

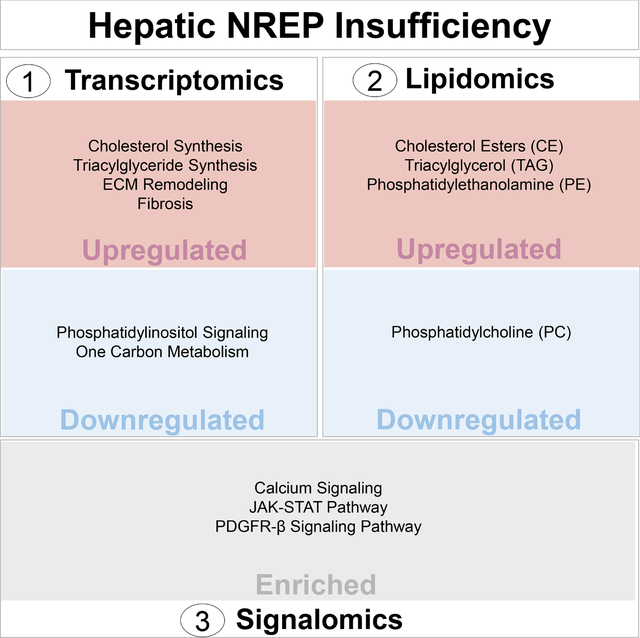
SUMMARY
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease. We recently discovered that neuronal regeneration-related protein (NREP/P311), an epigenetically regulated gene reprogrammed by parental metabolic syndrome, is downregulated in human NAFLD. To investigate the impact of NREP insufficiency, we used RNA-sequencing, lipidomics, and antibody-microarrays on primary human hepatocytes. NREP knockdown induced transcriptomic remodeling that overlapped with key pathways impacted in human steatosis and steatohepatitis. Additionally, we observed enrichment of pathways involving phosphatidylinositol signaling and one-carbon metabolism. Lipidomics analyses also revealed an increase in cholesterol esters and triglycerides and decreased phosphatidylcholine levels in NREP-deficient hepatocytes. Signalomics identified calcium signaling as a potential mediator of NREP insufficiency’s effects. Our results, together with the encouraging observation that several single nucleotide polymorphisms (SNPs) spanning the NREP locus are associated with metabolic traits, provide a strong rationale for targeting hepatic NREP to improve NAFLD pathophysiology.
Blurb:
“De Jesus et al. demonstrate that NREP deficiency in human hepatocytes leads to transcriptomic remodeling and metabolic dysregulation, resembling key pathways implicated in NAFLD. One carbon metabolism emerges as a potential mediator, highlighting NREP as a promising therapeutic target for improving NAFLD pathophysiology. These findings provide valuable insights into the molecular mechanisms underlying NAFLD progression.”
Graphical Abstract
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) can be characterized by simple hepatic lipid accumulation (steatosis) that can, in some cases, progress to increased inflammation and fibrosis and ultimately lead to non-alcoholic steatohepatitis (NASH), cirrhosis, and end-stage liver disease1–4.
One-carbon metabolism is among several pathways linked to the development of NAFLD. While choline can be synthesized endogenously from phosphatidylethanolamine (PE), then converted into phosphatidylcholine (PC)5,6, methionine is an essential amino acid and a precursor of PC and choline that is largely metabolized in hepatocytes6. This intrinsically connected pathway maintains a fine balance between PC and PE levels to carefully regulate the synthesis of very-low-density lipoproteins (VLDL) and the export of VLDL and triglycerides (TG)7. In NAFLD an imbalance between the levels of PC and PE due to a decrease in PC can lead to a deficient export of VLDL and accumulation of TG5,6,8,9.
Recently, calcium signaling is a modulator of ER stress response and an inducer of hepatic lipid accumulation10–12. Imbalance in the PC and PE composition of ER membranes associated with NAFLD can impair the function of the Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump that mediates ER calcium uptake13. The insulin-stimulated phosphoinositide 3-kinase (PI3K) pathway, which is involved in the recruitment of downstream signaling proteins with pleckstrin homology (PH) domains such as protein kinase B (AKT), is also affected by elevated cytosolic calcium levels14 that promote preferential binding of calcium to phosphoinositides, leading to insulin resistance12,14. Furthermore, chronic elevation of intracellular calcium levels can disrupt mitochondrial function by increasing mitochondrial calcium concentrations12,15 which in turn can increase the production of reactive oxygen species (ROS), mitochondrial depolarization, and a decrease in mitochondrial respiratory chain function12.
Previously, we reported the identification of neuronal regeneration-related protein (NREP) as an epigenetically regulated candidate with a regulatory role and the potential to act as a biomarker in the progression of NAFLD16. NREP, also known as P311, is a highly conserved 8 kD protein that has been associated with the TGF-β pathway17, wound healing18, nerve and lung regeneration19,20, and kidney fibrosis17. NREP silencing in HepG2 cells leads to steatosis with accumulation of cholesterol and triglycerides16.
We built on our previous observations by focusing on the transcriptomic, lipidomic, and signalomic alterations associated with altered NREP expression in primary human hepatocytes. We report that NREP silencing induces transcriptional remodeling that reflects downregulation of the phosphatidylinositol and one-carbon metabolism pathways and decreased phosphatidylcholine synthesis that is associated with upregulation of cholesterol synthesis and triglyceride accumulation. Our kinase inference analyses on phosphosites affected by NREP knockdown identified calcium/calmodulin-dependent protein kinase type II subunit alpha (CAMK2A). Our findings suggest that the reduction in NREP expression in primary human hepatocytes impacts hepatic intracellular calcium homeostasis, likely via an intimate relationship between the phosphatidylcholine and phosphatidylinositol pathways.
RESULTS
NREP silencing impacts gene expression networks that regulate cholesterol, phosphatidylinositol, and one-carbon metabolism in primary human hepatocytes
We first focused on the transcriptomic changes induced by NREP knock-down (KD) in primary human hepatocytes (Figure 1A). NREP silencing was efficient (Figure 1B), and principal component analyses showed sample segregation (Figure 1C). A total of 6442 genes were significantly altered in NREP KD compared to scramble (FDR<0.05) (Figure 1D). Enrichment pathway analyses of the upregulated genes (FDR<0.05) (Figure 1E) revealed enrichment for liver steatosis, cholesterol/steroid synthesis, and triglyceride synthesis (Figure 1E and 1F). On the other hand, analyses of the downregulated genes (FDR<0.05) (Figure 1G) revealed enrichment in phosphatidylinositol signaling and one-carbon metabolism pathways (Figure 1H).
Figure 1: NREP silencing impacts hepatic cholesterol and one-carbon metabolism gene expression network in primary human hepatocytes.
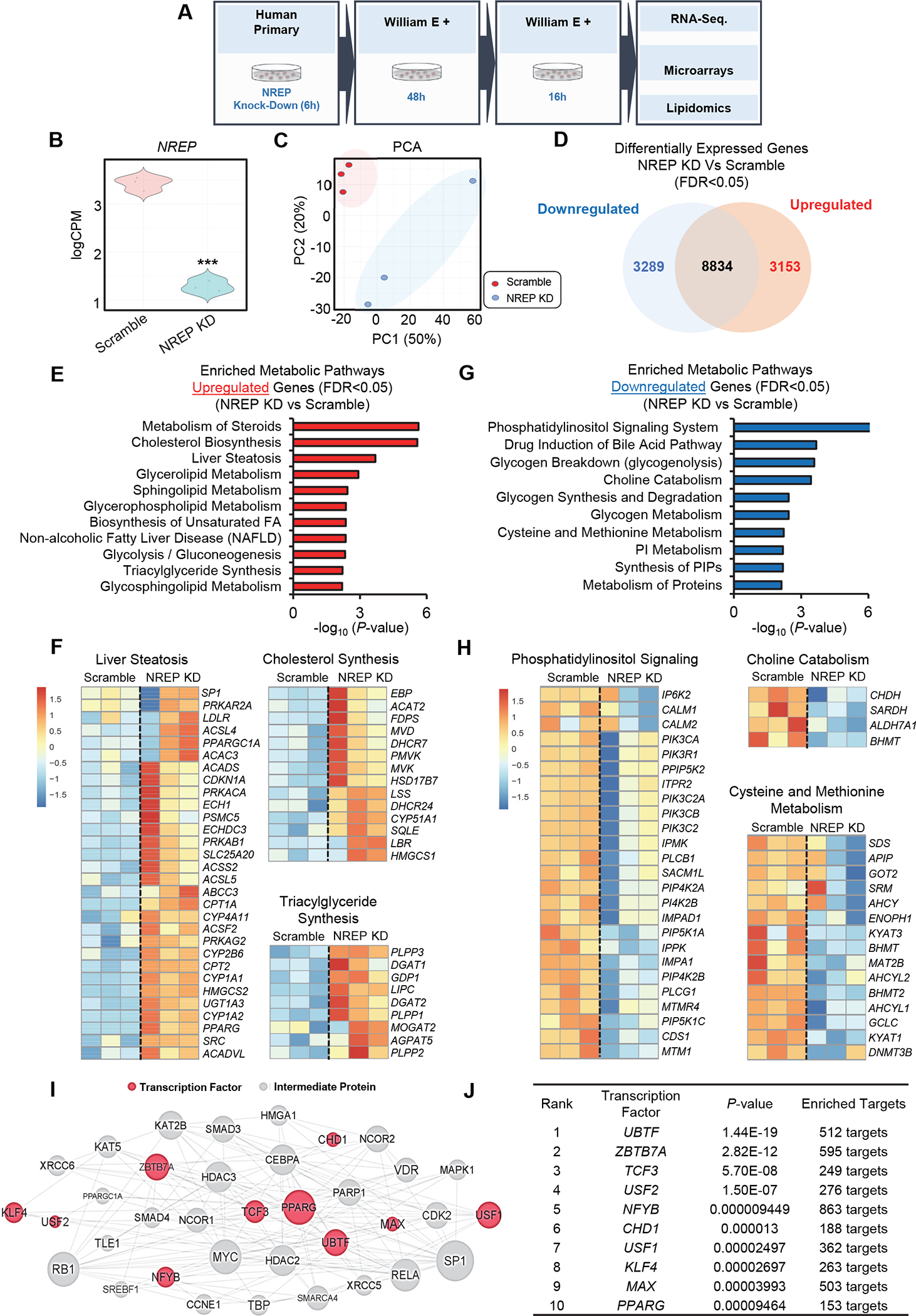
(A) Study overview. (B) Violin-plot representation of NREP mRNA levels in Scramble (represented in red) and NREP knock-down (KD) cells (represented in blue) measured by RNA-seq. (C) Principal component analyses (PCA). (D) Venn diagram of downregulated versus upregulated genes. (E) Pathway enrichment analyses on metabolism-associated upregulated genes. (F) Heat map representation of downregulated pathways and genes. (G) Pathway enrichment analyses on metabolic-associated downregulated genes. (H) Heat map representation of downregulated pathways and genes. (I) Induced network analyses of enriched transcription factors in NREP KD compared to Scramble. (J) Enriched transcription factors in NREP KD compared to Scramble. All, n=3 biological replicates/group. Statistical analyses were performed by Benjamini-Hochberg method (see Methods). All represented genes with FDR<0.05 in NREP KD compared to Scramble. Transcription factor upstream analyses with genes FDR<0.01 in NREP KD compared to Scramble.
Next, we compared the transcriptomic impact of NREP silencing in primary human hepatocytes with the data in HepG2 cells16. NREP downregulation induced profound transcriptomic remodeling in both cell systems (Supplementary Figure 1A). Enrichment pathway analyses of commonly upregulated genes revealed cholesterol metabolism (Supplementary Figure 1B). On the other hand, genes uniquely upregulated in primary hepatocytes upon NREP KD were enriched for focal adhesion and Rho GTPases (Supplementary Figure 1C) while those specific only to HepG2 cells identified P53 signaling and mitochondrial translation (Supplementary Figure 1D). We then performed similar analyses on the downregulated genes. Enriched pathways identified phosphatidylinositol signaling (Supplementary Figure 2E). On the other hand, genes specifically downregulated in primary hepatocytes were enriched for interferon signaling, mRNA splicing, and one-carbon metabolism (Supplementary Figure 2F). HepG2 in contrast revealed enrichment for peptide chain elongation, DNA damage response, and cell cycle pathways (Supplementary Figure 2G). Overall, these data reveal that NREP downregulation impacts cholesterol synthesis and phosphatidylinositol signaling in primary human hepatocytes and HepG2 cells. However, NREP silencing seems to impact one-carbon metabolism in primary hepatocytes particularly.
The robust and consistent transcriptomic changes induced by NREP silencing prompted us to hypothesize that NREP modulates the expression and/or activity of upstream transcriptional regulators. To test this we used de novo protein modeling software that uses machine learning21,22 to model NREP structure (Supplementary Figure 2, A and B) and predict its functions. Interestingly, the top biological process predicted for NREP molecular function was nucleic acid binding (Supplementary Figure 2C). Other predicted molecular functions included calmodulin-binding, suggesting a role for NREP in calcium signaling (Supplementary Figure 2C).
Next, we applied an in silico analysis that uses a signature of differentially expressed genes to create an interaction network while computing for transcription factors that act as mediators23. We observed several enriched transcription factors (Figure 1I, and 1J) with known roles in mediating hepatic lipid metabolism and NAFLD development including ZBTB7A24, USF225, NFYB26, USF127, and PPARG28. The latter is particularly interesting because NREP has been recently reported to interact physically with the PPARG promoter in adipocytes29. We used a specific double-stranded DNA (dsDNA) sequence containing the peroxisome proliferator response element (PPRE) to test PPARG DNA binding activity in nuclear extracts of HepG2. While NREP KD upregulated PPARG activity in basal state (BSA) there were no differences when cells were stimulated with palmitate (Supplementary Figure 2D). These data suggest that PPARG-independent transcription factors are involved in the transcriptomic remodeling associated with NREP downregulation in steatosis.
The fact that NREP was predicted to regulate metabolic processes, led us to further explore the genetic relationship with metabolic disease traits. Searching for NREP on the “The Common Metabolic Disease Knowledge Portal”30 that compiles data from several GWAS studies in metabolic-disease areas revealed NREP variants associated with metabolic phenotypes including BMI and HDL cholesterol (Supplementary Figure 2E and Supplementary Table 1). Several variants associated with HDL cholesterol were enriched near the 5’ UTR of NREP (Supplementary Figure 2F, 2G, and Supplementary Table 2). These results indicate that NREP is genetically linked to metabolic traits and its downregulation in primary human hepatocytes prompts a transcriptomic remodeling to downregulate phosphatidylinositol signaling and one-carbon metabolism that favors cholesterol synthesis and development of NAFLD.
Downregulation of genes associated with one-carbon metabolism is a common signature between NREP silencing in primary human hepatocytes and human NAFLD progression
Next, we sought to further explore the relevance of NREP in human NAFLD development. To this end, we reanalyzed a microarray dataset of samples of liver biopsy-proven patients with steatosis (n=14), steatohepatitis (n=8), and controls (n=10), transcriptionally well segregated by disease status26.
We began by first selecting the genes that were differentially expressed between NREP KD compared to scramble (FDR<0.05) and intersecting them with the differentially expressed genes (P<0.05) identified by comparing steatosis patients to controls (Figure 2A). Enriched pathway analyses of the 356 upregulated genes that were common between NREP KD and human steatosis identified pathways associated with apoptosis and cholesterol biosynthesis (Figure 2B). Analyses of the 587 downregulated genes that were common between NREP KD compared to scramble versus steatosis patients relative to controls identified several pathways involved in mRNA splicing, phosphatidylinositol signaling, and amino acid metabolism (Figure 2C). Among the commonly upregulated genes was mevalonate kinase (MVK), a key enzyme that acts early in sterol synthesis (Figure 2D). On the other hand, among the commonly downregulated genes, there were several related to the one-carbon metabolism pathways such as human betaine-homocysteine methyltransferase 2 (BHMT2) (Figure 2D). These data indicate that primary human hepatocytes merely harboring NREP KD and maintained in a basal state without exogenous stimuli share transcriptomic changes with human steatosis that mostly impact cholesterol synthesis and metabolism of amino acids.
Figure 2: Downregulation of one-carbon metabolism genes is a common signature between NREP silencing in primary human hepatocytes and human NAFLD.
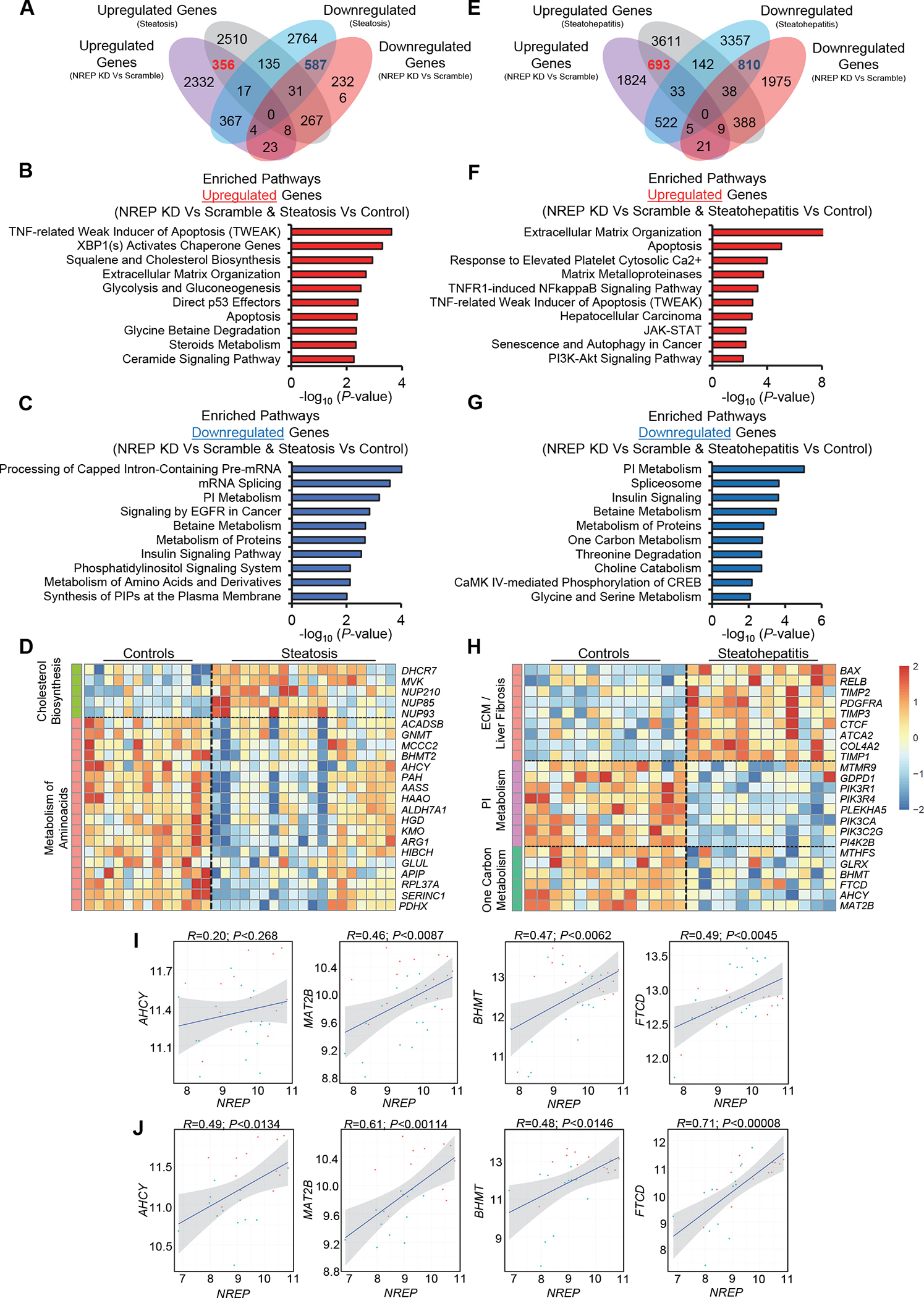
(A) Venn diagram of intersected genes between NREP knock-down (KD) versus Scramble and Steatosis versus Control patients reanalyzed from 31. (B) Pathway enrichment analyses on commonly upregulated genes between NREP KD versus Scramble and Steatosis versus Control patients. (C) Pathway enrichment analyses on commonly downregulated genes between NREP KD versus Scramble and Steatosis versus Control patients. (D) Heat-map of represented pathways and genes commonly altered between NREP KD versus Scramble and Steatosis versus Control patients. (E) Venn diagram of intersected genes between NREP KD versus Scramble and Steatohepatitis versus Control patients downloaded and reanalyzed from 31. (F) Pathway enrichment analyses on commonly upregulated genes between NREP KD versus Scramble and Steatohepatitis versus Control patients. (G) Pathway enrichment analyses on commonly downregulated genes between NREP KD versus Scramble and Steatohepatitis versus Control patients. (H) Heat-map of represented pathways and genes commonly upregulated genes between NREP KD versus Scramble and Steatohepatitis versus Control patients. (I) Pearson’s correlations between NREP and represented one-carbon metabolism genes in controls and Steatosis. (J) Pearson’s correlations between NREP and represented one-carbon metabolism genes in Controls and Steatohepatitis. Statistical analyses by the Benjamini-Hochberg method (see Methods). All represented genes with FDR<0.05 (our RNA-seq dataset) or P<0.05.
Next, we intersected the dataset of differentially expressed genes identified by comparing NREP KD with scramble versus the dataset identified by comparing human steatohepatitis with control patients (Figure 2E). Enriched pathway analyses of the 693 commonly upregulated genes (Figure 2F) showed several associated with ECM remodeling and fibrosis (Figure 2F). A similar analysis of the 810 commonly downregulated genes (Figure 2G) showed phosphatidylinositol metabolism and several pathways involved in amino acid metabolism (Figure 2G). Among the commonly upregulated genes were several associated with ECM remodeling and fibrosis such as platelet-derived growth factor receptor A (PDGFRA), alpha-smooth muscle actin (ACTA2), and connective tissue growth factor (CTGF) (Figure 2H). On the other hand, commonly downregulated genes included several rate-limiting enzymes involved in one-carbon metabolism, such as adenosylhomocysteinase (AHCY), methionine adenosyltransferase 2B (MAT2B), or 5-formyltetrahydrofolate cyclo-ligase (MTHFS). These data revealed that insufficiency of NREP in hepatocytes mimics the transcriptomic alterations that occur during the late stages of NAFLD, characterized by an upregulation of fibrotic genes and a decrease in one-carbon metabolism-related genes.
To explore the relevance of one-carbon metabolism further in our studies we performed correlation analyses between NREP mRNA levels and enzymes involved in one-carbon metabolism in human steatosis (Figure 2I), and steatohepatitis (Figure 2J) using the dataset described previously31. An obvious observation was a strong positive correlation between NREP mRNA and enzymes involved in one-carbon metabolism, suggesting the two increase concomitantly.
To further explore the relationship between NREP and one-carbon metabolism, we compared the downregulated genes (FDR<0.05) in primary human hepatocytes harboring NREP KD, with the downregulated genes (FDR<0.05) in an RNA-seq dataset from liver lysates of male mice fed a methionine-choline deficient diet (MCD) compared to chow diet-fed (Supplementary Figure 3A) (Tomohiko K & Kulkarni RN, unpublished). This revealed 418 commonly downregulates genes. Enriched pathway analyses identified metabolism, and several one-carbon metabolism-associated pathways (Supplementary Figure 3B and 3C). Importantly, Nrep was downregulated in livers of MCD-treated mice compared to chow, and its expression levels correlated positively with Ftcd and Ahcy (Supplementary Figure 3D), recapitulating the findings in human steatosis and NASH (Figure 2I and 2J). These data point to NREP as a bridge between one-carbon metabolism deficiency and the development of NAFLD in mice and humans.
NREP bridges one-carbon metabolism and NAFLD development by modulating phosphatidylcholine levels in primary human hepatocytes
To explore the impact of NREP deficiency in hepatic lipid metabolic we employed structural lipidomics analyses (study design in Figure 1A). NREP KD induced the upregulation of 214 and the downregulation of 161 lipid species (p<0.05) (Figure 3A). First, we classified the lipid species followed by further classifying each of the lipid classes (Figure 3B). Next, we used the Fry function of the Rotation Gene Set Test (Roast) in the limma32 R package to determine the overall direction (“UP” or “Down”) of lipid class using the proportion of lipids that are up or down at p<0.05. Finally, we calculated the percentage of lipids showing up- or downregulation within each lipid class (Figure 3B). Consistent with our gene expression data and previous data on hepatic cell lines16, NREP silencing led to an upregulation of cholesterol esters, free fatty acids (FFA), and triacylglycerol (TG) content in primary human hepatocytes harboring NREP KD (Figure 3B and 3C). Among the most significantly altered cholesterol ester species were 18:2, 20:0, and 22:1 (Figure 3C). Most significantly altered TAGs by NREP KD included TAG 48:7, 60:6, and 62:13, while the altered FFAs included 14:0, and 22:5 (Figure 3C).
Figure 3: NREP downregulation bridges one-carbon metabolism and NAFLD development by decreasing phosphatidylcholine levels in primary human hepatocytes.
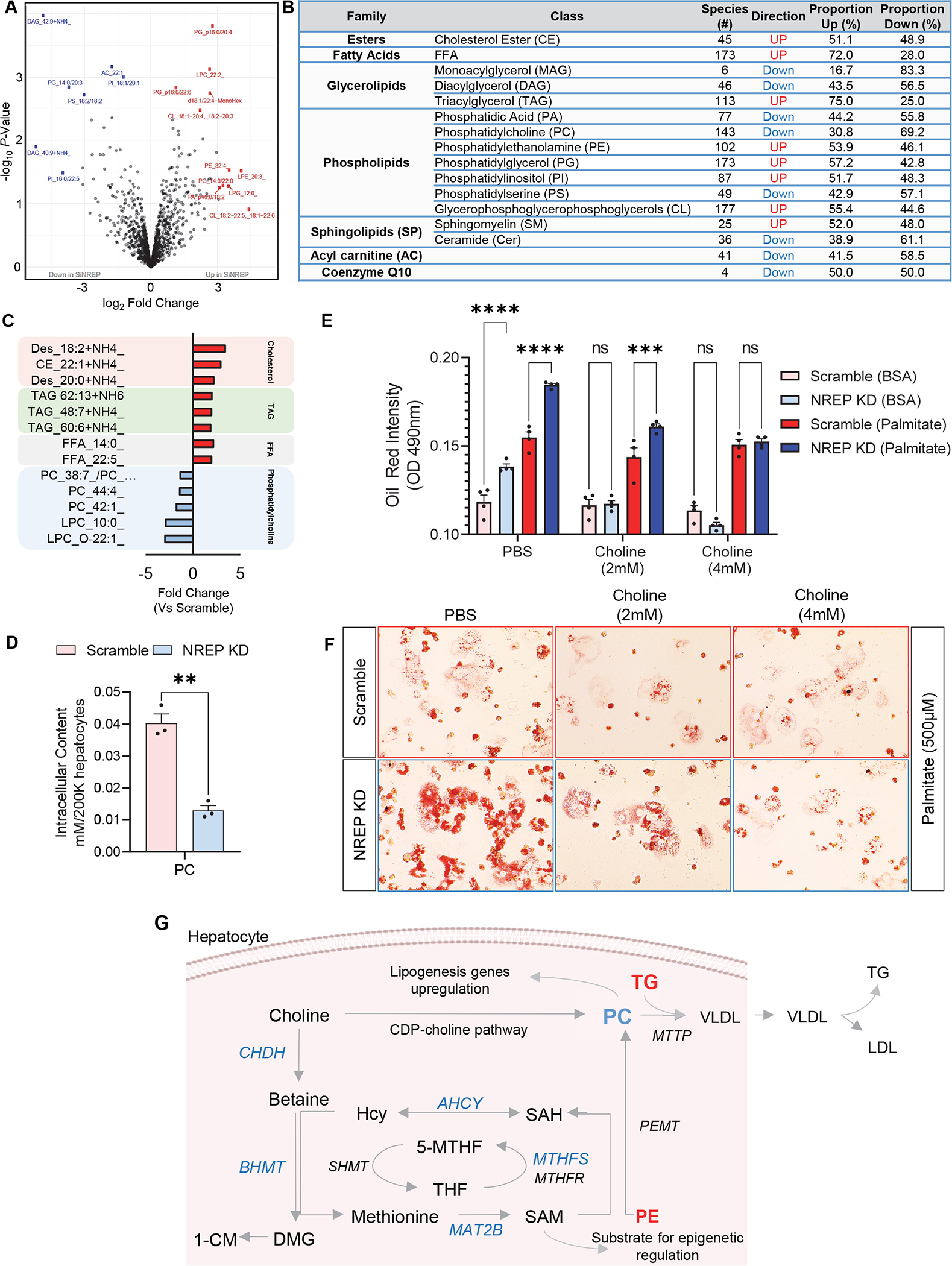
(A) Volcano-plot of lipidomics analysis in primary human hepatocytes harboring NREP KD compared to Scramble hepatocytes. (B) Effect of NREP KD on lipid class composition in primary human hepatocytes. (C) Cholesterol, free fatty acids, and PC lipid species most significantly impacted by NREP KD (P<0.05). (D) Phosphatidylcholine content levels in NREP KD and Scramble primary human hepatocytes (n=3). (E) Oil red intensity in Scramble or NREP KD primary human hepatocytes treated for 24h with PBS or choline chloride in the presence of BSA or 500 μM palmitate (n=4). (F) Representative oil red staining of Scramble or NREP KD cells challenged with 500 μM palmitate in the presence of PBS, 2mM choline chloride, or 4mM of choline chloride (n=4). (G) Scheme depicting one-carbon metabolism link with phosphatidylcholine synthesis and hepatic triglycerides export. PC, phosphatidylcholine; TG, triglycerides; Hcy, homocysteine; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; THF, tetrahydrofolate. Genes are depicted in italic. Downregulated genes in blue. Lipid classes in bold; Red represents upregulated and blue downregulated upon NREP KD compared to Scramble. All, n=3 (or otherwise stated) biological replicates/group. Statistical analyses by Benjamini-Hochberg method in A, B, and C. Unpaired Two-tailed t-test in D. Two-way ANOVA with Turkey’s multiple comparisons test in E.
NAFLD is characterized by structural lipid remodeling wherein phospholipids are particularly affected33. Several phospholipid classes were altered by NREP silencing (Figure 3B) including phosphatidylcholine (PC) (Figure 3C). We went on to first validate PC downregulation in NREP KD hepatocytes (Figure 3D). Next, we evaluated lipid accumulation upon NREP KD and the impact of choline supplementation (Figure 3E). NREP KD induced steatosis as previously described in hepatocytes16, and consistently, choline supplementation ameliorated lipid accumulation in NREP KD comparable to scramble cells, particularly when challenged with palmitate (Figure 3E and 3F).
Next, we integrated our RNA-sequencing data where we compared primary human hepatocytes harboring NREP deficiency to scramble, and visualized the transcriptomic changes associated with cholesterol and one-carbon metabolism. NREP silencing led to an upregulation of several enzymes involved in cholesterol (Supplementary Figure 4A) and steroid synthesis (Supplementary Figure 4A). Among the upregulated genes induced by NREP KD were enzymes important in regulating low-density lipoprotein (LDL) packaging and transport such as proprotein convertase subtilisin/kexin type 9 (PCSK9) (Supplementary Figure 4A).
Overall, these results demonstrate that NREP silencing in primary human hepatocytes decreases the expression of genes involved in the synthesis of one-carbon metabolites, and downregulation of PC, and the development of steatosis (Figure 3G).
NREP impacts calcium signaling networks in primary human hepatocytes
To dissect the pathways underlying the effects of NREP in an unbiased manner we employed phosphor-antibody microarrays. NREP KD-induced changes in the signalome are reflected by clear segregation among samples (Figure 4A). Among the most significantly altered phosphosites, we identified hyperphosphorylation of BRCA1 (S988), and PDGFRA (Y754), suggesting that hepatic NREP insufficiency activates DNA damage34, and fibrotic signaling pathways35 respectively. For a global perspective on the role of NREP in hepatocyte signaling, we performed enriched pathway analyses (Figure 4C) and identified erythropoietin, JAK-STAT, PDGFR-β, carbon metabolism in cancer, and calmodulin among other pathways associated with cancer and fibrosis (Figure 4C).
Figure 4: Signalome analyses of NREP-deficient primary human hepatocytes reveal pathways that are common to NAFLD and those associating NREP with calcium signaling.
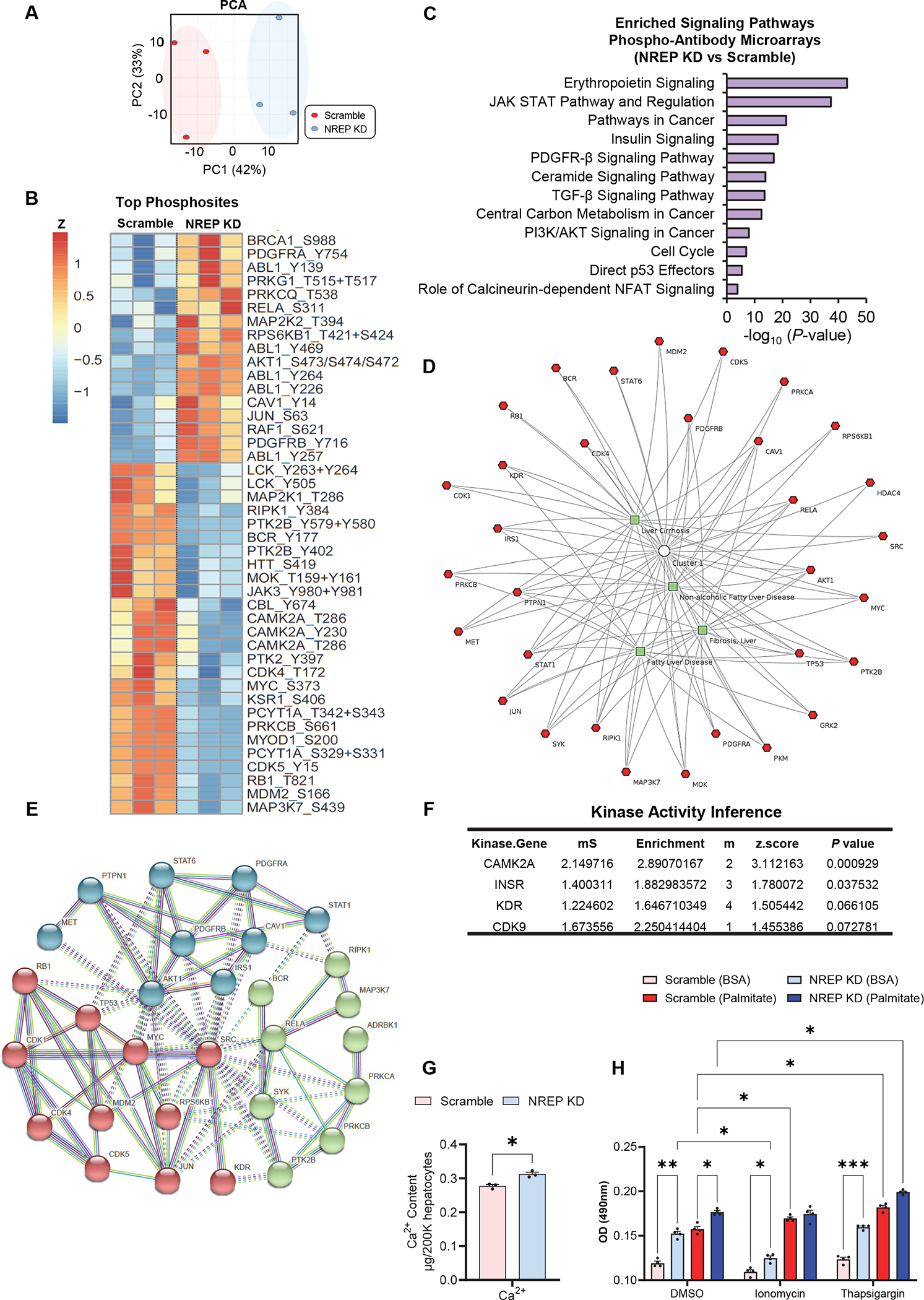
(A) Principal component analyses (PCA) of phospho-antibody microarrays performed in NREP KD primary human hepatocytes compared to Scramble. (B) Heat-map visualization of top altered phosphosites. (C) Pathway enrichment analyses on altered phosphosites. (D) Multi-cluster gene functional enrichment analyses identify several proteins associated with NAFLD, fibrosis, and cirrhosis. (E) Functional protein-protein interaction analyses on proteins presenting altered phosphosites. (F) Kinase substrate analyses on the differentially abundant phosphosites in NREP KD versus Scramble. (G) Calcium content levels in NREP KD and Scramble primary human hepatocytes. (H) Oil red intensity in Scramble or NREP KD primary human hepatocytes treated for 24h with DMSO, 1μg/mL ionomycin, or 1μM thapsigargin in the presence of BSA or 500 μM palmitate (n=4). All, n=3 biological replicates/group (or otherwise stated). Statistical analyses by Benjamini-Hochberg method (see Methods). Unpaired Two-tailed t-test in G. Two-way ANOVA with Turkey’s multiple comparisons test in H. All represented phosphosites with P<0.05 in NREP KD versus Scramble.
To narrow down the network and visualize the direct signaling link between NREP and the development of NAFLD we performed a multi-cluster gene functional enrichment analysis. Specifically, we examined proteins presenting differential phosphorylation in NREP KD compared to scramble that were directly associated with NAFLD, fibrosis, or cirrhosis terms (Figure 4D). Interestingly, the candidate list of phosphosites altered upon NREP KD in primary human hepatocytes presented several proteins that were associated with these disease terms to allow functional protein-protein analyses (Figure 4E). The three main interaction clusters included one that associated with cell cycle control (illustrated in red) such as MYC proto-oncogene, bHLH transcription factor (MYC), or retinoblastoma protein 1 (RB1). The second cluster was associated with fibrosis (illustrated in blue) and included PDGFRA or PDGFRB. Finally, a third cluster (illustrated in green) linked to proteins associated with the regulation of cell death by calcium and NF-kB signaling and included protein kinase C (PRKCA and PRKCB), receptor-interacting serine/threonine-protein kinase 1 (RIPK1), and nuclear factor NF-kappa-B p65 subunit (RELA) (Figure 4E).
Next, to identify an upstream kinase driving some of these phosphorylation changes induced by NREP we used an in silico approach36 to infer kinase activity among those phosphosites that were significantly different between NREP KD and scramble. This analysis identified calcium/calmodulin-dependent protein kinase II alpha (CAMK2A), and insulin receptor (INSR) among the most enriched kinases (Figure 4F). To further visualize the relationship between NREP and calcium signaling, we integrated our RNA-sequencing data, obtained by comparing NREP KD to scramble in primary human hepatocytes, with KEEG annotated pathways37 and used Pathview38 to visualize differentially expressed genes in our dataset and specifically those involved in calcium signaling (Supplementary Figure 4B). We identified several differentially expressed genes in this pathway, including the upregulation of several transmembrane calcium channels and receptors (Supplementary Figure 4B).
Finally, we explored the relationship between NREP downregulation and calcium mobilization in steatosis. First, we detected an increase in calcium content in primary human hepatocytes harboring NREP KD (Figure 4G). Next, we challenged hepatocytes with NREP insufficiency or scramble with palmitate or BSA (basal) and treated them with ionomycin39,40, thapsigargin 41, or DMSO (Figure 4H). Consistent with our previous experiments (Figure 3E), NREP KD increased steatosis both in the basal and upon palmitate treatment (Figure 4H). Ionomycin treatment decreased lipid accumulation in the basal state while it hampered steatosis in palmitate-treated NREP KD cells comparable to scramble. On the other hand, thapsigargin increased steatosis in palmitate-treated cells similarly between scramble and NREP KD compared to DMSO-treated (Figure 4H). These results suggest that NREP downregulation leads to mitochondrial calcium overload, which is transiently ameliorated with ionomycin but not with thapsigargin. This is consistent with the decreased oxygen consumption rate (OCR) seen in HepG2 cells harboring NREP KD16.
Overall, the signalome analyses of NREP downregulation in primary human hepatocytes support the notion that NREP acts on one-carbon metabolism to promote the development of NAFLD by impacting pathways that regulate cell death, inflammation, and fibrosis, while synergistically acting on calcium signaling12,42.
DISCUSSION
Previously, we reported the identification of NREP as a new epigenetically regulated gene that bridges TGF-β signaling and hepatic lipid metabolism16. Here, we adopted a comprehensive ‘omics’ approach by integrating the transcriptomic, lipidomic, and signalomic changes associated with NREP silencing. Our work métier relies on the usage of state-of-the-art techniques and the utilization of viable human hepatocytes that overcome many limitations associated with hepatic cell lines.
Silencing NREP in primary human hepatocytes revealed upregulated genes that are common with those induced in human steatosis and involve cholesterol synthesis. Furthermore, lipidomics analyses of NREP-deficient hepatocytes revealed accumulation of cholesterol esters. Examples of genes upregulated by NREP KD and increased in human steatosis included PCSK9. PCSK9 promotes LDLR degradation, leading to LDL accumulation in plasma, and is linked to familial hypercholesterolemia. PCSK9 chemical inhibitors are being explored to treat human NAFLD43. NREP insufficiency also favored the transcriptomic upregulation of several pathways that are hallmarks of NAFLD progression, including fibrosis, inflammation, and cell death.
The downregulation of NREP had a profound impact on the transcriptomics of gene networks associated with one-carbon metabolism and phosphatidylinositol signaling. This effect was further compounded by the observed downregulation of PC in NREP-deficient hepatocytes. Consistently, choline supplementation to NREP-deficient hepatocytes rescued steatosis. These findings have led us to investigate the potential involvement of calcium signaling, which has been increasingly recognized for its critical role in NAFLD research12,44. Specifically, the relationship between calcium signaling and the aforementioned metabolic pathways suggests a potentially important mechanistic link, which merits further exploration.
We provide several lines of evidence that NREP modulates calcium signaling. First, protein structure modeling and function prediction revealed calmodulin-binding by NREP. Second, enrichment analyses of the differentially expressed genes in NREP KD, which are common with human steatohepatitis identified pathways linked to calcium and CaMK IV-mediated phosphorylation of CREB (Figure 2G). Third, kinase activity inference based on the phosphosites altered in NREP KD hepatocytes identified CAMK2A as among the most enriched kinase. Fourth, calcium content levels were increased in human primary hepatocytes deficient for NREP. Fifth, ionomycin treatment reduced steatosis in palmitate-treated NREP deficient hepatocytes to a level comparable to that in scramble cells, while thapsigargin treatment had the opposite effect, exacerbating steatosis equally in both scramble and NREP KD cells. These observations suggest that NREP modulates calcium levels in hepatocytes, potentially leading to mitochondrial calcium overload. This conclusion is supported by the mechanism of action of the two agents: ionomycin increases cytosolic calcium by mobilizing calcium stores from the endoplasmic reticulum (ER) and mitochondria39,45, while thapsigargin specifically depletes ER calcium, leading to an increase in cytosolic calcium levels41.
In summary, the present study provides an unbiased characterization of the biological role of NREP in human hepatocytes and its relevance in mediating the development of NAFLD. We provide new insights into the role of NREP in regulating one-carbon metabolism and calcium signaling. These observations provide a strong rationale for the targeting of hepatic NREP to improve and/or block the progression of NAFLD.
LIMTATIONS OF THE STUDY
While our study provides valuable insights into the role of NREP in human hepatocytes and its potential involvement in the pathophysiology of NAFLD, there are some limitations that should be considered. One limitation is that our experiments were conducted in vitro using primary human hepatocytes. While this approach has the advantage of using viable human cells that overcome many limitations associated with cell lines, it may not fully reflect the complexity of the in vivo environment, including the influence of other cell types and tissue-specific factors.
Additionally, while we provide evidence for the involvement of NREP in calcium signaling and its potential role in NAFLD progression, further studies are needed to elucidate the exact mechanisms of NREP regulation of calcium homeostasis and how this may contribute to NAFLD development. Finally, while our findings suggest a potential therapeutic target in hepatic NREP, additional studies are required to determine the efficacy and safety of targeting NREP in human NAFLD. Further investigations are needed to establish the clinical relevance of NREP modulation in NAFLD and the potential for off-target effects of targeting NREP, including effects on other metabolic pathways and potential adverse effects.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rohit N Kulkarni (rohit.kulkanri@joslin.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
RNA-sequencing data in primary human hepatocytes have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus under accession code GSE229455. Antibody microarrays data in primary human hepatocytes have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus under accession code GSE229432.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Primary human hepatocyte culture
We used pure, highly viable, and plateable primary human hepatocytes from 5 pooled donors (ThermoFisher, #HMCPP5). Culture of plateable primary human hepatocytes was performed as previously reported16. Briefly, hepatocytes were seeded on 6-well collagen-treated plates containing William E media supplemented with primary hepatocyte supplements (ThermoFisher) and HepExtend supplement (ThermoFisher).
METHOD DETAILS
NREP knock-down in primary human hepatocytes
Hepatocytes were allowed to attach for 6h. Media was exchanged to eliminate non-viable/attached cells that were forward transfected with Lipofectamine RNAiMAX (ThermoFisher) and 100nM of siGENOME Non-Targeting siRNA Pool (Dharmacon, # D-001206–13-05), or siGENOME human NREP siRNA (Dharmacon, # M-019848–00-0005) in complete media. Media was then changed at 6h post-transfection with complete media. At 48h post-transfection cells were starved for 16h in William E media containing 0.1% BSA, washed three times with ice-cold DPBS, and snap-frozen and pelleted for further analyses.
Chemical treatments of primary human hepatocytes
For choline supplementation experiments hepatocytes were forward transfected and at 48h post-transfection were cultured for 24h in William E media containing BSA, or BSA + 500μM of palmitate (SIGMA) as previously described 16 supplemented with PBS, or 2mM choline chloride (SIGMA), or 4mM choline chloride (SGIMA). For calcium experiments, hepatocytes were forward transfected and at 48h post-transfection were cultured for 24h in William E media containing BSA, or BSA + 500μM of palmitate (SIGMA) as previously described 16 and were challenged with 1μg/mL ionomycin (SIGMA) or 1μM thapsigargin (SIGMA). Phosphatidylcholine levels were measured in 2×10^5 hepatocytes/sample using a colorimetric phosphatidylcholine assay kit (#10009926, Cayman) according to manufacturer guidelines. Calcium content was measured in 2×10^5 hepatocytes/sample using a colorimetric calcium assay kit (#ab102505, Abcam) according to manufacturer guidelines. Steatosis was measured using a steatosis colorimetric assay kit based on oil red staining (#10012643, Caymen) according to manufacturer guidelines.
RNA isolation
RNA isolation for RNA-seq. studies were performed as previously reported 46. In brief, total RNA (>200nt) was extracted using standard Trizol reagent (Invitrogen) according to manufacturer instructions. The resultant aqueous phase was mixed (1:1) with 70% RNA-free ethanol and added to Qiagen Rneasy mini kit columns (Qiagen) and the kit protocol was followed.
RNA-sequencing
Purified total RNA (500ng) was used for mRNA isolation and subsequent transcriptome library preparation using the TruSeq Stranded mRNA Library Preparation Kit (Illumina). Libraries were sequenced on the NovaSeq 6000 Sequencing System to obtain at least 50 million paired-end 100bp reads per sample. FASTQC was used to assess the quality of paired-end or single-end read sequences.
The count’s table tx2gene_counts.rds is generated by aligning reads to the mouse transcriptome (Ensembl version 94) using Kallisto and converting transcript counts to gene counts using tximport. To filter the low-expressing genes, we keep genes that have more than 0.5 counts per million (CPM) in at least 4 samples. We normalized the counts by weighted trimmed mean of M-values (TMM). To use linear models in the following analysis, we perform Voom transformation to transform counts into log CPM, where CPM = 1e+6 * count of a gene / (total counts of the sample * normalization factor of the sample).
Kinexus antibody-microarrays
Kinexus antibody microarrays were performed with modifications compared to the previous report 46. Briefly, protein lysates from 2×10^6 hepatocytes/sample were covalently labeled with biotin. Free biotin molecules were then removed after labeling reactions by gel filtration. After blocking non-specific binding sites on the array, an incubation chamber was mounted onto the Kinex™ KAM-1325 antibody microarrays to permit the loading of 2 samples side by side on the same chip. Following sample incubation, unbound proteins were washed away and the array was then probed with an anti-biotin antibody that is labeled with a proprietary fluorescent dye combination. Each array produced a pair of 16-bit images, which were captured with a Perkin-Elmer ScanArray Reader laser array scanner (Waltham, MA). Signal quantification was performed with ImaGene 9.0 from BioDiscovery (El Segundo, CA) with predetermined settings for spot segmentation and background correction.
For normalized phosphosite/protein differential abundance, we used limma, an R package that powers differential expression analyses49. We adjusted for the batch effects and performed moderated t-tests between NREP KD and scramble as previously described 46.
Lipidomics
Human primary hepatocyte cell pellets were resuspended in 500 μL of PBS. Sample aliquot volume was normalized by 1 mg protein determined by bicinchoninic acid (BCA) assay. Structural lipidomic analysis involved a modified Bligh and Dyer extraction utilizing a customized, automated liquid-liquid extraction sequence on a Hamilton Robotics StarLET Robot System. Four mL of chloroform:methanol (1:1, by vol) was added to the samples followed by the addition of 50 μL of a cocktail mixture of deuterium-labeled, odd chain, and extremely low naturally abundant internal standards. A front extraction was performed by adding 2 mL of 50mM lithium chloride to sample tubes, vortexed in Multi Tube Vortexer for 3 minutes, centrifuged at 1000 g for five minutes, and the bottom chloroform layer was carefully transferred to a new glass tube. An additional 1.8 mL of chloroform was added to source tubes, vortexed and centrifuged as described above, and transferred for a total of three chloroform transfers. The samples were dried down under a stream of N2 gas and resuspended in chloroform: methanol (1:1, by vol). A back extraction was performed for additional sample clean-up to remove contamination of protein, salts, and residual aqueous layer from front extraction. Two mL of 10mM lithium chloride was added to resuspended samples and the same protocol was followed as front extraction. Samples were dried down under N2, reconstituted in chloroform: methanol (1:1, by vol), and stored at −20°C. Samples were prepared to run on the mass spectrometer by diluting 250x in isopropanol:methanol:acetonitrile: water (3:3:3:1, by vol) with 2 mM ammonium acetate.
Electrospray ionization (ESI) MS was performed on a Sciex TripleTOF 5600+ coupled with a direct injection loop on an Eksigent Ekspert MicroLC 200 system. The ESI DuoSpray Ion Source parameter settings were set to ion source gas 1 (GS1) at 10, ion source gas 2 (GS2) at 10, curtain gas (CUR) at 20, temperature at 300°C, and Ion Spray Voltage Floating (ISVF) at 5500 V for positive mode and −4500 V for negative mode. The MS/MSALL acquisition method consisted of two experiments, first a TOF MS survey scan acquiring data from 200–1200 m/z. The second experiment in the acquisition method consisted of 1000 high-resolution production scans with precursors evenly spaced from m/z 200.05 to m/z 1200.05 with 300 ms of accumulation time for each scan. The collision energy was set to 35 eV and −35 eV for positive and negative modes, respectively, with a collision energy spread (CES) of ±15 eV. Cardiolipin and monoacylglycerol species were analyzed with unique acquisition methods as previously described 47. Mass shift was controlled by calibrant solution delivered every 10 samples at 500 μL/min by the atmospheric-pressure chemical ionization (APCI) probe. The MS/MSALL data was acquired by Analyst TF 1.7 software (Sciex) and processed with MultiQuant 1.2.2.5 (Sciex) using an in-house database of lipid species for identification and quantification.
After normalization to internal standards, there were 4743 missing values in the lipidomics data. We then checked the relationship between the logit missingness probability and the average of the observed lipid’s log abundance. They were negatively correlated to each other, which indicates the missing values abundance was below the detection limits. We kept lipids that have 50% valid values (1221 lipids were kept with 9.98% missing values). We then imputed the remaining missing lipid values half the minimum abundance of the corresponding lipid. To discover lipids that were differentially abundant between groups (i.e. NREP KD vs Scramble), we use limma 48 to perform moderated t-tests. We also estimate relative quality weights for each sample and use them in the tests. Next, we used the Fry function of the Rotation Gene Set Test (Roast) 32 in the limma R package 48 to determine the overall direction (“UP” or “Down”) of lipid class using the proportion of lipids that are up at p<0.05, and the proportion that are down at p<0.05. Finally, we calculated the proportion of lipids showing up- or downregulation within each lipid class.
Pathways and in silico analyses
NREP protein structure modeling and functional prediction were performed using The PSIPRED Workbench 49. Pathway enrichment analyses were performed using ConsensusPathDB using default settings 50. Pathway maps were generated using Pathview 38. Transcription factor enrichment analyses (TFEA) was performed using eXpression2Kinases (X2K) 23. Multi-cluster gene functional enrichment analyses was conducted on ToppGene 51 using our candidate protein list and the described disease terms. Kinase activity analyses were performed using The KSEA App 36 using statistically altered phosphosites between NREP KD compared to scramble. Functional protein-protein functional networks were constructed using STRING 52.
QUANTIFICATION AND STATISTICAL ANALYSIS
False discovery rates (FDRs) and P-values were calculated using the Benjamini-Hochberg method using the R software in the omics analyses. Pearson’s correlations were performed using the R software. Non-omics statistical analyses were performed using GraphPad Prism software version 8.0 (GraphPad Software, USA). To test whether the collected numerical data are normally distributed, the D’Agostino and Pearson was used. An FDR or P-value less than 0.05 was considered significant.
Supplementary Material
Supplementary Table 1: Variant associations with phenotypes, related to Figure 1
Supplementary Table 2: Variants in region_HDL cholesterol, related to Figure 1
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Human Plateable Hepatocytes, 5-Donor | ThermoFisher | Cat# HMCPP5 |
| Chemicals, peptides, and recombinant proteins | ||
| Choline chloride | Milipore-Sigma | Cat# C7017 |
| Thapsigargin | Milipore-Sigma | Cat# T9033 |
| Ionomycin | Milipore-Sigma | Cat# I9657–1MG |
| Bovine Serum Albumin (BSA), Fraction V | Roche | Cat# 10738328103 |
| Sodium palmitate | Milipore-Sigma | Cat# P9767 |
| Critical commercial assays | ||
| Steatosis Colorimetric Assay Kit | Cayman Chemical | Cat# 10012643 |
| Calcium Assay Kit (Colorimetric) | Abcam | Cat# ab102505 |
| Phosphatidylcholine Assay Kit (Colorimetric/Fluorometric) | Abcam | Cat# ab83377 |
| Deposited data | ||
| Microarray control, steatosis, steatohepatitis | Starmann et al.31 | GSE33814 |
| RNA-sequencing in primary human hepatocytes | This study | GSE229455 |
| Antibody-microarrays in primary human hepatocytes | This study | GSE229432 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| eXpression2Kinases (X2K) | Clarke et al.23 | |
| KEEG | Kanehisa et al.37 | |
| The KSEA App | Wiredja et al.36 | |
| limma | Ritchie et al.48 | |
| Pathview Web | Luo et al.38 | |
| The PSIPRED Protein Analysis Workbench | Buchan et al.22 | |
| ROAST | Wu et al.32 | |
| ToppGene Suite | Chen et al.51 | |
| fastp | Chen et al.53 | |
| ConsensusPathDB | Herwig et al.54 | |
| Other | ||
Highlights.
NREP deficiency mimics key pathways of human NAFLD.
Lack of NREP leads to increased cholesterol and decreased phosphatidylcholine.
Calcium signaling is a potential mediator of NREP deficiency.
Targeting hepatic NREP provides a promising strategy for improving NAFLD.
SIGNIFICANCE.
Nonalcoholic fatty liver disease (NAFLD) is a significant public health concern affecting millions of people worldwide. Despite its prevalence, the molecular mechanisms underlying its pathogenesis remain poorly understood. In this study, we investigated the impact of NREP insufficiency using RNA-sequencing, lipidomics, and antibody-microarrays on primary human hepatocytes. Our findings revealed that NREP knockdown-induced transcriptomic remodeling that overlapped with key pathways impacted in human steatosis and steatohepatitis. Furthermore, lipidomics analyses showed an increase in cholesterol esters and triglycerides and decreased phosphatidylcholine levels in NREP-deficient hepatocytes. Our results also identified calcium signaling as a potential mediator of NREP insufficiency’s effects. Taken together, our study provides novel insights into the molecular mechanisms underlying NAFLD pathogenesis and highlights NREP as a potential therapeutic target for NAFLD.
ACKNOWLEDGEMENTS
We thank Jonathan Dreyfuss PhD and Hui Pan PhD of Joslin Bioinformatics for assistance with the bioinformatics analyses. We thank Mike Kiebish PhD, Val Bussberg PhD, and Beverlyn Boateng PhD of BERG Health for their assistance with lipidomics experiments. We thank DNA Link for their assistance with RNA-sequencing and Kinexus for their help with antibody microarrays. RNK is supported by the US National Institutes of Health (NIH) grants R01 DK067536, R01 DK103215, P01 DK036836, and R01 DK117639. R.N.K. acknowledges support from the Margaret A. Congleton Endowed Chair. DFDJ acknowledges support by Mary K. Iacocca Junior Postdoctoral Fellowship and American Diabetes Association grant #7-21-PDF-140.
Footnotes
DECLARATION OF INTERESTS
R.N.K is on the scientific advisory council of Novo Nordisk, Biomea, and Inversago.
STUDY APPROVAL
Human plateable primary hepatocytes from 5-donors were purchased from Gibco (Thermo Fisher). All human studies and protocols used were approved by the Joslin Diabetes Center’s Committee on Human Studies (CHS#5–05).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Brunt EM, and Tiniakos DG (2010). Histopathology of nonalcoholic fatty liver disease. World journal of gastroenterology 16, 5286–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brumbaugh DE, and Friedman JE (2013). Developmental origins of nonalcoholic fatty liver disease. Pediatric Research 75, 140. 10.1038/pr.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, and Bugianesi E (2018). Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15, 11–20. 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 4.Loomba R, Friedman SL, and Shulman GI (2021). Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564. 10.1016/j.cell.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 5.Walker AK (2017). 1-Carbon Cycle Metabolites Methylate Their Way to Fatty Liver. Trends Endocrinol Metab 28, 63–72. 10.1016/j.tem.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radziejewska A, Muzsik A, Milagro FI, Martinez JA, and Chmurzynska A (2020). One-Carbon Metabolism and Nonalcoholic Fatty Liver Disease: The Crosstalk between Nutrients, Microbiota, and Genetics. Lifestyle Genom 13, 53–63. 10.1159/000504602. [DOI] [PubMed] [Google Scholar]
- 7.van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, and Jacobs RL (2017). The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr 1859, 1558–1572. 10.1016/j.bbamem.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Chiappini F, Coilly A, Kadar H, Gual P, Tran A, Desterke C, Samuel D, Duclos-Vallee JC, Touboul D, Bertrand-Michel J, et al. (2017). Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci Rep 7, 46658. 10.1038/srep46658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, and Sanyal AJ (2007). A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46, 1081–1090. 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 10.Bhowmick S, Singh V, Jash S, Lal M, and Sinha Roy S (2021). Mitochondrial metabolism and calcium homeostasis in the development of NAFLD leading to hepatocellular carcinoma. Mitochondrion 58, 24–37. 10.1016/j.mito.2021.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Ali ES, and Petrovsky N (2019). Calcium Signaling As a Therapeutic Target for Liver Steatosis. Trends in Endocrinology & Metabolism 30, 270–281. 10.1016/j.tem.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Oliva-Vilarnau N, Hankeova S, Vorrink SU, Mkrtchian S, Andersson ER, and Lauschke VM (2018). Calcium Signaling in Liver Injury and Regeneration. Front Med (Lausanne) 5, 192. 10.3389/fmed.2018.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, and Hotamisligil GS (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang JK, Kim OH, Hur J, Yu SH, Lamichhane S, Lee JW, Ojha U, Hong JH, Lee CS, Cha JY, et al. (2017). Increased intracellular Ca(2+) concentrations prevent membrane localization of PH domains through the formation of Ca(2+)-phosphoinositides. Proc Natl Acad Sci U S A 114, 11926–11931. 10.1073/pnas.1706489114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomar D, Jana F, Dong Z, Quinn WJ 3rd, Jadiya P, Breves SL, Daw CC, Srikantan S, Shanmughapriya S, Nemani N, et al. (2019). Blockade of MCU-Mediated Ca(2+) Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell Rep 26, 3709–3725 e3707. 10.1016/j.celrep.2019.02.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Jesus DF, Orime K, Kaminska D, Kimura T, Basile G, Wang CH, Haertle L, Riemens R, Brown NK, Hu J, et al. (2020). Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J Clin Invest 130, 2391–2407. 10.1172/JCI127502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao Z, Yang S, He W, Li L, Xu R, Zhang X, Li H, Zhan R, Sun W, Tan J, et al. (2015). P311 promotes renal fibrosis via TGFbeta1/Smad signaling. Sci Rep 5, 17032. 10.1038/srep17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan D, Zhe X, Jakkaraju S, Taylor GA, and Schuger L (2002). P311 induces a TGF-β1–independent, nonfibrogenic myofibroblast phenotype. The Journal of Clinical Investigation 110, 1349–1358. 10.1172/jci15614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujitani M, Yamagishi S, Che YH, Hata K, Kubo T, Ino H, Tohyama M, and Yamashita T (2004). P311 accelerates nerve regeneration of the axotomized facial nerve. Journal of Neurochemistry 91, 737–744. 10.1111/j.1471-4159.2004.02738.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhao L, Leung JK, Yamamoto H, Goswami S, Kheradmand F, and Vu TH (2006). Identification of P311 as a Potential Gene Regulating Alveolar Generation. American Journal of Respiratory Cell and Molecular Biology 35, 48–54. 10.1165/rcmb.2005-0475OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greener JG, Kandathil SM, and Jones DT (2019). Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat Commun 10, 3977. 10.1038/s41467-019-11994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchan DWA, and Jones DT (2019). The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 47, W402–W407. 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clarke DJB, Kuleshov MV, Schilder BM, Torre D, Duffy ME, Keenan AB, Lachmann A, Feldmann AS, Gundersen GW, Silverstein MC, et al. (2018). eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res 46, W171–W179. 10.1093/nar/gky458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou JP, Ren YD, Xu QY, Song Y, Zhou F, Chen MY, Liu JJ, Chen LG, and Pan JS (2020). Obesity-Induced Upregulation of ZBTB7A Promotes Lipid Accumulation through SREBP1. Biomed Res Int 2020, 4087928. 10.1155/2020/4087928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casado M, Vallet VS, Kahn A, and Vaulont S (1999). Essential role in vivo of upstream stimulatory factors for a normal dietary response of the fatty acid synthase gene in the liver. J Biol Chem 274, 2009–2013. 10.1074/jbc.274.4.2009. [DOI] [PubMed] [Google Scholar]
- 26.Takegoshi K, Honda M, Okada H, Takabatake R, Matsuzawa-Nagata N, Campbell JS, Nishikawa M, Shimakami T, Shirasaki T, Sakai Y, et al. (2017). Branched-chain amino acids prevent hepatic fibrosis and development of hepatocellular carcinoma in a non-alcoholic steatohepatitis mouse model. Oncotarget 8, 18191–18205. 10.18632/oncotarget.15304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Wang BF, Tong J, Chang B, and Wang BY (2015). USF-1 genetic polymorphisms confer a high risk of nonalcoholic fatty liver disease in Chinese population. Int J Clin Exp Med 8, 2545–2553. [PMC free article] [PubMed] [Google Scholar]
- 28.Lee YK, Park JE, Lee M, and Hardwick JP (2018). Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res 2, 209–215. 10.1016/j.livres.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nunez S, Young C, Adebayo O, Muppuru KM, and Badri KR (2019). P311, a novel intrinsically disordered protein, regulates adipocyte development. Biochem Biophys Res Commun 515, 234–240. 10.1016/j.bbrc.2019.05.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.CMDKP. Common Metabolic Diseases Knowledge Portal. https://hugeamp.org/region.html?chr=5&end=111383161&phenotype=PlatCount&start=110948318.
- 31.Starmann J, Falth M, Spindelbock W, Lanz KL, Lackner C, Zatloukal K, Trauner M, and Sultmann H (2012). Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS One 7, e46584. 10.1371/journal.pone.0046584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, and Smyth GK (2010). ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics 26, 2176–2182. 10.1093/bioinformatics/btq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall Z, Bond NJ, Ashmore T, Sanders F, Ament Z, Wang X, Murray AJ, Bellafante E, Virtue S, Vidal-Puig A, et al. (2017). Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 65, 1165–1180. 10.1002/hep.28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JS, Collins KM, Brown AL, Lee CH, and Chung JH (2000). hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404, 201–204. 10.1038/35004614. [DOI] [PubMed] [Google Scholar]
- 35.Olson LE, and Soriano P (2009). Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 16, 303–313. 10.1016/j.devcel.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiredja DD, Koyuturk M, and Chance MR (2017). The KSEA App: a web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 33, 3489–3491. 10.1093/bioinformatics/btx415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanehisa M, Sato Y, Kawashima M, Furumichi M, and Tanabe M (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–462. 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo W, Pant G, Bhavnasi YK, Blanchard SG Jr., and Brouwer C (2017). Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res 45, W501–W508. 10.1093/nar/gkx372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan AJ, and Jacob R (1994). Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem J 300 ( Pt 3), 665–672. 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kauffman RF, Taylor RW, and Pfeiffer DR (1980). Cation transport and specificity of ionomycin. Comparison with ionophore A23187 in rat liver mitochondria. J Biol Chem 255, 2735–2739. [PubMed] [Google Scholar]
- 41.Sehgal P, Szalai P, Olesen C, Praetorius HA, Nissen P, Christensen SB, Engedal N, and Møller JV (2017). Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. Journal of Biological Chemistry 292, 19656–19673. 10.1074/jbc.M117.796920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ali ES, and Petrovsky N (2019). Calcium Signaling As a Therapeutic Target for Liver Steatosis. Trends Endocrinol Metab 30, 270–281. 10.1016/j.tem.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Shafiq M, Walmann T, Nutalapati V, Gibson C, and Zafar Y (2020). Effects of proprotein convertase subtilisin/kexin type-9 inhibitors on fatty liver. World J Hepatol 12, 1258–1266. 10.4254/wjh.v12.i12.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen CC, Hsu LW, Chen KD, Chiu KW, Chen CL, and Huang KT (2021). Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease. Int J Mol Sci 23. 10.3390/ijms23010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaudhuri D, Artiga DJ, Abiria SA, and Clapham DE (2016). Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proceedings of the National Academy of Sciences 113, E1872–E1880. doi: 10.1073/pnas.1602264113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Jesus DF, Zhang Z, Kahraman S, Brown NK, Chen M, Hu J, Gupta MK, He C, and Kulkarni RN (2019). m(6)A mRNA Methylation Regulates Human beta-Cell Biology in Physiological States and in Type 2 Diabetes. Nat Metab 1, 765–774. 10.1038/s42255-019-0089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao F, McDaniel J, Chen EY, Rockwell HE, Nguyen C, Lynes MD, Tseng YH, Sarangarajan R, Narain NR, and Kiebish MA (2018). Adapted MS/MS(ALL) Shotgun Lipidomics Approach for Analysis of Cardiolipin Molecular Species. Lipids 53, 133–142. 10.1002/lipd.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buchan DWA, and Jones DT (2019). The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic acids research 47, W402–W407. 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herwig R, Hardt C, Lienhard M, and Kamburov A (2016). Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nature Protocols 11, 1889. 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Bardes EE, Aronow BJ, and Jegga AG (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–311. 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snel B, von Mering C, Jensen LJ, Krupp M, Huynen MA, Foglierini M, Jouffre N, Bork P, and Hooper SD (2005). STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Research 33, D433–D437. 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herwig R, Hardt C, Lienhard M, and Kamburov A (2016). Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nature Protocols 11, 1889–1907. 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Variant associations with phenotypes, related to Figure 1
Supplementary Table 2: Variants in region_HDL cholesterol, related to Figure 1
Data Availability Statement
RNA-sequencing data in primary human hepatocytes have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus under accession code GSE229455. Antibody microarrays data in primary human hepatocytes have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus under accession code GSE229432.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.