

SUMMARY
Liver mitochondria undergo architectural remodeling that maintains energy homeostasis in response to feeding and fasting. However, the specific components and molecular mechanisms driving these changes and their impact on energy metabolism remain unclear. Through comparative mouse proteomics, we found that fasting induces strain-specific mitochondrial cristae formation in the liver by upregulating MIC19, a subunit of the MICOS complex. Enforced MIC19 expression in the liver promotes cristae formation, mitochondrial respiration and fatty acid oxidation while suppressing gluconeogenesis. Mice overexpressing hepatic MIC19 show resistance to diet-induced obesity and improved glucose homeostasis. Interestingly, MIC19 overexpressing mice exhibit elevated energy expenditure and increased pedestrian locomotion. Metabolite profiling revealed that uracil accumulates in the livers of these mice due to increased Uridine phosphorylase UPP2 activity. Furthermore, uracil-supplemented diet increases locomotion in wild-type mice. Thus, MIC19-induced mitochondrial cristae formation in the liver increases uracil as a signal to promote locomotion, with protective effects against diet-induced obesity.
Keywords: Diabetes, obesity, brisk walking, fatty liver, mitochondrial cristae, uracil
Graphical Abstract
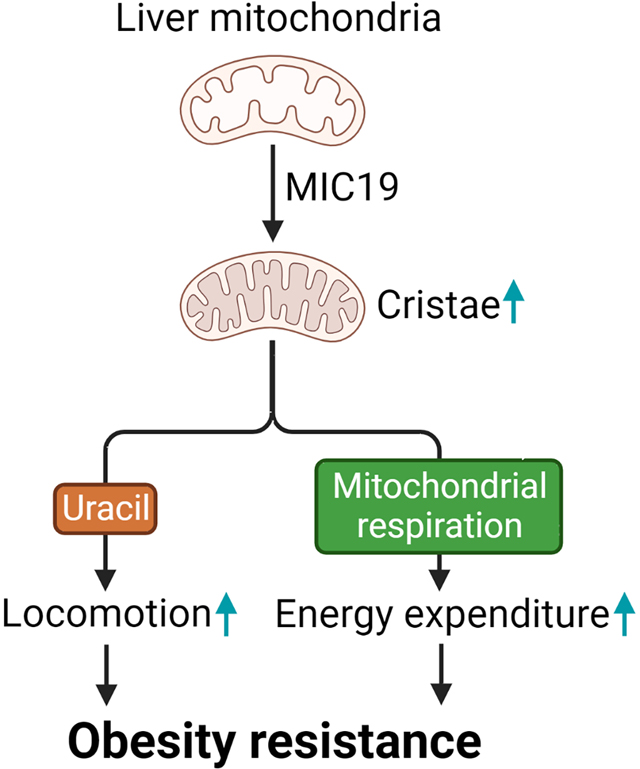
Sohn et al. investigate how cristae formation in liver is regulated upon nutritional changes and the effects of cristae on hepatic and whole-body energy metabolism. Hepatic MIC19 contributes to cristae remodeling and plays key roles in maintaining systemic energy balance by elevating energy expenditure and pedestrian locomotion.
INTRODUCTION
Overnutrition from an imbalanced diet coupled with low physical activity causes metabolic diseases that include obesity and type 2 diabetes (T2D). These metabolic diseases are associated with serious clinical complications including cardiovascular disease, viral infections, and cancer1,2. Moreover, accumulation of lipids in non-adipose tissues such as the liver can cause non-alcoholic fatty liver disease (NAFLD), which can progress to non-alcoholic steatohepatitis (NASH), liver fibrosis and cancer3–5. Strategies to combat overnutrition and clinical complications of obesity/T2D include personalized dietary regimens in combination with physical activity. For instance, brisk walking can improve maintenance of weight loss6. Even, small amounts of vigorous non-exercise physical activity are associated with substantially lower mortality risk of cancer and cardiovascular diseases7. However, failure to follow these recommendations leads to progression of these metabolic diseases in most of cases8–12. Bariatric surgery or anti-diabetic treatments have initial success, but management of body weight and long-term efficacy of diabetic therapies remains a challenging aspect of the global obesity and T2D epidemic13,14. While nutrient excess increases energy expenditure, this dissipation is not sufficient to prevent weight gain, leading to progression of obesity and T2D. Although much of the attention on energy expenditure has focused on thermogenic adipocytes15–18, liver energy metabolism also contributes to energy balance and controls whole body nutrient homeostasis19–22. Liver is a major metabolic organ that efficiently re-distributes metabolites between different tissues and becomes a central player during fasting/feeding transitions or physical activity22–25. During fasting, liver promotes mitochondrial activity to support glucose production and ketogenesis. These processes maintain blood glucose and supply ketone bodies to tissues including the brain26–28. These responses maintain fasting metabolic states and protect against obesity and T2D, as well as specific liver clinical pathologies including NAFLD29. However, the mechanisms that drive mitochondrial bioenergetics and dynamics in these conditions are largely unknown. Mitochondria have inner membrane invaginations called cristae that harbor functional respiratory complexes30,31. Cristae enhances efficiency of ATP production and accelerates nutrient oxidation by oxidative phosphorylation32,33. The formation and dynamics of these structures is regulated through different components including the mitochondrial contact site and cristae organizing system (MICOS) protein complex and optic atrophy 1 (OPA1) protein31,34. The MICOS complex is located at the cristae junctions, narrow tubules that connect cristae to the boundary membrane and is composed of two subcomplexes35–38. The MIC10-MIC26-MIC27 subcomplex is responsible for shaping and stabilizing cristae junctions39. The MIC60-MIC19-MIC25 subcomplex is essential for formation of contact sites between the inner and outer membranes and maintenance of cristae junctions that ensure mitochondrial integrity40. Through MIC19, two MICOS subcomplexes are interconnected41. In brown fat, mitochondrial import of MICOS complex subunit MIC19 is increased upon cold exposure to drive cristae formation38. The regulation of MICOS complex components and cristae in the liver during fasting remains unclear. Here, we show that MIC19, a limiting component of the MICOS complex, is increased in liver mitochondria during fasting conditions and promotes cristae formation and utilization of glucose and fatty acids. Ectopic expression of MIC19 in liver is sufficient to protect against obesity and T2D. We find that the protective effects of MIC19 overexpression are mediated through increased energy expenditure. Surprisingly, the increase in energy expenditure is not only regulated through rewiring of liver energy metabolism, but also increased movement of animals which likely involves signaling from liver mitochondria to the brain. This study elucidates nutrient-dependent regulatory molecular mechanisms of cristae formation and how it affects liver and whole-body energy metabolism in conditions of obesity and T2D.
RESULTS
The liver mitochondrial inner membrane proteome is remodeled upon fasting in CD-1 mice
CD-1 mice are resistant to developing hepatic steatosis in response to high-fat diet feeding, unlike the widely used C57BL/6 (BL6) mouse model for diet-induced fatty liver disease42. Given the association between dysfunctional mitochondria and hepatic metabolic diseases43, we hypothesized that liver mitochondria in these two genetic backgrounds responded differently to fasting. To test this, we took an unbiased proteomics approach to analyze the changes in liver mitochondrial content upon ad libitum feeding vs. 24 hours fasting in CD-1 vs. BL6 mice (Table S1). We identified a total of 839 mitochondrial proteins in CD-1 liver samples, where 145 (17.3%) of them were upregulated and 22 (2.6%) downregulated (Figure 1A). In BL6, out of 811 mitochondrial proteins identified, 16 were upregulated (2.0%) and 8 (1.0%) downregulated (Figure 1B). These results suggest more proteomic flexibility in CD1 livers in response to fasting. Absence of changes in the levels of AST and ALT in blood samples suggested that no detrimental effects occurred upon fasting (Figure S1A). While there were canonical proteins regulated by fasting in both strains (e.g., PDK4 upregulation), GO Term analysis revealed that there are differences between fasting-induced upregulated proteins of CD-1 and BL6 liver mitochondria. Upregulated proteins in CD-1 liver mitochondria upon fasting were associated with mitochondrial inner membrane structure and function, including respiratory chain complexes and cristae-related proteins (Figure 1C). By contrast, this signature that suggested mitochondrial inner membrane remodeling in the CD-1 strain upon fasting was absent from the BL6 strain (Figure 1D). Mitochondrial inner membrane forms invaginations named cristae, where oxidative phosphorylation (OxPHOS) complexes are concentrated. As shown in Figure 1E, the components of OxPHOS complexes I-V were induced by fasting in the CD-1 strain. Moreover, the MICOS complex subunit Chchd3/MIC19 was most strongly regulated when we analyzed the abundance of known mitochondrial cristae proteins (Figure 1F). Other MICOS complex subunits including Mic60, Mic26, Mic27 and mitochondrial dynamin Opa1 showed a trend towards an increase (Figure 1F). Interestingly, mitochondrial intermembrane space assembly (MIA) pathway for protein import into the mitochondria was affected. MIA40 serves as a receptor on the trans side of the Translocase of the Outer Membrane (TOM) complex on the outer membrane and is responsible for transporting cysteine rich proteins into the intermembrane space (IMS) and their oxidative folding44. MIC19 is also a target of MIA4045. MIA40 substrates were upregulated upon fasting in the CD-1 strain (Figure 1G). Because these results suggested that mitochondrial cristae were remodeled upon fasting in the CD-1 strain, we decided to focus on the MICOS complex subunit MIC19. Western blots validated the upregulation of MIC19 protein upon fasting in the CD-1 strain, but not the BL6 strain (Figure 1H). We noted that the TOM complex subunit TOM70, which is responsible for MIC19 protein import into mitochondria was also increased upon fasting in the CD-1 strain (Figure 1H). Along with induction of MIC19 protein expression upon fasting (Figure 1I), digitonin-based Blue-Native gel electrophoresis assays showed that more MIC19 was incorporated into the MICOS complex upon fasting in CD-1 livers (Figure 1J). The ratio between individual MIC19 protein and MIC19 incorporated into MICOS complex was not significantly changed upon fasting (Figure 1K), further supporting that fasting-induced MIC19 proteins are incorporated into the MICOS complex. Moreover, isolated mitochondria from the liver of fasted CD-1 mice showed increased import of 35S radiolabeled MIC19 compared to fed CD-1 mice (Figure 1L). Next, we determined whether MIC19 levels were transcriptionally regulated and quantified the mRNA levels in the liver upon fasting. Ppargc-1a was strongly induced by fasting in both strains as expected, but Chchd3 levels remained unchanged (Figures S1B and S1C). Regulation of MIC19 levels might depend on translation and/or protein import into mitochondria. In sum, the mitochondrial inner membrane proteome in the liver is remodeled by fasting in the CD-1 strain.
Figure 1. Fasting remodels the mitochondrial proteome in CD-1 livers, but not BL6.
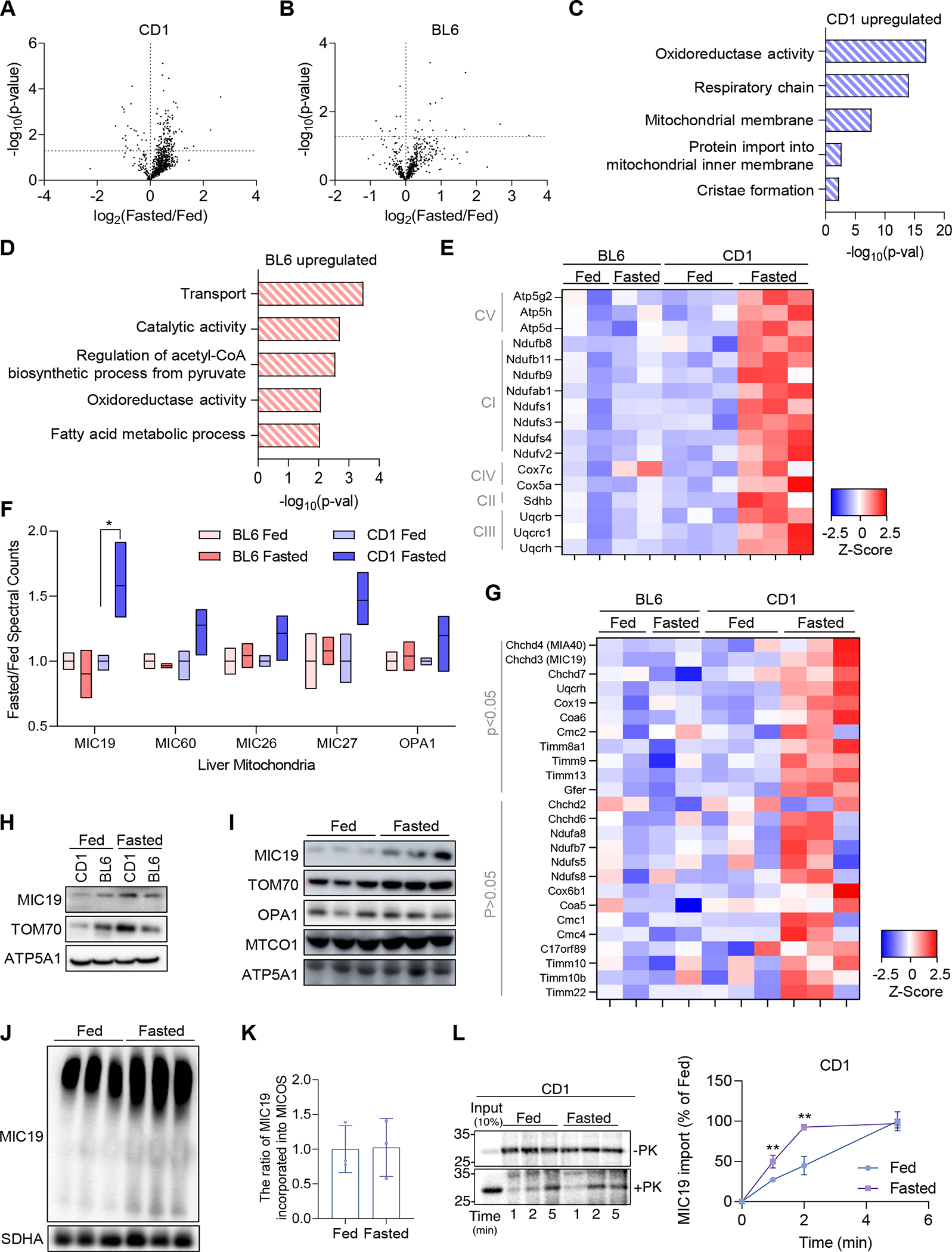
(A and B) Volcano plots showing changes in the mitochondrial proteome in 24 hour fasted vs. ad-libitum fed mice in CD-1 and BL6. n=3 for CD-1 and n=2 for BL6 in separate experiments. Dotted lines denote the significance cut-off (p<0.05).
(C and D) GO Term Analysis of upregulated genes in CD-1 and BL6 liver mitochondria.
(E) Heatmap showing fold change increase in specific subunits of the respiratory chain complexes I-V upon fasting in the CD-1 strain (p<0.05).
(F) Box plots extending from minimum to maximum values that show fold change in mitochondrial cristae proteins including OPA1 and MICOS complex components.
(G) Heatmap showing fold change increase in MIA40 substrates.
(H) Western blot validating MIC19 protein levels in isolated pure mitochondria of CD-1 and BL6 liver under fed or fasted conditions.
(I) Mitochondrial protein levels in the liver mitochondria of CD-1 upon fasting (n=3).
(J) Blue-Native Polyacrylamide Gel Electrophoresis blot showing more MIC19 incorporated into the MICOS complex upon 24 h fasting in the CD-1 strain. Lower band is around 700kDa, representing the MICOS complex and the upper band represents mitochondrial intermembrane bridging (MIB) supercomplex (n=3).
(K) The ratio of MIC19 incorporated into MICOS complex to individual MIC19 protein calculated based on (I) and (J).
(L) In vitro protein import of MIC19 using isolated liver mitochondria from CD-1 mice under fed or fasted conditions (n=3).
Data represented as mean±SD (K, L). *p< 0.05, **p< 0.01 by Student’s t-test (A, B, E-G, K, L). n represents the number of biological replicates. See also Figure S1.
Proteomics data suggested that CD-1 mice reorganize mitochondrial cristae upon fasting. To test this idea, we surveyed mitochondrial ultrastructure by transmission electron microscopy (TEM) upon fasting in CD-1 and BL6 mice livers. Consistent with the proteomics data and induction of MIC19, CD-1 liver mitochondria had more mitochondrial cristae per area, whereas mitochondrial cristae remained unaltered in BL6 livers (Figures 2A–2D). Next, we determined if mitochondrial respiration was increased upon fasting as more cristae formed. Seahorse assays were carried out with isolated mitochondria to measure oxygen consumption rates. We found that fasting promoted complex I (pyruvate+malate) and complex II (succinate) driven respiration in CD-1 liver mitochondria (Figure 2E), whereas BL6 did not (Figure 2F). Interestingly, mitochondrial shape was regulated by fasting in both strains, where fasting promoted elongation of mitochondria (Figures 2G and 2H). Previous results had indicated that mitochondria are elongated upon starvation, which is protective against autophagic degradation46. Thus, elongated mitochondria sustain cell viability by increasing cristae and ATP production47. Our results suggest that elongation of mitochondria can be uncoupled from cristae formation, since BL6 mitochondria are elongated, yet they don’t increase the number of cristae per mitochondrial area.
Figure 2. Fasting promotes mitochondrial cristae formation and mitochondrial respiration in CD-1 livers, but not BL6.
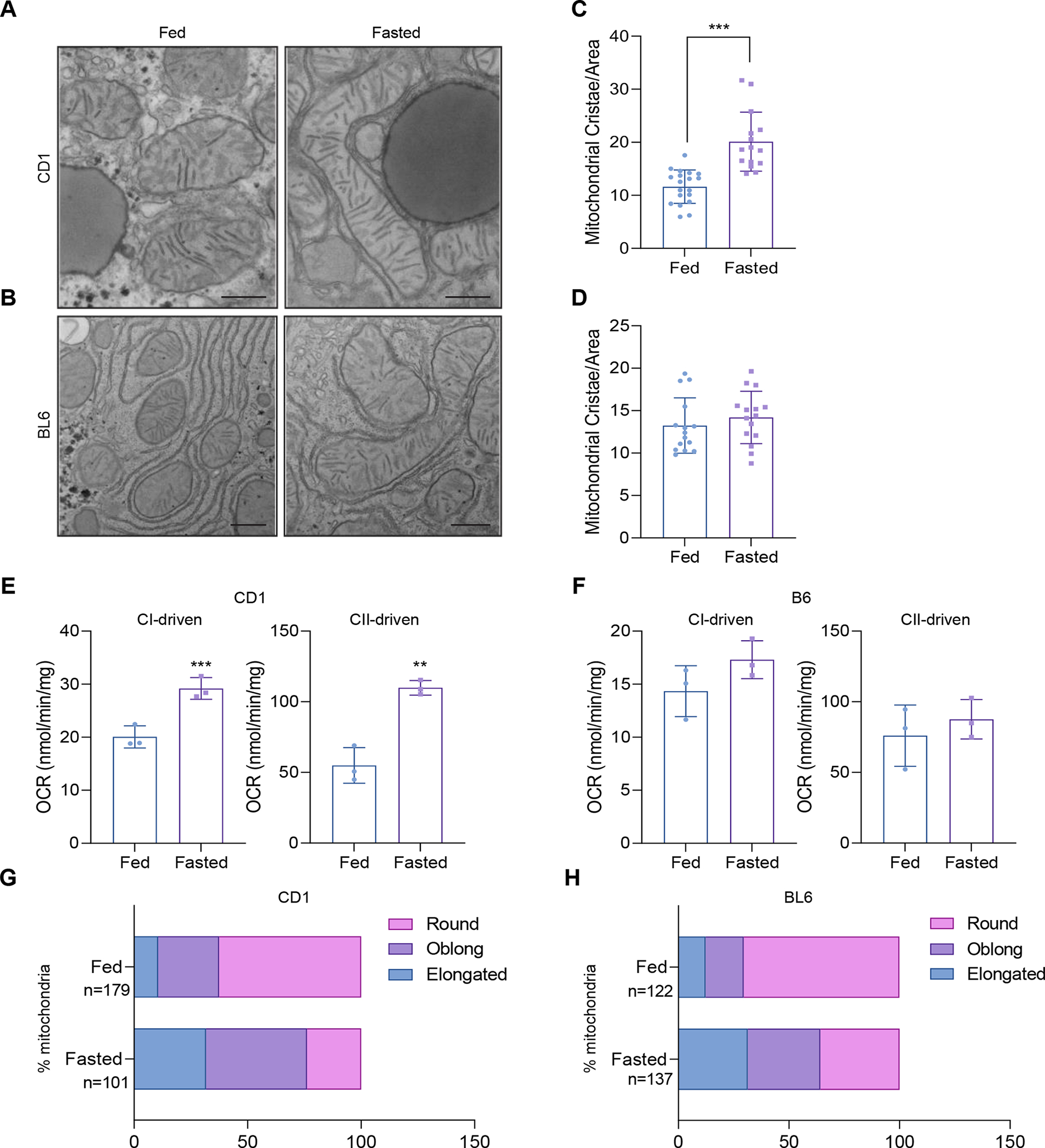
(A and B) Transmission Electron Microscopy (TEM) analysis of liver mitochondria in 24 hour fasted vs. ad libitum fed CD-1 and BL6 (n=2 mice, >5 section of images per animal).
(C and D) Number of mitochondrial cristae per mitochondrial area (a.u.) in fasted vs. fed animals in CD-1 and BL6 livers (n>14 mitochondria per group).
(E and F) Measurement of complex I- and complex II-linked oxygen consumption rate (OCR) in isolated liver mitochondria from fasted vs. fed CD-1 and BL6 (n=3).
(G and H) Percentage of mitochondria that are round, oblong, or elongated in fasted vs. fed animals in TEM images of CD-1 and BL6 mouse livers. Number of mitochondria analyzed are denoted in the figure.
Data represented as mean±SD (C-F). **p< 0.01, ***p< 0.001 by Student’s t-test (C-F). n represents the number of biological replicates.
MIC19 promotes fatty acid oxidation and suppresses glucose production in BL6 primary hepatocytes
Induction of the MICOS complex subunit MIC19 is a good candidate for driving mitochondrial cristae formation upon fasting. Cold promotes MIC19 import into brown fat mitochondria to form cristae38, and a similar mechanism might exist in the liver. We tested whether modulation of MIC19 in a BL6 background could drive cristae formation and alter energetic and metabolic processes in the liver that are normally regulated by fasting. Fasting promotes hepatic glucose production and fatty acid oxidation, and we determined if MIC19 had a role in regulating these processes. Primary hepatocytes were isolated from BL6 livers and used siChchd3 to downregulate MIC19 levels (Figures S2A and S2B) or overexpress MIC19 (Figures S2A and S2C). We used 100 nM glucagon stimulation to mimic fasting conditions in primary hepatocytes and measured hepatic glucose production from pyruvate and lactate. By using radioactively labeled substrates, we also quantified the utilization of fatty acids and glucose through the TCA cycle in primary hepatocytes (Figure S2D). Like fasting, glucagon stimulation induced glucose production and fatty acid oxidation (Figures S2E, S2F, S2I, and S2J). Surprisingly, unlike fasting or glucagon, MIC19 suppressed glucose production (Figures S2E and S2F). It is possible that MIC19 acts as part of a feedback loop that negatively regulates and attenuates glucose production during fasting. We quantified the oxidation of 14C-glucose and 14C-palmitate through the TCA cycle by measuring 14C-CO2 release. In line with the glucose production results, MIC19 boosted the oxidation of glucose through the TCA cycle (Figures S2G and S2H). Like fasting and glucagon treatment, MIC19 increased fatty acid oxidation (Figures S2I and S2J). These results suggest that overexpression of MIC19 can rewire the hepatic metabolic program to suppress glucose production and promote fatty acid oxidation. In addition, MIC19 downregulation reduced mitochondrial cristae abundance in the presence of glucagon without changes in total area of mitochondria (Figures S2K and S2L), implying that MIC19 autonomously regulate cristae formation in hepatocytes.
MIC19 is sufficient to drive cristae formation and promote mitochondrial respiration in the BL6 mouse liver
Our experiments with primary hepatocytes revealed that MIC19 promotes fatty acid oxidation and suppresses glucose production. Given these results, we hypothesized that MIC19 overexpression in the liver might be beneficial in the context of obesity/T2D/fatty liver disease, where there is lipid overload and unchecked glucose production by the liver. To overexpress MIC19 specifically in the mouse liver, we used AAVs with the serotype 2/8 and administered it via the tail vein. Western blot analysis revealed that GFP or MIC19 was expressed in the liver (Figure 3A). When we checked other tissues (brown fat, inguinal white adipose tissue (iWAT), brain, and skeletal muscle), MIC19 levels remained unchanged (Figures S3A–S3D). There was also no alteration in serum AST and ALT levels upon MIC19 expression (Figure S3E), implying that MIC19 overexpression does not induce liver toxicity. We first tested whether MIC19 overexpression in the liver was sufficient to alter mitochondrial ultrastructure and induce cristae formation. We found that MIC19 AAV liver mitochondria had more cristae per area without changes in total area of mitochondria (Figures 3B–3D). Blue Native Gel Electrophoresis analysis revealed that overexpressed MIC19 was incorporated into the MICOS protein complex and MIC19 overexpression induced dimerization of Complex V, a hallmark of increased cristae (Figure 3E). ATP synthase dimers are conserved feature of mitochondrial inner membrane and are required for cristae formation48. Although total amount of OxPHOS protein was not altered in the liver mitochondria by MIC19 overexpression, MIC19 upregulation induced the assembly of respiratory supercomplex associated with enhanced respiratory capacity49–51 (Figures 3F and 3G). Next, we determined if more cristae formation translated into more mitochondrial respiration. We used seahorse XF analyzer to measure complex I (pyruvate+malate) and complex II (succinate) driven respiration in isolated liver mitochondria and found that MIC19 AAV liver mitochondria displayed higher respiration rates, without changes in synthase activity (Figures 3H–3J). We also assessed cellular oxygen consumption rates in MIC19 overexpressed hepatocytes. MIC19 was overexpression in primary hepatocytes by AAV transduction. Upon MIC19 overexpression, basal and proton leak respiration were increased. Maximal respiration also appeared to be increased in MIC19 overexpressing hepatocytes (Figure S3F). On the other hand, mitochondrial DNA copy number in the liver were comparable between control and MIC19 overexpressing mice (Figure 3K). These data imply that MIC19 in the liver regulates mitochondrial respiration but not, mitochondrial biogenesis. Given the metabolic rewiring in BL6 primary hepatocytes by MIC19 overexpression, we determined how fatty acid oxidation and glucose homeostasis was affected in MIC19 AAV livers. We found that palmitate oxidation was increased in the MIC19 AAV livers, consistent with the data in primary hepatocytes (Figure 3L). Consistent with the increase in palmitate oxidation, malonyl-CoA levels were strongly reduced in MIC19 AAV livers (Figure 3M). Malony-CoA is an allosteric inhibitor of CPTI, an enzyme that controls the transfer of long-chain fatty acyl-coAs into the mitochondria for oxidation52. Our lipidomics analysis of MIC19 AAV livers revealed a decrease in acylcarnitines (6:0) (Figure S3G), which are tightly linked to FAO and thought to be markers of T2D and obesity53. On a chow diet, there was no significant alteration in the body weight, lean mass, fat mass, and the liver weight upon MIC19 overexpression (Figures 3N, S3H, and S3I). We injected a bolus of glucose to challenge the animals and found that the blood glucose levels in even lean MIC19 AAV animals recovered faster compared to GFP AAV (Figures 3O and 3P). However, fasting serum insulin levels of MIC19 overexpressing animals remained similar to that of wild type (Figure S3J). These results suggest that MIC19 overexpression in hepatocytes and in the liver in mice recapitulate similar metabolic effects: increased fatty acid oxidation and improved glucose homeostasis.
Figure 3. Overexpression of MIC19 in BL6 livers promotes mitochondrial cristae formation and respiration.
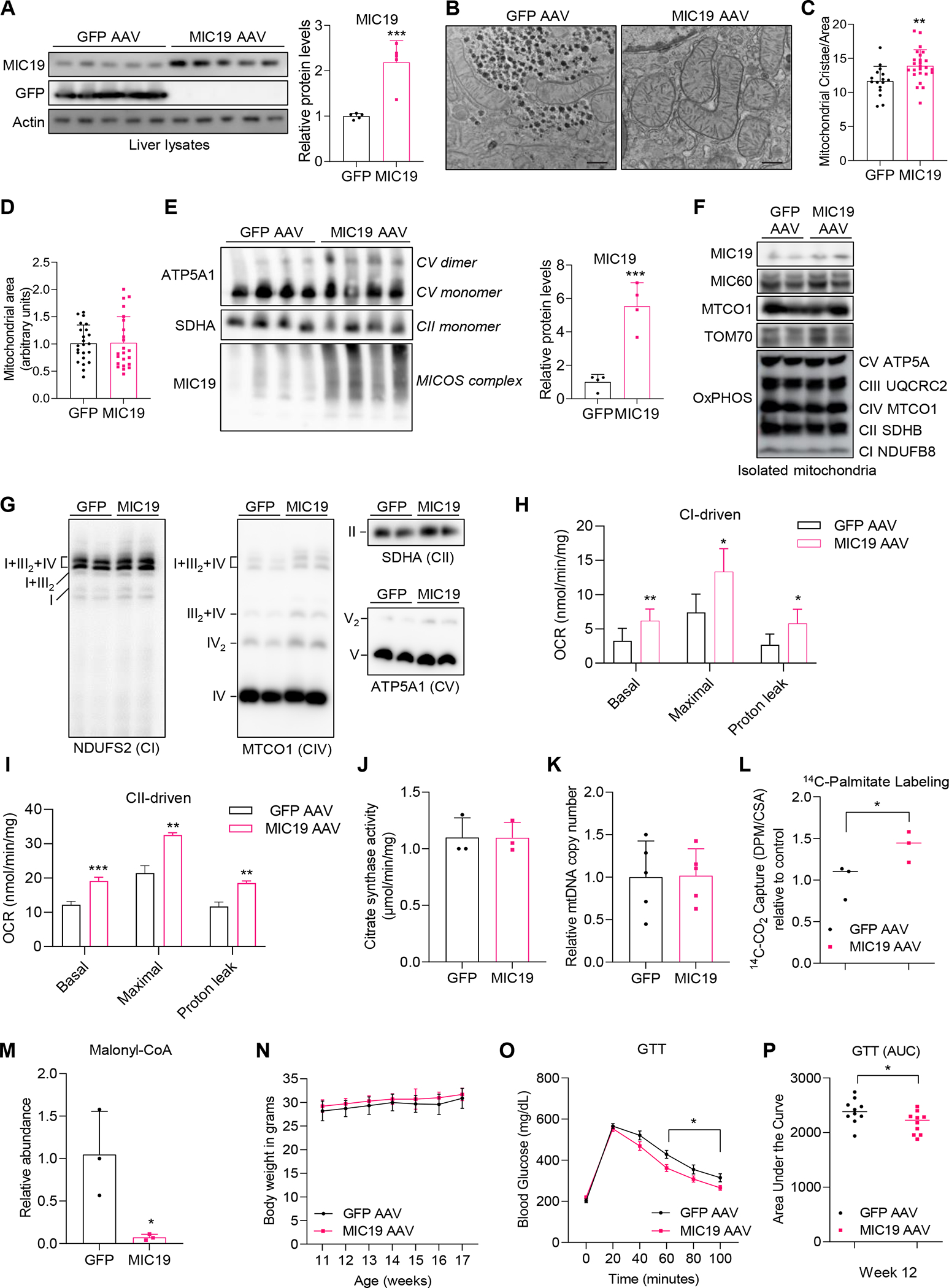
(A) Western blot showing overexpression of GFP or MIC19 in the livers of ad libitum fed BL6.
(B-C) TEM images of GFP AAV vs. MIC19 AAV mouse livers (n=2, >5 section of liver images per animal).
(D) The mitochondrial area (a.u.) in GFP vs. MIC19 AAV liver.
(E) Blue-Native Polyacrylamide Gel Electrophoresis showing the MICOS complex, monomers and dimers of CV (ATP5A1) and CII (SDHA) in GFP vs. MIC19 AAV liver mitochondria (n=4).
(F) Western blot showing expression of mitochondrial proteins (n=2).
(G) Respiratory complex assemblies in the liver mitochondria isolated from GFP or MIC19 AAV mice (n=2).
(H and I) Complex I- and Complex II-dependent OCR measured by Seahorse XF analyzer in isolated mitochondria from GFP AAV vs. MIC19 AAV livers (n=3).
(J) Citrate synthase activity (CSA) in the mitochondria isolated from GFP AAV vs. MIC19 AAV liver.
(K) Relative mitochondrial DNA content assessed by 16s rRNA levels normalized to HK2 mRNA level.
(L) 14C-palmitate oxidation in liver homogenates from GFP vs. MIC19 AAV livers normalized to CSA, shown relative to GFP AAV (n=3).
(M) Relative abundance of malonyl-CoA in the liver of GFP vs. MIC19 AAV mice (n=3). (N) Body weight of GFP AAV vs. MIC19 AAV animals on regular chow at weeks 11–17 (n=10).
(O) Blood glucose levels during a Glucose Tolerance Test (GTT) in GFP vs. MIC19 AAV animals (n=10). Data represented as mean±SEM.
(P) Area Under the Curve (AUC) for the GTT comparing GFP vs. MIC19 animals.
Data represented as mean±SD (A, C-E, H-N). *p< 0.05, **p< 0.01, ***p< 0.001 by Student’s t-test (A, C-E, H-M, O, P). n represents the number of biological replicates. See also Figures S2 and S3.
MIC19 overexpression in the liver is protective against weight gain on a high-fat diet (HFD)
MIC19 overexpression induced formation of mitochondrial cristae and fatty acid oxidation, while suppressing glucose production. We determined how MIC19 AAV animals would fare on an HFD. Strikingly, after 1 week on an HFD, MIC19 AAV animals gained significantly less weight compared to wild type (Figure 4A). These effects became more apparent over time, and after 8 weeks on an HFD, the fat mass was significantly less compared to GFP AAV animals (Figure 4B), while the food intake remained the same (Figure 4C). Liver triglycerides and fasting blood glucose was also decreased in MIC19 AAV animals (Figures 4D and 4E). Succinate driven mitochondrial respiration, but not citrate synthase activity, increased in MIC19 AAV animals on an HFD (Figures 4F–4H), similar to lean animals. These results demonstrate that MIC19 has beneficial effects on the liver and whole-body energy metabolism.
Figure 4. MIC19 overexpression in the liver has protective effects on an HFD.
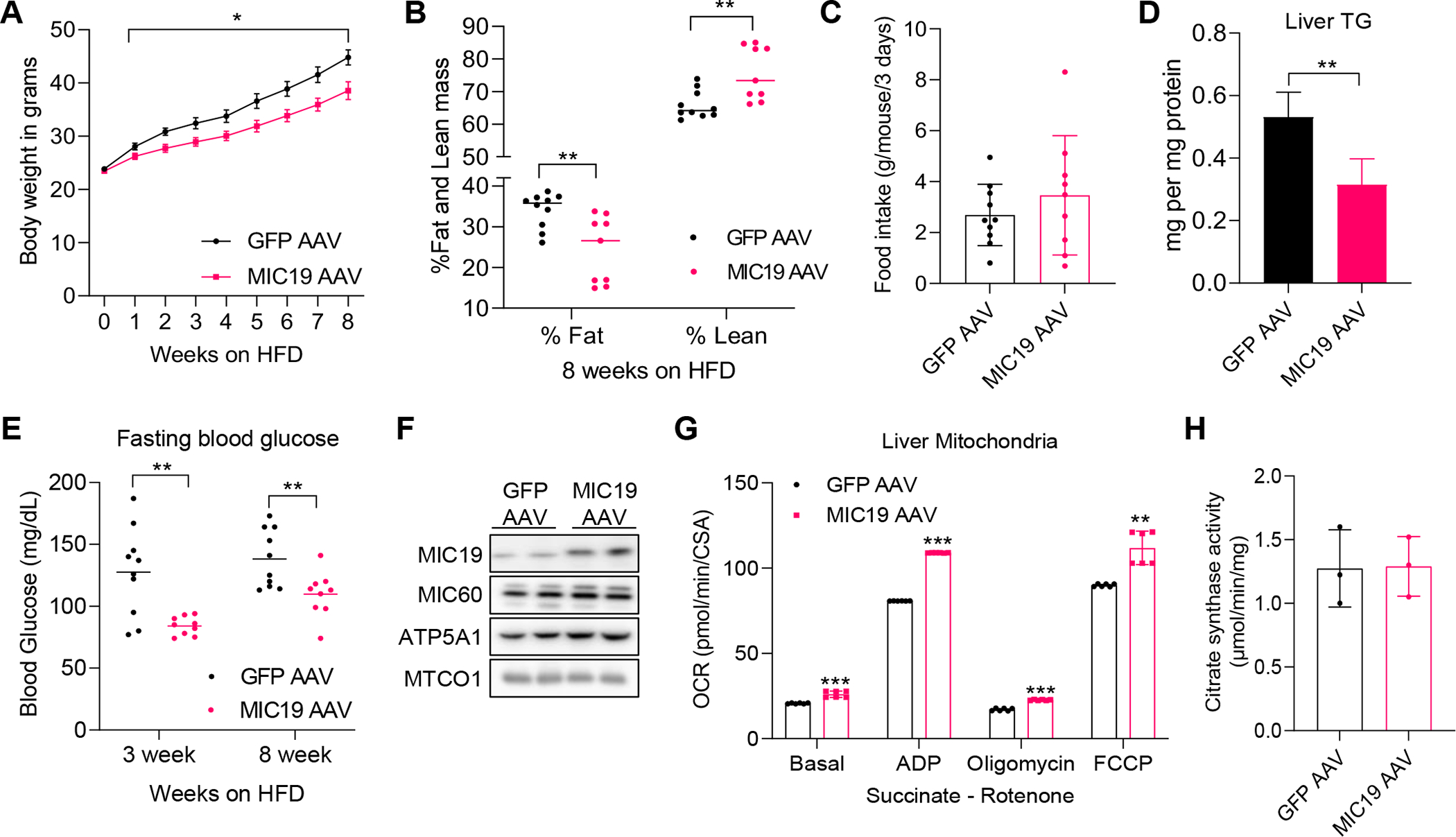
(A) Body weight of GFP (n=10) vs. MIC19 (n=9) AAV animals on an HFD. 6-week-old BL6 animals were administered GFP or MIC19 AAV via the tail vein and put on an HFD at 9 week-old. Data represented as mean±SEM.
(B) Fat and lean mass determined by Magnetic Resonance Imaging (MRI) imaging after 8 weeks on an HFD.
(C) Food intake in grams per mouse measured in metabolic cages across 3 days in GFP vs. MIC19 AAV animals.
(D) Liver triglyceride levels in GFP vs. MIC19 AAV animals on an HFD (n=4).
(E) Fasting blood glucose in GFP (n=10) vs. MIC19 (n=9) AAV animals after HFD.
(F) Western blot showing expression of mitochondrial proteins in samples used to measure mitochondrial respiration (n=2).
(G) OCR measured by Oroboros in isolated mitochondria from GFP AAV vs. MIC19 AAV livers (n=6). OCR was normalized to CSA.
(H) CSA in the mitochondria isolated from HFD-fed GFP AAV vs. MIC19 AAV liver (n=3).
Data represented as mean±SD (C, G, H). *p< 0.05, **p< 0.01, ***p< 0.001 by Student’s t-test (A-E, G, H). n represents the number of biological replicates.
Liver MIC19 overexpression leads to increased pedestrian locomotion
To investigate the effects of MIC19 overexpression on whole animal physiology, we used the Promethion Mouse Multiplexed Metabolic System (Sable Systems International). Mice were individually housed in metabolic cages and metabolic parameters, including locomotion, energy expenditure and respiratory exchange ratio (RER), were measured. To focus on the metabolic effects of liver, we chose to do this analysis at thermoneutrality (30°C), where thermogenic effects of brown fat are negligible. We first assessed if the increased fatty acid oxidation in the liver might shift RER in lean MIC19 AAV animals, since RER is an indicator of carbohydrate vs. fat utilization as a fuel source. Our results showed no changes in RER, suggesting that increased fatty acid oxidation in the liver is not sufficient to alter at the level of whole-body metabolism (Figures 5A and 5B). Total pedestrian locomotion (movement at speeds higher than 1cm/s) was increased significantly in lean MIC19 AAV animals compared to GFP AAV animals (Figures 5C and 5D), and MIC19 AAV mice had increased energy expenditure (EE) (Figures 5E and 5F). Further analysis revealed that MIC19 AAV mice moved more frequently and at higher speeds (Figure 5G).
Figure 5. Animals that overexpress MIC19 in the liver have increased pedestrian locomotion.
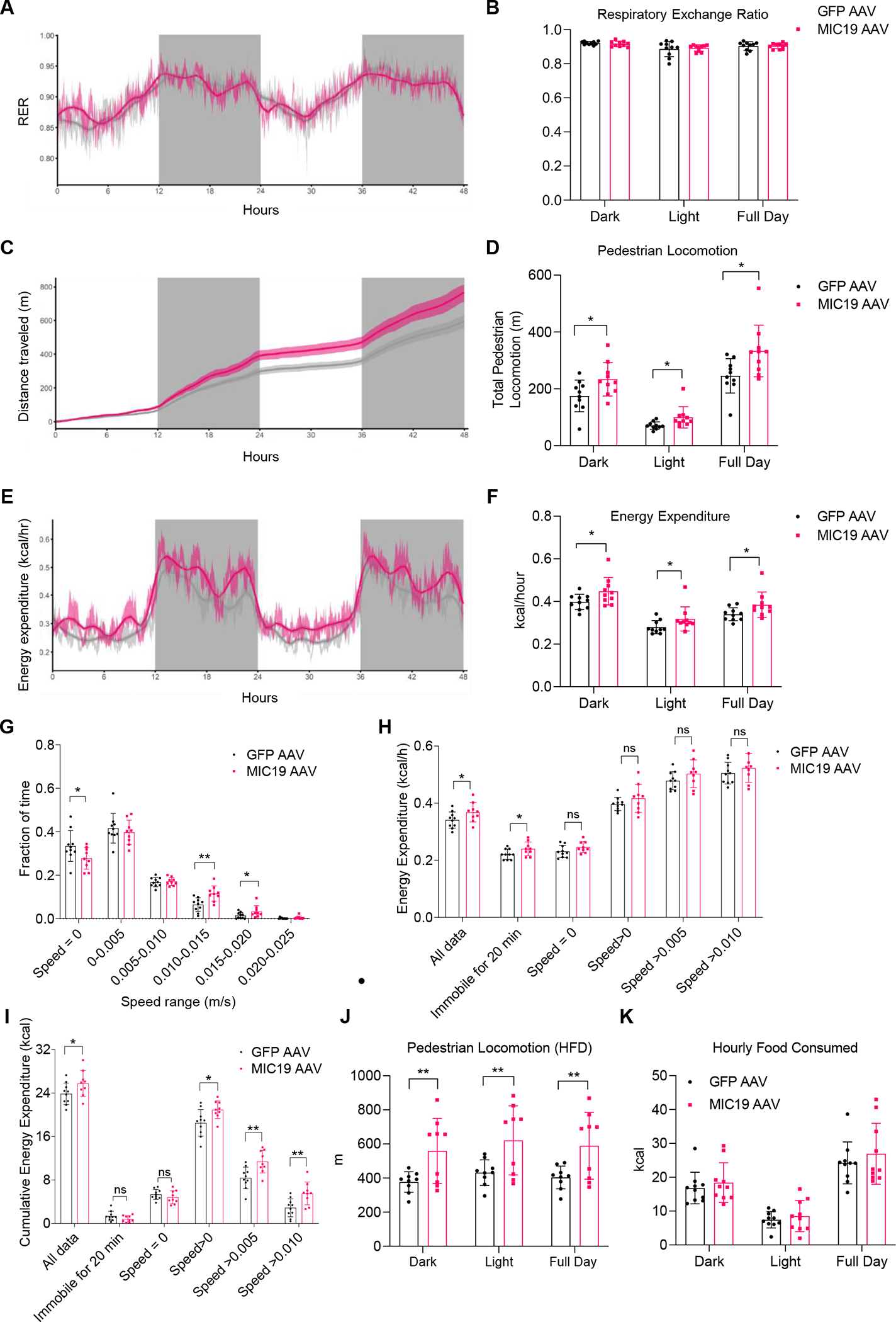
Metabolic parameters across light and dark cycles for GFP vs. MIC19 AAV mice at 30°C.
(A-F) Respiratory Exchange Ratio (RER), cumulative pedestrian locomotion, and energy expenditure (n=10).
(G-I) Fraction of time spent at certain speeds, mean energy expenditure at certain speeds, and cumulative energy expenditure at certain speeds (m/s). n=10 for GFP and n=9 for MIC19 AAV mice.
(J) Pedestrian locomotion in GFP vs. MIC19 AAV mice with a body weight difference on an HFD (n=9).
(K) Hourly Food Consumed in kcal (n=10).
Data represented as mean±SD (B, D, F-K) or as mean±SEM (A, C, E). *p< 0.05, **p< 0.01 by Student’s t-test (B, D, F-K). n represents the number of biological replicates. See also Figure S4.
We determined if the increase in EE could be explained by thermogenesis in brown fat in MIC19 mice. We quantified mRNA levels for thermogenic genes in brown fat and iWAT, and found that there was no change (Figures S4A and S4B). Although we cannot rule out the possibility that BAT activity could affect EE without regulation of thermogenic gene expression, the effect of thermogenic activity on EE in MIC19 mice would be not significant because the experiments were conducted under thermoneutral conditions. Next, we checked if the increased EE was due to increased locomotion or intrinsic changes to liver energy metabolism. We measured mean and sum EE during times when mice are immobile or move at certain speeds (Figures 5H and 5I). We found that mean energy expenditure increased in MIC19 AAV animals during times even when animals were immobile for 20 minutes (Figure 5H), suggesting that intrinsic liver metabolism contributes to this process. At higher speeds, mean EE increased compared to immobility, but was not different between GFP and MIC19 AAV animals (Figure 5H). Cumulative energy expenditure, on the other hand, was increased in MIC19 AAV animals at higher speeds since they spent more time moving at these speeds, whereas it didn’t change between the two groups when animals were immobile (Figure 5I). The increase in total pedestrian locomotion was also present in MIC19 AAV mice on an HFD (Figure 5J). Since food intake was not different in GFP vs. MIC19 animals (Figure 5K), these results suggest that the increase in EE is due to the combined effect of increased pedestrian locomotion and rewiring of liver energy metabolism.
MIC19 overexpression in the liver leads to accumulation of uracil, which promotes locomotion
Overexpression of MIC19 in liver mitochondria promoted locomotor activity in mice, raising the possibility that a liver secreted metabolite influenced neurons that regulate locomotion. Increased fatty acid oxidation in the liver can provide ketone bodies, which can be oxidized in extrahepatic mitochondria, as well as act as signaling molecules. A previous study had shown that fasting increased serum β-hydroxybutyrate (β-OHB), leading to food anticipation and increased movement54,55. Thus, we tested serum levels of ketone bodies, including β-OHB and acetoacetate, in GFP vs. MIC19 AAV mice. We found that there was no change in β-OHB and acetoacetate levels (Figures S5A and S5B). Given this result, we decided to take an unbiased approach and perform metabolomics to compare metabolite levels in GFP vs. MIC19 AAV livers (Figure 6A and Table S2). One of the most significantly enriched categories was ‘pyrimidine metabolism’ (Figure S5C). Upon closer analysis of this category, we found that uracil accumulated in MIC19 AAV livers, whereas uridine and cytidine were depleted (Figure 6B), suggesting their conversion into uracil. Uracil levels in the liver oscillate with the circadian clock, and levels reach maximum right before the dark phase when animals are more active56. We supplemented wild-type mice with exogenous uracil. Uracil supplementation was sufficient to increase total pedestrian locomotion and uracil supplemented animals moved at higher speeds (Figures 6C and 6D). Food intake remained the same (Figure S5D). When EE was analyzed, we did not observe any difference in mean EE but observed an increase in cumulative EE at higher speeds (Figures S5E and S5F). These data are consistent with a model where MIC19 overexpression in the liver has a two-pronged mechanism for increasing EE: i) rewiring of liver mitochondrial energy metabolism and ii) increased locomotion through accumulation of uracil. Interestingly, uracil levels in the brain were increased and serum uracil levels also tended to be increased in MIC19 AAV animals (Figures S5G and S5H). However, it remains unclear whether the increased locomotion is directly mediated by uracil or an uracil derived metabolite.
Figure 6. Uracil accumulates in MIC19 AAV livers and promotes locomotion.
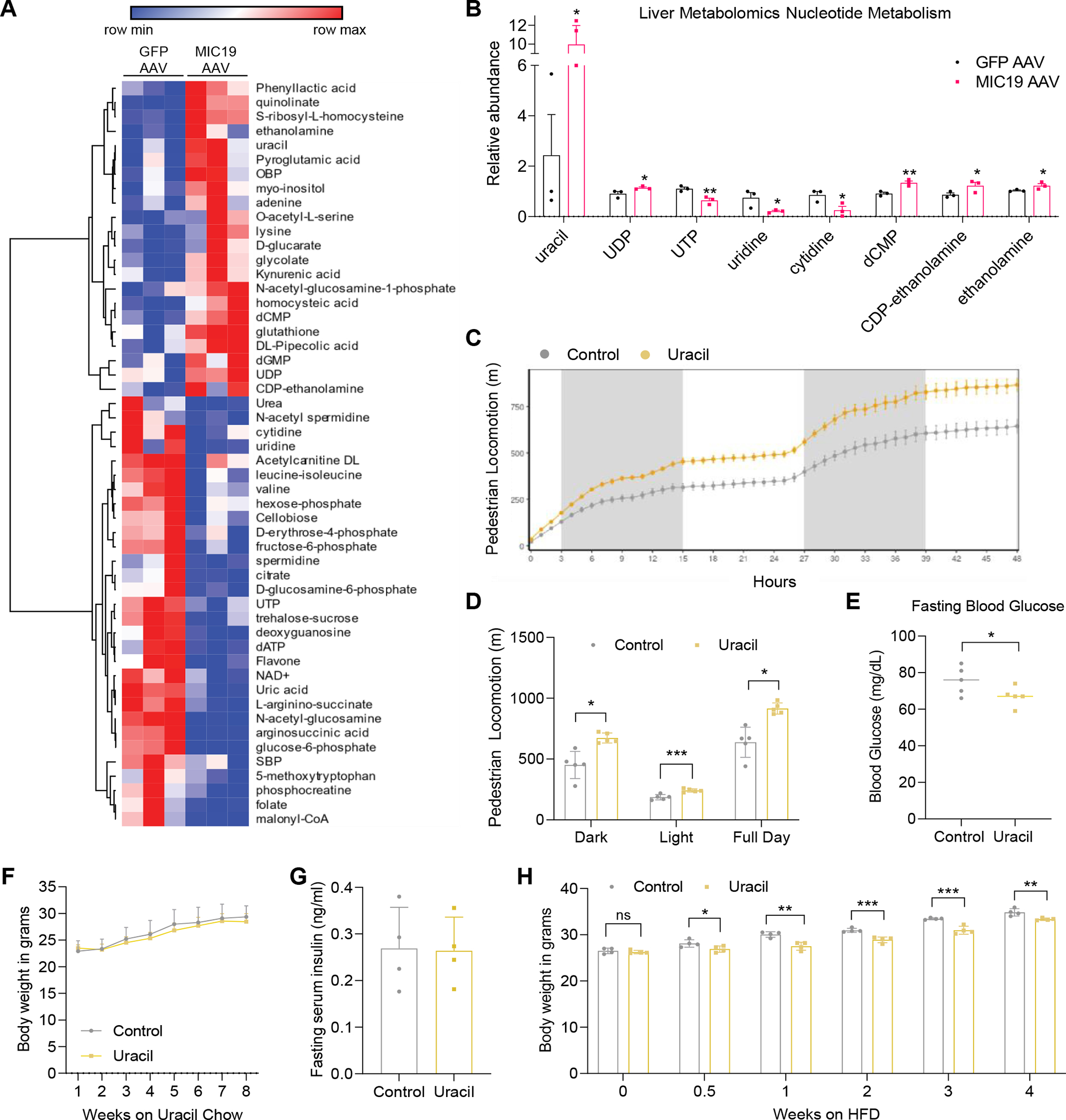
(A) Heatmap showing metabolites that change significantly between GFP vs. MIC19 AAV livers (p<0.05, n=3).
(B) Relative abundance of pyrimidine nucleotides in GFP vs. MIC19 AAV livers.
(C and D) Pedestrian locomotion in mice supplemented with uracil (0.1% in drinking water, n=5).
(E-G) Fasting blood glucose, body weight, and fasting serum insulin level of mice on a standard chow diet with or without 1% uracil supplementation (n=4~5).
(H) Body weight of mice on an HFD supplemented with uracil (0.1% in drinking water, n=4).
Data represented as mean±SD (D-H) or as mean±SEM (B, C). *p< 0.05, **p< 0.01, ***p< 0.001 by Student’s t-test (A, B, D-H). n represents the number of biological replicates. See also Figure S5.
Next, we investigated how uracil supplementation affected glucose homeostasis. Lean animals supplemented with uracil had higher uracil levels in serum and lower fasting blood glucose levels than control mice (Figures S5I and 6E). However, there was no changes in body weight and fasting serum insulin level (Figures 6F and S5J). Cristae formation and MIC19 protein expression in the liver also were not altered upon uracil supplementation, suggesting that exogenous uracil regulates locomotive activity directly without changes in hepatic MIC19 expression and cristae structure (Figures S5K and S5L). We tested if uracil supplementation could also be protective against weight gain on HFD and started feeding with this diet at the same time as uracil supplementation. Just after 3.5 days, uracil supplemented animals gained less weight compared to the controls and these effects became more apparent over time despite no changes in food intake (Figures 6H and S5M). These results suggest that uracil supplementation can recapitulate most pedestrian locomotion benefits provided by liver MIC19 overexpression.
MIC19 overexpression leads to increased UPP2 activity in the liver, leading to uracil accumulation
MIC19 overexpression led to uracil accumulation, which promoted locomotion (Figure 6). To investigate how uracil levels were regulated by MIC19, we tested if uridine, which can be converted into uracil, could also induce locomotion when exogenously supplemented to wild type animals. Addition of 3% uridine to the water caused increased pedestrian locomotion (Figures 7A and 7B), while food and water intake remained unchanged (Figures S6A and S6B). Uridine supplementation also protected against weight gain on an HFD (Figure S6C). A previous study had shown that during fasting, adipose tissue is the main source of uridine in the liver57. We found that uridine levels remained the same in iWAT of MIC19 animals (Figure 7C), suggesting that the liver might use local uridine or from other sources. Previous reports demonstrated that uridine is converted into uracil by UPP2, whose expression is regulated by the circadian clock56. The fact that uridine was decreased but uracil increased (Figure 6B), suggested that levels or activity of UPP2 might be increased in MIC19 AAV livers. We quantified the levels of UPP2 by qPCR and Western blot and found that there was no change (Figures 7D and 7E). We measured the conversion of 3H-uridine to 3H-uracil to quantify UPP2 activity in liver lysates. MIC19 AAV liver lysates had increased UPP2 activity, and treatment with the UPP2 inhibitor 5-benzylacyclouridine (1 mM) inhibited UPP2 activity as expected (Figure 7F).
Figure 7. Conversion of uridine to uracil is increased in MIC19 AAV animals in a redox sensitive manner.
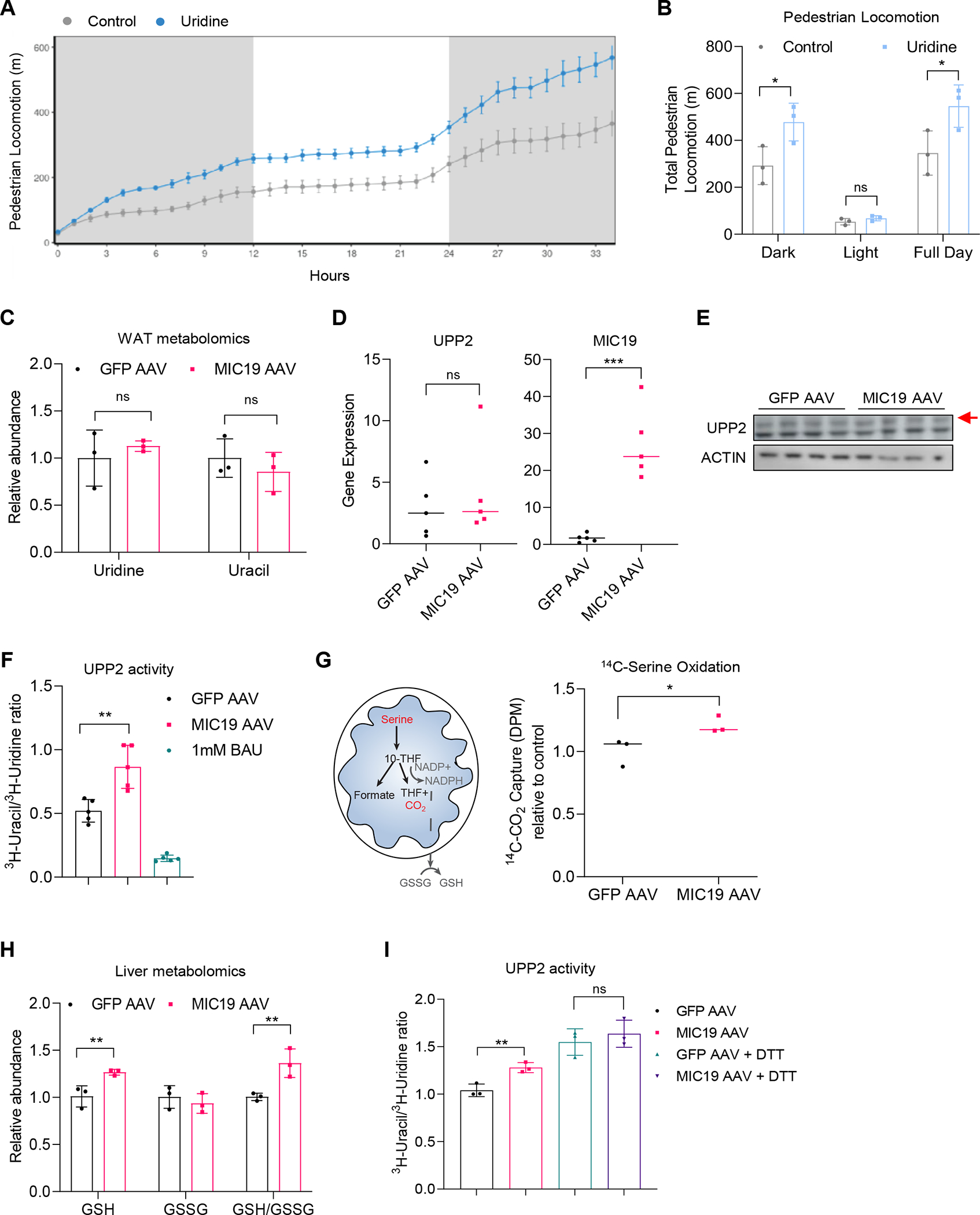
(A and B) Pedestrian locomotion in mice supplemented with uridine (3% drinking water, n=3 mice).
(C) Relative abundance of uridine and uracil in iWAT in GFP vs. MIC19 AAV mice from a metabolomics experiment (n=3).
(D) UPP2 and MIC19 mRNA expression levels in GFP vs. MIC19 AAV livers (n=5).
(E) Western blots showing UPP2 protein levels in GFP vs. MIC19 AAV livers. Red arrow denotes the size for UPP2.
(F) Ratio of 3H-uracil to 3H-uridine as a measure for UPP2 activity in GFP vs. MIC19 AAV liver lysates. Treatment of GFP AAV lysates with 1 mM BAU inhibits UPP2 activity (n=5).
(G) Left: Cartoon depicting the oxidation of serine through the mitochondrial one carbon pathway and its connection to glutathione. Right: 14C-serine oxidation in liver homogenates from GFP vs. MIC19 AAV livers normalized to CSA, shown relative to GFP AAV (n=3).
(H) Reduced and oxidized glutathione levels in GFP vs. MIC19 AAV livers (n=3). (I) UPP2 activity in GFP vs. MIC19 AAV liver lysates with or without 10 mM DTT treatment (n=3).
Data represented as mean±SD (B, C, F, H, I) or as mean±SEM (A). *p< 0.05, **p< 0.01, ***p< 0.001 by Student’s t-test (B-D, F-I). n represents the number of biological replicates. See also Figure S6.
Previously, we found that defects in mitochondrial OxPHOS were linked to compromised mitochondrial one carbon pathway and reduction in NADPH and GSH levels58. We assessed if remodeling of liver mitochondria by MIC19 could increase the mitochondrial one carbon pathway. We used isolated mitochondria from GFP vs. MIC19 AAV livers and tracked the fate of 14C-serine and its release as 14C-CO2. Liver mitochondria from MIC19 AAV animals had an increase in serine oxidation (Figure 7G), suggesting mitochondrial one carbon pathway activity is increased upon MIC19 overexpression. This could potentially lead to an increase in GSH levels in the liver and alter the activities of redox sensitive enzymes, such as UPP2. Formation of a intramolecular disulfide bridge between Cys95 and Cys102 within the protein can disable the UPP2 enzyme and reduced gluthathione (GSH) or DTT treatment can increase enzymatic activity59. GSH and GSH/GSSG ratios were increased in MIC19 AAV livers (Figure 7H), raising the possibility that redox balance is what drives UPP2 activity as previously reported. We obtained similar results, where the ratio of 3H-uracil to 3H-uridine was slightly increased after DTT treatment (Figure 7I). Consistent with our model that UPP2 is redox sensitive, differences in UPP2 activity between GFP vs. MIC19 AAV livers disappeared after DTT treatment (Figure 7I). These results suggest that UPP2 is the regulatory point for uracil accumulation in the liver.
DISCUSSION
Living organisms constantly need to adapt to nutritional changes in the environment and can switch between utilizing different fuel sources depending on their availability (e.g., glucose or fatty acids). The loss of adaptive capacity to nutritional changes is a typical hallmark of metabolic diseases including T2D and obesity60. Here, we show that overexpression of the MICOS complex subunit MIC19 and formation of mitochondrial cristae can rewire liver energy metabolism to suppress glucose production and promote fatty acid oxidation. Altered liver redox status after MIC19 overexpression increases UPP2 activity and conversion of uridine to uracil, which are naturally oscillating metabolites in the liver. Altered pyrimidine metabolism in the liver has far-reaching effects and promotes locomotor activity in mice. Rewiring the liver energy metabolism and promoting physical activity by MIC19 overexpression in the liver provides benefits during obesity and T2D.
Mitochondrial cristae formation and mitochondrial elongation
Elongation of mitochondria is a phenotype shared between the two mouse strains. Previous reports have suggested that starvation elongates mitochondria with a clear mechanism of action46,47. cAMP levels are increased upon starvation, which induces PKA and leads to DRP1 phosphorylation. Phosphorylated DRP1 is retained in the cytosol, favoring unopposed mitochondrial fusion, which elongates mitochondria. Elongated mitochondria possess more mitochondrial cristae. Our results show that BL6 strain can elongate mitochondria like the CD-1 strain. Yet, BL6 is unable to remodel the inner mitochondrial membrane (Figures 1 and 2). These results suggest that the two processes can be separated. Mitochondrial cristae formation is a complex process involving a coordinated effort of proteins like MICOS complex and lipids to establish the curvature of membranes and cristae junctions. MIC10 and MIC60, subunits of the MICOS complex, have the ability bend membranes for cristae biogenesis, whereas MIC19 was postulated to be a redox-dependent regulator of MICOS complex stability (reviewed in61). MIC19 shows two redox forms and only oxidized MIC19 can bind to MIC6062, potentially acting as a connector between two MICOS subcomplexes and leading to cristae formation41. In addition, interaction between MIC19 and MIC60 enhances the mitochondrial membrane-shaping activity and tetramerization of MIC60, thereby controlling structure and function of cristae junction63,64. Our work highlights MIC19 as a rate-limiting subunit of the MICOS complex that is sufficient to drive cristae formation in the liver and is differentially regulated in the CD-1 vs. BL6 livers upon fasting.
Mechanism of regulation of MIC19 levels by fasting in the CD-1 strain
Fasting leads to accumulation of MIC19 in mitochondria in the CD-1 strain, but not the BL6 strain. The layer of regulation on MIC19 could be transcriptional, translational, or based on protein degradation or mitochondrial import. We have ruled out transcriptional regulation in our studies. Future work will reveal if the exact mechanism by which CD-1 upregulates MIC19 upon fasting. We have seen an increase in MIA40 substrates (Figure 1G) and TOM70 protein levels (Figures 1H and 1I). Both pathways are responsible for protein import into mitochondria and may act as key regulators to remodel the mitochondrial inner membrane proteome upon fasting. Intriguingly, MIC19 was previously reported to be a substrate for the MIA pathway in yeast45. Previous studies have shown that post-translational modifications on TOM70 by OGT can directly influence the import efficiency of MIC19 into mitochondria in brown fat during cold exposure38. Briefly, cold stimulation activates PERK, a transmembrane kinase that localizes to the ER, which phosphorylates OGT. OGT glycosylates TOM70, enhancing MIC19 import into mitochondria. OGT also glycosylates CK2α and reduces its activity, which normally phosphorylates TOM70 to decrease mitochondrial import. The signature from our proteomics dataset raises the possibility that similar mechanisms that regulate mitochondrial import may exist in CD-1 liver. However, the regulatory mechanisms of fasting-induced MIC19 protein levels in the liver mitochondria could be different between CD-1 and BL6. The lack of change in MIC19 protein levels upon fasting in BL6 liver mitochondria might be due to already elevated expression under ad libitum condition (Figure 1H).
Contribution of intrinsic liver energy metabolism vs. locomotion to whole body energy expenditure upon MIC19 overexpression
The increase in energy expenditure in MIC19 overexpressing mice had 3 potential explanations: i) thermogenesis in the adipose tissue, ii) rewiring of liver energy metabolism and iii) increased locomotor activity. Most of the work on increasing EE to battle obesity and T2D in genetic and dietary mouse models has focused on thermogenic adipocytes and physical activity. The contribution of liver metabolism to energy expenditure remains largely underexplored. A previous study showed that the liver-targeted mitochondrial uncoupler DNP, which induces the conversion of energy derived from mitochondrial substrate oxidation into heat, has beneficial effects on metabolic disease65. Similarly, our results demonstrate that mitochondrial cristae formation driven by MIC19 overexpression in the liver can increase EE in mice (Figure 5). Our experiments were done at thermoneutrality (30°C), where contribution of adaptive thermogenesis to EE is negligible. Nor do we see upregulation of thermogenic markers in adipose tissue (Figure S4), ruling out adaptive thermogenesis. Physical activity also increases EE, and at 30°C accounts for 26% of total EE66. To dissect the contribution of movement vs. intrinsic energy metabolism to EE in our experiments, rather than immobilizing animals artificially, we mined the indirect calorimetry data to focus on time points where animals don’t move or move at low or high speeds. Our in-depth analysis of mice at 30°C shows that EE is increased even when MIC19 overexpressing animals are immobile, suggesting rewiring of liver energy metabolism is a crucial contributor. We show that MIC19 overexpressing animals spend more time moving at higher speeds, and as a result, the cumulative EE at higher speeds is increased in MIC19 overexpressing mice. These results suggest that both liver metabolism and physical activity contribute to increased EE.
Regulation of pedestrian locomotion by pyrimidine nucleotides
We find that the conversion of uridine to uracil by UPP2 is important for the accumulation of uracil in MIC19 overexpressing livers. Previously, uridine and uracil have been shown to oscillate with the circadian clock in the liver through transcriptional regulation of UPP256. The timing of uridine and uracil levels correlate with movement, where uracil reaches a peak right before the dark phase where nocturnal mice feed and move more. In our study, exogenous uridine or uracil supplementation was sufficient to promote locomotor activity in mice. It is intriguing that mice that overexpress MIC19 in the liver are tapping into a physiological response present in healthy animals to regulate physical activity. UPP2 gene or protein expression levels remain unchanged in MIC19 overexpressing animals, suggesting that the circadian rhythm is still intact in these mice. It is unclear whether a pyrimidine nucleotide is responsible for crossing the blood-brain-barrier (BBB) and directly inducing locomotion by binding purinergic receptors67. Circulating uridine is able to cross the BBB via the CNT2 transporter in rats68, but there is limited evidence for uracil uptake by the brain. The brain uptake index of [14C]-uracil and [14C]-uridine were estimated to be 0.83% and 4.18% respectively69,70. However, in our dataset, we observe a subtle but significant increase in uracil in the whole brains of MIC19 overexpressing mice, but not uridine (Figure S5G). It could be that these metabolites are enriched locally in specific subregions of the brain, missed by our analysis.
Interestingly, circulating uridine has been previously linked to regulation of feeding behavior and thermoregulation via the hypothalamus in the brain57,71. AgRP neurons that promote feeding express the purinergic receptor 6 (P2Y6), which is activated by UDP. The source of hypothalamic UDP is circulating uridine, which is elevated during obesity71. Plasma uridine levels are also tightly regulated by fasting/feeding cycles. In the fed state, the liver is the major organ responsible for circulating uridine. During fasting, the adipose tissue takes over uridine production and circulating uridine levels are increased, leading to a subtle drop in body temperature57. These studies raise the possibility that a pyrimidine nucleotide derived signal could mediate the locomotor effects as well. Alternatively, glutamatergic neurons in the midbrain could be the downstream and indirect targets of the pyrimidine metabolites to regulate locomotion72.
Redox regulation of UPP2
Our results show that UPP2 activity is increased in MIC19 animals in a redox sensitive manner (Figures 7H and 7I). Previous reports demonstrated that a conditional disulfide bridge forms within UPP2 (between C95 and C102), and dislocates the critical phosphate-coordinating arginine (R100) away from the active site, disabling UPP2 activity. Vice versa, treatment with reducing agents increases the activity of UPP259. The increase in GSH in MIC19 overexpressing livers likely explains the increase in UPP2 activity. Interestingly, we found that the one carbon metabolism was increased in mitochondria isolated from MIC19 overexpressing livers (Figure 7G), a pathway that can supply GSH to cells58.
In summary, these studies reveal that MIC19 in the liver provides metabolic benefits at the whole-body level in mice during T2D and obesity, through changes in liver energy metabolism and increased physical activity. Future work will further elucidate the signaling pathways that link the liver mitochondria to the brain and the downstream effects of pyrimidine nucleotides that are produced in the liver.
LIMITATIONS OF THE STUDY
This study suggested that enhanced UPP2 activity regulated by redox status promoted uracil accumulation in the livers of MIC19 overexpressing mice, resulting in the increased locomotion and energy expenditure. To clarify the roles of UPP2 in vivo, further analysis using liver-specific UPP2 knockout mouse model will be needed. In addition, to validate whether enzymatic activity of UPP2 is stimulated by increased GSH level in the liver of MIC19 overexpressing mice, it is required to assess UPP2 activity in the liver and locomotive activity upon manipulation of hepatic GSH level.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pere Puigserver (Pere_Puigserver@dfci.harvard.edu).
Material availability
All the materials generated in this study are available upon reasonable request to the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Procedures.
All C57BL6/J mice were purchased from Jackson laboratories (Stock: 000664). Mice were kept under a 12 h light/12 h dark cycle at 22 °C. Before handling, mice acclimated for at least 1 week at the animal facility. All studies were performed according to protocols approved by Beth Israel Deaconess Medical Center’s Animal Care and Use Committee. Mice were fed a standard chow diet (13% kcal fat, LabDiet, no. 5053) or an HFD (60% kcal fat, Research Diets, no. D12492i) for the indicated durations. For uracil supplementation, we used the Open Standard Diet (D11112201) vs. the same diet with 1% uracil supplementation. We also supplemented the drinking water of mice with 0.1% uracil due to low solubility in water. For uridine, 3% supplementation in drinking water was used. Glucose tolerance tests were performed with 2 g/kg glucose injections via I.P. for lean animals and 0.5 g/kg glucose for DIO mice on an HFD. Blood glucose was measured via tail bleed using a glucometer (OneTouch). For GTT and fasting blood glucose, animals were fasted overnight for 16 hours. For all experiments, age matched animals were used. AAV vector pAAV[Exp]-CAG>mChchd3[NM_001355667.1]:WPRE was purchased from VectorBuilder (Chicago, Illinois) and packaged into serotypes 2 and 8 for liver specific expression by Boston Children’s Hospital Viral Core (AAV2/8-CAG-GFP-WPRE and AAV2/8-CAG-mChchd3-WPRE-bGH). GFP or MIC19 AAVs were administered via the tail vein at 1.5×1011gc per mouse.
Hepatocyte Isolation.
Primary hepatocytes were isolated from 8- to 12-week-old male C57BL6/J mice. Livers of mice were perfused with liver digest medium (Invitrogen, 17703–034) and filtered through a 70 μm mesh. Primary hepatocytes were separated from other cell types and cell debris by a Percoll (Sigma, P7828) gradient centrifugation. Cells were seeded in Plating Medium (DMEM with 25 mM glucose, 10% FBS, 2 mM sodium pyruvate, 1% penicillin/streptomycin, 1 μM dexamethasone, and 100 nM insulin). After 4 h, the Plating Medium was removed and cells were incubated overnight in the Maintenance Medium (DMEM with 5 mM glucose, 0.2% BSA, 2 mM sodium pyruvate, 1% penicillin/streptomycin, 0.1 μM dexamethasone, and 1 nM insulin). Cells were collected within 48 h and medium was changed every day. For transient transfection, hepatocytes were transfected with DNA plasmids and siRNA (Origene) using Lipofectamine 3000 (Invitrogen). For AAV infection, cells were incubated for 16 h with AAV at MOI of 10000.
METHOD DETAILS
Indirect Calorimetry.
Metabolic parameters of mice were measured by using Sable System’s Promethion. Briefly, animals were housed in individual metabolic cages and oxygen consumption, carbondioxide production, food/water intake, body weight, physical activity was monitored for 48 hours. RER, Energy Expenditure and Pedestrian Locomotion was calculated as previously described 73 and data was analyzed using CalR software (https://calrapp.org/) 74. Pedestrian speed and energy expenditure was calculated at 5-minute intervals and used for speed cut-offs in analysis in Figures 5G, 5H, 5I, S5E, and S5F. To analyze energy expenditure at specific speeds, R software was used. Briefly, speeds were filtered according to the different criteria, and the corresponding energy expenditure at those time points were averaged or summed.
Glucose Production.
Hepatocytes were incubated for the last 3 hours in glucose-free medium (phenol-red/glucose free DMEM, 0.2% BSA, 2 mM sodium pyruvate and 20 mM sodium lactate). 200 nM glucagon was used for stimulation during the treatment and the cell medium was collected. The glucose level in the medium was measured using a Glucose (GO) Assay Kit from Sigma-Aldrich. 100 μl of assay buffer was added to 50 μl of cell medium. The mixture was incubated for 30 min at 37°C. 100 μl 6 M Sulfuric acid was added to the samples and absorbance was measured at 540 nm.
14C-Glucose and 14C-Palmitate Oxidation Assays.
Hepatocytes were incubated in Maintenance Medium for 2 days. On the day of the experiment, cells were treated with 14C substrates in DMEM without glucose, 0.2% BSA, 2 mM sodium pyruvate, 1% penicillin/streptomycin. 14C-glucose was added to 0.5 μC/well in 12-well plates.14C-palmitate was added to 0.4 μC/well in medium supplemented with 0.3% BSA and 100 μM unlabeled palmitate in each well as previously described 75. Circles of Whatman filter paper were inserted into the cap of 2 ml Eppendorf tubes and 400 μl of 1 M perchloric acid added to the bottom of the tube. 30 μl 2 M Sodium hydroxide was added to the filter paper to moisten it right before harvesting the medium from the cells. 800 μl medium was added on top of the perchloric acid to trap the 14C-CO2 on the filter paper. The samples were incubated for 4 hours and radioactive signal from the filter paper was measured in the scintillation counter for 3 minutes.
Proteomics.
Proteomics was performed as previously described 76. Briefly, livers were harvested and homogenized in MSHE Buffer (210 mM Mannitol, 70 mM Sucrose, 1 mM EGTA, 5 mM HEPES). Homogenized livers were centrifuged at 1000g for 5min at 4°C twice and the supernatant saved. The supernatant was centrifuged at 12000g for 20 minutes at 4°C to pellet mitochondria and mitochondria were washed with MSHE buffer 3 times. Mitochondria were solubilized in SDS lysis buffer (2% SDS, 250 mM NaCl, PhoStop Roche, phosphatase inhibitors, EDTA free protease inhibitor cocktail (Promega) and 100 mM HEPES, pH 8.5). Samples were reduced with 5 mM TCEP at 60°C for 30 minutes and alkylated with 14 mM iodoacetamide for 45 minutes. After chloroform/methanol precipitation, samples were solubilized in 8 M urea dissolved in 50 mM HEPES, pH 8.5 and protein content measured. 500 μg of protein for each samples was diluted to 4 M urea with digestion buffer (25 mM HEPES, pH 8.5). Proteins were digested with LysC (Wako, 2 μg/μl, 1:100) for 2 hours. Samples were then diluted to 2 M urea and further digested with LysC and Trypsin (0.5 μg/μl, 1:100) overnight at 37°C. In the morning, samples were diluted to 1 M urea and digested with Trypsin for 6 hours at 37°C. Samples were then acidified with 200 μl 25% acetic acid and centrifuged at maximum speed for 5 minutes and subjected to C18 solid-phase extraction (SPE) (50mg, Sep-Pak, Waters). 50 μg of digested peptides were labeled with tandem mass tag (TMT) reagents (Thermo Fisher Scientific), combined into one fraction, acidified with 20 μl of 20% FA (pH ~2) and subjected to C18 SPE on Sep-Pak cartridges (50 mg). Next, basic pH reversed-phase HPLC, followed by MS analysis was performed. Mitochondrial proteins were identified based on MitoCarta3.0. DAVID (https://david.ncifcrf.gov/tools.jsp) was used for Gene Ontology (GO) Term analysis.
Oxygen Consumption Measurements.
Isolation of mitochondria was performed as described in the proteomics section, with the addition of 1% BSA to MSHE to prevent clumping of mitochondria. Liver mitochondria were diluted in MAS Buffer (70 mM Sucrose, 220 mM Mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% BSA) and seeded on an XFe24 Seahorse plate (Seahorse Biosciences 102340–100) at 5–10 μg/well. The plate was centrifuged at 2000g for 20 minutes. For complex I-dependent OCR, the OCR was measured following treatment with 5 mM pyruvate+3 mM malate, 1 mM ADP, and 5 μM oligomycin+ 5 μM FCCP. For complex II-dependent OCR, the OCR was measured following treatment with 5 mM succinate+8 μM rotenone, 1 mM ADP, and 5 μM oligomycin+ 5 μM FCCP. OCRs (pmol/min) were assessed for each condition at 37°C using the Seahorse Bioanalyzer instrument as previously described 51. We also used the Oroboros O2K system to measure mitochondrial respiration. Injections were 1 mM ADP for maximal respiration, 1 μM oligomycin and 1 μM FCCP for maximal uncoupled respiration. Respiration rates were normalized to citrate synthase activity 77. Cellular OCR was analyzed using primary hepatocytes. Prior to analysis, primary hepatocytes were infected with AAV and were incubated in assay medium (25 mM glucose, 1 mM sodium pyruvate, 2 mM L-glutamine, and 0.2% fatty acid–free BSA in Seahorse XF base medium at pH 7.4). For mitochondrial stress tests, the OCR was measured following treatment with 3.75 μM oligomycin, 1 μM FCCP, and 4.5 μM antimycin A with 1.5 μM rotenone.
MIC19 Protein Import Assay.
Liver mitochondria were isolated and resuspended in the isolation buffer (20 mM HEPES-KOH, pH 7.4, 75 mM sucrose, 225 mM mannitol and 0.05 mM EGTA). The T7-based TnT reticulocyte system (Promega) was used to produce 35S-labeled MIC19 proteins in accordance with the manufacturer’s instructions. 20 μg of mitochondria were resuspended in 100 μL of Protein Import Buffer (50 mM HEPES-KOH, pH 7.4, 250 mM sucrose, 80 mM KOAc, 5 mM MgAOc, 0.1% BSA (fatty acid free), 2 mM ATP, 20 mM sodium succinate and 1 mM methionine). In order to eliminate non-imported precursors, proteinase K (50 μg/mL) was added to the TnT reticulocyte product and then incubated for 15 minutes on ice. To neutralize proteinase K, mitochondria were diluted in wash buffer with newly added 1 mM PMSF, and then centrifuged at 12,000 g for 3 minutes. There were two cycles of this wash.
Transmission Electron Microscopy (TEM).
Livers were perfused in 2.5% Glutaraldehyde 2.5% Paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) and cut into small pieces to store in the same buffer overnight at 4°C. Primary hepatocytes were fixed using a 1:1 mixture of cell media and the same buffer (2x) overnight at 4°C. After fixation the samples were submitted to Harvard Medical School Electron Microscopy Core. Liver tissue was shed in 0.1 M cacodylate buffer and post-fixed with 1% Osmiumtetroxide (OsO4)/1.5% Potassiumferrocyanide (KFeCN6) for 1 hour, washed in water 3x and incubated in 1% aqueous uranyl acetate for 1 hr followed by 2 washes in water and subsequent dehydration in grades of alcohol (10 min each; 50%, 70%, 90%, 2×10min 100%). The samples were then put in propyleneoxide for 1 hr and infiltrated ON in a 1:1 mixture of propyleneoxide and TAAB Epon (Marivac Canada Inc. St. Laurent, Canada). The following day the samples were embedded in TAAB Epon and polymerized at 60°C for 48 hrs. Ultrathin sections (about 80 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids, stained with lead citrate and examined in a JEOL 1200EX Transmission electron microscope or a TecnaiG2 Spirit BioTWIN. Images were recorded with an AMT 2k CCD camera and saved as TIFF files. For quantification, cristae numbers were normalized to mitochondrial area. Mitochondrial area was calculated in ImageJ by manually tracing the outer mitochondrial membrane for each mitochondrion.
Metabolomics.
500 μl of 80% Methanol (v/v) was added to approximately 10–15 mg of pulverized tissues and homogenized using metal beads in a shaker for 3 minutes. The samples were incubated at −80°C for 4 hours and centrifuged at 14000g for 10 minutes at 4°C. The supernatant was stored at −80°C and the pellet was used for re-extraction with 80% Methanol. 200 μl of 80% Methanol (v/v) was added to 50 μl of serum and incubated at −80°C for 6 hours. These were centrifuged twice at 14000g for 10 minutes at 4°C. Supernatants from both extractions were combined and lyophilized using a SpeedVac (Thermo Fisher Scientific) overnight at 4°C. Lyophilized samples were resuspended in 20 μl ultrapure water and subjected to metabolomics profiling using AB/SCIEX 5500 QTRAP triple quadrupole instrument as previously described 78,79. Data analysis was performed using Metaboanalyst software.
Lipidomics.
Lipidomic analysis was performed as previously described 80.10 mg pulverized liver tissue was suspended with 200 μl water and mixed with 1.5ml of HPLC-grade methanol and 5 ml of methyl tert-butyl ether (MTBE). Samples were agitated at room temperature for 1 hour. Next, 1.25 ml of water was added to each glass tube and centrifuged at 1000g for 10 minutes to promote phase separation. The upper MTBE phase was collected and dried in a SpeedVac at 4°C overnight. Dried samples were resuspended in 30 μl of 1:1 LC/MS grade isopropanol:acetonitrile methanol and subjected to lipidomics analysis using the QExactive Plus Orbitrat (Thermofisher Scientific) instrument. Data analysis was performed using Metaboanalyst software.
UPP2 Activity Assay.
Measurement of UPP2 enzymatic activity was adapted from 59. Briefly, Frozen pulverized livers were homogenized in 350 μl 50 mM Tris-HCl (pH 7.5 without DTT). Tissue homogenates were cleared by centrifugation at 14000g for 15 minutes at 4°C. The supernatant was used to measure UPP2 activity. The enzyme reaction was carried out in a total volume of 100 μl of 50 mM Tris Buffer (pH 7.5) containing 1 mM potassium phosphate, 10 μl tissue extract and 0.3 μCi [5-3H]uridine at 37°C for 30 minutes. 1 mM BAU was used to inhibit UPP2 activity. UPP2 activity was measured by monitoring conversion of [5-3H]uridine to [5-3H]uracil using TLC chromatographic separation. Aliquots of 10 μl from each sample were applied to TLC silica plates prespotted with 1 μl of uridine and 3 μl of uracil as markers. TLC plates were developed with chloroform:methanol:acetic acid (85:15:5, v/v). The unlabeled markers were visualized under UV light, and the radioactivity for uridine and uracil was measured in a scintillation counter by scraping off corresponding areas into scintillation liquid.
Blue-Native Gel Electrophoresis, SDS-PAGE and Western blot analysis.
Blue-Native Gel electrophoresis and western blot were performed as described previously 51. Only modification to the Blue-Native Gel Electrophoresis assay was that the solubilization of mitochondria was done at 4 g per g digitonin/protein ratio. The list of antibodies that were used: anti-SDHA (Abcam, catalog no. ab14715, 1:3,000), anti-ATP synthase alpha (Life Technologies, catalog no. 459240, 1:1,000), anti-MTCO1 (Abcam, catalog no. ab14705, 1:3,000), anti-actin (Cell Signaling Tech., catalog no. 4967 1:1000), anti-MIC60/Immt (Proteintech, catalog no. 10179-AP, 1:2000), anti-OPA1 (Cell Signaling Tech., catalog no. 67589S, 1:1000) anti-MIC19/Chcd3 (Life Technologies, catalog no. PA531578, 1:1000), anti-TOM70 (Santa Cruz Biotech., catalog no. SC-390545, 1:100), anti-UPP2 (Life Technologies, catalog no. PA550894, 1:100).
Serum and Liver Biochemistry Measurements.
Serum samples were gathered from blood collected from the orbital sinus, tail vein, or heart. To extract fats from liver, ~200 mg of pulverized liver tissue was homogenized in 800 μl PBS with a metal bead in a Tissuelyser for 3 minutes. Some homogenate was saved to measure protein concentration as a control. 400 μl of the homogenate was transferred to a new tube and 800 μl chloroform:methanol (2:1) added and vortexed for 3 minutes. The samples were incubated at 60°C for 15 minutes and centrifuged at maximum speed for 10 minutes at 4°C. 400 μl of the bottom layer was transferred to a new tube and dried overnight. The dried lipids were resuspended in 50 μl of 60% butanol, 40% solution of Triton-X:Methanol (2:1). Kits used to measure β-hydroxybutyrate (MAK041) and triglycerides (TR0100) were purchased from Sigma. Kits used to assess aspartate transaminase (EASTR100) and alanine transaminase (EALT100) were purchased from Bioassay Systems. Serum insulin was measured with Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, 90080).
RNA Isolation and qPCR.
Pulverized livers, brown fat, and iWAT were used to extract total RNA using TRIzol reagent (Ambion, Life Technologies. cDNA was prepared from 2 μg of total RNA with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). RT-qPCR reactions were ran using Sybr Green qPCR mastermix (Applied Biosystems) and analyzed by a CFX384 Real-Time system (Bio-Rad).
Supplementary Material
Table S1. Mitochondria proteome from BL6 and CD-1 liver, related to Figure 1
Table S2. Liver metabolomics from GFP and MIC19 overexpressing mice, related to Figure 6
Table S3. List of oligos used for qPCR assays, related to STAR Methods
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SDHA | Abcam | Cat#ab14715; RRID:AB_301433 |
| ATP5A1 | Life Technologies | Cat#459240; RRID:AB_2532234 |
| MTCO1 | Abcam | Cat#ab14705; RRID:AB_2084810 |
| Actin | Cell Signaling Tech. | Cat#4967; RRID:AB_330288 |
| MIC60 (Immt) | Proteintech | Cat#10179-AP; RRID:AB_2127193 |
| OPA1 | Cell Signaling Tech. | Cat#67589S; RRID:AB_2799728 |
| MIC19 (Chchd3) | Life Technologies | Cat#PA531578; RRID:AB_2549051 |
| TOM70 | Santa Cruz Biotech. | Cat#SC-390545; RRID:AB_2714192 |
| UPP2 | Life Technologies | Cat#PA550894; RRID:AB_2636346 |
| GFP | Abcam | Cat#ab290; RRID:AB_303395 |
| OXPHOS | Abcam | Cat#Ab110413; RRID:AB_2629281 |
| NDUFS2 | Abcam | Cat#ab192022; RRID:AB_2895019 |
| Tubulin | Cell Signaling Tech. | Cat#D3U1W; RRID:AB_2715541 |
| Bacterial and virus strains | ||
| AAV2/8-CAG-GFP-WPRE | Boston Children’s Hospital Viral Core |
N/A |
| AAV2/8-CAG-mChchd3-WPRE-bGH | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Oligomicyn A | Sigma Aldrich | Cat#75351 |
| FCCP | Sigma Aldrich | Cat#75351 |
| Rotenone | Sigma Aldrich | Cat#R8875 |
| Antimycin | Sigma Aldrich | Cat#A8674 |
| Uracil | Sigma Aldrich | Cat#U0750 |
| Uridine | Sigma Aldrich | Cat#U3750 |
| [5-3H]uridine | Moravek | Cat#MT602 |
| Benzylacyclouridine (BAU) | Med Chem Express | Cat#HY-106406 |
| 14C-glucose | Perkin Elmer | Cat#NEC042X |
| 35S-methionine | Perkin Elmer | Cat#NEG709A |
| 14C-palmitate | Perkin Elmer | Cat#NEC075H |
| 14C-serine | Perkin Elmer | Cat#NEC827 |
| Dexamethasone | Sigma Aldrich | Cat#D4902 |
| Insulin | Sigma Aldrich | Cat#I6634 |
| Percoll | Sigma Aldrich | Cat#P7828 |
| Perchloric acid | Sigma Aldrich | Cat#244252 |
| PhosStop phosphatase inhibitor | Roche | Cat#4906845001 |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | Cat#4693159001 |
| Digitonin | Sigma Aldrich | Cat#D141 |
| Critical commercial assays | ||
| Glucose (GO) Assay Kit | Sigma Aldrich | Cat#GAGO20 |
| β-hydroxybutyrate assay kit | Sigma Aldrich | Cat#MAK041 |
| Serum Triglyceride Determination Kit | Sigma Aldrich | Cat#TR0100 |
| Aspartate Transaminase Assay | Bioassay Systems | Cat#EASTR100 |
| Alanine Transaminase Assay Kit | Bioassay Systems | Cat#EALT100 |
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem. | Cat#90080 |
| NativePAGE 3–12% Bis-Tris Protein Gels | Thermo Fisher Sci. | Cat#BN1001BOX |
| Deposited data | ||
| Mitochondria proteome from mouse liver | This study | Table S1 |
| Mouse liver metabolomics | This study | Table S2 |
| Experimental models: Cell lines | ||
| Mouse primary hepatocytes | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL6 mice | Jackson Laboratory | Cat#000664 |
| CD-1 mice | Charles River | Cat#022 |
| Oligonucleotides | ||
| siRNA for M.musculus Mic19 (chchd3) | Origene | Cat#SR406213 |
| siRNA for negative control | Origene | Cat#SR30004 |
| qPCR oligos | Table S3, this paper | N/A |
| Recombinant DNA | ||
| pAAV[Exp]-CAG>mChchd3[NM 001355667.1]:WPRE | VectorBuilder | N/A |
| pET-T7-MIC19 (mouse) | (Latorre-Muro et al., 2021) | N/A |
| Software and algorithms | ||
| CalR | (Mina et al., 2018) | https://calrapp.org/ |
| R | R Development Core Team |
https://cran.r-project.org/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Graphpad Prism 9 | Graphpad | https://www.graphpad.com |
| Other | ||
| liver digest medium | Invitrogen | Cat#17703-034 |
| High fat diet | Research Diets | Cat#D12492i |
| Open Standard Diet | Research Diets | Cat#D11112201 |
| 1% uracil diet | Research Diets | N/A |
HIGHLIGHTS.
Fasting induces strain-specific cristae formation and MIC19 in liver mitochondria
MIC19 drives cristae formation in BL6 liver and protects against diet-induced obesity
Hepatic MIC19 promotes mitochondrial respiration and energy expenditure
MIC19 overexpression in liver promotes pedestrian locomotion through uracil production
ACKNOWLEDGMENTS
We acknowledge the Viral Core Facility at Boston Children’s Hospital for AAV generation. We thank the Electron Microscopy Facility at Harvard Medical School for assistance in the acquisition of data. NIH/NIDDK grants R01DK081418-12, R01DK089883-09, and R01DK117655-04 were awarded to P.P. In addition, J.H.S. was supported by Gottfried Family Women’s Health Fellowship and Friends of Dana-Farber Cancer Institute. Human Frontier Science Program Long-Term Fellowship (LT-000033-2019/L) was awarded to P.L.-M.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. Beste Mutlu is employed by Elsevier as a Scientific Editor in Cell Press. The work reported in the paper was completed before Dr. Mutlu joined Cell Press, and Dr. Mutlu was not involved in the peer-review process or the decision to accept the paper for publication.
DATA AND CODE AVAILABILITY
• All data points used to create the graphs and uncropped western blots are provided in Data S1.
• Original code used to analyze the data is available from the authors on request.
• Any additional information required to reanalyzed the data reported in this paper is available from the lead contact upon request.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Heymsfield SB, and Wadden TA (2017). Mechanisms, Pathophysiology, and Management of Obesity. N Engl J Med 376, 254–266. 10.1056/NEJMra1514009. [DOI] [PubMed] [Google Scholar]
- 2.Roden M, and Shulman GI (2019). The integrative biology of type 2 diabetes. Nature 576, 51–60. 10.1038/s41586-019-1797-8. [DOI] [PubMed] [Google Scholar]
- 3.Bence KK, and Birnbaum MJ (2021). Metabolic drivers of non-alcoholic fatty liver disease. Mol Metab 50, 101143. 10.1016/j.molmet.2020.101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, and Sanyal AJ (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357. 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 5.Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, and Ikramuddin S (2020). Nonalcoholic Steatohepatitis: A Review. JAMA 323, 1175–1183. 10.1001/jama.2020.2298. [DOI] [PubMed] [Google Scholar]
- 6.Browning RC, and Kram R (2005). Energetic Cost and Preferred Speed of Walking in Obese vs Normal Weight Women. Obesity Research 13, 891–899. [DOI] [PubMed] [Google Scholar]
- 7.Stamatakis E, Ahmadi MN, Gill JMR, Thøgersen-Ntoumani C, Gibala MJ, Doherty A, and Hamer M (2022). Association of wearable device-measured vigorous intermittent lifestyle physical activity with mortality. Nature medicine 28, 2521–2529. 10.1038/s41591-022-02100-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machado MV (2021). Aerobic Exercise in the Management of Metabolic Dysfunction Associated Fatty Liver Disease. Diabetes Metab Syndr Obes 14, 3627–3645. 10.2147/DMSO.S304357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrea L, Vetrani C, Caprio M, El Ghoch M, Frias-Toral E, Mehta RJ, Mendez V, Moriconi E, Paschou SA, Pazderska A, et al. (2021). Nutritional management of type 2 diabetes in subjects with obesity: an international guideline for clinical practice. Crit Rev Food Sci Nutr, 1–13. 10.1080/10408398.2021.1980766. [DOI] [PubMed] [Google Scholar]
- 10.Pillon NJ, Loos RJF, Marshall SM, and Zierath JR (2021). Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 184, 1530–1544. 10.1016/j.cell.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magkos F, Hjorth MF, and Astrup A (2020). Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat Rev Endocrinol 16, 545–555. 10.1038/s41574-020-0381-5. [DOI] [PubMed] [Google Scholar]
- 12.American Diabetes A (2018). 7. Obesity Management for the Treatment of Type 2 Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 41, S65–S72. 10.2337/dc18-S007. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell BG, and Gupta N (2021). Roux-en-Y Gastric Bypass. In StatPearls. [PubMed] [Google Scholar]
- 14.Hanipah ZN, and Schauer PR (2020). Bariatric Surgery as a Long-Term Treatment for Type 2 Diabetes/Metabolic Syndrome. Annu Rev Med 71, 1–15. 10.1146/annurev-med-053117-123246. [DOI] [PubMed] [Google Scholar]
- 15.Rothwell NJ, and Stock MJ (1979). A role for brown adipose tissue in diet-induced thermogenesis. Nature 281, 31–35. 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- 16.Chouchani ET, and Kajimura S (2019). Metabolic adaptation and maladaptation in adipose tissue. Nat Metab 1, 189–200. 10.1038/s42255-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowell BB, and Spiegelman BM (2000). Towards a molecular understanding of adaptive thermogenesis. Nature 404, 652–660. 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- 18.Chouchani ET, Kazak L, and Spiegelman BM (2019). New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab 29, 27–37. 10.1016/j.cmet.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Samuel VT, and Shulman GI (2018). Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab 27, 22–41. 10.1016/j.cmet.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goedeke L, Perry RJ, and Shulman GI (2019). Emerging Pharmacological Targets for the Treatment of Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Type 2 Diabetes. Annu Rev Pharmacol Toxicol 59, 65–87. 10.1146/annurev-pharmtox-010716-104727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, and Montminy M (2003). CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 426, 190–193. 10.1038/nature02110. [DOI] [PubMed] [Google Scholar]
- 22.Rodgers JT, and Puigserver P (2007). Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A 104, 12861–12866. 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rui L (2014). Energy metabolism in the liver. Compr Physiol 4, 177–197. 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geisler CE, Hepler C, Higgins MR, and Renquist BJ (2016). Hepatic adaptations to maintain metabolic homeostasis in response to fasting and refeeding in mice. Nutr Metab (Lond) 13, 62. 10.1186/s12986-016-0122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersen KF, Dufour S, Cline GW, and Shulman GI (2019). Regulation of hepatic mitochondrial oxidation by glucose-alanine cycling during starvation in humans. J Clin Invest 129, 4671–4675. 10.1172/JCI129913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandes GW, and Bocco B (2021). Hepatic Mediators of Lipid Metabolism and Ketogenesis: Focus on Fatty Liver and Diabetes. Curr Diabetes Rev 17, e110320187539. 10.2174/1573399816999201103141216. [DOI] [PubMed] [Google Scholar]
- 27.Perry RJ, Zhang D, Guerra MT, Brill AL, Goedeke L, Nasiri AR, Rabin-Court A, Wang Y, Peng L, Dufour S, et al. (2020). Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 579, 279–283. 10.1038/s41586-020-2074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rines AK, Sharabi K, Tavares CD, and Puigserver P (2016). Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov 15, 786–804. 10.1038/nrd.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loomba R, Friedman SL, and Shulman GI (2021). Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564. 10.1016/j.cell.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 30.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muñoz-Gómez SA, Slamovits CH, Dacks JB, Baier KA, Spencer KD, and Wideman JG (2015). Ancient homology of the mitochondrial contact site and cristae organizing system points to an endosymbiotic origin of mitochondrial cristae. Current biology : CB 25, 1489–1495. 10.1016/j.cub.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Cogliati S, Enriquez JA, and Scorrano L (2016). Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem Sci 41, 261–273. 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Kondadi AK, Anand R, and Reichert AS (2020). Cristae Membrane Dynamics - A Paradigm Change. Trends Cell Biol. 10.1016/j.tcb.2020.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Hu C, Shu L, Huang X, Yu J, Li L, Gong L, Yang M, Wu Z, Gao Z, Zhao Y, et al. (2020). OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell death & disease 11, 940. 10.1038/s41419-020-03152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacVicar T, and Langer T (2016). OPA1 processing in cell death and disease - the long and short of it. J Cell Sci 129, 2297–2306. 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 36.Kozjak-Pavlovic V (2017). The MICOS complex of human mitochondria. Cell Tissue Res 367, 83–93. 10.1007/s00441-016-2433-7. [DOI] [PubMed] [Google Scholar]
- 37.Khosravi S, and Harner ME (2020). The MICOS complex, a structural element of mitochondria with versatile functions. Biol Chem 401, 765–778. 10.1515/hsz-2020-0103. [DOI] [PubMed] [Google Scholar]
- 38.Latorre-Muro P, O’Malley KE, Bennett CF, Perry EA, Balsa E, Tavares CDJ, Jedrychowski M, Gygi SP, and Puigserver P (2021). A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab 33, 598–614 e597. 10.1016/j.cmet.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rampelt H, Zerbes RM, van der Laan M, and Pfanner N (2017). Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochimica et biophysica acta. Molecular cell research 1864, 737–746. 10.1016/j.bbamcr.2016.05.020. [DOI] [PubMed] [Google Scholar]
- 40.Tang J, Zhang K, Dong J, Yan C, Hu C, Ji H, Chen L, Chen S, Zhao H, and Song Z (2020). Sam50-Mic19-Mic60 axis determines mitochondrial cristae architecture by mediating mitochondrial outer and inner membrane contact. Cell death and differentiation 27, 146–160. 10.1038/s41418-019-0345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman JR, Mourier A, Yamada J, McCaffery JM, and Nunnari J (2015). MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. eLife 4. 10.7554/eLife.07739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fengler VHI, Macheiner T, Kessler SM, Czepukojc B, Gemperlein K, Müller R, Kiemer AK, Magnes C, Haybaeck J, Lackner C, and Sargsyan K (2016). Susceptibility of Different Mouse Wild Type Strains to Develop Diet-Induced NAFLD/AFLD-Associated Liver Disease. PLOS ONE 11, e0155163. 10.1371/journal.pone.0155163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morio B, Panthu B, Bassot A, and Rieusset J (2021). Role of mitochondria in liver metabolic health and diseases. Cell Calcium 94, 102336. 10.1016/j.ceca.2020.102336. [DOI] [PubMed] [Google Scholar]
- 44.Wrobel L, Trojanowska A, Sztolsztener ME, and Chacinska A (2013). Mitochondrial protein import: Mia40 facilitates Tim22 translocation into the inner membrane of mitochondria. Mol Biol Cell 24, 543–554. 10.1091/mbc.E12-09-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueda E, Tamura Y, Sakaue H, Kawano S, Kakuta C, Matsumoto S, and Endo T (2019). Myristoyl group-aided protein import into the mitochondrial intermembrane space. Sci Rep 9, 1185. 10.1038/s41598-018-38016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rambold AS, Kostelecky B, Elia N, and Lippincott-Schwartz J (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 108, 10190–10195. 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomes LC, Benedetto GD, and Scorrano L (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biology 13, 589–598. 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blum TB, Hahn A, Meier T, Davies KM, and Kühlbrandt W (2019). Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc Natl Acad Sci U S A 116, 4250–4255. 10.1073/pnas.1816556116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calvo E, Cogliati S, Hernansanz-Agustín P, Loureiro-López M, Guarás A, Casuso RA, García-Marqués F, Acín-Pérez R, Martí-Mateos Y, Silla-Castro JC, et al. (2020). Functional role of respiratory supercomplexes in mice: SCAF1 relevance and segmentation of the Q(pool). Science advances 6, eaba7509. 10.1126/sciadv.aba7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balsa E, Soustek MS, Thomas A, Cogliati S, García-Poyatos C, Martín-García E, Jedrychowski M, Gygi SP, Enriquez JA, and Puigserver P (2019). ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Molecular cell 74, 877–890.e876. 10.1016/j.molcel.2019.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett CF, O’Malley KE, Perry EA, Balsa E, Latorre-Muro P, Riley CL, Luo C, Jedrychowski M, Gygi SP, and Puigserver P (2021). Peroxisomal-derived ether phospholipids link nucleotides to respirasome assembly. Nature chemical biology 17, 703–710. 10.1038/s41589-021-00772-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paulson DJ, Ward KM, and Shug LA (1984). Malonyl CoA inhibition of carnitine palmityltransferase in rat heart mitochondria. FEBS Letters 176, 381–384. [DOI] [PubMed] [Google Scholar]
- 53.Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG, and DeLany JP (2010). Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 18, 1695–1700. 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chavan R, Feillet C, Costa SSF, Delorme JE, Okabe T, Ripperger JA, and Albrecht U (2016). Liver-derived ketone bodies are necessary for food anticipation. Nature Communications 7, 10580. 10.1038/ncomms10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puchalska P, and Crawford PA (2017). Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metabolism 25, 262–284. 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, and Sassone-Corsi P (2012). Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci U S A 109, 5541–5546. 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng Y, Wang ZV, Gordillo R, An Y, Zhang C, Liang Q, Yoshino J, Cautivo KM, De Brabander J, Elmquist JK, et al. (2017). An adipo-biliary-uridine axis that regulates energy homeostasis. Science 355. 10.1126/science.aaf5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balsa E, Perry EA, Bennett CF, Jedrychowski M, Gygi SP, Doench JG, and Puigserver P (2020). Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nature Communications 11. 10.1038/s41467-020-16423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roosild TP, Castronovo S, Villoso A, Ziemba A, and Pizzorno G (2011). A novel structural mechanism for redox regulation of uridine phosphorylase 2 activity. Journal of Structural Biology 176, 229–237. 10.1016/j.jsb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Begaye B, Vinales KL, Hollstein T, Ando T, Walter M, Bogardus C, Krakoff J, and Piaggi P (2020). Impaired Metabolic Flexibility to High-Fat Overfeeding Predicts Future Weight Gain in Healthy Adults. Diabetes 69, 181–192. 10.2337/db19-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anand R, Reichert AS, and Kondadi AK (2021). Emerging Roles of the MICOS Complex in Cristae Dynamics and Biogenesis. Biology (Basel) 10. 10.3390/biology10070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakowska P, Jans DC, Mohanraj K, Riedel D, Jakobs S, and Chacinska A (2015). The Oxidation Status of Mic19 Regulates MICOS Assembly. Molecular and cellular biology 35, 4222–4237. 10.1128/mcb.00578-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hessenberger M, Zerbes RM, Rampelt H, Kunz S, Xavier AH, Purfürst B, Lilie H, Pfanner N, van der Laan M, and Daumke O (2017). Regulated membrane remodeling by Mic60 controls formation of mitochondrial crista junctions. Nat Commun 8, 15258. 10.1038/ncomms15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bock-Bierbaum T, Funck K, Wollweber F, Lisicki E, von der Malsburg K, von der Malsburg A, Laborenz J, Noel JK, Hessenberger M, Jungbluth S, et al. (2022). Structural insights into crista junction formation by the Mic60-Mic19 complex. Science advances 8, eabo4946. 10.1126/sciadv.abo4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, et al. (2013). Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 18, 740–748. 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Virtue S, Even P, and Vidal-Puig A (2012). Below thermoneutrality, changes in activity do not drive changes in total daily energy expenditure between groups of mice. Cell Metab 16, 665–671. 10.1016/j.cmet.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zarrinmayeh H, and Territo PR (2020). Purinergic Receptors of the Central Nervous System: Biology, PET Ligands, and Their Applications. Mol Imaging 19, 1536012120927609. 10.1177/1536012120927609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cansev M (2006). Uridine and cytidine in the brain: their transport and utilization. Brain Res Rev 52, 389–397. 10.1016/j.brainresrev.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 69.Redzic ZB, Malatiali SA, Craik JD, Rakic ML, and Isakovic AJ (2009). Blood-brain barrier efflux transport of pyrimidine nucleosides and nucleobases in the rat. Neurochem Res 34, 566–573. 10.1007/s11064-008-9823-5. [DOI] [PubMed] [Google Scholar]
- 70.Cornford EM, and Oldendorf WH (1975). Independent blood-brain barrier transport systems for nucleic acid precursors. Biochimica et Biophysica Acta 394, 211–219. [DOI] [PubMed] [Google Scholar]
- 71.Steculorum SM, Paeger L, Bremser S, Evers N, Hinze Y, Idzko M, Kloppenburg P, and Bruning JC (2015). Hypothalamic UDP Increases in Obesity and Promotes Feeding via P2Y6-Dependent Activation of AgRP Neurons. Cell 162, 1404–1417. 10.1016/j.cell.2015.08.032. [DOI] [PubMed] [Google Scholar]
- 72.Caggiano V, Leiras R, Goni-Erro H, Masini D, Bellardita C, Bouvier J, Caldeira V, Fisone G, and Kiehn O (2018). Midbrain circuits that set locomotor speed and gait selection. Nature 553, 455–460. 10.1038/nature25448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cortopassi MD, Ramachandran D, Rubio WB, Hochbaum D, Sabatini BL, and Banks AS (2022). Analysis of Thermogenesis Experiments with CalR. Methods Mol Biol 2448, 43–72. 10.1007/978-1-0716-2087-8_3. [DOI] [PubMed] [Google Scholar]
- 74.Mina AI, LeClair RA, LeClair KB, Cohen DE, Lantier L, and Banks AS (2018). CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell Metab 28, 656–666.e651. 10.1016/j.cmet.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huynh FK, Green MF, Koves TR, and Hirschey MD (2014). Measurement of Fatty Acid Oxidation Rates in Animal Tissues and Cell Lines. In Methods in Enzymology, (Elsevier; ), pp. 391–405. 10.1016/b978-0-12-416618-9.00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balsa E, Soustek MS, Thomas A, Cogliati S, Garcia-Poyatos C, Martin-Garcia E, Jedrychowski M, Gygi SP, Enriquez JA, and Puigserver P (2019). ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Molecular cell 74, 877–890 e876. 10.1016/j.molcel.2019.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spinazzi M, Casarin A, Pertegato V, Salviati L, and Angelini C (2012). Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 7, 1235–1246. 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 78.Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7, 872–881. 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan M, Kremer DM, Huang H, Breitkopf SB, Ben-Sahra I, Manning BD, Lyssiotis CA, and Asara JM (2019). Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nat Protoc 14, 313–330. 10.1038/s41596-018-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Breitkopf SB, Ricoult SJH, Yuan M, Xu Y, Peake DA, Manning BD, and Asara JM (2017). A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics 13. 10.1007/s11306-016-1157-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Mitochondria proteome from BL6 and CD-1 liver, related to Figure 1
Table S2. Liver metabolomics from GFP and MIC19 overexpressing mice, related to Figure 6
Table S3. List of oligos used for qPCR assays, related to STAR Methods