raha T, Aiello DP, Green MR (2004) In vivo target of a transcriptional activator revealed by fluorescence resonance energy transfer. Genes Dev. 18: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhaumik SR (2006) Analysis of in vivo targets of transcriptional activators by fluorescence resonance energy transfer. Methods, 40: 353–9. [DOI] [PubMed] [Google Scholar]
- 34.Bhaumik SR (2021) Fluorescence resonance energy transfer in revealing protein-protein interactions in living cells. Emerging Topics in Life Sciences, 5: 49–59. [DOI] [PubMed] [Google Scholar]
- 35.Sutherland BW, Toews J, Kast J (2008) Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J Mass Spectrom, 43: 699–715. [DOI] [PubMed] [Google Scholar]
- 36.Kennedy-Darling J, Smith LM (2014) Measuring the formaldehyde Protein-DNA cross-link reversal rate. Anal Chem, 86: 5678–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metz B, Kersten GF, Baart GJ, de Jong A, Meiring H, ten Hove J, van Steenbergen MJ, Hennink WE, Crommelin DJ, Jiskoot W (2006) Identification of formaldehyde-induced modifications in proteins: reactions with insulin. Bioconjug Chem, 17: 815–22. [DOI] [PubMed] [Google Scholar]
- 38.Barman P, Sen R, Kaja A, Ferdoush J, Guha S, Govind CK, Bhaumik SR (2022) Genome-Wide Regulations of the Preinitiation Complex Formation and Elongating RNA Polymerase II by an E3 Ubiquitin Ligase, San1. Mol Cell Biol, 42: e0036821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malik S, Shukla A, Sen P, Bhaumik SR (2009) The 19 s proteasome subcomplex establishes a specific protein interaction network at the promoter for stimulated transcriptional initiation in vivo. J Biol Chem, 284: 35714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uprety B, Lahudkar S, Malik S, Bhaumik SR (2012) The 19S proteasome subcomplex promotes the targeting of NuA4 HAT to the promoters of ribosomal protein genes to facilitate the recruitment of TFIID for transcriptional initiation in vivo. Nucleic Acids Res 40:1969–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferdoush J, Sen R, Kaja A, Barman P, Bhaumik SR. (2018) Two Distinct Regulatory Mechanisms of Transcriptional Initiation in Response to Nutrient Signaling. Genetics, 208: 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barman P, Kaja A, Chakraborty P, Guha S, Roy A, Ferdoush J, Bhaumik SR (2023) A novel UPS regulation of Sgf73/ataxin-7 that maintains the integrity of the co-activator SAGA in orchestrating transcription. Genetics, Apr 19:iyad071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shukla A, Stanojevic N, Duan Z, Sen P, Bhaumik SR (2006) Ubp8p, a histone deubiquitinase whose association with SAGA is mediated by Sgf11p, differentially regulates lysine 4 methylation of histone H3 in vivo. Mol Cell Biol 26: 3339–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shukla A, Bajwa P, Bhaumik SR (2006) SAGA-associated Sgf73p facilitates formation of the preinitiation complex assembly at the promoters either in a HAT-dependent or independent manner in vivo. Nucleic Acids Res, 34: 6225–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shukla A, Lahudkar S, Durairaj G, Bhaumik SR (2012) Sgf29p facilitates the recruitment of TATA box binding protein but does not alter SAGA’s global structural integrity in vivo. Biochemistry, 51: 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durairaj G, Sen R, Uprety B, Shukla A, Bhaumik SR (2014) Sus1p facilitates pre-initiation complex formation at the SAGA-regulated genes independently of histone H2B de-ubiquitylation. J Mol Biol 426: 2928–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurdistani SK, Grunstein M (2003) In vivo protein-protein and protein-DNA crosslinking for genomewide binding microarray. Methods, 31: 90–5. [DOI] [PubMed] [Google Scholar]
- 48.Zeng Ping-Yao, Vakoc Christopher R., Chen Zhu-Chu, Blobel Gerd A., and Berger Shelley L. (2006) In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. BioTechniques, 41: 694–698. [DOI] [PubMed] [Google Scholar]
- 49.Lin Kui, Yan Qin, Mitchell Audrey, Funk Natasha, Lu Catherlin, and Xiao Hao (2020) A simple method for non-denaturing purification of biotin-tagged proteins through competitive elution with free biotin. BioTechniques, 68: 41–44. [DOI] [PubMed] [Google Scholar]
- 50.Bornhorst Joshua A. and Falke Joseph J. (2000) Purification of Proteins Using Polyhistidine Affinity Tags. Methods Enzymol, 326: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pingle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast, 14: 953–961. [DOI] [PubMed] [Google Scholar]
- 52.Fan X, Chou DM, Struhl K (2006) Activator-specific recruitment of Mediator in vivo. Nat Struct Mol Biol, 13: 117–20. [DOI] [PubMed] [Google Scholar]
- 53.Ferdoush J, Sen R, Durairaj G, Barman P, Kaja A, Guha S, Bhaumik SR (2020) An F-Box Protein, Mdm30, Interacts with TREX Subunit Sub2 To Regulate Cellular Abundance Cotranscriptionally in Orchestrating mRNA Export Independently of Splicing and Mitochondrial Function. Mol Cell Biol, 40: e00570–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaja Amala, Barman Priyanka, Guha Shalini and Bhaumik Sukesh R. (2023) Tandem affinity purification and mass-spectrometric analysis of FACT and associated proteins. Methods in Molecular Biology, 2701:209–227. [DOI] [PubMed] [Google Scholar]
- 55.Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, and Gardner RG (2011) Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell, 41: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selth LA, Close P, Svejstrup JQ (2011) Studying RNA-protein interactions in vivo by RNA immunoprecipitation. Methods Mol Biol, 791: 253–64. [DOI] [PubMed] [Google Scholar]
- 57.Gilbert C, Svejstrup JQ (2006) RNA immunoprecipitation for determining RNA-protein associations in vivo. Curr Protoc Mol Biol., Chapter 27:Unit 27.4. [DOI] [PubMed] [Google Scholar]