

Abstract
We report a strategy (called “tethering”) to discover low molecular weight ligands (≈250 Da) that bind weakly to targeted sites on proteins through an intermediary disulfide tether. A native or engineered cysteine in a protein is allowed to react reversibly with a small library of disulfide-containing molecules (≈1,200 compounds) at concentrations typically used in drug screening (10 to 200 μM). The cysteine-captured ligands, which are readily identified by MS, are among the most stable complexes, even though in the absence of the covalent tether the ligands may bind very weakly. This method was applied to generate a potent inhibitor for thymidylate synthase, an essential enzyme in pyrimidine metabolism with therapeutic applications in cancer and infectious diseases. The affinity of the untethered ligand (Ki≈1 mM) was improved 3,000-fold by synthesis of a small set of analogs with the aid of crystallographic structures of the tethered complex. Such site-directed ligand discovery allows one to nucleate drug design from a spatially targeted lead fragment.
The drug discovery process usually begins with massive screening of compound libraries (typically hundreds of thousands of members) to identify modest affinity leads (Kd≈1 to 10 μM). Although some targets are well suited for this screening process, most are problematic because moderate affinity leads are difficult to obtain. Identifying and subsequently optimizing weaker binding compounds would improve the success rate, but screening at high concentrations is generally impractical because of compound insolubility and assay artifacts. Moreover, the screening process does not target specific sites for drug design, only those sites for which a high-throughput assay is available. Finally, many traditional screening methods rely on inhibition assays that are often subject to artifacts caused by reactive chemical species or denaturants.
We have developed an alternative strategy to rapidly and reliably identify small soluble drug fragments (molecular weight ≈250 Da) that bind with low affinity to a specifically targeted site on a protein or macromolecule. This method relies on the formation of a disulfide bond between the ligand and a cysteine residue in the protein of interest (Fig. 1A). A library of disulfide-containing molecules (Fig. 1B) is allowed to react with a cysteine-containing target protein under partially reducing conditions that promote rapid thiol exchange. Most of these library members will show no intrinsic affinity for the protein, and therefore the associated disulfide bond to the protein will be easily reduced. However, if a molecule has even weak inherent affinity for the target protein, the disulfide bond will be entropically stabilized and the equilibrium will lie toward the modified protein. Tethered compounds can then be identified by MS. Furthermore, the tethered complex is amenable to analysis by x-ray crystallography, which greatly facilitates the optimization of affinity once the disulfide tether is removed.
Figure 1.
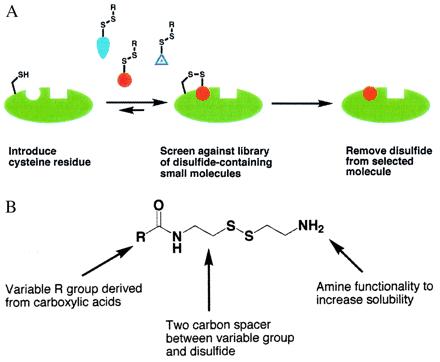
(A) Schematic illustration of the tethering approach: a cysteine-containing protein is equilibrated with a disulfide-containing library in the presence of a reducing agent such as 2-mercaptoethanol. Most of the library members will have little or no inherent affinity for the protein, and thus by mass action the equilibrium will lie toward the unmodified protein. However, if a library member does show inherent affinity for the protein, the equilibrium will shift toward the modified protein. (B) Schematic illustration of a generic disulfide library derived from carboxylic acids. Other functional groups have also been converted to disulfide libraries, as described in Materials and Methods. In the present case, 1,200 compounds were screened against TS in pools of 8 to 15 compounds.
As an initial test of this technology, we chose the enzyme thymidylate synthase (TS). TS is a critical component of the sole de novo pathway for synthesis of dTMP from dUMP (1, 2). Thus TS is a good anticancer and antiinfective drug target (3–6). TS has also previously been used as a model system with which to validate other methods of drug discovery (7–10).
Materials and Methods
Thymidylate Synthase.
Using the standard techniques of molecular biology, we have created three mutants of the unmodified or “wild-type” Escherichia coli TS enzyme, overexpressed them in E. coli strain χ2913 [in which the TS gene has been eliminated (11)], and purified them. The χ2913 strain requires a thymidine supplement because the (deleted) TS gene is essential for life. We have constructed mutants including one in which the active-site cysteine has been replaced by serine (abbreviated as C146S) and two in which, in addition to this change, a new cysteine residue has been introduced into the active site, denoted C146S/L143C or C146S/H147C. All proteins have been expressed, purified, and characterized by SDS/PAGE and MS. Enzymatic inhibition assays were performed largely as described previously (12, 13).
Reagents.
All commercially available materials were used as received. All synthesized compounds were characterized by 1H NMR [Bruker (Billerica, MA) DMX400 MHz Spectrometer] and HPLC-MS (Hewlett–Packard Series 1100 MSD).
Disulfide Libraries.
The disulfide-containing library members were made from commercially available carboxylic acids and mono-N-(tert-butoxycarbonyl)-protected cystamine (mono-BOC-cystamine) by adapting the method of Parlow and coworkers (14). Briefly, 260 μmol of each carboxylic acid was immobilized onto 130 μmol equivalents of 4-hydroxy3-nitrobenzophenone on polystyrene resin using 1,3-diisopropylcarbodiimide (DIC) in N,N-dimethylformamide (DMF). After 4 h at room temperature, the resin was rinsed with DMF (×2), dichloromethane (DCM, ×3), and tetrahydrofuran (THF, ×1) to remove uncoupled acid and DIC. The acids were cleaved from the resin via amide formation with 66 μmol of mono-BOC protected cystamine in THF. After reaction for 12 h at ambient temperature, the solvent was evaporated, and the BOC group was removed from the uncoupled half of each disulfide by using 80% trifluoroacetic acid (TFA) in DCM. The products were characterized by HPLC-MS, and those products that were substantially pure were used without further purification. After the initial discovery of the N-tosyl-d-proline lead (see below), a small number of other proline derivatives and N-tosyl-amino acids were added to the library. A total of 530 compounds were made by using this methodology.
Libraries (not shown) were also constructed from mono-BOC-protected cystamine and a variety of sulfonyl chlorides, isocyanates, and isothiocyanates. In the case of the sulfonyl chlorides, 10 μmol of each sulfonyl chloride was coupled with 10.5 μmol of mono-BOC-protected cystamine in THF (with 2% diisopropyl ethyl amine) in the presence of 15 mg of poly(4-vinyl pyridine). After 48 h, the poly(4-vinylpyridine) was removed via filtration, and the solvent was evaporated. The BOC group was removed by using 50% TFA in DCM. In the case of the iso(thio)cyanates, 10 μmol of each isocyanate or isothiocyanate was coupled with 10.5 μmol of mono-BOC-protected cystamine in THF. After reaction for 12 h at ambient temperature, the solvent was evaporated, and the BOC group was removed by using 50% TFA in DCM. A total of 212 compounds were made by using this methodology.
Finally, oxime-based libraries were constructed by reacting 10 μmol of specific aldehydes or ketones with 10.5 μmol of HO(CH2)2S-S(CH2)2ONH2 (to be described separately) in 1:1 methanol/chloroform (with 2% acetic acid added) for 12 h at ambient temperature to yield the oxime product. A total of 448 compounds were made by using this methodology.
Individual library members were redissolved in either acetonitrile or dimethyl sulfoxide to a final concentration of 50 or 100 mM. Aliquots of each of these were then pooled into groups of 8–15 discrete compounds, with each member of the pool having a unique molecular weight.
N-tosyl-d-proline Derivatives.
Synthesis of the N-tosyl-d-proline analogs began by reacting proline methyl ester hydrochloride with 4-(chlorosulfonyl)benzoic acid and sodium carbonate in water. The product was converted to the pentafluorophenyl ester by reacting it with pentafluorophenyl trifluoroacetate and pyridine in N,N-dimethylformamide and was purified via flash chromatography. This activated ester was then reacted with the methyl ester of glutamate (or any of the other amino acids tested) in the presence of triethylamine and dichloromethane, the product purified by flash chromatography and the methyl esters hydrolyzed with lithium hydroxide in water. The final products were purified via reverse-phase HPLC and lyophilized.
Alternatively, the above sequence was followed starting with proline t-butyl ester. After coupling of the amino ester to the benzoic acid, the t-butyl ester was removed with 50% TFA in DCM with triethylsilane as a scavenger. The free acid was then converted to a pentafluorophenyl ester as above and reacted with the appropriate amine. The methyl esters were hydrolyzed with lithium hydroxide in water, and the final products were purified via reverse-phase HPLC and lyophilized. Full synthetic details will be presented elsewhere.
Disulfide Library Screening.
In a typical experiment, 1 μl of a DMSO solution containing a library of 8–15 disulfide-containing compounds is added to 49 μl of protein-containing buffer. These compounds are chosen so that each has a unique molecular weight; ideally, these molecular weights differ by at least 10 atomic mass units so that deconvolution is unambiguous. Although we have typically chosen to screen pools of 8–15 disulfide-containing compounds for ease of deconvolution, larger pools can be used as discussed below and as shown in Fig. 2C. The protein is present at a concentration of ≈15 μM, each of the disulfide library members is present at ≈0.2 mM, and thus the total concentration of all disulfide library members is ≈2 mM. The reaction is done in a buffer containing 25 mM potassium phosphate (pH 7.5) and 1 mM 2-mercaptoethanol, although other buffers and reducing agents can be used. The reactions are allowed to equilibrate at ambient temperature for at least 30 min. These conditions can be varied considerably depending on the ease with which the protein ionizes in the mass spectrometer (see below), the reactivity of the specific cysteine(s), etc. In the case of TS, the conditions described above were found to be satisfactory. No special effort was made to exclude oxygen or adventitious metal ions; on the time scale of these reactions, there is sufficient free thiol to facilitate disulfide exchange.
Figure 2.
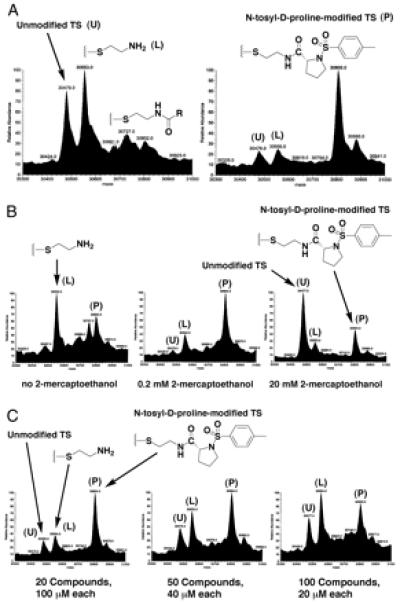
(A) Two representative tethering experiments. The protein TS is present at a concentration of 15 μM, and each of the 10 disulfide library members in each pool is present at 200 μM. The buffer contains 25 mM potassium phosphate (pH 7.5) and 1 mM 2-mercaptoethanol, and the samples were allowed to equilibrate at ambient temperature for 1 h before analysis. (B) Three tethering experiments in which the concentrations of 2-mercaptoethanol were varied as stated. The pool of disulfides is the same as in A Right, and the conditions (other than 2-mercaptoethanol concentration) were the same as above. (C) Three tethering experiments in which the pool size and concentrations were varied as stated. All other conditions were the same as above.
After equilibration, the reaction is injected onto an HP1100 HPLC and chromatographed on a C18 column attached to a mass spectrometer (Finnigan-MAT LCQ, San Jose, CA). The multiply charged ions arising from the protein are deconvoluted with available software (xcalibur) to arrive at the mass of the protein. The identity of any library member bonded through a disulfide bond to the protein is then easily determined by subtracting the known mass of the unmodified protein from the observed mass. This process assumes that the attachment of a library member does not dramatically change the ionization characteristics of the protein itself, a conservative assumption because in most cases the protein will be at least 20-fold larger than any given library member. This assumption was confirmed by demonstrating that small molecules selected by one protein are not selected by other proteins (data not shown).
Crystallography.
Crystals were grown as previously described (15), with the exception that for the noncovalent complexes, 1 mM compound was included in the crystallization buffer. Before data collection, crystals were transferred to a solution containing 70% saturated (NH4)2SO4, 20% glycerol, 50 mM K2HPO4, pH 7.0. For the noncovalent N-tosyl-d-proline complex, 10 mM compound was added to the soaking solution; for the other complexes, 1 mM compound was included. Diffraction data were collected at −170°C by using a Rigaku (Tokyo) RU-3R generator and an R-axis-IV detector and processed by using d*trek (16). As these crystals were isomorphous with previously described structures [Protein Data Bank code 1TJS (10) for the I213 form and 2TSC (17) for the P63 form], refinement began by rigid body refinement by using refmac [CCP4 (18)]. The protein model was adjusted by using o (19), compound model constructed in insight-ii (Molecular Simulations, Waltham, MA), and protin [CCP4 (18)] dictionary created by using makedic [CCP4 (18)]. Positional and individual isotropic temperature factor refinements were carried out with refmac [CCP4 (18)] by using all reflections in the indicated resolution ranges. Solvent molecules were placed automatically by using arpp (CCP4) and refinement continued until no interpretable features remained in Fo − Fc difference maps. Protein Data Bank accession numbers are 1F4B, 1F4C, 1F4D, 1F4E, 1F4F, and 1F4G for the native C146-tethered N-tosyl-d-proline, L143C-tethered N-tosyl-d-proline, N-tosyl-d-proline free acid soak, glutamate-N-tosyl-d-proline soak, and glutamate-N-tosyl-d-proline-β-alanine crystals, respectively.
Results and Discussion
The TS protein from E. coli contains an active-site cysteine as well as four nonconserved cysteine residues that are buried in the structure (15). Previous reports suggested that the active-site cysteine (C146) is particularly susceptible to modification (20). This was confirmed by reacting either wild-type or C146S TS (in which the active-site Cys-146 has been mutated to Ser) with cystamine, H2NCH2CH2S-SCH2CH2NH2. The results (not shown) revealed that whereas wild-type enzyme cleanly reacted with one equivalent of cystamine, the mutant TS did not react. This selectivity gave us confidence that in our screening experiments the site of modification would be the active-site cysteine.
Two representative tethering experiments are shown in Fig. 2A. On the left side is the deconvoluted mass spectrum of TS reacted with a pool of 10 different disulfide-containing molecules that do not specifically tether to TS. The large peak on the far left of the spectrum is unmodified TS (mass of 30,479). The slightly larger peak is TS disulfide-bonded to 2-aminoethanethiol (combined mass of 30,553). In the absence of any binding interactions, TS is statistically more likely to react with the 2-aminoethanethiol portion of the disulfide moiety than any individual library member because 2-aminoethanethiol is common to all of the library members. The small higher-mass peaks correspond to discrete library members, none of which is prominent. The spectrum on the right side of Fig. 2A shows the result of reacting TS with a different pool of 10 disulfide-containing molecules. In this case, the peaks corresponding to free TS and 2-aminoethanethiol-modified TS are dwarfed by a peak (mass of 30,805) that corresponds to TS disulfide bonded to the N-tosyl-d-proline compound shown.
For disulfide tethering to capture the most stable ligand, the reaction must be under rapid exchange to allow for equilibration. Moreover, it should be possible to increase the stringency of the selection to capture more stable ligands by addition of a reducing agent. Both these concepts can be tested by titration with 2-mercaptoethanol (Fig. 2B). In the absence of added reducing agent (far left), N-tosyl-d-proline is only moderately selected. This is because the initial reaction of the active-site cysteine with a disulfide is kinetically controlled. Under these conditions, most of the TS is modified with 2-aminoethanethiol. However, on addition of catalytic amounts of reducing agent (Middle), N-tosyl-d-proline is strongly selected. Even under strongly reducing conditions (Right), the covalent adduct with N-tosyl-d-proline disulfide is still prominent. These results demonstrate that the reaction with N-tosyl-d-proline disulfide is a thermodynamic rather than a kinetic process. Thus the selection is based in part on the actual binding energy of the N-tosyl-d-proline analog to TS.
To maximize throughput and efficiency, it is desirable to screen multiple compounds simultaneously. Because MS is used to deconvolute the pools, as long as each compound has a unique molecular weight, arbitrarily large pools can be screened. Fig. 2C shows three different tethering experiments in which N-tosyl-d-proline was selected from increasingly larger and more diverse pools at decreasing concentrations. As seen in the Fig. 2C Right, N-tosyl-d-proline is still readily selected even in the presence of 100 compounds. Each experiment requires only minutes to run, so in principle the technique is amenable to high-throughput screening.
After screening a library of 1,200 compounds, we started to observe some highly significant structure–activity relationships (SAR), as shown in Fig. 3. There is evidently a fair amount of flexibility around the phenyl ring, as shown by the fact that the methyl group can be replaced by a t-butyl group or removed entirely. However, the phenyl-sulfonamide core moiety appears to be essential, because methyl proline is not selected. Furthermore, the proline ring itself appears to be quite important; replacing it with a phenylalanine, phenylglycine, or pyrrole causes the resulting compound not to be selected. Of course, these data are qualitative and are not meant to be exhaustive. For example, although the pyrrole compound shown was not selected, this could be because of linker derivitization at the 3-position while proline is substituted at the 2-position. Although it would be possible to greatly expand the disulfide library to try to obtain detailed SAR, we felt it would be more efficient to remove the disulfide as soon as possible to determine the actual binding affinity of the untethered pharmacophore.
Figure 3.
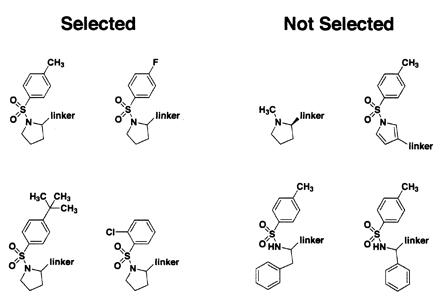
Compounds either selected by covalent tethering (Left) or present in the disulfide library but not selected (Right). Except in the case of N-tosyl-d-proline, all compounds tested were racemic unless otherwise indicated. For N-tosyl-d-proline, both stereoisomers were screened separately in different pools, and both were identified as hits, although the d-isomer appeared to be selected slightly more strongly than the l-isomer.
We determined the affinity of the tether-free analog, N-tosyl-d-proline, by Michaelis–Menten analysis. Enzymatic assays (12, 13) were performed to determine the inhibition constant (Ki). The Ki for the free acid was found to be 1.1 ± 0.25 mM and was competitive with respect to the substrate dUMP. Thus, N-tosyl-d-proline is a weak but competitive inhibitor of TS.
We next evaluated the set of ligands that could be captured by a nearby cysteine. To do this, we removed the active-site cysteine (C146S) and introduced a new cysteine nearby (L143C or H147C). Screening the disulfide-containing library against the C146S/L143C mutant TS produced similar results to the wild-type enzyme: the N-tosyl-d-proline analog was strongly selected (data not shown). In contrast, screening the disulfide-containing library against the C146S/H147C mutant TS did not result in selection of N-tosyl-d-proline. Several other molecules were selected by these mutants, but none were as prominent as N-tosyl-d-proline. These data demonstrate that the selection of N-tosyl-d-proline is not unique to the active-site cysteine, and that the exact placement of the reactive cysteine residue is not critical. As expected, however, there is some dependence on the position of the cysteine.
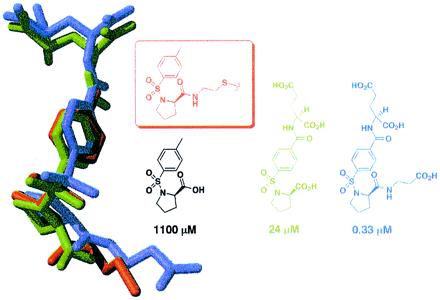
To determine the mode of tosyl-d-proline binding, either free or when tethered from C146 or L143C, we solved the x-ray crystallographic structures of the complexes (Table 1 and Fig. 4). Significantly, the location of the N-tosyl-d-proline moiety is very similar in all three cases (rms deviation of 0.55–1.88 Å, compared with 0.11–0.56 Å for all Cα carbons in the protein). The fact that the N-tosyl-d-proline substituents closely overlap while the alkyl-disulfide tethers converge onto this moiety from different cysteine residues supports the notion that the N-tosyl-d-proline moiety, not the tether, determines binding.
Table 1.
Crystallographic data and refinement parameters
| Data set | Space group* | Cell dimensions, Å | Resolution, Å | Reflections (no.)
|
Completeness,† % | Rsym (I),‡ % | I/σ | Rcryst,§ % | Rfree,¶ % | rms deviation bond lengths, Å | rms deviation bond angles, deg | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Unique | |||||||||||
| Native | I213 | a = 131.17 | 10 − 1.75 | 104,019 | 36,586 | 96.7 (91.6) | 4.9 (33.8) | 20.5 (4.0) | 19.8 | 24.4 | 0.010 | 2.30 |
| C146 tethered N-tosyl-d-proline | P63 | a = 126.22 c = 67.02 | 10 − 2.00 | 97,445 | 41,001 | 98.8 (94.5) | 4.4 (26.0) | 14.7 (4.1) | 19.8 | 26.8 | 0.010 | 2.59 |
| L143C tethered N-tosyl-d-proline | P63 | a = 126.33 c = 67.12 | 10 − 2.15 | 78,793 | 32,045 | 96.7 (92.1) | 8.1 (28.6) | 12.8 (4.5) | 19.6 | 26.7 | 0.014 | 3.06 |
| Noncovalent N-tosyl-d-proline | I213 | a = 131.88 | 10 − 1.90 | 202,300 | 31,422 | 100 (100) | 7.4 (28.2) | 19.7 (3.8) | 19.2 | 23.8 | 0.011 | 2.49 |
| Glu-TP | P63 | a = 126.14 c = 66.81 | 10 − 2.00 | 143,599 | 40,497 | 99.4 (96.9) | 8.5 (31.9) | 13.9 (4.0) | 19.4 | 25.1 | 0.007 | 2.15 |
| Glu-TP-β-Ala | P63 | a = 126.03 c = 66.84 | 10 − 1.75 | 142,016 | 58,487 | 95.8 (85.2) | 4.0 (22.5) | 17.1 (4.9) | 18.0 | 21.4 | 0.007 | 2.00 |
This is not a “true” free R factor because the starting model was a fully refined structure. However, the free R factor set of reflections was kept constant for each of the above refinements.
The I213 crystal contains one monomer per asymmetric unit. The P63 form contains the biologically relevant homodimer.
Values in parentheses are for the highest resolution bin.
Rsym (I) = Σhkl|Ihkl〈Ihkl〉|/ΣhklIhkl, where Ihkl is the intensity of reflection hkl.
Rcryst = Σhkl||Fobs| − |Fcalc||/|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively, for the data used in refinement.
Rfree = Σhkl||Fobs| − |Fcalc||/|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively, for 10% of the data omitted from refinement.
Figure 4.
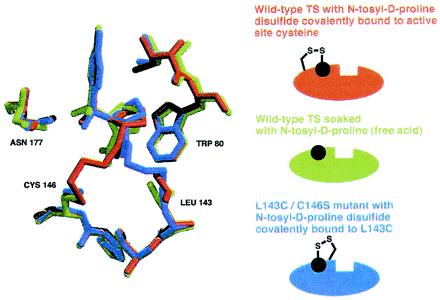
Overlay of three crystallographically determined structures. The structure in green was determined after soaking N-tosyl-d-proline (free acid) into crystals of unmodified TS. The structure in red is TS covalently modified by N-tosyl-d-proline disulfide-bonded to C146 (the active-site cysteine). Finally, the structure in blue is mutant TS (C146S/L143C) covalently modified by N-tosyl-d-proline disulfide bonded to L143C.
These results demonstrate that the tethering methodology can rapidly identify small low-affinity ligands. However, to be useful for drug discovery, the ligands identified must be amenable to medicinal chemistry and affinity maturation. In the case of N-tosyl-d-proline, examination of the bound structures revealed that the tosyl group is in roughly the same position and orientation as the benzamide moiety of methylenetetrahydrofolate, the natural cofactor for this enzyme (17, 21, 22). Given this positioning, we grafted the glutamate residue from methylenetetrahydrofolate onto the methyl group of N-tosyl-d-proline to try to enhance the affinity of the molecule (Fig. 5).
Figure 5.
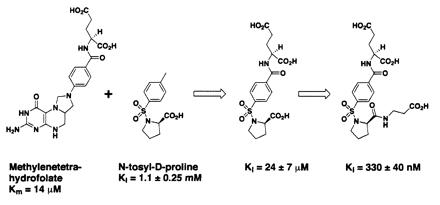
Grafting a glutamate residue onto N-tosyl-d-proline improves the affinity 50-fold, and adding a negatively charged appendage further increases the affinity by an additional 70-fold.
A series of compounds was synthesized and tested for TS inhibition. The parent compound, in which l-glutamate is grafted onto N-tosyl-d-proline, is almost 50-fold more active than N-tosyl-d-proline alone (Ki = 24 ± 7 μM). The α-carboxylate of the glutamate residue is very sensitive to modification, in that converting it to a primary amide reduces the affinity by an order of magnitude (not shown). Several substitutions were also incorporated at the proline carboxyl group; in one case, the displacement of the negatively charged carboxyl group away from the proline ring by two methylene units improves the inhibition constant by more than 70-fold, to Ki = 330 ± 40 nM (Fig. 5). Converting this β-alanine appendage to the isosteric isoamyl group [i.e., converting −NHCH2CH2CO2H to −NHCH2CH2CH(CH3)2, not shown] decreases the binding affinity by more than an order of magnitude (Ki = 12 ± 2.5 μM), suggesting that the displaced carboxylate is critical for improved binding. The crystallographic structures of TS complexed with two of the improved inhibitors (Table 1 and Fig. 6) reveal that the proline ring and the tosyl group overlap in all three structures, whereas the β-alanine and glutamate appendages make contacts with surrounding protein residues. The combined effects of these added substituents increase the affinity of one of the inhibitors for TS by more than 3,000-fold over N-tosyl-d-proline. The affinity of this inhibitor is submicromolar and thus well within the range of typical drug leads.
Figure 6.
Overlay of three crystallographically determined structures; only the inhibitor is shown for clarity. The inhibition constant (Ki) of each inhibitor is also shown.
In this study, we have focused on disulfide tethers. Although it is possible to use other types of reversible bonds, such as imines (23), the disulfide bond is well suited for tethering. Disulfide bonds can be formed and broken under very mild conditions with complete chemoselectivity. Moreover, the disulfide moiety itself can be easily introduced into a wide variety of small molecules at the end of a flexible and variable length tether. Finally, previous research has demonstrated that the redox potential of a disulfide bond in a macromolecule can vary with the conformation of the molecule, and that an intermolecular disulfide bond varies in stability as a function of the affinity of the two macromolecules (24–26). Thus, disulfide stability correlates with binding affinity.
A reasonable extension of the tethering technology would be to discover two weakly binding fragments that bind near one another and to link them to produce higher affinity compounds (27–31). Such a strategy has been applied in the technique “SAR by NMR” (32). In many cases, the new linked compounds bind to the target protein with much higher affinity than the precursors (33). Our tethering approach can also rapidly generate candidate molecules for linking.
In conclusion, we have developed an alternative and general approach for drug design that can be readily supplemented with existing technology. The approach allows us to identify ligands that bind very weakly to proteins and thus expands the discovery range for small molecule drugs. The weakly binding ligands can be rapidly optimized by using medicinal chemistry and structure-aided design. This approach is well suited to proteins containing a cysteine in the active site and is also expandable to proteins in which cysteines are introduced by mutagenesis. In principle, new cysteines can be placed anywhere; for example, if a cysteine is introduced onto the surface of a protein in an area known to be important for protein–protein interactions, small molecules could be selected that bind to and block this surface. We expect this covalent tethering methodology to be a powerful technique for generating drug leads.
Acknowledgments
We thank Dennis Lee and Jon Ellman for helpful discussions and Monya L. Baker for a critical reading of this manuscript. This research was supported in part by Small Business Innovation Research Grant No. 1 R43 CA85141–01.
Abbreviations
- TS
thymidylate synthase
- Ki
inhibition constant
- BOC
butoxycarbonyl
- DCM
dichloromethane
- TFA
trifluoroacetic acid
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1F4B, 1F4C, 1F4D, 1F4E, 1F4F, and 1F4G).
References
- 1.Santi D V, Danenberg P V. In: Chemistry and Biology of Folates. Blakley R L, Benkovic S J, editors. I. New York: Wiley; 1984. pp. 345–398. [Google Scholar]
- 2.Carreras C W, Santi D V. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 3.Kamen B. Semin Oncol. 1997;5 Suppl 18:S30–S39. [PubMed] [Google Scholar]
- 4.Jones T R, Calvert A H, Jackman A L, Brown S J, Jones M, Harrap K R. Eur J Cancer. 1981;17:11–19. doi: 10.1016/0014-2964(81)90206-1. [DOI] [PubMed] [Google Scholar]
- 5.Ivanetich K M, Santi D V. FASEB J. 1990;4:1591–1597. doi: 10.1096/fasebj.4.6.2180768. [DOI] [PubMed] [Google Scholar]
- 6.Vasquez J R, Gooze L, Kim K, Gut J, Petersen C, Nelson R G. Mol Biochem Parasitol. 1996;79:153–165. doi: 10.1016/0166-6851(96)02647-3. [DOI] [PubMed] [Google Scholar]
- 7.Webber S E, Bleckman T M, Attard J, Deal J G, Kathardekar V, Welsh K M, Webber S, Janson C A, Matthews D A, Smith W W, et al. J Med Chem. 1993;36:733–746. doi: 10.1021/jm00058a010. [DOI] [PubMed] [Google Scholar]
- 8.Shoichet B K, Stroud R M, Santi D V, Kuntz I D, Perry K M. Science. 1993;259:1445–1450. doi: 10.1126/science.8451640. [DOI] [PubMed] [Google Scholar]
- 9.Stout T J, Tondi D, Rinaldi M, Barlocco D, Pecorari P, Santi D V, Kuntz I D, Stroud R M, Shoichet B K, Costi M P. Biochemistry. 1999;38:1607–1617. doi: 10.1021/bi9815896. [DOI] [PubMed] [Google Scholar]
- 10.Stout T J, Sage C R, Stroud R M. Structure (London) 1998;6:839–848. doi: 10.1016/S0969-2126(98)00086-0. [DOI] [PubMed] [Google Scholar]
- 11.Climie S C, Carreras C W, Santi D V. Biochemistry. 1992;31:6032–6038. doi: 10.1021/bi00141a011. [DOI] [PubMed] [Google Scholar]
- 12.Chen S C, Daron H H, Aull J L. Int J Biochem. 1989;21:1217–1221. doi: 10.1016/0020-711x(89)90006-2. [DOI] [PubMed] [Google Scholar]
- 13.Wahba A L, Friedkin M. J Biol Chem. 1961;263:PC11–PC12. [PubMed] [Google Scholar]
- 14.Parlow J J, Normansell J E. Mol Diversity. 1995;1:266–269. doi: 10.1007/BF01715531. [DOI] [PubMed] [Google Scholar]
- 15.Perry K M, Fauman E B, Finer-Moore J S, Montfort W R, Maley G F, Maley F, Stroud R M. Proteins. 1990;8:315–333. doi: 10.1002/prot.340080406. [DOI] [PubMed] [Google Scholar]
- 16.Pflugrath J W. Acta Crystallogr D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 17.Montfort W R, Perry K M, Fauman E B, Finer-Moore J S, Maley G F, Hardy L, Maley F, Stroud R M. Biochemistry. 1990;29:6964–6977. doi: 10.1021/bi00482a004. [DOI] [PubMed] [Google Scholar]
- 18.Collaborative Computational Project N. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 19.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 20.Aull J L, Daron H H. Biochim Biophys Acta. 1980;614:31–39. doi: 10.1016/0005-2744(80)90164-3. [DOI] [PubMed] [Google Scholar]
- 21.Matthews D A, Appelt K, Oatley S J, Xuong N H. J Mol Biol. 1990;214:923–936. doi: 10.1016/0022-2836(90)90346-N. [DOI] [PubMed] [Google Scholar]
- 22.Kamb A, Finer-Moore J S, Stroud R M. Biochemistry. 1992;31:12876–12884. doi: 10.1021/bi00166a024. [DOI] [PubMed] [Google Scholar]
- 23.Huc I, Lehn J-M. Proc Natl Acad Sci USA. 1997;94:2106–2110. doi: 10.1073/pnas.94.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert H F. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 25.Stanojevic D, Verdine G L. Nat Struct Biol. 1995;2:450–457. doi: 10.1038/nsb0695-450. [DOI] [PubMed] [Google Scholar]
- 26.O'Shea E K, Rutkowski R, Stafford W F, III, Kim P S. Science. 1989;245:646–648. doi: 10.1126/science.2503872. [DOI] [PubMed] [Google Scholar]
- 27.Jencks W P. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Portoghese P S, Ronsisvalle G, Larson D L, Takemori A E. J Med Chem. 1986;29:1650–1653. doi: 10.1021/jm00159a014. [DOI] [PubMed] [Google Scholar]
- 29.Glick G D, Toogood P L, Wiley D C, Skehel J J, Knowles J R. J Biol Chem. 1991;266:23660–23669. [PubMed] [Google Scholar]
- 30.Wetterau J R, Gregg R E, Harrity T W, Arbeeny C, Cap M, Connolly F, Chu C-H, George R J, Gordon D A, Jamil H, et al. Science. 1998;282:751–754. doi: 10.1126/science.282.5389.751. [DOI] [PubMed] [Google Scholar]
- 31.Maly D J, Choong I C, Ellman J A. Proc Natl Acad Sci USA. 2000;97:2419–2424. doi: 10.1073/pnas.97.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shuker S B, Hajduk P J, Meadows R P, Fesik S W. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 33.Hajduk P J, Sheppard G, Nettesheim D G, Olejniczak E T, Shuker S B, Meadows R P, Steinman D H, Carrera G M, Marcotte P A, Severin J, et al. J Am Chem Soc. 1997;119:5818–5827. [Google Scholar]