

Abstract
Sympathetic activation during cold exposure increases adipocyte thermogenesis via the expression of mitochondrial protein uncoupling protein 1 (UCP1)1. The propensity of adipocytes to express UCP1 is under a critical influence of the adipose microenvironment and varies between sexes and among various fat depots2–7. Here we report that mammary gland ductal epithelial cells in the adipose niche regulate cold-induced adipocyte UCP1 expression in female mouse subcutaneous white adipose tissue (scWAT). Single-cell RNA sequencing shows that glandular luminal epithelium subtypes express transcripts that encode secretory factors controlling adipocyte UCP1 expression under cold conditions. We term these luminal epithelium secretory factors ‘mammokines’. Using 3D visualization of whole-tissue immunofluorescence, we reveal sympathetic nerve–ductal contact points. We show that mammary ducts activated by sympathetic nerves limit adipocyte UCP1 expression via the mammokine lipocalin 2. In vivo and ex vivo ablation of mammary duct epithelium enhance the cold-induced adipocyte thermogenic gene programme in scWAT. Since the mammary duct network extends throughout most of the scWAT in female mice, females show markedly less scWAT UCP1 expression, fat oxidation, energy expenditure and subcutaneous fat mass loss compared with male mice, implicating sex-specific roles of mammokines in adipose thermogenesis. These results reveal a role of sympathetic nerve-activated glandular epithelium in adipocyte UCP1 expression and suggest that mammary duct luminal epithelium has an important role in controlling glandular adiposity.
scWAT depots in female mice are mostly mammary gland white adipose tissue (mgWAT), which is a highly heterogenous tissue consisting of adipocytes, preadipocytes, mesenchymal stem cells, immune cells, endothelial cells, sympathetic nervous system (SNS) nerve fibres and mammary epithelial cells forming a ductal structure. The epithelial cells are divided into myoepithelial (basal) cells and luminal cells, which can be luminal hormone-sensing or alveolar8. In virgin female mice, the mammary gland already has ductal structures in the anterior and posterior scWAT, and metabolic cooperativity between luminal ductal cells and stroma is known to be important for mammary gland function and development8,9. Profound changes in mammary ducts and adipocytes are seen during gestation, pregnancy, lactation and post-involution10,11. The importance of adipocytes for mammary duct morphogenesis, the dedifferentiation of adipocytes during lactation and their reappearance during post-involution, suggest a dynamic homeostatic interplay between ductal luminal epithelial cells and adipocytes11–13. However, it is unclear what paracrine signalling programmes from mammary ducts regulate adipocyte metabolism and the thermogenic programme. Notably, our current-state-of knowledge of beige adipocytes, white adipose tissue thermogenesis and UCP1 expression is based mainly on male scWAT, which lack mammary glandular epithelial cells. Notably, besides the role of immune cells in the adipose microenvironment, the contribution of other cell types in controlling adipocyte UCP1 expression remains to be clarified.
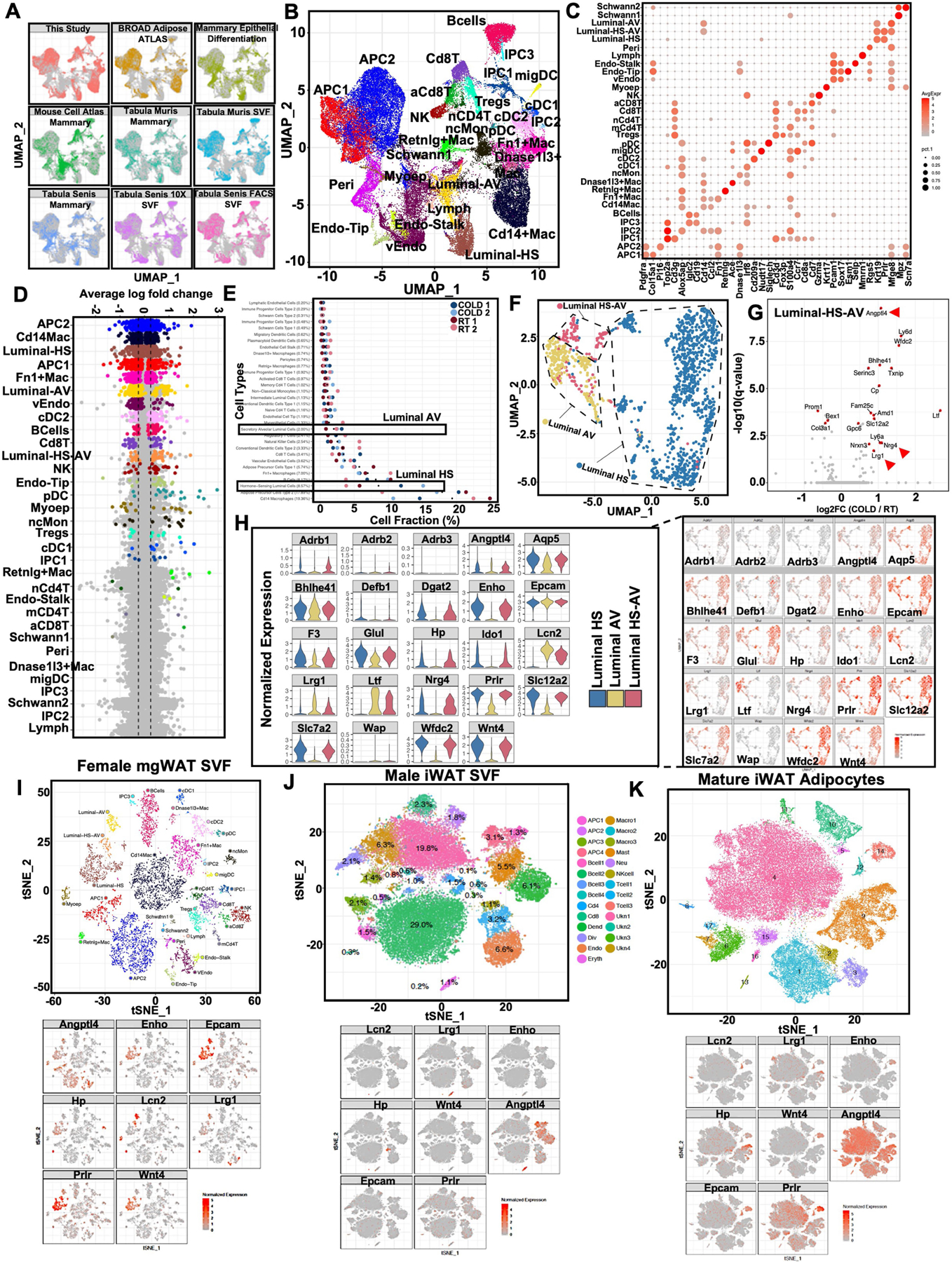
To study cellular heterogeneity, inter-tissue communication and cellular transcription dynamics in mgWAT in a thermogenic condition, we isolated the stromal vascular fraction (SVF) from the mgWAT of 10-week-old virgin female mice exposed to 24-h cold (cold, 4 °C) or room temperature (RT) and performed single-cell RNA sequencing (scRNA-seq) (Fig. 1a). The processed dataset consisted of 42,052 genes across 12,222 cells, with approximately 2,300–4,650 cells from each sample, and a median of 2,411 genes and 7,252 transcripts were detected per cell. We integrated our dataset with eight other publicly available single-cell datasets from mammary gland tissues14,15, including Broad Institute Adipose SVF Single Cell Atlas16, Tabula Muris and Tabula Muris Senis17,18: (1) for cell-type identification, (2) to increase confidence in the projected cell type, (3) for sex and age differences, and (4) for mammary gland development (Extended Data Fig. 1a and Supplementary Tables 1 and 2). This integrated dataset enabled us to precisely annotate various cell types in mgWAT of female mice. Further subclustering of our integrated dataset based on known cell-type marker genes identified clusters of adipocyte precursor cells (APCs), B cells, macrophages, T cells, endothelial cells, immune precursor cells, dendritic cells, Schwann cells, myoepithelial cells and luminal-hormone sensing (luminal-HS), luminal-alveolar (luminal-AV), luminal-hormone sensing alveolar (luminal-HS-AV) and myoepithelial cells (Fig. 1b and Extended Data Fig. 1b,c).
Fig. 1 |. Deconstruction of mgWAT shows cold-induced remodelling of mammary duct epithelium.
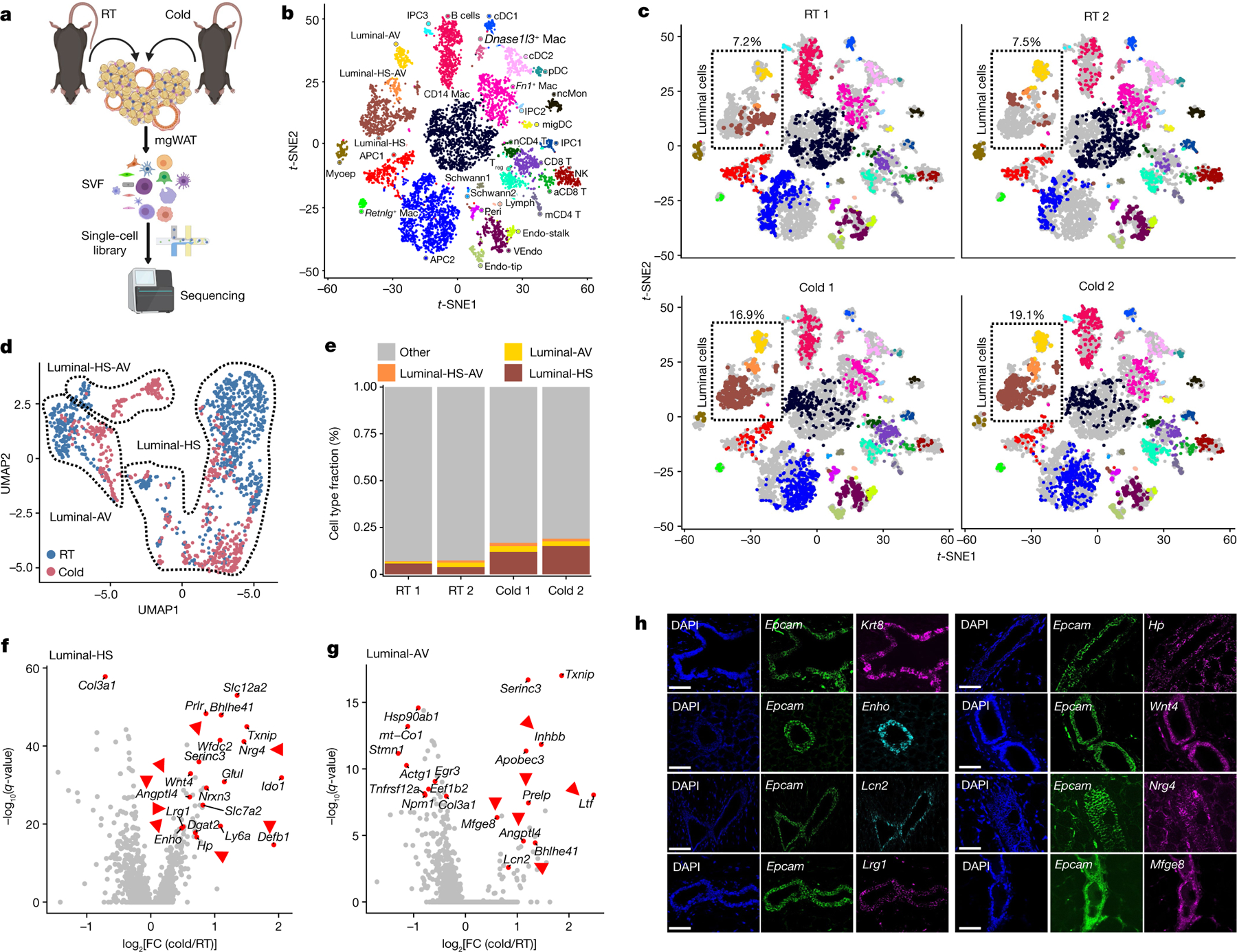
a, Cartoon depiction of the scRNA-seq experiment using the 10X Genomics (single-cell separation) workflow, showing isolation of SVF from mgWAT of 10-week-old female mice after 24 h RT or cold exposure. b, t-SNE plot of single cells from mammary gland and surrounding SVF from the present study coloured by cell type. Cell types were identified on the basis of expression of canonical marker genes. aCD8 T, activated CD8+ T cell; cDC, conventional dendritic cell; Endo-stalk, endothelial stalk cell; Endo-tip, endothelial tip cell; IPC, immune precursor cell; Lymph, lymphatic endothelial cell; Mac, macrophage; mCD4 T, memory CD4+ T cell; migDC, migratory dendritic cells; Myoep, myoepithelial cell; nCD4 T, naive CD4+ T cell; ncMon, non-classical monocytes; NK, natural killer cell; pDC, plasmacytoid dendritic cell; Peri, pericyte; Treg, T regulatory cell; VEndo, vascular endothelial cell. c, t-SNE plot of single cells from mammary gland and surrounding SVF coloured by cell type and separated by sample (RT or 4 °C (cold) exposure). Relative fractions of luminal cell type in each sample are indicated. Cell-type annotation as in b. d, Uniform manifold approximation and projection (UMAP) plot of luminal epithelial cell types from RT or cold-exposed mgWAT. e, The fraction of luminal cell subtypes from RT or cold conditions, derived from analysis in d. f,g, Differentially expressed genes between cold-treated mice and RT mice across luminal-HS (f) and luminal-AV (g) cells. Selected significant DEGs (adjusted P value < 0.05) are highlighted, with the average log fold change (FC) between cold and RT treatment indicated on the y axis. Genes indicated with red arrows encode secreted and extracellular signalling factors. f,g, Wilcoxon rank sum test for cold versus RT cells; adjusted P value based on Bonferroni correction. h, RNAscope FISH (see Methods) of indicated probes from mgWAT. Representative images from three independent RNAscope FISH experiments. Scale bars, 50 μm. Cartoon in a created using BioRender.com.
To gain insight into the remodelling of stromal cells under adrenergic stress, we segregated the cumulative t-distributed stochastic neighbour embedding (t-SNE) plot into RT and cold treatments by animal replicate. The t-SNE and dot plots reveal global changes in the relative proportions of SVF clusters between RT and cold treatments (Fig. 1c). Among all the clusters, luminal cells showed marked differences in cell-type percentages (RT 1: 7.2%, RT 2: 7.5%, cold 1: 16.9% and cold 2: 19.1%) and exhibited marked differences in their global transcriptomic profiles in the t-SNE two-dimensional projection in which cells from RT and cold treatments were segregated (Fig. 1c). To quantitatively determine the transcriptional effect of cold treatment on individual cell types, we characterized differentially expressed genes as a function of cluster types and found a high degree of transcriptional variation in luminal-HS and luminal-AV under the cold conditions (Extended Data Fig. 1d). Further subclustering of luminal epithelial cell types15 (luminal-HS, luminal-AV and luminal-HS-AV) revealed marked differences in clustering between RT and cold treatments (Fig. 1d,e and Extended Data Fig. 1e,f).
Luminal cell clusters showed remarkable cell fraction and transcriptional differences in cell clusters between RT and cold treatments, implicating a potential remodelling of the luminal epithelium upon cold exposure (Fig. 1c–e and Extended Data Fig. 1d–f). We performed differentially expressed gene (DEG) analysis on RT and cold-exposed luminal subclusters to probe for factors that are differentially expressed in luminal cells under cold exposure. We found upregulation of Wnt4, adropin (Enho), leucine rich α−2 glycoprotein (Lrg1), diglyceride acyltransferase (Dgat2), haptoglobin (Hp), β-defensin 1 (Defb1) and angiopoietin-like 4 (Angptl4) in luminal-HS cells, lipocalin 2 (Lcn2), Angptl4, lactotransferrin (Ltf ) and apolipoprotein B editing complex (Apobec3) in luminal-AV cells, and neuregulin 4 (Nrg4), ceruloplasmin (Cp) and Angptl4 in luminal-HS-AV cells (Fig. 1f,g and Extended Data Fig. 1g). Many of these genes (shown by red arrows in Fig. 1f,g and Extended Data Fig. 1g) encode secreted factors that have essential roles in local and systemic lipid and adipose metabolism19–29. We call these mammary duct secretory factors ‘mammokines’. t-SNE plots of normalized gene expression levels for cold-induced mammokines in mgWAT (this study), male scWAT SVF and mature adipocytes30 show that most of the cold-induced mammokine genes showed relatively localized expression in ductal epithelial cell adhesion molecule (Epcam)-positive cells (Extended Data Fig. 1h–k). Our RNAscope fluorescent in situ hybridization (FISH) analysis showed a highly localized expression of the mammokine genes Enho, Mfge8, Lrg1, Lcn2, Hp, Nrg4 and Wnt4 in Epcam+ and Krt8+ (a luminal epithelial marker) in mammary ductal luminal cells (Fig. 1h).
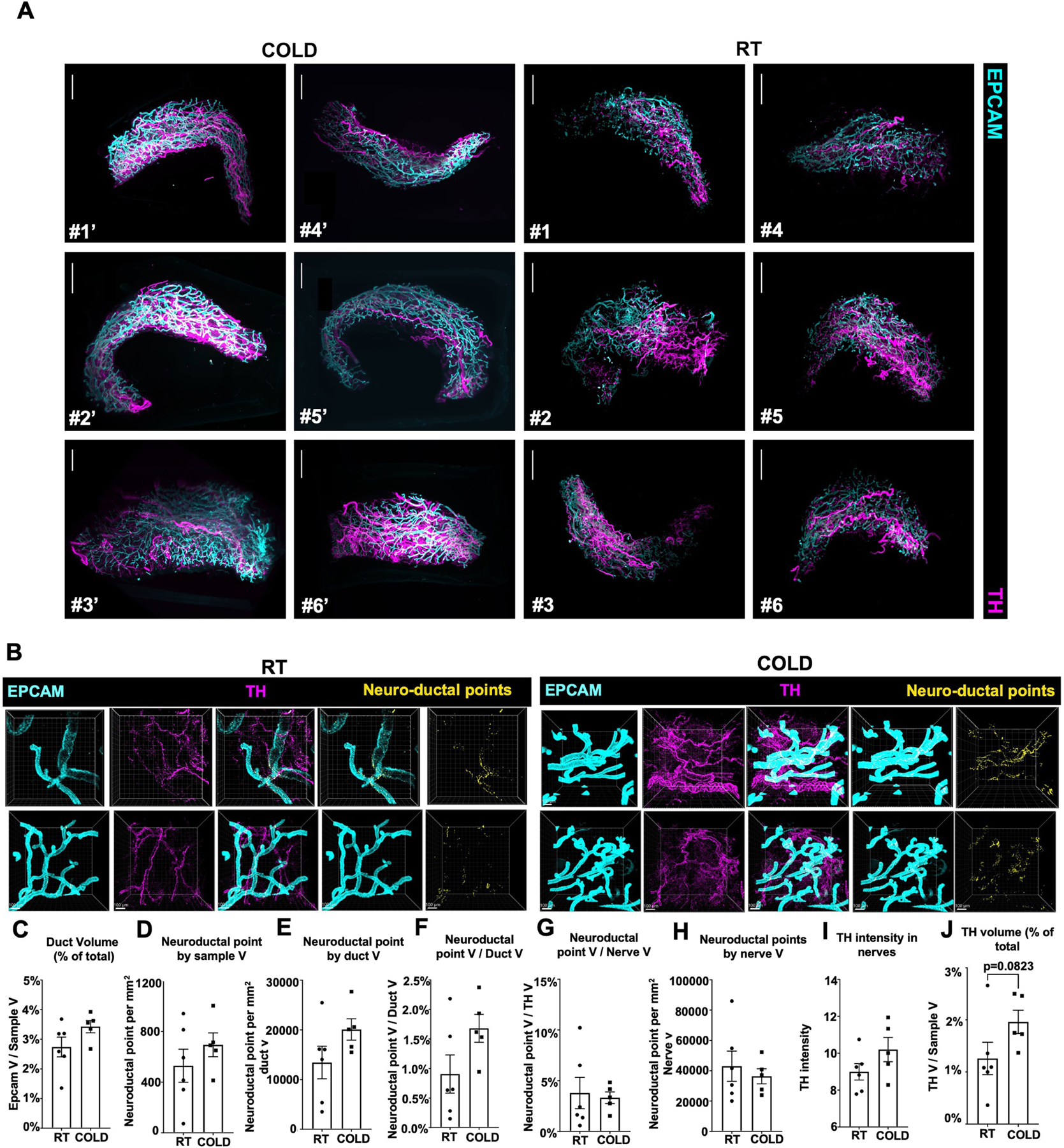
Shifts in the luminal epithelium transcriptomic state with the cold and localized expression of the β1AR gene Adrb1 in luminal cells, suggest that these cells may directly respond to cold-induced SNS activation31–33 (Fig. 1d and Extended Data Fig. 1h). To determine whether sympathetic nerves innervate ductal epithelial cells, we used Adipoclear, a robust protocol based on immunolabelling-enabled 3D imaging of solvent-cleared organ (iDISCO)1, for high-resolution 3D imaging of mammary tissue. We analysed the 3D distribution and density of a sympathetic marker, tyrosine hydroxylase (TH), and its relationship to EPCAM+ mammary ductal cells in mgWAT from mice exposed to RT or cold. Cold exposure produced noticeable morphological changes in mammary ducts, such as increased EPCAM+ branching and terminal ductal bifurcations (Fig. 2a and Extended Data Fig. 2a), consistent with data showing increased branch morphogenesis upon treatment with the β1AR and β2AR agonist isoproterenol31. Our data further revealed that nerve fibres are interwoven with mammary gland ducts and alveolar structures in mgWAT (Supplementary Videos 1 and 2). However, we did not see an effect of cold treatment on duct volume, nerve volume or the ratio of duct-to-nerve volume by light sheet fluorescence microscopy (LSFM) (Fig. 2b). To examine interactions between sympathetic innervation and mammary gland ducts in more detail, we performed confocal imaging in six regions of the mgWAT fat pad from each of six RT and five cold-treated mice. Consistent with our scRNA-seq data, we saw a significant increase in EPCAM staining and EPCAM+ cells in the ducts of cold-exposed mgWAT (Fig. 2c,d). We also identified contacts between TH+ nerve fibres and EPCAM+ ducts (neuroductal points) with a trend to increased volume of nerve contacts (normalized for duct volume) in cold-treated mice (P = 0.09; Extended Data Fig. 2e). Notably, TH intensity—which has been reported to increase with sympathetic activation—was significantly higher at the neuroductal points in cold-exposed mgWAT compared with controls (Fig. 2c,g and Supplementary Videos 3 and 4). Similarly, EPCAM intensity was significantly increased at neuroductal points in keeping with local induction of expression (Fig. 2c,e,f). There was a trend (P = 0.08) towards an increase in TH volume1,34,35 with 24 h cold exposure (Extended Data Fig. 2j), but no significant changes in duct or nerve volume or TH intensity across the fat pad (Extended Data Fig. 2c–i). Overall, our data show a substantial remodelling of mammary ducts and their contacts with sympathetic innervation upon 24 h of cold exposure.
Fig. 2 |. SNS fibres directly innervate mammary ductal epithelium.
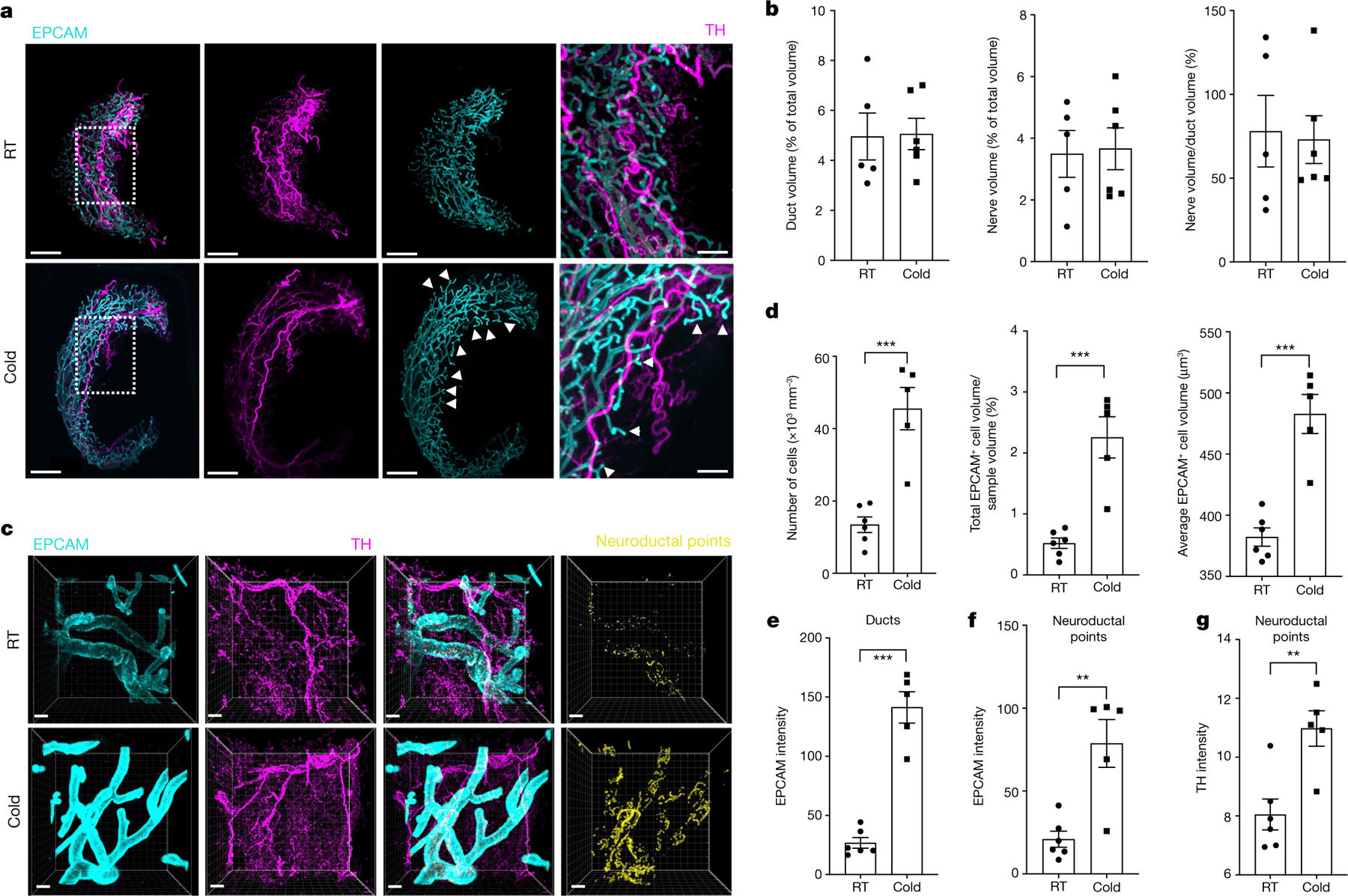
a, LSFM images of mgWAT isolated from female mice exposed to RT or cold for 24 h and stained with TH antibody (to mark SNS fibres) and EPCAM antibody (for ductal cells). Representative mgWAT images from six mice per condition. White arrows show terminal ductal bifurcations under the cold condition. Scale bars, 200 μm. b, Quantification of ductal volume and nerve volume as a percentage of total volume, and the ratio of nerve volume to ductal volume in RT or cold-exposed mgWAT. RT: n = 6; cold: n = 5. c, Confocal images of mgWAT isolated from female mice exposed to RT or cold for 24 h and stained with EPCAM and TH antibodies. Merged staining of EPCAM and TH represent neuroductal points. Representative image of six mice per condition. Scale bars, 100 μm. d–g, Quantification of EPCAM+ cells and cell volume (d), EPCAM intensity in ducts (e), EPCAM intensity at neuroductal points (f) and TH intensity at neuroductal points (g). RT: n = 6; cold: n = 5. b,d–g, Unpaired Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001. Data are mean ± s.e.m.
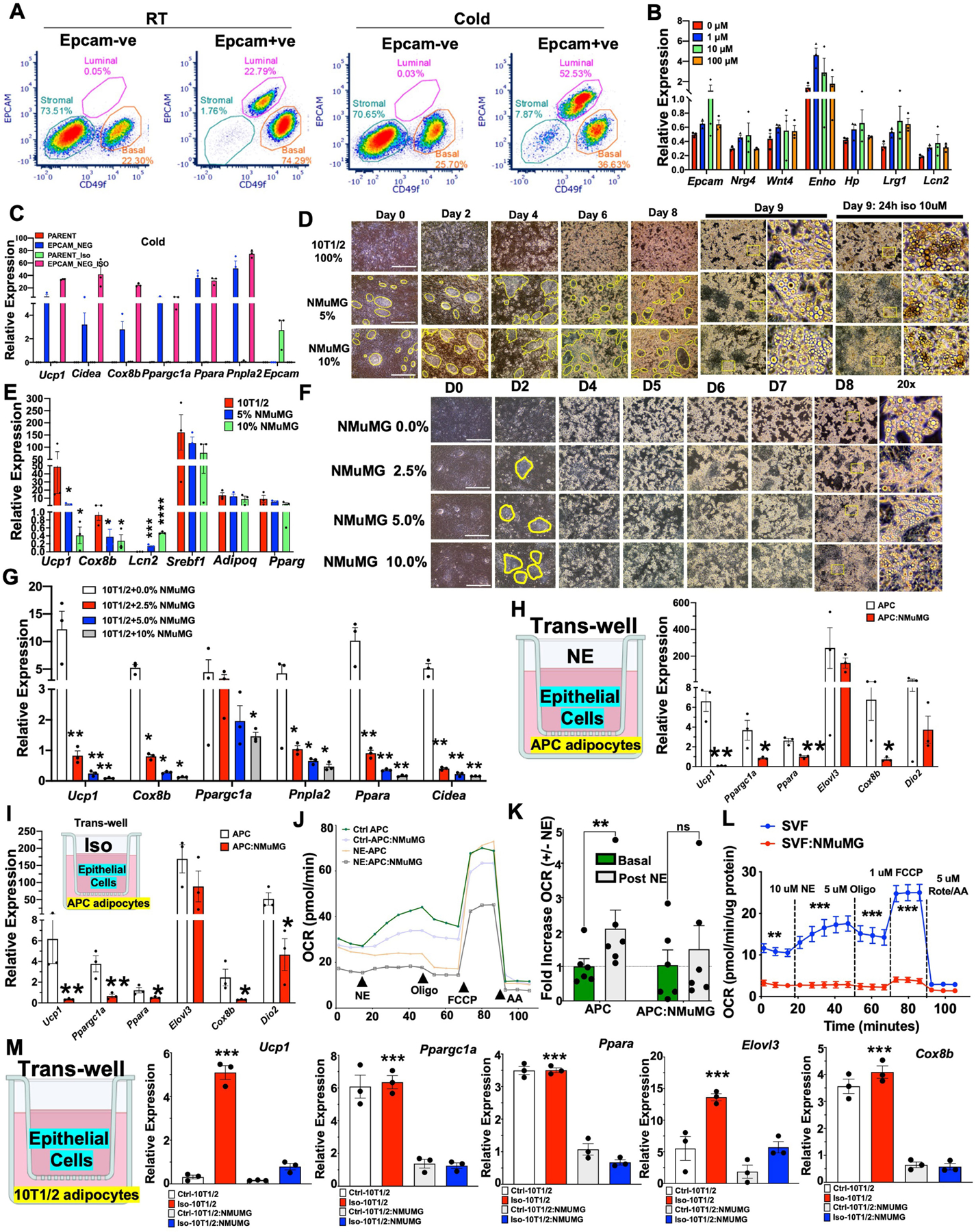
To explore the biological relevance of cold-induced increase in luminal population transcriptional state (Fig. 1) and EPCAM intensity and SNS innervation of mammary ducts (Fig. 2), we purified EPCAM+ and EPCAM− cells from mgWAT of RT and cold-exposed mice and stained them for EPCAM and CD49F for fluorescence-activated cell sorting (FACS) analysis to probe for changes in luminal cell populations under cold stress (Supplementary Fig. 2). We observed three distinct populations: of cells: EPCAM−/loCD49Flo (stromal), EPCAM+/hiCD49Fhi (luminal) and EPCAM−/loCD49Fhi (basal) (Extended Data Fig. 3a). Luminal cells were enriched only in EPCAM+ purified cells and showed a cold-dependent increase in cell population compared with RT conditions. We also observed a marked reduction in the basal cell population upon cold treatment in EPCAM+ purified cells, consistent with the scRNA-seq data in Fig. 1c (myoepithelial cluster: RT, 2.15%; cold, 0.65%). The presence of minute traces of stromal cells in the EPCAM+ population could indicate a non-specific interaction with the EPCAM antibody beads (Extended Data Fig. 3a). These data suggest a differential response to cold stress by luminal and basal cells in the ductal epithelium (Extended Data Fig. 3a). Since luminal epithelial cells expressed Adrb1, we used the β1AR and β2AR agonist isoproterenol to mimic the direct response of luminal cells to SNS catecholamine production under cold exposure. We tested mammokine expression in isolated primary EPCAM+ mgWAT with or without isoproterenol treatment; cold-induced mammokines showed increased expression upon isoproterenol treatment (Extended Data Fig. 3b). To determine whether adrenergic-activated luminal cells are involved in the mgWAT adipose thermogenic programme, we depleted EPCAM+ cells from mgWAT SVF by positive selection using magnetic cell sorting (MACS) and differentiated SVF cells ex vivo with and without ductal cells into beige adipocytes (Fig. 3a). Depletion of EPCAM+ cells from the SVF of RT mgWAT potentiated expression of thermogenic genes such as Ucp1, Cox8b, Ppargc1a and Cidea, and this potentiation of thermogenic genes was markedly amplified in the cold-exposed condition (Fig. 3a and Extended Data Fig. 3c). To further test the crosstalk of epithelial cells and adipocytes, we developed an in vitro co-culture system involving a controlled mixture of adipogenic 10T1/2 cells with nontransformed mouse mammary gland (NMuMG) cells, which are derived from ‘normal’ mammary epithelium with a luminal-specific phenotype36. A seeding density of as low as 2.5% NMuMG cells resulted in a significant reduction of Ucp1 and beiging potential of 10T1/2 cells compared with the pure culture (Extended Data Fig. 3d–g), with no change in general adipocyte differentiation. The higher the fraction of NMuMG cells in the co-culture, the lower the relative expression of Ucp1 and other thermogenic genes such as Cox8B and Ppargc1a (Extended Data Fig. 3d–g). The crosstalk and anti-thermogenic effect of luminal cells on adipocytes from beige differentiated SVF cells, 10T1/2 and APCs were further confirmed in a Transwell co-culture system (Fig. 3b and Extended Data Fig. 3h–m). Our mitochondrial respiration analysis using the Seahorse assay further confirmed the inhibition of the adipocyte thermogenic programme by luminal cells (Fig. 3b, inset and Extended Data Fig. 3j–l).
Fig. 3 |. Mammary ductal cells directly inhibit adipocyte thermogenesis.
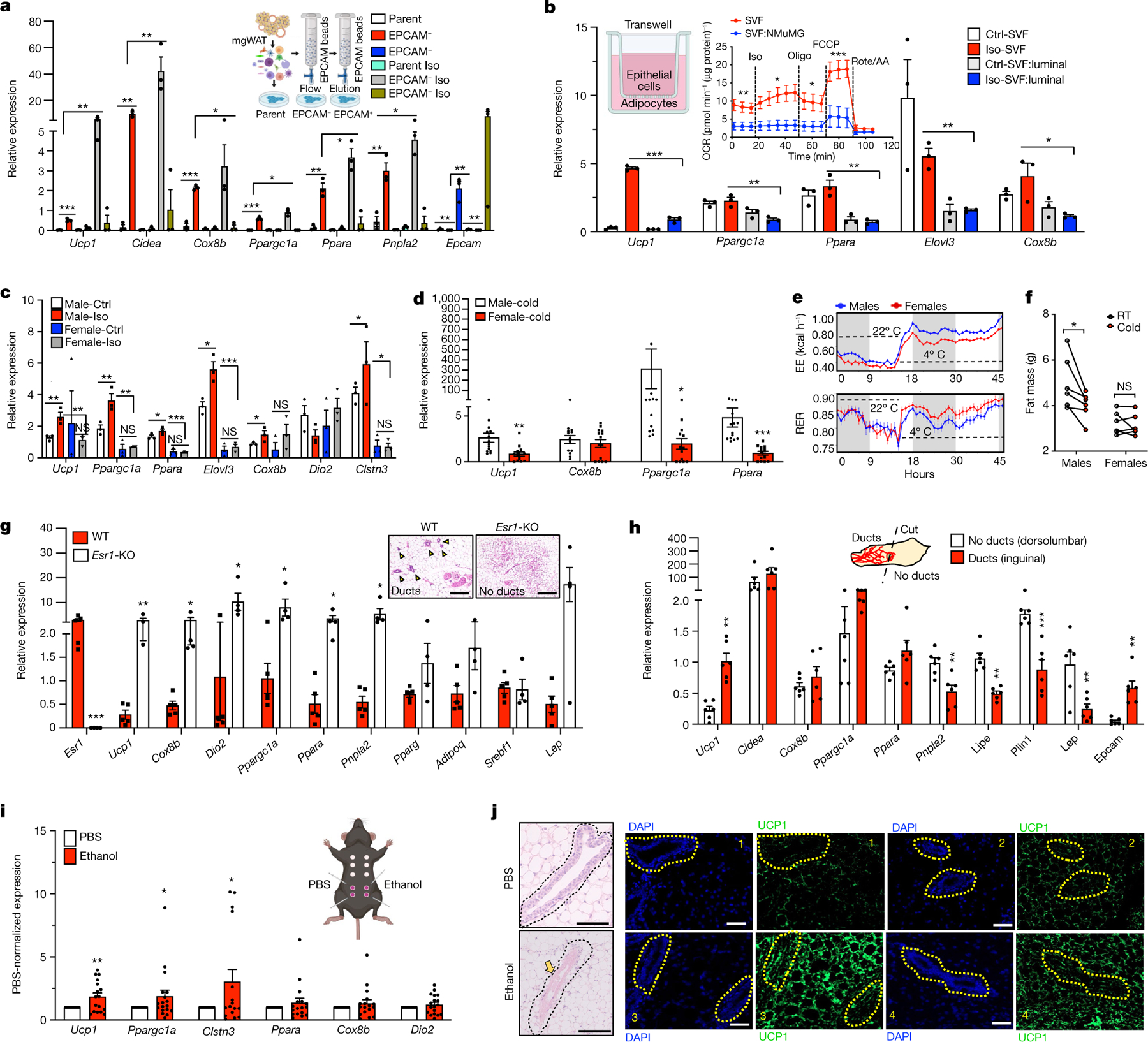
a, Quantitative PCR (qPCR) analysis of gene expression with beige differentiation of indicated cells treated with and without 10 μM isoproterenol (Iso) for 24 h. b, qPCR analysis of gene expression from a Transwell system of beige differentiated (Crtl-SVF) or isoproterenol-treated (Iso-SVF) SVF (SVF:NMuMG) in the presence of NMuMG cells with or without isoproterenol. Inset, average oxygen consumption rate (OCR) normalized to protein level in SVF and SVF:NMuMG (two-way ANOVA with Bonferroni post hoc test). SVF: n = 9; SVF:NMuMG: n = 9. Oligo, oligomycin; rote/AA, rotenone plus antimycin A. c, qPCR analysis of gene expression in beige differentiated SVF cells isolated from iWAT from male and female mice, treated with and without isoproterenol. Ctrl, control; NS, not significant. d, qPCR analysis of gene expression in iWAT from male and female mice, exposed to the cold condition for 24 h. Male: n = 13; female: n = 13. e, Energy expenditure (EE; in kcal h−1) and RER of male and female mice exposed to 22 °C (21 h) or 4 °C (24 h) (the white region represents the light cycle and the grey region represent the night cycle). RT: n = 6; cold: n = 6. f, Fat mass of mice from e. RT: n = 6; cold: n = 6. g, qPCR analysis of gene expression from wild-type and Esr1-KO mgWAT exposed to cold for 24 h. Wild type: n = 5; Esr1-KO: n = 5. Scale bars, 100 μm. h, qPCR analysis of gene expression in dorsolumbar or inguinal parts of cold-exposed five-week-old mice. dorsolumbar: n = 6; inguinal: n = 6. i,j, qPCR analysis of Ucp1 gene expression (i) and H&E staining and immunofluorescence microscopy of UCP1 (j) in mgWAT of wild-type female mice exposed to cold for 24 h and intraductally injected with PBS or 70% ethanol. j, The yellow arrow shows ablated ducts and yellow dotted outlines indicate ductal epithelium. mRNA expression was normalized to PBS control and represented as a fold change. n = 8 inguinal and n = 8 dorsolumbar mgWAT. Scale bars, 100 μm. a–c, Results are from three independent experiments. a–i, Unpaired Student’s t-test. Data are mean ± s.e.m. Cartoons in a,b,i created using BioRender.com.
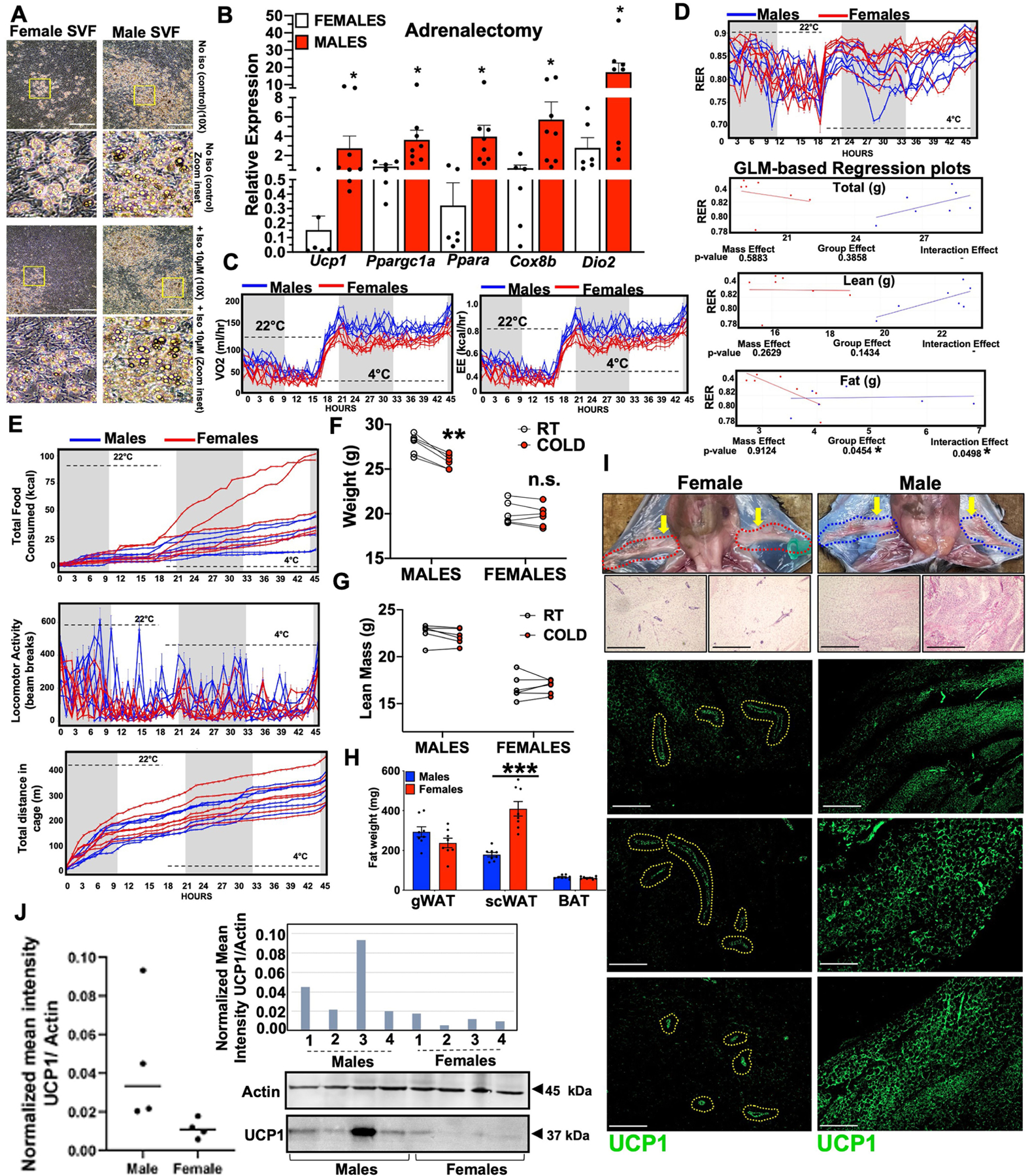
To test the role of ductal epithelial cells in adipose thermogenesis ex vivo and in vivo, we compared aged-matched female mgWAT with four complimentary ductal ablated models; (1) oestrogen receptor-α-knockout (Esr1-KO) mice37, (2) male mice lacking or possessing only rudimentary glandular ducts, (3) inguinal (ducts) or dorsolumbar (no ducts) portions of mgWAT from five-week-old female mice, and (4) genetic and chemical ablation of ducts. First, to test the role of ductal cells in adipose thermogenesis, we isolated SVF from male inguinal white adipose tissue (iWAT) and female mgWAT and differentiated them into beige adipocytes in the presence or absence of isoproterenol. We found that SVF cells from male iWAT show markedly higher beiging and isoproterenol-mediated Ucp1 expression compared with EPCAM+ SVF cells from female mgWAT (Fig. 3c and Extended Data Fig. 4a). In agreement with the ex vivo data, female mgWAT showed markedly less expression of cold-induced thermogenic genes such as Ucp1, Cox8b and Ppargc1a compared with male iWAT (Fig. 3d). Since adrenal glands also produce catecholamines, we performed adrenalectomy in male and female mice to confirm the local effect of SNS-derived catecholamines in the thermogenic phenotypes of male iWAT and female mgWAT. We continued to observe a markedly lower induction of the cold thermogenic gene programme in adrenalectomized female mice compared with adrenalectomized male mice (Extended Data Fig. 4b). Since mgWAT makes up almost all the subcutaneous fat mass in female mice, we reasoned that a highly reduced adipocyte thermogenic programme could potentially influence energy metabolism. To test this, we performed indirect calorimetry on age-matched male and female mice with RT and 24 h cold exposure using a metabolic chamber. We found that female mice showed highly reduced energy expenditure and oxygen consumption (VO2) during cold exposure compared with male mice (Fig. 3e and Extended Data Fig. 4c). Female mice showed markedly higher respiratory exchange ratio (RER) than males under cold exposure, indicating a possibility that mgWAT maintains adiposity under cold stress by inhibiting cold-induced lipid mobilization, fat oxidation and thermogenesis (Fig. 3e and Extended Data Fig. 4d). Generalized linearmodel(GLM)-based regression analyses showed a significant group and interaction effect in RER between males and females based on fat mass as a covariate (Extended Data Fig. 4d). We did not see significant differences in locomotor activities and food consumption between the sexes (Extended Data Fig. 4e). These RER data were further supported by our body composition analysis, which showed that male mice lose significant body weight and fat mass during cold stress, whereas female mice show no differences before and after cold stress (Fig. 3f and Extended Data Fig. 4f–h). We also observed markedly lower UCP1 protein levels in cold-exposed female mgWAT compared with male iWAT (Extended Data Fig. 4i,j). Next, we compared the cold-induced thermogenic gene expression programme between wild-type and Esr1-KO mice. Esr1-KO mice are known to possess hypoplastic mammary ducts that remain rudimentary throughout the life span of a female mouse37. As shown in Fig. 3g and Extended Data Fig. 5a, cold-exposed mgWAT from Esr1-KO mice showed markedly increased numbers of multilocular adipocytes and increased expression of Ucp1 and other thermogenic genes compared with wild-type controls. At five weeks after birth, the mammary ducts are concentrated in the inguinal portion closer to the nipple, confined near the lymph node, and almost completely absent toward the dorsolumbar region of the mgWAT, providing distinct anatomical areas within the mgWAT to test the role of the ductal epithelium in adipose thermogenesis38. We exposed five-week-old female mice to cold, and dissected the inguinal and dorsolumbar regions to assess thermogenic transcripts. Epcam transcripts were present only in the inguinal region; however, the expression of thermogenic genes was similar between the inguinal and dorsolumbar regions except for Ucp1 expression, which was increased in the inguinal part (Fig. 3h). Chi et al. previously reported regional differences between the inguinal and dorsolumbar regions1, and there could be a regional control of Ucp1 expression in five-week-old mice independent of ductal cells. However, consistent with our RER data (Fig. 3e and Extended Data Fig. 4d,h), we observed significantly lower expression of genes involved in lipid mobilization, such as Pnpla2 and Lipe, in inguinal regions compared with dorsolumbar regions, indicating that the ductal epithelium is potentially inhibiting cold-induced lipid mobilization. To further test the role of mammary ducts in adipose thermogenesis, we performed chemical ablation using 70% ethanol. Ethanol is a safe compound with multiple clinical applications as an ablative and sclerosing agent, and has been successfully used to ablate ducts in the mammary gland39–41. Intraductal injection of 70% ethanol effectively eliminates the mammary epithelium without damaging the surrounding stroma and vasculature in the mouse ductal tree42. To confirm a precise delivery of agents into the mammary duct, we validated our intraductal injection technique in the fourth and fifth nipples of mgWAT using Evan’s blue or GFP-expressing adenovirus (Extended Data Fig. 5b,c). We then performed intraductal ethanol or PBS control injections and, consistent with the data in Fig. 3d,g,h, intraductal injection of 70% ethanol caused site-specific ductal ablation and markedly increased thermogenic gene expression and protein levels of UCP1 compared with controls (Fig. 3i,j and Extended Data Fig. 5e,f). Similarly, diphtheria toxin A (DTA)-mediated ablation of luminal cells in a mouse model expressing diphtheria toxin receptor (DTR) under control of the Krt8 promoter (Krt8-DTR) showed increased UCP1 expression surrounding the ducts compared with PBS controls (Extended Data Fig. 5d). Unexpectedly, we noted prominent UCP1 staining in the ductal epithelium of control mice (Extended Data Fig. 5d). We also performed intraductal injection of 0.5% trypsin, which was shown to ablate the luminal cell population in the ducts of the prostate gland43. However, compared with ethanol, trypsin caused substantial damage to the stromal cells, including adipocytes, and thermogenic gene expression was similar to that in PBS controls (Extended Data Fig. 5e,g). Overall, our data suggest a unique role of the mammary duct epithelium in maintaining mgWAT adiposity by blocking adipose thermogenesis, beiging and energy utilization.
Our results demonstrate a possible SNS-mediated crosstalk between mammary ductal cells and adipocytes to control adipocyte thermogenesis. DEG analysis of mammary ductal cells under adrenergic stimulation showed upregulation of genes that encode factors such as adropin (Enho) and LRG1 (Lrg1) in luminal-HS (Fig. 1f) and LCN2 (Lcn2) in luminal-AV (Fig. 1g), which are known to have regulatory roles in adipose metabolism21,27,28. These factors were also shown to be highly enriched in mammary Epcam+ cells by scRNA-seq and RNAscope FISH (Fig. 1). Enho and Lrg1 showed a highly localized expression in mammary ductal cells upon cold exposure, but we also noted some signals outside of mammary ducts in RT samples (Extended Data Fig. 6a,b). The addition of recombinant adropin or LRG1 to beige differentiated mgWAT SVF did not alter the overall thermogenic gene programme compared with controls (Extended Data Fig. 6c), and mgWAT from cold-exposed LRG1 knockout (Lrg1-KO) and wild-type (WT) female mice showed similar thermogenic gene expression (Extended Data Fig. 6c,d). These data suggest that the expression of adropin and LRG1 by luminal-HS cells may not be directly involved in adipose thermogenesis and could have other metabolic roles in mgWAT. Lcn2 demonstrates a strong and significant downregulation in female gonadal white adipose tissue21 (gWAT) and concomitantly female gWAT, which lacks mammary ducts and express exceedingly high levels of Ucp1 compared to male gWAT21. To further study Lcn2 and Ucp1 expression correlation, we performed a meta-analysis on white adipose tissue data from the Hybrid Mouse Diversity Panel (HMDP)44, consisting of more than 100 strains and more than 300 mice, and found a highly significant inverse correlation of Lcn2 and Ucp1 expression in female mice but not in male mice (Extended Data Fig. 6e). Cold-stressed female mgWAT expresses markedly higher levels of Epcam and Lcn2 and lower levels of Ucp1 compared with male scWAT (Fig. 3d and Extended Data Fig. 6f), and scRNA-seq data from male and female SVF show that luminal cells in female mgWAT are the main source of LCN2 (Extended Data Fig. 6g). LCN2 protein was detected almost exclusively in the duct epithelial cells of mgWAT and showed a cold- and isoproterenol-mediated increase in protein and mRNA levels in the ducts of mgWAT (Fig. 4a,b and Extended Data Fig. 6h–j). Our gene regulatory network analysis in luminal cells using single-cell regulatory network inference and clustering (SCENIC)45 revealed highly dynamic cold-induced transcription factor activity (Extended Data Fig. 6k), regulon activity implicated BHLHE41 as a master regulator of genes in luminal-HS cells, and ELF5 showed a cold-specific gene transcriptional activity, which could be directly involved in the expression of mammokines such as Lcn2 and Igfbp5 (Extended Data Fig. 6k,l). ELF5 (ref. 46) is an important transcriptional regulator of mammary luminal alveolar development and our data implicate ELF5 as an adrenergic-activated master regulator in luminal cells that could enhance the expression of mammokines under cold stress, further supporting the luminal-specific activity of mammokine LCN2 under cold stress. In conjunction with our data in Fig. 3d–f and Extended Data Fig. 4e–g, Lcn2-KO female mice under cold exposure show a marked decrease in body weight and scWAT weight compared with wild-type controls and male Lcn2-KO mice (Extended Data Fig. 6m,n). The secretion of LCN2 by luminal epithelium could be a potential mechanism by which mammary ducts block excess thermogenesis and preserve adiposity and could also potentially explain conflicting results21,23,24 in adipose metabolic studies involving LCN2 in male and female mice. To test the cell-autonomous adrenergic ligand-dependent expression of LCN2 protein in luminal cells, we used organoids from a genetic mouse model based on a doxycycline (Dox)-based tet-inducible DTA system derived from interbreeding of mice expressing the tetracycline-on transcription factor rtTA under the control of the luminal epithelial cell-specific Krt8 gene promoter (Krt8-rtTA mice) with mice expressing tet-inducible DTA (tetO-DTA) that can be activated in the presence of Dox. This model enables the ablation of luminal cells in a temporally regulated manner. Mammary duct organoids isolated from Krt8-rtTA-DTA mice47 show increases in LCN2 protein expression on isoproterenol treatment, and these increases were diminished with Dox treatment (Fig. 4c and Extended Data Fig. 6o–t), showing a direct response of luminal cells producing LCN2. To test the physiological role of LCN2 in regulating mgWAT thermogenesis, we mimicked cold induction and adipocyte-specific uptake of LCN2 in mgWAT by inducing exogenous Lcn2 expression specifically in white adipose tissue of Lcn2-KO mice by injecting adeno-associated viruses (AAV) expressing LCN2 or GFP under the adipocyte-specific Adipoq promoter (adipoAAV-LCN2 or adipoAAV-GFP) (Methods). Our unbiased bulk RNA-sequencing data from mgWAT of adipoAAV-LCN2-injected mice show that Lcn2 expression was not supraphysiological compared with controls (Fig. 4d,e). The results shown in Fig. 4d,e demonstrated that exogenous expression of Lcn2 significantly decreased the expression of thermogenic genes such as Ucp1, Cidea and Ppara and increased the expression of adipogenic genes, including Lep, Mmp12 (refs. 48,49), Zfp423 (ref. 50) and Lbp51. Lcn2 overexpression also led to an increase in Aldh1a1, which was recently shown to inhibit adipose thermogenesis by downregulating Ucp1 expression52,53. We validated our RNA-sequencing data by directed qPCR in Lcn2-reconstituted mgWAT of Lcn2-KO and wild-type mice (Extended Data Fig. 7a) and in treated beige differentiated SVF cells isolated from Lcn2-KO mgWAT with and without recombinant LCN2 expression (Fig. 4f). To test the role of LCN2 under cold stress, we exposed male and female Lcn2-KO and wild-type control mice to low temperature for 24 h. Compared with RT controls, wild-type controls and male Lcn2-KO mice, mgWAT from cold-exposed female Lcn2-KO mice showed more beiging or browning and gene expression analysis showed an increase in thermogenic genes such as Ucp1, indicating that LCN2 is a potential limiting factor involved in regulating the propensity of mgWAT to beige (Extended Data Fig. 7b,c). Furthermore, reconstitution of LCN2 specifically in the mammary epithelium of Lcn2-KO mgWAT by intraductal injection of adenovirus encoding LCN2 rescued the anti-thermogenic effect seen in Lcn2-KO mgWAT (Fig. 4g and Extended Data Fig. 7d). Together, our scRNA-seq analysis and mgWAT mammary duct-specific experimental data demonstrate that LCN2 is a mammokine expressed in luminal-AV cells that could function to inhibit thermogenesis and maintain adiposity in mgWAT during cold stress.
Fig. 4 |. LCN2 preserves mgWAT adiposity.
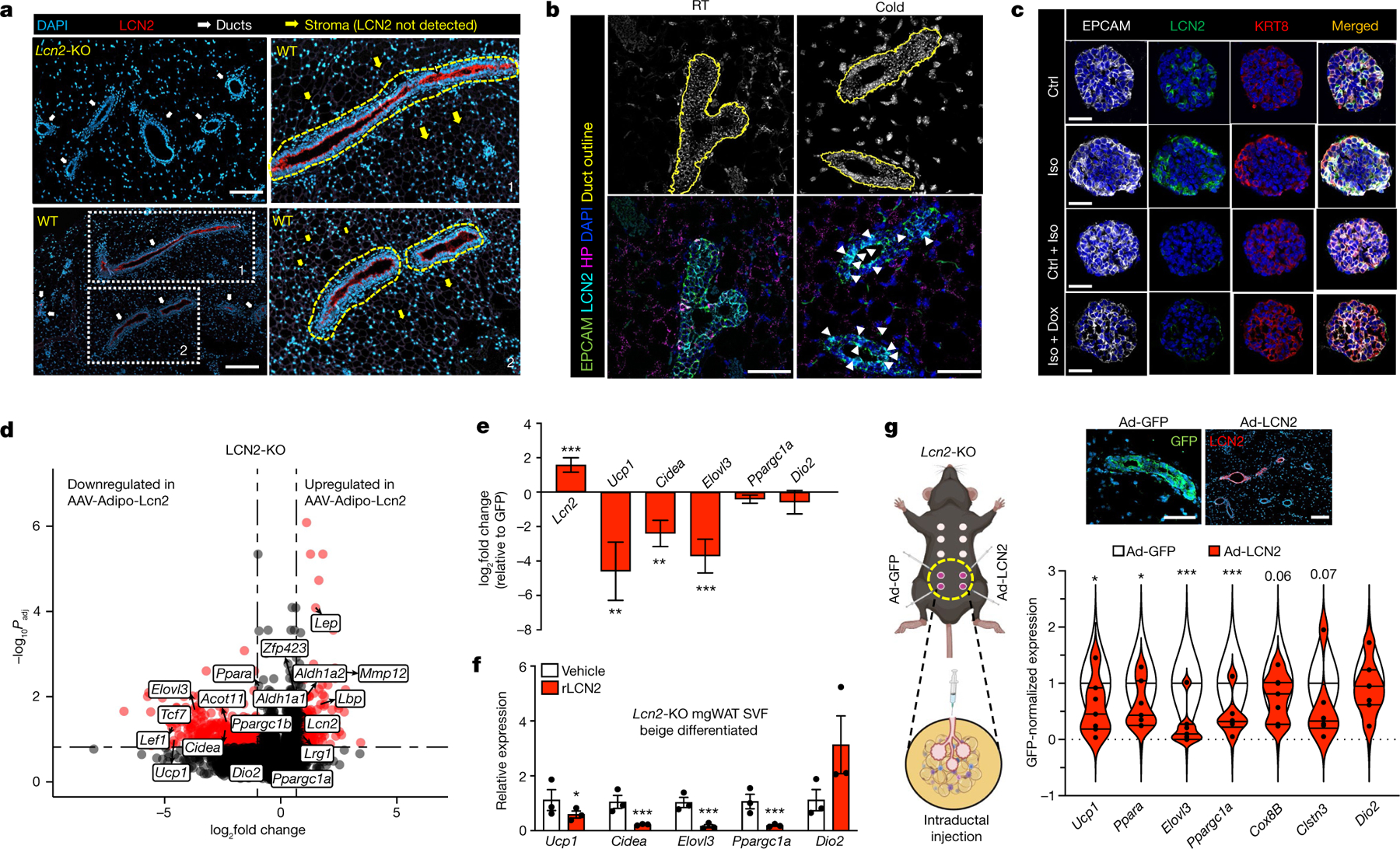
a, Immunostaining of LCN2 and DAPI in the mgWAT of Lcn2-KO and wild-type mice. Regions 1 and 2 are enlarged on the right. Dashed yellow outlines indicate LCN2+ ducts. Representative images from three experiments. Scale bars, 200 μm. b, RNAscope analysis of indicated probes from mgWAT of RT or 24 h cold-exposed mice. White arrowheads indicate increased LCN2 signals. Representative images from three experiments. Scale bars, 50 μm. c, Confocal images of immunofluorescence in organoids derived from Krt8-rtTA-DTA mice treated with and without isoproterenol and Dox. Representative images from three experiments. Scale bars, 10 μm. d,e, Volcano plot (d) and bar plot (e) from gene expression analysis in mgWAT of Lcn2-KO mice treated with adipose-specific AAV-LCN2 or AAV-GFP. Data are represented as fold change of LCN2/GFP ratio as a function of the P value. AAV-LCN2: n = 4; AAV-GFP: n = 4. d, Statistics for RNA-sequencing data derived using Wald test in DESeq2 Bioconductor. e, log2 fold change and statistics from DESeq2, which provides P values, mean and s.e.m. RNA-sequencing rlog values for GFP and LCN2 groups are provided in the Source Data. f, qPCR analysis of expression of indicated genes from beige differentiated mgWAT SVF derived from Lcn2-KO mice treated with and without recombinant LCN2 (rLCN2). Data from three independent experiments. g, Left, cartoon representing intraductal injection of Ad-GFP or Ad-LCN2 in the fourth and fifth nipple. Bottom right, qPCR analysis of expression of indicated genes in mgWAT of Lcn2-KO female mice reconstituted with adenovirus for LCN2 (Ad-LCN2) or GFP (Ad-GFP) expression by intraductal injection. Data were normalized to GFP and plotted as a violin plot. Top right, immunostaining for LCN2 and GFP show ductal-specific expression. n = 6 per group. Scale bars, 100 μm. f,g, Unpaired Student’s t-test. Data are mean ± s.e.m. Cartoon in g created using BioRender.com.
In summary, our studies uncover a direct inhibitory role of mammary duct luminal cells in adipocyte Ucp1 expression and the thermogenic programme. SNS fibres directly innervate EPCAM+ luminal cells, and adrenergic stimulation of luminal cells transduces mammokines such as LCN2, which regulates Ucp1 expression in mgWAT adipocytes under cold exposure. Depletion of EPCAM+ luminal epithelial cells potentiates the capacity of ex vivo differentiated mgWAT preadipocytes to express UCP1, and female mice with loss of ductal epithelium show higher cold-induced UCP1 expression compared to controls. Notably, female mice demonstrate significantly lower scWAT adipocyte UCP1 expression than male mice under cold exposure. Our findings highlight mammary gland epithelium as a highly active metabolic cell type and implicate a broad role of mammokines in controlling female adipose metabolism. Our results also provide a insights into mammary gland biology, expand our understanding of the role of sex, fat depots and the adipose microenvironment in adipocyte UCP1 expression, and reveal the potential of mammokines to regulate sex-specific local and systemic energy homeostasis.
Methods
Animal studies
C57BL/6 WT male and female mice (000664), female Esr1-KO (004744), and male and female Lcn2-KO (24630) mice were acquired from The Jackson Laboratory and maintained in a pathogen-free barrier-protected environment (12:12 h light:dark cycle, 22–24 °C and 40–60% humidity with ad libitum access to chow diet and water) at the Mount Sinai animal facilities. The female Krt8-DTR was from the laboratory of C.B. and the female Krt8-rtTA-TetO-DTA mouse model was described previously47. The female Lrg1-KO mouse model was a gift from P. Cohen28. All mice used in this study were 8 to 12 weeks old. For the timecourse cold exposure experiment, mice at 8 to 10 weeks of age were singly housed at 4 °C room in a non-bedded cage without food and water for first 6 h; thereafter food, water, and one cotton square were added. For the 24 h collection, 3 h before collection, food, water, and cotton square were removed and then mice were collected. At the end of the experiment, mgWAT was resected for analysis. Adrenalectomy was performed by The Jackson Laboratory Surgical Services. For overexpression studies, recombinant adeno-associated virus serotype 8 (AAV8) expressing LCN2 or GFP under the Adipoq promoter was generated and injected as described previously21. Indirect calorimetry was performed using Promethion Systems (Sable Systems). Mice were placed individually in chambers at ambient temperature (22.0 °C) for 21 h followed by 24 h cold (4.0 °C) with 12 h:12 h light:dark cycles. Animals had free access to food and water. Respiratory measurements were made in 5 min intervals after an initial 7–9 h acclimation period. Energy expenditure was calculated from VO2 and RER using the Lusk equation, energy expenditure (kcal h−1) = (3.815 + 1.232 × RER) × [VO2 (ml min−1)]. Indirect calorimetry data were analysed using CALR web-based software54. Body composition (fat mass) was determined using EchoMRI Body Composition Analyzer. Animal experiments were conducted in accordance with the Mount Sinai Institutional Animal Care and Use Committee (IACUC). Sample sizes were based on our previous experience in the field and past publications3,21. We did not use randomization in our allocation process since the experimental groups were primarily separated based on genotype or sex. For animal experiments, mice were separated based on genotype or sex, so the investigators were not blinded.
RNA sequencing
RNA isolation, library preparation, and analysis were conducted as previously described3,21. Flash-frozen mgWAT samples were homogenized in QIAzol (Qiagen), and following chloroform phase separation, RNA was isolated according to the manufacturer’s protocol using miRNeasy columns (Qiagen). Libraries were prepared from extracted mgWAT fat RNA (Agilent 2200 Tapestation eRIN >8.2) using KAPA Stranded mRNA-seq Kit (KK8421, KAPA Biosystems), per the manufacturers’ instructions. The pooled libraries were sequenced using an Illumina HiSeq 4000 instrument with SE50bp reads (Illumina). Reads were aligned to the mouse genome mm10 using STAR55 or HISAT2 (ref. 56) aligner and quantified using the Bioconductor R packages as described in the RNA-Seq workflow57. P values were adjusted using the Benjamini–Hochberg procedure of multiple hypothesis testing57.
Single-cell isolation from SVF
Single-cell SVF populations from adipose tissue were isolated as described previously3. The fourth iWAT depot mgWAT from mice exposed to cold stress (4 °C) or room temperature for 24 h was dissected and placed on a sterile 6-well tissue culture plate with ice-cold 1× DPBS. Excess liquid was removed from fat pads by blotting. Each tissue was cut and minced with scissors and then placed in 15 ml conical tubes containing digestion buffer (2 ml DPBS and Collagenase II at 3 mg ml−1 (Worthington Biochemical)) for 40 min of incubation at 37 °C with gentle shaking at 100 rpm.Followingtissuedigestion,enzymeactivitywas stopped with 8 ml of resuspension medium (DMEM/F12 with Glutamax supplemented with 15% FBS and 1% pen/strep (Thermo Scientific)). The digestion mixture was passed through a 100-μm cell strainer and centrifuged at 150g for 8 min at room temperature. To remove red blood cells, the pellet was resuspended and incubated in RBC lysis buffer (Thermo Scientific) for 3 min at room temperature, followed by centrifugation at 150g for 8 min. The pellet was then resuspended in resuspension medium and again spun down at 150g for 8 min. The cell pellet was resuspended in 1 ml of 0.01% BSA (in DPBS) and passed through a 40-μm cell strainer (Fisher Scientific) to discard debris. Cell number was counted for 10X Genomics single-cell application.
SVF single-cell barcoding and library preparation
To yield an expected recovery of 4,000–7,000 single cells, an estimated 10,000 single cells per channel were loaded onto Single Cell 3′ Chip (10X Genomics). The Single Cell 3′ Chip was placed on a 10X Genomics instrument to generate single-cell gel beads in emulsion. Chromium Single Cell 3′ v3 Library and Cell Bead Kits were used according to the manufacturer’s instructions to prepare scRNA-seq libraries.
Illumina high-throughput sequencing libraries
Qubit Fluorometric Quantitation (ThermoFisher) was used to quantify the 10X Genomics library molar concentration and a TapeStation (Aligent) was used to estimated library fragment length. Libraires were pooled and sequenced on an Illumina HiSeq 4000 (Illumina) with PE100 reads and an 8-bp index read for multiplexing. Read 1 contained the cell barcode and unique molecular identifier and read 2 contained the single-cell transcripts.
scRNA-seq pre-processing and quality control
To obtain digital gene expression matrices (DGEs) in sparse matrix representation, paired end reads from the Illumina HiSeq 4000 were processed and mapped to the mm10 mouse genome using 10X Genomics’ Cell Ranger v3.0.2 software suite. In brief, .bcl files from the UCLA Broad Stem Cell Research Center sequencing core were demultiplexed and converted to fastq format using the ‘mkfastq’ function from Cell Ranger. Next, the Cell Ranger ‘counts’ function mapped reads from fastq files to the mm10 reference genome and tagged mapped reads as either exonic, intronic, or intergenic. Only reads which aligned to exonic regions were used in the resulting DGEs. After combining all four sample DGEs into a single study DGE, we filtered out cells with (1) unique molecular identifier counts <700 or >30,000, (2) gene counts <200 or >8,000, and (3) mitochondrial gene ratio >10%. This filtering resulted in a dataset consisting of 42,052 genes across 12,222 cells, with approximately 2,300–4,650 cells from each sample. A median of 2,411 genes and 7,252 transcripts were detected per cell.
Identification of cell clusters
To achieve high-resolution cell-type identification and increased confidence in our cell-type clustering we brought in external publicly available single-cell data from SVF and mammary tissues. Specifically, we included single-cell data from 9 datasets comprising 91,577 single cells from the mammary gland and multiple adipose depots, across 4 different single-cell platforms (Supplementary Table 1). These external datasets and the SVF data from this study were all independently normalized using sctransform58 and integrated using Seurat59,60 v3.1.5. in R Studio 3.3.0. The single-cell expression profiles were projected into two dimensions using UMAP61 or t-SNE62 and the Louvain63 method for community detection was used to assign clusters. This integrated data was only used to identify and define the cell types. All plots which are not explicitly designated as integrated with at least one external dataset and all downstream analyses (for example, differential expression analyses) were conducted on non-integrated data to retain the biological effect of the cold treatment. Visualization of the non-integrated data was conducted on a subsampled dataset where all samples had the same number of cells to give an equal weight to each sample, however, all downstream analyses (for example, differential expression analyses) leveraged the full dataset.
Cell-type-specific marker gene signatures
Cell-type-specific marker gene signatures were generated by identifying genes with expression levels twofold greater (adjusted P values <0.05) than all other cell types. To ensure consistency across samples, Seurat’s FindConservedMarkers function (Wilcoxon rank sum test with a meta P value) was applied across each sample.
Resolving cell identities of the cell clusters
To identify the cell-type identity of each cluster, we used a curated set of canonical marker genes derived from the literature (Supplementary Table 2) to find distinct expression patterns in the cell clusters. Clusters which uniquely expressed known marker genes were used as evidence to identify that cell type. Cell subtypes that did not express previously established markers were labelled by both general cell-type markers and novel markers obtained with Seurat’s FindConservedMarkers function were used to define the cell subtype.
Differential gene expression analysis
Within each identified cell type or subtype, cold-treated and room temperature single cells were compared for differential gene expression using Seurat’s FindMarkers function (Wilcoxon rank sum test) in a manner similar to Li et al.15. DEGs were identified using two criteria: (1) an expression difference of ≥1.5-fold and adjusted P value <0.05 in a grouped analysis between room temperature (n = 2) and cold-treated (n = 2) mice; (2) an expression difference of ≥1.25-fold and consistent fold-change direction in all 4 possible pairwise combinations of cold-treated versus room temperature mice.
Gene regulatory network inference
Gene regulatory network inference was performed with pySCENIC45 following the workflow described by Van de Sande et al.64. In brief, starting with counts data, gene modules which are co-expressed with transcription factors were identified with GRNBoost2 (ref. 65). Next, candidate regulons were created from transcription factor–target gene interactions and indirect targets were pruned based on motif discovery with cisTarget45. Finally, regulon activity was quantified at cellular resolution with AUCell45 which allowed for the prioritization of regulons for each cell type based on the quantified activity.
Real-time qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and reverse transcribed with the iScript cDNA synthesis kit (Biorad). cDNA was quantified by real-time PCR using SYBR Green Master Mix (Diagenode) on a QuantStudio 6 instrument (Themo Scientific). Gene expression levels were determined by using a standard curve. Each gene was normalized to the housekeeping gene 36B4 and was analysed in duplicate. Primers used for real-time PCR are presented in Supplementary Table 3.
RNAscope FISH
mgWAT from RT or cold-exposed mice (Jackson Laboratory, 000664) was fixed in 10% formalin overnight, embedded with paraffin, and sectioned into unstained, 5-μm-thick sections. Sections were baked at 60 °C for 1 h, deparaffinized, and baked again at 60 °C for another hour prior to pre-treatment. The standard pre-treatment protocol was followed for all sectioned tissues. In situ hybridization was performed according to manufacturer’s instructions using the RNAscope Multiplex Fluorescent Reagent Kit v2 (323136, Advanced Cell Diagnostics,). Opal fluorophore reagent packs (Akoya Biosciences) for Opal 520 (FP1487A), Opal 570 (FP1488A), Opal 620 (FP1495A), and Opal 690 (FP1497A) were used at a 1:1,000 dilution in TSA buffer (322809, Advanced Cell Diagnostics). RNAscope probes from Advanced Cell Diagnostics were used for the following targets: EPCAM (catalogue no. 418151), ENHO (873251), LRG1 (423381), LCN2 (313971), HP (532711), WNT4 (401101), NRG4 (493731), KRT8 (424528), and MFGE8 (408778). Slides were mounted with ProLong Diamond Antifade Mountant with DAPI (P36962, Life Technologies). Fluorescent signals were captured with the 40x objective lens on a laser scanning confocal microscope LSM880, (Zeiss, White Plains, NY).
Fluorescence-activated cell sorting
mgWAT from RT or cold-exposed mice (Jackson Laboratory, 000664) was dissected, cut, minced and digested with collagenase D (5 mg ml−1, 11088882001) and dispase (2 mg ml−1, 17105041) over 40 min at 37 °C with gentle shaking at 100 rpm. Enzymatic digestion was stopped with DMEM/15% FBS and the cell suspension was filtered through a 100-μm nylon mesh cell strainer and centrifuged for 10 min at 700g. SVF pellet was resuspended in 1 ml Red Blood Cell lysis buffer (41027700) and incubated for 5 min at room temperature. Cell suspension was diluted in 4 ml DPBS and filtered through a 40-μm nylon mesh cell strainer and centrifuged for 10 min at 700g. Single-cell suspension was blocked for 10 min on ice in 500 μl DPBS/5% BSA (blocking buffer), centrifuged for 10 min at 700g, resuspended in 200 μl of DBPS/0.5% BSA (FACS buffer) solution containing the desired antibody mix, and incubated for 1 h at 4 °C in the dark with gentle rotation. Antibody-stained samples were washed with 800 μl FACS buffer, centrifuged 10 min at 700g, and resuspended in FACS buffer containing DAPI (at 1 μg ml−1). Flow cytometry analysis was performed on a BD FACS Canto II (BD Biosciences) and results analysed on FCS Express software (DeNovo Software). Fluorescently tagged anti-mouse antibodies (BioLegend) were used to label cell surface markers for flow cytometry analysis: EPCAM–FITC (clone G8.8, 118207, 1:250), Sca-1–APC (Ly6, clone E13–161.7, 122512, 1:500), CD49f–APC (clone GoH3, 313616, 1:250). For flow cytometry analysis, negative selection of CD45-expressing cells using CD45 microbeads (130052301) was performed immediately prior to the EPCAM positive selection protocol described above.
Isolation, selection and ex vivo treatment of EPCAM-positive cells
MACS microbeads (Miltenyi Biotec) were used for immuno-magnetic labelling positive selection of EPCAM-expressing cells (anti-CD326, 130105958). Before magnetic labelling, a single-cell suspension from the SVF of female mouse iWAT was prepared in MACS buffer (PBS, pH 7.2, 0.5% bovine serum albumin (A7030, Sigma) and 2 mM EDTA), filtered through a MACS pre-separation 30-μm nylon mesh (130041407) to remove cell clumps. Then, for magnetic labelling of EPCAM-expressing cells, 10 μl of EPCAM microbeads were added per 1 × 107 total cells in 100 μl buffer, incubated for 15 min with rotation at 4 °C, washed with 1 ml buffer, centrifuged at 700g for 5 min, resuspended in 500 μl buffer, and added to a pre-equilibrated MACS LS column (130042401) in the magnetic field of a MACS separator (130042302). Unlabelled EPCAM-negative cells were collected in the flow-through with three subsequent washes. The column was removed from the magnetic field, 5 ml of MACS buffer were added to the column, and the magnetically labelled EPCAM-positive cells retained in the column were collected by flushing the cells down the column with a plunger. Finally, EPCAM-negative and EPCAM-positive cell populations were centrifuged at 700g for 5 min, resuspended in DMEM/F12 with Glutamax supplemented with 15% FBS and 1% pen/strep (Thermo Scientific) and plated on collagen I-coated 12-well tissue culture plates (354500, Corning). Medium was replaced every other day for 6 days, followed by cell lysis with TriZol for phenol/chloroform RNA extraction, and real-time qPCR analysis.
Adipocyte differentiation and treatments
10T1/2 (Source: ATCC), SVF (from the laboratory of P.R.) or APCs isolated using APC isolation kit (Miltenyi Biotec,130-106-639) from the fourth and fifth mgWAT was isolated from 8-week-old wild-type or Lcn2-null female mice, respectively. 10T1/2 cells were maintained as previously described3. The SVF and APC cells were maintained in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F12 (DMEM/F12) supplemented with 1% Glutamax, 10% fetal calf serum and 100 U ml−1 of both penicillin and streptomycin (basal medium). Two days after plating (day 0), when the cells reached nearly 100% confluency, the cells were treated with induction medium containing basal medium supplemented with 4 μg ml−1 insulin, 0.5 mM IBMX, 1 μM dexamethasone, and 1 μM rosiglitazone. After 48 h, the cells were treated with a maintenance medium containing the basal medium supplemented with 4 μg ml−1 insulin, and 1 μM rosiglitazone, with a medium change every 2 days until day 10. For qPCR, differentiated cells were treated with 1 μg ml−1 recombinant LCN2 (Sino Biological), LRG1 (0–80 ng ml−1, R&D Systems), or Adropin (0–100 nM, Phoenix Pharmaceutical) for 24 h and then treated with isoproterenol (Sigma) or norepinephrine (Sigma) for 6 h after which RNA was collected.
iDISCO and Adipoclear tissue labelling and clearing
Sample collection.
Immediately after cold exposure mice were anaesthetized with isoflurane (3%) and perfused with heparinized saline followed by 4% paraformaldefyde (PFA) (Electron Microscopy Sciences). Fat pads were carefully dissected and postfixed overnight in 4% PFA at 4 °C. The following day the tissue was washed three times in PBS before proceeding with the optical clearing protocol.
Optical clearing.
Whole-mount staining and clearing was performed using the Adipoclear protocol as previously described1. Dissected fat pads were dehydrated at room temperature with a methanol/B1n buffer (0.3 M glycine, 0.1% Triton X-100 in H2O, pH 7) gradient (20%, 40%, 60%, 80%, 100%), incubated 3 times (1 h, overnight, 2 h) in 100% dichloromethane (Sigma-Aldrich) toremove hydrophobic lipids, washed twice in 100% methanol, and bleached in 5% H2O2 overnight at 4 °C to reduce tissue autofluorescence. Fat pads were then rehydrated with a methanol/B1n buffer gradient (80%, 60%, 40%, 20%) and then washed twice in 100% B1n buffer (1 h, overnight). Primary antibodies (EPCAM, ThermoFisher, 1:250; TH, Millipore Sigma, 1:1,000) were diluted in modified PTxwH (PBS with 0.5% Trixon X-100, 0.1% Tween-20, 2 μg ml−1 heparin, as previously described66) and applied for 6 days at 37 °C. Following 5 washes with modified PTxwH over 1 day with the last wash performed overnight, secondary antibodies (EPCAM: Donkey anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, DyLight 550, Invitrogen, 1:500; TH: Alexa Fluor 647 AffiniPure Donkey Anti-Rabbit IgG (H+L), Jackson Immunoresearch, 1:500) were diluted in modified PTxwH (1:500) and samples were incubated for 6 days at 37 °C. Samples were washed 5 times over 1 day in modified PTxwH at 37 °C, 5 times over 1 day in PBS at room temperature, embedded in 1% agarose, dehydrated with a methanol gradient in H2O (12%, 50%, 75%, 100%), washed 3 times for 1 h in 100% methanol followed by 3 times for 1 h in dichloromethane before being transferred to dibenzylether (Sigma-Aldrich) to clear.
Imaging.
Z-stacked optical sections of whole fat pads were acquired with an Ultramicroscope II (LaVision BioTec) at a ×1.3 magnification with a 4 μm step size and dynamic focus with a maximum projection filter. Samples were then imaged in glass-bottom μ-dishes (81158, Ibidi) using an inverted Zeiss LSM 710 confocal microscope with a ×10 (NA: 0.3) objective and a step size of 5 μm.
Image analysis.
Imaris versions 9.6.0–9.7.2 (Bitplane) were used to create digital surfaces covering ducts, TH+ innervation and total sample volume (×1.3 LSFM images and ×10 confocal images) to automatically determine volumes and intensity data. Volume reconstructions were performed using the surface function with local contrast background subtraction. For detection of EPCAM+ ducts in ×1.3 LSFM images, a smoothing factor of 5 μm was used and the threshold factor was set to correspond to the largest duct diameter in each sample. For detection of TH+ nerves in ×1.3 LSFM images, a smoothing factor of 3 μm and a threshold factor of 80 μm were used. For detection of EPCAM+ ducts in ×10 confocal images, a smoothing factor of 3.35 μm was used and a threshold factor corresponding to the diameter of the thickest duct wall in each sample was used. For detection of TH+ nerves in ×10 confocal images, a smoothing factor of 2 μm and a threshold factor of 5 μm were used. In ×10 confocal images, nerve–duct interactions were defined by masking the TH channel using the TH+ nerve surface to remove any background, and then masking it again using the EPCAM+ duct surface. This process revealed TH+ innervation overlapping with EPCAM+ staining. A new surface was created to cover this overlapping TH+ innervation using a smoothing factor of 2 μm and a threshold factor of 5 μm.
Mammary gland organoid culture
For organoid culture, we used a previously published protocol67. In brief, fat pads of 8- to 9-week-old K8rtTA/TetO-DTA47 virgin female mice were dissected, and the lymph nodes removed. Tissues were briefly washed in 70% ethanol and manually chopped into 1 mm3 pieces. The finely minced tissue was transferred to a digestion mix consisting of serum-free Leibovitz’s L15 medium (Gibco) containing 3 mg ml−1 collagenase A (Sigma) and 1.5 mg ml−1 trypsin (Sigma). This was incubated for 1 h at 37 °C to liberate epithelial tissue fragments (organoids). Isolated organoids were mixed with 50 μl of phenol-red-free Matrigel (BD Biosciences) and seeded in 24-well plates. The basal culture medium contained phenol-red-free DMEM/F12 with penicillin/streptomycin, 10 mM HEPES (Invitrogen), Glutamax (Invitrogen), N2 (Invitrogen) and B27 (Invitrogen). The basal medium was supplemented with Nrg1 (100 ng ml−1, R&D), Noggin (100 ng ml−1, Peprotech) and R-spondin 1 (100 ng ml−1, R&D). Then, 500 μl supplemented basal culture medium was added per well and organoids were maintained in a 37 °C humidified atmosphere under 5% CO2. After one week in culture, mammary organoids were released from the Matrigel by breaking the matrix with a P1000 pipette on ice. After 2–3 passages of washing and centrifugation at 1,500 rpm (140 g) for 5 min at 4 °C, mammary cells were resuspended in Matrigel, seeded in 24-well plates and exposed to the previously described culture conditions. Organoids were treated either with 10 μg ml−1 of Dox to promote luminal cell ablation or with 10 μM isoproterenol (Sigma) for 6 h. After 6 h treatment, organoids were collected from Matrigel as mentioned before to perform further analysis.
RNA extraction and real-time qPCR in organoid samples
To perform RNA extraction, isolated organoids were collected into kit lysis buffer. RNA was extracted with Qiagen RNeasy Micro Kit. After nanodrop RNA quantification and analysis of RNA integrity, purified RNA was used to synthesize the first-strand cDNA in a 30 μl final volume, using Superscript II (Invitrogen) and random hexamers (Roche). Genomic contamination was detected by performing the same procedure without reverse transcriptase. Quantitative PCR analyses were performed with 1 ng of cDNA as template, using FastStart Essential DNA green master (Roche) and a Light Cycler 96 (Roche) for real-time PCR system. Relative quantitative RNA was normalized using the house-keeping gene Gapdh. Analysis of the results was performed using Light Cycler 96 software (Roche) and relative quantification was performed using the ΔΔCt method using Gapdh as a reference.
Immunofluorescence in organoid samples
For immunofluorescence, collected organoids were pre-fixed in 4% PFA for 30 min at room temperature. Pre-fixed organoids were washed in 2% FBS-PBS, embedded in OCT and kept at −80 °C. Sections of 4 μm were cut using a HM560 Microm cryostat (Mikron Instruments). Tissue sections were incubated in blocking buffer (BSA 1%, HS 5%, Triton X-100 0.2% in PBS) for 1 h at room temperature. The different primary antibodies were incubated overnight at 4 °C. Sections were then rinsed in PBS and incubated with the corresponding secondary antibodies diluted at 1:400 in blocking buffer for 1 h at room temperature. The following primary antibodies were used: rat anti-K8 (1:1,000, Troma-I, Developmental Studies Hybridoma Bank, University of Iowa), rabbit anti-EPCAM (1:1,000, ab71916, Abcam), goat anti-LCN2 (1:50, AF1857, R&D). The following secondary antibodies, diluted 1:400, were used: anti-goat (A11055) conjugated to Alexa Fluor 488 (Invitrogen), anti-rat (712-295-155) rhodamine Red-X and anti-rabbit (711-605-152) Cy5 (Jackson ImmunoResearch). Nuclei were stained with Hoechst solution (1:2,000) and slides were mounted in DAKO mounting medium supplemented with 2.5% DABCO (Sigma).
Immunofluorescence microscopy of mgWAT sections
Fat pads were removed and fixed in 10% formalin and for 24 h then transferred to a 100% ethanol. Tissue sections were embedded in paraffin and sectioned at 5 μm using a Leica RM2125 microtome on glass slides for downstream processing. Inguinal fat samples were immunohistochemically stained to detect GFP (primary antibody: Rabbit anti-GFP, Invitrogen, CAT: A-11122, dilution 1:200; secondary antibody: Goat anti-Rabbit Alexa Fluor 488, Invitrogen, CAT: A-11008, dilution 1:200), UCP1 (primary antibody: Rabbit anti-UCP1, Abcam, CAT: ab10983, dilution 1:200; secondary antibody: Goat anti-Rabbit Alexa Fluor 488, Invitrogen, CAT: A-11008, dilution 1:200), or LCN2 (primary antibody: Goat anti-LCN2, R&D Systems, CAT: AF1857, dilution 1:150; secondary antibody: Donkey anti-Goat Alexa Fluor 568, Invitrogen, CAT: A-11057, dilution 1:200). All slides were and cover slipped with Vectasheild Antifade Mounting Medium containing DAPI. Tissue sections were analysed using a Zeiss AxioImager Z2M with ApoTome microscope. Exposure times for DAPI, GFP, and mCherry channels were kept consistent between samples. Images were subsequently analysed using Fiji image processing software (NIH).
Intraductal injection
Anaesthetized mice were injected with 10 μl viral suspension or ablation agent directly into the mammary ducts of the fourth and fifth nipple pairs, using Pasteur pipettes (World Precision Instruments) fitted into 10 μl or 50 μl gastight syringes (Hamilton). Luminal epithelial cells were ablated in WT mice with 70% molecular biology-grade ethanol in distilled water injected into the fourth and fifth nipple pairs of each mouse; PBS was injected of each mouse as a control. For LCN2 reconstitution experiment, LCN2 expression was reintroduced into fourth and fifth nipples of Lcn2-KO mice using Ad-CMV-LCN2 overexpression viruses at 2 × 1010 total plaque-forming units (PFU) per injection, diluted in PBS; GFP adenoviruses at 2 × 109 total PFU per injection were injected contralaterally as a control. Seventy-two hours after injection, mice were exposed to cold for 24 h and mgWAT containing the targeted ducts were collected. To reflect differing ductal densities, iWAT and dorsolumbar sections of mgWAT were separated for some analyses.
Adeno-associated virus generation
For overexpression studies, recombinant AAV8 expressing LCN2 or GFP was generated as described previously21. In brief, mouse Lcn2 cDNA or GFP was cloned into an AAV expression plasmid under an adiponectin promoter (AAV8-hAdp) with a liver-specific miRNA target sequence (miR122T) for adipose-specific expression. Viral packaging, propagation, and purification were carried out at the University of Pennsylvania Gene Therapy programme vector core.
Generation of adenoviruses
Ad-CMV-LCN2 plasmid was made using the pAd/CMV/V5-DEST Gateway recombination system (Life Technologies). For Ad-CMV-LCN2, the mouse full-length Lcn2 was amplified by PCR using primers; Lcn2 forward primer: tactac ggatcc ACCATGGCCCTGAGTGTCATGTGTCTG; Lcn2 reverse primer: TACTACGAATTCTCAGTTGTCAATGCATTGGTCGGT. The full-length Lcn2 was cloned into the pENTR vector by double digest with BamHI and EcoRI. PENTR vector containing full-length Lcn2 was recombined with pAd/CMV/V5-DEST. Adenoviruses were packaged and produced in 293A cells. Titres were determined by plaque assay.
Transwell experiments
APCs, SVF and 10T1/2 cells were seeded at density of 100,000 cells per well in 12-well plate format in Corning DMEM F12 or DMEM medium with initial beige differentiation drugs insulin (5 μg ml−1) and T3 (1 nM). Six Transwell (Corning, 3401) permeable polycarbonate membrane 0.4-μm pore size Transwell inserts were added to wells intended for co-culture. NMuMG cells (Source:ATCC) at density of 5,000 cells per well were seeded in DMEM medium to each Transwell insert with initial beige differentiation drugs insulin (5 μg ml−1) and T3 (1 nM). After two days of cells seeding, beige differentiation full cocktail (Insulin 5 μg ml−1, T3 1 nM, rosiglitazone 1 uM, IBMX 0.5 mM, Indomethacin 125 μM, dexamethasone 2 μg ml−1) were added in all cells including Transwell inserts with appropriate cell culture medium. After 48 h of full cocktail in all the cells for beige differentiation programme induction, Full cocktail were replaced with the beige maintenance medium (insulin 5 μg ml−1, T3 1 nM, rosiglitazone 1 μM). Beige maintenance medium were replaced on every two days. After significant density of lipid droplets formation at day 6 of the assay, Treatment wells were treated with 10 μM isoproterenol and control wells with none in beige maintenance medium. After 24 h of 10 uM isoproterenol treatment, all the control and isoproterenol-treated well cells were collected using Trizol reagent for real-time qPCR or Seahorse respirometry experiment.
Mitochondrial respirometry
OCR (in pmoles min−1 (μg protein)−1) was measured using a Seahorse XFe96 Extracellular Flux Analyzer (Agilent). The morning of the assay, SVF, SVF:NMuMG, APCs or APC:NMuMG cells were seeded into XFe96 plates (Agilent) at a density of 15,000 cells per well Following attachment, cells were washed twice with Seahorse XF Base Medium (Agilent) and cultured in XF Base Medium supplemented with 25 mM glucose, 2 mM sodium pyruvate and 2 mM glutamine (pH 7.4) for 1 h at 37 °C in a non-CO2 incubator. Following three baseline measurements, cells were sequentially exposed to isoproterenol (10 μM) or noradrenaline (10 μM), oligomycin (5 μM), FCCP (1 μM) and antimycin A (5 μM) as indicated. After completion of the assay, the microplate was saved, and protein isolated for normalization. In a certain set of experiments, cells were pre-treated with 10 μM noradrenaline or vehicle control for 5 h before switching cells to Seahorse base medium and measurement of OCR. The Seahorse XF Cell Mito Stress Test Report Generator (Agilent) was used to calculate the acute response to noradrenaline according to the manufacturer’s instructions. Statistical significance was based on two-way ANOVA with Bonferroni’s post hoc test.
Data and statistical analyses
Measurements were taken from distinct samples and data were analysed using Microsoft Excel and GraphPad Prism V8.4.3. Significance was determined by two-tailed unpaired t-test (parametric distribution), two-way ANOVA Bonferroni’s post hoc test, analysis of covariance (ANCOVA) or by a Mann–Whitney test (non-parametric distribution) or Bioconductor DSeq2 Wald test. Statistics for scRNA-seq data were derived using Wilcoxon rank sum test and adjusted P values were based on Bonferroni correction. Significance was set at an alpha level of 0.05.
Graphical and cartoon representation
Models were generated using BioRender.com under Icahn School of Medicine-Diabetes Institute’s Plan Academic Subscription, which includes journal publishing rights.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Cold-associated increase in cell percentages of luminal epithelium subtypes.
a) UMAP plots of integrated single cell data from this study and 8 external datasets (see Methods and Supplementary Table 1). Each point represents a single cell and are colored by dataset. b) UMAP plots of integrated single cell data from this study and 8 external datasets (see Methods). Each point represents a single cell and clusters are colored by cell type. c) Expression of known canonical markers for cell types in the SVF and mammary gland. Color corresponds to average expression level and size corresponds to percentage of cells which express the gene within the cluster. d) Differentially expressed genes between COLD (24 hr) treated mice and RT animals across all cell types. Significant DEGs (adjusted p-value < 0.05) are highlighted with the average log fold change between 4 degree and RT indicated on the y-axis. Cell types are ordered on the x-axis based on the number of significant DEGs. e) Relative fractions of cell types within each sample. Dots indicate average relative fractions across all samples. f) Aggregated UMAP plot of subclustering of luminal single cells from RT and cold-exposed mice. g) Differentially expressed genes between COLD treated mice and RT animals across Luminal-HS-AV. Select significant DEGs (adjusted p-value < 0.05) are highlighted with the average log fold change between 4 degree and RT indicated on the y-axis. Genes indicated by red arrows encode for secreted factors. h) UMAP and violin plots of normalized gene expression levels for genes of interest in luminal cells. Violin plots also show expression levels of Adrb1, Adrb2, and Adrb3 in luminal subtypes. i–k) tSNE plots of cell type clusters and normalized gene expression levels for genes of interest across multiple datasets including female mgWAT SVFs (i), male iWAT SVFs (j), and mature mgWAT adipocytes (k). Statistics for scRNA-seq data (d,g) was derived using Wilcoxon rank sum test between cold vs RT cells for cells and the adjusted p-value is based on Bonferroni correction.
Extended Data Fig. 2 |. SNS fibers directly innervate ductal epithelial cells.
a) Light sheet microscopy fluorescence (LSFM) images of mgWAT isolated from female mice exposed to RT or COLD for 24 h and stained with TH antibody (SNS fibers) and EPCAM antibody (ductal cells). N = 6,6. Scale bar at 200 μm. b) Confocal images of mgWAT isolated from female mice exposed to RT or COLD for 24 h and stained for EPCAM and TH antibodies. Merged staining of EPCAM and TH represent neuroductal points. Representative images of 6 mice per condition. Scale bar at 100 μm. c-j) Quantification of indicated parameters of images from (b). N = 6,5. Unpaired Student’s t Test (j). Data are represented as mean ± S.E.M.
Extended Data Fig. 3 |. Mammary gland ductal epithelium inhibits cold-induced adipocyte thermogenesis.
a) Representative FACS plot of CD49f and EPCAM expression in EPCAM bead selected EPCAM+ or EPCAM-ve epithelial cells from RT or cold mice. 2 mice per condition and 4 mammary fat pads per mouse. Representative data from 4 independent experiments. b) Real-time qPCR of indicated genes in beige differentiated SVFs from mgWAT treated with indicated concentration of Iso for 5 h. c) Real-time qPCR of indicated genes from beige differentiated primary mgWAT SVF (Parent) and EPCAM-ve (EPCAM-NEG) cells isolated from cold exposed mice treated with and without 10 μM isoproterenol (ISO) for 24 h ex vivo. d) Images showing cell morphology of D0-D9 beige differentiated 10T1/2 and NMuMG (0, 5, and 10%) mixture cells. Yellow enclosures show NMuMG epithelial cells surrounded by 10T1/2 cells. Scale bar at 100 μm. e) Real-time qPCR of indicated genes from beige differentiated and Iso treated 10T1/2 and 5–10% NMuMG mixture cells. f) Images showing cell morphology of D0-D8 beige differentiated 10T1/2 and NMuMG (0–10%) mixture cells. Yellow enclosures show NMuMG epithelial cells surrounded by 10T1/2 cells. Scale bar at 100 μm. g) Real-time qPCR of indicated genes from beige differentiated and Iso treated 10T1/2 and 2.5–10% NMuMG mixture cells. h–i) Real-time qPCR of indicated genes from a transwell system of beige differentiated norepinephrine (NE) treated (h) or Iso treated (APC) (i) APCs in the presence (APC:NMuMG) or absence (APC) of NMuMG. j) Average oxygen consumption rate (OCR) in APC and APC:NMuMG pretreated with (NE-APC and NE:APC:NMuMG) and without (Ctrl APC and Ctrl-APC:NMuMG) 10 μM norepinephrine (NE) followed by acute treatment with NE (10 μM), 5 μM oligomycin (Oligo), 1 μM FCCP, and 5 μM antimycin A (AA). Mean of N = 6,6. k) Fold increase in OCR from basal respiration (J) with and without acute NE injection. N = 6,6. l) Average OCR normalized to protein level (μg protein) in SVF and SVF:NMuMG with 10 μM NE, 5 μM Oligo, 1 μM FCCP, and 5 μM AA. N = 9,9. m) Real-time qPCR of indicated genes from a transwell system of beige differentiated (Ctrl 10T1/2) or Iso treated (Iso-10T1/2) 10T1/2 cells in the presence of NMuMG with (Iso-10T1/2:NMuMG) or without iso (Ctrl-10T1/2:NMuMG). Results are from three independent experiments. *, p < 0.05; **, p < 0.01, ***, p < 0.001 comparing Iso-10T1/2 and Iso-10T1/2:NMuMG. Unpaired Student’s t Test (E,G,H,I), 2way ANOVA Bonferroni posthoc test (k,l,m), *, p < 0.05, **, p < 0.01; ***, p < 0.001, ns, not significant. Data are represented as mean ± S.E.M. Cartoon models in h,i,m created using BioRender.
Extended Data Fig. 4 |. Maintenance of adiposity in female mice under cold exposure.
a) Images showing cell morphology of beige differentiated SVFs isolated from male and female iWATs. Scale bar at 100 μm. b) Real-time qPCR of indicated genes from the scWATs of 24 h cold exposed male and female adrenalectomized mice. N = 6,6. c–e) Individual mice data for oxygen consumption (VO2 ml/hr), and energy expenditure (EE kcal/hr) (c), respiratory exchange ratio (RER) and generalized linear model (GLM)-based regression plots of RER with total body weight (Total), lean mass (Lean) and fat mass (Fat) as co-variates (Two-way ANCOVA) showing p-value for Mass effect, Group effect, and Interaction effect (d), and total food consumed (kcal), locomotor activity (beam breaks), and total distance in cage (m) (e) of male and female mice exposed to 22 °C for 21 h and 4 °C for 24 h in Sable Promethion metabolic chambers (12 h light/dark cycle, 45 h total duration, white bar represents light cycle and grey bar represents night cycle). N = 6,6. f and g) Body weight (f) and lean mass (g) of male and female mice before (RT) and after 24 h cold exposure (COLD). N = 6,6. h) Weights of indicated fat depots in cold exposed male and female mice. N = 8,8. i and j) Gross appearance, H&E staining, and immunostaining of UCP1 of male dorsolumbar iWATs and female dorsolumbar mgWATs exposed to 24 h cold (i) and Western blot of UCP1 in iWAT from 24 h cold-exposed males and females (j). Actin was used as a loading control and UCP1 bands were quantified and normalized to Actin intensity. N = 4,4. Scale bar at 200 μm. Unpaired Student’s t Test (b,f,h) and ANCOVA (d) *, p < 0.05, **, p < 0.01; ***, p < 0.001, n.s., not significant. Data are represented as mean ± S.E.M.
Extended Data Fig. 5 |. Mammary duct ablation potentiates the beiging of mgWAT.
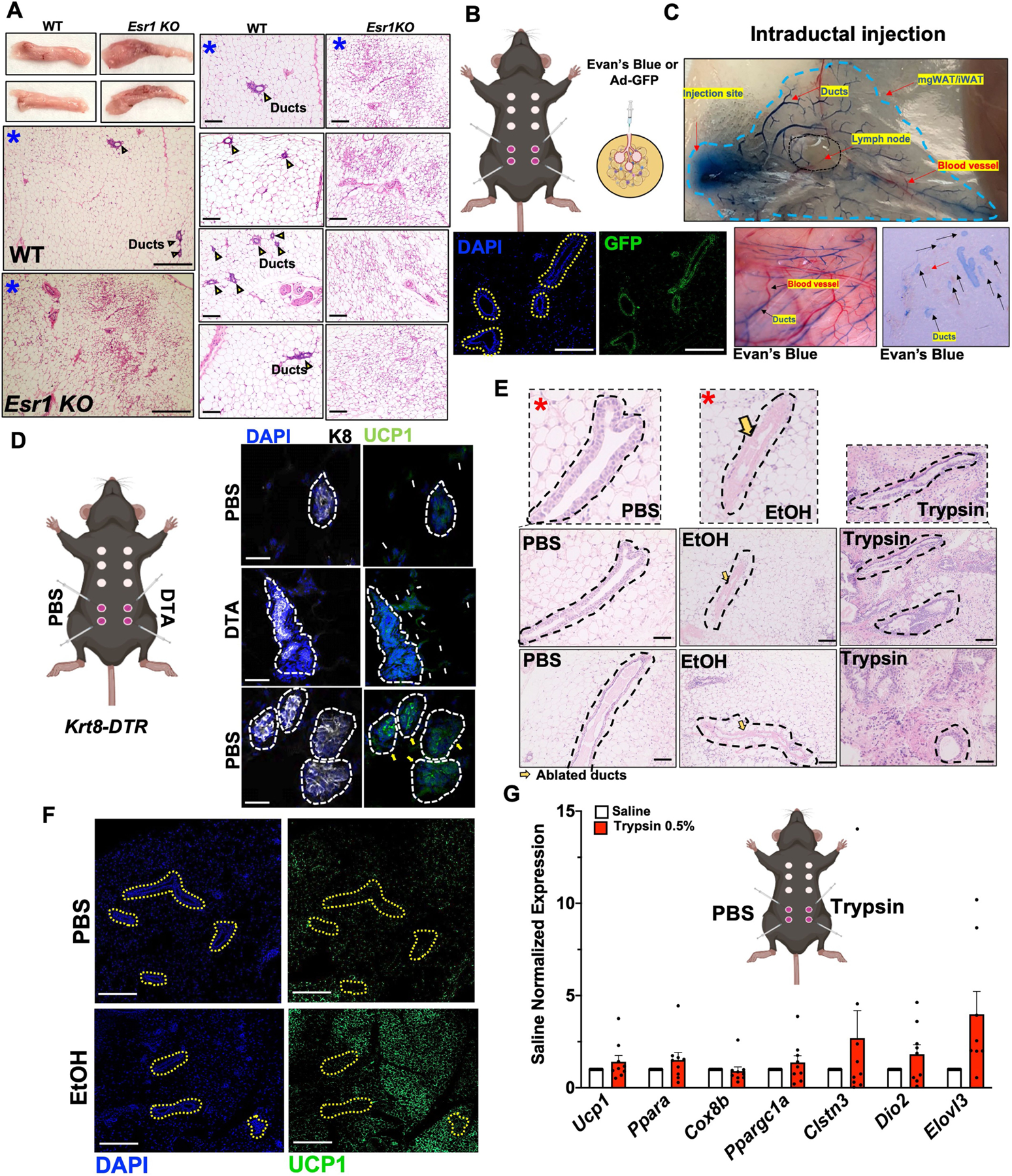
a) Whole tissue images and H&E sections of mgWATs of WT and Esr1KO cold exposed mice. Blue asterisks represent same samples at different magnifications. Scale bar at 100 μm. b and c) Cartoon showing intraductal injection into the 4th and 5th nipples of female mice (b) with adjacent cartoon showing injection of Evan’s blue or GFP expressing virus for confirmation ductal-specific injection (c). Scale bar at 100 μm d) Intraductal injection of Krt8-Dtr mice with contralateral injection of DTA or PBS and immunohistochemistry with DAPI or UCP1. White arrows indicated UCP1 signals in the stroma and yellow arrows indicated UCP1 signal in ducts. Scale bar at 50 μm e) H&E staining of mgWAT from mice intraductally injected with PBS, 70% EtOH, or 0.5% Trypsin. Yellow arrows indicate ablated ducts. Red asterixis indicate images used in Main Fig. 3j. Representative section from 7 mice. Scale bar at 50 μm. f) Immunofluorescence microscopy for UCP1 or DAPI in mice intraductally injected with PBS or EtOH. Dotted yellow outlines are ductal epithelium. Scale bar at 100 μm. g) Real-time qPCR analysis of indicated genes from mgWATs of mice intraductally injected with PBS or Trypsin. N = 9,9. Data are represented as mean ± S.E.M. Cartoon models in b,d,g created using BioRender.
Extended Data Fig. 6 |. LCN2 is a cold-induced mammokine involved in blocking cold-induced adipocyte UCP1 expression.
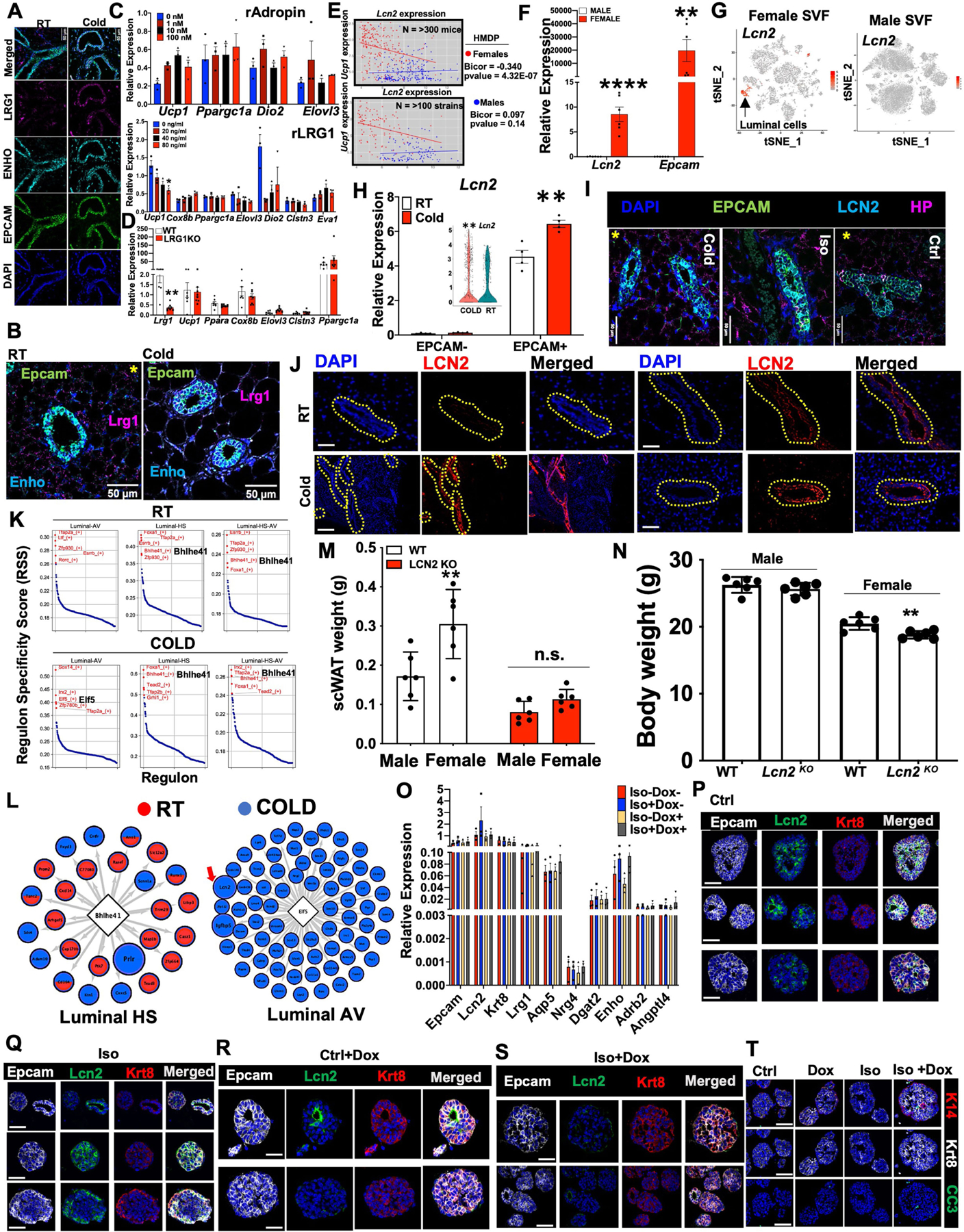
a and b) RNAScope FISH of indicated probes from mgWAT of RT or 24 h cold exposed (Cold) mice. Yellow asterisk indicate image used in Main Fig. 1f for Enho RNAScope. Scale bar at 50 μm. c) Real-time qPCR of indicated genes from mgWAT beige differentiated SVFs treated with 0–100 μm recombinant Adropin (Top) or 0–80 ng/ml recombinant LRG1 (Bottom) for 24 h. d) Real-time qPCR of indicated genes from mgWATs derived from 24 h cold-exposed WT or LRG1KO mice. N = 7,8. e) Gene correlation plots of Ucp1 and Lcn2 expression in WATs from male and female mice from the HMDP studies. f) Real-time qPCR of indicated genes from 24 h cold-exposed male and female iWATs. N = 6,6. g) tSNE plots of cell type clusters and normalized gene expression levels for Lcn2 across scRNA-seq datasets of female mgWAT SVFs and male iWAT SVFs. h) Real-time qPCR of Lcn2 in mgWAT EPCAM+ and EPCAM-cells from RT or 24 h cold-exposed mice. Inset shows violin plot of Lcn2 from scRNA-seq data. i) RNAscope FISH of indicated probes from mgWAT of 24 h cold exposed or Iso treated mice. Yellow asterisks indicate images used in Main Fig. 4b. Scale bar at 50 μm. j) Immunofluorescence microscopy for LCN2 or DAPI in mice exposed to 24 h cold or RT. Yellow outlines indicated ductal epithelium. Scale bar at 50 μm. k) The top five transcription factor regulons for luminal cell types at RT and COLD identified using SCENIC program. l) Subnetworks of transcription factor regulons of interest for Bhlhe41 in hormone sensing luminal cells (Up) and Elf5 in luminal alveolar cells (Down) derived using SCENIC. Transcription factors indicated in diamonds in the center and genes in the regulon in the surrounding circles. Genes in red were derived from the RT data, genes in blue were derived from the COLD data, and genes with both were present in the regulons in both conditions. Red arrow shows cold-induced mammokine Lcn2. m and n) Body weights (n) and scWAT weights (m) of 24 h cold exposed WT and Lcn2 KO male and female mice. N = 6,6 o) Real-time qPCR of indicated genes in organoids derived from K8rtTA-DTA mice treated with and without ISO and Doxycycline (Dox). Results from 3 organoid experiments. p-t) Confocal images of immunostaining of indicated antibodies in organoids derived from K8rtTA-DTA mice treated with and without Iso and Dox. Scale bar at 10 μm. Representative images from 3 organoid experiments. Each point represents three independent experiments (c, h, o). Unpaired two-tailed Student’s t test *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Data are represented as mean ± S.E.M.
Extended Data Fig. 7 |. LCN2 is a mammokine involved in blocking cold-induced adipocyte UCP1 expression.
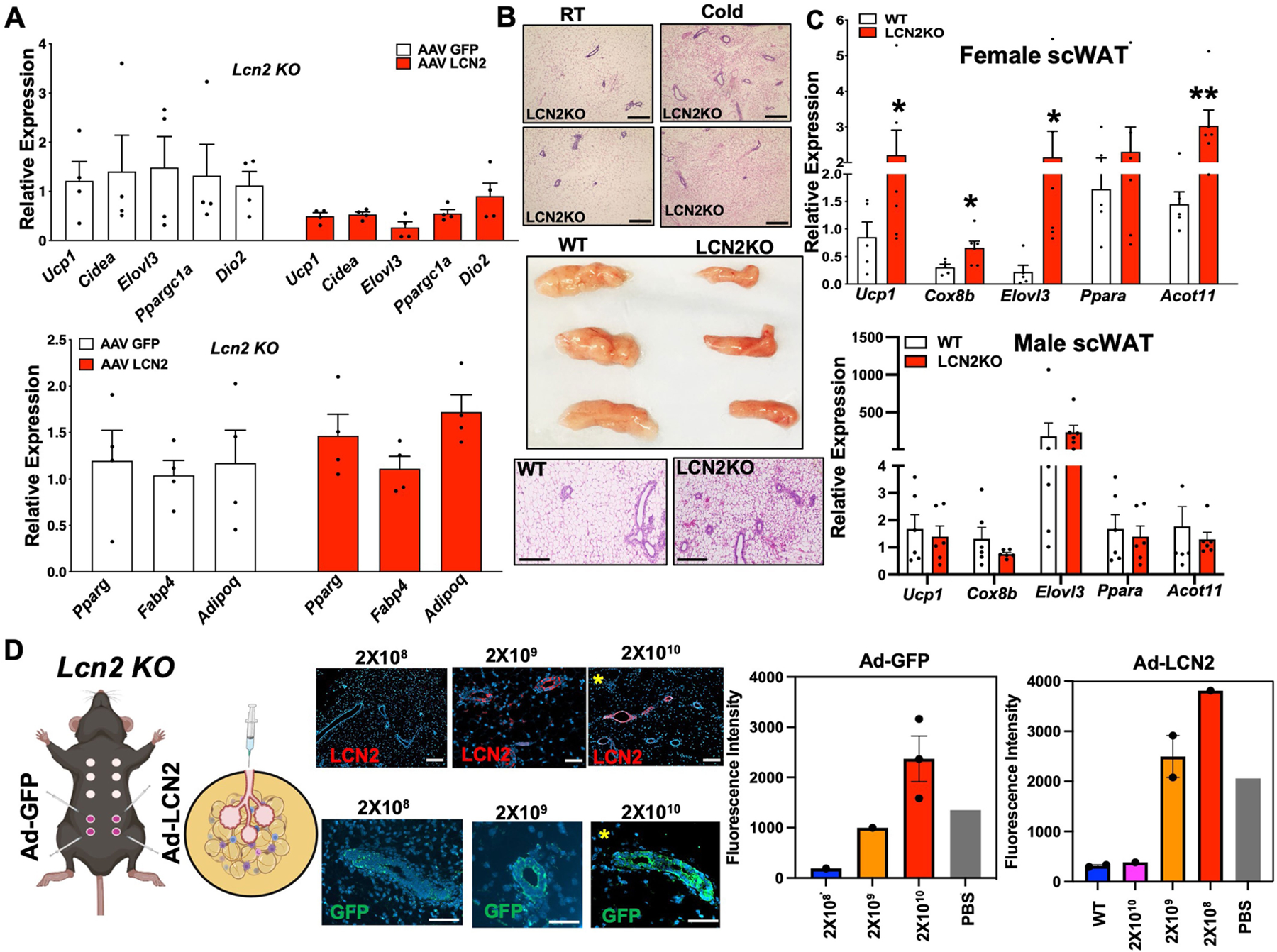
a) Real-time qPCR of indicated genes from mgWAT of AAV-GFP or AAV-LCN2 treated Lcn2 KO mice. N = 4,4. b) Gross appearance and H&E section of mgWAT from RT and cold-exposed LCN2KO mice (Top) and WT or LCN2KO mice exposed to 24 h cold (Bottom). Scale bar at 100 μm. c) Real-time qPCR of indicated genes from 24 h cold exposed females (Top) and males (bottom) WT and LCN2KO scWATs. N = 6,6. d) Intraductal injection of Ad-GFP or Ad-LCN2 contralaterally in the 4th and 5th nipple and immunofluorescence microscopy of GFP or LCN2 from Ad-GFP or Ad-LCN2 injected mice with indicated viral concentrations and barplot showing intensity of GFP or LCN2 from microscopy. Scale bar at 100 μm. Yellow asterisks indicate images also used in Main Fig. 4g. Representative image of two independent injections. For bar plots, quantification of intensity was calculated as a mean from multiple ducts from 1 image except for 2X1010 PFU for Ad-GFP (N = 3) and 2X109 for Ad-LCN2. (N = 2). Unpaired two-tailed Student’s t test *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data are represented as mean ± S.E.M. Cartoon model in D created using BioRender.
Supplementary Material
Acknowledgements
The authors thank former P.R. laboratory members S. Sadeh and S. Hart for technical assistance with metabolic chambers and RNAscope; P. Cohen and C. H. J. Choi at The Rockefeller University for the Lrg1-KO mice. LSFM and confocal microscopy were performed at the Microscopy and Advances Bioimaging Core at ISMMS. A.A. is supported by senior postdoctoral fellowship from the Charles H. Revson Foundation (grant no. 18–25), a fellowship from Sweden–America Foundation (Ernst O. Eks fond), and a postdoctoral scholarship from the Swedish Society for Medical Research (SSMF). S.A.S. is supported by American Diabetes Association Pathway to Stop Diabetes Grant ADA no. 1-17-ACE-31, NIH (R01NS097184, OT2OD024912 and R01DK124461) and Department of Defense (W81XWH-20-1-0345, W81XWH-20-1-0156). L.G. is supported by R00HL150234. A.J.L. is supported by NIH U01 AG070959 and U54 DK120342. X.Y. is supported by NIH R01 DK117850. P.R. is supported by R00DK114571, NIDDK-supported Einstein-Sinai Diabetes Research Center (DRC) Pilot and Feasibility Award, and Diabetes Action Research and Education Foundation (DREF) grant no. 501 (PR). P.W. is a member of the Human Islet and Adenovirus Core of the Einstein-Sinai Diabetes Research Center (ES-DRC) supported by NIHP30DK020541. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-023-06361-5.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-023-06361-5.
Peer review information Nature thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Data availability
Source data for all figures are provided with the paper. The scRNA-seq dataset (Fig. 1) generated for this paper has been uploaded to the Gene Expression Omnibus under accession number GSE231394. The bulk RNA-sequencing dataset (Fig. 4) generated for this paper has been uploaded to the GEO under accession number GSE121098 (GSM7287577–GSM7287584). Source data are provided with this paper.
References
- 1.Chi J et al. Three-dimensional adipose tissue imaging reveals regional variation in beige fat biogenesis and PRDM16-dependent sympathetic neurite density. Cell Metab 27, 226–236.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Zhang F et al. An adipose tissue atlas: an image-guided identification of human-like BAT and beige depots in rodents. Cell Metab 27, 252–262.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajbhandari P et al. IL-10 signaling remodels adipose chromatin architecture to limit thermogenesis and energy expenditure. Cell 172, 218–233.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knights AJ, Wu J & Tseng Y-H The heating microenvironment: intercellular cross talk within thermogenic adipose tissue. Diabetes 69, 1599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J et al. Neurotensin is an anti-thermogenic peptide produced by lymphatic endothelial cells. Cell Metab 33, 1449–1465.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu B et al. γδ T cells and adipocyte IL-17RC control fat innervation and thermogenesis. Nature 578, 610–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q et al. IL-27 signalling promotes adipocyte thermogenesis and energy expenditure. Nature 600, 314–318 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Gjorevski N & Nelson CM Integrated morphodynamic signalling of the mammary gland. Nat. Rev. Mol. Cell Biol 12, 581–593 (2011). [DOI] [PubMed] [Google Scholar]
- 9.McNally S & Martin F Molecular regulators of pubertal mammary gland development. Ann. Med 43, 212–234 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Inman JL, Robertson C, Mott JD & Bissell MJ Mammary gland development: cell fate specification, stem cells and the microenvironment. Development 142, 1028–1042 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Wang QA & Scherer PE Remodeling of murine mammary adipose tissue during pregnancy, lactation, and involution. J. Mammary Gland Biol. Neoplasia 24, 207–212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landskroner-Eiger S, Park J, Israel D, Pollard JW & Scherer PE Morphogenesis of the developing mammary gland: stage-dependent impact of adipocytes. Dev. Biol 344, 968–978 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang QA et al. Reversible de-differentiation of mature white adipocytes into preadipocyte-like precursors during lactation. Cell Metab 28, 282–288.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bach K et al. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun 8, 2128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li CM-C et al. Aging-associated alterations in mammary epithelia and stroma revealed by single-cell RNA sequencing. Cell Rep 33, 108566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emont MP et al. A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aryal B et al. Absence of ANGPTL4 in adipose tissue improves glucose tolerance and attenuates atherogenesis. JCI Insight 3, e97918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arner E et al. Ceruloplasmin is a novel adipokine which is overexpressed in adipose tissue of obese subjects and in obesity-associated cancer cells. PLoS ONE 9, e80274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chella Krishnan K et al. Sex-specific metabolic functions of adipose lipocalin-2. Mol. Metab 30, 30–47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun WY et al. Lipocalin-2 derived from adipose tissue mediates aldosterone-induced renal injury. JCI Insight 3, e120196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deis JA et al. Adipose lipocalin 2 overexpression protects against age-related decline in thermogenic function of adipose tissue and metabolic deterioration. Mol. Metab 24, 18–29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishii A et al. Obesity-promoting and anti-thermogenic effects of neutrophil gelatinase-associated lipocalin in mice. Sci. Rep 7, 15501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang GX et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat. Med 20, 1436–1443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maffei M, Barone I, Scabia G & Santini F The multifaceted haptoglobin in the context of adipose tissue and metabolism. Endocr. Rev 37, 403–416 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Kumar KG et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab 8, 468–481 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi CHJ et al. LRG1 is an adipokine that promotes insulin sensitivity and suppresses inflammation. eLife 11, e81559 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacCannell A, Futers S, Moran A, Whitehead A & Roberts L Lrg1 is a driver of brown adipose tissue dysfunction in obesity. FASEB J 10.1096/fasebj.2021.35.S1.00285 (2021). [DOI]
- 30.Rajbhandari P et al. Single cell analysis reveals immune cell-adipocyte crosstalk regulating the transcription of thermogenic adipocytes. eLife 8, e49501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gargiulo L et al. A novel effect of β-adrenergic receptor on mammary branching morphogenesis and its possible implications in breast cancer. J. Mammary Gland Biol. Neoplasia 22, 43–57 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Marchetti B et al. β-adrenergic receptors in the rat mammary gland during pregnancy and lactation: characterization, distribution, and coupling to adenylate cyclase. Endocrinology 126, 565–574 (1990). [DOI] [PubMed] [Google Scholar]
- 33.Nedvetsky PI, Kwon SH, Debnath J & Mostov KE Cyclic AMP regulates formation of mammary epithelial acini in vitro. Mol. Biol. Cell 23, 2973–2981 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao Q, Jing J, Cui X, Shi H & Xue B Sympathetic nerve innervation is required for beigeing in white fat. Physiol. Rep 7, e14031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang H, Ding X, Cao Y, Wang H & Zeng W Dense intra-adipose sympathetic arborizations are essential for cold-induced beiging of mouse white adipose tissue. Cell Metab 26, 686–692.e3 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Walker MR et al. Alveolar progenitor cells in the mammary gland are dependent on the β4 integrin. Dev. Biol 457, 13–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Y, Manka DR, Wagner K-U & Khan SA Estrogen receptor-α expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc. Natl Acad. Sci. USA 104, 14718–14723 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawson DA, Werb Z, Zong Y & Goldstein AS The cleared mammary fat pad transplantation assay for mammary epithelial organogenesis. Cold Spring Harb. Protoc 2015, pdb.prot078071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dompmartin A et al. Radio-opaque ethylcellulose-ethanol is a safe and efficient sclerosing agent for venous malformations. Eur. Radiol 21, 2647–2656 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Zhang WY, Li ZS & Jin ZD Endoscopic ultrasound-guided ethanol ablation therapy for tumors. World J. Gastroenterol 19, 3397–3403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuang M et al. Ethanol ablation of hepatocellular carcinoma up to 5.0 cm by using a multipronged injection needle with high-dose strategy. Radiology 253, 552–561 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Kenyon E et al. Ductal tree ablation by local delivery of ethanol prevents tumor formation in an aggressive mouse model of breast cancer. Breast Cancer Res 21, 129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horton C, Liu Y, Yu C, Xie Q & Wang ZA Luminal-contact-inhibition of epithelial basal stem cell multipotency in prostate organogenesis and homeostasis. Biol. Open 8, bio045724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parks BW et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17, 141–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oakes SR et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev 22, 581–586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Centonze A et al. Heterotypic cell–cell communication regulates glandular stem cell multipotency. Nature 584, 608–613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bauters D, Van Hul M & Lijnen HR Macrophage elastase (MMP-12) in expanding murine adipose tissue. Biochim. Biophys. Acta 1830, 2954–2959 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Lee JT et al. Macrophage metalloelastase (MMP12) regulates adipose tissue expansion, insulin sensitivity, and expression of inducible nitric oxide synthase. Endocrinology 155, 3409–3420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shao M et al. Zfp423 maintains white adipocyte identity through suppression of the beige cell thermogenic gene program. Cell Metab 23, 1167–1184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gavalda-Navarro A et al. Lipopolysaccharide-binding protein is a negative regulator of adipose tissue browning in mice and humans. Diabetologia 59, 2208–2218 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Sun W et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature 587, 98–102 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Kiefer FW et al. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med 18, 918–925 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mina AI et al. CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metab 28, 656–666.e1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Anders S, Kim V & Huber W RNA-seq workflow: gene-level exploratory analysis and differential expression. F1000Res 4, 1070 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McInnes L, Healy J & Melville J UMAP: uniform manifold approximation and projection for dimension reduction Preprint at https://arxiv.org/abs/1802.03426 (2018).
- 62.van der Maaten L & Hinton G Visualizing data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 63.Blondel VD, Guillaume J-L, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008 (2008). [Google Scholar]
- 64.Van de Sande B et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc 15, 2247–2276 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Moerman T et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Alvarsson A et al. Optical clearing and 3D analysis optimized for mouse and human pancreata. Bio Protoc 11, e4103 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jardé T et al. Wnt and neuregulin1/ErbB signalling extends 3D culture of hormone responsive mammary organoids. Nat. Commun 7, 13207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for all figures are provided with the paper. The scRNA-seq dataset (Fig. 1) generated for this paper has been uploaded to the Gene Expression Omnibus under accession number GSE231394. The bulk RNA-sequencing dataset (Fig. 4) generated for this paper has been uploaded to the GEO under accession number GSE121098 (GSM7287577–GSM7287584). Source data are provided with this paper.