

SUMMARY
Venezuelan equine encephalitis virus (VEEV) is an encephalitic alphavirus responsible for epidemics of neurological disease across the Americas. Low-density lipoprotein receptor class A domain-containing 3 (LDLRAD3) is a recently reported entry receptor for VEEV. Here, using wild-type and Ldlrad3-deficient mice, we define a critical role for LDLRAD3 in controlling steps in VEEV infection, pathogenesis, and neuro-tropism. Our analysis shows that LDLRAD3 is required for efficient VEEV infection and pathogenesis prior to and after central nervous system invasion. Ldlrad3-deficient mice survive intranasal and intracranial VEEV inoculation and show reduced infection of neurons in different brain regions. As LDLRAD3 is a determinant of pathogenesis and an entry receptor required for VEEV infection of neurons of the brain, receptor-targeted therapies may hold promise as countermeasures.
In brief
Kafai et al. demonstrate that the low-density lipoprotein receptor class A domain-containing 3 (LDLRAD3), a major VEEV receptor, is important at multiple stages of VEEV pathogenesis using a murine model and Ldlrad3-deficient mice. LDLRAD3 is particularly important for VEEV infection of neurons in the central nervous system.
Graphical Abstract
INTRODUCTION
Alphaviruses are mosquito-transmitted, enveloped, RNA viruses of the Togaviridae family that cause disease in millions of people worldwide.1–4 New World alphaviruses are zoonotic pathogens with the potential to cause severe neurological disease and include Eastern equine encephalitis (EEEV), Western equine encephalitis, and Venezuelan equine encephalitis (VEEV) viruses.5 In its epizootic cycle (subtypes IAB and IC), VEEV can have devasting consequences for equines (mortality rates of 50%–70%)5 with occasional spillover into humans.6–11 Enzootic VEE complex viruses (subtype ID-F and related species in subtypes II–VI) are less or non-virulent in equids but can still cause clinical disease in humans.12 Enzootic strains generally circulate between mosquito vectors and small mammals13–16 including spiny (Proechimys species) and cotton (Sigmodon species) rats.17–19 Laboratory studies have shown that a variety of wild rodents can survive experimental infection and develop strong antibody responses after infection with VEEV.17,19,20 Nonetheless, some wild rodents develop severe disease after VEEV infection, though responses vary among geographic host populations and virus strains.5,21 Additionally, VEEV poses a risk as an aerosolized bioweapon.22,23 Despite its potential to cause severe disease and death, there are no approved human vaccines or antiviral drugs against VEEV.24 A live-attenuated vaccine against VEEV (serially passaged strain TC-83 and boosters of C-84, an inactivated form of TC-83) is available as an investigational drug product in the United States through the Army Special Immunizations Program but only for at-risk laboratory workers and military personnel.25–31
Much of what is known about the VEEV infection life cycle is inferred from experiments with other alphaviruses. The alphavirus positive-sense RNA genome is approximately 11.5 kb and encodes four non-structural proteins, nsp1–4, and five structural proteins, capsid, E3, E2, 6K, and E1.32 The non-structural polyproteins are translated from genomic RNA in the host cell cytoplasm and regulate viral replication, protein processing, and immune evasion. A subgenomic 26S RNA encodes the structural polypeptide C-p62(E3-E2)-6K-E1, which is cleaved into proteins required for viral encapsidation, morphogenesis, and budding.33 The mature VEEV virion includes a nucleocapsid surrounded by a lipid envelope embedded with heterodimers of surface envelope glycoproteins E2 and E1. E2-E1 heterodimers assemble into trimeric spikes on the viral surface to create a virion with T = 4 quasi-icosahedral symmetry.34,35 The E1 protein lies beneath E2 at the base of each trimeric spike and mediates low-pH endosomal fusion before release of the viral nucleocapsid in the cytoplasm.33 The E2 protein, the target for most VEEV-specific neutralizing antibodies, protrudes centrally on each spike to facilitate host cell binding and entry.36–41
Recently, a VEEV-specific entry receptor, low-density lipoprotein receptor class A domain-containing 3 (LDLRAD3), was identified using a loss-of-infection-based CRISPR-Cas9 genome-wide screen.42 LDLRAD3 is a highly conserved type I membrane protein of the LDL scavenger receptor superfamily, found in mammals, birds, reptiles, amphibians, and fish,42,43 and has been reported to regulate amyloid precursor protein processing and auto-ubiquitination in neurons, although its endogenous ligand(s) are unknown.43,44 The most membrane-distal domain 1 (D1) of the three extracellular domains of LDLRAD3 was shown to be necessary and sufficient for VEEV infection, and a cryoelectron microscopy structure showed that LDLRAD3 D1 binds in a cleft formed between adjacent VEEV E2 and E1 proteins on the virion surface.45,46 The horse (Equus caballus) LDLRAD3 ortholog is 100% identical in the D1 ligand-binding domain to human LDLRAD3.
In humans, most VEEV infections are self-limiting and manifest as a flu-like illness. A small percentage of cases develop central nervous system (CNS) infection, and the ensuing neurological signs can progress to coma or death.14,47–50 Although the pathogenesis of VEEV in humans is not fully characterized, it is believed to share some features of disease observed in horses, namely a biphasic febrile illness with damage to lymphoid tissues and CNS involvement.51–53 Because VEEV infects rodents in its enzootic cycle, laboratory mice have been used as experimental models, and different VEEV exposures can be modeled by the inoculation route, including naturally via mosquito bite through subcutaneous injections and via aerosol exposure through intranasal or aerosolized inoculation.51,54 Using these models, VEEV cellular tropism and routes of infection leading to neuroinvasion and encephalitis have been studied.55–57
After subcutaneous inoculation in mice, VEEV first infects myeloid cells in the skin, which transit to draining lymph nodes where VEEV replicates and then disseminates to the blood-stream.51,58,59 Although VEEV targets multiple peripheral organs, it preferentially infects lymphoid tissues.51,58–60 VEEV can rapidly infect the brain as early as 12 h after inoculation in some mouse models.61,62 Several routes of entry into the CNS are proposed for VEEV, including by hematogenous routes or via olfactory sensory neurons of the olfactory neuroepithelium to the olfactory bulb.61–65 Although VEEV can cross an intact blood-brain barrier (BBB), permeability increases approximately 3 days after infection, which allows for infiltration of immune cells and more virus from the circulation.66–68 Once in the brain, VEEV primarily infects neurons but also targets other supporting cells, including astrocytes upon initial invasion via the BBB.62 This infection ultimately leads to neuronal cell death, gliosis, and neuroinflammation.62,69–71 Although a prior study showed that Ldlrad3-deficient mice survived subcutaneous challenge with pathogenic VEEV strains,42 the role of LDLRAD3 in VEEV tropism and pathogenesis was not explored.
Here, we used Ldlrad3-deficient mice to delineate further the role of LDLRAD3 in VEEV pathogenesis. Our experiments showed that LDLRAD3 expression is required for VEEV infection and disease in mice after peripheral or direct CNS infection and that it has key roles at multiple stages in viral pathogenesis. In the brain, LDLRAD3 serves as a primary determinant of VEEV infection of neurons.
RESULTS
LDLRAD3 has a role in VEEV infection immediately after subcutaneous inoculation
We previously generated C57BL/6J mice with frameshift deletions lacking 14 (Δ14) nucleotides in exon 2 of Ldlrad3 (Figure S1A), a region corresponding to D1, which mediates VEEV binding and infection.42,45 Male and female Ldlrad3Δ14/Δ414 mice survived subcutaneous challenge with epizootic (IAB, Trinidad donkey [TrD]) or enzootic (ID, ZPC738) strains, whereas congenic wild-type mice of both sexes did not.42 To determine whether the resistance of Ldlrad3Δ14/Δ14 mice to VEEV could be overcome by a 1,000-fold-higher inoculating dose, wild-type and Ldlrad3Δ14/Δ14 mice were administered 105 focus-forming units (FFU) of VEEV ZPC738 (herein, VEEV) and monitored for weight loss and survival (Figure S1B). In contrast to wild-type mice, Ldlrad3Δ14/Δ14 mice survived the higher dose VEEV challenge with minimal to no weight loss, corroborating a key role for LDLRAD3 in VEEV pathogenesis (Figure S1B). We confirmed that naive wild-type and Ldlrad3Δ14/Δ14 mice had similar numbers of circulating B cells, CD4+ T cells, CD8+ T cells, neutrophils, eosinophils, natural killer (NK) cells, and Ly6Chi or Ly6Clo monocytes (Figures S1C and S1D). To demonstrate the specificity of the resistance phenotype for VEEV, we subcutaneously inoculated Ldlrad3Δ14/Δ14 mice with 103 FFU of Madariaga virus (MADV), a South American lineage of EEEV72,73 that does not use LDLRAD3 as an entry receptor.42,74 Ldlrad3Δ14/Δ14 mice lost weight and succumbed to MADV infection at levels comparable to wild-type mice (Figure S1E). This experiment confirms that the resistance of Ldlrad3Δ14/Δ14 mice to alphavirus pathogenesis in vivo is specific to VEEV and does not extend to other encephalitic alphaviruses.
To understand how LDLRAD3 expression affects VEEV pathogenesis, we compared the kinetics of VEEV infection and dissemination in wild-type and Ldlrad3Δ14/Δ14 mice. We first assessed the contribution of LDLRAD3 within 6 and 12 h of subcutaneous virus inoculation by comparing viral RNA levels in the skin, the underlying tissue of the foot, serum, peripheral blood leukocytes (PBLs), and other target tissues of wild-type and Ldlrad3Δ14/Δ14 mice (Figure 1A). At 6 h post-infection (hpi), in both wild-type and Ldlrad3Δ14/Δ14 mice, viral RNA levels remained at or near the limit of detection in the skin of the inoculated foot, the underlying tissue of the foot, and distal organs including the thymus, lung, and brain (Figure 1A). However, viral RNA was detected in the draining lymph nodes (DLNs; popliteal and inguinal), serum, and spleen of both wild-type and Ldlrad3Δ14/Δ14 mice (Figure 1A), which is consistent with rapid lymphatic transport of viruses from the skin.75–78 Nonetheless, at 6 hpi, Ldlrad3Δ14/Δ14 mice had lower levels of viral RNA in the popliteal DLNs (5-fold, **p < 0.01), inguinal DLNs (7-fold, *p < 0.05), and serum (3-fold, *p < 0.05), suggesting that the earliest stages of VEEV infection and dissemination depend in part on LDLRAD3 expression. At 12 hpi, lower levels of VEEV RNA were detected in the underlying tissue of the foot (5-fold, **p < 0.01), peripheral visceral organs (lungs, [10-fold, *p < 0.05], spleen [19-fold, ***p <0.001], and thymus [10-fold, ***p < 0.001), DLNs (popliteal [5-fold, ***p < 0.001] and inguinal [17-fold, **p < 0.01]), serum (23-fold, ***p < 0.001), and PBLs (16-fold, ***p < 0.001) of Ldlrad3Δ14/Δ14 than wild-type mice, although infection generally was increased in both cohorts of animals compared with 6 hpi (Figure 1A). At 12 hpi, low levels of viral RNA were detected in the olfactory bulb and the cerebral cortex of wild-type, but not Ldlrad3Δ14/Δ14, mice. To corroborate our measurements of viral RNA, we quantitated infectious virus levels in selected peripheral organs (inoculated foot skin, popliteal DLN, and lung) at 6 and 12 hpi using focus-forming assays (Figure 1B). Similar patterns were observed, with lower levels of infectious virus detected in Ldlrad3−Δ 14/Δ14 than wild-type mice. Even though viral RNA and infectious virus levels were lower in Ldlrad3Δ14/Δ14 mice, VEEV replication still occurred in the absence of LDLRAD3 expression, indicating the possible existence of alternative, yet subordinate, entry receptor pathway(s).
Figure 1. Ldlrad3Δ14/Δ14 mice show decreased VEEV infection as early as 6 and 12 hpi.
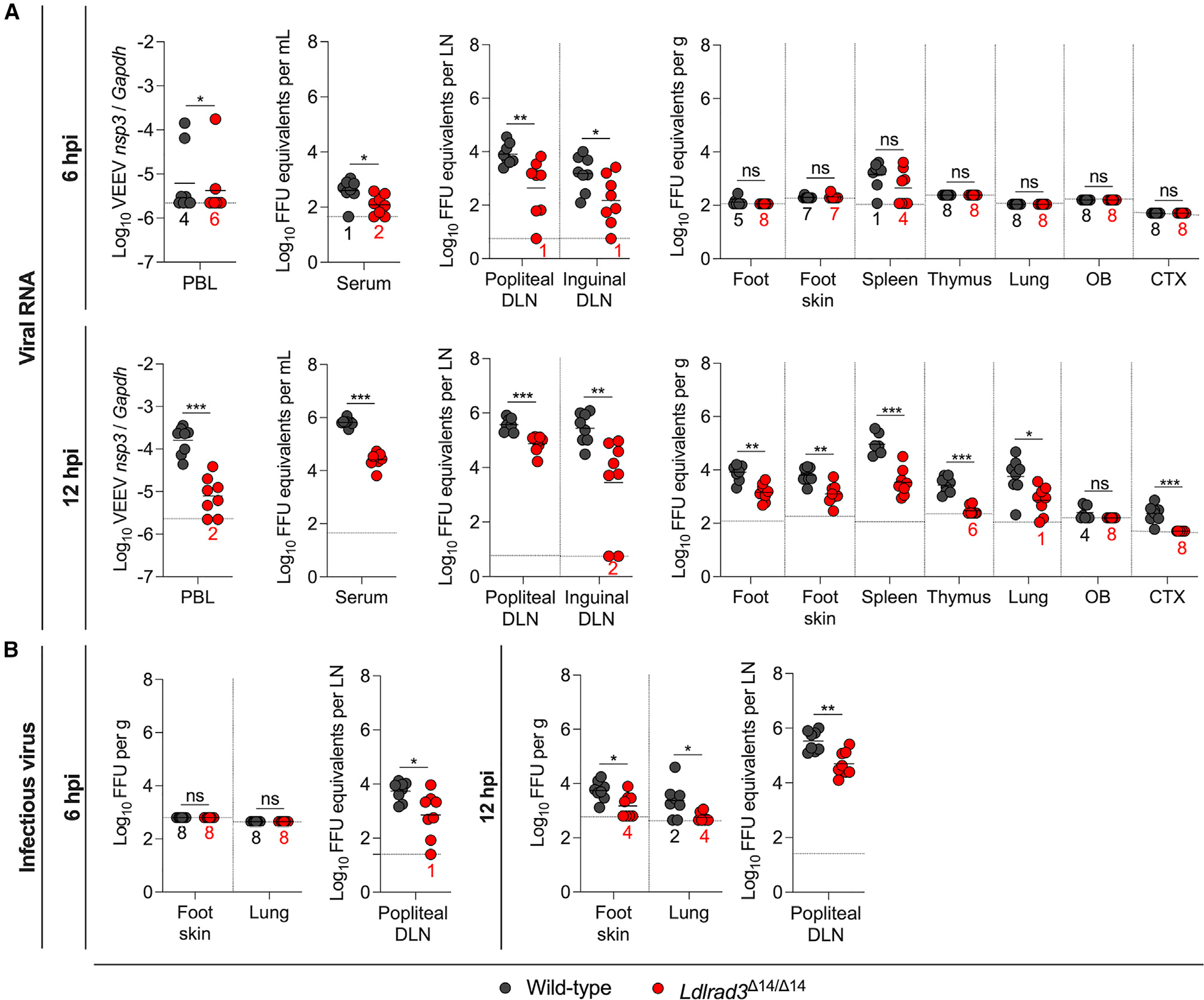
(A and B) Wild-type and Ldlrad3Δ14/Δ14 mice (n = 7–8) were inoculated subcutaneously in the footpad with 102 FFU of VEEV ZPC738. At 6 or 12 hpi, the indicated tissues and samples were collected (peripheral blood leukocytes [PBLs], draining lymph node [DLN], olfactory bulb [OB], cerebral cortex [CTX], gram [g]).
(A) VEEV RNA levels were quantified by qRT-PCR and normalized to a standard curve of infectious virus (all tissues and serum) or Gapdh (PBLs).
(B) Infectious virus was determined by focus-forming assay (FFA) for selected tissues collected at 6 (left panels) or 12 hpi (right panels). Mean values are shown. The limit of detection (LOD) for each tissue is indicated by a dashed line, and numbers in black or red enumerate samples with titers at the LOD. Data are from three independent experiments per time point and were analyzed by Mann-Whitney test (ns, not significant; *p < 0.05; **p < 0.01**, and ***p < 0.001).
See also Figure S1.
VEEV dissemination in Ldlrad3Δ14/Δ14 mice after subcutaneous inoculation
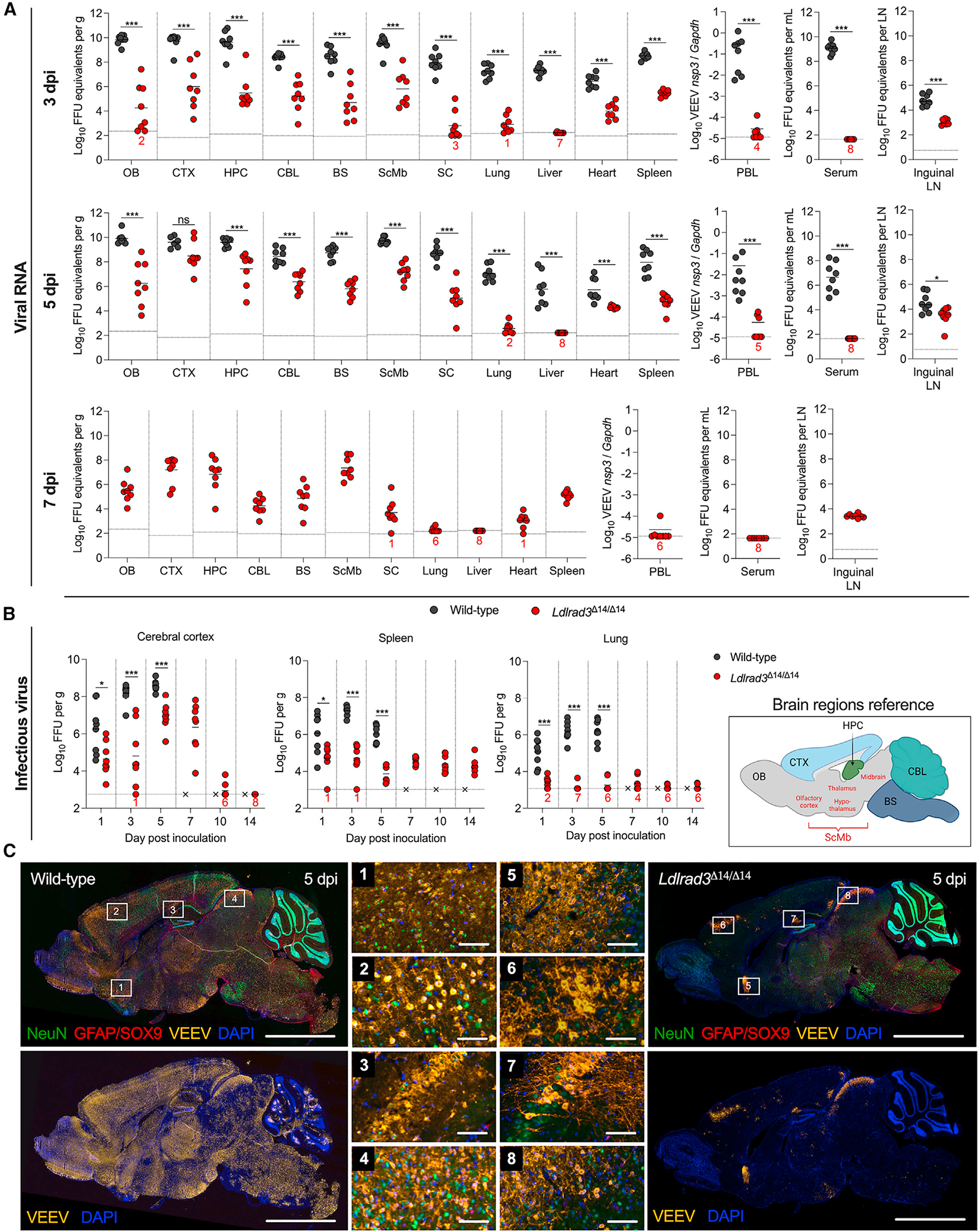
To define further the impact of LDLRAD3 expression on virus dissemination, we performed a more extended kinetic analysis of infection in wild-type and Ldlrad3Δ14/Δ14 mice after subcutaneous inoculation of VEEV. We quantified viral RNA levels at 1, 3, 5, 7, or 14 days post-infection (dpi) in serum, PBLs, peripheral organs (inguinal DLN, liver, heart, spleen, and lung), and compartments of the CNS (spinal cord, olfactory bulb, cerebral cortex, hippocampus, cerebellum, and brainstem) (Figures 2A and S2A). Tissues from wild-type mice were only collected through 5 dpi, as animals became moribund soon thereafter. Whereas VEEV RNA levels increased to 106–108 FFU equivalents/mL or gram in serum and peripheral organs, respectively, and 1010 FFU equivalents/gram in the CNS of wild-type mice, substantially less viral RNA was detected in Ldlrad3Δ14/Δ14 mice at each time point (Figures 2A and S2A). At 5 dpi, VEEV RNA was virtually undetectable in serum and PBL samples of Ldlrad3Δ14/Δ14 mice (Figure 2A). In the CNS of Ldlrad3Δ14/Δ14 mice, viral RNA was detectable by 1 dpi and increased steadily over time, with levels in some mice reaching 107–108 FFU equivalents/gram in the olfactory bulb, cerebral cortex, and hippocampus by 7 dpi, approximately 100- to 1,000-fold lower than peak levels detected in wild-type mice. By day 14, viral RNA was cleared from most peripheral organs and CNS tissues. However, lymphoid tissues (inguinal DLN and spleen) from Ldlrad3Δ14/Δ14 mice maintained measurable levels of viral RNA (104–105 FFU equivalents/gram) through 14 days (Figures 2A and S2A).
Figure 2. Kinetics of VEEV infection in Ldlrad3Δ14/Δ14 mice after subcutaneous inoculation.
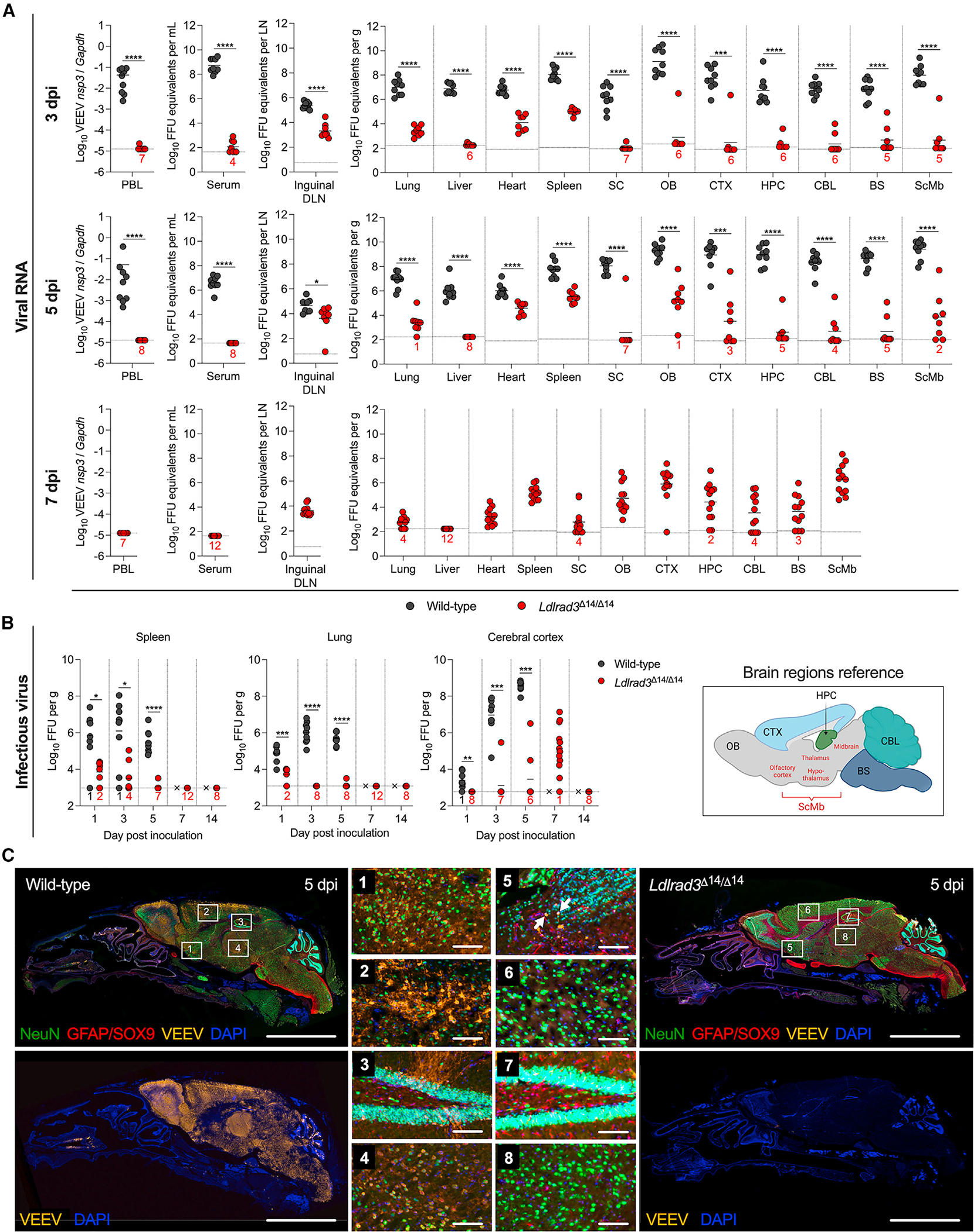
(A and B) Wild-type (n = 7–9) or Ldlrad3Δ14/Δ14 (n = 7–12) C57BL6/J mice were inoculated subcutaneously with 102 FFU of VEEV ZPC738.
(A) At 3, 5 (for wild-type and Ldlrad3Δ14/Δ14 mice), or 7 dpi (Ldlrad3Δ14/Δ14 mice only), indicated tissues and samples were assessed for VEEV viral RNA as described in Figure 1 (PBLs, DLN, spinal cord [SC], OB, CTX, hippocampus [HPC], cerebellum [CBL], brainstem [BS], subcortical/midbrain regions [ScMbs]).
(B) For spleen, lung, and CTX samples, infectious virus was determined at each time point. Mean values are shown. The LOD for each tissue is indicated by a dashed line, and numbers in black or red enumerate samples with titers at the LOD. Data are from two or three independent experiments per time point and were analyzed by Mann-Whitney test (*p < 0.05; **p < 0.01, ***p < 0.001, and ****p < 0.001).
(C) Representative images of sagittal skull and brain sections from wild-type or Ldlrad3Δ14/Δ14 mice (n = 4) 5 days after subcutaneous inoculation with 102 FFU of VEEV ZPC738 (scale bars: 5 mm) and combination fluorescence in situ hybridization (FISH) for VEEV RNA, immunohistochemical staining for cell-type-specific antigens, and DAPI counterstaining for nuclei visualization. White boxes indicate enlarged insets: (1)–(4) insets are for wild-type and (5)–(8) insets are for Ldlrad3Δ14/Δ14 brains (scale bars: 100 μm). White arrows indicate NeuN+VEEV+ cells. Data are representative of images from two experiments. Cartoon schematic of an annotated sagittal mouse brain section is included for reference and was generated using BioRender.
See also Figures S2 and S3.
We also measured infectious virus levels in representative lymphoid (e.g., spleen), visceral (e.g., lung), and CNS (e.g., cerebral cortex) tissues (Figure 2B). Whereas amounts of infectious virus in tissues from wild-type mice generally correlated with viral RNA levels, we observed some discrepancies in those from Ldlrad3Δ14/Δ14 mice. Whereas the spleens of VEEV-infected Ldlrad3Δ14/Δ14 mice had low yet detectable levels (~104 FFUs) of infectious virus at 1 and 3 dpi, all other samples through 14 dpi were negative despite persistence of viral RNA. Thus, over time, the spleens of Ldlrad3Δ14/Δ14 mice may retain viral RNA in a non- or poorly replicating form. The lungs of Ldlrad3Δ14/Δ14 mice also showed little to no infectious virus at any time point. In comparison, infectious virus was detected in the cerebral cortex of Ldlrad3Δ14/Δ14 mice, peaking at 7 dpi, which corresponded to the kinetics of viral RNA accumulation (Figure 2B). To visualize VEEV infection patterns in the brain at 3 and 5 dpi after subcutaneous inoculation, we performed fluorescence in situ hybridization (FISH) of VEEV RNA (Figures 2C, S3A, and S3B) and, additionally, immunohistochemistry (IHC) staining for neuron (NeuN+) and astrocyte (combination GFAP/SOX9+) markers on sagittal skull and brain tissue sections of wild-type and Ldlrad3Δ14/Δ14 mice at 5 dpi (Figure 2C). Staining of VEEV-infected samples with a negative control probe or mock-infected samples stained with a VEEV-specific probe showed no background staining for viral RNA (Figures S2B and S3C). Wild-type mice infected with VEEV showed extensive foci of viral RNA throughout the brain, including the olfactory bulb, lateral olfactory tracts, cerebral cortex, hippocampus, hypothalamus, thalamus, brainstem, and cerebellum (Figures 2C, S3A, and S3B). VEEV localized primarily to NeuN+ neurons (Figure 2C) and did not appear to co-localize with GFAP/SOX9+ astrocytes (Figure S2C). In comparison, brain tissues from Ldlrad3Δ14/Δ14 mice showed little VEEV RNA staining (Figures S3A and S3B), with scattered VEEV+ neurons detected within and near the olfactory bulb (Figure 2C). This delayed peak of VEEV infection in the cerebral cortex of Ldlrad3Δ14/Δ14 mice relative to wild-type mice might represent slower spread to the CNS, possibly due to less viral replication in peripheral organ tissues through LDLRAD3-independent pathways.
LDLRAD3 expression on radio-resistant cells is important for VEEV pathogenesis
The bone marrow hematopoietic stem cell (HSC) compartment gives rise to circulating leukocytes (e.g., monocytes, T cells, B cells, and granulocytes) and many tissue-resident myeloid cells (e.g., macrophages and dendritic cells) that could be targeted by VEEV in a LDLRAD3-dependent manner.59 To begin to define whether LDLRAD3-expressing cell types arising from HSCs contribute to VEEV pathogenesis, we infected bone marrow chimeric mice in which LDLRAD3 was absent from the radiation-sensitive HSC compartment (Ldlrad3Δ14/Δ14 bone marrow transferred to irradiated wild-type recipients) or the radiation-resistant non-hematopoietic cell types (wild-type bone marrow transferred to irradiated Ldlrad3Δ14/Δ14 recipients). We utilized congenic leukocyte markers CD45.1 and CD45.2 to differentiate between donor and recipient cells by flow cytometry. Wild-type bone marrow transplanted into irradiated wild-type recipients and Ldlrad3Δ14/Δ14 bone marrow transplanted into irradiated Ldlrad3Δ14/Δ14 recipients served as controls for effects of irradiation and engraftment (Figure 3A). At 5 weeks after reconstitution, chimerism was confirmed for recipient mice by flow cytometry of PBLs (Figure 3B). Chimeric mice were inoculated subcutaneously with 102 FFU of VEEV (Figure 3A) and analyzed for weight loss (Figure 3C) and viral infection (Figure 3D). Recipient Ldlrad3Δ14/Δ14 mice receiving wild-type bone marrow experienced an approximately 7% weight decline by 2 dpi before recovering to levels closer to those seen with control-infected Ldlrad3Δ14/Δ14 mice that received Ldlrad3Δ14/Δ14 bone marrow, indicating a minor role for LDLRAD3-expressing cells arising from HSCs in promoting VEEV pathogenesis. Recipient wild-type mice, regardless of whether they received wild-type or Ldlrad3Δ14/Δ14 bone marrow, lost ~25% of their body weight over 5 days (Figure 3C). Thus, expression of LDLRAD3 on immune cells arising from HSCs did not alter VEEV pathogenesis as judged by weight loss.
Figure 3. VEEV pathogenesis is dependent on LDLRAD3-expressing radio-resistant cells.
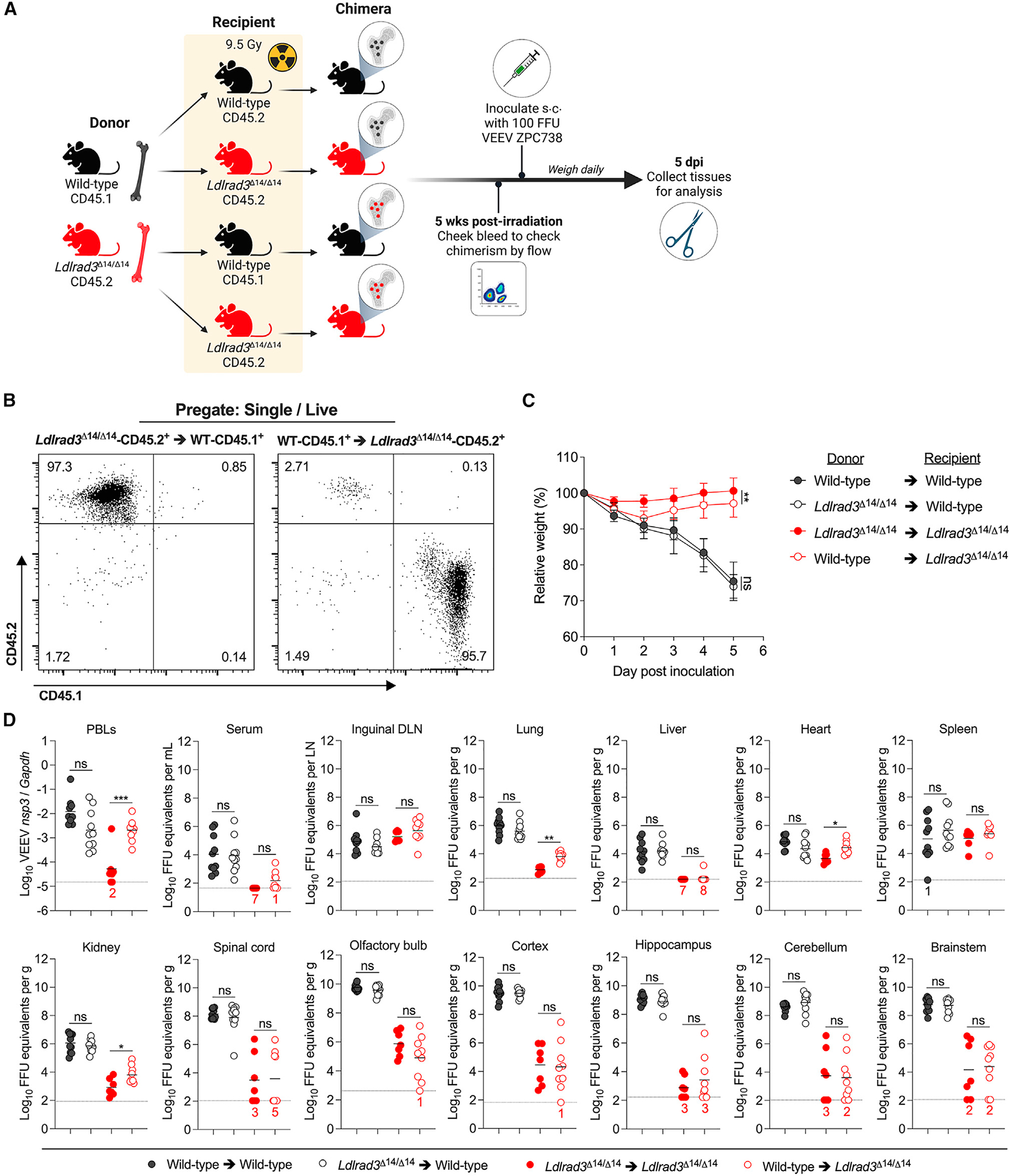
(A) Experimental scheme for generating wild-type and Ldlrad3Δ14/Δ14 bone marrow chimeric mice. Six weeks after irradiation and reconstitution with donor bone marrow hematopoietic stem cells, recipient mice were subcutaneously (s.c.) inoculated with 102 FFU of VEEV ZPC738, weighed, and sacrificed after 5 days.
(B) Representative flow cytometry plots are shown analyzing the percentage of CD45.1+ and CD45.2+ cells of peripheral blood from recipient wild-type-CD45.1+ cells reconstituted with donor Ldlrad3Δ14/Δ14-CD45.2+ cells and recipient Ldlrad3Δ14/Δ14-CD45.2+ mice reconstituted with donor wild-type-CD45.1+ cells 5 weeks after reconstitution and before inoculation with VEEV.
(C) Relative weight change with symbols representing mean ± SD. Significance is indicated in black for wild-type bone marrow (donor) → wild-type (recipient) relative to Ldlrad3Δ14/Δ14 (donor) → wild-type (recipient) and in red for Ldlrad3Δ14/Δ14 (donor) → Ldlrad3Δ14/Δ14 (recipient) relative to wild-type (donor) → Ldlrad3Δ14/Δ14 (recipient) cohorts.
(D) At 5 dpi, indicated tissues and samples were assessed for viral RNA as described in Figure 1 (PBLs, DLN). Mean values are shown. The LOD for each tissue is indicated by a dashed line, and numbers in black or red enumerate samples with titers at the LOD. Data are from two independent experiments (wild-type → wild-type [n = 10]; Ldlrad3Δ14/Δ14 → wild-type [n = 10]; Ldlrad3Δ14/Δ14 → Ldlrad3Δ14/Δ14 [n = 7]; wild-type → Ldlrad3Δ14/Δ14 [n = 9]). For comparison of weight changes, area under the curve analysis was performed. To avoid survival bias in weight curves, statistical significance was only calculated at time points when all mice were alive.
Data were analyzed by unpaired t test (C) and Mann-Whitney test (D) (ns, not significant; *p < 0.05, **p < 0.01, and ***p < 0.001).
Virological analysis of serum, PBLs, peripheral organs, and CNS tissues at 5 dpi was performed (Figure 3D). Viral RNA levels in wild-type mice that received Ldlrad3Δ14/14 bone marrow were comparable to those that received wild-type bone marrow, with titers ranging from 104 to 107 FFU equivalents/gram in the inguinal DLN, spleen, and visceral organs and from 108 to 1010 FFU equivalents/gram in CNS tissues. In Ldlrad3Δ14/14 recipient mice, viral RNA levels were similar in animals with wild-type or Ldlrad3Δ14/14 transplanted bone marrow in lymphoid tissues, serum, and CNS tissues but were comparatively lower than in all wild-type recipient mice. Nonetheless, Ldlrad3Δ14/14 recipient mice receiving wild-type bone marrow had higher levels of infection in PBLs and selected visceral organs including the lung (10-fold, **p < 0.01), heart (8-fold, *p < 0.05), and kidney (9-fold, **p < 0.05) than mice reconstituted with Ldlrad3Δ14/14 bone marrow, indicating that LDLRAD3 expression on cells arising from HSCs contributes to VEEV infection in blood and visceral organs but not in the CNS (Figure 3D). Overall, the infection data in bone marrow chimeric mice suggest a lesser contribution of LDLRAD3 expression on radiation-sensitive cells and a more dominant contribution on radiation-resistant cells to VEEV pathogenesis. Furthermore, expression of LDLRAD3 on infiltrating leukocytes does not contribute to increases in CNS infection of VEEV.
LDLRAD3 has a CNS-specific role in VEEV pathogenesis
We used FISH of Ldlrad3 mRNA and immunostaining of NeuN+ neurons to visualize neuron-specific expression of LDLRAD3 in brain tissue sections of adult naive wild-type mice (Figure S4A). Ldlrad3 mRNA was expressed in NeuN+ neurons across all brain regions but was also expressed in many other unidentified non-neuronal cells (Figure S4A). Using an available Mouse Cell Atlas database (https://bis.zju.edu.cn/MCA/), we observed that Ldlrad3 mRNA expression in the adult mouse brain extends to oligodendrocytes, oligodendrocyte precursors, astrocytes, monocytes, and neurons (Figure S4B). Based on these data and our bone marrow chimera experiments, we hypothesized that LDLRAD3 might be required for VEEV neuropathogenesis.
To test this hypothesis, we evaluated the effect of inoculation route (subcutaneous, intranasal, or intracranial) on VEEV pathogenesis in wild-type and Ldlrad3Δ14/Δ14 mice. All wild-type mice rapidly lost weight and uniformly succumbed to infection by 9 dpi after subcutaneous, intranasal, or intracranial inoculation of 102 FFU of VEEV (Figures 4A–4C). In comparison, Ldlrad3Δ14/Δ14 mice inoculated via intranasal (Figure 4B) or intracranial routes (Figure 4C) lost approximately 20% of their body weight within 1 week but then recovered weight and survived.
Figure 4. Ldlrad3Δ14/Δ14 mice survive intranasal and intracranial VEEV inoculation.
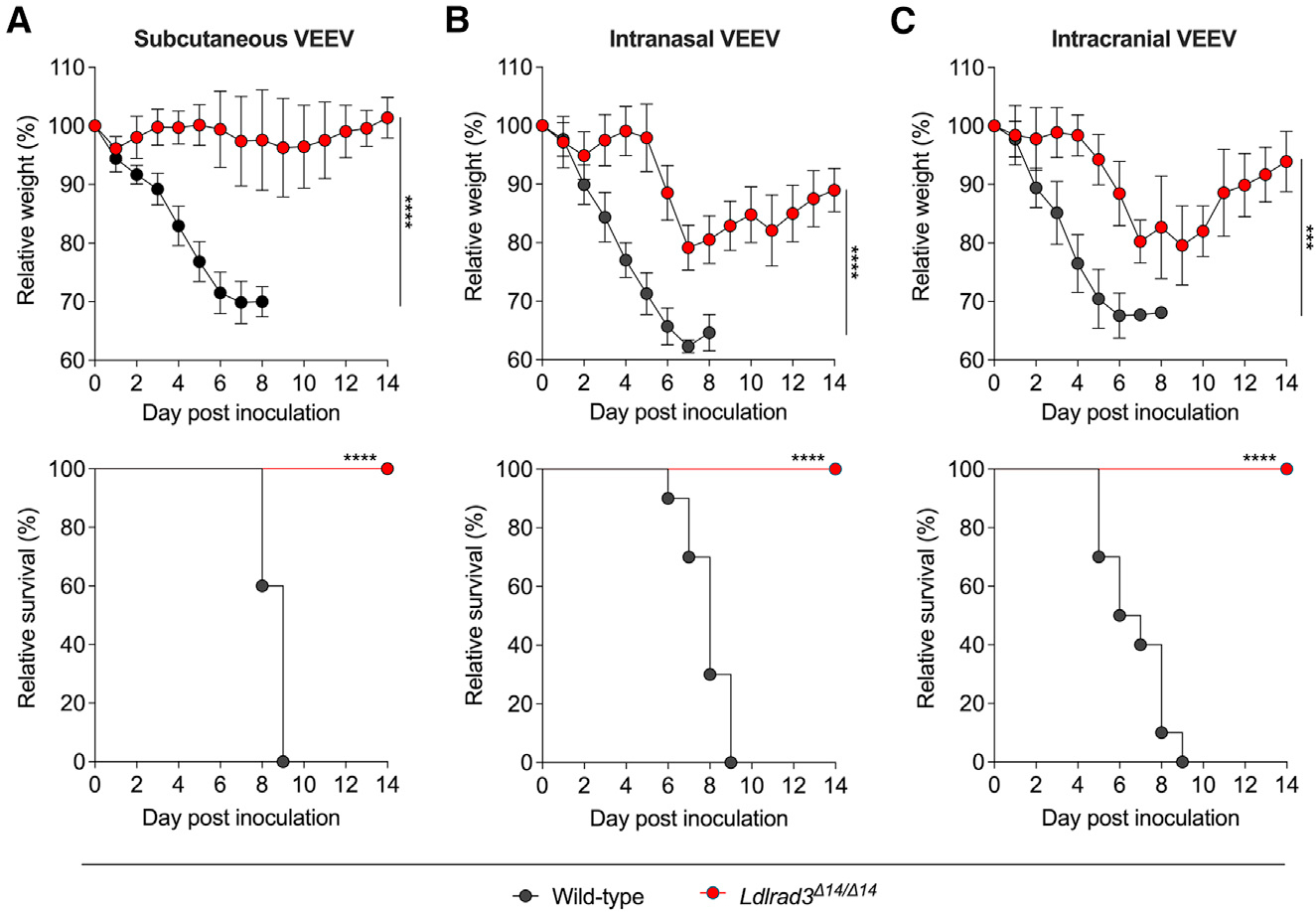
Wild-type or Ldlrad3Δ14/Δ14 mice were inoculated with 102 FFU of VEEV ZPC738 via subcutaneous route (A, wild-type [n = 10]; Ldlrad3Δ14/Δ14 [n = 8]), intranasal route (B, wild-type [n = 10]; Ldlrad3Δ14/Δ14 [n = 8]), or intracranial route (C, wild-type [n = 10]; Ldlrad3Δ14/Δ14 [n = 9]) and monitored daily for weight change (mean ± SD) and survival. For comparison of weight changes, area under the curve analysis was performed. To avoid survival bias in weight curves, statistical significance was only calculated at time points when all mice were alive. Data were analyzed by unpaired t test (top panels) or log-rank test (bottom panels) (ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). See also Figure S4.
To assess the direct impact of LDLRAD3 expression on CNS infection, we performed a kinetic analysis in wild-type and Ldlrad3Δ14/Δ14 mice after intracranial inoculation with 102 FFU of VEEV. We quantified viral RNA levels over time in CNS tissues and visceral organs (Figures 5A and S5A). Tissues from wild-type mice post intracranial inoculation were collected only through 5 dpi, as animals became moribund soon after. As expected, VEEV RNA levels rapidly increased in CNS tissues of wild-type mice (106–108 FFU/gram equivalents at 1 dpi with peak levels of 109–1010 FFU/gram equivalents at 5 dpi), and this was associated with rapid spread to blood, lymphoid tissues, and visceral organs. In Ldlrad3Δ14/Δ14 mice, levels of VEEV RNA in the CNS were lower (10- to 1,000-fold, depending on the day and brain region) and waned over the course of 14 days. Although viral spread to peripheral tissues after intracranial inoculation also was observed in VEEV-infected Ldlrad3Δ14/Δ14 mice, levels were lower than in wild-type mice and, in some organs (e.g., liver and lung), were at or near the limit of detection (Figures 5A and S5A).
Figure 5. Kinetics of VEEV infection in Ldlrad3Δ14/Δ14 mice after intracranial inoculation.
(A and B) Wild-type or Ldlrad3Δ14/Δ14 (n = 8) mice were inoculated intracranially with 102 FFU of VEEV ZPC738.
(A) At 3, 5 (for wild-type and Ldlrad3Δ14/Δ14 mice), or 7 dpi (Ldlrad3Δ14/Δ14 mice only), indicated tissues and samples were assessed for VEEV RNA as described in Figure 1 (OB, CTX, HPC, CBL, BS, ScMbs, SC, PBLs, LN).
(B) For CTX, spleen, and lung samples, infectious virus was determined at each time point. Mean values are shown. The LOD for each tissue is indicated by a dashed line, and numbers in black or red enumerate samples with titers at the LOD. Data are from three independent experiments per time point and were analyzed by Mann-Whitney test (ns, not significant; *p < 0.05 and ***p < 0.001).
(C) Representative images of sagittal brain sections from wild-type or Ldlrad3Δ14/Δ14 mice (n = 4) harvested 5 days after intracranial inoculation with 102 FFU of VEEV ZPC738 (scale bars: 5 mm) and combination FISH for VEEV RNA, immunohistochemical staining for cell-type-specific antigens, and DAPI counterstaining for nuclei visualization. White boxes indicate enlarged insets: (1)–(4) insets are for wild-type and (5)–(8) insets are for Ldlrad3Δ14/Δ14 brains (scale bars: 100 mm). Data are representative of images from two experiments. Cartoon schematic of an annotated sagittal mouse brain section is included for reference and was generated using BioRender.
See also Figures S5 and S6.
We also measured infectious virus levels after intracranial inoculation across all time points in representative CNS (e.g., cerebral cortex), visceral (e.g., lung), and lymphoid (e.g., spleen) tissues (Figure 5B). In wild-type mice, levels of infectious virus in tissues were lower than, but correlated with, their viral RNA levels. In Ldlrad3Δ14/Δ14 mice, VEEV infection in the cerebral cortex increased through 7 dpi and then declined, with no infectious virus detected at 14 dpi, which generally paralleled viral RNA measurements. The spleens of Ldlrad3Δ14/Δ14 mice consistently had low levels of infectious virus through 14 dpi, whereas the lungs did not. These virological data after intracranial inoculation suggest that LDLRAD3-independent VEEV infection can occur in the CNS, albeit to a lower level, which likely explains the weight loss and survival phenotypes in infected Ldlrad3Δ14/Δ14 mice (Figures 4B and 4C).
To assess directly how LDLRAD3 affects VEEV tropism in the CNS, we performed FISH for VEEV RNA and IHC for staining of neurons (NeuN+) and astrocyte (combination GFAP/SOX9+) markers in brain tissue sections of wild-type and Ldlrad3Δ14/Δ14 mice at 3 and 5 days after intracranial inoculation (Figures 5C, S6A, and S6B). Staining of VEEV-infected samples with a negative control probe or mock-infected samples stained with the VEEV-specific probe again showed no background signal (Figures S5B and S6C). In brains from wild-type mice, viral RNA staining was extensive (Figures 5C, S6A, and S6B), with substantial co-localization with NeuN+ neurons in the olfactory bulb, cortex, thalamus, hypothalamus, midbrain, and pons (Figure 5C). VEEV RNA was not readily apparent in GFAP/SOX9+ cells at this time point (Figure S5C). Brains from Ldlrad3Δ14/Δ14 mice showed more limited infection (Figures 5C, S6A, and S6B), with co-localization in NeuN+ neurons only in focal regions of the cortex, midbrain, and hypothalamus (Figure 5C). In Ldlrad3Δ14/Δ14 mice, VEEV RNA was largely absent from the olfactory bulb, cerebellum, brainstem, and dentate gyrus (Figures 5C, S6A, and S6B). These direct inoculation experiments in the brain suggest that LDLRAD3 expression is a critical determinant for brain infection of most NeuN+ neurons.
The absence of LDLRAD3 impairs VEEV infection of neurons and glia in primary cultures
To corroborate the effects of LDLRAD3 expression on VEEV tropism, we cultivated mixed primary neuron-glia cultures from cortices of wild-type or Ldlrad3Δ14/Δ14 fetuses harvested at embryonic day 17. Using immunostaining and confocal microscopy, we confirmed that these cultures contained NeuN+ neurons (Figure 6A). We inoculated these cells with VEEV ZPC738-EGFP at a multiplicity of infection (MOI) of 20 before fixation and staining at 7 hpi (Figure 6A). Notably, we observed significantly fewer infected neurons (GFP+NeuN+ cells) in cultures derived from Ldlrad3Δ14/Δ14 fetuses than wild-type (Figures 6A and 6B), indicating that VEEV principally utilizes LDLRAD3 to target NeuN+ neurons in these cultures. We also inoculated neuron-glia cultures with a virus (Sindbis [SINV]-VEEV TrD-GFP) that encodes the structural genes of the epizootic strain VEEV TrD and observed similar reductions in GFP+ signal in NeuN+ neurons derived from Ldlrad3Δ14/Δ14 fetuses (Figure 6C).
Figure 6. VEEV infection of neurons in primary mixed neuron-glia cultures.
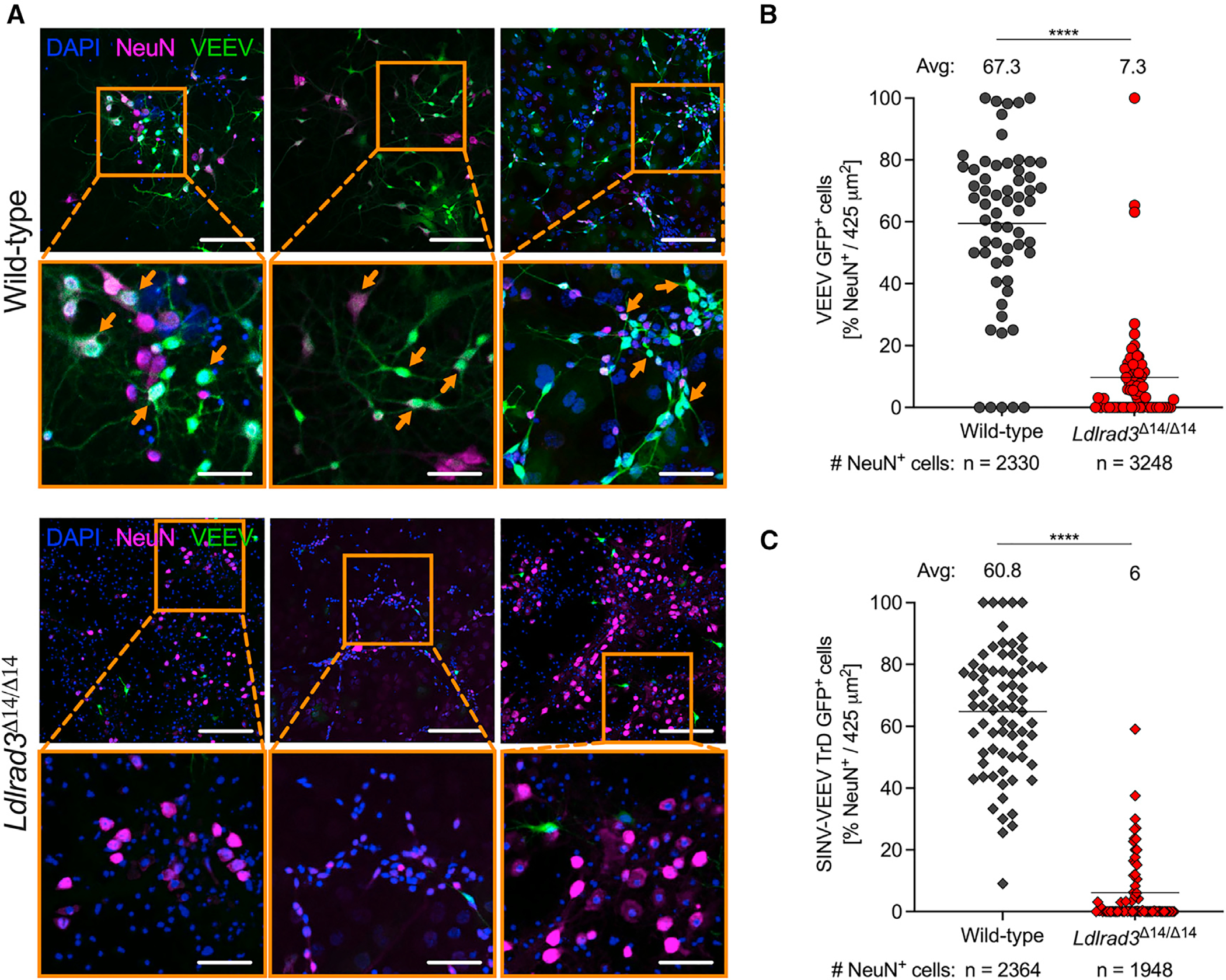
(A–C) Immunofluorescence analysis of VEEV-EGFP- or SINV-VEEV TrD-GFP-infected NeuN+ neurons in mixed neuron-glia cultures isolated from embryonic day 17 (E17) embryos from wild-type and Ldlrad3Δ14/Δ14 mice and infected 11–14 days after plating at MOI 20 for 7 h.
(A) Representative confocal microscopy images of mixed neuron-glia cultures from wild-type (top panels) and Ldlrad3Δ14/Δ14 (bottom panels) fetuses highlighting nuclei (DAPI+), neurons (NeuN+), and VEEV infection (GFP+). Orange boxes indicate enlarged insets, and orange arrows indicate examples of infected neurons (NeuN+GFP+ co-localization) (low magnification, scale bars: 100 μm; high magnification, scale bars: 270 μm).
(B and C) Quantification of VEEV-infected neurons is represented per image area (425 μm2) as the percentage of NeuN+ cells that are GFP+ and infected with (B) VEEV ZPC738-EGFP or (C) SINV-VEEV TrD-GFP. The mean percentage of infected neurons is indicated above each dataset, and the total number of NeuN+ cells counted is indicated below. Data are from two independent experiments, each with three technical replicates, and were analyzed by Mann-Whitney test (****p < 0.0001).
See also Figure S7.
In comparison, in cultures from Ldlrad3Δ14/Δ14 mice, we observed preferential infection of NeuN− cells with distinct morphologies, suggesting these cells become infected with VEEV through a LDLRAD3-independent pathway. We identified these cells as Olig2+, a transcription factor expressed in oligodendrocyte lineage cells79,80 and in subpopulations of astrocytes in the adult CNS81,82 (Figure S7A). Quantification of VEEV ZPC738-EGFP- (Figure S7B) or SINV-VEEV TrD-GFP-infected (Figure S7C) Olig2+ cells in wild-type and Ldlrad3Δ14/Δ14 cultures indicates infection is also decreased in the absence of LDLRAD3 (Figures S7B and S7C). Overall, these results indicate that LDLRAD3 expression impacts VEEV tropism in the CNS in a cell-type-specific manner, predominantly targeting VEEV to neurons and, possibly, other glial cells.
DISCUSSION
In this study, we investigated the role of the entry receptor LDLRAD3 in VEEV pathogenesis using Ldlrad3-deficient mice. We found consistently lower levels of VEEV infection in all target tissues of Ldlrad3-deficient mice after subcutaneous inoculation, as early as 6 hpi and at every time point tested thereafter. While VEEV entry into the brain occurred in the absence of LDLRAD3 expression, spread was delayed, infection accumulated at substantially lower levels, and animals did not sustain weight loss or lethality. Bone marrow chimera studies established that VEEV pathogenesis was largely dependent on LDLRAD3 expression in radio-resistant stromal cells. Direct inoculation of VEEV into the brain via intracranial or intranasal inoculation resulted in uniform lethality in wild-type mice, whereas in Ldlrad3-deficient mice, animals lost weight but survived infection. This phenotype was associated with comparatively lower CNS viral burdens in Ldlrad3-deficient mice. Also, the absence of LDLRAD3 was associated with marked decreases in infection of neurons in adult mouse brains by FISH and IHC and in mixed primary neuron cultures isolated from embryos. Overall, these experiments establish a key role for LDLRAD3 in the infection, dissemination, and pathogenesis of VEEV in peripheral and CNS tissues.
Although the endogenous role of LDLRAD3 is poorly understood, it has been reported to regulate amyloid processing in neurons43 and auto-ubiquitination to promote the activity of E3-ubiquitin ligases.44 According to publicly available transcriptomics data,83–87 Ldlrad3 mRNA expression is found in several human and mouse tissues including the brain, respiratory tract, gastrointestinal tract, reproductive tracts, and connective and soft tissues. As it relates to VEEV tropism in mice, Ldlrad3 mRNA expression is detected in myeloid cells such as monocytes and macrophages and in neurons, astrocytes, and oligo-dendrocytes of the CNS.83–87 Given the overlap in predicted cell-type-specific expression of LDLRAD3 and known features of VEEV tropism,88–90 we hypothesized that LDLRAD3 might be important at several steps in VEEV pathogenesis. For this reason, we extended our infection analysis to include lymphoid and visceral organs, in addition to CNS tissues in each of our virological time courses.
Natural or mosquito-transmitted infection by VEEV is modeled by subcutaneous inoculation in mice and results initially in infection of dermal dendritic cells and macrophages.59,90–92 DCs rapidly emigrate from the skin to carry virus to the DLNs, where VEEV replication can occur as early as 4 hpi, resulting in spread into circulation and to distant tissues.90,91,93 Though it has not been shown for VEEV, free virus can be transported to the DLNs through lymphatic fluid.76 In the absence of LDLRAD3 expression, VEEV exhibited an early defect, at 6 and 12 hpi, in DLN infection relative to wild-type mice, which suggested a possible role for LDLRAD3 in infection of tissue-resident and/or circulating myeloid cells. Our bone marrow chimera studies confirm a contributing role for LDLRAD3-expressing cells arising from the HSC compartment for VEEV infectivity in the peripheral blood and visceral organs, as Ldlrad3-deficient mice reconstituted with wild-type cells showed a small but significant decrease in weight at 2 dpi and increases in viral RNA levels in PBLs, lung, heart, and kidneys at 5 dpi relative to Ldlrad3-deficient mice reconstituted with LDLRAD3-expressing cells. However, irradiated Ldlrad3-deficient mice reconstituted with wild-type bone marrow did not show the substantial weight loss or VEEV infection seen in wild-type recipient mice after subcutaneous inoculation. Likely, LDLRAD3-expressing radiation-resistant cells in tissue stroma and/or the CNS are readily infected by VEEV in wild-type recipient mice reconstituted with donor HSCs from wild-type or Ldlrad3-deficient mice. Future experiments with conditional deletions in LDLRAD3 will be required to identify the specific cell types that contribute to VEEV infection and pathogenesis in a LDLRAD3-dependent manner.
The lower level of viral infection in Ldlrad3-deficient mice at early time points after subcutaneous inoculation resulted in less infection in every tissue tested, including those in the CNS. To study the CNS-specific effects of LDLRAD3 more directly, we evaluated infection in Ldlrad3-deficient mice after intracranial inoculation. Compared with wild-type mice, infection in the brain and spinal cord of Ldlrad3-deficient mice occurred at much lower levels (approximately 100- to 1,000-fold). This result likely explains the decreased weight loss and lethality seen in VEEV-infected Ldlrad3-deficient mice after direct CNS infection. Nonetheless, infection did progress in the CNS of Ldlrad3-deficient mice, with peak tissue titers in the olfactory bulb and cerebral cortex occurring between 5 and 7 dpi before declining through 14 dpi. This result establishes a dominant role for LDLRAD3 for VEEV infection in the CNS in mice but also highlights the existence of additional, uncharacterized subordinate entry pathways. This does not include other recently characterized alphavirus receptors such as MXRA8,94 VLDLR, or ApoER274 since VEEV does not utilize any of these molecules to enter and infect cells.74,94 Although no other physiologically relevant entry receptor has been described for VEEV, laminin-binding protein reportedly enhances infection of mosquito and human cell lines,95,96 and C-type lectins can promote VEEV infection of cells by mosquito-derived alphaviruses.97 Moreover, infection of cell-culture-adapted VEEV strains is increased by binding to heparan sulfate proteoglycans, though its significance in vivo remains uncertain.98 Future screening campaigns are needed to identify additional VEEV receptors that contribute to infection of peripheral and CNS tissues in mammals, or in mosquito vectors, which lack an apparent LDLRAD3 ortholog.42,74
Using FISH and IHC of brains from mice intracranially infected with VEEV, we observed that neurons are primary targets of VEEV infection in wild-type mice, as reported previously.99 However, in Ldlrad3-deficient mice, at the peak time point of infection, neurons also appear to be targeted, albeit at much lower levels. This may indicate differential expression of LDLRAD3 or other entry ligands in neuron subpopulations. We also were unable to identify VEEV-infected GFAP/SOX9+ astrocytes at this time point. This result is consistent with data from others showing that astrocytes may not be primary targets of VEEV infection in vivo, although they can still be infected.62,69 In mixed neuron-glia cell cultures, we observed a substantial impact of LDLRAD3 expression on VEEV infection of NeuN+ neurons and on Olig2+ gliogenic progenitor cells. This effect was comparable for viruses displaying structural proteins from enzootic or epizootic strains indicating that LDLRAD3-dependent tropism in the CNS likely is similar among different clades of VEEV. While mature oligodendrocytes have been proposed as targets of VEEV infection in the CNS,100 our neuron-glia cultures are not definitive since Olig2 expression is reportedly found in subpopulations of neurons and in all gliogenic precursors.79,80,101–103 Studies that delete LDLRAD3 from specific neuronal cell subpopulations will be needed to complete our understanding of the role of LDLRAD3 in VEEV neuropathogenesis.
Limitations of the study
(1) While we measured weight loss and survival differences in VEEV-infected wild-type and Ldlrad3Δ14/Δ14 mice, we did not systematically examine signs of neurological disease (e.g., tremors, ataxia, seizures, and/or behavioral changes). (2) Although we observed no differences in the numbers of circulating immune cells in the peripheral blood of naive wild-type and Ldlrad3Δ14/Δ14 mice, additional immunophenotyping may be warranted to determine effects on immune system function. (3) Our bone marrow chimera studies suggest that radio-resistant cells expressing LDLRAD3 are key drivers of VEEV pathogenesis in mice. However, the precise cell types were not defined. Future studies with Ldlrad3fl/fl conditional knockout mice are required to address which cell lineages are targeted by VEEV in a LDLRAD3-dependent manner and contribute to severe disease. (4) Because of an absence of reagents, we did not perform LDLRAD3 antigen staining in tissue sections, so its protein expression in vivo remains to be determined. (5) As our infection studies are restricted to mice, experiments in other reservoir animals are needed to corroborate our findings and confirm the contribution of LDLRAD3 to VEEV pathogenesis.
Our study demonstrates the importance of LDLRAD3 expression at many key stages of VEEV pathogenesis including in the CNS. Because of its importance for VEEV infection and pathogenesis, blockade of receptor antibodies,45 the use of receptor fusion decoy proteins,42 or small molecules that disrupt VEEV-LDLRAD3 engagement45,46 could represent strategies to prevent or treat VEEV infection. Ultimately, given that VEEV can infect some cells using a LDLRAD3-independent entry pathway, strategies targeting multiple cellular receptors may be required to completely limit the cellular entry of pathogenic VEEV strains.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Michael S. Diamond (mdiamond@wustl.edu).
Materials availability
All requests for resources and reagents should be directed to the Lead Contact author. This includes viruses, primer-probe sets, and mice. All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and code availability
All data supporting the findings of this study are available within the paper and from the corresponding author upon request. This paper does not include original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cells
Vero (ATCC #CCL-81) cells were propagated in Dulbecco’s Modified Eagle Medium (DMEM; Gibco #11995–040) supplemented with 5% heat-inactivated FBS (Omega Scientific #FB-01), 100 U/ml penicillin-streptomycin (Gibco #15140–122), and 10 mM HEPES (Gibco #15630–080). Cells were maintained at 37°C in the presence of 5% CO2 and confirmed as mycoplasma-negative by the Genome Engineering and iPSC Center at Washington University.
Viruses
The construction and generation of VEEV subtype ID strain ZPC738105,106, recombinant GFP reporter viruses (VEEV ZPC738-eGFP62,107 and SINV-VEEV TrD-GFP42,108), and infectious cDNA clones have been described previously. The non-select agent Madariaga virus (MADV) strain Arg LL was isolated from a horse in 1936 and provided by the World Reference Center for Emerging Viruses and Arboviruses (S. Weaver and K. Plante, University of Texas Medical Branch). Viral stocks were titered by focus-forming assay on Vero cells and stored at −80°C as single-use aliquots. All work with full-length VEEV ZPC738, VEEV ZPC738-EGFP, and MADV was performed in BSL3 and A-BSL3 facilities at Washington University School of Medicine in accordance with approved Institutional Biosafety protocols.
Mice
Animal studies were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (assurance number A3381–01). Mice were housed in groups and fed standard chow diets. Virus inoculations and sample collections were performed under anesthesia and induced and maintained with ketamine hydrochloride and xylazine. All efforts were made to minimize animal suffering.
Ldlrad3Δ14/Δ14 mice on a C57BL/6J background were generated by CRISPR-Cas9 gene editing and described previously.42 Wild-type C57BL/6J (RRID:IMSR_JAX:000664) or C57BL/6J congenic CD45.1 mice (RRID:IMSR_Jac:002014) were purchased from Jackson Laboratories. Female or male and age-matched (6–11-week-old, depending on the experiment) Ldlrad3Δ14/Δ14 and wild-type C57BL/6J or CD45.1 C57BL/6J mice were used.
METHOD DETAILS
Mouse experiments
For VEEV ZPC738 infections, 7 to 11-week-old male and female Ldlrad3Δ14/Δ14 and wild-type C57BL/6J mice were anesthetized as described above and inoculated with 102 focus-forming units (FFU) of virus diluted in sterile PBS (Gibco #14190–136) via a subcutaneous, intranasal, or intracranial routes. For subcutaneous inoculations, 50 μL of diluted virus was injected into the left rear footpad. For intranasal inoculations, 20 μL of diluted virus was delivered dropwise into the nares (10 μL per nare) using a micropipettor. For intracranial inoculations, 10 μL of diluted virus was injected into the left hind cortex using a pre-measured needle guide and a 0.3 mL 29G × ½” insulin syringe (Exel Int #26018). For MADV infections, six to seven-week-old male and female Ldlrad3Δ14/Δ14 and wild-type C57BL/6J mice were inoculated via a subcutaneous route with 103 FFU of virus diluted in sterile PBS as described above. Survival and body weight were measured on the day of inoculation and daily thereafter for 14 days.
For sample collection, peripheral blood was collected via cardiac puncture and added to serum-separating tubes (BD Microtainer #365967) or tubes with K2EDTA (BD Microtainer #365974) supplemented with 30 μL of 0.5 M EDTA (Corning #46–034-CI) for peripheral blood leukocyte (PBL) collection. After obtaining blood samples, mice were perfused with 20 mL of PBS. Depending on the experiment, tissues in the periphery (foot skin, underlying tissue of the foot, popliteal or inguinal DLN, spleen, liver, heart, lung, thymus) and CNS (olfactory bulb, cortex, cerebellum, hippocampus, brainstem, other brain, and spinal cord) were collected at 6 h, 12 h, day 1, 3, 5, 7, 10, or 14 after inoculation. Samples were immediately placed on dry ice. PBLs were further processed to remove red blood cells with ACK lysing buffer (Gibco #A10492–01). PBLs were then washed twice with 15 mL of iced FACS buffer (2% FBS, 2 mM EDTA in PBS) and resuspended in 5X MagMax-96 Viral RNA Isolation Kit (Applied Biosystems #AMB18365) lysis buffer. All samples were stored at −80°C before viral RNA and infectious virus quantification. During the kinetic analysis of viral burden after intracranial inoculation, two Ldlrad3Δ14/Δ14 mice died before the timepoint of tissue collection and were necessarily excluded from analysis.
Flow cytometric analysis of PBLs
PBLs were isolated from peripheral blood of naive Ldlrad3Δ14/Δ14 and wild-type C57BL/6J mice by cardiac puncture and processed into single cell suspensions as described above in ‘Mouse experiments.’ In a 96-well plate, single-cell suspensions were first treated for 30 min at 4°C with TruStain FcX anti-mouse CD16/32 (diluted 1:200; BioLegend #101320) to block Fcγ receptor binding and fixable viability dye ViaDye Red (Cytek #R7–60008) to exclude dead cells. Cells were washed twice in cold FACS buffer and centrifuged at 350 × g for 4 min at 4°C. For staining of surface antigens, cells were incubated for 30 min at 4°C with panels of fluorescent-dye conjugated antibodies diluted 1:200 in FACS buffer. PBLs were stained with the following antibodies from BioLegend: CD45 BV421 (clone 30-F11 #103133), CD19 BV650 (clone 6D5 #115541), TCR-β BV421 (clone H57–597 #109229), CD8α BV570 (clone 53–6.7 #100739), CD4 PerCP/Cyanine5.5 (clone RM4–5 #100540), NK1.1 PE/Cy7 (clone PK136 #108713), CD11b APC (clone M1/70 #101211), Siglec-F PE (clone S17007L #155505), Ly6G BV605 (clone 1A8 #127639), and Ly6C AF700 (clone HK1.4 #128023). Subsequently, all cells were washed twice by resuspension in cold FACS buffer and centrifugation at 350 × g for 4 min at 4°C. After fixation with 4% paraformaldehyde (PFA; EMS #15713-S) diluted in PBS (PFA/PBS), cells were resuspended in 200 μL of FACS buffer and analyzed by spectral flow cytometry on a Cytek Aurora. For each experiment unmixing was performed using single stained and unstained cells or AbC Total Antibody Compensation beads (Life Technologies #A10497). Cell counts were determined using Precision Count Beads (BioLegend #424902). All analysis was conducted using FlowJo software (v10, BD Biosciences).
Viral RNA measurements
Tissues were weighed and homogenized with ~200 μL of zirconia/silica beads (BioSpec #11079110) and DMEM supplemented with 2% heat-inactivated FBS in a MagNA Lyser instrument. Tissue homogenates were clarified by centrifugation at 9,000 × g for 5 min. Viral RNA was extracted from tissues using the MagMax-96 Viral RNA Isolation Kit and the KingFisher Flex Purification System (Thermo Scientific #5400610) and quantified by qRT-PCR using a Taqman RNA-to-CT 1-Step Kit (Applied Biosystems #4392938). Reverse transcription was carried out using a QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher #4485691) at 48°C for 15 min followed by a 10 min incubation step at 95°C to inactivate the enzyme. DNA amplification was accomplished over 40 cycles as follows: 95°C for 15 s and 60°C for 1 min. A standard curve was generated using serial 10-fold dilutions of VEEV ZPC738 extracted from a viral stock of known titer. Serum and organs were expressed on a log10 scale as FFU equivalents per mL of serum or per g of tissue. For lymph nodes, infection was expressed as FFU equivalents per whole tissue. PBL viral RNA levels were normalized to Mus musculus Gapdh levels and expressed as fold change determined using the 2-ΔCt method. Primer and probe sequences used to determine RNA levels are published42 and as follows: VEEV ZPC738 nsP3 forward: 5′-CAAGTCGAGGCAGACATTCA-3′; VEEV ZPC738 nsP3 reverse: 5′-CAGGGTGTCAAGGATGGATAAA-3′; VEEV ZPC738 nsP3 probe: 5′-/56-FAM/TGGTCCATT/ZEN/CCTCATGCATCCGAC/3IABkFQ/−3′ (Integrated DNA Technologies, Custom Primetime Standard qPCR assay); and M. musculus Gapdh (Integrated DNA Technologies, predesigned set Mm.PT.39a.1).
Focus-forming assays
Vero cells were seeded in flat-bottom 96-well plates (TPP #92696) at 3 × 104 cells/well in a volume of 100 μL per well. The next day, virus stocks or pre-weighed homogenized and clarified tissue supernatants were serially diluted in infection media (DMEM with 2% heat-inactivated FBS, 100 U/ml penicillin-streptomycin, and 10 mM HEPES). 100 μL of the diluted samples were added to Vero cell monolayers and incubated for 1 h at 37°C in 5% CO2. Subsequently, cells were overlaid with 100 μL of 1% methylcellulose in Minimum Essential Medium (MEM; Sigma #M0275) supplemented with 100 U/ml penicillin-streptomycin, 10 mM HEPES, and GlutaMAX (Gibco #35050–061). Plates were fixed 18 to 20 h after virus inoculation. For samples infected with VEEV ZPC738, VEEV ZPC738-EGFP, and MADV, the methylcellulose overlay was first gently removed with a multichannel pipette and then, 300 μL of 4% PFA/PBS was added to each well for 1 h at room temperature. After three washes in PBS with 0.05% Tween 20 (Sigma #P1379) (PBS-T), samples were incubated on a plate rocker with 1 μg/mL of mouse anti-VEEV monoclonal antibody (mAb) 1A4A-1104 diluted in permeabilization buffer (PBS, 0.1% saponin [Sigma #S7900], and 0.1% bovine serum albumin [BSA; Sigma #A2153]) for 2 h at room temperature or overnight at 4°C. Primary mAb was removed after three washes with PBS-T, and samples were incubated with secondary peroxidase-conjugated goat anti-mouse IgG (Sigma #A5278) diluted 1:500 in permeabilization buffer for 1 h at room temperature on a rocker. After three washes with PBS-T, virus-infected foci were developed using KPL TrueBlue peroxidase substrate (SeraCare #5510–0050), washed twice with Milli-Q water, and counted using an CTL-S6 Universal Analyzer (ImmunoSpot). Viral titers were expressed as FFU per mL for viral stocks, FFU per gram for tissues, or FFU per lymph node.
Bone marrow chimeras
Chimeric mice were generated using modifications to a published protocol.110 Bone marrow ablation was achieved by irradiating four to six-week-old wild-type C57BL/6J (CD45.1) and Ldlrad3Δ14/Δ14 mice (CD45.2) with 9.5 Gy (X-ray) total body irradiation. The next day, bone marrow cells were collected from the tibias and femurs of five-week-old CD45.1 or CD45.2 wild-type mice, or CD45.2 Ldlrad3Δ14/Δ14 mice. Each irradiated, recipient mouse was administered 3 × 106 sex-matched bone marrow cells via retro-orbital injection. Five weeks after bone marrow transplantation, chimerism of each mouse (>88% CD45.1 or CD45.2 reconstitution) was confirmed by flow cytometric analysis of PBLs. Erythrocytes were lysed as described above in ‘Mouse experiments,’ and cells were processed in a round-bottom 96-well plate format (TPP #92697). Single-cell suspensions were treated for 30 min at 4°C with TruStain FcX anti-mouse CD16/32 (diluted 1:200; BioLegend #101320) to block Fcγ receptor binding and fixable viability dye ViaDye Red (Cytek #R7–60008) to exclude dead cells. Cells were washed twice, resuspended in iced FACS buffer, and then stained for 30 min at 4°C with the following panel of BioLegend antibodies diluted 1:200 in FACS buffer: CD45.1 FITC (clone A20 #110705), CD45.2 APC (clone 104 #109813) before fixation with 2% PFA/PBS for 20 min at room temperature. After two washes with FACS buffer, cells were resuspended in FACS buffer and analyzed using spectral flow cytometry on a Cytek Aurora. For each experiment unmixing was performed using single stained and unstained cells or AbC Total Antibody Compensation beads (Life Technologies #A10497). All analysis was conducted using FlowJo software (v10, BD Life Sciences). Six weeks after bone marrow transfer, chimeric mice were inoculated with 102 FFU of VEEV ZPC738 via a subcutaneous route. Weight loss was monitored for five days, and then serum, PBLs, and selected tissues were collected to determine viral RNA levels by qRT-PCR as described in ‘Mouse experiments.’
Tissue fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC)
Analysis of brains of VEEV-infected or mock-infected Ldlrad3Δ14/Δ14 and wild-type mice was performed at 3 or 5 days after subcutaneous or intracranial inoculation. Mice were anesthetized as described above, perfused with 20 mL of PBS and 20 mL of 4% PFA/PBS at 4°C. Brains were immersion-fixed in 40 mL of 4% PFA/PBS for 24 h at 4°C. For mice that were inoculated via a subcutaneous route, intact brains with the encasing skulls were decalcified for 7 days with daily exchanges of 30 mL of 0.5 M EDTA in water titrated to pH 7.4 at 4°C. This was followed by two exchanges of 30 mL of PBS on day 8 and 9 at 4°C, and for cryoprotection, two exchanges of 30 mL of filtered 30% sucrose diluted in PBS (sucrose/PBS) on day 10 and 11 at 4C. Tissues were sliced midsagittally. The skull and brain were incubated at room temperature with increasing concentrations of optimal cutting temperature compound (OCT) in 30% sucrose/PBS before embedding: 20% OCT in 30% sucrose for 1 h, 40% OCT in 30% sucrose for 2 h, and two washes of 100% OCT for 10 min. For mice that were inoculated via an intracranial route, brains were briefly washed with PBS before two exchanges of 30 mL of filtered 30% sucrose/PBS and incubation for two days. On the day of embedding, brains were subjected to two washes with 100% OCT for 10 min at room temperature. Tissues were then embedded in OCT as full sagittal sections in 24 × 24 × 5 mm disposable base molds (Fisher Healthcare #22363554). Freezing of OCT blocks was performed on dry ice in aluminum dishes containing Cytocool II aerosol freezing spray (Epredia #8323). Tissue was sectioned into 10 μm sections for brains and mounted by the Washington University Musculoskeletal Histology and Morphometry Core and stored at −80°C before staining.
Tissue staining was performed directly on microscope slides. Tissue sections were stained using the Advanced Cell Diagnostics (ACD) Integrated RNAscope Multiplex Fluorescent v2 Assay combined with RNA-Protein Co-detection Ancillary Kit for fresh frozen tissue (ACD #323180). Reagents included RNA-Protein Co-Detection Ancillary kit (ACD #323180), RNAscope H2O2 and Protease Reagents (ACD #322381), RNAscope Multiplex Fluorescent Reagent Kit v2 (ACD #323100), RNAscope Multiplex TSA Buffer (ACD #322809), and RNAscope 50X Wash Buffer (ACD #310091). Briefly, slides were pre-treated to promote tissue adherence by incubation in PBS for 5 min before baking for 30 min at 60°C and then post-fixed in 4% PFA/PBS for 15 min at 4°C. To dehydrate samples, slides were immersed in increasing concentrations of ethanol (EtOH) at room temperature (50% EtOH for 5 min; 70% EtOH for 5 min; 2 × 100% EtOH for 5 min) and allowed to air dry. After tissue blocking and target retrieval, primary antibodies diluted 1:500 (guinea pig anti-NeuN [Millipore, clone A60 #ABN90P], rat anti-GFAP [Thermo Fisher, clone 2.2B10 #130300], and rabbit anti-SOX9 [Millipore #AB5535]) were applied to samples and incubated overnight in a dark humidified chamber. The next day, samples underwent post-primary fixation, hybridization with a VEEV ZPC738 probe (ACD #87638142) or a Ldlrad3 probe (ACD #87210142), signal amplification, and secondary antibody staining with an HRP-conjugated secondary Opal 650 Dye diluted 1:750 (Akoya Biosciences #OP-001005). Tissues were then incubated with fluorophore conjugated secondary antibodies for detection of cell-specific antigens diluted 1:400 (donkey anti-guinea pig IgG AF488 [Jackson ImmunoResearch #AB_2340472], donkey anti-rat IgG AF555 [Thermo Fisher #A48270], and donkey anti-rabbit IgG AF555 [Thermo Fisher #A31572]), and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Tissues on slides were mounted using rectangular cover glass (22 mm × 50 mm, thickness #1.5 [Fisher Scientific #12544D]) and Prolong Gold Antifade Mountant (Thermo Fisher #P36930) before drying overnight in the dark at room temperature and subsequent storage at 4°C. Tissues were imaged on a NanoZoomer 2.0-HT system (Hamamatsu) at the Washington University Alafi Neuroimaging Laboratory and analyzed using NDP.view2 software (Hamatsu).
Neuron and glia cell infection
Glass cover slips (12 mm, thickness #1.5 [Electron Microscopy Sciences #72290–04]) were prepared with acetone and 100% ethanol washes under sterile conditions. Autoclaved coverslips were distributed into wells of 24-well plates (TPP #92424). Coverslips were coated with 20 μg/mL of poly-D-lysine (Sigma #P7280) and 20 μg/mL of laminin (Sigma #L2020) diluted in sterile water overnight at 37°C in 5% CO2. Coverslips were washed four times with sterile water and air dried in a tissue culture hood. Dried plates were sealed with Parafilm (Parafilm M #PM-996) and stored at room temperature in preparation for cell plating. Mixed primary neuron-glia cell cultures were prepared from E16.5-E17 mouse embryos. To limit fibroblast contamination, the meninges and choroid plexus were removed from each brain under a stereomicroscope (Olympus #SZX7). The cerebellum and olfactory bulb also were removed. Cortices and hippocampi from a single pregnancy (4–8 embryos) were pooled in filtered HBSS (Gibco #25200–056) supplemented with 1% glucose (Millipore #G8270) on ice, centrifuged at 20 × g for 5 min at 4°C, and each embryo was dissociated under sterile conditions in 1 mL of 0.05% trypsin-EDTA (Gibco #25300–054) and 0.3 mL of 2,000 KU/mL of DNase I (Sigma #D4263) for 15 min at room temperature. To halt digestion, 3 mL of DMEM with 10% FBS was added, and cells were centrifuged at 500 × g for 5 min at 4°C. After decanting the supernatant, the cells were resuspended in 1 mL of B27-media (Neurobasal media [Gibco #21103–049] with B27 supplement [Gibco #17504044], GlutaMax, and 100 U/ml penicillin-streptomycin). The cell suspension was filtered using a 70 μm cell strainer (NEST #258368) and rinsed with B27-media to give a final volume of 1 mL per embryo. Cells were seeded in the center of the pre-coated coverslips in a 24-well plate at 5 × 104 cells in a total volume of 400 μL per well. B27-media was refreshed every 2–3 days by removing and replacing 200 μL of media per well. On day 11–12 post plating, cultures were inoculated with VEEV ZPC738-EGFP or SINV-VEEV TrD-GFP at a MOI of 20, diluted in 400 μL per well of pre-warmed B27-media at 37°C in 5% CO2. Infected coverslip samples were transferred to a new 24-well plate for a 7 h incubation. Subsequently, cells were fixed with 1 mL per well of 4% PFA/PBS for 20 min at room temperature. Samples then were washed three times with 1 mL of PBS and stored in 1 mL of PBS supplemented with 0.02% NaN3 at 4°C until staining.
Cell culture immunofluorescence staining and quantification
Fixed primary neuron and glia cells on glass coverslips were incubated with blocking solution (1% BSA, 0.1% Triton X-, and 3% donkey serum diluted in PBS) for 1 h at room temperature. Coverslips were incubated with primary antibodies (guinea pig anti-NeuN [1:500; Millipore, clone A60 #ABN90P]), rat anti-GFAP [Thermo Fisher, clone 2.2B10 #130300], and rabbit anti-Olig2 [1:1000; Millipore #AB9610]) diluted in blocking solution overnight at 4°C in the dark. The next day, antibodies were rinsed off with three PBS washes at room temperature and incubated with secondary antibodies (donkey anti-guinea pig AF647 [Jackson ImmunoResearch #AB_2340476], donkey anti-rat IgG AF555 [Thermo Fisher #A48279], and anti-rabbit AF546 [Thermo Fisher #A10040]) diluted 1:1000 in blocking solution for 2 h at room temperature in the dark. After another three PBS washes, nuclei were counterstained with DAPI (Thermo Fisher #D1306) diluted 1:10,000 in PBS for 10 min at room temperature and again washed three times with PBS. Coverslips were mounted on glass microscope slides (Fisher Scientific #12–550-343) using Prolong Glass Antifade Mountant and dried overnight in the dark at room temperature before storage at 4°C. Images were captured as above using an LSM 880 confocal laser scanning microscope (Zeiss) at the Washington University Molecular Microbiology Imaging Facility at 20X (NA 0.8) objective. Z steps were acquired through the entire cell monolayer (2–3 z-layers). Image analyses were performed blinded to the investigator, using Fiji software (https://fiji.sc/Fiji).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was assigned when p values were <0.05 using Prism version 8 (GraphPad). Tests, number of animals (n), mean values, and statistical comparison groups are indicated in the Figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Monoclonal mouse anti-VEEV antibody 1A4A-1 | Roehrig and Mathews104 | N/A |
| TruStain FcX monoclonal rat anti-mouse CD16/32 | BioLegend | Cat#101320; RRID:AB_1574975 |
| FITC anti-mouse CD45.1 clone A20 | BioLegend | Cat#110705; RRID:AB_313494 |
| APC anti-mouse CD45.2 clone 104 | BioLegend | Cat#109813; RRID:AB_389210 |
| BV421 anti-mouse CD45 clone 30-F11 | BioLegend | Cat#103133; RRID:AB_10899570 |
| BV650 anti-mouse CD19 clone 6D5 | BioLegend | Cat#115541; RRID:AB_11204087 |
| BV421 anti-mouse TCR-β clone H57-597 | BioLegend | Cat#109229; RRID:AB_10933263 |
| BV570 anti-mouse CD8α clone 53-6.7 | BioLegend | Cat#100739; RRID:AB_10897645 |
| PerCP/Cyanine5.5 anti-mouse CD4 clone RM4-5 | BioLegend | Cat#100540; RRID:AB_893326 |
| PE/Cyanine7 anti-mouse NK1.1 clone PK136 | BioLegend | Cat#108713; RRID:AB_389363 |
| APC anti-mouse/human CD11b clone M1/70 | BioLegend | Cat#101211; RRID:AB_312794 |
| PE anti-mouse CD170 (Siglec-F) clone S17007L | BioLegend | Cat#155505; RRID:AB_2750234 |
| BV605 anti-mouse Ly6G clone 1A8 | BioLegend | Cat#127639; RRID:AB_2565880 |
| AF700 Ly6C clone HK1.4 | BioLegend | Cat#128023; RRID:AB_10640119 |
| Guinea pig polyclonal anti-NeuN clone A60 | Millipore | Cat#ABN90P; RRID:AB_2341095 |
| Rat monoclonal anti-GFAP clone 2.2B10 | Thermo Fisher | Cat#130300; RRID:AB_2532994 |
| Rabbit polyclonal anti-SOX9 | Millipore | Cat#AB5535; RRID:AB_2239761 |
| Rabbit polyclonal anti-Olig2 | Millipore | Cat#AB9610; RRID:AB_570666 |
| AF488 Donkey anti-guinea pig IgG | Jackson ImmunoResearch | Cat#706-545-148; RRID:AB_2340472 |
| AF647 Donkey anti-guinea pig IgG | Jackson ImmunoResearch | Cat#706-605-148; RRID:AB_2340476 |
| AF555 Donkey anti-rat IgG | Thermo Fisher | Cat#A48270; RRID:AB_2896336 |
| AF555 Donkey anti-rabbit IgG | Thermo Fisher | Cat#A31572; RRID:AB_162543 |
| AF546 Donkey anti-rabbit IgG | Thermo Fisher | Cat#A10040; RRID:AB_2534016 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| VEEV ZPC738 | Anishchenko et al.,105 Hydeetal.,106 Ma et al.42 | N/A |
| VEEV ZPC738-EGFP | Sun et al.,107 Salimi et al.,62 | N/A |
| SINV-VEEV TrD-GFP | Paessler et al.,108 Ma et al.42 | N/A |
| Madariaga Arg LL | UTMB Arbovirus Reference Collection (Casals et al.,109 this paper) | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| MagMAX-96 Viral RNA Isolation Kit | Applied Biosystems | Cat#AM1836 |
| Taqman RNA-to-Ct 1-Step Kit | ThermoFisher | Cat#4392939 |
| RNA-Protein Co-Detection Ancillary kit | Advanced Cell Diagnostics | Cat#323180 |
| RNAscope H2O2 and Protease Reagents | Advanced Cell Diagnostics | Cat#322381 |
| RNAscope Multiplex Fluorescent Reagent Kit v2 | Advanced Cell Diagnostics | Cat#323100 |
| RNAscope Multiplex TSA Buffer | Advanced Cell Diagnostics | Cat#322809 |
| RNAscope 50X Wash Buffer | Advanced Cell Diagnostics | Cat#310091 |
| HRP-conjugated secondary Opal 650 Dye | Akoya Biosciences | Prod#FP1496A; Cat#OP-001005 |
| ViaDye Red Fixable Viability Dye Kit | Cytek | Cat#R7-60008 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Vero cells | ATCC | Cat#CCL-81; RRID:CVCL_0059 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson Laboratories | Strain#000664; RRID:IMSR_JAX:000664 |
| C57BL/6J CD45.1 mice | Jackson Laboratories | Strain#002014; RRID:IMSR_JAX:002014 |
| Ldlrad3Δ14/Δ14 mice | Ma et al.42 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| VEEV ZPC738 nsP3 forward: 5’-CAAGTCGAGGCAGACATTCA-3’ | IDT PrimeTime Assay | Custom (1 probe/2 primers) |
| VEEV ZPC738 nsP3 reverse: 5’-CAGGGTGTCAAGGATGGATAAA-3’ | IDT PrimeTime Assay | Custom (1 probe/2 primers) |
| VEEV ZPC738 nsP3 probe: 5’-/56-FAM/TGGTCCATT/ZEN/CCTCATGCATCCGAC/3IABkFQ/-3’ | IDT PrimeTime Assay | Custom (1 probe/2 primers) |
| Mus musculus Gapdh primer + probe | IDT PrimeTime Assay | Mm.PT.39a.1 |
| VEEV ZPC738 RNAscope Probe | ACD (Ma et al.42) | Cat#876381 |
| Ldlrad3 RNAscope Probe | ACD (Ma et al.42) | Cat#872101 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | BD Life Sciences | V10.7.2; RRID:SCR_008520 |
| GraphPad Prism | GraphPad | V9.5.0; RRID:SCR_002798 |
| FIJI | ImageJ2 | V2.9.0/1.53t; RRID:SCR_002285 |
| NDP.view2 | Hamamatsu | V2 #U12388-01 |
| BioRender | BioRender | RRID:SCR_018361 |
|
| ||
| Other | ||
|
| ||
| Gibco 0.05% trypsin-EDTA | Thermo Fisher | Cat#25300054 |
| DNase I | Sigma Aldrich | Cat#D4263 |
Highlights.
LDLRAD3 is important for VEEV dissemination and central nervous system infection in mice
Ldlrad3-deficient mice survive lethal VEEV challenge with substantially less virus infection
LDLRAD3 expression is required for efficient VEEV infection of primary neuron cultures
VEEV replication can occur in mice independently of LDLRAD3
ACKNOWLEDGMENTS
We thank Crystal Idleburg and Samantha Coleman Cathcart at the Washington University Musculoskeletal Histology and Morphometry Core for providing tissue sectioning services, Dr. Krzysztof Hyrc at the Washington University Alafi Neuroimaging Laboratory for imaging and scanning services, and Dr. Wandy Beatty at the Washington University Molecular Microbiology Imaging Facility for use of the laser scanning confocal microscope. We also thank members of the Diamond and Klein laboratories for sharing reagents and providing technical guidance. This study was supported by National Institutes of Health grants (T32 AI007172, F30AI164842, R01AI164653, and R01AI14367) and the Defense Threat Reduction Agency (W15QKN1691002). Some of the figures were created with BioRender, as is indicated in the relevant figure legends.
Footnotes
DECLARATION OF INTERESTS
M.S.D. is a consultant for Inbios, Vir Biotechnology, Ocugen, Topspin Therapeutics, Moderna, and Immunome. The Diamond laboratory has received unrelated funding support in sponsored research agreements from Moderna, Vir Biotechnology, Generate Biomedicines, and Emergent BioSolutions.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112946.
REFERENCES
- 1.Strauss JH, and Strauss EG (1994). The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58, 491–562. 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver SC, and Barrett ADT (2004). Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2, 789–801. 10.1038/nrmicro1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryman KD, and Klimstra WB (2008). Host responses to alphavirus infection. Immunol. Rev. 225, 27–45. 10.1111/j.1600-065X.2008.00670.x. [DOI] [PubMed] [Google Scholar]
- 4.Weaver SC, Winegar R, Manger ID, and Forrester NL (2012). Alphaviruses: population genetics and determinants of emergence. Antiviral Res. 94, 242–257. 10.1016/j.antiviral.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzmán-Terán C, Calderón-Rangel A, Rodriguez-Morales A, and Mattar S (2020). Venezuelan equine encephalitis virus: the problem is not over for tropical America. Ann. Clin. Microbiol. Antimicrob. 19, 19. 10.1186/s12941-020-00360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi AL (1967). Rural epidemic encephalitis in Venezuela caused by a group A arbovirus (VEE). Prog. Med. Virol. 9, 176–203. [PubMed] [Google Scholar]
- 7.Weaver SC, Salas R, Rico-Hesse R, Ludwig GV, Oberste MS, Boshell J, and Tesh RB (1996). Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. VEE Study Group. Lancet 348, 436–440. 10.1016/s0140-6736(96)02275-1. [DOI] [PubMed] [Google Scholar]
- 8.Oberste MS, Fraire M, Navarro R, Zepeda C, Zarate ML, Ludwig GV, Kondig JF, Weaver SC, Smith JF, and Rico-Hesse R (1998). Association of Venezuelan equine encephalitis virus subtype IE with two equine epizootics in Mexico. Am. J. Trop. Med. Hyg. 59, 100–107. 10.4269/ajtmh.1998.59.100. [DOI] [PubMed] [Google Scholar]
- 9.Forrester NL, Wertheim JO, Dugan VG, Auguste AJ, Lin D, Adams AP, Chen R, Gorchakov R, Leal G, Estrada-Franco JG, et al. (2017). Evolution and spread of Venezuelan equine encephalitis complex alphavirus in the Americas. PLoS Neglected Trop. Dis. 11, e0005693. 10.1371/journal.pntd.0005693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zacks MA, and Paessler S (2010). Encephalitic alphaviruses. Vet. Microbiol. 140, 281–286. 10.1016/j.vetmic.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrera JP, Pittí Y, Molares-Martínez JC, Casal E, Pereyra-Elias R, Saenz L, Guerrero I, Galué J, Rodriguez-Alvarez F, Jackman C, et al. (2020). Clinical and serological findings of Madariaga and Venezuelan equine encephalitis viral infections: a follow-up study 5 years after an outbreak in Panama. Open Forum Infect. Dis. 7, ofaa359. 10.1093/ofid/ofaa359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Salazar D, Estrada-Franco JG, Weaver SC, Carrara AS, and Aronson JF (2003). Weaver SC. Equine amplil’ieation and virulenee of subtype IE Venezuelan equine encephalitis viruses isolated during the 1993 iind 1996 Mexican epizootics. Emerg. Infect. Dis. 9, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deardorff ER, Estrada-Franco JG, Freier JE, Navarro-Lopez R, Travassos Da Rosa A, Tesh RB, and Weaver SC (2011). Candidate vectors and rodent hosts of Venezuelan equine encephalitis virus, Chiapas, 2006–2007. Am. J. Trop. Med. Hyg. 85, 1146–1153. 10.4269/ajtmh.2011.11-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver SC, Ferro C, Barrera R, Boshell J, and Navarro J-C (2004). Venezuelan equine encephalitis. Annu. Rev. Entomol. 49, 141–174. 10.1146/annurev.ento.49.061802.123422. [DOI] [PubMed] [Google Scholar]
- 15.Barrera R, Ferro C, Navarro JC, Freier J, Liria J, Salas R, Ahumada M, Vasquez C, Gonzalez M, Kang W, et al. (2002). Contrasting sylvatic foci of Venezuelan equine encephalitis virus in northern South America. Am. J. Trop. Med. Hyg. 67, 324–334. 10.4269/ajtmh.2002.67.324. [DOI] [PubMed] [Google Scholar]
- 16.Salas RA, Garcia CZ, Liria J, Barrera R, Navarro JC, Medina G, Vasquez C, Fernandez Z, and Weaver SC (2001). Ecological studies of enzootic Venezuelan equine encephalitis in north-central Venezuela, 1997–1998. Am. J. Trop. Med. Hyg. 64, 84–92. 10.4269/ajtmh.2001.64.84. [DOI] [PubMed] [Google Scholar]
- 17.Carrara AS, Coffey LL, Aguilar PV, Moncayo AC, Da Rosa APAT, Nunes MRT, Tesh RB, and Weaver SC (2007). Venezuelan equine encephalitis virus infection of cotton rats. Emerg. Infect. Dis. 13, 1158–1165. 10.3201/eid1308.061157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson KM, and Martin DH (1974). Venezuelan equine encephalitis. Adv. Vet. Sci. Comp. Med. 18, 79–116. [PubMed] [Google Scholar]
- 19.Young NA, Johnson KM, and Gauld LW (1969). Viruses of the Venezuelan equine encephalomyelitis complex. Experimental infection of Panamanian rodents. Am. J. Trop. Med. Hyg. 18, 290–296. [PubMed] [Google Scholar]
- 20.Bowen GS (1976). Experimental infection of North American mammals with epidemic Venezuelan encephalitis virus. Am. J. Trop. Med. Hyg. 25, 891–899. 10.4269/ajtmh.1976.25.891. [DOI] [PubMed] [Google Scholar]
- 21.Aguilar PV, Estrada-Franco JG, Navarro-Lopez R, Ferro C, Haddow AD, and Weaver SC (2011). Endemic Venezuelan equine encephalitis in the Americas: hidden under the dengue umbrella. Future Virol. 6, 721–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bronze MS, Huycke MM, Machado LJ, Voskuhl GW, and Green-field RA (2002). Viral agents as biological weapons and agents of bioterrorism. Am. J. Med. Sci. 323, 316–325. 10.1097/00000441-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Croddy E, Perez-Armendariz C, and Hart J (2002). Chemical and Biological Warfare: A Comprehensive Survey for the Concerned Citizen (Springer). [Google Scholar]
- 24.Paessler S, and Weaver SC (2009). Vaccines for Venezuelan equine encephalitisD80-D85. Vaccine 27 (Suppl 4), S0264–410X(09)01132–3 [pii]. 10.1016/j.vaccine.2009.07.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinney RM, Chang GJ, Tsuchiya KR, Sneider JM, Roehrig JT, Woodward TM, and Trent DW (1993). Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5’-noncoding region and the E2 envelope glycoprotein. J. Virol. 67, 1269–1277. 10.1128/JVI.67.3.1269-1277.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelman R, Ascher MS, Oster CN, Ramsburg HH, Cole FE, and Eddy GA (1979). Evaluation in humans of a new, inactivated vaccine for Venezuelan equine encephalitis virus (C-84). J. Infect. Dis. 140, 708–715. 10.1093/infdis/140.5.708. [DOI] [PubMed] [Google Scholar]
- 27.Berge TO, Banks IS, and Tigertt WD (1961). Attenuation of Venezuelan equine encephalomyelitis virus by in vitro cultivation in guinea pig heart cells. Am. J. Epidemiol. 73, 209–218. 10.1093/oxfordjournals.aje.a120178. [DOI] [Google Scholar]
- 28.Kitchen LW, and Vaughn DW (2007). Role of U.S. military research programs in the development of U.S.-licensed vaccines for naturally occurring infectious diseases. Vaccine 25, 7017–7030. 10.1016/j.vaccine.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 29.Grabenstein JD, Pittman PR, Greenwood JT, and Engler RJM (2006). Immunization to protect the US Armed Forces: heritage, current practice, and prospects. Epidemiol. Rev. 28, 3–26. 10.1093/epirev/mxj003. [DOI] [PubMed] [Google Scholar]
- 30.Hoke CH Jr. (2005). History of U.S. military contributions to the study of viral encephalitis. Mil. Med. 170, 92–105. 10.7205/milmed.170.4s.92. [DOI] [PubMed] [Google Scholar]
- 31.Pittman PR, Makuch RS, Mangiafico JA, Cannon TL, Gibbs PH, and Peters CJ (1996). Long-term duration of detectable neutralizing antibodies after administration of live-attenuated VEE vaccine and following booster vaccination with inactivated VEE vaccine. Vaccine 14, 337–343. 10.1016/0264-410x(95)00168-z. [DOI] [PubMed] [Google Scholar]
- 32.Leung JY-S, Ng MM-L, and Chu JJH (2011). Replication of Alphaviruses: A Review on the Entry Process of Alphaviruses into Cells. Adv. Virol. 2011, 249640. 10.1155/2011/249640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Wengler G, and Rey FA (2001). The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105, 137–148. 10.1016/S0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, and Chiu W (2011). 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 30, 3854–3863. 10.1038/emboj.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voss JE, Vaney M-C, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, and Rey FA (2010). Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468, 709–712. 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 36.Roehrig JT, Day JW, and Kinney RM (1982). Antigenic analysis of the surface glycoproteins of a Venezuelan equine encephalomyelitis virus (TC-83) using monoclonal antibodies. Virology 118, 269–278. [DOI] [PubMed] [Google Scholar]
- 37.Mathews JH, and Roehrig JT (1982). Determination of the protective epitopes on the glycoproteins of Venezuelan equine encephalomyelitis virus by passive transfer of monoclonal antibodies. J. Immunol. 129, 2763–2767. [PubMed] [Google Scholar]
- 38.Porta J, Jose J, Roehrig JT, Blair CD, Kuhn RJ, Rossmann MG, and Dermody TS (2014). Locking and blocking the viral landscape of an alphavirus with neutralizing antibodies. J. Virol. 88, 9616–9623. 10.1128/JVI.01286-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillpotts RJ (2006). Venezuelan equine encephalitis virus complex-specific monoclonal antibody provides broad protection, in murine models, against airborne challenge with viruses from serogroups I, II and III. Virus Res. 120, 107–112. 10.1016/j.virusres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Burke CW, Froude JW, Rossi F, White CE, Moyer CL, Ennis J, Pitt ML, Streatfield S, Jones RM, Musiychuk K, et al. (2019). Therapeutic monoclonal antibody treatment protects nonhuman primates from severe Venezuelan equine encephalitis virus disease after aerosol exposure. PLoS Pathog. 15, e1008157. 10.1371/journal.ppat.1008157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kafai NM, Williamson LE, Binshtein E, Sukupolvi-Petty S, Gardner CL, Liu J, Mackin S, Kim AS, Kose N, Carnahan RH, et al. (2022). Neutralizing antibodies protect mice against Venezuelan equine encephalitis virus aerosol challenge. J. Exp. Med. 219, e20212532. 10.1084/jem.20212532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma H, Kim AS, Kafai NM, Earnest JT, Shah AP, Case JB, Basore K, Gilliland TC, Sun C, Nelson CA, et al. (2020). LDLRAD3 is a receptor for Venezuelan equine encephalitis virus. Nature 588, 308–314. 10.1038/s41586-020-2915-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ranganathan S, Noyes NC, Migliorini M, Winkles JA, Battey FD,Hyman BT, Smith E, Yepes M, Mikhailenko I, and Strickland DK (2011). LRAD3, a novel low-density lipoprotein receptor family member that modulates amyloid precursor protein trafficking. J. Neurosci. 31, 10836–10846. 10.1523/jneurosci.5065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noyes NC, Hampton B, Migliorini M, and Strickland DK (2016). Regulation of Itch and Nedd4 E3 Ligase Activity and Degradation by LRAD3. Biochemistry 55, 1204–1213. 10.1021/acs.biochem.5b01218. [DOI] [PubMed] [Google Scholar]
- 45.Basore K, Ma H, Kafai NM, Mackin S, Kim AS, Nelson CA, Diamond MS, and Fremont DH (2021). Structure of Venezuelan equine encephalitis virus in complex with the LDLRAD3 receptor. Nature 598, 672–676. 10.1038/s41586-021-03963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma B, Huang C, Ma J, Xiang Y, and Zhang X (2021). Structure of Venezuelan equine encephalitis virus with its receptor LDLRAD3. Nature 598, 677–681. 10.1038/s41586-021-03909-1. [DOI] [PubMed] [Google Scholar]
- 47.de la Monte S, Castro F, Bonilla NJ, Gaskin de Urdaneta A, and Hutchins GM (1985). The systemic pathology of Venezuelan equine encephalitis virus infection in humans. Am. J. Trop. Med. Hyg. 34, 194–202. 10.4269/ajtmh.1985.34.194. [DOI] [PubMed] [Google Scholar]
- 48.Bowen GS, Fashinell TR, Dean PB, and Gregg MB (1976). Clinical aspects of human Venezuelan equine encephalitis in Texas. Bull. Pan Am. Health Organ. 10, 46–57. [PubMed] [Google Scholar]
- 49.Quiroz E, Aguilar PV, Cisneros J, Tesh RB, and Weaver SC (2009). Venezuelan equine encephalitis in Panama: fatal endemic disease and genetic diversity of etiologic viral strains. PLoS Neglected Trop. Dis. 3, e472. 10.1371/journal.pntd.0000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ronca SE, Dineley KT, and Paessler S (2016). Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 7, 959. 10.3389/fmicb.2016.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Charles PC, Trgovcich J, Davis NL, and Johnston RE (2001). Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 284, 190–202. 10.1006/viro.2001.0878. [DOI] [PubMed] [Google Scholar]
- 52.Jackson AC, SenGupta SK, and Smith JF (1991). Pathogenesis of Venezuelan equine encephalitis virus infection in mice and hamsters. Vet. Pathol. 28, 410–418. 10.1177/030098589102800509. [DOI] [PubMed] [Google Scholar]
- 53.Grieder FB, Davis BK, Zhou XD, Chen SJ, Finkelman FD, and Gause WC (1997). Kinetics of cytokine expression and regulation of host protection following infection with molecularly cloned Venezuelan equine encephalitis virus. Virology 233, 302–312. 10.1006/viro.1997.8617. [DOI] [PubMed] [Google Scholar]
- 54.Ronca SE, Smith J, Koma T, Miller MM, Yun N, Dineley KT, and Paessler S (2017). Mouse model of neurological complications resulting from encephalitic alphavirus infection. Front. Microbiol. 8, 188. 10.3389/fmicb.2017.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charles PC, Walters E, Margolis F, and Johnston RE (1995). Mechanism of neuroinvasion of Venezuelan equine encephalitis virus in the mouse. Virology 208, 662–671. 10.1006/viro.1995.1197. [DOI] [PubMed] [Google Scholar]
- 56.Taylor A, Herrero LJ, Rudd PA, and Mahalingam S (2015). Mouse models of alphavirus-induced inflammatory disease. J. Gen. Virol. 96, 221–238. 10.1099/vir.0.071282-0. [DOI] [PubMed] [Google Scholar]
- 57.Reyna RA, and Weaver SC (2023). Sequelae and Animal Modeling of Encephalitic Alphavirus Infections. Viruses 15. 10.3390/v15020382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, and Frolov I (2007). The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 81, 2472–2484, JVI.02073–06 [pii]. 10.1128/JVI.02073-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gardner CL, Burke CW, Tesfay MZ, Glass PJ, Klimstra WB, and Ryman KD (2008). Eastern and Venezuelan equine encephalitis viruses differ in their ability to infect dendritic cells and macrophages: impact of altered cell tropism on pathogenesis. J. Virol. 82, 10634–10646. 10.1128/JVI.01323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grieder FB, Davis NL, Aronson JF, Charles PC, Sellon DC, Suzuki K, and Johnston RE (1995). Specific restrictions in the progression of Venezuelan equine encephalitis virus-induced disease resulting from single amino acid changes in the glycoproteins. Virology 206, 994–1006. 10.1006/viro.1995.1022. [DOI] [PubMed] [Google Scholar]
- 61.Cain MD, Salimi H, Gong Y, Yang L, Hamilton SL, Heffernan JR, Hou J, Miller MJ, and Klein RS (2017). Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 308, 118–130. 10.1016/j.jneuroim.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salimi H, Cain MD, Jiang X, Roth RA, Beatty WL, Sun C, Klimstra WB, Hou J, Klein RS, and Duggal NK (2020). Encephalitic Alphaviruses Exploit Caveola-Mediated Transcytosis at the Blood-Brain Barrier for Central Nervous System Entry. mBio 11, e02731–19-e02719. 10.1128/mBio.02731-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryzhikov AB, Tkacheva NV, Sergeev AN, and Ryabchikova EI (1991). Venezuelan equine encephalitis virus propagation in the olfactory tract of normal and immunized mice. Biomed. Sci. 2, 607–614. [PubMed] [Google Scholar]
- 64.Ryzhikov AB, Ryabchikova EI, Sergeev AN, and Tkacheva NV (1995). Spread of Venezuelan equine encephalitis virus in mice olfactory tract. Arch. Virol. 140, 2243–2254. 10.1007/bf01323243. [DOI] [PubMed] [Google Scholar]
- 65.Phillips AT, Rico AB, Stauft CB, Hammond SL, Aboellail TA, Tjalkens RB, and Olson KE (2016). Entry Sites of Venezuelan and Western Equine Encephalitis Viruses in the Mouse Central Nervous System following Peripheral Infection. J. Virol. 90, 5785–5796. 10.1128/JVI.03219-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schä fer A, Brooke CB, Whitmore AC, and Johnston RE (2011). The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J. Virol. 85, 10682–10690. 10.1128/jvi.05032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keogh B, Sheahan BJ, Atkins GJ, and Mills KHG (2003). Inhibition of matrix metalloproteinases ameliorates blood-brain barrier disruption and neuropathological lesions caused by avirulent Semliki Forest virus infection. Vet. Immunol. Immunopathol. 94, 185–190. 10.1016/s0165-2427(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 68.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, and Tsukita S (2003). Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 161, 653–660. 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schoneboom BA, Fultz MJ, Miller TH, McKinney LC, and Grieder FB (1999). Astrocytes as targets for Venezuelan equine encephalitis virus infection. J. Neurovirol. 5, 342–354. 10.3109/13550289909029475. [DOI] [PubMed] [Google Scholar]
- 70.Jackson AC, and Rossiter JP (1997). Apoptotic cell death is an important cause of neuronal injury in experimental Venezuelan equine encephalitis virus infection of mice. Acta Neuropathol. 93, 349–353. 10.1007/s004010050626. [DOI] [PubMed] [Google Scholar]
- 71.Kehn-Hall K, and Bradfute SB (2022). Understanding host responses to equine encephalitis virus infection: implications for therapeutic development. Expert Rev. Anti Infect. Ther. 20, 1551–1566. 10.1080/14787210.2022.2141224. [DOI] [PubMed] [Google Scholar]
- 72.Arrigo NC, Adams AP, and Weaver SC (2010). Evolutionary patterns of eastern equine encephalitis virus in North versus South America suggest ecological differences and taxonomic revision. J. Virol. 84, 1014–1025. 10.1128/jvi.01586-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gil LHVG, Magalhaes T, Santos BSAS, Oliveira LV, Oliveira-Filho EF, Cunha JLR, Fraiha ALS, Rocha BMM, Longo BC, Ecco R, et al. (2021). Active Circulation of Madariaga Virus, a Member of the Eastern Equine Encephalitis Virus Complex, in Northeast Brazil. Pathogens 10, 983. 10.3390/pathogens10080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clark LE, Clark SA, Lin C, Liu J, Coscia A, Nabel KG, Yang P,Neel DV, Lee H, Brusic V, et al. (2022). VLDLR and ApoER2 are receptors for multiple alphaviruses. Nature 602, 475–480. 10.1038/s41586-021-04326-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reynoso GV, Gordon DN, Kalia A, Aguilar CC, Malo CS, Aleshnick M, Dowd KA, Cherry CR, Shannon JP, Vrba SM, et al. (2023). Zika virus spreads through infection of lymph node-resident macrophages. Cell Rep. 42, 112126. 10.1016/j.celrep.2023.112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reynoso GV, Weisberg AS, Shannon JP, McManus DT, Shores L, Americo JL, Stan RV, Yewdell JW, and Hickman HD (2019). Lymph node conduits transport virions for rapid T cell activation. Nat. Immunol. 20, 602–612. 10.1038/s41590-019-0342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, and von Andrian UH (2010). Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature 465, 1079–1083. 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, et al. (2007). Subcapsular sinus macrophages in lymph nodes clear lymphborne viruses and present them to antiviral B cells. Nature 450, 110–114. [DOI] [PubMed] [Google Scholar]
- 79.Dittmer M, Young A, O’Hagan T, Eleftheriadis G, Bankhead P, Dombrowski Y, Medina RJ, and Fitzgerald DC (2018). Characterization of a murine mixed neuron-glia model and cellular responses to regulatory T cell-derived factors. Mol. Brain 11, 25. 10.1186/s13041-018-0367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang J, Cheng X, Shen J, Xie B, Zhao X, Zhang Z, Cao Q, Shen Y, and Qiu M (2016). A Novel Approach for Amplification and Purification of Mouse Oligodendrocyte Progenitor Cells. Front. Cell. Neurosci. 10, 203. 10.3389/fncel.2016.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang H, Xu L, Lai C, Hou K, Chen J, Guo Y, Sambangi A, Swaminathan S, Xie C, Wu Z, and Chen G (2021). Region-specific distribution of Olig2-expressing astrocytes in adult mouse brain and spinal cord. Mol. Brain 14, 36. 10.1186/s13041-021-00747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marshall CAG, Novitch BG, and Goldman JE (2005). Olig2 Directs Astrocyte and Oligodendrocyte Formation in Postnatal Subventricular Zone Cells. J. Neurosci. 25, 7289–7298. 10.1523/jneurosci.1924-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17. 10.1016/j.cell.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 84.Fei L, Chen H, Ma L, E, W., Wang, R., Fang, X., Zhou, Z., Sun, H., Wang, J., Jiang, M., et al. (2022). Systematic identification of cell-fate regulatory programs using a single-cell atlas of mouse development. Nat. Genet. 54, 1051–1061. 10.1038/s41588-022-01118-8. [DOI] [PubMed] [Google Scholar]
- 85.Wang R, Zhang P, Wang J, Ma L, E W, Suo S, Jiang M, Li J, Chen H, Sun H, et al. (2023). Construction of a cross-species cell landscape at single-cell level. Nucleic Acids Res. 51, 501–516. 10.1093/nar/gkac633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karlsson M, Zhang C, Méar L, Zhong W, Digre A, Katona B, Sjöstedt E, Butler L, Odeberg J, Dusart P, et al. (2021). A single-cell type transcriptomics map of human tissues. Sci. Adv. 7, eabh2169. 10.1126/sciadv.abh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 88.Johnson KM, Shelokov A, Peralta PH, Dammin GJ, and Young NA (1968). Recovery of Venezuelan equine encephalomyelitis virus in Panama. A fatal case in man. Am. J. Trop. Med. Hyg. 17, 432–440. 10.4269/ajtmh.1968.17.432. [DOI] [PubMed] [Google Scholar]
- 89.Steele KE, and Twenhafel NA (2010). REVIEW PAPER:Pathology of Animal Models of Alphavirus Encephalitis. Vet. Pathol. 47, 790–805. 10.1177/0300985810372508. [DOI] [PubMed] [Google Scholar]
- 90.MacDonald GH, and Johnston RE (2000). Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J. Virol. 74, 914–922. 10.1128/jvi.74.2.914-922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nishimoto KP, Laust AK, Wang K, Kamrud KI, Hubby B, Smith JF, and Nelson EL (2007). Restricted and Selective Tropism of a Venezuelan Equine Encephalitis Virus-Derived Replicon Vector for Human Dendritic Cells. Viral Immunol. 20, 88–104. 10.1089/vim.2006.0090. [DOI] [PubMed] [Google Scholar]
- 92.Tonkin DR, Whitmore A, Johnston RE, and Barro M (2012). Infected dendritic cells are sufficient to mediate the adjuvant activity generated by Venezuelan equine encephalitis virus replicon particles. Vaccine 30, 4532–4542. 10.1016/j.vaccine.2012.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Konopka JL, Thompson JM, Whitmore AC, Webb DL, and Johnston RE (2009). Acute infection with venezuelan equine encephalitis virus replicon particles catalyzes a systemic antiviral state and protects from lethal virus challenge. J. Virol. 83, 12432–12442. 10.1128/JVI.00564-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, et al. (2018). Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557, 570–574. 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ludwig GV, Kondig JP, and Smith JF (1996). A putative receptor for Venezuelan equine encephalitis virus from mosquito cells. J. Virol. 70, 5592–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malygin AA, Bondarenko EI, Ivanisenko VA, Protopopova EV, Karpova GG, and Loktev VB (2009). C-terminal fragment of human laminin-binding protein contains a receptor domain for Venezuelan equine encephalitis and tick-borne encephalitis viruses. Biochemistry 74, 1328–1336. 10.1134/S0006297909120050. [DOI] [PubMed] [Google Scholar]
- 97.Klimstra WB, Nangle EM, Smith MS, Yurochko AD, and Ryman KD (2003). DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J. Virol. 77, 12022–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bernard KA, Klimstra WB, and Johnston RE (2000). Mutations in theE2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 276, 93–103. 10.1006/viro.2000.0546. [DOI] [PubMed] [Google Scholar]
- 99.Kafai NM, Diamond MS, and Fox JM (2022). Distinct Cellular Tropism and Immune Responses to Alphavirus Infection. Annu. Rev. Immunol. 40, 615–649. 10.1146/annurev-immunol-101220-014952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gorelkin L (1973). Venezuelan equine encephalomyelitis in an adult animal host. An electron microscopic study. Am. J. Pathol. 73, 425–442. [PMC free article] [PubMed] [Google Scholar]
- 101.Jakovcevski I, and Zecevic N (2005). Olig transcription factors are expressed in oligodendrocyte and neuronal cells in human fetal CNS. J. Neurosci. 25, 10064–10073. 10.1523/jneurosci.2324-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu R, Brawner AT, Li S, Liu JJ, Kim H, Xue H, Pang ZP, Kim WY, Hart RP, Liu Y, and Jiang P (2019). OLIG2 Drives Abnormal Neurodevelopmental Phenotypes in Human iPSC-Based Organoid and Chimeric Mouse Models of Down Syndrome. Cell Stem Cell 24, 908–926.e8. 10.1016/j.stem.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Szu J, Wojcinski A, Jiang P, and Kesari S (2021). Impact of the Olig Family on Neurodevelopmental Disorders. Front. Neurosci. 15, 659601. 10.3389/fnins.2021.659601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roehrig JT, and Mathews JH (1985). The neutralization site on the E2 glycoprotein of Venezuelan equine encephalomyelitis (TC-83) virus is composed of multiple conformationally stable epitopes. Virology 142, 347–356. [DOI] [PubMed] [Google Scholar]
- 105.Anishchenko M, Paessler S, Greene IP, Aguilar PV, Carrara A-S, and Weaver SC (2004). Generation and characterization of closely related epizootic and enzootic infectious cDNA clones for studying interferon sensitivity and emergence mechanisms of venezuelan equine encephalitis virus. J. Virol. 78, 1–8. 10.1128/JVI.78.1.1-8.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, Takeda K, et al. (2014). A viral RNA structural element alters host recognition of nonself RNA. Science (New York, N.Y.) 343, 783–787. 10.1126/science.1248465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun C, Gardner CL, Watson AM, Ryman KD, and Klimstra WB (2014). Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J. Virol. 88, 2035–2046. 10.1128/JVI.02990-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Paessler S, Fayzulin RZ, Anishchenko M, Greene IP, Weaver SC, and Frolov I (2003). Recombinant Sindbis/Venezuelan equine encephalitis virus Is highly attenuated and immunogenic. J. Virol. 77, 9278–9286. 10.1128/JVI.77.17.9278-9286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Casals J (1964). Antigenic variants of Eastern equine encephalitis virus. J Exp Med 119, 547–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schlüter D, Meyer T, Strack A, Reiter S, Kretschmar M, Wiestler OD, Hof H, and Deckert M (2001). Regulation of microglia by CD4+ and CD8+ T cells: selective analysis in CD45-congenic normal and Toxoplasma gondii-infected bone marrow chimeras. Brain Pathol. 11, 44–55. 10.1111/j.1750-3639.2001.tb00380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and from the corresponding author upon request. This paper does not include original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.