

Abstract
Recent Alzheimer’s research has shown increasing interest in the caspase-2 (Casp2) enzyme. However, the available Casp2 inhibitors, which have been pentapeptides or peptidomimetics, face challenges for use as CNS drugs. In this study, we successfully screened a 1920-compound chloroacetamide-based, electrophilic fragment library from Enamine. Our two-point dose screen identified 64 Casp2 hits, which were further evaluated in a ten-point dose-response study to assess selectivity over Casp3. We discovered compounds with inhibition values in the single-digit micromolar and sub-micromolar range, as well as up to 32-fold selectivity for Casp2 over Casp3. Target engagement analysis confirmed the covalent-irreversible binding of the selected fragments to Cys320 at the active site of Casp2. Overall, our findings lay a strong foundation for the future development of small-molecule Casp2 inhibitors.
Graphical Abstract
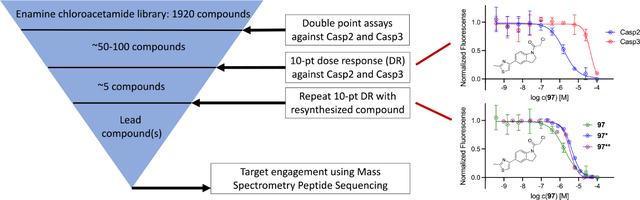
Introduction
Finding new lead structures with improved physicochemical properties is a key issue in modern drug discovery, particularly for targets with peptidic leads. The high polarity of these compounds often hinders their bioavailability, as predicted by their poor performance in Lipinski’s Rule of Five (R05)[1] and their unfavorable physicochemical properties characterized by high topological polar surface areas (TPSA) and a large number of rotatable bonds. These properties hamper compounds from crossing the blood-brain barrier to reach targets in the central nervous system (CNS). While medicinal chemists tend to focus on biological activity and selectivity, they often overlook the importance of a drug’s pharmacokinetics, including absorption, distribution, metabolism, and excretion (ADME).
The evolution of modern drug discovery may be summarized by decade: natural product synthesis in the 1960s, quantitative structure-activity relationships (QSAR) in the 1970s, structure-based drug discovery (SBDD) in the 1980s), combinatorial chemistry in the 1990s, and fragment-based drug discovery (FBDD) in the 2000s.[2] FBDD has emerged as a valuable approach, prioritizing drug-like properties and addressing the aforementioned neglected aspects. FBDD libraries typically contain a few thousand fragments with a molecular weight of 150–250 daltons and emphasize the diversity of chemical scaffolds and functionalities present in approved drugs.[3] Success is achieved with biological activities in the low micromolar range, aiming for further optimization to achieve nanomolar range activities and maintain drug-like properties while increasing molecular weight.
Caspase-2 (Casp2), a cysteine protease,[4,5] has primarily been targeted with peptidic and peptidomimetic ligands. It plays multiple roles in the CNS,[6–14] and has gained attention in Alzheimer’s research due to its involvement in synaptic dysfunction through the cleavage of the neuronal scaffold-protein tau.[12] Unlike Casp3 and Casp6, which cleave tau leading to neurofibrillary tangles,[15,16] Casp2 cleavage of tau leads to a reversible form of synaptic dysfunction,[12] making it a promising therapeutic target for restoring synaptic function. The tau fragment Δtau314, exclusively formed by Casp2,[12] is associated with synaptic dysfunction in various diseases, including Alzheimer’s disease (AD)[12,17], Parkinson’s disease with dementia (PDD)[18], Lewy body dementia (LBD)[18], Huntington’s disease (HD)[19] and frontotemporal dementia (FTD)[12,20]. These findings support further drug development efforts to target Casp2 and restore synaptic dysfunction in Alzheimer’s disease and related dementias (ADRD).[21]
Starting from the canonical but non-selective, peptidic Casp2 inhibitor AcVDVAD-CHO (Figure 1), numerous further developments have been published in recent years with a focus on both covalent reversible and covalent irreversible binding modes.[22–25] By incorporating non-natural amino acids into the pentapeptide inhibitor scaffold, highly potent Casp2 inhibitors in the single-digit nanomolar range could be prepared.[25] Selectivity within the caspases, particularly over the homologous caspase-3 (Casp3), was achieved by modifying amino acid positions P2 and P4 leading to compound 1 and 3 (Figure 1).[25] To overcome the challenges associated with developing bioavailable peptidomimetic drugs that penetrate the brain, alternative strategies based on non-peptide structures are needed. One possible approach is to develop small molecule fragments obtained by identifying Casp2 inhibitors in FBDD libraries of molecules < 300 daltons that have favorable physicochemical properties and adhere to the Rule of Three (R03).[26,27] These types of fragments can serve as starting points for optimization of pharmacokinetic and pharmacodynamic properties, although to our knowledge this approach has not yet been applied to Casp2.
Figure 1.
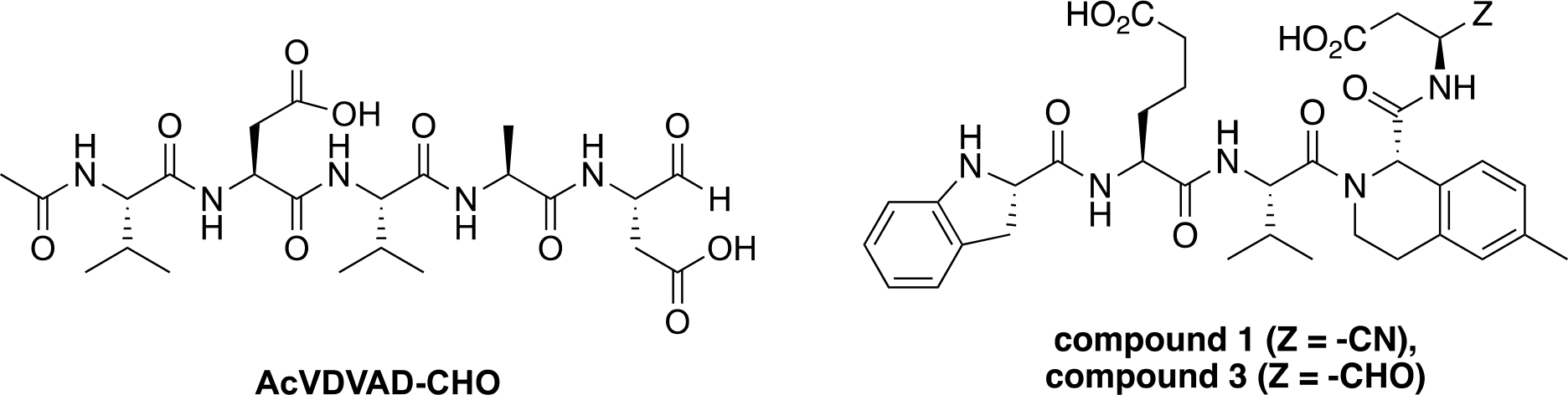
Chemical structures of canonical, non-selective, peptidic Casp2 inhibitor AcVDVAD-CHO and selective, peptidomimetic Casp2 inhibitors compound 1 and compound 3.
Similarly, researchers have prepared libraries of small electrophilic compounds that target nucleophilic amino acids, including cysteine, in the binding pocket. Disulfide tethering, using disulfide-containing compounds,[28] has been used successfully to inhibit cysteine proteases, such as the G12C K-Ras oncoprotein.[29,30] Covalent tethering, a newer technique utilizing electrophilic moieties such as the widely used acrylamide and vinyl sulfone adducts,[31] has also shown promise in reversible [32–35] and irreversible [36–38] inhibition of cysteine proteases.
Therefore, we decided to conduct a fragment-based, electrophilic screening campaign for the development of small molecule caspase-2 inhibitors, since electrophilic screens have already been successful for various cysteine proteases and specifically against caspases.[35–40]
Results and Discussion
The compound library
An α-chloroacetamide library from Enamine containing 1920 compounds was acquired and initially tested for inhibitory effects on Casp2 with Casp3 as a counterscreen in a two-point assay (two concentrations per compound in duplicates). Despite different evolutionary classifications, Casp2 and Casp3 have similar substrate recognition sequences. The canonical Casp2 inhibitors AcVDVAD-CHO and AcVDVAD-pNA inhibit both Casp3 and Casp7, with greater potency against Casp3 as reported by our group[23] and others.[41,42] In particular, selectivity against Casp3 is critically important because of the high concentration of active Casp3 relative to other caspases in almost all tissues and of its promiscuous nature.[42,43] Thus, it is important to develop a Casp2 tool with minimal or no Casp3 activity in order to be able to specifically evaluate the role of Casp-2 in neurodegenerative diseases.
The decision on the electrophilic covalent reactive group (CRG) was made in favor of the irreversible α-chloroacetamides since they a) react strongly with thiols under physiological aqueous conditions,[44,45] b) are readily available from commercial sources, c) show selectivity in chemical-proteomic screens,[46] and because d) have recently been successfully applied to other cysteine proteases.[38,39] The distribution of physicochemical properties of the chloroacetamide library is shown in the Supporting Information (SI) (Figure S1–S9).
Two-point dose screening against Casp2 and Casp3
To ensure meaningful results, it was crucial to select appropriate concentrations for testing, achieve good signal-to-noise ratios, and clearly distinguish between baseline values and hits. In our laboratory, we opted for a fluorometric enzyme assay for Casp2 and Casp3, a well-established system for both routine and high-throughput testing (cf. Experimental Section). A screening method to test each compound with two concentrations in duplicates resulted in the use of 12.5 and 6.25 μM for compounds at Casp2 and 125 and 62.5 μM at Casp3. The plots in Figure 2 and Figure S10 (SI) show the distribution of inhibition values at Casp2 (in these figures a value of 1.0 corresponds to 100% inhibition), with compounds at 12.5 μM shown in Figure 2 and at 6.25 μM in Figure S10 (S10). The three-sigma rule (μ + 3σ) was used as a measure for the decision to classify a compound as a hit with μ as the mean and σ as the standard deviation. As expected, the mean and standard deviation increased with higher concentration (μ = 0.102, σ = 0.165, Figure 2), resulting in better differentiation between hit compounds and the baseline. Thus, at a concentration of 12.5 μM, 64 hit compounds of Casp2 were obtained with a Z’ factor of 0.79, indicating the assay’s high quality according to Zhang et al.[47] The most promising results, with Casp2 inhibition values ≥ 90%, were observed with compounds 17, 46, 196, 294, and 1532, summarized in Table1. Structures of the 64 Casp2 hits can be found in Figure S12 (SI).
Figure 2.
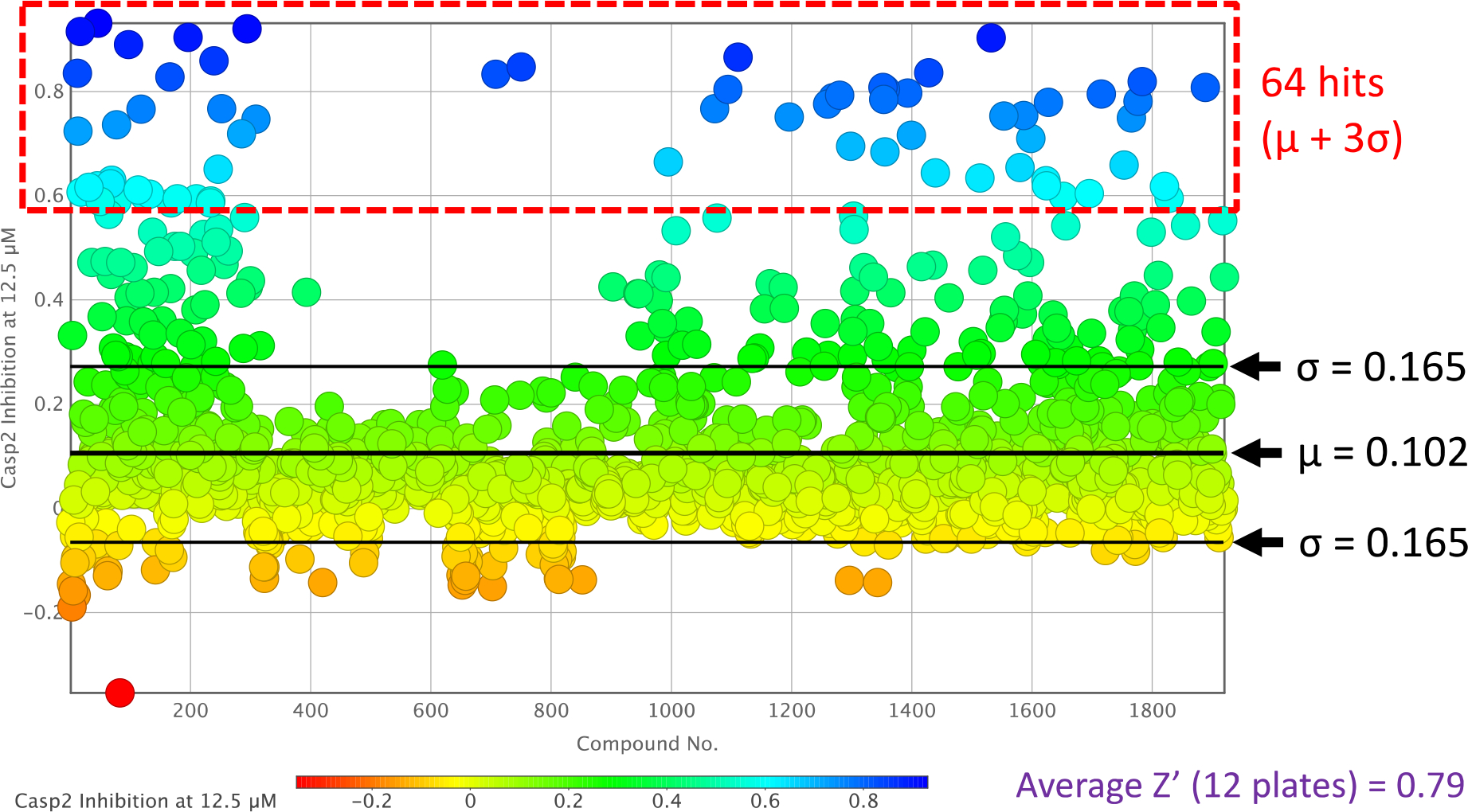
Caspase-2 inhibition plot of the two-point dose screening assay of compounds 1–1920 at 12.5 μM.
Table 1.
Enzyme inhibition activities from two-point and ten-point dose response screenings of selected electrophilic fragments at caspase-2 and caspase-3.
| Cmp d. | Casp2 Inhibiti on at 12.5 μM [%]a | Casp2 Inhibiti on at 6.25 μM [%]a | Casp3 Inhibiti on at 125 μM [%]a | Casp3 Inhibiti on at 62.5 μM [%1a | pIC50 ± SEMa | selectivity: | kinact/Ki (Casp2) [M−1 s−1]b | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Casp2 | N | Casp3 | N | IC50 (Casp3 ) / IC50 (Casp2) | ||||||
|
| ||||||||||
| 12 | 84 | 62 | 91 | 80 | 5.57 ± 0.01 | 2 | < 4.30 | 2 | > 19 | n.d. |
| 17 | 92 | 86 | 90 | 59 | 5.97 ± 0.03 | 2 | 4.54 ± 0.01 | 2 | 27 | 4.49 × 104 |
| 46 | 93 | 92 | 30 | 24 | 5.89 ± 0.01 | 2 | 5.02 ± 0.08 | 2 | 7 | 5.86 × 104 |
| 97 | 89 | 79 | 16 | 10 | 5.74 ± 0.03 | 2 | 4.45 ± 0.05 | 2 | 19 | 7.97 × 104 |
| 118 | 77 | 56 | 0 | 5 | 5.43 ± 0.01 | 2 | < 4.20 | 2 | > 17 | 4.60 × 101 |
| 166 | 83 | 68 | 92 | 32 | 5.56 ± 0.04 | 2 | 4.41 ± 0.05 | 2 | 14 | n.d. |
| 196 | 90 | 86 | 96 | 96 | 6.15 ± 0.06 | 2 | 5.14 ± 0.05 | 2 | 10 | 4.37 × 102 |
| 239 | 86 | 70 | 80 | 36 | 5.50 ± 0.01 | 2 | < 4.30 | 2 | > 16 | n.d. |
| 294 | 92 | 82 | 88 | 53 | 5.66 ± 0.01 | 2 | 4.50 ± 0.03 | 2 | 14 | n.d. |
| 750 | 85 | 70 | 86 | 46 | 5.49 ± 0.02 | 2 | < 4.00 | 2 | > 31 | n.d. |
| 1269 | 79 | 62 | 14 | 0 | 5.50 ± 0.04 | 2 | < 4.30 | 2 | > 16 | 6.32 × 105 |
| 1532 | 90 | 79 | 59 | 18 | 5.50 ± 0.02 | 2 | < 4.00 | 2 | > 32 | n.d. |
| 1598 | 71 | 53 | 0 | 1 | 5.31 ± 0.01 | 2 | < 4.00 | 2 | > 20 | 5.21 × 104 |
| 1715 | 60 | 62 | 69 | 23 | 5.31 ± 0.01 | 2 | < 4.00 | 2 | > 20 | n.d. |
| 1765 | 66 | 57 | 52 | 6 | 5.38 ± 0.01 | 2 | < 4.00 | 2 | > 24 | n.d. |
pIC50 values and single dose inhibition values were obtained based on measurements at 40 min. Data shown are mean values ± SEM of N independent experiments, each performed in duplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration-response curves (variable slope, four parameter fit).
kinact/Ki values were calculated in two-steps by obtaining kobs values first (a) Irreversible Inactivation and b) Two Step Irreversable Secondary Regression (Copeland 9.2)) and using kinetic data of at least four different inhibitor concentrations. n.d. = not determined.
A similar distribution was observed when Casp3 was used as the target protein, but with 10-fold higher compound concentrations compared to Casp2 (Figure 3 and Figure S11, SI). The higher compound concentration of 125 μM at Casp3 (Figure 3) achieved an optimal separation of hit compounds, indicated by a Z’ factor of 0.85, compared to the results at 62.5 μM (Figure S11, SI, Table 1, and Table S1, SI). Applying the three-sigma rule, 72 hit compounds were identified for Casp3 (μ = 0.067, σ = 0.175, Figure 3). When tested at the concentration range used for Casp2 (12.5 μM and 6.25 μM), however, these compounds exhibited significantly lower inhibition values and signal-to-noise ratios. Table 1 and Table S1 (SI) display the inhibition values from the two-point dose screen at Casp3 for the selected compounds (64 Casp2 hits).
Figure 3.
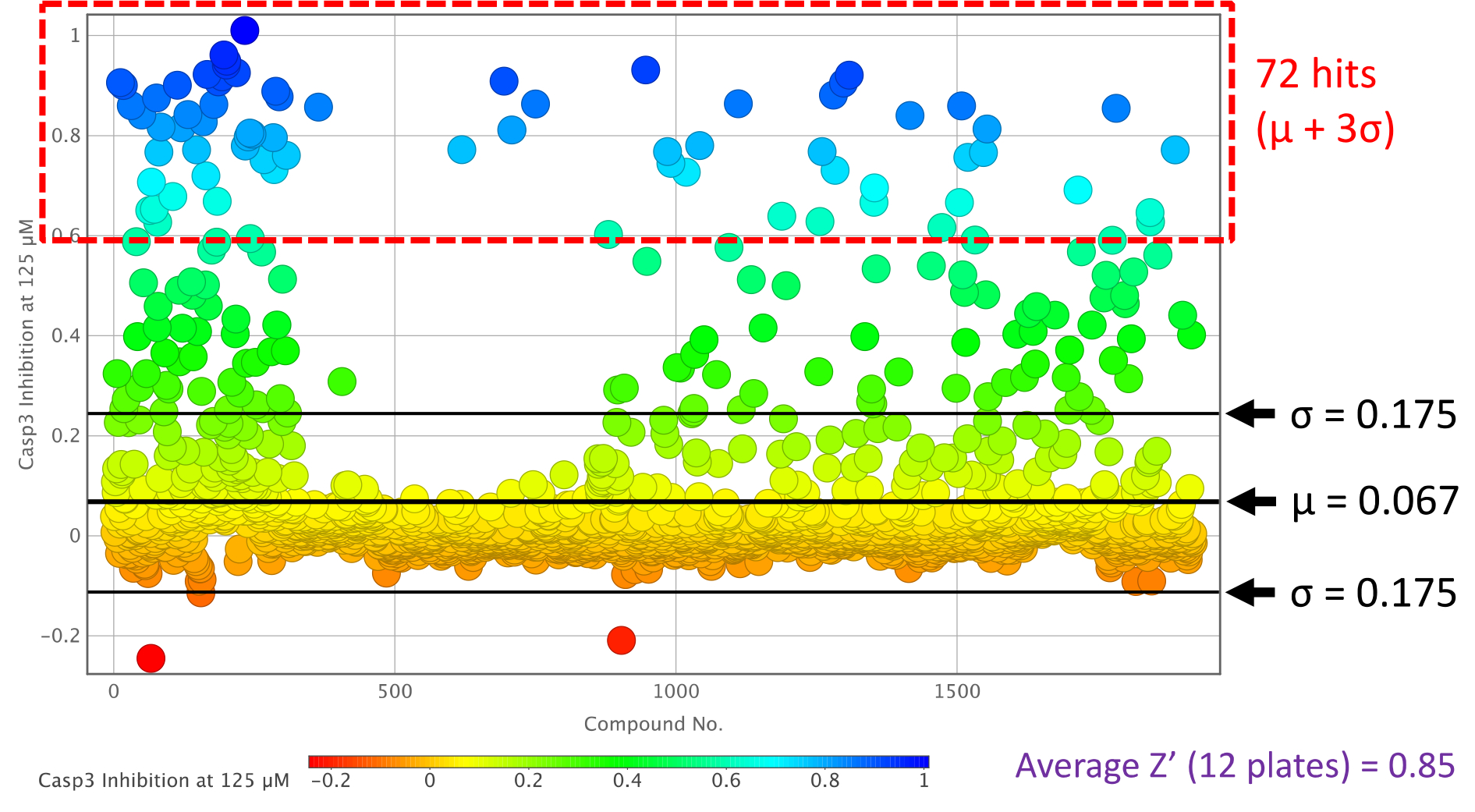
Caspase-3 inhibition plot of the two-point dose screening assay of compounds 1–1920 at 125 μM.
Since our main focus is on the discovery of a small molecule inhibitor for Casp2, a cross comparison with Casp3 data had to be made for a final assessment of the individual Casp2 hits in the two-point dose screening. Figure 4 therefore shows the previously discussed distribution of Figure 2 (12.5 μM compound concentration at Casp2) but taking into account the Casp3 inhibition values, which are color-coded in the form of a heat map. Even though all 64 Casp2 hits were considered for a more advanced selectivity study against both proteins, some compounds already suggest a tendency toward Casp2 selectivity over Casp3 (Figure 4). Overall, these results thus confirm the successful development of a screening assay for both Casp2 and Casp3.
Figure 4.
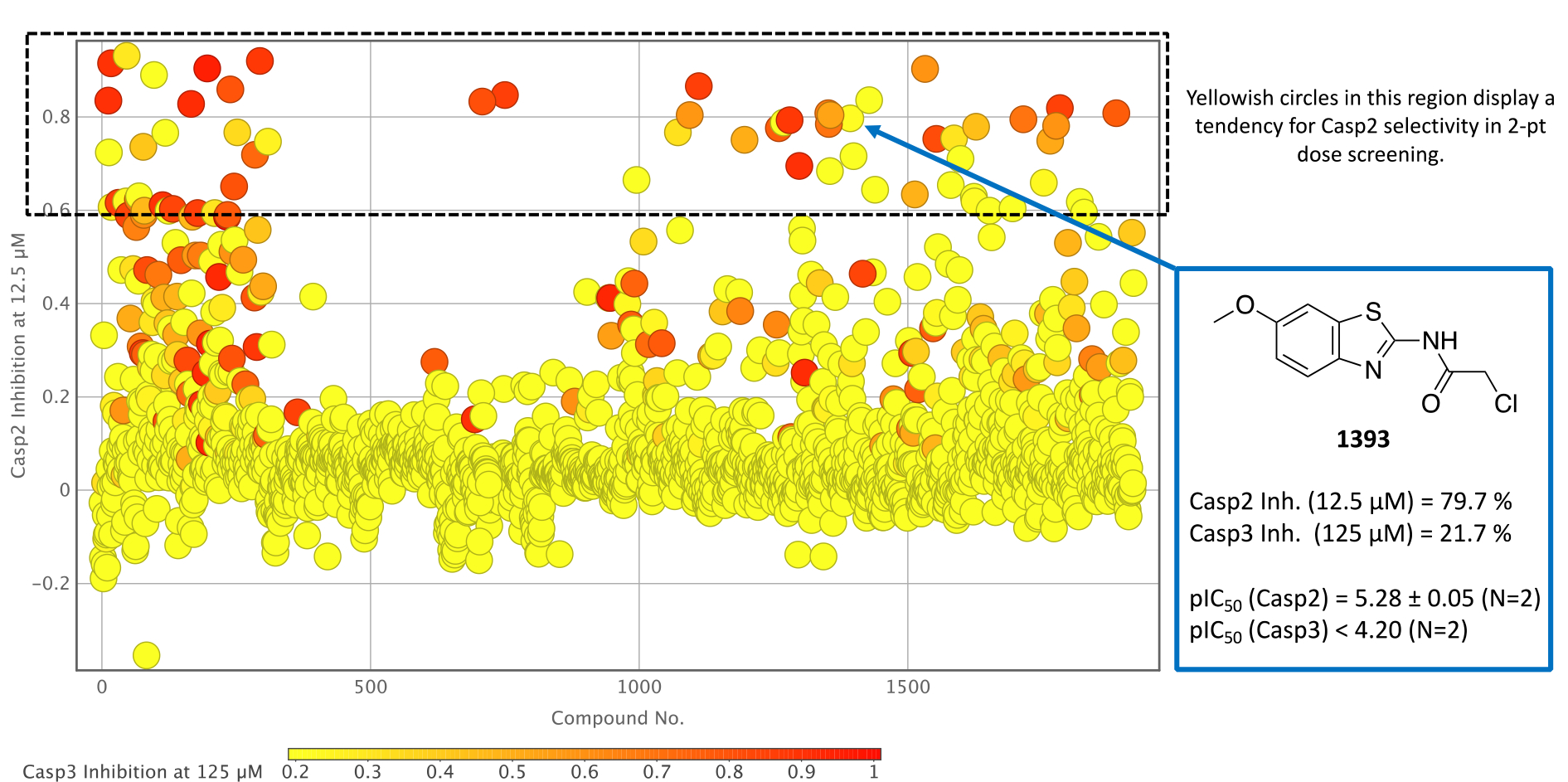
Caspase-2 inhibition plot of the two-point dose screening assay of compounds 1–1920 at 12.5 μM with associated caspase-3 data incorporated as a color-coded heat map. Exemplary enzyme inhibition data are shown from benzothiazole-containing compound 1393.
Ten-point dose response study against Casp2 and Casp3
The selectivity study mentioned above was subsequently performed with all Casp2 hits and ten concentrations in duplicates. The resulting ten-point dose response curves allowed a much more accurate analysis and calculation of pIC50 values, allowing a selectivity profile to be established for the selected compounds (Table 1 and Table S1, SI). One of these compounds is the benzothiazole-containing compound 1393 (Figure 4) with a Casp2 inhibition in the single-digit micromolar range (pIC50 (Casp2) = 5.28, Table S1, SI) and a selectivity factor > 12 (Table S1, SI). Even higher affinities and/or selectivities were observed for compounds 17, 46, 97, 118, 196, 1269, and 1598 (Table 1), for which the concentration-response curves (CRC) for Casp2 and Casp3 are exemplarily shown in Figure 5A–D and Figure S13–S15 (SI). Additionally, the CNS Multiparameter Optimization (MPO) scores[48,49] for these compounds were all ≥ 4 (Table S2, SI). Compound 196, the only compound showing sub-micromolar activity (pIC50 (Casp2) = 6.15, Table 1), stands out with regard to Casp2 inhibition, while the highest Casp3 inhibition could also be detected here (10-fold selectivity, Table 1). Compounds 750 and 1532 (Figure S1, SI) showed the highest selectivity for Casp2 with a selectivity factor of at least 31- and 32-fold, respectively (Table 1). For the seven compounds shown in Figure 5 and Figure S13–S15 (SI), the kinact/KI values frequently used for irreversible inhibitors were calculated and shown in Table 1. The study confirmed the tendency for Casp2 selectivity of almost all selected fragments and provided satisfactory inhibition values in the single-digit micromolar or even sub-micromolar range. The concentration-response curves show sigmoidal and concentration-dependent effects and no abnormally high slopes, ruling out artifact effects. The selected fragments therefore provide a great foundation for further development of small molecule caspase-2 inhibitors.
Figure 5.
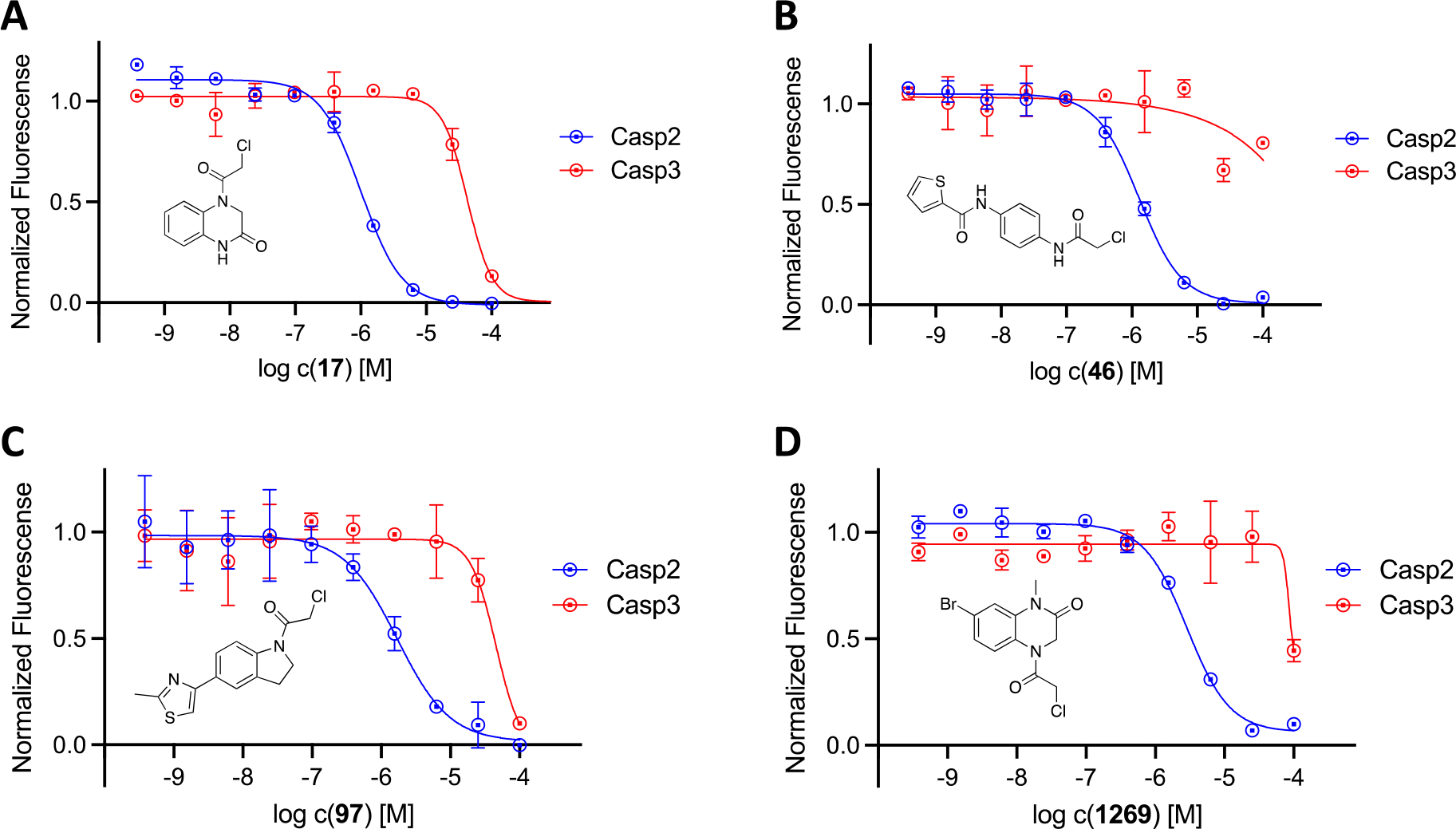
Concentration-response curves of 17 (A), 46 (B), 97 (C), and 1269 (D) at caspase-2 (blue curves) and caspase-3 (red curves) in the fluorometric enzyme assay. Data points represent mean values ± SD from representative experiments, each performed in duplicate.
Target engagement of selected fragments using mass spectrometry peptide sequencing
In order to further investigate the covalent tethering of our fragments to Casp2, a target engagement study was carried out using mass spectrometry peptide sequencing (MSPS) including downstream LC-MS/MS analysis according to publications from Carlson[50] and Jackson[51]. For demonstrating the irreversible binding of the fragments to the active site cysteine Cys320, the selected compounds 17, 46, 97, 196, 1269, and 1598 or the reference compound iodoacetamide (IAD), a nonselective thiol alkylator, were first incubated with the target enzyme Casp2. After protein denaturation, a trypsin digest of the protein/fragment adduct was performed. Following trypsin cleavage, analysis of the cleavage fragments was completed by MS/MS, with particular attention paid to the protein fragment MFFIQAC320R, as it contains cysteine Cys320, the active site of the caspase-2 enzyme (Figure 6A).
Figure 6.
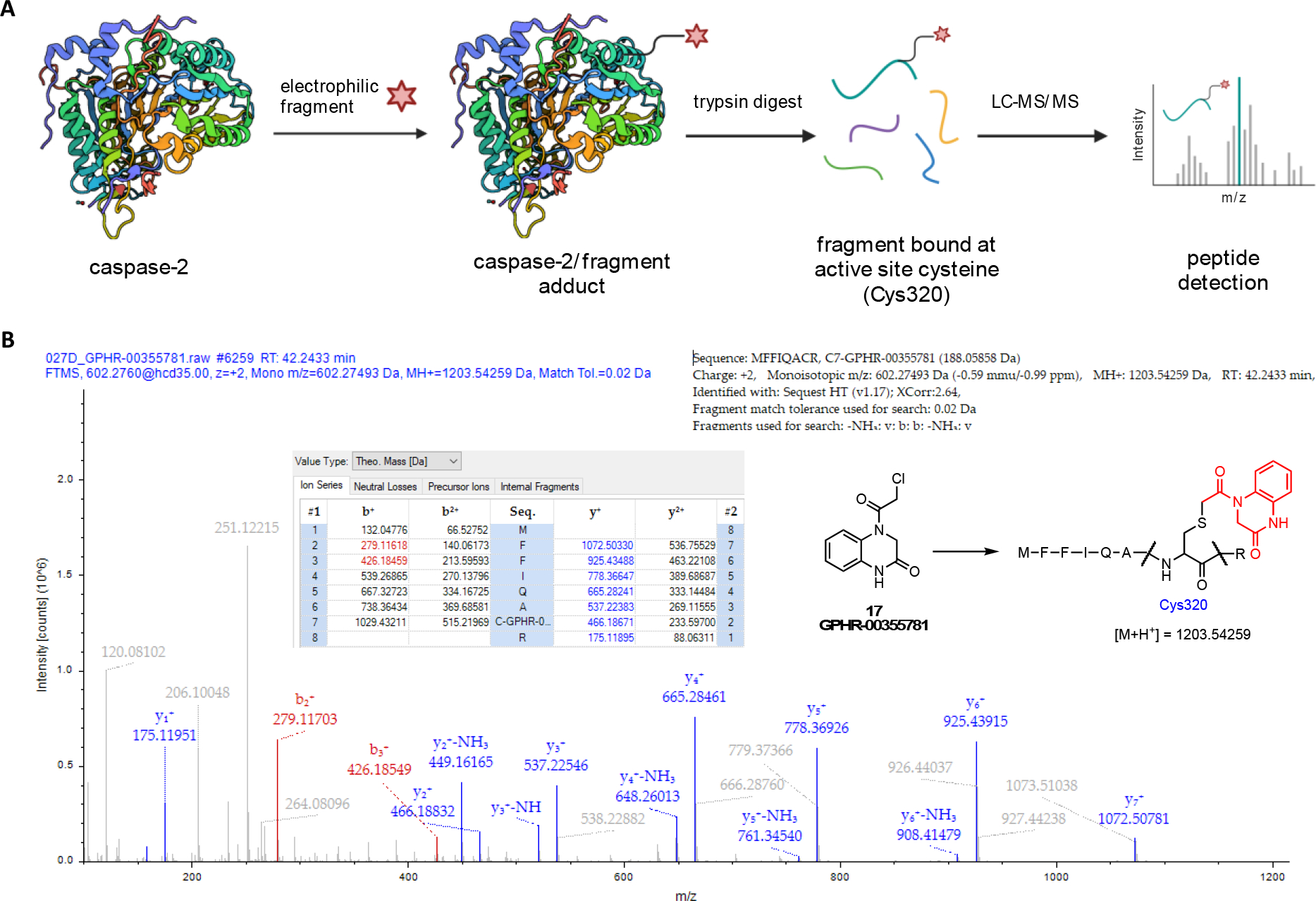
A: Schematic overview of the target engagement study of electrophilic fragments with Casp2. After incubation of the fragment compound with the target enzyme, a trypsin digest was performed with downstream LC-MS/MS analysis to detect the bound fragment at the active site cysteine Cys320. Created with BioRender.com. B: Mass spectrometry (MS) chromatogram of the adducted peptide MFFIQACR with electrophilic fragment 17 (GPHR-00355781). Sequence analysis by MS confirms fragment binding at active site Cys320. Columns b+/b2+ and y+/y2+ (single/double charge) represent the MS results of stepwise fragmentation of peptide “MFFIQA(C-fragment)R” (b: C-terminal fragmentation; y: N-terminal fragmentation).
The analysis of MS chromatograms confirmed the covalent-irreversible binding of all tested electrophilic fragments to Cys320. Figure 6B illustrates the MS chromatogram and sequence analysis for fragment 17 (GPHR-00355781), highlighting the specific mass increase (Cys[+188]) associated with fragment 17. Additional chromatograms and sequence analyses for the reference compound IAD (Cys[+57], Figure S16, SI), as well as fragments 46 (Cys[+258], Figure S17, SI), 97 (Cys[+256], Figure S18, SI), 196 (Cys[+203], Figure S19, SI), 1269 (Cys[+280], Figure S20, SI), and 1598 (Cys[+247], Figure S21, SI) can be found in the Supporting Information, along with a table summarizing key peaks from the MS study associated with each tested hit compound (Table S3, SI). The Supporting Information also provides a summary of off-target cysteines labeled by the tested chloroacetamides. Notably, compound 1269 exhibited the most favorable off-target labeling to cysteines at Casp2 in this study.
Furthermore, we conducted reactivity measurements of our hits (17, 97, 1269, and 1921) against glutathione, since glutathione depletion can lead to oxidative stress and cell death. In parallel, we performed a cell viability assay. For compounds 17, 1269, and 1921, no glutathione (GSH) depletion was observed at concentrations below 10 μM (refer to Figure S22–S23 and S25–S28, SI). At higher concentrations, only minimal levels of GSH were measured by luminescence, and a corresponding decrease in the viability assay signal was observed in all cases (Figure S22–S23 and S25–S28, SI). It is likely that an artifact is detected in the GSH assay, indicated by very steep hill slopes, which could be attributed to cell death at high compound concentrations. While the possibility of GSH depletion being associated with loss of cell viability cannot be entirely ruled out, the unusual curve profiles make it less likely from our perspective. However, in the case of compound 97, the curve closely resembles a dose-dependent concentration-response curve, with a pIC50 of 5.75 in the GSH assay and pIC50 = 5.65 in the cell viability assay (Figure S24, SI).
Hit validation of selected fragments from electrophilic screening at Casp2
In an attempt to validate some of the most promising hit compounds from our electrophilic fragment screen, the chloroacetamide compounds 97 and 1269 were resynthesized in-house in order to confirm their activity in the fluorometric enzyme assay. To differentiate the resynthesized fragments, we marked them with an asterisk (*) or double asterisk (**). For compound 97*, Suzuki-Miyaura coupling strategy was employed to build the core thiazoloindoline structure. The aryl bromide component A1 was synthesized via selective halogen-metal exchange followed by methylation, whereas the aryl pinacol borate substrate A3 was generated via Boc-protection of 5-bromoindoline and subsequent Miyaura borylation of A2. The coupling product A4 was then subject to Boc-deprotection and acylation of chloroacetyl chloride warhead to give the final compound 97*.
For the synthesis of 7-bromo-4-(2-chloroacetyl)-1-methyl-3,4-dihydroquinoxalin-2(1H)-one (1269*) (Scheme 2), 3-bromo-N-methylaniline and 2-cyanoacetic acid were used as starting materials to provide B1 in a coupling reaction using EDC-HCl. In a tandem nitrosylation/oxidation intramolecular ring closure reaction the N-oxide B2 was obtained, whereby the quinoxalinone backbone of the final compound could be formed. Two successive reductions, first with sodium dithionite gave B3 and subsequently, using sodium borohydride, the desired dihydroquinoxalinone structure B4. In a final step, as described above, the chloroacetamide warhead was implemented using chloroacetyl chloride to yield the desired product 1269*.
Scheme 2.
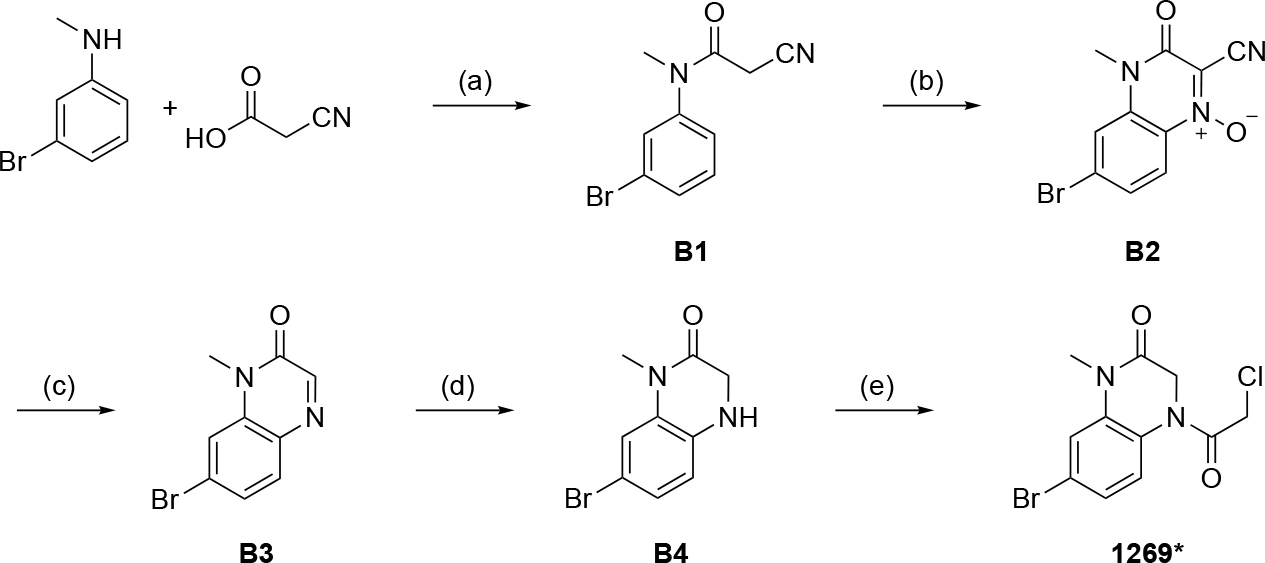
Synthesis of 7-bromo-4-(2-chloroacetyl)-1-methyl-3,4-dihydroquinoxalin-2(1H)-one (1269*). Reagents and conditions: a) EDC hydrochloride, DCM, rt, 1h; b) NaNO2, H2SO4, O2, MeCN, rt, overnight; c) Na2S2O4, EtOH, H2O, reflux, 2h; d) NaBH4, THF, reflux, 30 min. e) chloroacetyl chloride, Et3N, DCM, 0 °C to rt, 1h.
In a collaboration with NCATS (National Center for Advancing Translational Sciences), fragments 17*, 97**, and 1921, a bromo-derivative of 196 (structure is shown in the SI) that was not included in the original Enamine library, were also synthesized. The synthesis and analytical data for these compounds can be found in the SI.
For further evaluation, the resynthesized compounds were tested against Casp2 and Casp3 in the fluorometric enzyme assay. Fragments 97* and 1269* again showed Casp2 affinities in the single-digit micromolar range (pIC50 = 5.31 and 5.22, respectively), with no significant Casp3 inhibition (Table 2). Comparison with the CRCs from the ten-point dose response screening of the Enamine library shows minimal differences in pIC50 values (Figure 7, Table 2), with 2.64-fold lower Casp2 inhibition for 97* (Figure 7A, Table 2) and only 1.89-fold lower for 1269* (Figure 7B, Table 2). In order to further validate our hit fragments, compounds 17*, 97**, and the hit derivative 1921 were synthesized and tested for their caspase-2 activity in an external laboratory in collaboration with NCATS. Compared to the screening results, the fragments showed very similar activities with only minor differences in their inhibition values (Table 2). Fragment 1921 also showed Casp2 activity in the single-digit micromolar range (pIC50 = 5.26, Table 2) and can be considered as a possible scaffold for further development. All in all, the results confirm both activity and selectivity of the investigated compounds and thus also the success of the presented study of electrophilic fragment screening against Casp2.
Table 2.
Enzyme inhibition activities from ten-point dose response assays of selected resynthesized electrophilic fragments at caspase-2 and caspase-3.
| pIC50 ± SEMa | |||||
|---|---|---|---|---|---|
| Cmpd. | Casp2 | N | Casp3 | N | IC50 („cmp“ @Casp2) / IC50 („cmp*“@Casp2) |
|
| |||||
| AcVDVAD-CHO b | 7.32 ± 0.01d | 3 | n.d. | - | - |
| AcDEVD-CHO c | n.d. | - | 8.86 ± 0.01 | 3 | - |
| 17* | 5.48 | 1 | n.d. | - | 3.06 |
| 97* | 5,31 ± 0.01d | 3 | < 4.50 | 3 | 2.64 |
| 97** | 5.47 | 1 | n.d. | - | 1.85 |
| 1269* | 5,22 ± 0.01d | 3 | < 4.50 | 3 | 1.89 |
| 1921 | 5.27 | 2 | n.d. | - | - |
pIC50 values were obtained based on measurements at 40 min. Data shown are mean values ± SEM of N independent experiments, each performed in duplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration-response curves (variable slope, four parameter fit).
AcVDVAD-CHO was used as a reference compound against caspase-2.
AcDEVD-CHO was used as a reference compound against caspase-3.
Assayed with cpCasp2.[23]
Figure 7.
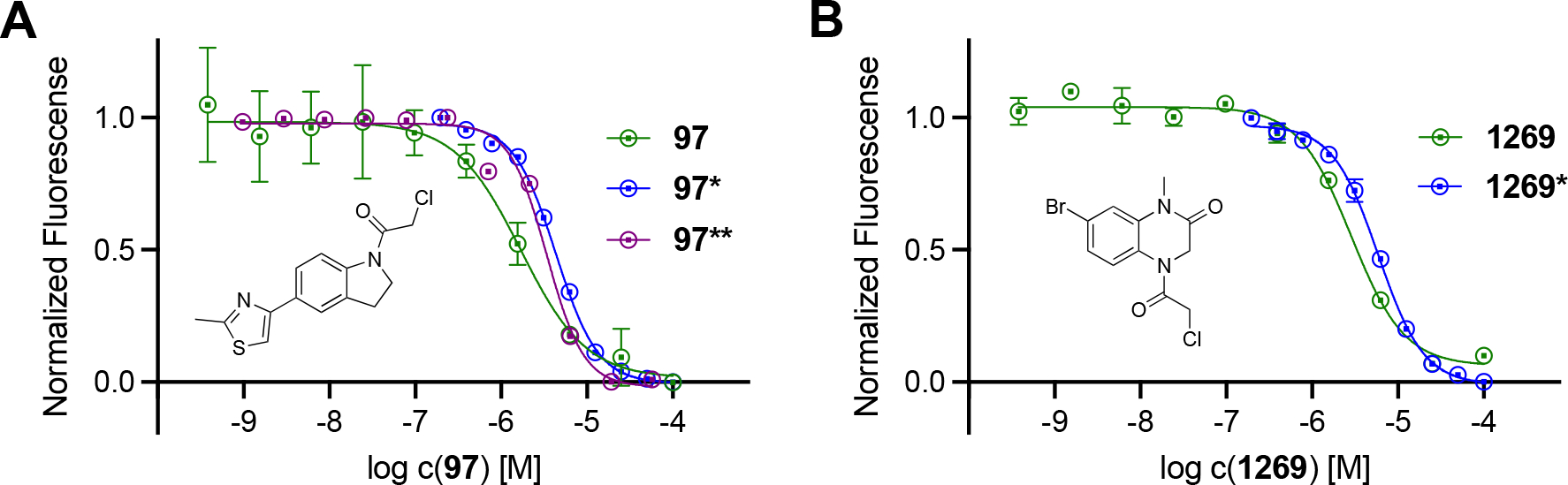
Concentration-response curves of 97 (green curve), 97* (blue curve), 97** (purple curve), (A) and 1269 (green curves), 1269* (blue curve) (B) at caspase-2 in the fluorometric enzyme assay. Data points represent mean values ± SD from representative experiments, each performed in duplicate.
Summary and Conclusion
In this work, we have successfully established an electrophilic fragment screening targeting Casp2. The testing of a 1920 large chloroacetamide compound library from Enamine was initiated in a two-point dose screen, with excellent parameters in terms of Z’, hit separation and favorable signal-to-noise ratios. The subsequent selection of 64 Casp2 hit compounds led to the performance of an extended selectivity study with a ten-point dose response screen on both enzymes. The results showed that the majority of the fragments not only demonstrated inhibition of Casp2 but also exhibited a selective preference for Casp2 over Casp3. Notably, compound 196 displayed sub-micromolar Casp2 inhibition (pIC50 (Casp2) = 6.15), while compounds 750 and 1532 showed the highest selectivity factor (31- and 32-fold, respectively). A target engagement study of selected fragments by MSPS also confirmed covalent tethering to cysteine Cys320 in the active site of caspase-2 for all compounds, validating Casp2 as a target for our fragment compounds. In order to complete this study, most promising compounds were hit validated by resynthesis and retesting in different laboratories, which was also successful with comparable inhibition values for both Casp2 and Casp3.
Overall, our electrophilic fragment screening yielded a significant number of diverse fragment scaffolds with notable affinity and selectivity for Casp2. The observed inhibition concentrations predominantly fell within the desired low micromolar range, providing an excellent starting point for further development of selective small molecule caspase-2 inhibitors.
Experimental Section
Fluorometric enzyme assay
The fluorometric enzyme assay was conducted according to already described protocols with minor modifications.[23] The electrophilic fragment compounds were added to the assay plate (Corning 4514) in duplicate at ten sets of concentrations (100 μM, 25 μM, 6.2 μM, 1.6 μM, 0.4 μM, 100 nM, 25 nM, 6.2 nM, 1.6 nM, and 0.4 nM). The control compounds (AcVDVAD-FMK for caspase-2, AcDEVD-FMK for caspase-3) were added to the assay plate in duplicate at the same sets of concentrations. The corresponding assay buffer was added to the assay plate followed by the corresponding recombinant caspase protein (5 nM caspase-2/cp-caspase-2, 2 nM caspase-3). The assay plate was sealed and incubated at 37 °C for 40 min. The corresponding AFC containing inhibitor peptide (5 μM Z-VDVAD-AFC for caspase-2/cp-caspase-2, 2.5 μM Ac-DEVD-AFC for caspase-3) was added to the plate and the fluorescence of the AFC was recorded (Excitation = 400 nm, Emission = 505 nm) every 3 minutes for 1 hour. The average fluorescence of the samples containing only buffer and protein was used to correct the raw fluorescence values recorded. The IC50 of the corrected fluorescence values at the 40 minute time point was calculated using Prism. Two-point dose screening was conducted following the same protocol but with different concentrations for the electrophilic fragment compounds (12.5 μM, 6.25 μM for caspase-2 and 125 μM, 62.5 μM for caspase-3). In addition, the reaction was stopped after incubation (40 minutes, 37 °C) with AFC containing peptides by adding 5 M acetic acid and the fluorescence was recorded.
Caspase-2/cp-caspase-2 assay buffer: 100 mM MES (pH 6.5), 150 mM NaCl, 0.1% CHAPS, 1.5% sucrose, 10 mM DTT. Caspase 3 assay buffer: 100 mM HEPES (pH 7.0), 150 mM NaCl, 0.1% CHAPS, 1.5% sucrose, 10 mM DTT.
Mass spectrometry peptide sequencing
Caspase-2 MS Sample Preparation
The tryptic digestion of purified recombinant protein was adapted from a previously published protocols.[50,51] All solutions utilized in this section were purified by centrifugation with a 3 kDa molecular weight cutoff (MWCO) filter (Amicon, MilliporeSigma) and collection of the flow-through. Recombinant Caspase-2 (15 μL; 34 μM) was added to a solution of the test compound (1 μL of 10 mM DMSO stock). The sample was further diluted to 50 μL with 100 mM MES (pH 6.5), 150 mM NaCl, 0.1% CHAPS, 1.5% sucrose, 10 mM DTT and incubated for 1 h at 37 °C. Next, iodoacetamide (IAD) was added to a final concentration of 62 mM and the solution was incubated in the dark for 1 h at room temperature. The protein was then isolated by spinning down the samples in a 3 kDa MWCO filter, diluting the sample with distilled and deionized H2O (ddH2O, 500 μL) and isolating the protein again through the MWCO filter (3× total). The protein was then evaporated to dryness overnight (SpeedVac). The dried protein-compound samples were resuspended in 20 μL of a freshly prepared aqueous denaturing solution (8 M urea, 50 mM ammonium bicarbonate, 5 mM DTT) and incubated at 37 °C for 1 h. A solution of 30 mM IAD in ddH2O was prepared and 20 μL was added to each sample. The samples were incubated for 1 h in the dark at room temperature. Next, 120 μL of 50 mM ammonium bicarbonate aqueous solution was added to each sample in order to dilute the urea concentration for trypsin digestion. A 1 μg/μL solution of trypsin (Promega, catalog #V5280) in 50 mM acetic acid was diluted to 100 ng/μL with 100 mM aqueous ammonium bicarbonate and 10 μL of the resulting solution was added to each sample. Acetonitrile was immediately added to each sample to a final concentration of 10% (v/ v). Samples were incubated in a rotating (800 rpm) heat block overnight at 37 °C. Glacial acetic acid was added until the pH of each sample was less than 4.0 (~5 μL) and then the samples were desalted using C18 resin pipette tips according to the manufacturer’s protocol (Pierce C18 Tips 10 μL bed, ThermoFisher). Desalted samples were evaporated to dryness (SpeedVac) and then reconstituted in 20 μL of 98:2 LC–MS-grade H2O/MeCN containing 0.1% formic acid (v/v).
Caspase-2 MS Analysis
Peptides within each sample were separated with a front-end Dionex UltiMate 3000 ultrahigh performance liquid chromatography instrument. Peptides were separated using a home-packed analytical Luna C18 (100 Å pore, 5 μm particles) reverse-phase column (75 μm ID × 200 mm, 10 μm emitter orifice, Phenomenex), where MS-grade water with 0.1% formic acid was eluent A and acetonitrile with 0.1% formic acid was eluent B at room temperature. Initially, an isocratic 2% B elution was run for 5 min (1 μL/min flow rate). Then, the gradient elution (0.3 μL/min flow rate for all gradient steps) began by running from 2% B to 10% B over 5min, followed by 10% B to 25% B over 40min and then 25% B to 40% B over 10 min. The elution gradient further increased from 40% B to 90% B over 1 min, then held at 90% B for 4 min before decreasing from 90% B to 2% B over 0.5 min and re-equilibrated at 2% B for 4.5 min (1 μL/min flow rate). Eluted peptides were analyzed with an LTQ Orbitrap Velos (ThermoFisher) in the Nth Order Double Play mode. The mass spectrometer utilized an electrospray ionization source with a source voltage of +2.5 kV. MS1 scan range was m/z 220.0–1800.0. For MS2, the precursor ion mass of peptides containing the target cysteines in caspase-2 were predicted using Proteome Discoverer software. The m/z of the double and triple- charged peptides were calculated for peptide MFFIQAC320R. All possible cysteine modifications were evaluated for the following seven compounds: carbamidomethylation (+57.0215 Da), 17/GPHR-00355781 (+188.0586 Da), 46/GPHR-00355810 (+258.0463 Da), 97/GPHR-00355861 (+256.0670 Da), 196/GPHR-00355959 (+203.0577 Da), 1269/GPHR-00357032 (+279.9847 Da), and 1598/GPHR-00357358 (+247.1208 Da).
GSH-Glo™ Glutathione Assay:
HepG2 cells (5 μL, 250 cells/well) were seeded in a 1536-well white solid bottom plate (Greiner Bio One, Monroe, NC) and incubated at 37 °C and 5 % CO2 for four hours. Compounds (50 nL) were transferred into each well by Echo 650 Acoustic Liquid Handlers (Beckman Coulter, Brea, CA) and incubated at 37 °C and 5 % CO2 for 22 hours. The media was subsequently removed by centrifugation (1000 rpm, 15 sec) and 5 μL 1X GSH-Glo™ Reagent (Promega, Madison, WI) was added to each well and incubated for 30 minutes at room temperature. 5 μL of Luciferin Detection Reagent (Promega, Madison, WI) was finally added into each well and was incubated for 15 minutes at room temperature before luminescence was measured by ViewLux™ ultra HTS Microplate Imager (PerkinElmer, Waltham, MA).
CellTiter-Glo™ Luminescent Cell Viability Assay:
HepG2 cells (5 μL, 250 cells/well) were seeded in a 1536-well white solid bottom plate (Greiner Bio One, Monroe, NC) and incubated at 37 °C and 5 % CO2 for four hours. Compounds (50 nL) were transferred into each well by Echo 650 Acoustic Liquid Handlers (Beckman Coulter, Brea, CA) and incubated at 37 °C and 5 % CO2 for 22 hours. 5 μL of CellTiter-Glo™ Reagent (Promega, Madison, WI) was finally added into each well and was incubated for 15 minutes at room temperature before luminescence was measured by ViewLux™ ultra HTS Microplate Imager (PerkinElmer, Waltham, MA).
Chemistry
General Methods for Chemistry:
All air- or moisture-sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane, N,N-dimethylformamide (DMF), acetonitrile, methanol and triethylamine were purchased from Sigma-Aldrich. All starting materials, solvents, and reagents used in solution phase syntheses were purchased from commercial sources and used without further purification. Flash chromatography using 230–400 mesh silica gel was applied as needed in purifying intermediates and final products. Preparative purification was performed on a Waters semi-preparative HPLC system. The column used was a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. Purification was performed using Method Acidic Standard Gradient (10:90 MeCN with 0.1% TFA/deionized water with 0.1% TFA ramped to 100% deionized water with 0.1% TFA) over 8 minutes unless otherwise noted. Fraction collection was triggered by UV detection (220 nM). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Purity analysis was determined using a 7 minute gradient of 4% to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) with an 8 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50 °C using an Agilent Diode Array Detector. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz and Bruker 400 MHz spectrometers. Chemical shifts are reported in ppm with non-deuterated solvent (DMSO-d5 peak at 2.50 ppm, CHCl3 peak at 7.26 ppm) as internal standard for DMSO-d6 and CDCl3 solutions, respectively. All of the analogs tested in the biological assays have a purity greater than 95% based on LCMS analysis. High resolution mass spectrometry was recorded on Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formulae was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02). NMR spectra and RP-HPLC chromatograms of 97*and 1269* (Figure S29–S34) are shown in the SI.
Synthesis and analytical data
4-Bromo-2-methylthiazole (A1)
4-Bromo-2-methylthiazole (A1) was synthesized following procedures reported by Karama et. al.[52] Starting from 4 g of 2,4-dibromothiazole, 2.29 g of the product was obtained as a light yellow oil in 76% yield. 1H NMR (400 MHz, CDCl3) δ 7.06 (s, 1H), 2.72 (s, 3H).
tert-Butyl 5-bromoindoline-1-carboxylate (A2)
N-Boc-5-bromoindoline (A2) was synthesized using methods by Axten et. al.[53] Starting from 1.98 g of 5-bromoindoline, 2.83 g of the product was isolated as a white solid in 95% yield. 1H NMR (400 MHz, CDCl3) δ 7.90 – 7.18 (m, 3H), 3.97 (t, J = 8.7 Hz, 2H), 3.07 (t, J = 8.7 Hz, 2H), 1.56 (s, 9H).
tert-Butyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indoline-1-carboxylate (A3)
Compound A3 was synthesized using methods by Axten et. al.[53] Starting from 268 mg of A2, 189 mg of the product was isolated as a white solid in 61% yield. 1H NMR (400 MHz, DMSO) δ 7.79 – 7.40 (m, 3H), 3.90 (t, J = 8.7 Hz, 2H), 3.05 (t, J = 8.7 Hz, 2H), 1.50 (s, 9H), 1.27 (s, 12H).
tert-Butyl 5-(2-methylthiazol-4-yl)indoline-1-carboxylate (A4)
Compound A1 (178 mg, 1 mmol), Compound A3 (345mg, 1mmol), Pd2(dba)3 (92 mg, 0.1 mmol), Cs2CO3 (977 mg, 3 mmol), and xPhos (95 mg, 0.2 mmol) were dissolved in a mixture of 3 mL dioxane and 1 mL H2O. The biphasic solution was purged with N2 and heated to 80°C overnight. The reaction was diluted with H2O and extracted with DCM. The organic phase was dried over anhydrous Na2SO4 and concentrated. The mixture was purified via flash chromatography with 4:1 hexanes:EtOAc as eluting solvent to give 283 mg of product A4 as a colorless oil. Yield: 89.4% 1H NMR (400 MHz, CDCl3) δ 8.00 – 7.60 (m, 3H), 7.18 (s, 1H), 4.00 (t, J = 8.8 Hz, 2H), 3.12 (t, J = 8.8 Hz, 2H), 2.76 (s, 3H), 1.57 (s, 9H).
2-Chloro-1-(5-(2-methylthiazol-4-yl)indolin-1-yl)ethan-1-one (97*)
227 mg (0.717 mmol) of A4 was dissolved in 4 mL DCM and 1 mL of TFA was added. The deprotection reaction was carried out at room temperature for 1 hour. Upon completion, the reaction was diluted with 10 mL DCM and washed with saturated NaHCO3 solution repeatedly until no bubbles were formed. The organic phase containing compound A5 was dried over anhydrous Na2SO4, DIPEA (0.249 mL, 1.43 mmol) was added and cooled to 0°C. 0.113 g (1 mmol) of chloroacetyl chloride in 5 mL DCM was added dropwise. The reaction was allowed to warm to room temperature, stirred for 1 hour and was monitored with TLC. Once completed, the reaction was washed with 0.1 M hydrochloric acid, dried over anhydrous Na2SO4 and rotavaped to give crude 97*. The crude product was purified via flash chromatography using 2:1 hexanes:EtOAc as eluent to give 107 mg of pure 97* as white solid. Yield: 51%. 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 1.8 Hz, 1H), 7.70 (dd, J = 8.4, 1.8 Hz, 1H), 7.24 (s, 1H), 4.23 – 4.14 (m, 4H), 3.27 (t, J = 8.4 Hz, 2H), 2.76 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 165.88, 163.92, 154.71, 142.25, 131.81, 131.16, 125.85, 122.71, 117.38, 111.61, 48.11, 43.08, 28.10, 19.33. HRMS: m/z of [M+H]+ calculated for molecular formula C14H14ClN2OS: 293.0515, found: 293.0498. HPLC purity: 99.3%.
N-(3-Bromophenyl)-2-cyano-N-methylacetamide (B1)
To a solution of 1 mmol (186.05 mg) 3-bromo-N-methylaniline and 1.1 mmol (93.57 mg) of 2-cyanoacetic acid in 30 mL DCM a solution of 1.1 mmol (210.87 mg) EDC hydrochloride in 10 mL DCM was added. The reaction was stirred at room temperature for 1 hour. Upon completion, DCM was evaporated and the crude mixture was loaded onto a silica plug and eluted with 2:1 hexanes:EtOAc as eluent to give 250 mg of the desired product B1 as a white solid. Yield: 98.8%. 1H NMR (400 MHz, CDCl3) δ 7.62 – 7.55 (m, 1H), 7.42 (t, J = 2.0 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.21 (ddd, J = 7.9, 2.1, 1.0 Hz, 1H), 3.31 (s, 3H), 3.25 (s, 2H).
6-Bromo-2-cyano-4-methyl-3-oxo-3,4-dihydroquinoxaline 1-oxide (B2)
Compound B2 was synthesized according to the procedure reported by Kobayashi et. al.[54] Starting from 253 mg of B1, 212 mg of the N-oxide product was obtained as a bright yellow solid in 75.7% yield. 1H NMR (400 MHz, DMSO) δ 8.13 (d, J = 9.0 Hz, 1H), 8.06 (d, J = 1.9 Hz, 1H), 7.68 (dd, J = 8.9, 1.9 Hz, 1H), 3.62 (s, 3H).
7-Bromo-1-methylquinoxalin-2(1H)-one (B3)
A suspension of 210 mg (0.75 mmol) of B2 and 391 mg (2.25 mmol) of sodium dithionite in a mixture of 10 mL EtOH and 20 mL H2O was heated to reflux for 2 hours. Once cooled to room temperature, the reaction was extracted with EtOAc three times. The combined organic phase was dried over anhydrous Na2SO4 and rotavaped to give 0.168 g of B3 as a light yellow solid. Yield: 93.7%. 1H NMR (400 MHz, DMSO) δ 8.25 (s, 1H), 7.82 (d, J = 2.0 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.55 (dd, J = 8.5, 2.0 Hz, 1H), 3.59 (s, 3H).
7-Bromo-1-methyl-3,4-dihydroquinoxalin-2(1H)-one (B4)
To a solution of 143 mg (0.6 mmol) B3 in 5 mL THF 114 mg (3 mmol) of NaBH4 was added. The reaction was heated to reflux for 30 min. Upon completion, the reaction was poured onto an ice-water mix to quench the NaBH4 excess and stirred for approximately 30 min until no bubbles were formed. This suspension was extracted with EtOAc three times. The combined organic phase was dried over anhydrous Na2SO4 and rotavaped to give 0.131 g of B4 as a light yellow solid. Yield: 90.56%. 1H NMR (400 MHz, DMSO) δ 7.09 (d, J = 2.2 Hz, 1H), 7.00 (dd, J = 8.3, 2.1 Hz, 1H), 6.67 (d, J = 8.3 Hz, 1H), 6.24 (s, 1H), 3.79 (d, J = 1.8 Hz, 2H), 3.22 (s, 3H). HRMS: m/z of [M+H]+ calculated for molecular formula C11H11BrClN2O2: 316.9692, found: 316.9682.
7-Bromo-4-(2-chloroacetyl)-1-methyl-3,4-dihydroquinoxalin-2(1H)-one (1269*)
A solution of 96 mg (0.4 mmol) of B4 and 0.111 mL (0.8 mmol) Et3N in 10 mL DCM was cooled to 0°C. A solution of chloroacetyl chloride in 5 mL DCM was added dropwise to the prior solution. The reaction was allowed to warm to room temperature and stirred for 1 hour and monitored with TLC. Once completed, the reaction was washed with 0.1 M hydrochloric acid, dried over anhydrous Na2SO4 and rotavaped to give crude 1269*. The crude product was recrystallized from DCM and hexanes to give 50 mg of pure 1269* as white crystalline solid. Yield, 39.4%. 1H NMR (400 MHz, CDCl3) δ 7.33 – 7.27 (m, 2H), 7.25 (s, 1H), 4.49 (s, 2H), 4.20 (s, 2H), 3.36 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.35, 165.20, 135.68, 126.52, 126.29, 124.27, 120.87, 119.05, 46.66, 40.26, 29.34.. HRMS: m/z of [M+H]+ calculated for molecular formula C11H11BrClN2O2: 316.9692, found: 316.9682. HPLC purity: 99.4%.
Supplementary Material
Scheme 1.
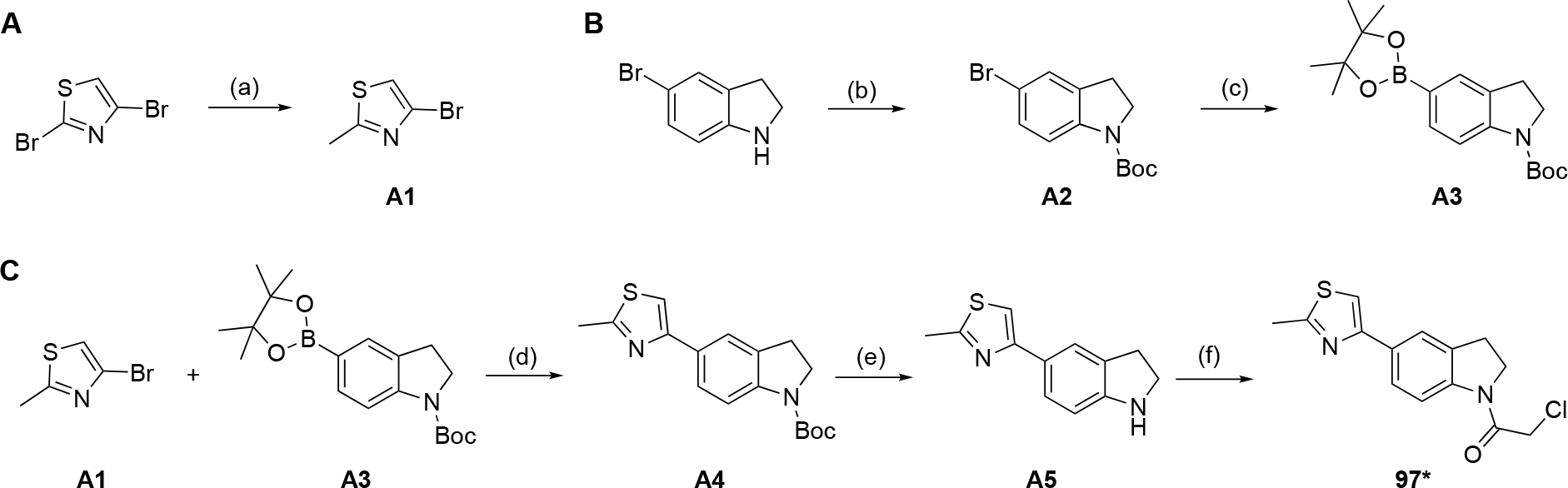
Synthesis of 2-chloro-1-(5-(2-methylthiazol-4-yl)indolin-1-yl)ethan-1-one (97*). Reagents and conditions: A: a) nBuLi, Et2O, −78 °C, 4h; Me2SO4, −78 °C, overnight; B: b) Boc2O, DMAP, MeCN, rt, 1h; c) B2pin2, Pd(dppf)Cl2, KOAc, DMSO, 80°C, overnight; C: d) Pd2(dba)3, xPhos, Cs2CO3, dioxane, H2O, 80 °C, overnight; e) TFA, DCM, rt, 1h, f) chloroacetyl chloride, Et3N, DCM, 0°C, 1h.
Highlights:
Electrophilic fragment screening for caspase-2 (Casp2) identified 64 hit compounds
Single-digit micromolar affinity for Casp2 and up to 32-fold selectivity over Casp3
Successful hit validation for selected fragments
Target engagement study using mass spectrometry peptide sequencing (MSPS)
Covalent-irreversible fragment binding to cysteine-320 at the active site of Casp2
Acknowledgments
The work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; 436921318, PO 2563/1–1, Pockes) and NIH Grant R01AG0623199 (Ashe, Walters). Figure 6A was created with BioRender.com. Plots in Figure 2, 3, 4, S10, and S11 were created with DataWarrior.
Abbreviations
- ADRD
Alzheimer’s disease and related dementias
- AFC
7-amido-4-trifluoromethylcoumarin
- Casp2
caspase-2
- Casp3
caspase-3
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- CNS
central nervous system
- CRC
concentration-response curve
- CRG
covalent reactive group
- Cys320
cysteine at position 320 of the caspase-2 enzyme
- EDC-HCl
N-Ethyl-N′-carbodiimide hydrochloride
- FTD
frontotemporal dementia
- G12C K-Ras
mutation of the K-Ras protein
- HD
Huntington’s disease
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IAD
iodoacetamide
- LBD
Lewy body dementia
- MES
2-(N-morpholino)ethanesulfonic acid
- MSPS
mass spectrometry peptide sequencing
- MWCO
molecular weight cutoff
- NCATS
National Center for Advancing Translational Sciences
- PDD
Parkinson’s disease with dementia
- R03
Rule of Three
- TPSA
topological polar surface area
- xPhos
dicyclohexyl[2′,4′,6′-tris(propan-2-yl)[1,1′-biphenyl]-2-yl]phosphane
- Z
benzyloxycarbonyl
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article. K.H.A., M.A.W. and S.P. are founders and shareholders of Myriel, Inc.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Steffen Pockes reports a relationship with Myriel, Inc. that includes: consulting or advisory and equity or stocks. Karen H. Ashe, Michael A. Walters and Steffen Pockes are founders and shareholders of Myriel, Inc.
Supporting Information
The Supporting Information is available free of charge at “link” DOI: XXX/number Hit fragments from two-point dose screening at caspase-2, Fluorometric enzyme assay at Casp2 and Casp3, Distribution of physicochemical properties of the chloroacetamide library, Target engagement study using MSPS, GSH-Glo™ Glutathione and CellTiter-Glo™ Luminescent Cell Viability Assay, Synthesis for hit validation, NMR spectra and RP-HPLC chromatograms of 97* and 1269* (PDF).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Advanced Drug Delivery Reviews. 23 (1997) 3–25. [DOI] [PubMed] [Google Scholar]
- [2].Murray CW, Rees DC, The rise of fragment-based drug discovery, Nature Chem. 1 (2009) 187–192. 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- [3].Fattori D, Molecular recognition: the fragment approach in lead generation., Drug Discov Today. 9 (2004) 229–238. 10.1016/S1359-6446(03)03007-1. [DOI] [PubMed] [Google Scholar]
- [4].Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA, Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme., Genes & Development. 8 (1994) 1613–1626. [DOI] [PubMed] [Google Scholar]
- [5].Wang L, Miura M, Bergeron L, Zhu H, Yuan J, Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death., Cell. 78 (1994) 739–750. 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- [6].Kinoshita M, Tomimoto H, Kinoshita A, Kumar S, Noda M, Up-regulation of the Nedd2 gene encoding an ICE/Ced-3-like cysteine protease in the gerbil brain after transient global ischemia., J Cereb Blood Flow Metab. 17 (1997) 507–514. 10.1097/00004647-199705000-00004. [DOI] [PubMed] [Google Scholar]
- [7].Niizuma K, Endo H, Nito C, Myer DJ, Kim GS, Chan PH, The PIDDosome mediates delayed death of hippocampal CA1 neurons after transient global cerebral ischemia in rats, Proceedings of the National Academy of Sciences. 105 (2008) 16368–16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Carroll JB, Southwell AL, Graham RK, Lerch JP, Ehrnhoefer DE, Cao L-P, Zhang W-N, Deng Y, Bissada N, Henkelman RM, Mice lacking caspase-2 are protected from behavioral changes, but not pathology, in the YAC128 model of Huntington disease, Molecular Neurodegeneration. 6 (2011) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tiwari M, Herman B, Morgan WW, A knockout of the caspase 2 gene produces increased resistance of the nigrostriatal dopaminergic pathway to MPTP-induced toxicity, Experimental Neurology. 229 (2011) 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M, Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice., Nat Commun. 4 (2013) 1939. 10.1038/ncomms2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vigneswara V, Ahmed Z, Long-term neuroprotection of retinal ganglion cells by inhibiting caspase-2., Cell Death Discov. 2 (2016) 16044. 10.1038/cddiscovery.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, Cleary J, Ashe KH, Caspase-2 cleavage of tau reversibly impairs memory, Nature Medicine. 22 (2016) 1268. [DOI] [PubMed] [Google Scholar]
- [13].Xu Z-X, Tan J-W, Xu H, Hill CJ, Ostrovskaya O, Martemyanov KA, Xu B, Caspase-2 promotes AMPA receptor internalization and cognitive flexibility via mTORC2-AKT-GSK3β signaling, Nature Communications. 10 (2019) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vigneswara V, Ahmed Z, The role of caspase-2 in regulating cell fate, Cells. 9 (2020) 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC, Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease., Am J Pathol. 165 (2004) 523–531. 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT, Propagation of tau pathology in a model of early Alzheimer’s disease., Neuron. 73 (2012) 685–697. 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu P, Smith BR, Montonye ML, Kemper LJ, Leinonen-Wright K, Nelson KM, Higgins L, Guerrero CR, Markowski TW, Zhao X, A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals, Scientific Reports. 10 (2020) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Smith BR, Nelson KM, Kemper LJ, Leinonen-Wright K, Petersen A, Keene CD, Ashe KH, A soluble tau fragment generated by caspase-2 is associated with dementia in Lewy body disease, Acta Neuropathologica Communications. 7 (2019). 10.1186/s40478-019-0765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu P, Smith BR, Huang ES, Mahesh A, Vonsattel JPG, Petersen AJ, Gomez-Pastor R, Ashe KH, A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients, Acta Neuropathologica Communications. 7 (2019) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Steuer EL, Kemper LJ, Hlynialuk CJ, Leinonen-Wright K, Montonye ML, Lapcinski IP, Forster CL, Ashe KH, Liu P, Blocking site-specific cleavage of human tau delays progression of disease-related phenotypes in genetically matched tau-transgenic mice modeling frontotemporal dementia, Journal of Neuroscience. 42 (2022) 4737–4754. 10.1523/JNEUROSCI.0543-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pockes S, Walters MA, Ashe KH, Targeting caspase-2 interactions with tau in Alzheimer’s disease and related dementias., Transl Res. (2022) S1931–5244(22)00241–9. 10.1016/j.trsl.2022.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Poreba M, Rut W, Groborz K, Snipas SJ, Salvesen GS, Drag M, Potent and selective caspase-2 inhibitor prevents MDM-2 cleavage in reversine-treated colon cancer cells, Cell Death & Differentiation. 26 (2019) 2695–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bresinsky M, Strasser JM, Vallaster B, Liu P, McCue WM, Fuller J, Hubmann A, Singh G, Nelson KM, Cuellar ME, Wilmot CM, Finzel BC, Ashe KH, Walters MA, Pockes S, Structure-Based Design and Biological Evaluation of Novel Caspase-2 Inhibitors Based on the Peptide AcVDVAD-CHO and the Caspase-2-Mediated Tau Cleavage Sequence YKPVD314, ACS Pharmacology & Translational Science. 5 (2022) 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bresinsky M, Strasser JM, Hubmann A, Vallaster B, McCue WM, Fuller J, Singh G, Nelson KM, Cuellar ME, Finzel BC, Ashe KH, Walters MA, Pockes S, Characterization of caspase-2 inhibitors based on specific sites of caspase-2-mediated proteolysis., Arch Pharm (Weinheim). 355 (2022) e2200095. 10.1002/ardp.202200095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Singh G, Liu P, Yao KR, Strasser JM, Hlynialuk C, Leinonen-Wright K, Teravskis PJ, Choquette JM, Ikramuddin J, Bresinsky M, Nelson KM, Liao D, Ashe KH, Walters MA, Pockes S, Caspase-2 Inhibitor Blocks Tau Truncation and Restores Excitatory Neurotransmission in Neurons Modeling FTDP-17 Tauopathy, ACS Chem. Neurosci. 13 (2022) 1549–1557. 10.1021/acschemneuro.2c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Congreve M, Carr R, Murray C, Jhoti H, A ‘Rule of Three’ for fragment-based lead discovery?, Drug Discovery Today. 8 (2003) 876–877. 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- [27].Jhoti H, Williams G, Rees DC, Murray CW, The “rule of three” for fragment-based drug discovery: where are we now?, Nat Rev Drug Discov. 12 (2013) 644–644. 10.1038/nrd3926-c1. [DOI] [PubMed] [Google Scholar]
- [28].Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA, Site-directed ligand discovery., Proc Natl Acad Sci U S A. 97 (2000) 9367–9372. 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Erlanson DA, McDowell RS, O’Brien T, Fragment-based drug discovery., J Med Chem. 47 (2004) 3463–3482. 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]
- [30].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM, K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions., Nature. 503 (2013) 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kathman SG, Statsyuk AV, Covalent Tethering of Fragments For Covalent Probe Discovery., Medchemcomm. 7 (2016) 576–585. 10.1039/c5md00518c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frödin M, Taunton J, Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles., Nat Chem Biol. 8 (2012) 471–476. 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermančič P, Bonnet R, Shoichet BK, Covalent docking of large libraries for the discovery of chemical probes, Nature Chemical Biology. 10 (2014) 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J, Prolonged and tunable residence time using reversible covalent kinase inhibitors., Nat Chem Biol. 11 (2015) 525–531. 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, Song H, Shi Y, Li X, Tan W, Qi J, Gao GF, Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease, Nat Commun. 11 (2020) 4417. 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kathman SG, Xu Z, Statsyuk AV, A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases., J Med Chem. 57 (2014) 4969–4974. 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McShan D, Kathman S, Lowe B, Xu Z, Zhan J, Statsyuk A, Ogungbe IV, Identification of non-peptidic cysteine reactive fragments as inhibitors of cysteine protease rhodesain, Bioorganic & Medicinal Chemistry Letters. 25 (2015) 4509–4512. 10.1016/j.bmcl.2015.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Resnick E, Bradley A, Gan J, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D, Bennett J, Fairhead M, Fedorov O, Gabizon R, Gan J, Guo J, Plotnikov A, Reznik N, Ruda GF, Díaz-Sáez L, Straub VM, Szommer T, Velupillai S, Zaidman D, Zhang Y, Coker AR, Dowson CG, Barr HM, Wang C, Huber KVM, Brennan PE, Ovaa H, von Delft F, London N, Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening, J. Am. Chem. Soc. 141 (2019) 8951–8968. 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gao S, Song L, Claff T, Woodson M, Sylvester K, Jing L, Weiße RH, Cheng Y, Sträter N, Schäkel L, Gütschow M, Ye B, Yang M, Zhang T, Kang D, Toth K, Tavis J, Tollefson AE, Müller CE, Zhan P, Liu X, Discovery and Crystallographic Studies of Nonpeptidic Piperazine Derivatives as Covalent SARS-CoV-2 Main Protease Inhibitors, J. Med. Chem. 65 (2022) 16902–16917. 10.1021/acs.jmedchem.2c01716. [DOI] [PubMed] [Google Scholar]
- [40].Murray J, Giannetti AM, Steffek M, Gibbons P, Hearn BR, Cohen F, Tam C, Pozniak C, Bravo B, Lewcock J, Jaishankar P, Ly CQ, Zhao X, Tang Y, Chugha P, Arkin MR, Flygare J, Renslo AR, Tailoring small molecules for an allosteric site on procaspase-6., ChemMedChem. 9 (2014) 73–77, 2. 10.1002/cmdc.201300424. [DOI] [PubMed] [Google Scholar]
- [41].Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW, Substrate specificities of caspase family proteases, Journal of Biological Chemistry. 272 (1997) 9677–9682. [DOI] [PubMed] [Google Scholar]
- [42].Maillard MC, Brookfield FA, Courtney SM, Eustache FM, Gemkow MJ, Handel RK, Johnson LC, Johnson PD, Kerry MA, Krieger F, Meniconi M, Muñoz-Sanjuán I, Palfrey JJ, Park H, Schaertl S, Taylor MG, Weddell D, Dominguez C, Exploiting differences in caspase-2 and −3 S2 subsites for selectivity: Structure-based design, solid-phase synthesis and in vitro activity of novel substrate-based caspase-2 inhibitors, Bioorganic & Medicinal Chemistry. 19 (2011) 5833–5851. 10.1016/j.bmc.2011.08.020. [DOI] [PubMed] [Google Scholar]
- [43].Toulmond S, Tang K, Bureau Y, Ashdown H, Degen S, O’Donnell R, Tam J, Han Y, Colucci J, Giroux A, Neuroprotective effects of M826, a reversible caspase-3 inhibitor, in the rat malonate model of Huntington’s disease, British Journal of Pharmacology. 141 (2004) 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lu W, Kostic M, Zhang T, Che J, Patricelli MP, Jones LH, Chouchani ET, Gray NS, Fragment-based covalent ligand discovery., RSC Chem Biol. 2 (2021) 354–367. 10.1039/d0cb00222d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang F, Han X, Xiao X, Zhou J, Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development, Molecules. 27 (2022) 7728. 10.3390/molecules27227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF, Proteome-wide covalent ligand discovery in native biological systems., Nature. 534 (2016) 570–574. 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang J-H, Chung TDY, Oldenburg KR, A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays, J Biomol Screen. 4 (1999) 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- [48].Wager TT, Hou X, Verhoest PR, Villalobos A, Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery, ACS Chemical Neuroscience. 7 (2016) 767–775. 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- [49].Wager TT, Hou X, Verhoest PR, Villalobos A, Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties, ACS Chemical Neuroscience. 1 (2010) 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Carlson TL, Moini M, Eckenrode BA, Allred BM, Donfack J, Protein extraction from human anagen head hairs 1-millimeter or less in total length., Biotechniques. 64 (2018) 170–176. 10.2144/btn-2018-2004. [DOI] [PubMed] [Google Scholar]
- [51].Jackson PA, Schares HAM, Jones KFM, Widen JC, Dempe DP, Grillet F, Cuellar ME, Walters MA, Harki DA, Brummond KM, Synthesis of Guaianolide Analogues with a Tunable α-Methylene-γ-lactam Electrophile and Correlating Bioactivity with Thiol Reactivity., J Med Chem. 63 (2020) 14951–14978. 10.1021/acs.jmedchem.0c01464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Karama U, Höfle G, Synthesis of Epothilone 16, 17-Alkyne Analogs by Replacement of the C13− C15 (O)-Ring Segment of Natural Epothilone C, European Journal of Organic Chemistry. 2003 (2003) 1042–1049. [Google Scholar]
- [53].Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT, Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}−2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK)., J Med Chem. 55 (2012) 7193–7207. 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- [54].Kobayashi Y, Kuroda M, Toba N, Okada M, Tanaka R, Kimachi T, Highly efficient synthesis of quinoxalinone-N-oxide via tandem nitrosation/aerobic oxidative C-N bond formation., Org Lett. 13 (2011) 6280–6283. 10.1021/ol202760c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.