

SUMMARY:
Changes in the function of inhibitory interneurons (INs) during cortical development could contribute to the pathophysiology of neurodevelopmental disorders. Using all-optical in vivo approaches, we find that parvalbumin (PV) IN and their immature precursors are hypoactive and transiently decoupled from excitatory neurons in postnatal mouse somatosensory cortex (S1) of Fmr1 KO mice, a model of Fragile X Syndrome (FXS). This leads to a loss of parvalbumin INs (PV-INs) in both mice and humans with FXS. Increasing the activity of future PV-INs in neonatal Fmr1 KO mice restores PV density and ameliorates transcriptional dysregulation in S1, but not circuit dysfunction. Critically, administering an allosteric modulator of Kv3.1 channels after the S1 critical period does rescue circuit dynamics and tactile defensiveness. Symptoms in FXS and related disorders could be mitigated by targeting PV-INs.
Keywords: In vivo calcium imaging, Autism spectrum disorders, Intellectual disability, Interneuron, Kv3.1, Medial ganglionic eminence, Parvalbumin, Ribotag, RNAseq, Somatosensory cortex, Transcriptomics, Two-photon
Graphical Abstract
In brief:
Kourdougli et al. use all-optical in vivo approaches to show that parvalbumin interneurons are hypoactive and decoupled from excitatory partners in the developing neocortex of Fragile X Syndrome model mice. Restoring activity of remaining PV cells after the second postnatal week (not earlier) ameliorates sensory hypersensitivity in the mouse model.
INTRODUCTION:
Neurodevelopmental disorders (NDDs) arise due to changes in developmental trajectories of neurons during the early stages of circuit assembly in the brain. Although symptoms of NDDs, such as intellectual disability and autism, are first recognized in the toddler stage, circuit differences are likely present at birth and may begin even earlier.1 From a therapeutic perspective identifying the earliest circuit changes in NDDs is critical because early interventions are more likely to redirect the trajectory of neural development before it is irreversibly changed as a consequence of genetic and/or environmental factors.
Differences in GABAergic inhibition and excitability have been implicated in the origins of NDDs and autism, and proposed as targets for therapy.2-5 However, the prevalent notion that an imbalance in excitatory and inhibitory signaling is associated with NDDs is principally based on observations in adulthood. In the last decade, as our understanding of cortical development grew significantly, there has been increased awareness about the important developmental role of inhibitory interneurons (INs) in shaping neuronal circuits.6,7 The typical density, function, and integration of INs into cortical networks all depend on genetic and activity-dependent programs.8 Deviations from the usual trajectory of these developmental programs in NDDs could have an impact on functional circuit assembly.4,5 For example, hypofunction of cortical INs has been described in multiple models of autism and other psychiatric conditions,4,9-11 but the nature of GABAergic population dynamics throughout neonatal development in NDDs remains an unexplored territory.
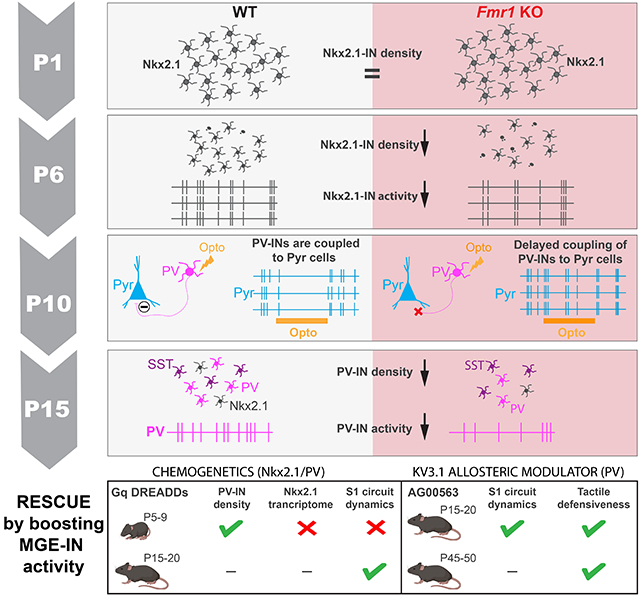
To investigate this, we focused on Fragile X Syndrome (FXS) because it is the most common single gene cause of intellectual disability and autism,12 and because hypoactivity of fast-spiking PV-INs has been observed repeatedly in Fmr1 knockout mice (Fmr1 KO, referring to both male Fmr1−/y and females Fmr1−/− mice) the principal animal model of FXS.13-16 Additionally, reciprocal connectivity of fast-spiking INs with excitatory cells is altered in layer (L) 4 of S1 in developing Fmr1 KO mice.15,17,18 In this study, we examined the developmental origins of PV-IN hypoactivity in L2/3 of S1 in vivo because we previously identified early postnatal circuit changes in Fmr1 mice (excessive network synchrony and reduced adaptation to repetitive tactile stimulation) that could be associated with reduced PV-IN activity.19,20 We show that PV-INs and their immature counterparts from the medial ganglionic eminence (MGE), which express the transcription factor Nkx2.1, are hypoactive in S1 as early as postnatal day (P) 6 and fail to modulate excitatory neurons in Fmr1 mice before P15. Their density is also reduced from early postnatal development to adulthood in Fmr1 KO mice and, remarkably, also in humans with FXS. Interestingly, an early chemogenetic intervention to increase Nkx2.1-IN firing in Fmr1 KO mice at P5-P9 failed to restore circuit dysfunction despite partially correcting the FXS S1 transcriptome, whereas a delayed intervention at P15-P20 (post S1 critical period) was more successful. Finally, boosting PV-IN activity more globally with a Kv3.1 channel modulator at P15-P20 significantly improved both circuit and behavioral sensory phenotypes of Fmr1 KO mice. Thus, circuit changes in FXS (and perhaps in other NDDs) can be reversed by targeting PV-INs, but the timing of circuit interventions may be critical.
RESULTS:
Reduced activity of cortical PV- INs and MGE-derived INs in early postnatal Fmr1 KO mice.
Several in vivo studies have shown that the activity of cortical PV-INs is reduced in adult Fmr1 KO mice.14,21 Moreover, we previously discovered pronounced circuit changes in S1 during the critical period at P14-P16 that could be due to reduced inhibition, including fewer whisker-responsive excitatory pyramidal (Pyr) neurons and lack of neuronal adaptation to repeated stimulation. To assess whether PV-INs were also hypoactive in S1 earlier in development, we used in vivo two-photon calcium imaging at P15 to record from them in S1 of PV-Cre;WT or Fmr1 KO mice injected with AAV1-CAG-Flex-GCaMP6s at P10 (Fig 1A-C). This approach led to GCaMP6s expression in ~80% of PV-INs in L2/3 of the transfected region (Fig. S1A,B). We found that both spontaneous and whisker-evoked activity of PV-INs were significantly reduced by ~35% in Fmr1 KO mice (n=7) compared to WT mice (n=6) (Fig. 1D-F). The percentage of PV-INs that were spontaneously active was also significantly reduced (Fig. 1E), though the percentage of whisker-responsive PV-INs was not different from controls (Fig. 1F).
Figure 1: PV-INs and their MGE-derived immature INs are hypoactive in S1 of developing Fmr1 KO mice.

A. Cartoon of experimental design.
B. Example field of view of PV-INs expressing AAV1-flex-GCaMP6s in S1 cortex of PV-Cre mice (scale=25μm).
C. Representative traces of PV-IN calcium transients in WT and Fmr1 KO mice. Whisker stimulation (blue bars, 1s at 10 Hz, 3 s I.S.I).
D. Mean Z-scores of PV-INs at P15 are significantly lower in Fmr1 KO than in WT mice. In panels D-F and J-L, symbols represent individual mice (sample size in parenthesis, females=circles, males=squares. (spontaneous: 1.98±0.15 for WT vs. 1.28±0.17 for Fmr1 KO; whisker-evoked: 2.49±0.29 for WT vs. 1.61±0.21 for Fmr1 KO, respectively; p=0.008 and p=0.046, M-W t-test, n=6 WT and n=7 Fmr1 KO mice).
E. Percentage of active PV-INs in WT and Fmr1 KO mice (spontaneous: 98.9±0.8% for WT vs. 74.2±8.9% for Fmr1 KO; p=0.049, whisker-evoked: 95.5±3.2% for WT vs. 83.6±6.1% for Fmr1 KO, p=0.444, M-W t-test).
F. Percentage of stimulus-locked PV-INs (61.4±6.9% for WT vs. 45.2±7.9% for Fmr1 KO; p=0.126, M-W t-test).
G. Experimental design for in vivo recordings at P6.
H. Example field of view of Nkx2.1-INs expressing AAV1-flex-GcaMP6s in S1 cortex Nkx2.1-Cre mice (scale =25μm).
I. Example calcium traces of Nkx2.1-INs in WT and Fmr1 KO mice. Inset shows expanded traces for representative synchronous network events.
J. The percentage of active Nkx2.1-INs at P6 was significantly lower in Fmr1 KO mice than in WT controls. (99.2±0.6% for WT and 89.8±3.5% for Fmr1 KO, n=9 and 8, respectively; p=0.026, MW t-test).
K. The frequency of synchronous network events for Nkx2.1-INs was significantly lower in Fmr1 KO mice. (0.32±0.09 events per min for WT vs. 0.11±0.04 for Fmr1 KO, n=9 WT and n=8 Fmr1 KO, p=1.4x10−3, MW t-test).
L. The amplitude of calcium transient events of Nkx2.1-INs was not different between genotypes (15.5±1.8 for WT vs. 16.2±3.7 for Fmr1 KO, p=0.541, MW t-test).
Somatostatin-expressing INs (SST-INs) and PV-INs both arise from a common precursor in the MGE.22-24 Therefore, we also recorded the activity of SST-INs in vivo at P15 in Sst-FlpO+/− mice (Fig. S2A,B). We did not find significant differences between genotypes in either spontaneous or whisker-evoked activity, although the percentage of whisker-responsive SST-INs was lower in Fmr1 KO mice (Fig. S2C-E). Thus, PV-INs in S1, but not SST-INs, manifest significant hypoactivity in two-week-old Fmr1 KO mice.
During the period that spans from the establishment of barrels to the closure of S1 critical period, GABAergic assemblies display synchronous dynamics that help shape developing circuits.25,26 We next investigated whether differences in MGE-IN activity during the first postnatal week contributes to previously reported signs of early cortical circuit dysfunction.19,27,28Cortical PV-INs do not express PVALB before P10,24,29,30 but their immature precursors from the MGE can be identified through their expression of the transcription factor Nkx2.1. We used in vivo calcium imaging to record from MGE-INs at P6 in Nkx2.1-Cre mice injected with AAV1-CAG-Flex-GCaMP6s into S1 at P1 (Fig. 1G-I), which led to 43% of Nkx2.1-INs expressing the indicator in the transfected region (Fig. S1C,D); a small fraction of Nkx2.1-INs also express SST at P6 (Fig. S1E-H). Early cortical network activity is dominated by large and infrequent synchronous network events that propagate as waves.31,32 We confirmed that MGE-INs participated in synchronous cortical activity, as previously reported.33 However, we found a significantly lower proportion of active Nkx2.1-INs and a lower frequency of synchronous MGE-IN network events in Fmr1 KO mice (n=8) compared to WT controls (n=9), even though the amplitude of these events was similar (Fig. 1J-L). Hence, Nkx2.1-INs (which give rise to PV- and SST-INs) are hypoactive at P6 and less likely to participate in synchronous network activity.
Future PV-INs within the Nkx2.1-IN population fail to modulate Pyr cells in neonatal Fmr1 KO mice.
The hypoactivity of Nkx2.1-INs could account for the previously reported hypersynchrony and hyperactivity of Pyr neurons in early postnatal Fmr1 KO mice.19,27,28 Indeed, the maturation of cortical networks depends on the proper integration and function of GABAergic INs7,34 and co-activation of MGE-INs and Pyr cells during the first postnatal week is thought to restrict the spread of spontaneous synchronous network events.33,35 However, even though GABAergic perisomatic axons are observed in the first postnatal days, functional synaptic inhibition in S1 does not emerge until P8-10.36,37 Because fast-spiking INs in S1 of Fmr1 KO mice seem significantly immature relative to those in WT controls,15 we sought to determine whether or not Nkx2.1-INs in developing Fmr1 KO mice are properly integrated into the S1 network. We used an all-optical two-photon optogenetic approach in vivo38,39 to specifically increase the firing of putative future PV-INs within this population, since SST-INs are not hypoactive at P15 (Fig. S2). Because SST-INs and PV-INs arise from a common MGE-derived precursor, they cannot be distinguished in Nkx2.1-Cre mice at P6. However, SST-INs begin to express SST before PV-INs express PVALB;22-24 the SST mRNA is active even during embryonic development.40 Thus, we used an intersectional genetic strategy 41 to optogenetically activate putative future PV-INs at P10 without activating SST-INs. We generated Nkx2.1-Cre;Sst-FlpO mice and injected rAAV8-nEF1-Con/Foff-ChRmine-oScarlet into S1 at P1 (Fig. 2A) allowing expression of ChRmine exclusively in Nkx2.1-Cre+/Sst-FlpO− cells (Fig. S3A-D). This allowed us to record from INs and Pyr cells simultaneously at P10 while we delivered pulses of laser light (1 s pulses at 1,040 nm every 3 s) (Fig. 2C,D, Video S1). Although laser stimulation significantly drove activity of Nkx2.1+;Sst−-INs in both WT and Fmr1 KO mice (Fig. 2E), it only reduced Pyr cell activity in WT mice, as manifested by a lower frequency of their synchronous events (Fig. 2F), as well as a lower proportion of active cells per event (Fig. S3E). Laser stimulation also significantly decorrelated activity in both WT mice and Fmr1 KO mice, but the magnitude of the effect was negligible in the latter (Fig. 2G; Fig. S3F). There was no effect on network activity in control mice that did not express ChRmine (Fig. S3G). Incidentally, L2/3 Pyr cell activity at P10 was hypersynchronous in Fmr1 KO mice (Fig. 2G; Fig. S3F, Fig. S4), as previously reported.19,28 Taken together, these optogenetic results show that putative future PV-INs in Fmr1 KO mice at P10 are functionally decoupled from Pyr cells and fail to properly modulate network activity.
Figure 2: Nkx2.1-INs form a weak functional network with Pyr cells in neonatal Fmr1 KO mice.

A. Experimental design for optogenetic experiments.
B. Example field of view of Pyr cells expressing GCaMP6s in Nkx2.1-Cre;SST-FlpO mice at P10.
C. Representative calcium traces for 5 presumed Nkx2.1+/Sst−-INs (magenta) and 4 Pyr cells (black).
D. Raster plot of neuronal activity in a representative WT mouse.
E. Mean z-score of activity in Nkx2.1+/SST−-INs before (pre) and during optogenetic stimulation (laser). Each line represents an individual mouse (mean normalized z-score increased by 63.7%±25.5 for WT, p=0.015, and 79.9%±15.3, p=0.031, for Fmr1 KO mice upon laser-on stimulation; Wilcoxon matched-pairs signed rank test; n=6 and 5, respectively).
F. Mean frequency of Pyr cell calcium transients was significantly lower during optogenetic stimulation in WT mice but was unchanged in Fmr1 KO mice. (0.63±0.09 events.min−1 pre vs. 0.38±0.11 with laser; p=0.029 in WT; 1.06±0.21 events.min−1 pre vs. 1.06±0.43 with laser for Fmr1 KO, p> 0.99; two-way ANOVA, post-hoc Tukey).
G. Pair-wise correlation coefficients of Pyr cells were significantly modulated by optogenetic stimulation in both WT and Fmr1 KO mice but the magnitude of the effect was greater in WT (mean Corr. Coeff. WT: 0.52±0.0035 for pre vs. 0.45±0.0044 for laser; p=2.2x10−16; Fmr1 KO: 0.66±0.0024 for pre vs. 0.64±0.0025 for laser, p=3.3x10−9; and p<0.001 for WT pre vs. Fmr1 KO pre; Kolmogorov-Smirnov test).
What are the consequences of having Nkx2.1-INs that are hypoactive and largely decoupled from Pyr cells during neonatal cortical development? It is known that a wave of programmed cell death affects MGE-INs from ~P5 to P10 in mice,42 which is regulated by cortical network activity33,43. Decreasing or increasing the activity of INs in the neonatal period results in lower or higher density of INs in juvenile mice, respectively.44,45 We considered the possibility that the significant hypoactivity of Nkx2.1-INs in Fmr1 KO mice might lead to their excessive cell death and, eventually, a reduced density of PV-INs. Previous reports of reduced PV-IN density in adult S1 and primary auditory cortex of Fmr1 KO mice had used PVALB immunoreactivity.46,47 However, because PVALB expression correlates with PV-IN activity levels,48 we chose instead to quantify their density in PV-Cre;tdTom mice (see Methods). We found a drastic reduction in the density of PV-Cre;tdTom+ in Fmr1 KO mice at P15 throughout the cortex (Fig. S5A), including a 65% reduction in S1 across all layers (Fig. 3A,B). A significant reduction in PV-IN density was also observed in S1 of 9-10 month-old Fmr1 KO mice (Fig. S5B,C), suggesting their loss is permanent. Interestingly, the density of SST and calretinin-INs was higher, while calbindin-IN density was similar in adult Fmr1 KO mice (Fig. S6A-G), consistent with the large overlap between these two IN subclasses.49
Figure 3: Reduced density of PV-INs and their MGE-derived precursors in mice and humans with FXS.

A. Example images from coronal sections through barrel field of S1BF from PV-Cre;tdTom mice at P15. (DAPI in blue). Scale=5μm.
B. Mean density of PV-tdTom+ INs is significantly lower in Fmr1 KO mice overall (top) and across cortical layers (bottom). Sample size in parenthesis, diamonds indicate the sex was not recorded. (total density: 154.1±8.0 cells per mm2 for WT vs. 58.5±15.1 for Fmr1 KO; n=9 and n=8, respectively; p=0.0007, unpaired t-test; L2/3: 56.1±9.4 vs 12.7±4.5, p=0.018; L4: 322.0±57.4 vs. 71.7±21.1, p=0.016; L5/6: 227.3±41.5 vs. 68.7, p=0.018 two-way ANOVA with post-hoc Holm-Sidak test).
C. Example images from coronal sections through BA3 from neurotypical (NT) and FXS human tissue.
D. Mean density of PVALB+ cells in BA3 is significantly lower in FXS cases overall (top) and in supragranular cortical layers (bottom) (170.6±26.0 PVALB+ cells per bin for NT vs. 98.1±17.4 for FXS cases, n=8 cases each; p=0.038, unpaired t-test; supragranular layer: 83.6±18.6 for neurotypical vs 35.5±7.0, p=0.0294; infragranular: 87.0±23.2 vs. 62.6±14.3, p=0.386, unpaired t-tests). See also Table S1.
E. Coronal sections through S1 showing Nkx2.1-Cre;tdTom+ INs (red) in WT and Fmr1 KO mice at P1 and P10 (DAPI: blue). Scale=100 μm.
F. Mean density of Nkx2.1;tdTom+ INs is lower in Fmr1 KO mice at P6 and P10, but not at P1. (P1: 3,408±354 cells/mm2 for WT vs. 3,472±548 for Fmr1 KO, p=0.907; P6: 10,200±1,027 cells/mm2 for WT vs. 8,035±860 for Fmr1 KO, p=0.056; P10: 8,633±318 for WT vs. 6,650±279 for Fmr1 KO, p=0.029; n=6 per genotype at P1 and P10, n=7 at P6, two-way ANOVA with post-hoc Holm-Sidak).
To determine whether this phenotype is perhaps unique to Fmr1 KO mice, we also quantified the density of three major types of INs in human autopsy material from adult FXS cases and age-matched neurotypical (NT) controls (n=8 each) using immunohistochemistry (see Methods). Remarkably, we found that the density of PVALB-expressing INs in Brodmann area 3 (equivalent to S1 in mice) was significantly lower (by 42%) in FXS cases compared to NT controls (Fig. 3C,D). In contrast, the density of calretinin-expressing and calbindin-expressing IN subclasses was not different between FXS cases and NT controls (Fig. S6H,I). This suggests that PV-IN density in S1 is similarly reduced in both Fmr1 KO mice and humans with FXS.
We also observed a reduced density of Nkx2.1-Cre;tdTom+ cells in Fmr1 KO mice at both P6 and P10 (Fig. 3E), but not at P1, suggesting that the lower density of PV-INs is not caused by reduced migration and/or neurogenesis of MGE-INs. Next, we quantified the expression of the cell death marker cleaved Caspase-3 within Nkx2.1Cre;tdTom+-INs and found an increased density of apoptotic MGE-INs at P6 in Fmr1 KO mice (Fig. S7A,B). To further confirm that loss of MGE-INs was due to apoptosis, we performed longitudinal in vivo imaging of Nkx2.1-Cre;tdTom+ IN at P6 and P7 in Fmr1 KO mice. We found several examples of pyknotic Nkx2.1-Cre;tdTom+ INs, and evidence that they disappeared within the short interval between imaging sessions (Fig. S7C). Considering findings from previous developmental studies,33,43 and the fact that MGE-INs complete their migration by the end of the first postnatal week,50 our data suggest that the reduced density of Nkx2.1-INs at P6-P10 is due to apoptosis, and that those cells are irreversibly lost (Fig. S7D).
Activating of Nkx2.1-INs in neonatal Fmr1 KO mice partially rescues PV cell density.
Our findings thus far are consistent with a model in which hypoactivity of the Nkx2.1-IN population and their uncoupling from Pyr cells in neonatal Fmr1 KO mice results in excess developmental cell death of future PV-INs.43 We reasoned that artificially increasing the activity of Nkx2.1-INs during the apoptosis window (P5-P10) in Fmr1 KO mice might rescue the density of PV-INs and other circuit phenotypes.20 To test this, we used Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) and injected rAAV5-DIO-hM3Dq-mCherry (or a rAAV5-DIO-mCherry control virus) in Nkx2.1-Cre;Fmr1 KO mice at P1 (Fig. 4A) and performed immunohistochemistry for PVALB at P15 (Fig. 4B). This virus approach led to only a small proportion (25%) of PVALB+ cells expressing the Gq-DREADD (Fig. S8A,B). Next, we administered a DREADD agonist, compound 21 (C21; 1 mg/kg, s.c., twice daily), from P5 to P9 (Fig. 4A).51 We found that the density of PVALB-expressing INs in L2/3 of Fmr1 KO;hM3Dq mice at P15 was fully restored to WT levels, but remained significantly below WT levels in Fmr1 KO;mCherry controls treated with C21 (Fig. 4B,C). The effect of DREADDs on PVALB density was less pronounced in deeper layers (Fig. S8C). The fact that increasing MGE-IN activity can prevent their death in Fmr1 KO mice further confirms our theory that many MGE-INs eventually die because of their hypoactivity.
Figure 4: Chronic chemogenetic activation of Nkx2.1-INs in S1 restores PV-IN density in L2/3 of neonatal Fmr1 KO mice.

A. Experimental design for chronic chemogenetic activation of Nkx2.1-INs in Fmr1 KO mice.
B. Representative images of PVALB immunostaining. Scale=5μm
C. Quantification of total PVALB+ cell density at P15 in S1BF. (160.9±6.5 cells/mm2, 87.4±5.5, and 118.6±9.0, n=6, 7, and 8, respectively, two-way ANOVA with post-hoc Holm-Sidak).
Neonatal chemogenetic activation of Nkx2.1-INs partially rescues the global cortical transcriptome but further derails the Nkx2.1-specific translatome.
Having restored the density of PV-INs to WT levels by boosting Nkx2.1-IN activity with Gq-DREADDS in neonatal Fmr1 KO mice, we hypothesized that this strategy might have been sufficient to restore the developmental trajectory of the entire local cortical circuit. To test this, we first investigated transcriptional differences in S1 between WT and Fmr1 KO mice, and whether expressing Gq-DREADDs in Nkx2.1-INs might reduce those differences. Previous studies in Fmr1 KO mice had shown that loss of Fragile X messenger ribonucleoprotein (FMRP), which is translational suppressor of key proteins involved in brain maturation and plasticity,52-54 causes transcriptional dysregulation in the adult hippocampus and cerebellum.55-57 But to date, there are no mRNA sequencing (RNAseq) studies specific to MGE-INs in the neocortex during early postnatal development. Moreover, neuronal activity is known to regulate multiple aspects of IN development, including their transcriptional program.34,58-60 Thus, the changes in the activity of both MGE-INs and Pyr cells of neonatal Fmr1 KO mice (Figs. 1-2) are likely to impact transcription both globally in the cortex but also specifically within Nkx2.1-INs. Comparing the S1 cortical transcriptome of WT and Fmr1 KO mice at P15 would also help us uncover potential molecular mechanisms underlying MGE-IN hypofunction.
We used a Ribotag RNAseq approach,61 which allowed us to assess both the global transcriptome in S1 and the Nkx2-1-IN-specific translatome (see Methods).62 First, we generated Nkx2.1-Cre;Rpl22HA/− [WT or Fmr1 KO] triple transgenic mice and virally expressed hM3Dq (or mCherry) in Nkx2.1-INs from P1 onward (Fig. 5A,B). We identified 2,898 differentially expressed (DE) genes in the bulk RNA samples (before RiboTag pulldown) of Fmr1 KO;mCherry mice (n=8 mice) compared to WT;mCherry mice (n=7 mice; Fig. 5C). Gene ontology (GO) analysis (see Methods) revealed an enrichment in genes involved in Synapse organization and Neuronal apoptosis among other categories (Fig. 5D). Notably, we found that several MGE-INs expressing genes (or transcription factors)60,63,64 were significantly downregulated in Fmr1 KO mice, including several PV-enriched genes (e.g., Pvalb, Bcan, Syt2, Kcnab3), while other MGE-IN genes were upregulated, including some SST-enriched genes (Sst, Pcdh18) (Fig. S9A).
Figure 5: Bulk transcriptome and Nkx2.1-IN specific translatome is altered in P15 Fmr1 KO mice.

A. Experimental design for transcriptomic analysis at P15.
B. Cartoon of Ribotag approach for RNA-seq in S1 at P15.
C. Volcano plot of differentially expressed (DE) genes in the bulk RNA of Fmr1 KO-mCherry mice (n=8) compared to WT-mCherry (n=7).
D. Top 10 ‘GO terms’ of the bulk transcriptome in Fmr1 KO-mCherry mice using the biological process package (see Methods).
E. Number of DE genes in Fmr1 KO-mCherry mice within the bulk transcriptome dataset that are known targets of FMRP, based on previously published database (see Methods). Heatmaps represent the changes of expression as Z-scores among the top 25 upregulated and downregulated genes.
F. Volcano plot of DE genes in the Nkx2.1-IN-specific translatome of Fmr1 KO-mCherry mice (n=8) compared to WT-mCherry control (n=3).
G. Top 10 ‘GO terms’ in Nkx2.1-IN-specific translatome in Fmr1 KO-mCherry mice.
H. Number of DE genes in Fmr1 KO-mCherry mice within the Nkx2.1-specific translatome dataset that are known targets of FMRP (shown as in panel E).
We also compared the DE genes from the bulk transcriptome of P15 Fmr1 KO-mCherry mice to a list of genes that are bound to FMRP (presumably regulated by it) in developing or adult mice.65,66 Strikingly, 266 of the DE genes were known FMRP targets (Fig. 5E), which implies that in the absence of FMRP, its own mRNA targets are transcriptionally dysregulated from early developmental stages. Furthermore, our list of DE genes from the bulk cortical transcriptome also partially overlaps with previously published lists of DE genes in the hippocampus and cerebellum of adult Fmr1 KO mice (Fig. S9B).55,56
We next analyzed the Nkx2.1-specific translatome (RiboTag ‘pull-down’) and identified 2,039 DE genes in Fmr1 KO;mCherry mice, including down-regulated Pvalb, Pink1, and Gabra2 (Fig. 5F). GO analysis revealed an enrichment of upregulated genes in Synapse organization, Axonogenesis, and Neural migration, and downregulated genes in the Lipid metabolism and Neuron proliferation/morphogenesis (Fig. 5G). Furthermore, we found that several Neuronal apoptosis DE genes in the Nkx2.1 translatome (Fig. S10B), which may relate to the excess death of MGE-INs (Fig. 3, Fig. S7). Additionally, 279 of the DE genes in the Nkx2.1 translatome are FMRP targets (Fig. 5H), and most of these (242/279) are upregulated in Fmr1 KO mice. This again highlights the fact that loss of FMRP has hereto unrecognized consequences on Nkx2.1-IN gene expression that are already apparent at 2 weeks of age.
Our principal goal had been to determine whether a DREADD-mediated boost of Nkx2.1-IN firing in Fmr1 KO mice might reduce their dysregulated gene expression (as an indication that it could restore their identity and function). We first compared the DE genes from the bulk transcriptome of Fmr1 KO;hM3Dq mice (n=7 mice) treated with C21 from P5-P9 to those of similarly treated Fmr1 KO;mCherry controls (n=8 mice) and found 15% fewer DE genes (2,463 vs. 2,898; Fig. 6A-C,F). Even though our chemogenetic intervention targeted INs, DE genes associated with excitatory neurons showed surprisingly more improvement in expression than those of inhibitory neurons (Fig. 6D). The expression of several genes enriched in MGE-INs was restored to WT levels, including genes expressed in basket and chandelier cells (Pvalb, Kcnn2, Wasf3, Sox11), as well as SST-INs (Cnst) (Fig. 6E,H, Fig. S9A). DE genes in the bulk transcriptome whose expression was rescued by Gq-DREADDs belonged primarily to Synapse organization and Neuronal apoptosis GO categories (Figs. S9C, Fig. S10A,B).
Figure 6: Neonatal chemogenetic activation of Nkx2.1-INs partially rescues the bulk S1 transcriptome but further derails the Nkx2.1-IN translatome.

A. Experimental design for chronic chemogenetic activation of Nkx2.1-INs (from P5 to P9) in Fmr1 KO mice at P15.
B. Top: The total number of down- and up-regulated genes in the bulk transcriptome was 15% smaller in Fmr1 KO-hM3Dq mice than in Fmr1 KO-mCherry. Bottom: The total number of down- and up-regulated genes in the translatome.
C. Density plot showing how the DREADD manipulation led to fewer DE genes in the Fmr1 KO-hM3Dq group compared to WT mice.
D. Density plot showing how the DREADD manipulation affected expression of genes specific to excitatory cells (left) or interneurons (right). Note that fewer genes are different from WT mice in the Fmr1 KO-hM3Dq group, particularly for excitatory cell markers.
E. Volcano plot of DE genes in the bulk RNA of Fmr1 KO-hM3Dq mice (n=7) compared to WT-mCherry controls (n=8). DE genes that had been labeled in Fig. 5C and were fully or partially corrected by the DREADD manipulation are shown in charcoal or lighter color font, respectively.
F. Correlation plot of genes affected in the Fmr1 KO-mCherry and Fmr1 KO-hM3Dq. Red and purple dots denote DE genes that are uniquely and significantly different from WT controls in Fmr1 KO-mCherry and Fmr1 KO-hM3Dq mice, respectively. Grey dots are shared DE genes in both groups (unaffected by DREADDs), whereas purple genes are less different from WT than red genes.
G. Volcano plot of DE genes in the Nkx2.1-IN -specific translatome of Fmr1 KO-mCherry mice (n=8) compared to WT-mCherry controls (n=3).
H. Representative examples of how DREADDs affected genes for the “Synaptic organization” GO term (gray), or different subclasses of MGE-INs. Scale bar represents the log2 CPM (count per millions).
I. List of DE genes associated with different subclasses of MGE-INs for which expression was changed by DREADDs (same groups as in panel H).
When we turned to the Nkx2.1-specific translatome, we found a dramatic and unexpected increase in the number of DE genes (6,105) in chemogenetically treated Fmr1 KO-hM3Dq mice compared to WT controls (Fig. 6B,G). Although some GO analysis categories were improved in Fmr1 KO;hM3Dq mice (Fig. S9E), many others were newly dysregulated. Moreover, within the Nkx2.1-IN translatome, the chemogenetic intervention had only a modest effect on DE genes associated with MGE-INs (Fig. 6I).
In summary, our Ribotag RNAseq approach uncovered previously unknown dysregulation in the neocortical transcriptome and Nkx2.1-specific translatome of Fmr1 KO mice at a very early stage of development. Chronically increasing the activity of Nkx2.1-INs in Fmr1 KO mice from P5 to P9 modestly restored the global cortical transcriptome, while further derailing the translatome of Nkx2.1-INs.
Post critical period, but not neonatal, chemogenetic activation of Nkx2.1-INs partially rescues S1 circuit deficits in Fmr1 KO mice.
We next tested whether the same neonatal DREADD manipulation of Nkx2.1-INs could restore circuit changes of Fmr1 KO mice. We focused on two robust S1 phenotypes we previously reported in Fmr1 KO mice at P15, namely the low percentage of whisker-responsive Pyr cells in L2/3 and their lack of adaptation to repetitive whisker stimulation.20 Once again, we virally expressed hM3Dq (or mCherry) in Fmr1 KO mice and administered C21 from P5 to P9. We then implanted cranial windows (and injected rAAV1-Syn-GCaMP6s) at P15 and performed in vivo calcium imaging of Pyr cells at P21 (Fig. 7A,B). This neonatal DREADD activation of Nkx2.1-INs did not significantly change the proportion of whisker-responsive Pyr neurons, nor their adaptation to repetitive whisker stimulation in Fmr1 KO mice (Fig. 7C,D). We hypothesized that this failure to restore Pyr cell responsivity and adaptation was due to the fact that immature Nkx2.1-INs are decoupled from Pyr cells (Fig. 2; Fig. S3). To further test this theory, we used the same hM3Dq-DREADD approach in a different group of Fmr1 KO mice (n=4), but acutely administered C21 at P10 to activate Nkx2.1-INs and monitored with calcium imaging the activity of both Nkx2.1-INs and Pyr cells (Fig. S11A,B, Video S2). We discovered that C21 could reliably increase the activity of Nkx2.1-INs (Fig. S11C), but it failed to suppress Pyr cell firing in Fmr1 KO mice (Fig. S11D). Not surprisingly, when we administered C21 chronically from P5 to P9 in a separate cohort of Fmr1 KO mice (n=4) and recorded Pyr cell activity at P10, we found it did not change the synchronous network events (Fig. S11E,F). Thus, activating Nkx2.1-INs neonatally fails to modulate sensory-evoked activity in Fmr1 KO mice because MGE-INs are not yet functionally connected with Pyr cells before P10.
Figure 7: Post-critical period but not neonatal chronic chemogenetic activation of Nkx2.1-INs improves S1 circuit deficits.

A. Experimental design for chronic chemogenetic activation of Nkx2.1-INs in neonatal Fmr1 KO mice from P5 to P9 to assess circuit deficits using in vivo two-photon calcium imaging at P21.
B. The proportion of whisker-responsive Pyr cells was not changed by DREADDs in Fmr1 KO mice. (14.4±4.4% vs. 15.3±3.3%, p=0.875, unpaired t-test)
C. The neuronal adaptation index of Pyr cells to repetitive whisker stimulation was not changed by chemogenetics (0.02±0.004 for Fmr1 KO mCherry vs. 0.03±0.01 for Fmr1 KO; p=0.652, unpaired t-test).
D. Experimental design for chronic chemogenetic activation of Nkx2.1-INs in juvenile (post-critical period) Fmr1 KO mice.
E. The proportion of whisker-responsive Pyr cells was significantly changed by DREADDs in Fmr1 KO mice. (10.4±3.35% vs. 21.6±6.37%, p=0.0229, MW t-test)
F. The neuronal adaptation index of Pyr cells to repetitive whisker stimulation was ~340% higher in the Fmr1 KO-hM3Dq group compared to Fmr1 KO-mCherry controls, though the difference was not significant (0.0016±0.03 for Fmr1 KO-mCherry vs. 0.058±0.015 for Fmr1 KO; p=0.114, MW test).
Several aspects of cortical maturation are known to be transiently delayed in Fmr1 KO mice, but eventually catch up to WT levels.19,67,68 We wondered whether the coupling of Nkx2.1-INs (or eventually PV-INs) to Pyr cells is similarly delayed in in vivo in Fmr1 KO mice and therefore tested whether PV INs might be able to modulate Pyr cell activity in L2/3 of S1 by the end second postnatal week. We first used an acute chemogenetic approach to transiently increase PV-IN activity by expressing hM3Dq (or mCherry) and GCaMP6s at P10 in PV-Cre;Fmr1 KO mice and then recorded from L2/3 Pyr neurons in vivo at P15, before and 30-40 min after administering a single dose of C21 (1 mg/kg, s.c.) (Fig. S12A). Compared to vehicle injection, acute C21 injection resulted in a significant reduction of spontaneous activity of Pyr cells in Fmr1 KO mice expressing hM3Dq (n=10), but not in mCherry controls (n=6) (Fig. S12B). We also repeated the all-optical approach with ChRmine optogenetics in Nkx2.1-Cre;Sst-FlpO;Fmr1 KO mice at P15 (n=3) and discovered that, unlike at P10, optogenetic activation of putative future PV-INs significantly reduced Pyr cell activity (Fig. S12C-F). As expected, no effect was observed in mCherry-expressing controls (n=2).
Knowing that PV-INs are capable of modulating Pyr cells in Fmr1 KO mice by P15, we tested the effect of chronic chemogenetic activation of Nkx2.1-INs from P15 to 20 on S1 circuit deficits (Fig. 7E). Post critical period DREADD activation of Nkx2.1-INs significantly increased the proportion of whisker-responsive neurons (Fig. 7F) and modestly increased their adaptation to repetitive whisker stimulation, though this did not reach significance (Fig. 7G). Similarly, using an acute chemogenetic approach at P15 but targeting PV-INs (instead of Nkx2.1-INs) in PV-Cre;Fmr1 KO mice at P15, we observed that a single dose of C21 also caused a significant increase in the percentage of whisker-responsive Pyr cells in the hM3Dq group compared to vehicle injection, but no change to their adaptation (Fig. S12G-I). Altogether, these results suggest that by the end of second postnatal week, putative future PV-INs within the Nkx2.1 population in Fmr1 KO mice are finally functionally integrated in S1 and that boosting their firing at this age (but not earlier) can lessen whisker-evoked circuit deficits.
Boosting PV-IN activity using a Kv3.1 modulator ameliorates S1 circuit deficits and tactile defensiveness in juvenile Fmr1 KO mice.
Our chronic chemogenetic activation of Nkx2.1-INs from P15 to P20 only partially restored S1 circuit dysfunction in Fmr1 KO mice. One possibility is that, because the local viral injection only infects a subset of our targeted neurons (Fig. S8A,B), we could not drive the activity of a sufficient number of Nkx2.1-INs with DREADDs. We therefore tested a chronic pharmacologic approach to achieve a more global and longer-lasting increase of PV cell activity in Fmr1 KO mice after the critical period. After ~P10, cortical PV-INs assume their fast-spiking characteristics due to their expression of Kv3.1b channels. This subclass of voltage-gated potassium channels, responsible for rapid repolarization that enables their fast-spiking behavior, is almost exclusively expressed in PV-INs.69,70 We reasoned that targeting Kv3.1b channels pharmacologically could be used to modulate the firing of PV-INs as a potential treatment for various NDDs. We used the compound AG00563 (1-(4-methylbenzene-1-sulfonyl)-N-[(1,3-oxazol-2-yl)methyl]-1H-pyrrole-3-carboxamide), a Kv3.1 positive allosteric modulator (Fig. 8A). Patch-clamp recordings of identified PV-INs in acute slices from P14-P16 PV-Cre;tdTom;Fmr1 KO mice (see Methods), showed that bath application of AG00563 (10 μM) significantly increased their excitability (Fig. 8B,C). Of note, AG000563 did not affect the membrane potential or excitability of Pyr cells, nor the input resistance of PV-INs or Pyr cells (Fig. S13A-E).
Figure 8: A Kv3.1 allosteric modulator, AG00563, ameliorates circuit function and tactile defensiveness in juvenile and young adult Fmr1 KO mice.

A. Chemical structure of the AG00563 compound and experimental design for the in vitro patch-clamp recordings of PV-INs.
B. Example traces of action potential trains evoked by 250 ms current injection in a PV-Cre-tdTom+ cell from a Fmr1 KO mouse at P15 in S1.
C. Cumulative input-output curves during baseline (red) or AG00563 (gray) (n=15 cells from 6 PV-Cre;tdTom;Fmr1 KO mice, two-way RM ANOVA).
D. Experimental design for chronic AG00563 vs. vehicle treatment (3 mg/kg, s.c, twice daily) from P15-20 or P45-50 in Fmr1 KO mice.
E. Example traces of L2/3 Pyr cell calcium transients in Fmr1 KO mouse. Whisker stimulation (1 s at 10 Hz, 3 s I.S.I, blue bars).
F. The percentage of whisker-responsive Pyr cells was significantly higher in Fmr1 KO mice treated with AG00563 than in vehicle controls. (20.3±3.7% vs. 8.7±1.5%, p=0.012; unpaired t-test, n=10 and 13 mice, respectively).
G. The adaptation index of Pyr cells was also significantly higher in Fmr1 KO mice treated with AG00563- compared to vehicle controls. (0.01±0.03 vs. 0.08±0.03; p=0.0438; unpaired t-test).
H. Cartoon of tactile defensiveness behavioral assay.
I. Left: The proportion of time spent grabbing the stimulator was significantly lower in AG00563-treated mice than in vehicle controls (0.62%±0.28 s vs. 3.75%±1.33 s, p=0.0343, MW t-test, n=15 and 13 Fmr1 KO mice, respectively). Right: The proportion of time spent grooming was higher in AG00563-treated group, but not significant (6.25%±1.9 s vs. 1.06%±0.53 s, p=0.094, MW t-test).
J. Left: The proportion of time spent grabbing the stimulator was significantly lower in AG00563-treated mice than in controls (5.0%±1.38 s vs. 15.45%±4.9 s, p=0.037, MW t-test, n=10 and 11 mice, respectively). Right: The proportion of time spent grooming (1.02%±0.57 s vs. 0.92%±0.79 s, p=0.58, MW t-test).
Using in vivo calcium imaging, we found that acute administration of AG00563 (3 mg/kg, s.c.) at P15 significantly increased the fraction of whisker-responsive Pyr cells but did not change their adaptation index (Fig. S13F-H), which matches our results with acute excitatory DREADDs in PV-INs (Fig. S12G-I). We next administered AG00563 (or vehicle) chronically (3 mg/kg, s.c., twice daily) to Fmr1 KO mice from P15 to P20, and then performed calcium imaging the following day (Fig. 8D,E). We found that compared to vehicle controls (n=11 mice) the proportion of whisker-responsive Pyr cells in S1 was significantly higher in AG00563-treated mice (n=13 mice) (Fig. 8F), reaching near WT levels.20 Moreover, the adaptation index of Pyr cells was also significantly increased by AG00563 (Fig. 8G).
The absence of neuronal adaptation in certain brain circuits is one potential reason why children with NDDs/autism exhibit sensory hypersensitivity, because they are unable to ‘tune out’ non-threatening or non-salient stimuli.71 We used a tactile defensiveness assay based on repetitive whisker stimulation20 (see Methods; Fig. 8H) to test whether AG006563 might lessen the maladaptive avoidance/defensive behaviors previously observed in Fmr1 KO mice. We found that mice chronically treated with AG00563 from P15 to P20 manifested significantly less grabbing of the stimulator than vehicle-treated Fmr1 KO controls, and they also spent more time demonstrating healthy adaptive behaviors, such as grooming (Fig. 8I). Overall, more AG00563-treated juvenile Fmr1 KO mice displayed grooming and fewer showed grabbing during whisker stimulation, while the opposite was true in vehicle-treated Fmr1 KO mice (Fig. S13I). To determine whether this pharmacologic approach could also be beneficial in older animals, we also treated young adult Fmr1 KO mice with AG00563 (from P45 to P50) and found a significant reduction in the proportion of time spent grabbing the stimulator (Fig. 8J). Therefore, chronic pharmacological activation of PV-INs any time after P15 can ameliorate S1 sensory circuit deficits and rescue behavioral manifestations of tactile defensiveness in Fmr1 KO mice.
DISCUSSION
We set out to identify when IN hypofunction first begins in Fmr1 KO mice to better understand how developmental trajectories of cortical circuits are changed in FXS and in other NDDs that share a cortical IN hypofunction phenotype.4 We used an intersectional strategy to express chemo- and optogenetic tools to manipulate Nkx2.1-INs, as well as in vivo calcium imaging to record from them and their excitatory Pyr cell partners. We discovered that: 1. PV-INs and their immature precursors from the MGE in Fmr1 KO mice are hypoactive as early as P6 and their density is reduced in both mice and humans with FXS, due to an excess of developmental apoptosis; 2. The bulk S1 cortical transcriptome and the MGE IN-specific translatome are drastically changed in Fmr1 KO mice at the closure of the S1 critical period; 3. Increasing the activity of MGE-INs during the apoptosis window (P5-P10) restores the density of PV-INs and a subset of DE cortical genes, but fails to rescue circuit dynamics because MGE-INs are decoupled from Pyr cells at that stage; 4. In contrast, boosting PV-IN activity after the S1 critical period (P15-P20) does restore network activity in S1, especially when using a Kv3.1 allosteric modulator, which also ameliorate tactile defensiveness in Fmr1 KO mice.
Critical developmental role of Nkx2.1-INs in sensory circuits in FXS:
GABAergic INs govern crucial steps in the maturation of brain circuits and have been hypothesized to play a key role in NDDs.4,5 We confirm previous observations that, during the early postnatal period, spontaneous Pyr cell activity is hypersynchronous in Fmr1 KO mice.19,28 Additionally, we now demonstrate that Nkx2.1-INs are hypoactive in neonatal Fmr1 KO mice during a time that coincides with the emergence of perisomatic GABAergic inhibition onto Pyr cells.37 Considering the emerging knowledge about IN-Pyr connectivity in neonatal cortex,23,72 this hypoactivity likely has drastic consequences for both structural and functional connectivity,73 and sensory processing. Inhibition is likely necessary for the desynchronization of network activity at around P12.31 Indeed, our all-optical two-photon optogenetic approach demonstrated that activation of Nkx2.1-INs can drive the decorrelation of Pyr cells in WT mice at P10 (Fig. 2F). The previously reported developmental delay in this desynchronization in Fmr1 KO mice19,28 is probably due to the fact that Nkx2.1-INs are hypoactive and functionally decoupled from Pyr cells. Thus, early changes in cortical connectivity could have profound effects on the developmental trajectory of PV-INs.
Our Ribotag RNaseq reveals that several PV-IN genes are dysregulated in Fmr1 KO mice at P15, which is consistent with prior studies showing developmental delays in the maturation of the intrinsic properties and morphogenesis of fast-spiking INs,15 as well as their excitatory inputs.17,18 Our new data suggests that the developmental trajectory of cortical INs in FXS begins to deviate very early in development, when highly synchronous neural activity dominates in neocortex, a stage that roughly corresponds to the late 3rd trimester of gestation in humans.74 Importantly, our results are not consistent with major disruptions in the generation or migration of MGE-INs.
Excess developmental death of PV-INs in FXS:
There has been some controversy about whether the lower density of PV-INs reported in several models of NDDs/autism represents an artifact.4 The argument is that those studies had relied on PVALB immunohistochemistry to identify PV-INs but the problem is that PVALB expression levels are known to correlate with PV-IN activity;48 therefore, low PV-IN density in autism models might just reflect their hypoactivity.75 In the present study, we circumvent this potential confound in Fmr1 KO mice by counting tdTom+ cells in PV-Cre;Ai9 mice and confirm a marked reduction in PV-IN density. In some mice, only rare, scattered cortical PV-INs could be identified (Fig. S5A, example 2). It may seem counterintuitive that PV-IN density in both WT and Fmr1 KO mice was higher at 9 months of age than at P15 (Fig. 3A,B; Fig. S5B). Our results match those of several other studies that quantified PV-IN density using immunohistochemistry or in situ hybridization.43,47,76-81 This is almost certainly due to the fact that, even by P15, not all future PV-INs have turned on the Pvalb promoter.
We also found strong evidence that Nkx2.1-INs succumb to programmed cell death (Fig. S7). The increased density of SST-INs in Fmr1 KO mice (Fig. S6), and their higher thalamocortical afferent connectivity,24 may represent compensatory phenomena for the loss of PV-INs (and their hypoactivity). It is possible that some MGE-INs that would have become PV-INs actually differentiated into SST-INs (Fig. S7D).
We report, for the first time, that the density of PV-INs (but not calretinin- or calbindin-expressing INs) is also reduced in human FXS cases (Fig. 3C,D). This apparent loss of PVALB+ INs in human tissue could be a result of their hypoactivity, or perhaps their unique vulnerability of these neurons to the post-mortem delay in FXS. Still, the parallels between mouse and human data with regard to reduced PV-IN density remain striking. Interestingly, organoids derived from human induced pluripotent stem cells from patients with FXS had a lower density of GABAergic neurons,82 which was felt to be due to reduced neurogenesis. Because we found a normal density of Nkx2.1-INs at P1 in Fmr1 KO mice, we do not believe that the reduced PV-IN density is caused by differences with their birth or migration. Additional studies will be needed to confirm these results in a larger number of human cases, and ideally with attempts to correlate age and symptom severity with PV-IN density in various brain regions.
Insights from RNAseq studies in developing Fmr1 KO mice.
FMRP is an mRNA binding protein that, in response to neuronal activity, negatively regulates the translation of many mRNAs that encode proteins important for neuronal development and synaptic function.65,83 Our bulk RNAseq dataset identified dozens of DE genes (including Chrm4, Syt2, Nedd4) that had been previously found in RNAseq screens from the hippocampus of adult Fmr1 KO mice,55,56 suggesting that the dysregulation of those genes is affected by loss of FMRP from early developmental stages. The relatively small overlap (72 DE genes) between our study and previous ones may be due to differences in techniques, brain regions, or mouse age. Our RiboTag study is the first to show that loss of FMRP causes a profound dysregulation of the translatome of MGE-INs during development. This data set will hopefully provide an opportunity for future comparisons of developmental RNAseq data sets. Surprisingly, boosting the activity of Nkx2.1-INs, which significantly reduced their developmental apoptosis, did not reduce the number of MGE-IN-specific DE genes; in fact, it caused a massive dysregulation of their translatome. Perhaps this is a cautionary note that intervening at early stages of cortical development to modulate activity of immature neurons, could have unintended consequences. Thus, further investigations are needed to fully understand the effects of early developmental IN hypoactivity on gene expression, circuit plasticity, and sensory processing.
Why did chronic chemogenetic activation of Nkx2.1-INs at P5-P9 fail to fully rescue S1 circuit dynamics in Fmr1 KO mice?
One of our most striking observations was that raising the excitability of INs in neonatal Fmr1 KO mice was not sufficient to restore sensory-evoked network function in S1. Our data provide strong evidence that the reason is that MGE-INs are decoupled from their Pyr cell partners during neonatal development (Fig. 2). This could reflect changes in INs themselves (e.g., a delay in synaptogenesis) or in post-synaptic Pyr cells (e.g., changes in post-synaptic GABA receptor expression). We also find that eventually PV-IN → Pyr cell connectivity is established (Fig. S12), which is consistent with the known delayed maturation PV-INs in Fmr1 KO mice.15,18 This likely explains why we could restore circuit dynamics in S1 and ameliorate sensory avoidance behaviors in Fmr1 KO mice by boosting PV-IN firing after P15, but not earlier.
Implications for treatment of FXS:
Because symptoms of NDDs are first recognized in toddlers, it is generally understood that they arise because of changes in the brain that likely occur in utero. This makes sense for FXS because expression of FMRP in the brain starts prenatally, so its absence could change the typical developmental trajectory of brain maturation in the third trimester of gestation or earlier. Therapeutic interventions for intellectual disability or autism that begin at the earliest stages of brain development should therefore be the most effective.84,85 However, we found that increasing the activity of Nkx2.1-INs in neonatal mice did not fully rescue Fmr1 KO circuit phenotypes in S1, whereas increasing PV-IN firing after P15 did and also mitigated tactile defensiveness. This offers hope in FXS, because it means that interventions to boost IN activity need not start at birth (this may not be in fact desirable), but could be efficacious if started in childhood, adolescence, or even adulthood.14 Atypical sensory perception is present in toddlers with FXS and other NDDs and is believed to contribute to social communication differences, repetitive behaviors and learning disability in adulthood.86,87 Our encouraging preclinical results with the Kv3.1 activator AG00563 suggest that increasing activity of PV-INs is a plausible strategy to lessen certain symptoms in children and adults with NDDs.
STAR METHODS
Detailed methods are provided in the online version of this paper.
Experimental Model and Study Participant Details:
Mice lines
All experiments followed the U.S. National Institutes of Health guidelines for animal research, under an animal use protocol (ARC #2007-035) approved by the Chancellor's Animal Research Committee and Office for Animal Research Oversight at the University of California, Los Angeles. All mice were housed in a vivarium with a 12/12 h light/dark cycle and experiments were performed during the light cycle. Mice of both sexes were used in this study (males are represented by a square and females by a circle). Because we use both sexes, we use Fmr1 KO to describe both male (Fmr1−/y) and female (Fmr1−/−) knockout mice. No sex differences were found in this study. The mouse lines used in this study were obtained from Jackson laboratory: PV-Cre mice (JAX 008069), Ai9;WT (JAX 007909), Nkx2.1-Cre;WT (JAX 008661), Ai14;WT (JAX 007914), Sst-FlpO (JAX 031629), Ai65F;WT (JAX 032864), Rpl22HA/HA (JAX0110029). Mouse lines were crossed to WT (JAX line 000664) or Fmr1 KO female mice (JAX 003025). PV-Cre mice and Ai9 reporter lines (both on C57BL/6J background) were back crossed to FVB WT and Fmr1 KO mice for 10-12 generations.14 For this study, the following mouse lines were generated: Nkx2.1-Cre+/−;Fmr1 KO, Ai14+/+;Fmr1 KO, Sst-FlpO+/+;Fmr1 KO, Nkx2.1-Cre+/−;Sst-FlpO+/+ WT, Nkx2.1-Cre+/−;Sst-FlpO+/+;Fmr1 KO, Nkx2.1-Cre+/−;Rpl22HA/HA;WT, Nkx2.1-Cre+/−;Rpl22HA/HA;Fmr1 KO. PV-Cre, Nkx2.1-Cre, SST-FlpO, Rpl22 HA, Ai9 and Ai14 transgenic lines were used as heterozygotes mice in this study.
Study Participant Details
We collected post-mortem cortical samples of Brodmann Area 3 from 8 FXS cases and 8 neurotypical, sex and age-matched cases (Table S1). The formalin-fixed tissue was obtained from CENE (Hispano-American brain bank of Neurodevelopmental Conditions, https://health.ucdavis.edu/mindinstitute/research/cene-brain-bank/cene-about.html). The neurotypical cases were determined to be free of neurological disorders, including autism or intellectual disability, based on medical records and information gathered at the time of death from next of kin. All cases were males, except for one control case and one FXS case, and no gender difference were found.
Methods details
Viral injections:
Depending on the experiment, stereotaxic viral injections were done using glass micropipettes (Sutter Instrument, 1.5 mm outer diameter, 0.86 mm inner diameter) through burr holes (at P0-P1 or P6) or at the time of the cranial window (at P10 or P15; see below). All mice were anesthetized with isoflurane (5% induction, 1-1.5% maintenance via a nose cone, v/v) and placed in a stereotaxic frame with rubber ear bars. Pups were allowed to recover on a warm water circulation blanket before being returned to the dam. For injections at P0-P1, we made a small skin incision on the scalp over the right hemisphere under sterile conditions and drilled a small burr hole over the right S1 cortex using a dental drill, as previously described.88 A single injection of 250-350 nL of rAAV was done using a Picospritzer (General Valve, 30 pulses of 6 ms, 30 psi). For injections at P10 or P15, we typically pressure-injected 250-350 nL of rAAV at 4-6 different locations over S1 cortex at a depth of 0.2 mm below the dura (30 pulses, 6 ms each, 30 psi). The following viruses were used: AAV1-Syn-GCaMP6s-WPRE-SV40 (Addgene 100843), AAV1-CAG-Flex-GCaMP6s-WPRE.SV40 (Addgene 100842), AAV8-Ef1a-fDIO-GCaMP6s (Addgene 105714), AAV1-hSyn-DIO-hM3D(Gq)-mCherry (Addgene 44361), AAV-hSyn-DIO-mCherry (Addgene 50459-AAV1, 50459-AAV8), AAV8-nEF-Con/Foff-ChRmine-oScarlet, AAV8-EF1a-Con/Foff-2.0-mCherry (Addgene 137133).
Cranial windows:
Pups were anesthetized with isoflurane (5% induction, 1.5–2% maintenance via a nose cone, v/v) and secured in a stereotaxic frame with rubber ear bars. A 3.0–3.5 mm diameter craniotomy was performed over the right S1 cortex, under sterile conditions, by removing the skull without disturbing the pia, as described previously.20,32 The craniotomy was then covered with a 3 mm glass coverslip and secured by cyanoacrylate glue and dental cement. An aluminum head bar was attached to the skull contralaterally to the window with dental cement to secure the animal to the microscope stage. Within 1 h after surgery, the pups appeared fully recovered from the effects of anesthesia and could be returned to their dam. For in vivo imaging at P6 and P10, cranial windows were performed on the day of the recording under 1-1.5% isoflurane anesthesia (5% induction) and we allowed mice to recover for 4-6 h with their dam prior to calcium imaging. For in vivo calcium imaging at P15 or P21, cranial windows were implanted at P10 and P15, respectively, during which we performed GCaMP6s viral injections.
Intrinsic signal imaging:
For animals undergoing calcium imaging at P15 or P21, we previously mapped the location of the barrel field of S1 cortex (S1BF), with intrinsic signal imaging, 1-3 d after cranial window surgery, as described previously.89 The cortical surface was illuminated by green LEDs (535 nm) to visualize the superficial vasculature. The macroscope was then focused ~300 μm below the cortical surface and red LEDs (630 nm) were used to record intrinsic signals, with frames collected at 30 Hz 0.9 s before and 1.5 s after stimulation, using a fast CCD camera (Teledyne Dalsa Pantera 1M60), a frame grabber (64 Xcelera-CL PX4, Dalsa), and custom routines written in MATLAB (version 2009a). Thirty trials separated by 20 s were conducted for each imaging session. The whiskers contralateral to the cranial window were bundled together using bone wax and gently attached to a glass microelectrode coupled to a ceramic piezo-actuator (PI127-Physik Instrumente). Each stimulation trial consisted of a 100 Hz sawtooth stimulation lasting 1.5 s. The response signal was divided by the averaged baseline signal, summed for all trials, with a threshold at a fraction (65%) of maximum response to delineate the cortical representation of stimulated whiskers and guide viral injections to S1BF.
In vivo two-photon (2P) calcium imaging in head-restrained mice:
Calcium imaging was performed on one of two microscopes. For Figs. 1, 8 and Supplementary Figs. S12A-B, and S12G-I, we used a custom-built 2P microscope and acquired frames (1024x128 down sampled to 256 x 128 pixels, 210x105 μm) at 7.8 Hz with a 20x objective (0.95 NA, Olympus) and ScanImage software.90 For Figs. 2, 7 and Figs. S2, S3E-G, S4, S5C, S11, S12C-F, we used a Bergamo II (Thorlabs) at 15.13 Hz (512 x 512 pixels, or 667 x 667 μm) with the same 20x objective. Both microscopes were coupled to a Chameleon Ultra II Ti:sapphire laser (Coherent) tuned to 930 nm (average power at the sample was <90 mW). We imaged at a depth of 170-190 μm or 220-250 μm below the dura for P6-P10 mice and P15-P21 mice, respectively, corresponding to L2/3. At P15 and P21, mice were lightly sedated with chlorprothixene (2 mg/kg, i.p.) and isoflurane (0.1-0.5%) and kept at 37°C using a temperature control device and heating blanket (Harvard Apparatus). The isoflurane level was manually adjusted to maintain a constant breathing rate (100-150 breaths/min for P15-P16 mice and 140-150 breaths/min for P21 mice). For P6 and P10 experiments mice were unanesthetized and placed in a cotton pad and kept at 37°C using a temperature control device and heating blanket (Harvard Apparatus).32 Whisker stimulation was achieved by bundling the contralateral whiskers (typically all macrovibrissae of at least 1 cm in length) with soft bone wax and attaching them to a glass capillary coupled to a piezo-actuator.
In vivo two-photon optogenetic stimulation:
Optogenetic photostimulation of ChRmine-expressing neurons91 was performed by scanning (at 15.1 fps) a separate 1,040 nm femtosecond-pulsed laser (Fidelity, Coherent) using the same 2P microscope (Bergamo II). Laser power was typically 50-60 mW at the objective. Photostimulation consisted of twenty 1 s-long stimulation pulses, with an inter-stimulation interval of 3 s, and was controlled by an optical beam shutter controller (SC10, Thorlabs) triggered via custom-built MATLAB code.
Nkx2.1-Cre-specific Ribotag RNA extraction and sequencing:
Aged matched (P15), Nkx2.1-Cre+/−;Rpl22HA/−;WT or Nkx2.1-Cre+/−;Rpl22HA/−;Fmr1 KO, injected at P1 with AAV virus expressing mCherry or hM3Dq were used for the Ribotag RNA sequencing experiments. For each RNA isolation experiment, coronal sections (1.5 mm thick) containing S1 (A/P −0.5 to + 1.82mm, M/L 3.0 to 4.0 mm) were collected and placed in chilled choline-ACSF (132 mM choline, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 7 mM MgCl2, 0.5 mM CaCl2, and 8 mM D-glucose), from which we dissected S1 bilaterally. Samples were independently homogenized with a 2 mL Dounce homogenizer in 1mL of supplemented homogenization buffer containing: 1% w/v, HB-S; 1 mM DTT, Protease inhibitors (1X), RNAsin (200 units/mL), Cycloheximide (2 mg/l), Heparin (1mg/mL). 100 μL of the homogenate was used for isolating bulk RNA from S1 cortex. To isolate mRNA of HA-tagged Nkx2.1+ IN-specific ribosomes from non-HA tagged ribosomes, samples were incubated for 4 h with an anti-HA antibody (1:180, 5 μL in 900 μL of homogenate; 901513 Covance Anti-HA antibody), followed by overnight incubation with magnetic beads (Pierce Protein A/G Magnetic Beads 88802, 200 μL) at 4°C at 15 rpm centrifugation. The supernatant was washed thrice with 800 μL of high salt buffer (Tris 50mM, KCl 300mM, MgCl2 12mM, 10% NP-40 (1%), DTT 1mM, Cycloheximide 100μg/ml, dH2O), centrifuged at 15 rpm for 10 min at 4°C. For RNA extraction, we used Zymo Research Direct-zol RNA Miniprep kit (R2051) for the homogenate (input), and the Zymo Research Direct-zol RNA Microprep kit (R2061) for the bead-antibody-protein sample (IP); RNA was eluted with DNAase/RNAse-free water. RNA Integrity Number (RIN) scores were used to evaluate the integrity of RNA through the ratio of 28S:18S ribosomal RNA (Agilent TapeStation 4200).92 We set a minimum of RIN score 7 for our samples.93 RNA sequencing libraries were prepared using Ovation® RNA-Seq System V2. Libraries were multiplexed for paired-end 50 bp sequencing on NovaSeq S2, with read depth of 50 million reads on average. Demultiplex reads were aligned using STAR to the mouse genome (reference genome ID: mm10) and fragment counts were derived using HTS-seq.94 Outliers were calculated by measuring connectivity between samples, using the WCGNA package95 and removing samples >2 SD from the mean for Bulk and IP samples. All analysis was performed using R4.0.3. Differential expression analysis was performed using the Bioconductor DESEQ2 package 96 with FDR of 0.1. Go enrichment analysis was performed using Bioconductor clusterProfiler 4.0.97 GoTerms with greater then 70% overlap of genes was combined into one category. The GoTerm with the highest category was chosen as the name of the category. Non-brain related pathways were excluded with the gene enrichment analysis. For comparisons with previously published RNAseq data, we compiled all DE genes from those studies55-57 and compared the compiled list with our DE genes. Statistical significance was tested using the Fisher exact Test in R4.0.3.
Data analysis for calcium imaging:
Calcium imaging data were analyzed using custom-written MATLAB routines (MATLAB version 2020a). X–Y drift in the movies was first corrected using either a frame-by-frame, hidden Markov model-based registration routine98 or a cross-correlation-based, nonrigid alignment algorithm.99 The choice of registration algorithm did not affect the data analysis since the fluorescence data for each neuron was always normalized to its own baseline. For ROI segmentation, we used either EZcalcium100 (for Figs. 1, 8E-G, Fig. S12B, Fig. S12G-I) or Suite2P (for Figs. 2, 7, Fig. S2, S3E-G, S4, S11, S12C-F). To quantify activity levels, a “modified Z score” Z_F vector (an epoch was 8 consecutive frames wherein Z_F> 3 for every frame of that epoch) for each neuron was calculated as Z_F [F(t) mean(quietest period)]/SD(quietest period), where the quietest period is the 10 s period with the lowest variation (standard deviation) in dF/F, as described previously.20 All subsequent analyses were performed using the Z_F vectors. To define whether an individual cell showed responses that coincided with epochs of whisker stimulation (“stimulus-locked”), a probabilistic bootstrapping (10,000 scrambles, threshold p<0.01) method was implemented as previously described;20 among the ROI comprised in the Z_F vector. To assess adaptation of neuronal activity to repetitive whisker stimulation we calculated an adaptation index: [(Z score during first five stimulations) − (Z score during last five stimulations)]/[(Z score during first five stimulations) + (Z score during last five stimulations)]. The percentage of active cells was based on the Z_F vector, calculated as of proportion of cells reaching the criterion of having at least one activity epoch with Z_F> 3 for 8 consecutive frames, amongst the total number of segmented ROIs. A Pearson’s correlation coefficient was calculated for all neuron pairs in each calcium imaging movie using the Z_F vector.
Brain slice electrophysiology
PV-Cre;Ai14;Fmr1 KO mice at P15 were deeply anesthetized using 5% isoflurane prior to decapitation and 400 μm-thick coronal slices were prepared with a vibratome (Leica VT1200S, Germany). Slices were prepared in ice-cold oxygenated (95% O2 and 5% CO2) modified artificial cerebrospinal fluid (ACSF) containing (in mM): 92 NMDG, 5 Na+-ascorbate, 3 Na+-Pyruvate 2.5 KCl, 10 MgSO4, 2 CaCl2, 1.2 Na2HPO4, 24 NaHCO3, 5 HEPES, 25 Glucose. Slices were then left to recover in the modified ACSF at ~33°C for 30 min before being transferred to oxygenated regular ACSF at room temperature (RT) for storage for at least 1 h prior to recording. Recordings were made at 32-33°C in oxygenated ACSF containing (in mM) 124 NaCl, 2.5 KCl, 2 MgSO4, 2 CaCl2, 1.2 Na2HPO4, 24 NaHCO3, 5 HEPES, 13 Glucose, and the perfusion rate was set to 5 mL/min. Whole-cell patch-clamp recordings were performed in cortical L2/3 PV-Cre-tdTom+ cells. Recordings relied on fluorescence visualization using a Zeiss AxioSKOp FS+ microscope and additional verification by their intrinsic electrophysiological properties. For current-clamp recordings, the intracellular solution consisted of (in mM) 100 K-gluconate, 20 KCl, 4 ATP-Mg, 10 phosphocreatine, 0.3 GTP-Na, 10 HEPES (adjusted to pH 7.3 and 300 mOsm). Intrinsic excitability was measured as the number of action potentials evoked during a 250 ms current step at intensities of 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5 nA. Wash-on of AG00563 (10 μM) was performed at 32-33°C for a minimum of 5 min before assessing PV-IN intrinsic excitability. Series resistance was monitored, and recordings were discarded if series resistance changed by more than 15%, or if apparent loss of current clamp control occurred as reflected by a sudden change in the recording stability.
Immunohistochemistry and IN quantification in mouse tissue:
Mice were anesthetized with 5% isoflurane and transcardially perfused with ice cold phosphate buffer saline PBS (0.1 M) followed by ice cold 4% paraformaldehyde in PBS and post-fixed overnight at 4°C. Coronal sections (60 μm) were cut on a vibratome (Leica VT1200S). Sections were permeabilized with 0.3% Triton X-100 and blocked with 5% normal goat serum (NGS) for 1 h at RT. Sections were then incubated overnight at 4°C with the primary antibody diluted in PBS with 5% NGS and 0.1% Triton X-100. After rinsing in PBS for 5-10 min 3 times, sections were incubated for 1 h at RT with the corresponding Alexa Fluor-conjugated secondary antibody diluted in PBS (1:1000, Invitrogen). After rinsing in PBS for 5-10 min 3 times, sections were mounted onto Superfrost Plus glass slides (Vectashield Vibrance) with DAPI mounting medium and stored in the dark at RT. The following primary antibodies were used: mouse anti-parvalbumin (1:1,000, SAB4200545, Sigma Aldrich), rat anti-somatostatin (1:300, ab108456, Abcam), mouse anti-calretinin (1:500, MAB1568, Millipore), goat anti-mCherry (1:1000, M11240, Millipore), chicken anti-GFP (1:2000, 600-901-215, Thermo Fischer), rabbit anti cleaved-Caspase-3 (1:400, D175-5A1E, Cell Signalling).
To quantify IN density, sections were imaged on an ApoTome2 microscope (Zen2 software, Zeiss; 10X objective, 0.3 NA), and analyzed with ImageJ (https://imagej.nih.gov/ij/). The section sampling fraction (ssf=1/6) for S1BF was determined within [A/P: bregma −0.94mm to −1.94mm]. Composite images spanning the S1BF were acquired by stitching a grid of 4-6 confocal images (2754x2061 pixels). For Nkx2.1Cre+;tdTom+ and Pv-Cre+;tdTom+ cell count was performed by automated detection using ImageJ plug-ins (Filter minima=1.5 pixel, Process maxima prominence>200pixels). PVALB+ and Cleaved-Caspase3+ cell count was performed using Cell counter plug-in. Cortical layers were determined using DAPI counterstaining and cell counts were subdivided into layers 1-3, 4 and 5-6. The size of the total cortical area and layer-specific ROIs were measured using ImageJ. Cell counts were divided by ROI area and reported as cells/mm2. For the quantification of Nkx2.1-Cre;tdTom+ or PV-Cre;tdTom+ overlap with GCaMP6s expression, we first applied a filter minima=2 pixels, and process maxima prominence>200pixels for both channels using ImageJ. We next used Cellpose for the segmentation of the cells in each channel (following automated diameter calibration and using the “cyto” model).101 The generated masks for each channel were then superimposed to quantify the overlap.
Histology and IN quantification in human tissue:
Tissue blocks were fixed in 10% buffered formalin, cryoprotected in a 30% sucrose solution in 0.1 M phosphate-buffered saline with 0.1% sodium azide. Tissue was embedded in optimum cutting temperature compound, and frozen at −80 °C. A cryostat was used to cut 14 μm-thick slide-mounted sections, stored at −80 °C until use. Based on Brodmann cortical neuroanatomy, blocks containing area BA3 (primary somatosensory cortex) were isolated from each case. We cut 14 μm sections on a cryostat, stained 1 section with Nissl, and chose sections that exactly matched the von Economo descriptions for BA3. On adjacent sections we performed triple immunostaining for PV, Calbindin (CB), and Calretinin (CR). We used the following primary antibodies for our quantification experiments: monoclonal mouse anti-CB-D28k (1:500, Swant 300, Switzerland), polyclonal rabbit anti-CR (1:500, Swant 7697), and monoclonal mouse anti-PV (1:500, Swant 235). Secondary antibodies were donkey anti-mouse conjugated with biotin, amplified with avidin-biotin complex (ABC) and developed with diaminobenzidine (DAB) or Vector NovaRED substrates (all from Vector, USA) for CB and PV detection, respectively. Donkey anti-rabbit antibody conjugated with alkaline phosphatase and Vector Blue substrate (Vector) was used for CR detection. Tissue was pretreated in Diva decloaker (DV2004 LX, MX, Biocare medical, USA) in a decloaking chamber (Biocare medical, USA) at 110°C for 6 min. Subsequently, immunostained tissues were immersed in 0.1% Nissl for 1 min and then dehydrated in successive baths of 50% ethanol (30 s), 70% ethanol (30 s), 95% ethanol (10 min), and 100% ethanol (5 min), isopropanol (5 min), followed by xylene (15 min). Sections were then mounted and coverslipped with Permount (Fisher). The PV-red staining was visualized as pink following Nissl staining. We quantified the number of immunopositive cells for each interneuron subtype (CB+, CR+, or PV+ cells). We processed 1 section of each case and chose a 3 mm wide bin parallel to the pial surface that extended through the cortical gray matter to include all cortical layers. We imaged the tissue a 100X oil objective (Olympus BX61 microscope Hamamatsu Camera) and quantified IN density using MBF Bioscience Stereoinvestigator V.9 Software (MicroBrightField, Williston, VT), on a Precision PWS 690, Intel Xeon CPU computer (Dell) with Windows XP Professional V.2002 system (Microsoft).
DREADD agonist (C21) administration in vivo:
For DREADD experiments, the agonist C21 (HelloBio) was dissolved in 0.9% saline to 2 mg/mL. For acute DREADD experiments at P15 in Pv-Cre;Fmr1 KO mice, we injected C21 at 1 mg/kg, s.c.51 For the chronic DREADDs experiments in Nkx2.1 pups we injected C21 twice daily from P5 to P9 at a dose of 1 mg/kg, s.c.
AG00563 treatment in vivo:
AG00563 (1-(4-methylbenzene-1-sulfonyl)-N-[(1,3-oxazol-2-yl)methyl]-1H-pyrrole-3-carboxamide) was synthesized by Lundbeck. Purified AG00563 was shipped to UCLA for reconstitution and experiments. AG00563 was dissolved in a solution of 10% of hydroxypropyl-β-cyclodextrine (Sigma) in 0.9% saline. For the Kv3.1 pharmacological manipulation, mice were injected with AG000563 (3 mg/kg, s.c.) either once acutely at P15, or chronically (twice daily) from P15 to P20 or P45 to P50. Control animals were injected with a vehicle solution containing 10% of hydroxypropyl-beta cyclodextrine in 0.9% saline.
Tactile defensiveness assay in head-restrained mice:
We used a paradigm we previously established to assess tactile defensiveness in juvenile and adult mice.20 Briefly, titanium head bars were implanted at P15 and then mice were habituated to head restraint and to running on an air-suspended 200 mm polystyrene ball. For habituation, mice were placed on the ball for 20 min/d for 5 consecutive days before testing at P21 or P50. On the test day, each animal was first placed on the ball for a 3 min baseline period. Next, we performed a sham stimulation trial in which the whisker stimulator was visibly moving, but just out of the reach of the whiskers on the animal’s left side. The stimulator consisted of a long, narrow comb of five slightly flexible von Frey nylon filaments that were attached to a piezoelectric actuator (Physik Instrumente). During the stimulation trial, the filaments were intercalated between the whiskers. Bundling of whiskers onto a glass capillary (as was done for calcium imaging) was not feasible for these awake experiments because the mouse could have damaged the capillary or unbundled some of its whiskers with its forepaw. The stimulation protocol consisted of a 10 s baseline followed by 20 sequential bouts of whisker deflections along the anterior–posterior direction (1 s long at 10 Hz), with a 3 s interstimulus interval, and ending with another 10 s post-stimulation baseline. A custom-written semiautomated video analysis was implemented in MATLAB to score defensive behaviors (grabbing the stimulator) and adaptive healthy behaviors (grooming), in each 1s increment of the videos during the 20 stimulations.20
Quantification and Statistical analysis:
Unless otherwise specified, results were plotted and tested for statistical significance using Prism 9. Statistical analyses of normality (Lilliefors and Shapiro Wilk tests) were performed on each data set; depending on whether the data significantly deviated from normality (p<0.05) or not (p>0.05), appropriate non-parametric or parametric tests were performed. The statistical tests performed are mentioned in the text and the legends. For parametric two-group analyses, a Student’s t-test (paired or unpaired) was used. For non-parametric tests, we used Mann-Whitney test (two groups) and the Friedman test (repeated measures). Multiple comparisons with single variables were analyzed using one-way ANOVA with post-hoc Bonferroni’s test (comparing the mean of each column with the mean of every other column) or Dunnett’s test (comparing the mean of each column with the mean of a control column) for normally distributed samples. For multiple comparisons with more than one variable, a two-way ANOVA with post hoc Sidak’s test was used. No statistical methods were used to predetermine sample size. Sample sizes were calculated based on similar published studies. In the figures, significance levels are represented with the following convention: * for p<0.05; ** for p<0.01, *** for p<0.001. All experiments were replicated at least in 3 different litters. In all the figures, we plot the standard error of the mean (s.e.m.). Graphs either show individual data points from each animal or group means (averaged over different mice) superimposed on individual data points.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact: Dr. Carlos Portera-Cailliau CPCailliau@mednet.ucla.edu
Materials availability
This study did not generate new unique reagents.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Parvalbumin | Sigma-Aldrich | SAB4200545, RRID:AB_2857970 |
| Mouse anti-Parvalbumin | Swant | 235, RRID:AB_10000343 |
| Rat anti-Somatostatin | Abcam | Ab108456, RRID:AB_11158517 |
| Mouse anti-Calretinin | Milllipore | MAB1568, RRID:AB_94259 |
| Rabbit anti-Calretinin | Swant | 7697, RRID:AB_2721226 |
| Mouse anti-Calbindin | Swant | 300, RRID:AB_10000347 |
| Rabbit anti-Calbindin | Thermo Fischer Scientific | PA5-85669, RRID:AB_2792808 |
| Rabbit anti-cleaved caspase3 | Cell signaling | D175-%A1E, RRID:AB_2070042 |
| Chicken anti-GFP | Thermo Fischer Scientific | 600-901-215, RRID:AB_1537402 |
| Rat anti-mCherry | Thermo Fischer Scientific | M11240, RRID:AB_2536614 |
| Bacterial and virus strains | ||
| AAV1-Syn-GCaMP6s-WPRE-SV40 | Addgene | Cat #100843 |
| AAV1-CAG-Flex-GCaMP6s-WPRE.SV40 | Addgene | Cat #100842 |
| AAV8-Ef1a-fDIO-GCaMP6s | Addgene | Cat #105714 |
| AAV1-hSyn-DIO-hM3D(Gq)-mCherry | Addgene | Cat #44361 |
| AAV5-hSyn-DIO-mCherry | Addgene | Cat #50459-AAV5 |
| AAV8-hSyn-DIO-mCherry | Addgene | Cat #50459-AAV8 |
| Biological samples | ||
| Human brain samples | CENE | https://health.ucdavis.edu/mindinstitute/research/cene-brain-bank/cene-about.html |
| Deposited data | ||
| RNAseq transcriptomic and translatome data at P15 | This paper | https://github.com/porteralab/MGERNAseq |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J (WT) | Jackson Laboratories | Cat #000664 |
| Mouse: B6.129P2-Fmr1tm1Cgr/J (C57BL/6J-Fmr1−/−) | Jackson Laboratories | Cat # 003025 |
| Mouse: FVB.129P2-Pde6b+ Tyrc-ch/AntJ (FVB) | Jackson Laboratories | Cat #004828 |
| Mouse: FVB.129P2-Pde6b+ Tyrc-ch Fmr1tm1Cgr/J (FVB-Fmr1−/−) | Jackson Laboratories | Cat #004826 |
| Mouse: B6;129P2-Pvalbtm1(cre)Arbr/J (PV-Cre) | Jackson Laboratories | Cat #008069 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Ai9) | Jackson Laboratories | Cat #007909 |
| Mouse: C57BL/6J-Tg(Nkx2-1-cre)2Sand/J (Nkx2.1-Cre) | Jackson Laboratories | Cat #008661 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Ai14) | Jackson Laboratories | Cat #007914 |
| Mouse: B6J.Cg-Ssttm3.1(flpo)Zjh/AreckJ (SST-FLP) | Jackson Laboratories | Cat #031629 |
| Mouse: B6N.129-Rpl22tm1.1Psam/J | Jackson Laboratories | Cat #011029 |
| Software and algorithms | ||
| ImajeJ-Fiji 2.0.0 | NIH | https://imagej.nih.gov/ij/ |
| MATLAB 2009a/ 2018b/2020a/2022a | MathWorks | https://www.mathworks.com/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/features |
| ThorImageLS 4 | Thorlabs | https://www.thorlabs.com/ |
| Python 3.8.5 | Python Software Foundation | https://www.python.org/ |
| R 4.0.3 | R package | https://www.npackd.org/p/r/4.0.3 |
| Suite2P | Pachitariu et al., 2016 | https://github.com/MouseLand/suite2p |
| Cellpose2.0 | Stringer et al., 2021 | https://github.com/MouseLand/cellpose |
| EZCalcium | Cantu et al., 2021 | https://github.com/porteralab/EZcalcium |
| DESEQ2 | Love et al., 2014 | https://github.com/porteralab/EZcalcium |
| HTseq -count | Anders et al 2015 | N/A |
| Bioconductor clusterProfiler 4.0 | Wu et al., 2014 | N/A |
Highlights:
PV interneurons and MGE precursors are hypoactive in developing S1 of Fmr1 KO mice
PV neurons are decoupled from pyramidal cells until 2nd postnatal week
Lower density of PV cells in S1 of Fmr1 KO mice and in postmortem FXS human tissue
Boosting PV activity in S1 ameliorates tactile defensiveness in Fmr1 KO mice
Acknowledgments:
The authors thank Anis Contractor, Aaron McGee for feedback on the manuscript, and Fuying Gao, Riki Kawaguchi from the bioinformatics core at UCLA.
Funding:
this work was supported by the following grants: R01NS117597 (NIH-NINDS), R01HD054453 (NIH-NICHD), Department of Defense (DOD, 13196175) awarded to C.P-C, R01NS116589 (NIH-NINDS) awarded to D.V.B, R01MH094681 (NIH-NIMH) awarded to V.M.-C, a grant from the FRAXA foundation awarded to N.K.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Competing interests: A.G.S., C.M. and B.J.H. are employees of H. Lundbeck A/S. who developed the AG00563 compound. N.K., A.S., A.L, D.T.C., P.J., B.L. M.G., V.M.-C., D.B., and C.P.-C. declare no competing interests. A patent was filled by H. Lundbeck A/S (WO 2020/089262)
Data and code availability
This paper does not report original code. Previously published codes are publicly available at https://github.com/porteralabas listed in the key resources table.
The RNAseq data generated in this study is publicly available at https://github.com/porteralab/MGERNAseq as listed in the key resources table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robertson CE, and Baron-Cohen S (2017). Sensory perception in autism. Nat Rev Neurosci 18. [DOI] [PubMed] [Google Scholar]
- 2.Rubenstein JLR, and Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2, 255–267. 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Contractor A, and Klyachko VA (2015). Altered neuronal and circuit excitability in Fragile X syndrome. Neuron 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Contractor A, Ethell IM, and Portera-Cailliau C (2021). Cortical interneurons in autism. Nat Neurosci 24, 1648–1659. 10.1038/s41593-021-00967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marín O (2016). Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Medicine 22, 1229–1238. 10.1038/nm.4225. [DOI] [PubMed] [Google Scholar]
- 6.Dorrn AL, Yuan K, Barker AJ, Schreiner CE, and Froemke RC (2010). Developmental sensory experience balances cortical excitation and inhibition. Nature 465, 932–936. 10.1038/nature09119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fishell G, and Kepecs A (2020). Interneuron Types as Attractors and Controllers. Annu Rev Neurosci 43, 1–30. 10.1146/annurev-neuro-070918-050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wamsley B, and Fishell G (2017). Genetic and activity-dependent mechanisms underlying interneuron diversity. Nature Reviews Neuroscience 18, 299–309. 10.1038/nrn.2017.30. [DOI] [PubMed] [Google Scholar]
- 9.Salazar RF, Bertollini C, Mutel S, Roo M, and Muller D (2018). Restoring wild-type-like CA1 network dynamics and behavior during adulthood in a mouse model of schizophrenia. Nat Neurosci 21, 1–14. 10.1038/s41593-018-0225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Deister CA, Gao X, Guo B, Lynn-Jones T, Chen N, Wells MF, Liu R, Goard MJ, Feng S, et al. (2020). Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a Shank3 mouse model of ASD. Nat Neurosci 23, 1–18. 10.1038/s41593-020-0598-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marín O (2012). Interneuron dysfunction in psychiatric disorders. Nature Reviews Neuroscience 13, 107–120. 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 12.Rifé M, Badenas C, Mallolas J, Jiménez L, Cervera R, Maya A, Glover G, Rivera F, and Milà M (2003). Incidence of fragile X in 5,000 consecutive newborn males. Genet. Test 7, 339–343. 10.1089/109065703322783725. [DOI] [PubMed] [Google Scholar]
- 13.Antoine MW, Langberg T, Schnepel P, and Feldman DE (2019). Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 101, 648–661.e4. 10.1016/j.neuron.2018.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goel A, Cantu DA, Guilfoyle J, Chaudhari GR, Newadkar A, Todisco B, Alba D, NAZIM K, Schmitt LM, Pedapati E, et al. (2018). Impaired perceptual learning in a mouse model of Fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat Neurosci 21, 1–14. 10.1038/s41593-018-0231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nomura T, Musial TF, Marshall JJ, Zhu Y, Remmers CL, Xu J, and Nicholson DA (2017). Delayed Maturation of Fast-Spiking Interneurons is Rectified by Activation of the TrkB Receptor in the Mouse Model of Fragile X Syndrome. J. Neurosci, 2893-16–37. 10.1523/jneurosci.2893-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berzhanskaya J, Phillips MA, Shen J, and Colonnese MT (2017). Sensory hypoexcitability in a rat model of fetal development in Fragile X Syndrome. Nature Publishing Group; 6, 1–12. 10.1038/srep30769. [DOI] [Google Scholar]
- 17.Gibson JR, Bartley AF, Hays SA, and Huber KM (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. Journal of Neurophysiology 100, 2615–2626. 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domanski APF, Booker SA, Wyllie DJA, Isaac JTR, and Kind PC (2019). Cellular and synaptic phenotypes lead to disrupted information processing in Fmr1-KO mouse layer 4 barrel cortex. Nature Communications 10, 4814–4818. 10.1038/s41467-019-12736-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonçalves JT, Anstey JE, Golshani P, and Portera-Cailliau C (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci 16, 903–909. 10.1038/nn.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He CX, Cantu DA, Mantri SS, Zeiger WA, Goel A, and Portera-Cailliau C (2017). Tactile Defensiveness and Impaired Adaptation of Neuronal Activity in the Fmr1 Knock-Out Mouse Model of Autism. The J. Neurosci 37, 6475–6487. 10.1523/jneurosci.0651-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antoine MW, Schnepel P, Langberg T, and Feldman DE (2018). Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. 1–33. 10.1101/317693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuncdemir SN, Wamsley B, Stam FJ, Osakada F, Goulding M, and Rudy B (2016). Early Somatostatin Interneuron Connectivity Mediates the Maturation of Deep Layer Cortical Circuits. Neuron 89, 521–535. 10.1016/j.neuron.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marques-Smith A, Lyngholm D, Kaufmann A-K, Stacey JA, Hoerder-Suabedissen A, Becker EBE, and Wilson MC (2016). A Transient Translaminar GABAergic Interneuron Circuit Connects Thalamocortical Recipient Layers in Neonatal Somatosensory Cortex. Neuron 89, 536–549. 10.1016/j.neuron.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pouchelon G, Dwivedi D, Bollmann Y, Agba CK, Xu Q, Mirow AMC, Kim S, Qiu Y, Sevier E, Ritola KD, et al. (2021). The organization and development of cortical interneuron presynaptic circuits are area specific. CellReports 37, 109993. 10.1016/j.celrep.2021.109993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mòdol L, Bollmann Y, Tressard T, Baude A, Che A, Duan ZRS, Babij R, García NVDM, and Cossart R (2020). Assemblies of Perisomatic GABAergic Neurons in the Developing Barrel Cortex. Neuron 105, 93–105.e4. 10.1016/j.neuron.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stern EA, Maravall M, and Svoboda K (2001). Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron 31, 305–315. 10.1016/s0896-6273(01)00360-9. [DOI] [PubMed] [Google Scholar]
- 27.Fata GL, Gärtner A, Domínguez-Iturza N, Dresselaers T, Dawitz J, Poorthuis RB, Averna M, Himmelreich U, Meredith RM, Achsel T, et al. (2014). FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. 17, 1693–1700. 10.1038/nn.3870. [DOI] [PubMed] [Google Scholar]
- 28.Cheyne JE, Zabouri N, Baddeley D, and Lohmann C (2019). Spontaneous Activity Patterns Are Altered in the Developing Visual Cortex of the Fmr1 Knockout Mouse. Front Neural Circuits 13, 57. 10.3389/fncir.2019.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Lecea L, del Río JA, and Soriano E (1995). Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res Mol Brain Res 32, 1–13. 10.1016/0169-328x(95)00056-x. [DOI] [PubMed] [Google Scholar]
- 30.Butt SJB, Sousa VH, Fuccillo MV, Hjerling-Leffler J, Miyoshi G, Kimura S, and Fishell G (2008). The requirement of Nkx2-1 in the temporal specification of cortical interneuron subtypes. Neuron 59, 722–732. 10.1016/j.neuron.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rochefort NL, Garaschuk O, Milos R-I, Narushima M, Marandi N, Pichler B, Kovalchuk Y, and Konnerth A (2009). Sparsification of neuronal activity in the visual cortex at eye-opening. Proceedings of the National Academy of Sciences 106, 15049–15054. 10.1073/pnas.0907660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golshani Peyman, Gonçalves, and Tiago J (2009). Internally Mediated Developmental Desynchronization of Neocortical Network Activity. J. Neurosci 29, 10890–10899. 10.1523/jneurosci.2012-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan ZRS, Che A, Chu P, Mòdol L, Bollmann Y, Babij R, Fetcho RN, Otsuka T, Fuccillo MV, Liston C, et al. (2020). GABAergic Restriction of Network Dynamics Regulates Interneuron Survival in the Developing Cortex. Neuron 105, 75–92.e5. 10.1016/j.neuron.2019.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.García NVDM, Karayannis T, and Fishell G (2011). Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature 472, 351–355. 10.1038/nature09865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leighton AH, Cheyne JE, Houwen GJ, Maldonado PP, Winter FD, Levelt CN, and Lohmann C (2021). Somatostatin interneurons restrict cell recruitment to retinally driven spontaneous activity in the developing cortex. CellReports 36, 109316. 10.1016/j.celrep.2021.109316. [DOI] [PubMed] [Google Scholar]
- 36.Favuzzi E, Marques-Smith A, Deogracias R, Winterflood CM, Sánchez-Aguilera A, Maeso P, Fernandes C, and Ewers H (2017). Activity-Dependent Gating of Parvalbumin Interneuron Function by the Perineuronal Net Protein Brevican. Neuron 95, 639–655.e10. 10.1016/j.neuron.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 37.Gour A, Boergens KM, Heike N, Hua Y, Laserstein P, Song K, and Helmstaedter M (2021). Postnatal connectomic development of inhibition in mouse barrel cortex. Science 371. 10.1126/science.abb4534. [DOI] [PubMed] [Google Scholar]
- 38.Rickgauer JP, Deisseroth K, and Tank DW (2014). Simultaneous cellular-resolution optical perturbation and imaging of place cell firing fields. Nat. Neurosci 17, 1816–1824. 10.1038/nn.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Packer AM, Russell LE, Dalgleish HWP, and Häusser M (2015). Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nat Meth 12, 140–146. 10.1038/nmeth.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim L, Pakan JMP, Selten MM, Marques-Smith A, Llorca A, Bae SE, Rochefort NL, and Marín O (2018). Optimization of interneuron function by direct coupling of cell migration and axonal targeting. Nat Neurosci 21, 920–931. 10.1038/s41593-018-0162-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenno LE, Ramakrishnan C, Kim YS, Evans KE, Lo M, Vesuna S, Inoue M, Cheung KYM, Yuen E, Pichamoorthy N, et al. (2020). Comprehensive Dual- and Triple-Feature Intersectional Single-Vector Delivery of Diverse Functional Payloads to Cells of Behaving Mammals. Neuron 107, 836–853.e11. 10.1016/j.neuron.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Southwell DG, Paredes MF, Galvao RP, Jones DL, Froemke RC, Sebe JY, Alfaro-Cervello C, Tang Y, Garcia-Verdugo JM, Rubenstein JL, et al. (2012). Intrinsically determined cell death of developing cortical interneurons. Nature 491, 109–113. 10.1038/nature11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong FK, Bercsenyi K, Portalés A, and Fernández-Otero M (2018). Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557, 668–673. 10.1038/s41586-018-0139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Priya R, Paredes MF, Karayannis T, Yusuf N, Liu X, Jaglin X, Graef I, and Alvarez-Buylla A (2018). Activity Regulates Cell Death within Cortical Interneurons through a Calcineurin-Dependent Mechanism. CellReports 22, 1695–1709. 10.1016/j.celrep.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denaxa M, Neves G, Rabinowitz A, Kemlo S, Liodis P, Burrone J, and Pachnis V (2018). Modulation of Apoptosis Controls Inhibitory Interneuron Number in the Cortex. CellReports 22, 1710–1721. 10.1016/j.celrep.2018.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selby L, Zhang C, and Sun Q-Q (2007). Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neuroscience Letters 412, 227–232. 10.1016/j.neulet.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen TH, Afroz S, Reinhard SM, Palacios AR, Tapia K, Binder DK, Razak KA, and Ethell IM (2018). Genetic Reduction of Matrix Metalloproteinase-9 Promotes Formation of Perineuronal Nets Around Parvalbumin-Expressing Interneurons and Normalizes Auditory Cortex Responses in Developing Fmr1 Knock-Out Mice. Cereb Cortex 28, 3951–3964. 10.1093/cercor/bhx258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donato F, Rompani SB, and Caroni P (2013). Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 504, 272–276. 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- 49.Rudy B, Fishell G, Lee S, and Hjerling-Leffler J (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71, 45–61. 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bortone D, and Polleux F (2009). KCC2 Expression Promotes the Termination of Cortical Interneuron Migration in a Voltage-Sensitive Calcium-Dependent Manner. Neuron 62, 53–71. 10.1016/j.neuron.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson KJ, Khajehali E, Bradley SJ, Navarrete JS, Huang XP, Slocum S, Jin J, Liu J, Xiong Y, Olsen RHJ, et al. (2018). DREADD Agonist 21 Is an Effective Agonist for Muscarinic-Based DREADDs in Vitro and in Vivo. ACS Pharmacol Transl Sci 1, 61–72. 10.1021/acsptsci.8b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darnell JC, and Klann E (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16, 1530–1536. 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, and Eberwine J (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 54.Sharma SD, Metz JB, Li H, Hobson BD, Hornstein N, Sulzer D, Tang G, and Sims PA (2019). Widespread Alterations in Translation Elongation in the Brain of Juvenile Fmr1 Knockout Mice. CellReports 26, 3313–3322.e5. 10.1016/j.celrep.2019.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC, et al. (2017). Cell-Type-Specific Translation Profiling Reveals a Novel Strategy for Treating Fragile X Syndrome. Neuron 95, 550–563.e5. 10.1016/j.neuron.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Driesche SJV, Kang JJ, Darnell JC, and Darnell RB (2019). FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. Elife 8. 10.7554/elife.46919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ceolin L, Bouquier N, Vitre-Boubaker J, Rialle S, Severac D, Valjent E, Perroy J, and Puighermanal E (2017). Cell Type-Specific mRNA Dysregulation in Hippocampal CA1 Pyramidal Neurons of the Fragile X Syndrome Mouse Model. Front Mol Neurosci 10, 340. 10.3389/fnmol.2017.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chattopadhyaya B, Cristo GD, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, and Huang ZJ (2004). Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. Journal of Neuroscience 24, 9598–9611. 10.1523/jneurosci.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dehorter N, Ciceri G, Bartolini G, Lim L, del Pino I, and Marín O (2015). Tuning of fast-spiking interneuron properties by an activity-dependent transcriptional switch. Science 349, 1216–1220. 10.1126/science.aab3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahadevan V, Mitra A, Zhang Y, Yuan X, Peltekian A, Chittajallu R, Esnault C, Maric D, Rhodes C, Pelkey KA, et al. (2021). NMDARs Drive the Expression of Neuropsychiatric Disorder Risk Genes Within GABAergic Interneuron Subtypes in the Juvenile Brain. Front Mol Neurosci 14, 712609. 10.3389/fnmol.2021.712609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences 106, 13939–13944. 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahadevan V, Peltekian A, and McBain CJ (2020). Translatome Analyses Using Conditional Ribosomal Tagging in GABAergic Interneurons and Other Sparse Cell Types. Curr Protoc Neurosci 92, e93. 10.1002/cpns.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paul A, Crow M, Raudales R, He M, Gillis J, and Huang ZJ (2017). Transcriptional Architecture of Synaptic Communication Delineates GABAergic Neuron Identity. Cell 171, 522–539.e20. 10.1016/jcell.2017.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Favuzzi E, Deogracias R, Marques-Smith A, Maeso P, Jezequel J, Exposito-Alonso D, Balia M, Kroon T, Hinojosa AJ, Maraver EF, et al. (2019). Distinct molecular programs regulate synapse specificity in cortical inhibitory circuits. Science 363, 413–417. 10.1126/science.aau8977. [DOI] [PubMed] [Google Scholar]
- 65.Darnell JC, Driesche SJV, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maurin T, Lebrigand K, Castagnola S, Paquet A, Jarjat M, Popa A, Grossi M, Rage F, and Bardoni B (2018). HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res 46, 6344–6355. 10.1093/nar/gky267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cruz-Martín A, Crespo M, and Portera-Cailliau C (2010). Delayed stabilization of dendritic spines in fragile X mice. Journal of Neuroscience 30, 7793–7803. 10.1523/jneurosci.0577-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Q, Nomura T, Xu J, and Contractor A (2014). The Developmental Switch in GABA Polarity Is Delayed in Fragile X Mice. Journal of Neuroscience 34, 446–450. 10.1523/jneurosci.4447-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du J, Zhang L, Weiser M, Rudy B, and McBain CJ (1996). Developmental expression and functional characterization of the potassium-channel subunit Kv3.1b in parvalbumin-containing interneurons of the rat hippocampus. J. Neurosci 16, 506–518. 10.1523/jneurosci.16-02-00506.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rudy B, and McBain CJ (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends in Neurosciences 24, 517–526. 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- 71.Green SA, Hernandez L, Tottenham N, Krasileva K, Bookheimer SY, and Dapretto M (2015). Neurobiology of Sensory Overresponsivity in Youth With Autism Spectrum Disorders. JAMA Psychiatry 72, 778–786. 10.1001/jamapsychiatry.2015.0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anastasiades PG, Marques-Smith A, Lyngholm D, Lickiss T, Raffiq S, Kätzel D, Miesenböck G, and Butt SJB (2016). GABAergic interneurons form transient layer-specific circuits in early postnatal neocortex. Nature Communications 7, 10584–13. 10.1038/ncomms10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haberl MG, Zerbi V, Veltien A, Ginger M, Heerschap A, and Frick A (2015). Structural-functional connectivity deficits of neocortical circuits in the Fmr1−/y mouse model of autism. Sci Adv 1, e1500775–e1500775. 10.1126/sciadv.1500775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Molnár Z, Luhmann HJ, and Kanold PO (2020). Transient cortical circuits match spontaneous and sensory-driven activity during development. Science 370. 10.1126/science.abb2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Filice F, Vörckel KJ, Sungur AÖ, Wöhr M, and Schwaller B (2016). Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Molecular Brain 9, 10–17. 10.1186/s13041-016-0192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gonchar Y, Wang Q, and Burkhalter A (2007). Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front Neuroanat 1, 3. 10.3389/neuro.05.003.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, and Tonegawa S (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 78.Tomassy GS, Morello N, Calcagno E, and Giustetto M (2014). Developmental abnormalities of cortical interneurons precede symptoms onset in a mouse model of Rett syndrome. Journal of Neurochemistry 131, 115–127. 10.1111/jnc.12803. [DOI] [PubMed] [Google Scholar]
- 79.del Río JA, de Lecea L, Ferrer I, and Soriano E (1994). The development of parvalbumin-immunoreactivity in the neocortex of the mouse. Brain Res Dev Brain Res 81, 247–259. 10.1016/0165-3806(94)90311-5. [DOI] [PubMed] [Google Scholar]
- 80.Mukhopadhyay A, McGuire T, Peng C-Y, and Kessler JA (2009). Differential effects of BMP signaling on parvalbumin and somatostatin interneuron differentiation. Development 136, 2633–2642. 10.1242/dev.034439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patz S, Grabert J, Gorba T, Wirth MJ, and Wahle P (2004). Parvalbumin expression in visual cortical interneurons depends on neuronal activity and TrkB ligands during an Early period of postnatal development. Cerebral Cortex 14, 342–351. 10.1093/cercor/bhg132. [DOI] [PubMed] [Google Scholar]
- 82.Kang Y, Zhou Y, Li Y, Han Y, Xu J, Niu W, Li Z, Liu S, Feng H, Huang W, et al. (2021). A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat Neurosci 24, 1377–1391. 10.1038/s41593-021-00913-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bassell GJ, and Warren ST (2008). Fragile X Syndrome: Loss of Local mRNA Regulation Alters Synaptic Development and Function. Neuron 60, 201–214. 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Veenstra-VanderWeele J, and Warren Z (2015). Intervention in the context of development: pathways toward new treatments. 40, 225–237. 10.1038/npp.2014.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robertson CE, and Baron-Cohen S (2017). Sensory perception in autism. Nat Rev Neurosci 18. [DOI] [PubMed] [Google Scholar]
- 86.Rogers SJ, Hepburn S, and Wehner E (2003). Parent reports of sensory symptoms in toddlers with autism and those with other developmental disorders. J Autism Dev Disord 33, 631–642. 10.1023/b:jadd.0000006000.38991.a7. [DOI] [PubMed] [Google Scholar]
- 87.Robertson CE, and Baron-Cohen S (2017). Sensory perception in autism. Nature Reviews Neuroscience 18, 671–684. 10.1038/nrn.2017.112. [DOI] [PubMed] [Google Scholar]
- 88.He CX, Arroyo ED, Cantu DA, and Goel A (2018). A Versatile Method for Viral Transfection of Calcium Indicators in the Neonatal Mouse Brain. Front Neural Circuits 12, 12851–13. 10.3389/fncir.2018.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeiger WA, Marosi M, Saggi S, Noble N, Samad I, and Portera-Cailliau C (2021). Barrel cortex plasticity after photothrombotic stroke involves potentiating responses of pre-existing circuits but not functional remapping to new circuits. Nature Communications 12, 3972–14. 10.1038/s41467-021-24211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pologruto TA, Sabatini BL, and Svoboda K (2003). ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2, 13–19. 10.1186/1475-925x-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marshel JH, Kim YS, Machado TA, Quirin S, Benson B, Kadmon J, Raja C, Chibukhchyan A, Ramakrishnan C, Inoue M, et al. (2019). Cortical layer-specific critical dynamics triggering perception. Science 365. 10.1126/science.aaw5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, and Ragg T (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7, 3–14. 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362. 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559–13. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550–21. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y) 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dombeck DA, Khabbaz AN, Collman F, Adelman TL, and Tank DW (2007). Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron 56, 43–57. 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mineault PJ, Tring E, Trachtenberg JT, and Ringach DL (2016). Enhanced spatial resolution during locomotion and heightened attention in mouse primary visual cortex. J. Neurosci 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cantu DA, Wang B, Gongwer MW, He CX, Goel A, Suresh A, NAZIM K, Arroyo ED, Zeiger W, and Portera-Cailliau C (2020). EZcalcium: Open-Source Toolbox for Analysis of Calcium Imaging Data. Front Neural Circuits 14, 25. 10.3389/fncir.2020.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stringer C, Wang T, Michaelos M, and Pachitariu M (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat Meth 18, 100–106. 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.