

Abstract
Background:
Filamin C (FLNC), a member of the filamin family predominantly expressed in striated muscles, plays a crucial role in bridging the cytoskeleton and extracellular matrix (ECM) in cardiomyocytes, thereby maintaining heart integrity and function. While genetic variants within the N-terminal actin-binding domain (ABD) of FLNC have been identified in patients with cardiomyopathy, the precise contribution of the actin-binding capability to FLNC’s function in mammalian hearts remains poorly understood.
Methods:
We conducted in silico analysis of the three-dimensional structure of mouse FLNC to identify key amino-acid residues within the ABD that are essential for FLNC’s actin-binding capacity. Subsequently, we performed co-immunoprecipitation (co-IP) and immunofluorescent assays to validate the in silico findings and assess the impact of these mutations on the interactions with other binding partners and the subcellular localization of FLNC. Additionally, we generated and analyzed knock-in mouse models in which the FLNC-actin interaction was completely disrupted by these mutations.
Results:
Our findings revealed that F93A/L98E mutations completely disrupted FLNC-actin interaction while preserving FLNC’s ability to interact with other binding partners β1 integrin (ITGB1) and γ-Sarcoglycan (γ-SAG), as well as maintaining FLNC subcellular localization. Loss of FLNC-actin interaction in embryonic cardiomyocytes resulted in embryonic lethality and cardiac developmental defects, including ventricular wall malformation and reduced cardiomyocyte proliferation. Moreover, disruption of FLNC-actin interaction in adult cardiomyocytes led to severe dilated cardiomyopathy, enhanced lethality and dysregulation of key cytoskeleton components.
Conclusion:
Our data strongly support the crucial role of FLNC as a bridge between actin filaments and ECM through its interactions with actin, ITGB1, γ-SAG and other associated proteins in cardiomyocytes. Disruption of FLN-actin interaction may result in detachment of actin filaments from the extracellular matrix, ultimately impairing normal cardiac development and function. These findings also provide insights into mechanisms underlying cardiomyopathy associated with genetic variants in FLNC ABD and other regions.
Keywords: Cardiomyopathy, Basic Science Research, Myocardial Biology, filamin C, actin-binding activity, cardiac defects
Graphical Abstract
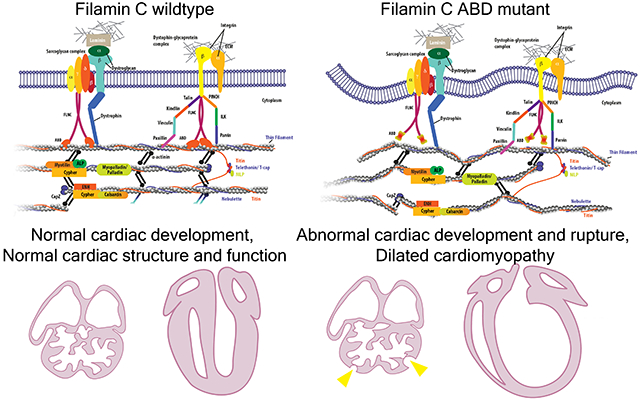
Introduction
The proper arrangement of the cardiomyocyte cytoskeleton, including the organization and stabilization of the sarcomere and its connection to the sarcolemma, is essential for maintaining cardiac structure and contractile function1–3. A variety of protein families contribute to the scaffolding and signaling functions of the cardiomyocyte cytoskeleton through protein-protein interaction. Mutations in genes encoding these proteins are associated with human cardiomyopathies, which are the leading cause of death worldwide3–8. One important component among these proteins is filamin C (FLNC), a member of the filamin protein family.
Filamins (FLNs) are large, homo-dimeric actin-binding and -crosslinking proteins that play a crucial role in connecting the cytoskeleton and plasma membrane, which is important for cardiac development and function9–11. Unlike the other two filamin family members in vertebrate cells, filamin A and B (FLNA and FLNB), which are ubiquitously expressed12, filamin C (FLNC) is predominantly expressed in skeletal and cardiac muscles. An increasing number of genetic variants in FLNC have been found to be associated with a variety of familial cardiomyopathies, including hypertrophic, dilated, and restrictive cardiomyopathy (HCM, DCM, and RCM, respectively), as well as arrhythmogenic right ventricular cardiomyopathy (ARVC)13–19.
Structurally, FLNC consists of an N-terminal actin-binding domain (ABD), as well as 24 Immunoglobulin (Ig)-like repeat domains (R1-R24) that form two rod structures named ROD1 and ROD2 subdomains, which are interrupted by two hinge regions20. The C-terminal part of FLNC is responsible for protein homo-dimerization and the interaction with many binding partners, including proteins at Z-disc and costameres3,21–23. Importantly, previous studies have identified numerous genetic variants associated with cardiomyopathy within the N-terminal actin-binding domain (ABD) of FLNC24, highlighting the crucial role of the N-terminal ABD in FLNC function in the heart. Nevertheless, our understanding of how the ABD of FLNC specifically contributes to its function in cardiomyocytes remains limited.
To address this question, we performed in silico analysis on the three-dimensional structure of mouse FLNC protein using cryo-electron microscopy-derived F-actin and FLNA molecular structure (pdb:6d8c) and identified two amino-acid residues, F93 and L98, which are localized in ABD and predicted to be essential for the actin-binding capacity of FLNC25. We then experimentally validated that F93 and L98 are required for the interaction between cardiac α-actin and FLNC ABD. To study the importance of the actin-binding activity of FLNC in vivo, we generated a global Flnc knock-in mouse model, in which two missense mutations F93A and L98E were introduced to ABD of FLNC. Global homozygous FLNC F93A/L98E mutant (gMut) mice died before E11.5 and exhibited profound cardiac developmental defects, resembling the phenotypes of Flnc knockout mice26. Moreover, we crossed the FLNC F93A/L98E mutant mice with Flnc floxed mice, and the constitutive or tamoxifen (TAM)-inducible cardiac-specific Cre lines (Xmlc2-Cre and Myh6-MerCreMer, respectively), to generate the Flnc cMut and icMut mouse lines, in which only the FLNC F93A/L98E mutant allele was retained in cardiomyocytes at embryonic and adult stage respectively. Strikingly, the Flnc cMut mouse line showed cardiac abnormalities during embryonic development, while the icMut mouse line displayed cardiac defects in adulthood after TAM induction. Our findings demonstrated that the actin-binding activity is indispensable for the functional role of FLNC in both developing and adult cardiomyocytes and also provide insights into mechanisms underlying cardiomyopathy associated with genetic variants in FLNC ABD and other regions.
Methods
Data Availability:
The data supporting the conclusions of this study are available from the corresponding author upon reasonable requests. A Major Resources Table is available in the Supplemental Material.
Design of actin binding defective mutations in FLNC:
Previously determined structure of filamin A ABD bound to actin (PDB code: 6d8c) was used to examine the filamin A/actin interface using PyMOL 1.3 (Schrödinger, LLC). Critical interface residues F99 and L104 were identified in filamin A, which correspond to F93 and L98 in FLNC based on the sequence homology. F93 and L98 were then selected for mutations to disrupt the FLNC binding to actin.
Animals:
FLNC F93A/L98E knock-in mice were generated using a cloning-free strategy as previously described27. Briefly, the CRISPR RNA (crRNA) targeting the genomic region encoding FLNC F93 and L98, and the single strand oligodeoxynucleotide (ssODN) containing F93A/L98E mutations were designed and then injected together with tracrRNA and Cas9 protein (New England BioLabs, M0386S) into the zygotes from C57BL/6N mice. The heterozygous FLNC F93A/L98E knock-in mice in F0 generation were identified by PCR analysis using mouse tail extracts and mutation-specific primers (listed in Supplementary Table 1), and further confirmed by sequencing. The heterozygous mice were subsequently intercrossed to generate the gMut mice. The cMut, icMut and the corresponding control mice were generated by crossing the FLNC F93A/L98E mutant mice with Flnc floxed mice and the constitutive and TAM-inducible cardiac-specific Cre lines (Xmlc2-Cre and Myh6-MerCreMer, respectively) using the breeding strategies shown in Supplementary Figure 1 and 2. For the treatment of TAM, 8-10 weeks old male mice were assigned randomly to different cages during the weaning process and intraperitoneally injected with TAM (Sigma-Aldrich, T5648, 40 mg/kg/day) for 3 consecutive days28, and the performers were blinded to the genotypes. All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Fig. 2. Generation of FLNC F93A/L98E knock-in mice.
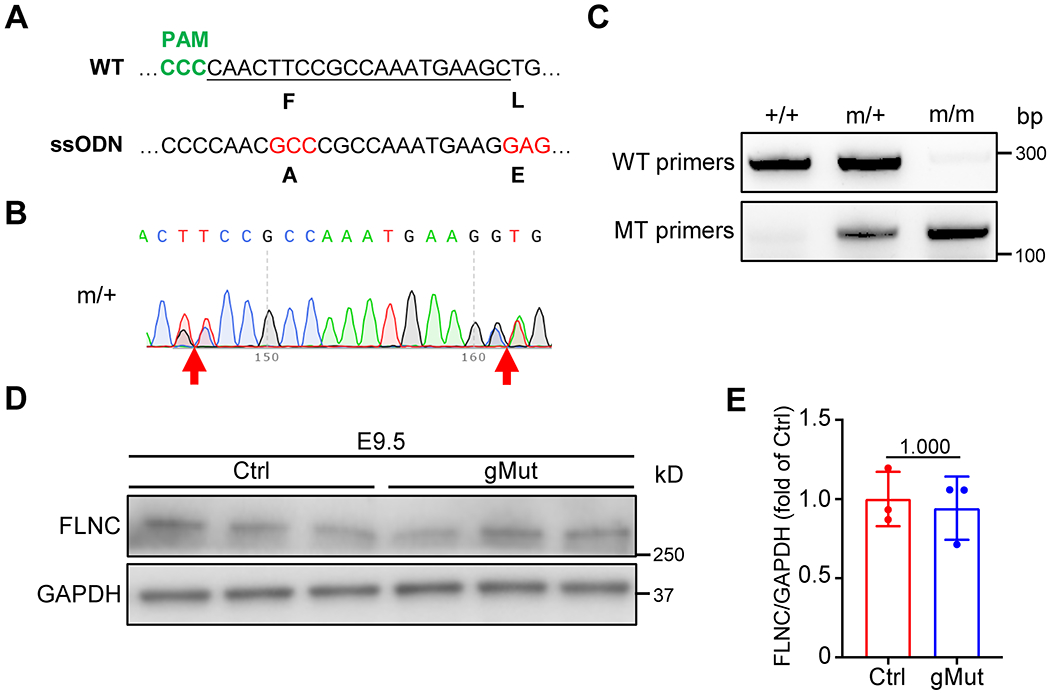
(A) Sequence of single-guide RNA (sgRNA) to target the Cas9 nuclease to the F93/L98E region in FLNC actin binding domain (ABD). WT, wild type; ssODN, single-stranded oligodeoxynucleotides; PAM, protospacer adjacent motif. (B) Heterozygous F93/L98E mutation shown by sequencing results. (C) Genotyping of WT (+/+), heterozygous (m/+) and homozygous (m/m) F93A/L98E mutant mice by PCR analysis of DNA isolated from tails using WT and mutant specific primers. (D-E) Western blot (D) and corresponding quantitative analysis (E) of FLNC from wild-type control (Ctrl) and global homozygous FLNC F93/L98E mutant (gMut) embryos at E9.5. n = 3 biologically independent samples/group. All quantitative data shown in this figure are presented as mean values ± SD. Mann–Whitney U test was used to calculate the presented p-values in E.
Echocardiography:
Echocardiography was performed as previously described29. Briefly, mice were anesthetized with 1% isoflurane and underwent echocardiography using a FUJIFILM VisualSonics SonoSite Vevo 2100 ultrasound system with a 32- to 55-MHz linear transducer. The percentage of FS was used as an indicator of systolic cardiac function. Measurements of heart rate (HR), LVIDs, and LVIDd were determined from the LV M-mode tracing. The operators were blinded to the genotypes.
Protein isolation and western blot analysis:
Protein isolation and western blot analysis were performed as described in our previous publication29. Briefly, embryo and adult cardiac tissues were harvested and snap-frozen in liquid nitrogen, and then homogenized in urea lysis buffer (8 M urea, 2 M thiourea, 3% SDS, 75 mM DTT, 0.03% bromophenol blue, 0.05 M Tris-HCl, pH 6.8). Protein lysates were separated on 4-12% SDS-PAGE gels (Thermo Fisher, NW04125BOX) and transferred onto PVDF membranes (Bio-Rad, 1620177) overnight at 4 °C. After blocking in blocking buffer (TBS containing 0.1% Tween-20 and 5% dry milk) for 30 min, the membranes were incubated overnight at 4°C with primary antibodies diluted in blocking buffer. Blots were washed and incubated with the corresponding HRP-conjugated secondary antibodies for 1 hour at room temperature. Immunoreactive protein bands were visualized using ECL reagent (Thermo Fisher, 34096). The primary and secondary antibodies used for western blot analysis were listed in Supplementary Table 2.
Co-Immunoprecipitation (Co-IP) analysis:
The cDNA sequences encoding 3 x FLAG tagged wild type or F93A/L98E mutant FLNC ABD (1-263 aa, uniprot: Q8VHX6), Human influenza hemagglutinin (HA) tagged full length mouse cardiac α-actin (ACTC1, uniprot: P68033), as well as V5 tagged full length mouse γ-Sarcoglycan (γ-SAG, uniprot: P82348) were inserted into the mammalian expression vector pcDNA3 by using T4 DNA Ligase (New England BioLabs, M0202S). The vectors encoding full-length mouse FLNC tagged with TurboGFP (tGFP) and full-length mouse β1 integrin (ITGB1, uniprot: P09055) tagged with FLAG were obtained from Origene (Origene, MG222854) and Addgene (Addgene, 30153), respectively. A vector containing tGFP tagged F93A/L98E mutant FLNC was generated by introducing site-directed mutagenesis into the TurboGFP-FLNC sequence. Vectors were co-transfected into HEK 293 cells (ATCC, CRL-1573™) using FuGENE HD Transfection Reagent (Promega, E2311). Protein extracts were prepared with Pierce™ IP Lysis Buffer (Thermo Fisher, 87787) and Halt™ Protease & Phosphatase Inhibitor Cocktail (Thermo Fisher, 1861284). 10% of the supernatant was taken out as input, and the remaining supernatant was incubated with the corresponding antibodies and Dynabeads™ Protein G (Thermo Fisher, 10003D) on rotation overnight at 4 °C. After washing 5 times with Pierce™ IP Lysis Buffer, the beads were resuspended in IP Elution Buffer (50 mM Glycine, pH 2.8), NuPAGE LDS Sample Buffer (Thermo Fisher, NP0007) and NuPAGE Sample Reducing Agent (Thermo Fisher, NP0009), then heated on a thermomixer for 10 min at 70 °C to elute the captured proteins. The eluted and input samples were examined by western blot analysis (refer to “Protein isolation and western blot analysis”). All antibodies used for IP and western blot analyses were listed in Supplementary Table 2.
Isolation and transfection of neonatal mouse cardiomyocytes:
Neonatal mouse cardiomyocytes were isolated as previously described30 and cultured with Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher, 11995-065) supplemented with 20% Medium 199 (Thermo Fisher, 11150-059), 10% horse serum (Thermo Fisher, 26050-088), 5% fetal bovine serum (FBS, Thermo Fisher, 10100-147) and 100 U/mL penicillin/streptomycin (Thermo Fisher, 15140-122) for 24 hours prior to treatment. Transfection of vectors containing tGFP-tagged wild-type and F93A/L98E mutant mouse FLNC was performed using Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher, L3000001) according to the manufacturer’s instructions.
Quantitative RT-PCR:
Total RNA was isolated from frozen embryo and adult cardiac tissues using TRIzol™ Reagent (Thermo Fisher, 15596026) and purified using RNeasy mini kit (Qiagen, 74106). ~1 μg RNA was used for reverse transcription with M-MLV Reverse Transcriptase (Promega, M1701). Quantitative real-time PCR reactions were performed using iTaq Universal SYBR Green Supermix (Bio-Rad, 1725120) on CFX Opus 96 Real-Time PCR System (Bio-Rad). All data were quantified by using the comparative CT method and normalized to the mRNA levels of the housekeeping gene Polr2a. The primer sequences are listed in Supplementary Table 1.
Histology:
Embryos and adult hearts were fixed overnight in 4% paraformaldehyde, and then dehydrated with gradient concentrations of sucrose and embedded in Tissue-Tek OCT Compound (Sakura, 4583). The tissue blocks were sectioned at 5 μm on Leica CM3050S Cryostat. H&E staining was performed as previously described31. Picrosirius red staining was performed using Sirius Red/Fast Green Collagen Staining Kit (Chondrex, 9046) according to the manufacturer’s instructions.
Immunofluorescence:
Immunofluorescence was performed as previously described32 with minor modifications. Cryosections were fixed in 4% paraformaldehyde for 10 min at room temperature, and then washed in TBS containing 0.01% Triton X-100. For antigen retrieval, the sections were immersed in pre-heated Antigen Unmasking Solution (Vector laboratories, H-3300) and boiled in a microwave for 10 min. After washing in TBS containing 0.01% Triton X-100, the sections were blocked in antibody dilution buffer (PBS with 0.1% Triton X-100, 1% bovine serum albumin and 0.1% Saponin) for 30 min, and then incubated with primary antibodies overnight at 4 °C and the corresponding secondary antibodies for 2 h at room temperature. After washing with TBS containing 0.01% Triton X-100, the sections were mounted with Fluoromount-G with DAPI (Invitrogen, 00-49590-52). Images were captured using Leica SP8 Lightning Confocal Microscope. The primary and secondary antibodies used for immunofluorescence were listed in Supplementary Table 2. The validation of all primary antibodies for immunofluorescence was conducted through the parallel stainings using isotype IgG and/or no primary antibody control. The representative images were selected to display an approximation of the mean value of the data.
TUNEL assay:
TUNEL assays were executed on heart sections with an In Situ Cell Death Detection Kit (Roche, 11684795910) that labeled the nuclei of dying cells with green fluorescence. Cardiomyocytes were co-stained by immunostaining with an Nkx2.5 antibody.
Statistical analysis:
The quantitative analyses of immunofluorescence and western blot results were performed by an investigator who was blinded to the conditions of the samples. Image-Pro Plus and ImageJ software were utilized for the analyses. All statistics were calculated using Graphpad Prism 8 and IBM SPSS Statistics 28.0.1. The data are reported as mean ± SD. The significance of differences between two groups was determined using the Mann-Whitney U test. 2-sided asymptotic p-value vas reported for the results of Mann-Whitney U test. For survival analysis, the Log-rank (Mantel-Cox) test was utilized. We did not adjust for multiple comparisons in our statistical analysis, a decision we recognize as a potential limitation of our study. The relative values and fold changes displayed in each bar chart were calculated by normalizing the data with the absolute mean value of the corresponding control group. A p-value less than 0.05 is referred to as statistically significant.
Results
Identification and validation of amino-acid residues essential for FLNC-actin interaction
The ABDs of the three filamin paralogs in mammals are more than 70% identical in amino acid sequences. Similarly, many actin proteins found in mammalian genomes are more than 95% identical. We therefore used the F-actin and FLNA ABD structure (pdb: 6d8c) to identify residues in murine FLNC ABD that are essential for its interaction with F-actin. Filamin ABD links adjacent actin monomers in the F-actin bound state. In the F-actin FLNA ABD structure, F99 and L104 are two residues that glue the adjacent actin monomers by extensive interactions (Figure 1A), which correspond to F93 and L98 in murine FLNC. The targeting of filamin to F-actin was compromised when these two residues were mutated to alanine, as assessed by GFP-actin colocalization25. F99 is lodged between two helices in the minor actin interface of FLNA ABD complex (Figure 1A). The L104 makes extensive contacts with loop residues V45-G48 in the major actin binding interface of ABD. Therefore, we hypothesized that a double mutation of the corresponding F93 and L98 in the mouse FLNC should greatly reduce filamin ABD actin interaction. To test this hypothesis, we mutated F93 of mouse FLNC to a smaller alanine and L98 to a negatively charged glutamine to perturb actin binding. By performing co-immunoprecipitation (co-IP) assays, we found that F93A/L98E mutant FLNC ABD failed to co-precipitated with cardiac muscle alpha actin (ACTC1) (Figure 1B), indicating that F93 and L98 are required for the interaction between FLNC and cardiac actin.
Fig. 1. F93A/L98E mutations impairs the interaction between FLNC ABD and actin.
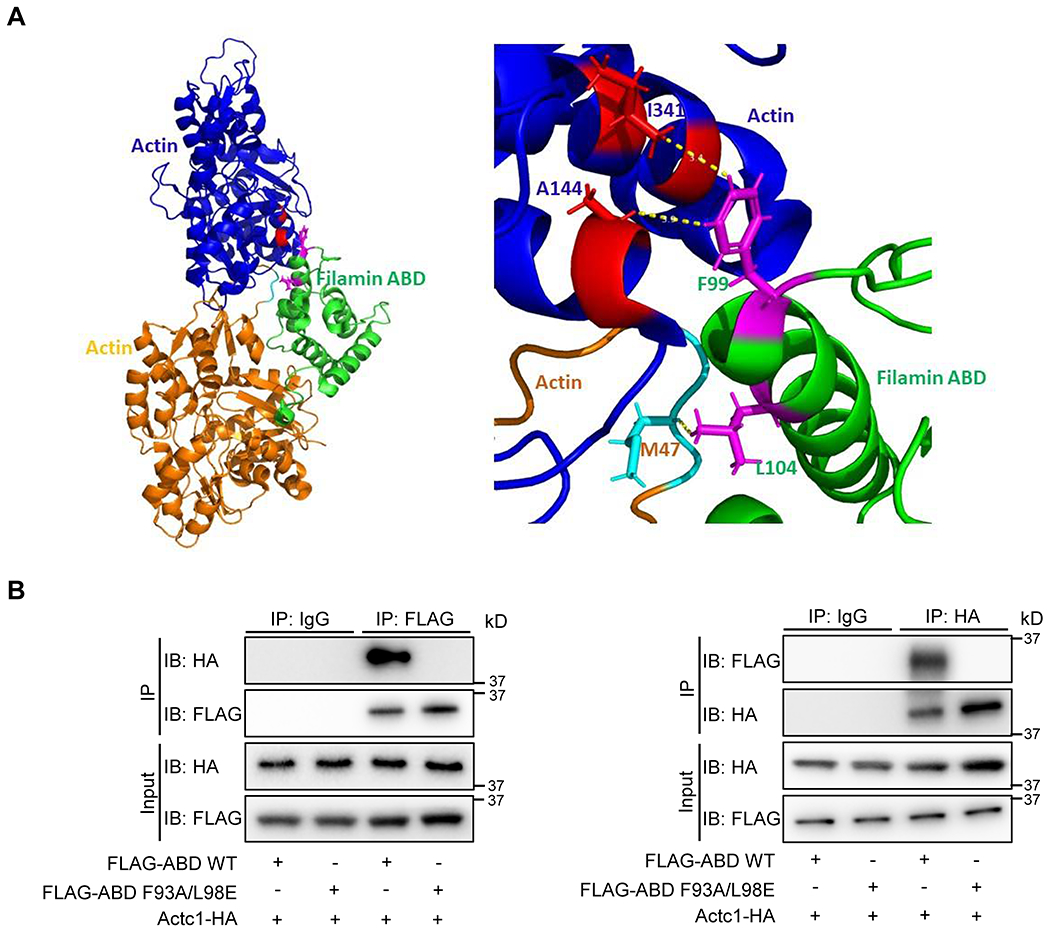
(A) Filamin ABD (green) binds to F-actin at a junction between adjacent actin monomers (orange and blue) in F-actin (pdb: 6d8c). The residues corresponding to F93 and L98 of mouse FLNC (F99 and L104 in human FLNC) are highlighted in magenta. F99 contacts in actin are in red and L104 contacts are in cyan. Inset: Selection of close contacts to F99 and L104 are highlighted. The double mutant F93A and L99E in mouse FLNC was hypothesized to substantially disrupt FLNC ABD interaction to F-actin. (B) Co-immunoprecipitation (Co-IP) and reverse Co-IP of Human influenza hemagglutinin (HA) tagged full length cardiac α-actin (ACTC1) with 3 x FLAG tagged wild type (WT) or F93A/L98E mutant FLNC actin binding domain (ABD). The cDNA sequences encoding all fusion proteins were inserted into pcDNA3 vector and co-transfected into HEK 293 cells for overexpression. n = 3 independently repeated experiments.
The F93A/L98E mutations maintain FLNC’s ability to interact with ITGB1 and γ-SAG and preserve its subcellular localization.
We conducted co-IP and immunofluorescent assays to assess the impact of F93A/L98E mutations on FLNC’s interaction with other binding partners and subcellular localization. We observed that the FLNC F93A/L98E mutations do not impair FLNC’s ability to interact with ITGB1 and γ-SAG, both of which are known to bind to the C-terminal Ig repeat domains of FLNC21,33 (Supplementary Figure 3A). Additionally, both wild-type and F93A/L98E mutant FLNC exhibited similar subcellular localization, characterized by striated pattern and colocalization with the Z-line protein α-actinin (Supplementary Figure 3B). Taken together, these findings suggest that FLNC F93A/L98E mutations do not affect its interaction with binding partners other than actin, or FLNC subcellular localization.
FLNC-actin interaction is indispensable for cardiac development
To assess the functional consequences of impaired actin-binding activity of FLNC in vivo, we generated a knock-in mouse line using CRISPR/Cas9 technology27. Two missense mutations, F93A and L98E, were introduced into the N-terminal actin-binding domain (ABD) of the FLNC protein (Figure 2A). Verifications through DNA sequencing for mouse tail samples confirmed the presence of the desired F93A/L98E mutations in the mutant mice (Figure 2B).
The heterozygous FLNC F93A/L98E mutant mice exhibited normal growth and did not show signs of cardiac dysfunction up to 6 months of age (Supplementary Figure 4). Subsequently, we performed a crossbreeding experiment between the heterozygous FLNC F93A/L98E mutant mice to obtain the global homozygous FLNC F93A/L98E mutant (gMut) mice (Figure 2C). Western blot analyses demonstrated that the levels of FLNC protein were comparable between the gMut and wild-type control embryos at embryonic day (E) 9.5 (Figure 2D–E), suggesting that the F93A/L98E mutations do not affect the expression or stability of FLNC protein.
The gMut mice displayed smaller body sizes, cardiac morphological defects, and pericardial effusion from E9.5 onwards and all died before E11.5 (Figure 3A and Supplementary Figure 5A). However, no developmental abnormalities or cardiac defects were observed in the gMut embryos at E8.5 (Supplementary Figure 6A). These findings indicate that the cardiac defects, as well as developmental abnormalities and embryonic lethality observed began to appear after E8.5 in the gMut mice.
Fig. 3. Disruption of FLNC-actin binding leads to cardiac defects in developing hearts.
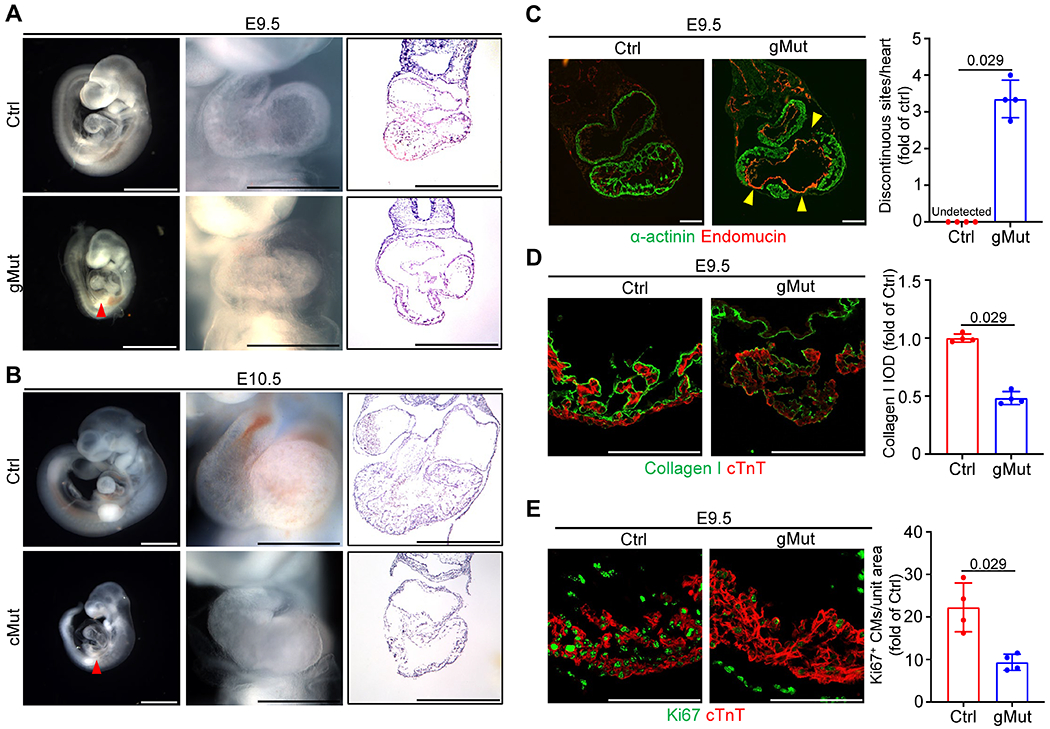
(A) Representative images of whole embryos (scale bar: 1 mm), whole hearts (scale bar: 0.5 mm) and H&E stained heart sections (scale bar: 0.5 mm) from wild-type control (Ctrl) and global FLNC F93/L98E mutant (gMut) mice at E9.5. n ≥ 5 biologically independent samples/group. (B) Representative images of whole embryos (scale bar: 1 mm), whole hearts (scale bar: 0.5 mm) and H&E stained heart sections (scale bar: 0.5 mm) from Xmlc2-Cre+;Flncmutant/flox (cMut) and control (Ctrl) mice at E10.5. n ≥ 5/group. The red arrowheads in A and B indicate the pericardial effusion. (C) Representative immunofluorescence of α-actinin and Endomucin, as well as the quantitative analysis of discontinuous sites on the ventricular wall, on wild-type control (Ctrl) and gMut heart sections at E9.5. n = 4 biologically independent samples/group. Scale bar: 100 μm. The yellow arrowheads indicate the discontinuous sites on the ventricular wall. (D) Representative immunofluorescence and corresponding quantitative analysis of Collagen I and cardiac troponin T (cTnT) on heart sections from wild-type control (Ctrl) and gMut embryos at E9.5. n = 4 biologically independent samples/group. Scale bar: 100 μm. IOD: integrated optical density. (E) Representative immunofluorescence and corresponding quantitative analysis of Ki67 and cardiac troponin T (cTnT) on heart sections from wild-type control (Ctrl) and gMut embryos at E9.5. n = 4 biologically independent samples/group. Scale bar: 100 μm. All quantitative data shown in this figure are presented as mean values ± SD. Mann–Whitney U test was used to calculate the presented p-values in C, D and E.
To further investigate whether the observed phenotypes in the gMut mice are resulted by the disruption of FLNC-actin interaction in a cardiomyocyte cell-autonomous manner, we conducted cross-breeding experiments involving the heterozygous FLNC F93A/L98E mutant mice, Flnc floxed mice26,28, and Xmlc2-Cre transgenic mice. The Xmlc2-Cre transgenic mice have been widely used to selectively ablate genes in cardiomyocytes as early as E7.5 without causing cardiac toxicity34. This breeding strategy resulted in the generation of Flnc cMut mice (Xmlc2-Cre+;Flncmutant/flox[m/f]), where the wild-type Flnc allele was specifically deleted in cardiomyocytes. As a result, only the F93A/L98E mutant FLNC protein was expressed in cardiomyocytes (Supplementary Figure 1). Phenotypic analysis of the Flnc cMut mice revealed identical cardiac morphological abnormalities to those observed in the gMut mice from E10.5, and these mice died before E11.5 (Figure 3B and Supplementary Figure 5B). These results indicate that the cardiac defects in gMut mice are primarily due to the disruption of FLNC-actin interaction in cardiomyocytes.
We previously reported the malformation of ventricular wall, displayed by the impairment of myocardial integrity, in Flnc knockout embryos26. To investigate whether the loss of actin-binding activity in FLNC can also result in the malformation of ventricular wall, we conducted immunofluorescence (IF) assays using α-actinin and endomucin to label the myocardium and endocardium in sections of gMut and control hearts at E8.5 and E9.5. Remarkably, the gMut hearts exhibited multiple discontinuous sites on the ventricular wall, rather than the endocardium, at E9.5 but not E8.5 (Figure 3C and Supplementary Figure 6B), resembling the phenotypes observed in Flnc knockout mice26.
In a previous study, we also demonstrated that extracellular matrix (ECM) disorganization contributes to the loss of ventricular wall integrity in Flnc knockout mice26. To assess ECM organization in gMut hearts, we performed IF staining for collagen I, along with the myocardial marker cardiac troponin T (cTnT), and observed a significant reduction in collagen I levels in the gMut hearts at E9.5 (Figure 3D). This suggests that the loss of ventricular wall integrity in gMut hearts is in part due to ECM disorganization.
We subsequently evaluated whether gMut mice exhibit impaired cardiomyocyte proliferation or apoptosis. Immunofluorescent (IF) analysis using antibodies against cTnT and Ki67, an indicator of active cell proliferation, revealed a substantial reduction in cardiomyocyte proliferation in the gMut hearts at E9.5 (Figure 3E). However, cardiomyocyte proliferation was comparable between control and mutant hearts at E8.5 (Supplementary Figure 6C), a stage when the gMut hearts did not display overt phenotypes. Additionally, apoptotic cardiomyocytes were absent in both control and mutant hearts as revealed by TUNEL assay (Supplementary Figure 7). These findings suggest that the decreased cardiomyocyte proliferation rate observed at E9.5 was likely secondary to the overall pathophysiological state of cardiomyocytes in gMut mice.
We previously observed a reduced level of active β1 integrin in the myocardial layer of Flnc knockout embryonic hearts26. However, when we performed the same analysis using the antibody 9EG7, which specifically recognizes the active form of β1 integrin35, we found that the level of active β1 integrin was comparable between control and gMut hearts at E9.5 (Figure 4). These results indicate that the actin binding capacity of FLNC is not required for the activation of β1 integrin in cardiomyocytes.
Fig. 4. Loss of FLNC-actin binding has no effect on the level of active β1 integrin in cardiomyocytes.
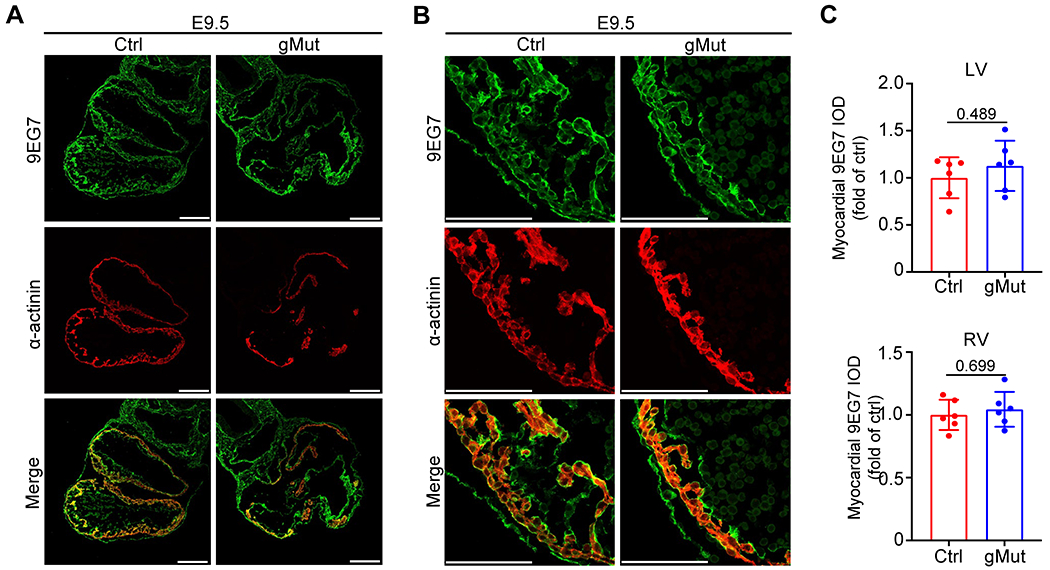
(A-B) Representative whole heart scale (A) and zoom-in (B) images for immunofluorescence of active β1 integrin and α-actinin on heart sections from wild-type control (Ctrl) and global FLNC F93/L98E mutant (gMut) embryos at E9.5. n = 4 biologically independent samples/group. Scale bar: 100 μm. (C) Quantitative analysis of active β1 integrin levels in left and right ventricles (LV and RV) from (A) and (B). n = 6 unit areas/group. IOD: integrated optical density. All quantitative data shown in this figure are presented as mean values ± SD. Mann–Whitney U test was used to calculate the presented p-values in C.
Loss of FLNC-actin interaction in adult cardiomyocytes leads to DCM
We previously reported that loss of FLNC in adult cardiomyocytes leads to DCM28. Therefore, we aimed to investigate whether the actin-binding activity of FLNC is also critical for the structure and function of adult hearts. To this end, we crossed the heterozygous FLNC F93A/L98E mutant mice with Flnc floxed mice and Myh6-MerCreMer transgenic mice36, to generate the Myh6-MerCreMer+;Flncm/f (icMut) mice, in which only the FLNC F93A/L98E mutant allele was retained in cardiomyocytes after TAM treatment (Supplementary Figure 2).
The icMut mice began to exhibit mortality within 10 days after TAM induction, with a 50% mortality rate by 20 days after TAM induction (Figure 5A). Analysis of heart weight (HW) to body weight (BW) ratios and HW to tibia length (TL) ratios showed significantly increased heart mass in icMut mice compared to control mice after 2 weeks of TAM treatment (Figure 5B). Morphological and histological analysis revealed severe ventricular dilation and fibrosis in icMut hearts (Figure 5C). Furthermore, echocardiography revealed reduced left ventricular (LV) systolic function (fractional shortening), accompanied by LV chamber dilation 2 weeks after TAM treatment, as evidenced by significantly increased end-diastolic and end-systolic LV internal diameter (LVIDd and LVIDs), as well as reduced LV posterior wall thickness (LVPWd) in icMut mice (Figure 5D). The cardiac stress markers Anf, Bnp, and Myh7, as well as the profibrotic genes Col1a1 and Col3a1, were also upregulated in icMut hearts after 2 weeks of TAM treatment (Figure 5E), indicating an ongoing pathological remodeling in icMut hearts.
Fig. 5. Impairment of FLNC-actin interaction in adult cardiomyocytes leads to dilated cardiomyopathy.
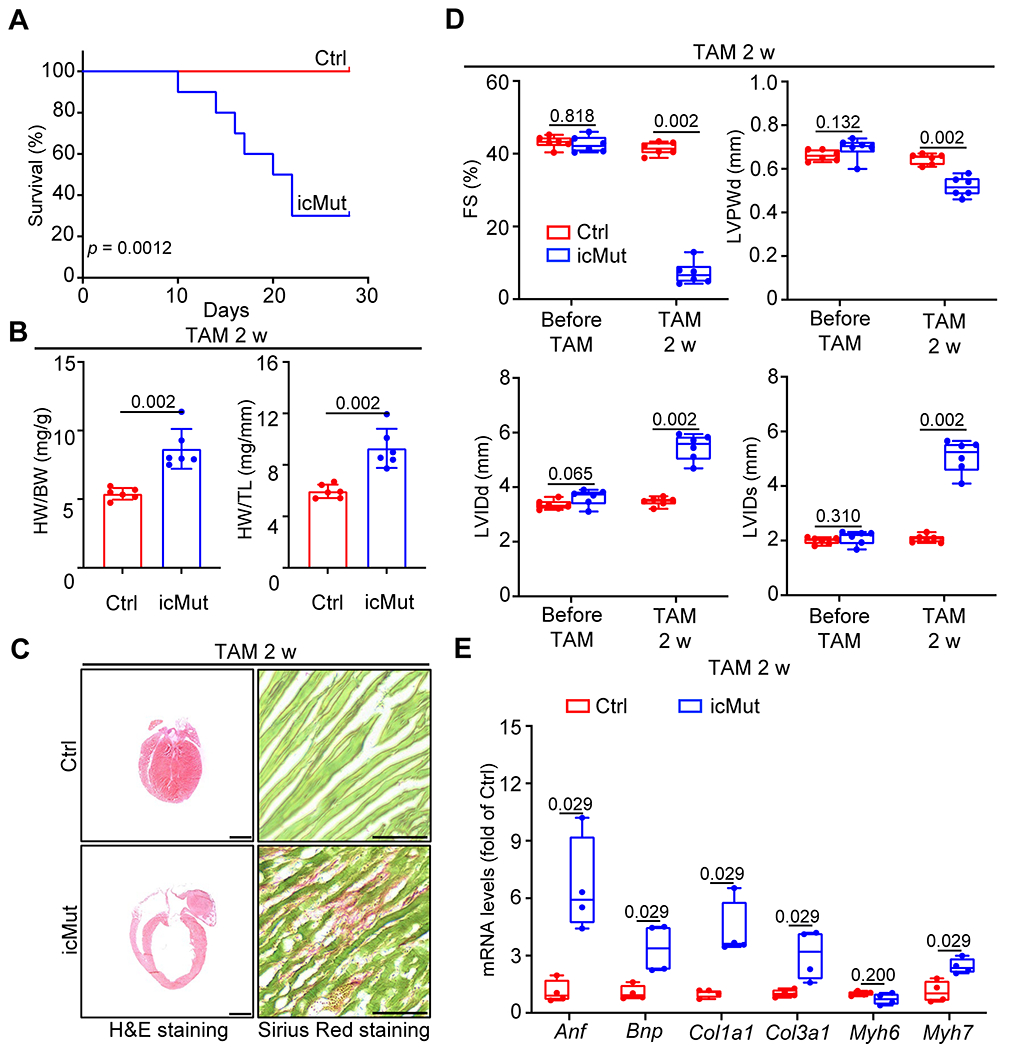
(A) Kaplan-Meier survival curves of Myh6-MerCreMer+;Flncmutant/flox (icMut) and control (Ctrl) mice up to 30 d after TAM injection. n = 10 animals/group. (B) The ratios of heart weight (HW) to body weight (BW) and HW to tibia length (TL) in icMut and control mice at 2 w after TAM injection. n = 6 animals/group. (C) Representative images of H&E staining (scale bar: 1 mm) and Picro-Sirius Red staining (scale bar: 500 μm) for heart sections from icMut and control mice at 2 w after TAM injection. Hearts were arrested at diastole by intraventricular injection of 10% KCl prior to fixation. n = 6 biologically independent samples/group. (D) Measurements of cardiac function index FS, as well as the left ventricular dilatation indexes LVIDd, LVIDs and LVPWD from echocardiographic assays on icMut and control mice at 3 w after TAM injection. n = 6 animals/group. FS: fractional shortening; LVIDd: left ventricular internal diameter, end diastole; LVIDs: left ventricular internal diameter, end systole; LVPWD: left ventricle posterior wall thickness, end diastolic. (E) qRT-PCR analysis of Anf, Bnp, Col1a1, Col3a1, Myh6 and Myh7 in icMut and control hearts at 2 w after TAM injection. Polr2a was used as an internal control gene. n = 4 biologically independent samples/group. All quantitative data shown in this figure are presented as mean values ± SD. Log-rank (Mantel-Cox) test (for A) and Mann–Whitney U test (for B, D and E) were used to calculate the presented p-values.
In previous studies, we have also demonstrated that loss of FLNC in adult cardiomyocytes leads to increased levels of various major cytoskeleton proteins28. Similarly, we conducted western blot analyses and observed upregulation of these proteins in icMut hearts at 2 weeks after TAM treatment. These include talin1, β1D-integrin, and integrin-linked kinase (ILK), which are components of the integrin complex, as well as dystrophin and γ-sarcoglycan (γ-SAG) from the dystrophin-associated glycoprotein complex (DGC). Additionally, the Z-disc protein desmin, and the intercalated disc (ICD) proteins xirp2 and junction plakoglobin (JUP) were also upregulated. Notably, the level of FLNC was comparable between icMut and control hearts (Figure 6).
Fig. 6. Loss of FLNC-actin interaction in adult cardiomyocytes results in excessive levels of major cytoskeleton components.
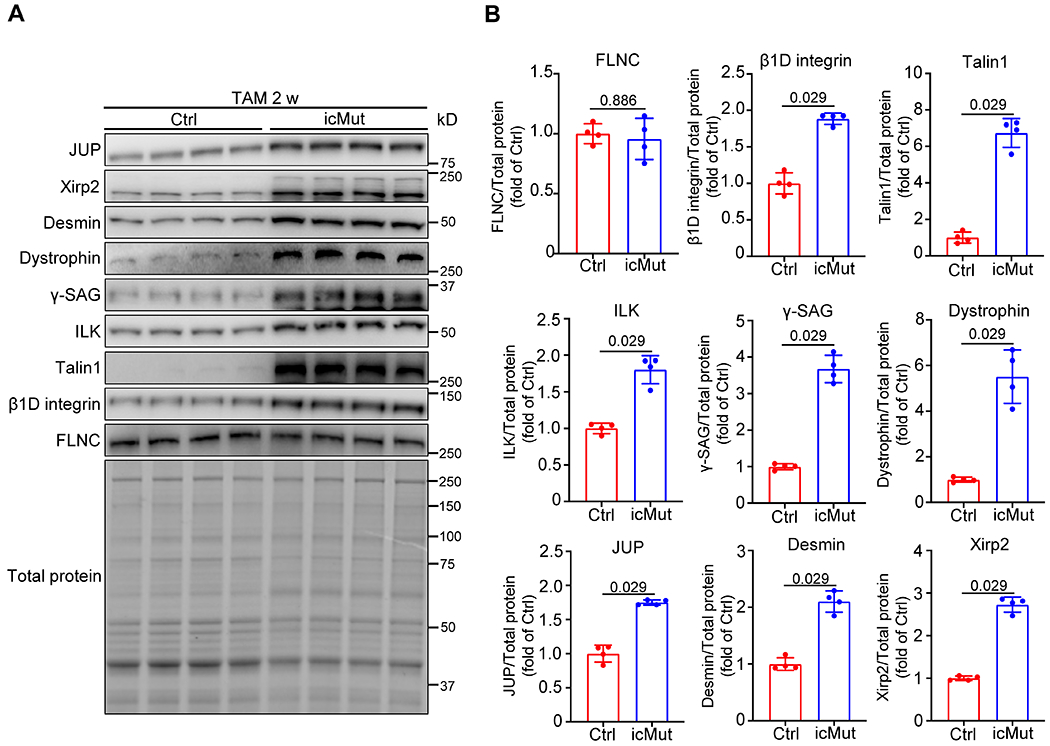
(A-B) Western blot (A) and corresponding quantitative analysis (B) of key cytoskeleton proteins, including integrin complex components β1D integrin, Talin1 and ILK, Z-disc protein Desmin, dystrophin-associated glycoprotein complex proteins Dystrophin and γ-Sacroglycan (γ-SAG), intercalated disc proteins Xirp2 and JUP, as well as FLNC, in Myh6-MerCreMer+; Flncmutant/flox (icMut) and control (Ctrl) mice at 2 w after TAM injection. n = 4 biologically independent samples/group. All quantitative data shown in this figure are presented as mean values ± SD. Mann–Whitney U test was used to calculate the presented p-values in B.
Discussion
In previous studies, a cohort of FLNC binding proteins has been identified, revealing its diverse interaction network3. For instance, it was reported that FLNC binds Z-disc proteins, such as myotilin and myozenin-1, and costamere proteins such as ITGB1 and sarcoglycans via its C-terminal region facilitating the connection between Z-disc proteins and the sarcolemma3,21,33.
While previous studies have primarily focused on the interaction of FLNC with its binding partners through the C-terminal Ig-like repeat regions, limited attention has been given to the importance of the ABD of FLNC. A series of genetic variants associated with cardiomyopathy have been identified within N-terminal ABD of FLNC, highlighting the critical role of this region in FLNC’s function24. However, the specific contribution of the N-terminal ABD to the function of cardiac myocytes remains poorly understood. This knowledge gap warrants the need for further investigation of the importance of ABD in FLNC’s function in cardiomyocytes.
In this study, we have demonstrated that FLNC F93A/L98E mutations completely disrupted the interaction between FLNC and actin while still maintaining its ability to interact with other binding partners, such as ITGB1 and γ-SAG, and also preserving its subcellular localization. Additionally, we found that FLNC F93A/L98E mutations did not affect the protein level of FLNC. Moreover, contrary to our previously published results in Flnc knockout mice, in which the activated ITGB1 levels were reduced compared to control hearts26, we observed comparable levels of activated ITGB1 between control and gMut myocardium at E9.5. These findings suggest that FLNC’s actin-binding capacity does not affect ITGB1 activation in cardiomyocytes. However, considering the crucial roles of both FLNC and ITGB1 in myocardial integrity26, the exclusive involvement of the C-terminal R20 domain of FLNC in interacting with β1 integrin26, and that the FLNC F93A/L98E mutations do not impair its ability to interact with ITGB1, it is plausible that FLNC plays a critical role in integrin-mediated outside-in signaling events following integrin activation as indicated in a previous study on filamin A37.
We also demonstrated that loss of FLNC actin-binding activity in embryonic cardiomyocytes led to smaller embryonic body size, abnormal cardiac morphology, impaired cardiomyocyte proliferation, loss of ventricular wall integrity and eventual embryonic lethality. Furthermore, our data demonstrated comparable embryonic body sizes cardiomyocyte proliferation between gMut and control embryos at E8.5, a developmental period when the loss of ventricular wall integrity are not observed in gMut embryos. However, as the loss of ventricular wall integrity occurs, a reduction in body size and a decrease in cardiomyocyte proliferation become evident in gMut embryos during later stages. These findings are consistent with our previously published data, which indicated that Flnc global and conditional knockout embryos exhibit a similar body size to controls during early embryonic development but display a reduced body size after E9.526. Therefore, we interpret the decreased body size and reduced cardiomyocyte proliferation in later stages of gMut development as a manifestation of the overall impaired pathophysiological state of cardiomyocytes due to the complete loss of FLNC-actin interaction in the gMut embryos.
Moreover, the icMut mice had severe dilated DCM and mortality, accompanied by elevated protein levels of key cytoskeletal components in cardiomyocytes. These phenotypes resemble those observed in our previous studies, where we reported TAM-inducible cardiomyocyte-specific Flnc knockout mice as we previously reported28. Collectively, these findings highlight the indispensable role of actin-binding activity of FLNC in the maintenance of the cytoskeleton in adult cardiomyocytes. Furthermore, the upregulation of cytoskeletal proteins in icMut hearts further emphasizes the critical function of FLNC’s actin-binding activity in facilitating proper cardiac contraction in adult hearts.
In summary, our study presents compelling new evidence from both in vitro and in vivo experiments that strongly support the crucial role of FLNC as a pivotal bridge between actin filaments and the ECM through its interactions with actin, ITGB1, γ-SAG and other associated proteins via distinct functional domains. Disruption of FLNC’s interaction with actin in developing or adult cardiomyocytes may lead to the detachment of actin filaments from the ECM, resulting in profound impairments in normal cardiac development and severe DCM in adulthood. These findings also provide insights into mechanisms underlying cardiomyopathy associated with genetic variants in FLNC ABD and other regions.
Supplementary Material
Novelty and Significance.
What is known?
Filamin C (FLNC), a member of the actin-binding filamin protein family predominantly expressed in striated muscles, plays a vital role in bridging the cytoskeleton and extracellular matrix (ECM) in cardiomyocytes.
Numerous genetic variants within the N-terminal actin-binding domain (ABD) of FLNC have been identified in patients with cardiomyopathy.
What new information does this article contribute?
We identified the essential F93 and L98 amino acid residues in the ABD of FLNC, and introduced F93A/L98E missense mutations to disrupt FLNC’s actin-binding capacity in a newly generated mutant mouse line.
Loss of FLNC-actin interactions in embryonic cardiomyocytes leads to cardiac developmental defects and embryonic lethality.
Disruption of the FLNC-actin interaction in adult cardiomyocytes resulted in the development of dilated cardiomyopathy (DCM), accompanied by the dysregulation of key cytoskeletal components.
FLNC, a vital actin-binding protein, plays a critical role in connecting the cytoskeleton to ECM. Previous studies identified various genetic variants associated with cardiomyopathy within FLNC’s N-terminal ABD. Therefore, understanding the contribution of FLNC’s ABD to its function in cardiomyocytes is crucial. We first conducted an in silico analysis of the three-dimensional structure of the mouse FLNC protein, which revealed that the F93 and L98 amino acid residues are essential for FLNC’s actin-binding capacity. Subsequently, we confirmed the requirement of F93 and L98 for the interaction between cardiac α-actin and FLNC’s ABD. To further investigate the impact of disrupted FLNC-actin interaction, we generated and analyzed FLNC mutant mice in which the interaction between FLNC and actin was completely disrupted by introducing F93A/L98E missense mutations into the ABD of FLNC. Our findings demonstrate that the loss of FLNC-actin interaction in embryonic cardiomyocytes results in cardiac developmental defects and embryonic lethality. Additionally, disrupting the FLNC-actin interaction in adult cardiomyocytes leads to fatal DCM, accompanied by the dysregulation of key cytoskeletal components. These findings demonstrate the crucial role of FLNC as a bridge between actin filaments and the ECM through its interactions with actin and other associated proteins within cardiomyocytes.
Acknowledgments
The authors thank the Transgenic Core at UCSD for generating the FLNC F93A/L98E knock-in mice, and Jennifer Santini and all staffs of University of California San Diego Microscopic Core Facility (supported by US National Institutes of Health grant P30 NS047101).
Sources of Funding
J. Chen is funded by National Heart, Lung, and Blood Institute (NHLBI) Grants and holds an American Heart Association Endowed Chair in Cardiovascular Research. J. Qin is funded by NHLBI and holds a Bonnie & Eunice Collins endowed chair for innovative diabetes research.
Nonstandard Abbreviations and Acronyms
- ABD
actin-binding domain
- ACTC1
cardiac muscle alpha actin
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- DCM
dilated cardiomyopathy
- FLNC
filamin C
- FS
fractional shortening
- HCM
hypertrophic cardiomyopathy
- ITGB1
β1 integrin
- LVIDd
left ventricular internal diameter end diastole
- LVIDs
left ventricular internal diameter end systole
- LVPWd
left ventricle posterior wall thickness end diastolic
- RCM
restrictive cardiomyopathy
- TAM
tamoxifen
- γ-SAG
γ-Sarcoglycan
Footnotes
Disclosures
J. Chen consults for LEXEO Therapeutics and Morphic Therapeutic.
References
- 1.Chen J, Chien KR. Complexity in simplicity: monogenic disorders and complex cardiomyopathies. J Clin Invest. 1999;103:1483–1485. doi: 10.1172/JCI7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peter AK, Cheng H, Ross RS, Knowlton KU, Chen J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog Pediatr Cardiol. 2011;31:83–88. doi: 10.1016/j.ppedcard.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bang ML, Bogomolovas J, Chen J. Understanding the molecular basis of cardiomyopathy. Am J Physiol Heart Circ Physiol. 2022;322:H181–H233. doi: 10.1152/ajpheart.00562.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knöll R, Hoshijima M, Chien K. Cardiac mechanotransduction and implications for heart disease. J Mol Med (Berl). 2003;81:750–756. doi: 10.1007/s00109-003-0488-x [DOI] [PubMed] [Google Scholar]
- 5.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–526. doi: 10.1172/JCI24351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng M, Cheng H, Banerjee I, Chen J. ALP/Enigma PDZ-LIM domain proteins in the heart. J Mol Cell Biol. 2010;2:96–102. doi: 10.1093/jmcb/mjp038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stroud MJ, Banerjee I, Veevers J, Chen J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ Res. 2014;114:538–548. doi: 10.1161/CIRCRESAHA.114.301236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pecorari I, Mestroni L, Sbaizero O. Current Understanding of the Role of Cytoskeletal Cross-Linkers in the Onset and Development of Cardiomyopathies. Int J Mol Sci. 2020;21. doi: 10.3390/ijms21165865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr. 2011;5:160–169. doi: 10.4161/cam.5.2.14401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razinia Z, Mäkelä T, Ylänne J, Calderwood DA. Filamins in mechanosensing and signaling. Annu Rev Biophys. 2012;41:227–246. doi: 10.1146/annurev-biophys-050511-102252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutherland-Smith AJ. Filamin structure, function and mechanics: are altered filamin-mediated force responses associated with human disease? Biophys Rev. 2011;3:15–23. doi: 10.1007/s12551-011-0042-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maestrini E, Patrosso C, Mancini M, Rivella S, Rocchi M, Repetto M, Villa A, Frattini A, Zoppè M, Vezzoni P. Mapping of two genes encoding isoforms of the actin binding protein ABP-280, a dystrophin like protein, to Xq28 and to chromosome 7. Hum Mol Genet. 1993;2:761–766. doi: 10.1093/hmg/2.6.761 [DOI] [PubMed] [Google Scholar]
- 13.Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, Pytel P, McNally EM. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:751–759. doi: 10.1161/CIRCGENETICS.113.000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, Reguero JR, Álvarez V, Morís C, León D, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326. doi: 10.1038/ncomms6326 [DOI] [PubMed] [Google Scholar]
- 15.Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, Gibson WT, McKinnon ML, McGillivray B, Alvarez N, et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum Mutat. 2016;37:269–279. doi: 10.1002/humu.22942 [DOI] [PubMed] [Google Scholar]
- 16.Reinstein E, Gutierrez-Fernandez A, Tzur S, Bormans C, Marcu S, Tayeb-Fligelman E, Vinkler C, Raas-Rothschild A, Irge D, Landau M, et al. Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. Eur J Hum Genet. 2016;24:1792–1796. doi: 10.1038/ejhg.2016.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padrón-Barthe L, Duro-Aguado I, Jiménez-Jáimez J, Hidalgo-Olivares VM, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. doi: 10.1016/j.jacc.2016.09.927 [DOI] [PubMed] [Google Scholar]
- 18.Roldán-Sevilla A, Palomino-Doza J, de Juan J, Sánchez V, Domínguez-González C, Salguero-Bodes R, Arribas-Ynsaurriaga F. Missense Mutations in the FLNC Gene Causing Familial Restrictive Cardiomyopathy. Circ Genom Precis Med. 2019;12:e002388. doi: 10.1161/CIRCGEN.118.002388 [DOI] [PubMed] [Google Scholar]
- 19.Verdonschot JAJ, Vanhoutte EK, Claes GRF, Helderman-van den Enden ATJM, Hoeijmakers JGJ, Hellebrekers DMEI, de Haan A, Christiaans I, Lekanne Deprez RH, Boen HM, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat. 2020;41:1091–1111. doi: 10.1002/humu.24004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao Z, Nakamura F. Structure and Function of Filamin C in the Muscle Z-Disc. Int J Mol Sci. 2020;21. doi: 10.3390/ijms21082696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson TG, Chan YM, Hack AA, Brosius M, Rajala M, Lidov HG, McNally EM, Watkins S, Kunkel LM. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148:115–126. doi: 10.1083/jcb.148.1.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Ven PF, Obermann WM, Lemke B, Gautel M, Weber K, Fürst DO. Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil Cytoskeleton. 2000;45:149–162. doi: [DOI] [PubMed] [Google Scholar]
- 23.van der Ven PF, Wiesner S, Salmikangas P, Auerbach D, Himmel M, Kempa S, Hayess K, Pacholsky D, Taivainen A, Schröder R, et al. Indications for a novel muscular dystrophy pathway. gamma-filamin, the muscle-specific filamin isoform, interacts with myotilin. J Cell Biol. 2000;151:235–248. doi: 10.1083/jcb.151.2.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eden M, Frey N. Cardiac Filaminopathies: Illuminating the Divergent Role of Filamin C Mutations in Human Cardiomyopathy. J Clin Med. 2021;10. doi: 10.3390/jcm10040577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwamoto DV, Huehn A, Simon B, Huet-Calderwood C, Baldassarre M, Sindelar CV, Calderwood DA. Structural basis of the filamin A actin-binding domain interaction with F-actin. Nat Struct Mol Biol. 2018;25:918–927. doi: 10.1038/s41594-018-0128-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T, Xu Y, Zhang L, Liang Z, Zhou X, Evans SM, Chen J. Filamin C is Essential for mammalian myocardial integrity. PLoS Genet. 2023;19:e1010630. doi: 10.1371/journal.pgen.1010630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma X, Chen C, Veevers J, Zhou X, Ross RS, Feng W, Chen J. CRISPR/Cas9-mediated gene manipulation to create single-amino-acid-substituted and floxed mice with a cloning-free method. Sci Rep. 2017;7:42244. doi: 10.1038/srep42244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Chen Z, Zhang L, Zhu M, Tan C, Zhou X, Evans SM, Fang X, Feng W, Chen J. Loss of Filamin C Is Catastrophic for Heart Function. Circulation. 2020;141:869–871. doi: 10.1161/CIRCULATIONAHA.119.044061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. doi: 10.1172/JCI94310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang X, Stroud MJ, Ouyang K, Fang L, Zhang J, Dalton ND, Gu Y, Wu T, Peterson KL, Huang HD, et al. Adipocyte-specific loss of PPARγ attenuates cardiac hypertrophy. JCI Insight. 2016;1:e89908. doi: 10.1172/jci.insight.89908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu T, Liang Z, Zhang Z, Liu C, Zhang L, Gu Y, Peterson KL, Evans SM, Fu XD, Chen J. PRDM16 Is a Compact Myocardium-Enriched Transcription Factor Required to Maintain Compact Myocardial Cardiomyocyte Identity in Left Ventricle. Circulation. 2022;145:586–602. doi: 10.1161/CIRCULATIONAHA.121.056666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thanisch K, Song C, Engelkamp D, Koch J, Wang A, Hallberg E, Foisner R, Leonhardt H, Stewart CL, Joffe B, et al. Nuclear envelope localization of LEMD2 is developmentally dynamic and lamin A/C dependent yet insufficient for heterochromatin tethering. Differentiation. 2017;94:58–70. doi: 10.1016/j.diff.2016.12.002 [DOI] [PubMed] [Google Scholar]
- 33.Gontier Y, Taivainen A, Fontao L, Sonnenberg A, van der Flier A, Carpen O, Faulkner G, Borradori L. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J Cell Sci. 2005;118:3739–3749. doi: 10.1242/jcs.02484 [DOI] [PubMed] [Google Scholar]
- 34.Breckenridge R, Kotecha S, Towers N, Bennett M, Mohun T. Pan-myocardial expression of Cre recombinase throughout mouse development. Genesis. 2007;45:135–144. doi: 10.1002/dvg.20275 [DOI] [PubMed] [Google Scholar]
- 35.Lenter M, Uhlig H, Hamann A, Jenö P, Imhof B, Vestweber D. A monoclonal antibody against an activation epitope on mouse integrin chain beta 1 blocks adhesion of lymphocytes to the endothelial integrin alpha 6 beta 1. Proc Natl Acad Sci U S A. 1993;90:9051–9055. doi: 10.1073/pnas.90.19.9051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687 [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Lu F, Ithychanda SS, Apostol M, Das M, Deshpande G, Plow EF, Qin J. A mechanism of platelet integrin αIIbβ3 outside-in signaling through a novel integrin αIIb subunit-filamin-actin linkage. Blood. 2023;141:2629–2641. doi: 10.1182/blood.2022018333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the conclusions of this study are available from the corresponding author upon reasonable requests. A Major Resources Table is available in the Supplemental Material.