

Graphical Abstract
Keywords: Nitric oxide, carbon monoxide, hydrogen sulfide, crosstalk, reactive oxygen species, cancer, cell signaling, iNOS, CBS, CSE, HO-1
Abstract
Nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) are three endogenously produced gases with important functions in the vasculature, immune defense, and inflammation. It is increasingly apparent that, far from working in isolation, these three exert many effects by modulating each other’s activity. Each gas is produced by three enzymes, which have some tissue specificities and can also be non-enzymatically produced by redox reactions of various substrates. Both NO and CO share similar properties, such as activating soluble guanylate cyclase (sGC) to increase cyclic guanosine monophosphate (cGMP) levels. At the same time, H2S both inhibits phosphodiesterase 5A (PDE5A), an enzyme that metabolizes sGC and exerts redox regulation on sGC. The role of NO, CO, and H2S in the setting of cancer has been quite perplexing, as there is evidence for both tumor-promoting and pro-inflammatory effects and anti-tumor and anti-inflammatory activities. Each gasotransmitter has been found to have dual effects on different aspects of cancer biology, including cancer cell proliferation and apoptosis, invasion and metastasis, angiogenesis, and immunomodulation. These seemingly contradictory actions may relate to each gas having a dual effect dependent on its local flux. In this review, we discuss the major roles of NO, CO, and H2S in the context of cancer, with an effort to highlight the dual nature of each gas in different events occurring during cancer progression.
1. Introduction
Nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) are gases that have been present in the Earth’s atmosphere from the beginning of time. Historically, these three gases have been considered pollutants from industrial activity, such as oil refineries, paper mills, and tanneries (Li, Hsu et al. 2009). However, it is now understood that these gases are endogenously produced and have important biological roles in most mammalian tissues. The term gasotransmitter was introduced in 2002 to describe NO, CO, and H2S (Wang 2002), particularly in relation to their regulatory functions within the nervous system (Nowaczyk, Kowalska et al. 2021, Siracusa, Schaufler et al. 2021).
Jan Baptista van Helmont first observed the formation of a red gas when aqua fortis (nitric acid) reacted with silver. This red gas is of course nitrogen dioxide, which is formed when NO reacts with oxygen; thus, van Helmont is credited for discovering NO (Butler and Nicholson 2003). However, it was Joseph Priestly who, in 1772, published a paper in the Philosophical Transactions (Priestley 1772) where he recognized NO as a distinct entity and gave it the name ‘nitrous air’ (Brennan, Thomas et al. 2003, McEvoy 2015, Lancaster 2020). The important role of NO in human biology was first recognized in 1992 when the journal Science introduced NO as the “Molecule of the Year” (Culotta and Koshland 1992), followed by the awarding of the Nobel Prize in Physiology and Medicine in 1998 to Robert F. Furchgott, Louis J. Ignarro, and Ferid Murad for the major discoveries surrounding NO and establishing its role as a messenger molecule. In the same year as his discovery of NO, Joesph Priestly produced CO via heating chalk to produce CO2 and reduced it to CO over a hot iron; thus, he is also credited with the formal discovery of CO (Schofield 1967, Hopper, Zambrana et al. 2021). The endogenous formation of CO was demonstrated in the early 1950s (Sjostrand 1951); but it was not until the mid-1990s that it was acknowledged as the second gasotransmitter (Verma, Hirsch et al. 1993). The discovery of the last gasotransmitter, H2S, was made in 1775, credited to the Swedish-German chemist Carl Wilhelm who produced this gas by heating sulfur in hydrogen gas (Nicholls and Kim 1981). French chemist Claude Louis Bethollet then determined the chemical composition of H2S in 1789 (Mitchell and Davenport 1924). In a landmark study in 1996, Abe and Kimura demonstrated the physiological importance of H2S as a neuromodulator (Abe and Kimura 1996), followed by Rui Wang’s proposal in 2002 that this was the third gasotransmitter to be added to the list alongside NO and CO (Wang 2002). For detailed historical perspectives on NO, CO, and H2S, please see (Lancaster 2020, Ghasemi and Kashfi 2022), (Kashfi and Patel 2022), (Szabo 2018) respectively.
Although these gasotransmitters can be toxic, they are now recognized as having multiple roles in normal physiology. NO is important in the vasculature for vasorelaxation (Ignarro, Buga et al. 1987) and modulation of platelet and leukocyte activation, adhesion, and aggregation (Wallace, Viappiani et al. 2009), in controlling inflammation through modulation of NF-κB activity (Katsuyama, Shichiri et al. 1998, Hattori, Kasai et al. 2004), and suppressing the expression of pro-inflammatory mediators in mast cells, macrophages, and vascular smooth muscle (Hogaboam, Befus et al. 1993, Huang, Sridhar et al. 1998, Naseem 2005), and in the nervous system through functions in non-adrenergic non-cholinergic (NANC) signaling (Król and Kepinska 2020) The second gasotransmitter, CO, has earned itself a reputation as a ‘silent killer’ because as a colorless, odorless gas, it is hard to detect and its intoxication can be fatal. Despite its known toxic effects, however, CO is an important endogenous signaling molecule with many physiological roles, including smooth muscle relaxation at low concentrations through ATP-dependent potassium (KATP) channel activation, which lowers blood pressure (Motterlini and Otterbein 2010), as well as effects on mitogen-activated protein kinase (MAPK) signaling pathways (Szabo 2016). As for H2S, functions include vasorelaxation through KATP channel activation through S-sulfhydration (Meng, Zhao et al. 2018), and maintenance of antioxidant defense (Xie, Feng et al. 2016). The biological half-lives of these gasotransmitters are variable; for NO, it is quite short, in the order of a few seconds; for CO, it is relatively long, in the order of minutes; and for H2S, it is somewhere in between, seconds to minutes (Szabo 2016).
The previously mentioned actions of the gasotransmitters have important implications in their effects in the context of cancer. NO, CO, and H2S have a dichotomous role in cancer, with some studies suggesting anti-inflammatory, anti-cancer effects of these gasotransmitters, while others suggest these gasotransmitters as players contributing to immune-mediated tissue injury and tumor promotion (Bhatia, Wong et al. 2005, Li, Bhatia et al. 2006, Tamizhselvi, Moore et al. 2007, Wallace 2007, Bhatia, Sidhapuriwala et al. 2008, Kashfi and Duvalsaint 2017, Szabo 2017). These controversies may relate to each having a dual effect dependent on the local flux of each gas (Ridnour, Thomas et al. 2006, Szabo 2016, Kashfi and Esmaili 2017, Szabo 2017). In this review, we present the current evidence surrounding the potential role of NO, CO, and H2S in the cancer setting, highlighting the paradoxical effects of each seen in cancer cell proliferation and apoptosis, invasion and metastasis, angiogenesis, immunomodulation, as well as consideration of the potential of these gases in the prevention/management of cancer immunosuppression-related infectious complications.
2. NO, CO, and H2S biosynthesis, metabolism, and signaling
NO is produced from the metabolism of L-arginine by the enzyme nitric oxide synthase (NOS) (Ignarro 1989, Moncada and Higgs 1993), which exists as three different isoforms with some differences in tissue specificity (Figure 1). Neuronal (nNOS or NOS1) and endothelial (eNOS or NOS3) are both constitutive, calcium-dependent forms of the enzyme that undergo negative feedback regulation (Stuehr 1997), in addition to regulatory phosphorylation (Dimmeler, Fleming et al. 1999), and interaction with other regulatory molecules (Sessa 2004). These isoforms produce nanomolar concentrations of NO for seconds or minutes to regulate neural and vascular function, respectively (Geller and Billiar 1998, Alderton, Cooper et al. 2001). The last NOS isoform, inducible NOS (iNOS or NOS2), is calcium-independent and transcriptionally regulated, induced by the presence of oxidative stress, inflammatory cytokines, hypoxia, and endotoxins (Nathan and Xie 1994, Kleinert, Schwarz et al. 2003). iNOS produces micromolar to low millimolar levels of NO, which may continue for hours or days (Michel and Feron 1997, Goligorsky, Brodsky et al. 2004, Kolios, Valatas et al. 2004) and plays a role involved in immune defense and inflammation. Aligning with its pro-inflammatory role, this isoform is highly expressed in many cancers, including glioma, breast, gastric, colon, leukemia, melanoma, ovarian, prostate, renal, and squamous carcinoma (Radomski, Jenkins et al. 1991, Jenkins, Charles et al. 1995, Thomsen and Miles 1998, Jahani-Asl and Bonni 2013, Heinecke, Ridnour et al. 2014, Granados-Principal, Liu et al. 2015, Vannini, Kashfi et al. 2015, Szabo 2016). Interestingly, the other two isoforms have also been documented to be upregulated in certain cancers; for instance, nNOS is induced in glioma, melanoma, and myeloma, and eNOS was found induced in pancreatic cancer, sarcoma, and renal cell carcinoma (Thomsen and Miles 1998, Vannini, Kashfi et al. 2015, Szabo 2016). In addition to its enzymatic production, NO can also be produced through the reduction of nitrate/nitrite, the so-called ‘nitrate-nitrite-NO pathway,’ or the non-enzymatic pathway under low oxygen conditions, as reviewed in (Kashfi 2018). NO signaling involves diffusion of the gasotransmitter across the plasma membrane into the cytoplasm, where it binds to the heme group of soluble guanylate cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP). The downstream effects of this signaling are mediated through cGMP-dependent Protein Kinase (PK) G (Friebe and Koesling 2003). Most of the actions of NO are a result of this signaling pathway, including, for instance, its inhibitory effects on leukocyte and platelet adhesion and aggregation and vasorelaxation (Lincoln, Cornwell et al. 1996, Dangel, Mergia et al. 2010). NO also participates in signaling independent of the sGC-cGMP pathway through direct protein modifications, including S-nitrosylation and tyrosine nitration, as well as effects mediated by peroxynitrite, a radical produced by the reaction of NO with superoxide (Martínez-Ruiz, Cadenas et al. 2011, Chiesa, Baidanoff et al. 2018). Increased activity of iNOS during periods of inflammation as well as eNOS uncoupling arising from endothelial damage, can lead to increased formation of peroxynitrite (Guzik, West et al. 2002, Münzel, Daiber et al. 2005, Li, Witte et al. 2006, Santhanam, d’,Uscio et al. 2012), which further promotes inflammation and is important in the defense against pathogens.
Figure 1. NO, H2S, CO biosynthesis and crosstalk.
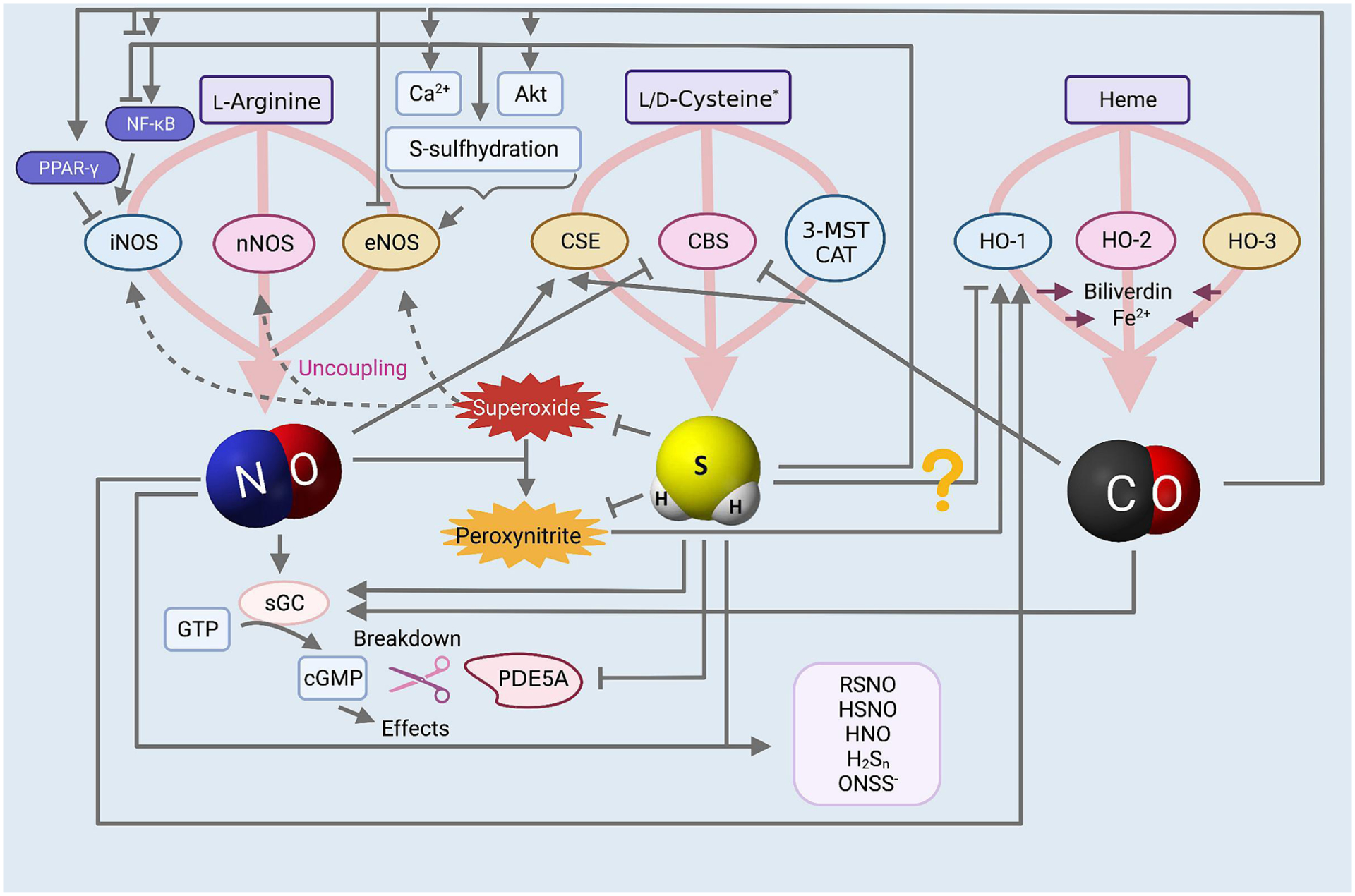
There is an extensive level of crosstalk between the gasotransmitters. NO is enzymatically produced from L-arginine by nNOS, eNOS, and iNOS, mediates many effects through the sGC-cGMP pathway, and this signaling is terminated by the degradation of cGMP by PDE5A. Superoxide anions can reduce NO bioavailability by causing NOS uncoupling and by reacting with NO to form peroxynitrite. H2S is enzymatically produced from L-cysteine by CBS, CSE, and 3-MST in conjunction with CAT. CO is enzymatically produced from the heme breakdown by the enzymes HO-1, HO-2, and HO-3, which additionally produce biliverdin and Fe2+. NO and H2S crosstalk: NO upregulates CSE expression while it inhibits the activity of CBS. H2S can upregulate eNOS expression and activity through intracellular Ca2+ release, Akt-mediated activating phosphorylation of eNOS, and eNOS S-sulfhydration, which inhibits the negative feedback regulation of NO on eNOS activity and maintains the active dimerized state. H2S has a dual effect on iNOS activity, and is capable of both increasing and decreasing its activity through similar dual effects on activating or inhibiting the iNOS transcription factor NF-κB. H2S reduces oxidative stress which increases NO bioavailability, and is a scavenger of peroxynitrite. H2S augments downstream NO signaling by inhibiting PDE5A activity to allow increased cGMP accumulation, and exerting redox regulation rendering sGC more responsive to NO. NO and H2S may also react with each other to form new chemical species such as nitrosothiols, nitroxyl, and others. NO and CO crosstalk: CO may have a dual effect on eNOS activity, both inhibiting it and augmenting it, the latter through Ca2+ release, Akt-mediated activating phosphorylation of eNOS, and protection of the enzyme from downregulation in inflammatory conditions. CO has dual effects on iNOS activity as well by either increasing or decreasing NF-κB transcription factor activity; additionally, CO has been found to activate PPAR-γ to downregulate iNOS as well. NO and peroxynitrite have both been seen to upregulate HO-1 expression. CO and H2S crosstalk: CO may inhibit CBS activity but upregulate CSE expression, while H2S may inhibit CO-1, although more investigation into these relationships is necessary.
The breakdown of heme is the major endogenous source of CO (Figure 1). Physiological degradation of heme is tightly controlled and involves the enzyme heme oxygenase (HO) (Tenhunen, Marver et al. 1969); the CO-producing reaction also releases free Fe2+ and biliverdin, the latter of which is then reduced to yield bilirubin. The enzyme HO has three isoforms, similar to the NOS enzymes. HO-1, also known as heat-shock protein 32 (Hsp32), is an inducible isoform that responds to oxidative stress, hypoxia, hyperoxia, UV irradiation, heat shock, ischemia, hyperthermia, and NO (Applegate, Luscher et al. 1991, Foresti and Motterlini 1999, Otterbein and Choi 2000, Liu, Mukosera et al. 2020). HO-2 is constitutively expressed in the brain, kidney, liver, and sleep; its function is closely associated with neurotransmission and regulation of vascular tone (Maines 1997, Foresti and Motterlini 1999). The third isoform, HO-3, is also a constitutive isoform of the enzyme, but cannot degrade heme, and thus its function remains to be elucidated (Wu and Wang 2005, Mann and Motterlini 2007). The enzymatic activity of heme oxygenase depends on nicotinamide adenine dinucleotide phosphate (NADPH) and molecular oxygen. Like NO, CO signaling involves the activation of guanylyl cyclase to produce cGMP, but it is approximately only 1/80th as effective in doing so as NO (Mann and Motterlini 2007). Low concentrations of CO also activate KATP channels and affect MAPK signaling pathways (Szabo 2016). The toxicity of CO that is seen with higher concentrations, earning it the name ‘silent killer,’ arises from its relatively high affinity for hemoglobin (Hb), which is 250 times greater than that of oxygen, resulting in the displacement of oxygen from Hb and preventing sufficient delivery to tissues. Furthermore, CO can also inhibit mitochondrial electron transport by irreversibly inhibiting cytochrome c oxidase (complex IV), contributing to CO inhalation poisoning (Motterlini and Otterbein 2010).With regards to the last of the known gasotransmitters, most of the biosynthesis of H2S has been attributed to three enzymes (Kashfi and Olson 2012): cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) and the tandem enzymes cysteine aminotransferase (CAT) and 3-mercaptopyruvate sulfurtransferase (3-MST) (Figure 1). CBS, CSE, and CAT metabolize L-cysteine in an enzymatic reaction requiring pyridoxal 5´-phosphate (PLP) as a cofactor (Erickson, Maxwell et al. 1990, Bukovska, Kery et al. 1994). 3-MST produces H2S using 3-mercaptopyruvate (3-MP), a metabolite of L-cysteine produced by CAT (Shibuya, Tanaka et al. 2009), (reviewed in (Kashfi 2018)). CBS, CSE, and MST are regarded as constitutive enzymes (Szabo 2016), although there is some evidence suggesting that CBS and CSE may be inducible (Paul and Snyder 2015, Phillips, Zatarain et al. 2017, Czikora, Erdélyi et al. 2022). The expression of CSE can be modulated by tumor necrosis factor-α (TNF-α), lipopolysaccharides (LPS), glucocorticoids, glucose, as well as endoplasmic reticulum (ER) stress (Zhu, Liu et al. 2010, Krishnan, Fu et al. 2011, Sen, Paul et al. 2012, Hine, Harputlugil et al. 2015, Paul and Snyder 2015). CBS expression has been found to be upregulated in myeloma, breast, colon, prostate, biliary tract, renal, and ovarian cancers compared to adjacent normal tissue or non-cancerous cells (De Vos, Thykjaer et al. 2002, Guo, Gai et al. 2012, Bhattacharyya, Saha et al. 2013, Szabo, Coletta et al. 2013, Sen, Kawahara et al. 2015, Szabo 2016, Vellecco, Mancini et al. 2016). CSE has also been reported to have a high expression in colon, melanoma, lung, and prostate cancers (Hellmich, Coletta et al. 2015, Hellmich and Szabo 2015, Panza, De Cicco et al. 2015), while MST is reported to be high in glioma and melanoma (Jurkowska, Placha et al. 2011, Hellmich and Szabo 2015, Szabo 2016). In addition to the well-known CBS, CSE, 3-MST/CAT enzymes, which metabolize L-amino acids, H2S can also be synthesized from D-cysteine (Shibuya and Kimura 2013, Shibuya, Koike et al. 2013) by the peroxisomal enzyme D-amino acid oxidase (DAO) (Gould, Keller et al. 1988) to 3-MP, the substrate for the mitochondrial enzyme 3-MST (Shibuya, Tanaka et al. 2009). However, as DAO is only localized to the brain and kidneys, the DAO/3-MST pathway is exclusively active in these two organs. Interestingly, D-cysteine is preferentially used by the kidney to produce H2S (Kimura 2015). H2S can also be endogenously produced through nonenzymatic pathways involving the reduction of cysteine (Yang, Minkler et al. 2019) and elemental sulfur in the blood (Westley and Westley 1991) using reducing equivalents from glycolysis (Searcy and Lee 1998), and from sulfur storage forms including thiosulfate, thiocysteine and sulfite (Kolluru, Shen et al. 2013), reviewed in (Kashfi and Olson 2012, Olson 2017). Furthermore, H2S levels can be indirectly increased through the activity of cysteinyl t-RNA synthetase (CARS) enzymes, which have been shown to produce cysteine persulfide that can then be metabolized to H2S and other sulfur species (Akaike, Ida et al. 2017). There are two known CARS enzymes, cytosolic CARS1 and mitochondrial CARS2; cysteine persulfide produced by the latter is important as an electron acceptor in the mitochondrial electron transport chain, a process during which H2S is created (Sawa, Takata et al. 2022).
H2S signaling involves S-sulfhydration of its protein targets, which is responsible for some of its physiological effects, including vasorelaxation (Zhao, Zhang et al. 2001) eNOS activity upregulation (Altaany, Ju et al. 2014), as well as activation of several signaling pathways including Keap1-Nrf2 (Yang, Zhao et al. 2012) and NF-κB (Sen, Paul et al. 2012). Recent studies have highlighted that a wide range of biological activity attributed to H2S results from other sulfur species, such as hydropersulfides and related polysulfides (Bianco, Akaike et al. 2019). For instance, cysteine persulfide, which can be produced by the CARS enzymes as previously mentioned (Akaike, Ida et al. 2017), and to a lesser extent, by CBS and CSE from serine (Bianco, Akaike et al. 2019): such hydropersulfides can exist in equilibrium with H2S in the presence of an oxidized thiol, and can serve to liberate H2S. There is also evidence to suggest that these sulfur species are responsible for the S-sulfhydration and thiol reduction thus far credited to H2S (Kolluru, Shen et al. 2020). Therefore, it is unclear whether H2S or these separate sulfur species are genuinely responsible for the actions attributed thus far to H2S. This is a point to consider when interpreting studies that assess the actions of H2S on various physiological and pathological models, as it is unclear whether H2S is always the bioactive molecule behind the effects observed. For more information on the biological roles of additional related sulfur species, please see (Ono, Akaike et al. 2014, Fukuto, Ignarro et al. 2018).
2.1. NO, CO, and H2S interactions and crosstalk
There is a vast level of interaction between the three gasotransmitters, which includes modulation of each other’s synthesis, downstream signaling, and direct chemical reactions producing intermediates with either enhanced or entirely different actions compared to the individual gasotransmitters (Figure 1). For instance, NO, CO, and H2S all bind avidly to hemoglobin. NO and hemoglobin interact to form nitrosyl hemoglobin, H2S and hemoglobin together form green sulfhemoglobin, and CO combines with hemoglobin to form scarlet carboxyhemoglobin (Arp, Childress et al. 1987, Wang 1998, Wang 2002). The competition between the three gasotransmitters for binding to hemoglobin is one manner in which each of the gasotransmitters can influence the activity of the others. Understanding of these interactions can be implemented to tailor combination therapies with either two or all three of these gasotransmitters in order to enhance their individual activities while curtailing unwanted effects of any one gasotransmitter. The specifics of these interactions are discussed in the following sections.
2.1.1. Nitric oxide and hydrogen sulfide interactions
Several studies report the modulation of H2S bioavailability by NO. Increasing NO levels using NO donors and the administration of the NOS substrate L-arginine has been found to increase H2S production from vascular tissue (Zhao, Zhang et al. 2001, Monti, Hyseni et al. 2018) and upregulate CSE expression (Yanfei, Lin et al. 2006, Lucetti, Silva et al. 2017), whereas inhibiting NOS using non-specific NOS inhibitor L-NG-Nitro arginine methyl ester (L-NAME) yields the opposite results (Zhong, Chen et al. 2003). Thus, exogenous NO administration may serve to both increase NO, as well as increase CSE-derived H2S. On the other hand, NO has been reported to bind and inhibit CBS (Taoka and Banerjee 2001), and reduce H2S formation in the liver and kidneys in a murine lipopolysaccharide (LPS) endotoxic shock model (Anuar, Whiteman et al. 2006), suggesting that the effects of NO on H2S production may be enzyme- and context-specific. When considering exogenous NO administration, carefully taking advantage of these interactions can allow efficient modulation of two of these gasotransmitters simultaneously.
Increasing H2S levels has also been found to augment NO bioavailability in mice with CSE knockout (King, Polhemus et al. 2014) and angiotensin II-induced hypertension (Al-Magableh, Kemp-Harper et al. 2015). This effect arises at least partly from H2S increasing eNOS expression and activity (King, Polhemus et al. 2014, Lucetti, Silva et al. 2017), which involves mechanisms including intracellular Ca2+ release (Kida, Sugiyama et al. 2013), Akt-mediated activating phosphorylation of eNOS (Cardounel, Julian et al. 2011), and S-sulfhydration of eNOS, which prevents the negative feedback regulation of NO (Altaany, Ju et al. 2014). H2S may also downregulate iNOS-derived NO in inflammatory conditions by inhibiting its transcription factor, nuclear factor kappa light chain enhancer of activated B cells (NF-κB), as seen in LPS-stimulated macrophages; interestingly, this effect also involved the HO-1/CO pathway, demonstrating the intertwined nature of all three gasotransmitters (Oh, Pae et al. 2006). A similar finding was also reported in another study, in which CSE inhibition allowed for increased LPS-induced macrophage production of NO, whereas overexpression of this enzyme curtailed NO release (Zhu, Liu et al. 2010). This effect may depend on the specifics of the experimental model, as highlighted by one study in which H2S was seen to enhance interleukin (IL)-1β-induced NO production, iNOS expression, and NF-κB activation but had no effect in the absence of IL-1β stimulation (Jeong, Pae et al. 2006). Furthermore, the effects of H2S differ depending on its concentration and the release kinetics of the donor molecules used; for instance, the slow release of H2S using GYY4137 suppresses LPS-induced NO release, whereas fast-releasing donor NaHS was only inhibitory at lower concentrations (200 μM) (Whiteman, Li et al. 2009). In addition to affecting the production of NO, H2S also increases the effects of NO signaling through redox regulation of sGC, which enhances the response of sGC to NO (Zhou, Martin et al. 2016), inhibition of PDE5A, which allows prolonged effects of NO (Bucci, Papapetropoulos et al. 2010), and antioxidant activity, which decreases oxidative and nitrosative stress, thus protecting against NOS uncoupling (Drachuk, Kotsjuruba et al. 2015).
In addition to modulating one another’s synthesis and downstream signaling, the direct reaction of NO and H2S and their derivatives can produce compounds such as nitrosothiols (RSNO), thionitrous acid (HSNO), polysulfides (H2Sn), nitroxyl (HNO), and nitropersulfide (ONSS−) (Cortese-Krott, Fernandez et al. 2014, Cortese-Krott, Kuhnle et al. 2015, Miyamoto, Koike et al. 2017), and these reaction products have been found to have either different, enhanced, or diminished effects compared to the gasotransmitters in isolation (Whiteman, Li et al. 2006, Yong, Hu et al. 2010, Berenyiova, Grman et al. 2015). These reaction products are reviewed in detail (Kevil, Cortese-Krott et al. 2017). Several NO- and H2S-co-releasing therapies, such as NOSHnonsteroidal anti-inflammatory drugs (NSAIDs), have already been designed, taking advantage of the extensive crosstalk between these two gasotransmitters (Kashfi, Chattopadhyay et al. 2015).
2.1.2. Carbon monoxide and nitric oxide interactions
Similar to the level of interaction between H2S and NO, extensive crosstalk also exists between CO and NO. The vasodilator effects of CO are largely suppressed by NOS inhibition (Foresti, Hammad et al. 2004), and CO has been found to increase steady-state NO levels in vitro by competing for endothelial intracellular binding sites (Thom, Xu et al. 1997). Taken together, these findings suggest the possibility that the vasodilator effects of CO rely at least partly on NOS-dependent NO production. Evidence suggests that the NO-upregulating effects of CO may be concentration-dependent, as in one study, low levels of CO induced NO release from intracellular storage pools. In contrast, higher concentrations reduced NO release due to eNOS inhibition in vitro (Thorup, Jones et al. 1999). In contrast to the eNOS inhibition seen in this study, CO has elsewhere been found to induce eNOS activation both in vitro (Yang, Huang et al. 2016, Choi, Kim et al. 2017) and in vivo (Fujimoto, Ohno et al. 2004). Mechanistically, this effect was attributed to the stimulation of intracellular Ca2+ release through inositol triphosphate (IP3) signaling, activating phosphorylation of Akt, and eNOS phosphorylation and dimerization (Yang, Huang et al. 2016). In addition to stimulating eNOS activity, CO has also been seen to inhibit pro-inflammatory TNF-α-mediated eNOS downregulation by inhibiting NF-κB and thus its downstream microRNA (miR)-155–5p (Choi, Kim et al. 2017). Therefore, in inflammatory states, CO may be protective towards eNOS. CO can also inhibit iNOS in states of inflammation, as seen in LPS-activated macrophages; mechanistically, this effect has been found to involve peroxisome proliferator-activated receptor (PPAR)-γ activation (Tsoyi, Ha et al. 2009) and NF-κB inhibition (Oh, Pae et al. 2006). Interestingly, there is evidence that these effects may be tissue-dependent, as in one study, CO prevented LPS-induced lung iNOS upregulation while supporting liver iNOS upregulation, both in vitro and in vivo (Sarady, Zuckerbraun et al. 2004). The effects of CO on iNOS are less well described. In the neuronal system, CO was seen to block NO-mediated increases in cGMP, suggesting the possibility that CO can inhibit nNOS-derived NO. However, no specific enzyme isoform was identified in this study (Ingi, Cheng et al. 1996).
The effects of NO on CO are less extensively characterized. However, there is evidence that exogenous NO administration can upregulate HO-1 expression and that this enzyme is responsible for the cytoprotective actions of NO in the endothelium (Polte, Abate et al. 2000). Additionally, the NO derivative peroxynitrite has also demonstrated the ability to upregulate HO-1, indicating this upregulation as a defense mechanism against NO-mediated oxidative and nitrosative stress (Foresti, Sarathchandra et al. 1999). Mechanistically, these effects are through the stabilization of HO-1 mRNA (Bouton and Demple 2000). Further study with the specific effects of NO on CO rather than its enzymatic source would be beneficial to inform future possibilities of combined use of these gasotransmitters, as a combined exogenous administration of the two has already demonstrated promising synergistic effects in the context of infection (Gao, Cheng et al. 2022).
2.1.3. Carbon monoxide and hydrogen sulfide interactions
Like with CO and NO, the interactions between CO and H2S are not as well characterized as those between NO and H2S. With the heme-binding activity of CO and the heme-containing structure of CBS, CO has been found to bind to CBS and induce its inactivation in vitro (Taoka, West et al. 1999, Puranik, Weeks et al. 2006). These results are supported by in vivo findings, where CO overproduction in the liver has been seen to downregulate H2S production and stimulate choleresis. In contrast, in CBS-knockout mice, CO overproduction could not cause either of those effects (Shintani, Iwabuchi et al. 2009). CO can also exert additional effects by inhibiting CBS, one of which includes activation of global protein methylation (Yamamoto, Takano et al. 2010). Additionally, there is evidence that CBS activity can inhibit H2S production by CSE, such that CO inhibition of the former can lead to an upregulation of H2S production by CSE (Kabil, Yadav et al. 2016). Thus, CO can exert direct actions on H2S-producing enzymes, while also indirectly affecting their activity by interfering with their own interactions. Correspondingly, in aortic smooth muscle cells, inhibition of endogenous CO production led to the upregulation of H2S content and CSE expression (Jin, Du et al. 2006), and in a murine model of gastric ulcers, exogenous CO administration increased endogenous H2S production and mRNA expression of CSE and CBS (Magierowski, Magierowska et al. 2018). The upregulation of CSE in both studies aligns with the previous discussion of CO enhancing CSE activity by relieving the negative feedback of CBS; however, the upregulation of CBS in the latter is a curious finding. It is possible that the inhibition of CBS activity by CO may activate compensatory mechanisms, which upregulate CBS production to restore H2S production by this enzyme.
There is also some evidence that H2S can impact CO availability. In the same in vitro study with aortic smooth muscle cells, inhibition of endogenous H2S production increased CO levels and HO-1 expression, whereas both decreased with the administration of NaHS (Jin, Du et al. 2006). These gasotransmitters can work together in different pathological contexts to exert a protective effect against various pathologies. In a murine model of chronic kidney disease-induced cognitive impairment, both NaHS and CO-releasing molecule (CORM)-3 reversed cognitive impairment and reduced oxidative stress; the protective effects of NaHS were abrogated by HO inhibition and CO depletion, and similarly, the effects of CORM-3 were abrogated by inhibition of endogenous H2S production (Hamidizad, Kadkhodaee et al. 2022). Thus, both gasotransmitters were interdependently neuroprotective. Yet in the gastric mucosa, whereas the protective effects of H2S have been found to be dependent upon endogenous CO, CO could exert protective effects independent of endogenous H2S (Magierowski, Magierowska et al. 2016, Magierowski, Magierowska et al. 2018). Thus, there seems to be a significant amount of crosstalk between these two gasotransmitters, and further characterization of the precise interactions may be useful so that combination therapies with CO and H2S can be designed, similar to the NO- and H2S-donating NSAIDs which have already demonstrated promising effects in several contexts (Kashfi, Chattopadhyay et al. 2015).
3. The Triple Crown: NO, CO, and H2S in carcinogenesis
There is significant evidence to suggest dual pro- and anti- cancer effects of NO, CO, and H2S due to their roles in apoptosis, cancer invasion and metastasis, angiogenesis, and immunomodulation (Figures 2,3,4). In addition, each of these gasotransmitters may have use in the prevention and/or management of severe infection in cancer patients, who are often predisposed to this complication. The actions of the gasotransmitters in each of these arenas are dependent upon the physiological or pathological model, dosage, release kinetics, and the mode of gasotransmitter delivery.
Figure 2. NO effects in the context of cancer.
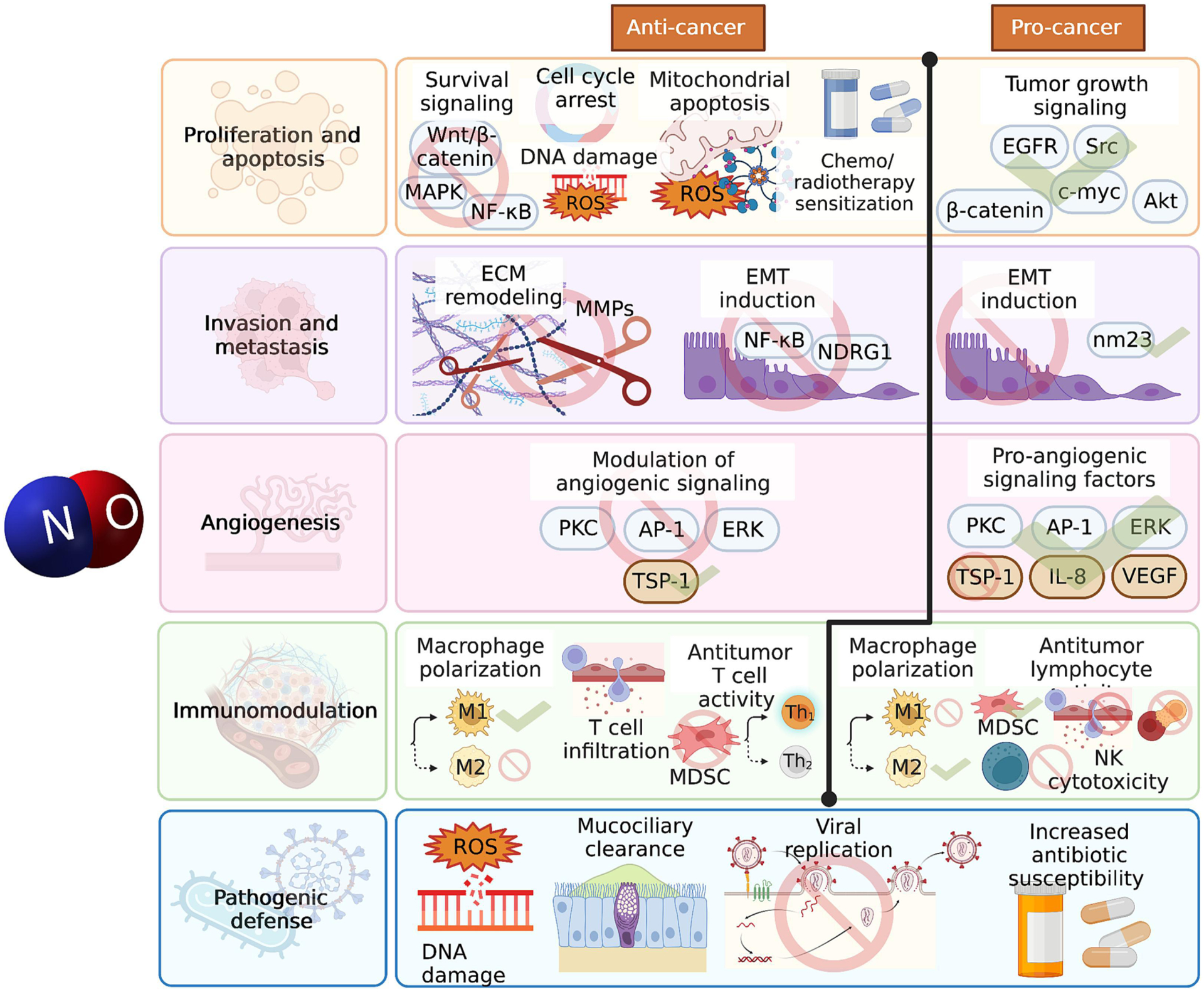
NO has both cancer-combatting and cancer-promoting effects that may depend on factors such as concentration, flux, and physiological or pathological setting. NO can reduce cancer cell proliferation by inhibiting several cell survival signaling pathways, causing cell cycle arrest, and increasing cancer cell apoptosis by causing DNA damage, increasing oxidative stress, and shifting the balance of pro- and anti- apoptotic proteins in favor of mitochondrial apoptosis; NO has also been found to increase the sensitivity of cancer cells to existing chemotherapeutic and radiation treatments. On the other hand, NO may also have pro-proliferative effects in the context of cancer by enhancing several other tumor growth signaling pathways. NO can inhibit EMT through inhibition of signaling pathways implicated in this phenomenon, and can inhibit ECM remodeling by downregulating MMPs. Both of these reduce the ability of the cancer to invade and metastasize. Yet NO has also been found to induce EMT in separate studies through the activation of different pathways than those implicated in inhibiting EMT. In the context of angiogenesis, NO may either facilitate angiogenesis or inhibit it through the modulation of different signaling pathways involved in this process. NO can enhance the anticancer immune response by enhancing M1 macrophage polarization, enhancing T cell infiltration, selectively inducing Th1 polarization, and inhibiting the immunosuppressive action of MDSCs. On the other hand, NO can suppress the host anticancer immune response by enhancing immunosuppressive MDSC accumulation, increasing M2 macrophage polarization, reducing NK cell cytotoxicity, and inhibiting cytotoxic T cell activity by reducing T cell activation and inducing apoptosis, hindering T cell infiltration, and facilitating the development of T cell tolerance. In addition to these actions, NO can play an important role in the increased infectious complications associated with cancer; this gasotransmitter has been shown to directly combat pathogens through ROS and DNA damage induction, increase mucociliary clearance, inhibit various steps of viral entry and replication, and increase the antibiotic susceptibility of bacteria.
Figure 3. CO effects in the context of cancer.
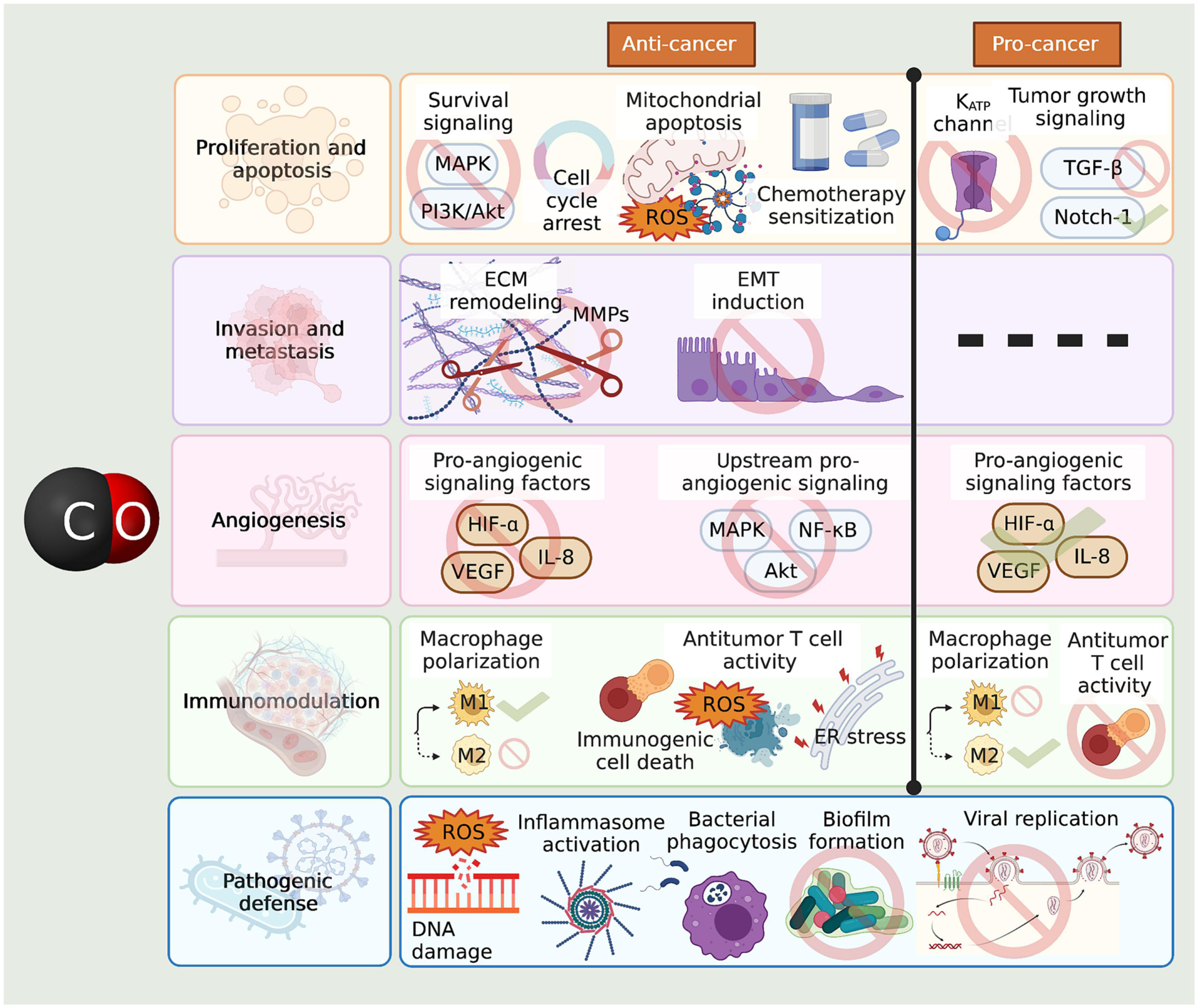
CO has widespread effects on cancer proliferation and apoptosis, invasion and metastasis, angiogenesis, and host anticancer immunity. The gasototransmitter has both cancer-combatting and cancer-promoting effects in these arenas, which may depend on factors such as concentration, flux, and physiological or pathological setting. CO can reduce cancer cell proliferation by inhibiting several cell survival pathways, inducing cell cycle arrest, favoring mitochondrial apoptosis, and increasing cancer sensitivity to chemotherapy. On the other hand, CO has also been found to increase cancer cell proliferation by increasing the activity of different growth signaling pathways, and to increase resistance to oxidant-induced apoptosis through a mechanism involving the inhibition of KATP channels. CO can suppress cancer cell invasion and metastasis by inhibiting ECM remodeling through MMP downregulation, and by suppressing EMT. With regards to angiogenesis, the gasotransmitter can either increase or decrease angiogenesis by different, likely context-dependent effects on several angiogenic signaling pathways. CO can increase antitumor host immunity by inducing M1 macrophage polarization, inducing immunogenic cell death through ROS accumulation, and inducing moderate ER stress, which leads to protective autophagy and epigenetic reprogramming of T cell into a stronger antitumoral phenotype. Yet CO may also suppress host antitumor immunity by inducing M2 macrophage polarization and suppressing T cell activity. In the context of infection, CO can cause DNA damage by ROS induction, cause bactericidal inflammasome activation and phagocytosis of bacteria, inhibit biofilm formation, and inhibit viral replication.
Figure 4. H2S effects in the context of cancer.
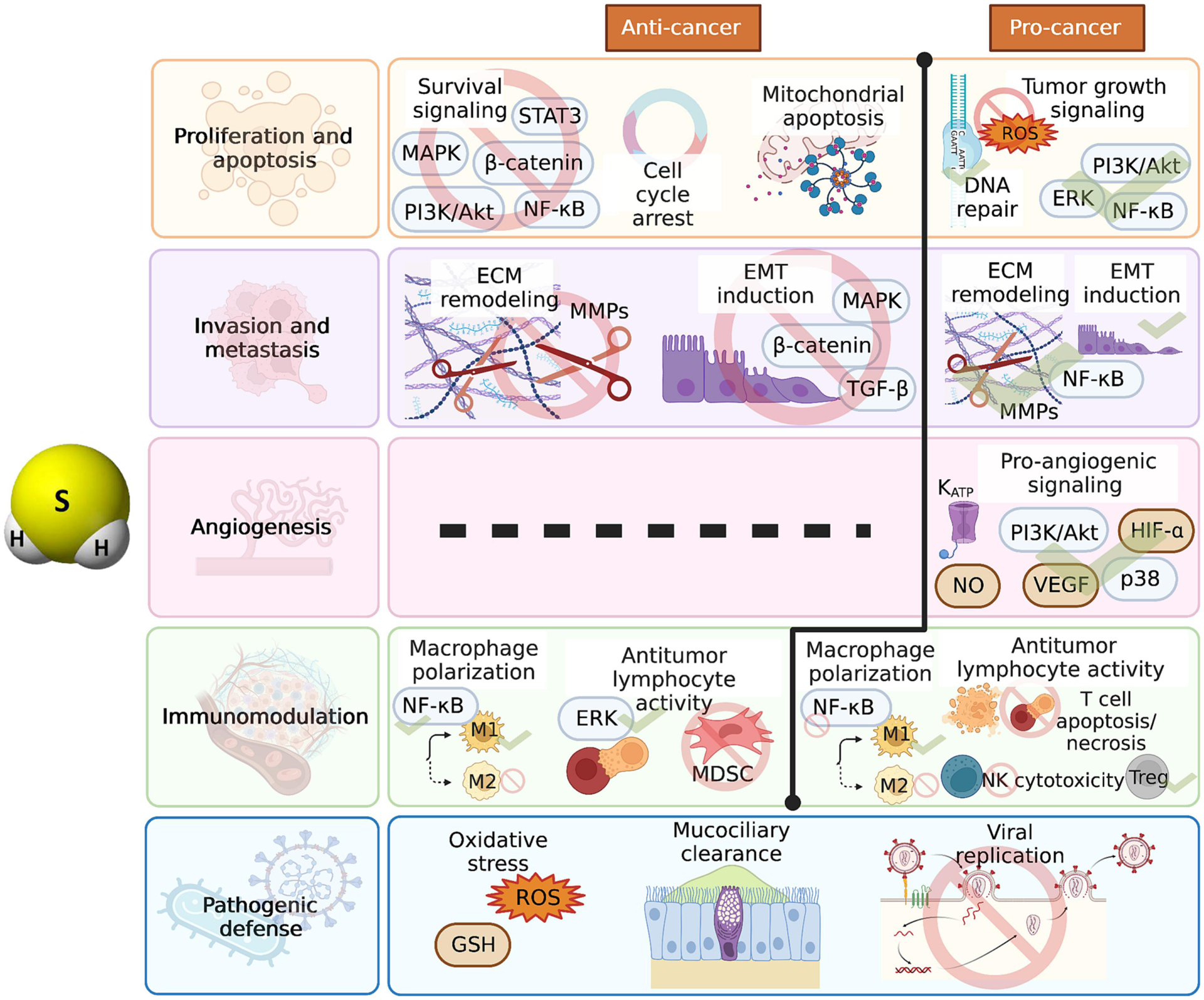
H2S also has both cancer-promoting and cancer-combatting effects resulting from its impact on cell proliferation and apoptosis, invasion and metastasis, angiogenesis, and antitumoral immunity. H2S can inhibit tumor proliferation by inhibiting various growth signaling pathways, inducing cell cycle arrest, and increasing cancer cell apoptosis. Contrarily, H2S can increase cancer proliferation and protect against apoptosis by activating various growth signaling pathways, maintaining DNA repair mechanisms, and suppressing ROS production. The gasotransmitter has been seen to both increase and suppress EMT induction and ECM remodeling through modulation of several different signaling pathways. In the context of angiogenesis, only a pro-angiogenic effect has been reported through increased pro-angiogenic signaling; mechanistically, NO and KATP channels have been found to be involved in the pro-angiogenic effect of H2S. H2S can boost anticancer immunity by increasing M1 macrophage polarization, enhancing T cell activation, and suppressing the immunosuppressive MDSCs; yet immunosuppressive activity of H2S has also been described, including M2 macrophage polarization, T cell death, impaired NK cell cytotoxicity, and enhanced Treg differentiation. In the context of infection, H2S exerts antimicrobial actions through increased oxidative stress upregulation of the antioxidant and antiviral agent GSH, increased mucociliary clearance, and inhibition of viral replication.
3.1. Role in cancer cell proliferation and apoptosis
Inhibition of the growth and proliferation of cancer cells is an effective mechanism to disrupt cancer progression and is the goal of a range of cancer therapies, such as radiation therapy and cytotoxic chemotherapeutic agents. There are several mechanisms to halt cancer growth, including induction of cell cycle arrest, inhibition of cell survival and growth signaling pathways such as phosphoinositide 3-kinase (PI3-K)/Akt, c-Jun N-terminal kinases (JNK), and p38 MAPK pathways—which are often aberrant in cancer (Wagner and Nebreda 2009, Samatar and Poulikakos 2014, Noorolyai, Shajari et al. 2019)—and induction of apoptotic cell death. Through the course of their development, cancer cells acquire many mutations that enable their uncontrolled proliferation and resistance to many cancer therapies (Fernald and Kurokawa 2013). These mutations encompass, among others, (1) mutations in tumor suppressors including p53, which stops cell cycle progression and activates the transcription of pro-apoptotic proteins (Rivlin, Brosh et al. 2011), (2) mutations in caspase genes (Soung, Lee et al. 2004, Sun, Gao et al. 2007) (3) overexpression of anti-apoptotic B-cell lymphoma 2 (Bcl-2) protein (Campana, Coustan-Smith et al. 1993), (4) increased levels of inhibitor of apoptosis proteins (IAP), including survivin (Small, Keerthivasan et al. 2010, Hernandez, Farma et al. 2011), X-linked inhibitor of apoptosis protein (XIAP) (Yu, Jin et al. 2018), and cellular inhibitor of apoptosis protein 1 (cIAP1) (Esposito, Kleeff et al. 2007), and (5) increased activation of cell survival signaling pathways such as the PI3-K/Akt pathway (Noorolyai, Shajari et al. 2019), MAPK pathway (Braicu, Buse et al. 2019), and Wingless/Integrated (Wnt)/β-catenin pathway (Jackstadt, Hodder et al. 2020). NO, H2S. and CO have all separately demonstrated the ability to either induce or protect against cancer cell growth and apoptosis through impacts on some of these survival adaptations.
3.1.1. Nitric oxide in cancer cell proliferation and apoptosis
NO has both pro- and anti-proliferative effects (Figure 2), which may depend on the physiological or pathological system in which its effects are observed. NO has shown pro-apoptotic activity against many cancers, including colon cancer (Oláh, Módis et al. 2018), pre-B acute lymphoblastic leukemia (Khan, Cisterne et al. 2012), and bladder carcinoma (Fabbri, Brigliadori et al. 2005), among others. Experimental upregulation of NO production through NOS2 transduction was found to almost completely inhibit the growth of prostate, gastric, colon, fibrosarcoma, breast, ovarian, renal cell carcinoma, and bladder tumor cell lines, which was linked to the induction of apoptosis (Le, Wei et al. 2005).
The cancer-targeting apoptotic effects of NO have been linked to multiple mechanisms, one of which is the induction of oxidative stress either through increased ROS production (Khan, Cisterne et al. 2012) or depletion of antioxidant machinery such as glutathione stores (Gao, Liu et al. 2005), leading to subsequent caspase activation and the mitochondrial apoptotic pathway. Increased oxidative stress can induce apoptosis via its known ability to cause DNA damage and impair DNA repair mechanisms. Indeed, there is evidence that NO can induce double-stranded DNA breaks in multiple myeloma cells, and its cytotoxic effects have been associated with the activation of the JNK pathway and activation of both intrinsic and extrinsic apoptotic pathways (Kiziltepe, Hideshima et al. 2007). In addition, there is evidence to suggest the ability of NO to affect the Bax/Bcl-2 ratio in favor of a pro-apoptotic balance in experimental cancer models (Singh, Chaudhary et al. 2012); in one study, the treatment of both non-metastatic and metastatic melanoma cells with NO donors resulted in dose-dependent apoptotic cell death of both cell populations, an effect that was associated with the downregulation of Bcl-2, and which was significantly reduced with Bcl-2 transfection (Xie, Wang et al. 1997). Interestingly, more aggressive cancer cells may be able to adopt mechanisms to resist the anticancer effects of NO by suppressing its release; in one study with melanoma cells, while IL-1α and interferon (IFN)-γ were able to induce NO production, Bcl-2 downregulation, and subsequent apoptotic cell death in nonmetastatic cells, these effects were minimal in the metastatic cells (Xie, Wang et al. 1997). In other words, the more aggressive metastatic cells were resistant to an increase in NO production and subsequent apoptosis upon exogenous stimulation. Correspondingly, in a murine model, endogenous NO production was inversely correlated with the survival of melanoma cells to produce metastases (Dong, Staroselsky et al. 1994). These findings suggest the downregulation of NO production as a pro-survival mechanism of aggressive cancer cells, which indicates a benefit to exogenous NO administration, especially against more aggressive cancers.
In addition to the direct induction of apoptosis, the suppression of several pro-survival signaling pathways is another mechanism through which NO inhibits cancer growth. The S-nitrosylating NO donor GSNO has, for instance, been found to inhibit the proliferation of both chemoresponsive and chemoresistant ovarian cancer cell lines by abolishing growth factor signaling, and was found to potentiate the toxicity of cisplatin therapy in vivo (Giri, Rattan et al. 2014). Furthermore, NO has been reported to induce apoptosis through the suppression of NF-κB (Williams, Nath et al. 2003, Khan, Cisterne et al. 2012), a transcription factor known to participate in carcinogenesis (Khan, Lopez-Dee et al. 2013) and whose suppression using agents other than NO has also been found to be sufficient to induce apoptosis in acute lymphoblastic leukemia cells (Meng, Martinez et al. 2010, Khan, Cisterne et al. 2012). Along with the suppression of NF-κB, the inhibition of colon cancer and breast cancer cell growth by NO-donating aspirin was associated with inhibition of the β-catenin/T-cell factor (Tcf) signaling pathway and upregulation of cyclooxygenase (COX)-2 (Williams, Nath et al. 2003, Nath, Vassell et al. 2009). The Wnt/β-catenin pathway has been identified as a pro-survival pathway implicated in cancer progression (Khan, Bradstock et al. 2007), and its inhibition is thus a mechanism for combatting cancer. The inhibition of Wnt signaling by NO has been further elucidated as having a dual mechanism based on a murine model of colon cancer – low concentrations of NO were seen to block the formation of β-catenin/Tcf complexes, and the additional mechanism of β-catenin cleavage was seen at higher concentrations (Gao, Liu et al. 2005). In addition to its impact on NF-κB and Wnt/β-catenin pathways, NO has also been seen to modulate MAPK signaling. Classical MAPK signaling involves three pathways—p38 MAPK, JNK, and extracellular signal-regulated kinase (ERK)1/2 (Zhang and Liu 2002). In a model of photo-induced carcinogenesis, topical NO administration using NO-exisulind reduced UVB-induced phosphorylation of the MAPK proteins ERK 1/2 and p38 (Singh, Chaudhary et al. 2012). Inhibition of these pro-survival pathways is thus a major mechanism through which NO can combat cancer growth.
NO may also inhibit cancer growth by directly influencing cell cycle progression. There is evidence from a range of cancer cell lines, including pancreatic, colon, prostate, lung, tongue, skin, cervix, and breast cancer cells that NO induces cell cycle arrest in G2/M (Kashfi, Rayyan et al. 2002, Gao and Williams 2012). Further investigation has revealed that NO can impact cell cycle progression by modulating the expression and activity of cell cycle regulatory proteins: increasing cyclin B1 expression, decreasing cyclin D1 and CDC25C expression, and increasing the phosphorylation of cyclin-dependent kinase (CDK)1 (Gao and Williams 2012). Considering the reversal of these effects with the administration of the antioxidant N-acetyl-cysteine, redox signaling was identified as the likely mediator of these effects (Gao and Williams 2012).
Aligning with its growth-inhibitory effects, NO has been found to increase the effects of various anticancer therapies. iNOS transfection has been demonstrated to enhance cisplatin toxicity in radiation-induced fibrosarcoma cells (Adams, McCarthy et al. 2009). Similarly, the treatment of prostate cancer cells with NO donors sensitized them to apoptosis by TNF-related apoptosis-inducing ligand (TRAIL) through NF-κB inactivation and inhibition of Bcl-related gene expression (Huerta-Yepez, Vega et al. 2004). These studies indicate that both endogenous and exogenous sources of NO can increase the pro-apoptotic effects of other anticancer agents. There have also been reports of radiosensitizing effect of NO and iNOS on colorectal cancer cells (Chung, Cook et al. 2003, Wang, Cook et al. 2004). In one of these studies, increased NO was associated with increased tumor vascularity, which although generally considered as a cancer-promoting hallmark, was suggested as a mechanism to increase the radioresponsiveness of the tumor (Wang, Cook et al. 2004). A second mechanism for the enhanced radiosensitivity was also found, involving NO-induced increase in the expression and activation of p53 (Wang, Zalcenstein et al. 2003, Cook, Wang et al. 2004). It seems that a functional p53 protein would be key to this effect, which may introduce a problem in the large proportion of p53-mutant tumors. Based on a separate study, the effect of NO on cancer growth may indeed depend on whether the tumor cells have functional or mutated p53; this study demonstrated that NO reduced the growth of colorectal cancer cell lines expressing wild-type p53, whereas it accelerated growth in their mutant p53 counterparts (Ambs, Merriam et al. 1998). These results suggest that the genotype of the cancer is an important consideration and may affect whether NO exerts the desired anticancer effects or does the opposite.
While the above studies indicate very promising, pro-apoptotic anticancer effects of NO, there is also evidence to the contrary. In a murine model of renal cell carcinoma, NO administration was found to be unable to affect the primary tumor burden in the mice, although it was able to reduce metastases and therefore improve survival (Weiss, Ridnour et al. 2010). Thus, in this case NO was unable to induce sufficient apoptosis to reduce the tumor burden, contrary to the previous studies demonstrating significant pro-apoptotic effects of the gasotransmitter; still, the beneficial effects of NO remained due to its ability to suppress the spread of the cancer. On the other hand, in a model of human breast cancer, NO induced the activation of epidermal growth factor receptor (EGFR) and steroid receptor coactivator (Src) through a mechanism involving S-nitrosylation which led to further activation of oncogenic pathways including cellular myelocytomatosis (c-Myc), Akt, and β-catenin, inhibited the tumor suppressor protein phosphatase 2A (PP2A)-c, and thus increased cancer cell proliferation and led to increased resistance of the cancer cells to adriamycin and paclitaxel chemotherapies (Switzer, Glynn et al. 2012). This directly contradicts prior studies in which NO was seen to inhibit c-Myc, Akt, and β-catenin signaling. Similarly, exogenous NO administration has been shown to protect neuroblastoma cells against H2O2-induced apoptosis (Yoo, Jung et al. 2018). A separate study with several different tumor models in vitro and in vivo also demonstrated that iNOS-derived NO production by tumor-associated M2-polarized macrophages protected tumor cells against apoptosis induced by cisplatin therapy by inhibiting acid sphingomyelinase (Perrotta, Cervia et al. 2018), whereas iNOS inhibition suppressed melanoma growth and synergized with cisplatin therapy in vivo (Sikora, Gelbard et al. 2010). The reason for these conflicting study results may be attributable to various factors, including method of NO augmentation, specific cancer model used, release kinetics, and concentration of gasotransmitter used. A bell-shaped effect of NO on the growth of colon cancer cells has been reported; whereas endogenous NO promoted colon cancer cell proliferation, the inhibition of endogenous CO production as well as exogenous administration suppressed cancer cell proliferation (Oláh, Módis et al. 2018).
3.1.2. Carbon monoxide in cancer cell proliferation and apoptosis
There is a wide range of evidence to support an anti-proliferative, pro-apoptotic effect of CO in different cancer models (Figure 3). A series of studies with photoactive CORMs have demonstrated their ability to induce apoptosis in the malignant cell lines HeLa and MDA-MB-231 human breast cancer cells with an efficacy comparable to approximately 3-day incubation with 10–25 μM 5-fluorouracil, a widely used cytotoxic chemotherapy (Carrington, Chakraborty et al. 2013, Carrington, Chakraborty et al. 2014, Chakraborty, Carrington et al. 2015). Recently, the antigen-specific delivery of CO using photoactivatable antibody-photoCORM has also demonstrated promise in selective CO delivery and cytotoxicity towards cancer cells in an ovarian cancer model (Kawahara, Gao et al. 2020).
Further exploration with another photoactive CORM, [Mn(CO)3Br(μ-bpcpd)]2 (MnCORM) with lung, cervical, breast, and colon cancer cell lines revealed that apoptosis was induced through the intrinsic pathway by increasing ROS, resulting in the loss of mitochondrial membrane potential and corresponding increases in Bax, cytochrome c release, caspases −3 and −9, and a decrease in anti-apoptotic Bcl-2 levels (P, D et al. 2018). These findings have found confirmation in other studies using CORM-2 with non-small cell lung cancer (Shao, Gu et al. 2018) and pancreatic cancer (Yan, Du et al. 2018). CO was also found to induce cell death in human lung carcinoma cells through mitochondrial exhaustion (Wang, Kan et al. 2021) and in breast cancer cells through metabolic starvation by inhibiting aerobic glycolysis (Guan, Zhou et al. 2019). Thus, CO may induce apoptosis through direct targeting of cancer cell metabolism and mitochondrial apoptosis. CO may also inhibit the proliferation of cancer cells through modulation of cell cycle regulatory proteins, demonstrated by the downregulation of cyclin D1 and CDK4 upon CORM treatment of MCF7 and MDA-MB-231 breast cancer cells (Lee, Chen et al. 2014). Cyclin D1 and CDK4 are both involved in promoting cell cycle progression and their upregulation in different cancers has been associated with poorer prognosis (Kenny, Hui et al. 1999, Dong, Sui et al. 2001, Bahnassy, Zekri et al. 2004, Wu, Wu et al. 2011), thus the ability of CO to downregulate these proteins can exert anticancer effects by suppressing cell cycle progression. CO was also able to downregulate human telomerase reverse transcriptase (hTERT) (Lee, Chen et al. 2014), which is implicated in conferring cancer cells with replicative immortality, motility, and a stem cell phenotype (Hannen and Bartsch 2018). Importantly, this gasotransmitter has also demonstrated the ability to modulate the expression of p53, a tumor suppressor protein responsible for cell cycle arrest and apoptosis where needed, mutations of which can promote cancer development and resistance to cancer drug resistance (Hientz, Mohr et al. 2017, Hu, Cao et al. 2021). Interestingly, CORM treatment was found to increase p53 expression in MCF7 cells, which had wild-type p53, while it downregulated p53 expression via ubiquitination in MDA-MB-231 cells, which had mutant p53 (Lee, Chen et al. 2014). This indicates that CO therapy may both, boost the tumor suppressive activity of p53 in cancer cells where p53 function remains normal, while eliminating mutant p53 that may promote cancer cell growth and drug resistance (Hientz, Mohr et al. 2017, Hu, Cao et al. 2021). An earlier study with chronic lymphocytic leukemia cells showed a similar pattern, where inhibition of heat shock protein 90 (Hsp90) downregulated mutant p53 while upregulating wild-type p53 (Lin, Rockliffe et al. 2008). This suggests the possibility that CO may affect p53 through an Hsp90-dependent mechanism; indeed, both CORM treatment and HO-1 induction resulted in downregulation of Hsp90-client proteins, including Akt, estrogen receptor alpha (Erα), and CDK4, indicating inhibition of Hsp90 by this gasotransmitter (Lee, Chen et al. 2014). Akt is a serine/threonine kinase whose aberrant activation is oncogenic and has been seen in many cancers, and its functions are reviewed in detail in (Revathidevi and Munirajan 2019). This highlights another avenue through which CO can inhibit cancer proliferation: through downregulation of growth signaling pathways that can increase tumor growth. The modulation of Akt activity by CO was also reported in a model of pancreatic cancer, where decreased activating phosphorylation of Akt was seen in association with CO (Vítek, Gbelcová et al. 2014). Other findings that have been linked with the CO-mediated induction of apoptosis in lung tumors were increased expression of CD86, a molecule necessary for antigen presenting cells (Axelsson, Magnuson et al. 2020), and activation of MAPK/ERK1/2-c-myc pathway in the tumor microenvironment (TME) (Nemeth, Csizmadia et al. 2016), a pathway which has also been identified to be important in solid tumor growth (Zuo, Liu et al. 2023).
Importantly, CO administration may also increase the cytotoxic effects of various existing chemotherapies. In two separate studies, CO exposure through administration of either HisAgCCN, a photocatalytic nanomaterial that can convert endogenous CO2 to CO, or air exposure at 250 ppm, was found to synergize DNA-damaging doxorubicin and camptothecin chemotherapies in inhibiting prostate cancer growth; these studies reported increased oxidative stress (Zheng, Li et al. 2017) and mitotic catastrophe followed by mitochondrial collapse (Wegiel, Gallo et al. 2013) as the mechanisms of apoptosis induction. Interestingly, both studies also demonstrated protective actions of CO for normal cells against chemotherapy-induced cell death (Wegiel, Gallo et al. 2013, Zheng, Li et al. 2017), an indication that CO therapy have a use in the selective targeting of cancer cells. A separate study with doxorubicin-resistant MCF-7/ADR tumors showed that thermal-induced CO-releasing platforms reversed the resistance to therapy by simultaneously causing mitochondrial exhaustion, resulting in ATP depletion and blocking ATP-dependent efflux of doxorubicin, while also inducing apoptosis through caspase-3 upregulation (Li, Dang et al. 2019). CO treatment has also been found to synergize with a multitude of other chemotherapeutic agents, including the experimental chemotherapeutic agent tirapazamine by inducing mitochondrial exhaustion apoptosis in breast cancer (Li, Dang et al. 2019), the microtubule-targeting paclitaxel by inhibiting CYP3A4/2C8 (Kawahara, Faull et al. 2021), and, in ovarian cancer, the cytotoxic DNA-damaging agent cisplatin by inhibiting CBS, thus reducing glutathione and metallothionein, which may be implicated in cisplatin resistance (Kawahara, Ramadoss et al. 2019). Interestingly, while CO may increase the sensitivity of cancer cells to cisplatin-induced apoptosis, it was protective against cisplatin-induced apoptosis in non-cancerous renal tubular cells (Tayem, Johnson et al. 2006); these opposing effects in cancerous vs non-cancerous cells may indicate a beneficial selectivity of this gasotransmitter’s pro-apoptotic effects. In contrast to the anti-proliferative and pro-apoptotic effects of CO on various cancer models as described above, exogenous administration of CO has been found to increase the resistance of medulloblastoma cells to oxidant-induced apoptosis (Al-Owais, Scragg et al. 2012), and protect hepatocellular cancer cells against cell cycle arrest induced by the anti-proliferative cytokine transforming growth factor (TGF)-β1 (Park, Lee et al. 2018); mechanistically, these findings were linked to an inhibitory effect of CO on potassium (K+) channels, and CO-induced Smad3 phosphorylation through the ERK 1/2 pathway, respectively. Similarly, inhibition of HO-1 was found to reduce, whereas HO-1 inducers increased, the size and number of tumorspheres in breast cancer stem cells (Kim, Yoon et al. 2018). These effects were attributed to HO-1-derived CO, as CORM-2 treatment increased the proportion of MDA-MB-231 retaining cancer stem cell properties, and increased the expression of neurogenic locus notch homolog protein 1 (Notch-1), which was accompanied by an increase in tumorsphere formation (Kim, Yoon et al. 2018). The reason behind these conflicting effects of CO is not entirely clear, although it is possible that the experimental setup, including the CO administration strategy, concentration, route, release kinetics, and the type of cancer may play a role. Interestingly, one in vitro study demonstrated a bell-shaped effect of CO on the proliferation of colon cancer cells: endogenous CO production was found to promote their proliferation, whereas both inhibition of endogenous CO production and exogenous administration suppressed proliferation (Oláh, Módis et al. 2018). Thus, it is possible that while physiologic H2S levels promote cancer growth, both supraphysiological and below physiological levels causes cancer growth inhibition.
3.1.3. Hydrogen sulfide in cancer cell proliferation and apoptosis
Exogenous H2S administration using H2S-releasing NSAIDs has demonstrated the potential to exert growth inhibitory effects against several cancer cell lines in vitro, including colon, breast, T-cell leukemia, pancreatic, prostate, and lung cancers (Chattopadhyay, Kodela et al. 2012). There is evidence to suggest a protective role of CSE, in particular, against cancer. For instance, the analysis of human melanoma samples revealed that the highest levels of CSE were found in primary tumors, whereas metastases, especially those involving the lymph nodes, had severely reduced CSE (Panza, De Cicco et al. 2015), raising the possibility of CSE being a protective factor whose loss allows increased tumor aggressiveness. Correspondingly, in further investigation with melanoma cell lines both CSE overexpression and exogenous H2S administration inhibited cell proliferation and induced apoptosis; likewise, L-cysteine administration to mice bearing melanoma tumors was protective against tumor growth, whereas CSE inhibition abolished this protective effect (Panza, De Cicco et al. 2015). Interestingly, H2S donor S-propargyl-cysteine (SPRC), which was seen to have significant anti-cancer effects both in vitro and in vivo in gastric cancer, was also seen to upregulate CSE expression and activity (Ma, Liu et al. 2011). This suggests the possibility that exogenous administration of H2S can be used to upregulate the innate antitumor defense provided by endogenous H2S production.
Mechanistically, H2S employs both pro-apoptotic and anti-proliferative effects to combat cancer growth (Figure 4). Exogenous H2S administration using various H2S donors in vitro has been seen to cause cell cycle arrest in the G0/G1 phase in melanoma cells (Panza, De Cicco et al. 2015), gastric cancer cells (Ma, Liu et al. 2011), urothelial bladder carcinoma cells (Panza, Bello et al. 2022), colon cancer cells (Chattopadhyay, Kodela et al. 2012, Wu, Wang et al. 2012), cisplatin-resistant lung cancer cells (Ma, Yan et al. 2018), and breast cancer cells (Chattopadhyay, Kodela et al. 2012). Separate studies also reported H2S to cause G2/M arrest in breast cancer cells (Lee, Zhou et al. 2011) and pancreatic adenocarcinoma cells, the latter of which was also found to have S phase arrest (Citi, Piragine et al. 2019). Cell cycle arrest by exogenous H2S administration has been associated with the upregulation of the cyclin-dependent kinase inhibitor p21cip (Takeuchi, Setoguchi et al. 2008, Wu, Wang et al. 2012), suppression of cyclin D1 and CDK4 expression Panza, Bello et al. 2022) and inhibition of inactivation phosphorylation of the cell cycle regulatory Retinoblastoma (Rb) protein (Takeuchi, Setoguchi et al. 2008).
In combination with cell cycle arrest, H2S also induces apoptosis to further inhibit cancer growth (Lee, Zhou et al. 2011, Ma, Liu et al. 2011, Chattopadhyay, Kodela et al. 2012, Chattopadhyay, Kodela et al. 2012, Panza, De Cicco et al. 2015). The pro-apoptotic effects of H2S involve several mechanisms, including induction of a shift to a pro-apoptotic state by affecting the balance of pro- and anti-apoptotic proteins. In vitro, the H2S donor diallyl trisulfide (DATS) has been found to downregulate the anti-apoptotic proteins XIAP and Bcl-2 in urothelial bladder carcinoma cells (Panza, Bello et al. 2022), and similarly, NaHS downregulated Bcl-xL in lung cancer cells (Ma, Yan et al. 2018). In addition to the downregulation of anti-apoptotic proteins, exogenous H2S administration using NaHS also upregulated pro-apoptotic and cell cycle inhibitory proteins p53 and its activated phosphorylated form, p21, caspase-3, and Bax (Ma, Yan et al. 2018). These findings have been supported in vivo in mice bearing gastric cancer tumors, where SPRC was seen to increase Bax and p53 expression while decreasing the expression of Bcl-2 (Ma, Liu et al. 2011). The downregulation of anti-apoptotic proteins has been attributed to H2S-induced inhibition of NF-κB activation, which plays an anti-apoptotic role through upstream upregulation of these proteins (Panza, De Cicco et al. 2015, De Cicco, Panza et al. 2017). A separate study using H2S-releasing aspirin further elucidated that the inhibition of NF-κB DNA binding activity is associated with prevention of IkappaKinase activation, leading to inhibition of phosphorylation-mediated degradation of IκBα, ultimately preventing the translocation of NF-κB-p65 into the nucleus (Chattopadhyay, Kodela et al. 2012). Additionally, reduced activation of the β-catenin (Huang, Yang et al. 2015), signal transducer and activator of transcription (STAT)3 (Lu, Gao et al. 2014), PI3-K/Akt pathways and MAPK/ERK pathways (Panza, De Cicco et al. 2015, De Cicco, Panza et al. 2017), as well as the other two MAPKs, JNK and p38 (Pei, Wu et al. 2011) have been implicated in H2S-mediated apoptosis. Interestingly, one study attributed the anti-proliferative effect of H2S to induction of autophagy rather than apoptosis; mechanistically, this induction of autophagy was through H2S stimulation of AMP-activated protein kinase (AMPK) phosphorylation, and inhibition of mammalian target of rapamycin (mTOR) phosphorylation (Wu, Wang et al. 2012). Yet another study identified both, apoptosis and autophagy, as the mechanism of anticancer activity exerted by DATS (Panza, Bello et al. 2022).
A drawback of many currently available cancer therapies is a lack of selectivity for cancerous tissue, leading to significant damage to normal tissue. In one study, H2S donor NaHS demonstrated a similar drawback, being non-selective in its growth inhibitory effects on colon cancer cells and non-cancerous colon epithelium (Wu, Wang et al. 2012). The possibility of H2S damaging normal non-cancerous tissues aligns with the known cellular cytotoxicity of H2S (Jiang, Chan et al. 2016) that for so long led to it being known as nothing more than a toxic gas. On the other hand, however, several studies have demonstrated selectivity in the growth-inhibitory actions of various H2S donors; for instance, H2S-releasing aspirin was seen to preferentially inhibit the growth of breast cancer cells compared to normal mammary epithelial cells (Chattopadhyay, Kodela et al. 2012), and GYY4137 was able to inhibit the growth of seven different cancer cell lines without any effect on non-cancer fibroblast cells (Lee, Zhou et al. 2011). Additionally, in one study, the continuous exposure of cancer cells, but not non-cancerous cells, to GYY4137 resulted in increased glycolysis and lactate overproduction alongside reduced pH regulatory activity, leading to intracellular acidification and cell death (Lee, Teo et al. 2014). Thus, there may be an optimal therapeutic administration strategy that maximizes cancer cell damage without allowing H2S to exert toxic effects on the host. Considering the promise that H2S may potentially act as a selective cancer therapy that minimizes damage to non-cancerous tissue, further investigation to elucidate the extent of H2S selectivity is worthwhile.
Interestingly, an anti-apoptotic, pro-proliferative effect of H2S has also been described in the context of cancer (Figure 4). NaHS has been found to induce proliferation of colon cancer cells (Cai, Wang et al. 2010) and oral squamous cell carcinoma cells (Ma, Bi et al. 2015), the former through Akt and ERK phosphorylation and p21 inhibition to allow cell cycle progression (Cai, Wang et al. 2010), and the latter through downregulation of cell cycle regulatory genes RPA70 and Rb1, and upregulation of proliferating cell nuclear antigen (PCNA) and CDK4 (Ma, Bi et al. 2015). Many of same targets have been previously reported to be affected by H2S in a completely opposite fashion, as highlighted in preceding sections. In addition to enabling proliferation itself, NaHS has also demonstrated the ability to protect colon cancer cells from apoptosis induced by the potential anti-tumor agent β-phenylethyl isothiocyanate (PEITC) in vitro (Rose, Moore et al. 2005).
One possibility accounting for the opposite effects of H2S on cancer cell growth may be differences in the dosage and H2S release kinetics of the exogenous donor used. In one study, whereas low dose NaHS (10–100 μM) enhanced the proliferation and viability of hepatocellular carcinoma cells, the same donor at higher concentrations (600–1000 μM) dose-dependently inhibited their proliferation (Wu, Li et al. 2017). This supports the suggestion of a biphasic effect of this gasotransmitter. In addition to dose-dependent effects, release kinetics may also play a role in these conflicting results. The slow donor GYY4137 has been seen to significantly reduce the growth of several cancer cell lines in vitro—30–70% reduction at 400 μM, and 75–95% reduction at 800 μM—including breast adenocarcinoma, acute promyelocytic leukemia, myelomonocytic leukemia, cervical carcinoma, colorectal carcinoma, hepatocellular carcinoma, and osteosarcoma. Yet in the same study, quick-releasing donor NaHS had no effects on cancer cell survival at the lower concentration, and yielded only a small reduction (15–30%) in the viability of three of the seven cell lines at the higher concentration (Lee, Zhou et al. 2011). In a separate study using several H2S donors, NaHS was the only one of all compounds tested that failed to demonstrate melanoma cell proliferation in vitro (Panza, De Cicco et al. 2015), raising the possibility that the release kinetics and properties of this particular H2S donor is the reason behind the discrepancy observed here, rather than an anti-apoptotic effect of H2S itself. Additionally, as H2S is a highly volatile compound, it is likely that exposure times were short, further impacting the results seen.
Yet other studies have implicated not only NaHS administration, but also CSE and CBS expression and activity in the pro-proliferative effects on cancer cells, indicating a more complex relationship than proposed above. Exogenous H2S administration using NaHS as well as increased endogenous production through CBS overexpression was seen to protect neuroblastoma cells from apoptosis induced by a dopaminergic neurotoxin; these effects were associated with activation of the protein kinase C (PKC)/PI3K/Akt pathway (Tiong, Lu et al. 2010). Thus, the overexpression of endogenous H2S-producing enzymes may contribute to cancer growth and progression, contrary to the benefits observed previously with exogenous H2S supplementation. Correspondingly, CBS overexpression has been documented in ovarian carcinoma (Bhattacharyya, Saha et al. 2013) and both CSE and CBS overexpression has been reported in hepatocellular carcinoma (Pan, Ye et al. 2014, Zhen, Pan et al. 2015). Inhibition of these enzymes has been found to inhibit tumor growth, increase apoptosis, and sensitize cancer cells chemotherapy (Bhattacharyya, Saha et al. 2013, Pan, Ye et al. 2014). Differences between in vitro and in vivo anticancer activity of H2S may at least partly contribute to these contradictory findings, as in one study NaHS was seen in vitro to increase responsiveness of tumor cells to radiotherapy, whereas no effects on tumor growth was seen in vivo (De Preter, Deriemaeker et al. 2016). Still, given that both anti- and pro- apoptotic effects have been seen in vitro and in vivo, other factors as donor molecule chosen, concentrations tested, release kinetics, etc, may together be responsible for such alternating findings.
Mechanistically, the demonstrated anti-apoptotic and pro-proliferative effects of H2S observed from these studies have been attributed to a variety of mechanisms, including activation of NF-κB (Zhen, Pan et al. 2015) possibly through sulfhydration of the enzyme (Sen, Paul et al. 2012), and activation of Akt alongside suppression of ROS production (Taniguchi, Kang et al. 2011), and stimulation and maintenance of DNA damage repair mechanisms (Zhao, Ju et al. 2014, Szczesny, Marcatti et al. 2016). NaHS administration has also been seen to induce survivin expression (Cai, Wang et al. 2007), a known inhibitor of apoptosis and downstream product in the PI3-K/Akt pathway. Inhibition of H2S-producing enzymes, on the other hand, has been found to be responsible for increasing ROS and disruption of mitochondrial activity (Bhattacharyya, Saha et al. 2013), suppressing PI3K/Akt/mTOR signaling (Khan, Wang et al. 2022), activating cell cycle regulatory proteins p53 and p21, decreasing Bcl-2/Bax ratio, and inhibiting of ERK1/2, leading to suppression of EGFR survival signaling (Pan, Ye et al. 2014). These findings are surprising in that many of these very same pathways were found to be utilized by endogenous and exogenous H2S to exert anti-tumor activity, raising questions as to the reason behind these findings.
3.1.4. Gasotransmitters in combination in cancer cell proliferation and apoptosis
In addition to the individual potential benefits of NO, H2S, and CO in various cancer models, the development of dual gasotransmitter-releasing compounds, mainly NO- and H2S- releasing NOSH-aspirin, illustrate that combination therapy with two or more of these gasotransmitters with other pharmacological agents is another possible strategy. NOSH-aspirin has been found to inhibit tumor growth in xenograft colon cancer murine models (Chattopadhyay, Kodela et al. 2012, Kodela, Chattopadhyay et al. 2015), with a growth reduction of 85% reported (Chattopadhyay, Kodela et al. 2012). Another NO- and H2S- donating compound, NOSH-sulindac, has also been seen to inhibit the growth of 12 cancer cell lines from 6 different tissues by inhibiting cell proliferation, inducing apoptosis, and causing G2/M cell cycle block (Kashfi, Chattopadhyay et al. 2015).
The anticancer effects of NOSH-aspirin have been attributed to growth inhibition due to cell cycle arrest in G0/G1, which both decreases cell proliferation and contributes to increased induction of apoptosis, as seen in pancreatic and colon cancer cells (Chattopadhyay, Kodela et al. 2012, Chattopadhyay, Kodela et al. 2020). Interestingly, NOSH-aspirin was found to be 9000 times more potent in inhibiting colon cancer growth than the sum of its parts, determined through comparison with combinations of aspirin, NO- and H2S- donating compounds, which suggested that the biological activity of NOSH-aspirin may not be entirely attributed to the synergistic effects of its individual components (Chattopadhyay, Kodela et al. 2012). Interestingly, the same group also found that the growth-inhibiting effect of NOSH-aspirin on colon cancer cells significantly differs based on its positional isomers and the presence of different electron -donating or -withdrawing groups on its benzoate moieties (Vannini, MacKessack-Leitch et al. 2015), suggesting an ability to fine-tune and enhance the anti-proliferative and pro-apoptotic effects of the individual gasotransmitters through synthesis of hybrid compounds. Other mechanisms through which NOSH-aspirin induces apoptosis include increased expression of iNOS and p53 – both involved in ROS- and reactive nitrogen species (RNS)- induced apoptosis (Banerjee, Ganguly et al. 2014), and decreased expression of NF-κB and Forkhead box (Fox)-M1 (Chattopadhyay, Kodela et al. 2020).
Whereas pro-apoptotic effects are widely described with the use of NOSH-aspirin in different cancer models, it seems that normal, non-cancerous cells are protected from these effects. In fact, whereas NOSH-aspirin significantly induced apoptosis in pancreatic cancer cells, the treatment had no inhibitory effects on the regular pancreatic epithelial cells at the same concentration (Chattopadhyay, Kodela et al. 2020). These selective cancer cell-targeting actions are promising in their ability to combat the cancer while minimizing treatment-related toxicities, which continues to be a widespread issue in many current cancer therapy regimens.
3.2. Role in tumor invasion and metastasis
The ability to invade surrounding tissue and spread, or metastasize, to distant sites is identified as a hallmark of cancer (Fouad and Aanei 2017). Cancer metastasis is an event that is associated with significantly high mortality (Dillekås, Rogers et al. 2019, Globus, Sagie et al. 2021) and is thus an important target for improving prognosis among cancer patients.
For cancerous cells to metastasize, they must first detach from the original mass, invade the extracellular matrix (ECM) and break through the basement membrane, after which they can enter into a circulatory vessel, travel to a new location, extravasate from the vessel, and establish a new niche in which to grow (Fouad and Aanei 2017). For these steps to occur and allow tumor metastasis, cancer cells take advantage of several molecular changes that are not available to normal cells.
Starting from the first step, in the case of normal epithelial cells, detachment from the ECM results in a process of programmed cell death termed anoikis (Taddei, Giannoni et al. 2012). Invasive cancerous cells can acquire changes that render them resistant to anoikis, through mechanisms such as a switch in the expression of cell adhesion molecules including integrins, increased ROS production, and other changes reviewed in (Taddei, Giannoni et al. 2012). The collection of changes that confer cancer cells with mesenchymal properties and the abilities for detachment, resistance of anoikis, motility, production of their own ECM, and ultimately invasion is termed epithelial-mesencymal transition (EMT) (Kalluri and Weinberg 2009). EMT is characterized by the downregulation of epithelial markers such as E-cadherin and upregulation of mesenchymal markers in their place, such as N-cadherin (Loh, Chai et al. 2019).
Remodeling of the ECM is another process taken advantage of by cancer cells. Cancer cells can hijack the degradation of the ECM by matrix metalloproteinase (MMP) enzymes. In the physiologic system, MMPs are important for cell proliferation, tissue remodeling, wound healing, the immune response, and angiogenesis (Yan and Boyd 2007). Cancer cells can take advantage of MMPs to invade through the ECM and ultimately metastasize. Increased levels of different MMPs have been reported in multiple different cancer types, along with an association to poorer prognosis (Hadler-Olsen, Winberg et al. 2013). Therefore, inhibition of MMPs is a possible therapeutic avenue in combatting cancer. There is evidence that NO, H2S, and CO may target some of these mechanisms of invasion and metastasis, as discussed in the following sections.
3.2.1. Nitric oxide in cancer cell invasion and metastasis
There is a wide range of evidence to suggest a role of NO in suppressing cancer invasion and metastasis (Figure 2), indicating the importance of endogenous NO production in the host defense against cancer. In fact, the lack of host iNOS, as investigated in one study using iNOS-null mice, was associated with increased experimental metastases of three iNOS-null fibrosarcoma cell lines (Wei, Richardson et al. 2003). The anticancer defense of iNOS-derived NO is further supported by reports that the upregulation of iNOS inhibited the migration and metastasis of several cancer models both in vitro and in vivo, including oral cancer (Harada, Supriatno et al. 2004), ovarian cancer (Giri, Rattan et al. 2014), as well as orthotopic xenograft models of renal cell carcinoma, pancreatic cancer, colon cancer, and prostate cancer (Le, Wei et al. 2005). In addition to exerting anti-migratory and anti-invasive effects on the primary tumor, endogenous NO production also defends secondary sites from the establishment of a new metastasis, as seen in a murine model of lymphoma, in which NO produced by the liver endothelial cells was credited with the resistance against liver metastasis in a T-cell-dependent manner (Rocha, Krüger et al. 1995).
Mechanistically, there are several ways in which NO exerts its anti-invasive and anti-metastatic effects. As previously mentioned, EMT is one of the major events in metastasis of carcinomas, and NO has demonstrated the potential to curtail this transition. In a murine model of UVB-induced photo-carcinogenesis, topical treatment of the mice with the NO donor NO-exisulind reduced the overall aggressiveness of the tumor, increased the expression of the epithelial marker E-cadherin, reduced levels of various EMT markers, including fibronectin and N-cadherin, and downregulated EMT inducers such as Snail, Slug, and Twist (Singh, Chaudhary et al. 2012). These EMT inducers have been associated with cancer chemoresistance (Haslehurst, Koti et al. 2012), reduced overall survival (Wushou, Hou et al. 2014), and recurrence (Moody, Perez et al. 2005), thus their downregulation by NO would be greatly beneficial in hindering cancer spread. A separate study exploring the mechanism of EMT inhibition by NO in prostate metastatic cell lines further elucidated that NO inhibits the transcription factor NF-κB, of which Snail is one downstream product; in this way, NO has two major effects – it downregulates the EMT-inducer Snail, and also upregulates the metastasis suppressor Raf-1 kinase inhibitor protein (RKIP), which is typically inhibited by Snail (Baritaki, Huerta-Yepez et al. 2010). A separate pathway through which NO has been suggested to suppress EMT is through the upregulation of the metastasis-suppressing protein N-Myc-downstream-regulated gene 1 (NDRG1), an effect associated with the interaction of NO with the chelatable iron pool, as seen in a model of triple negative breast cancer (Hickok, Sahni et al. 2011). The EMT-suppressing effects of NO may not just be limited to cancer models, as exogenous NO administration was seen to suppress EMT in alveolar epithelial cells exposed to the EMT-inducing cytokine TGF-β, whereas inhibition of NOS enzymes by L-NAME treatment resulted in spontaneous EMT (Vyas-Read, Shaul et al. 2007).
In addition to preventing EMT, NO can reduce tumor invasion and metastasis by directly interfering with ECM remodeling. MMP-9, a protease involved in ECM remodeling, is one of the most widely investigated MMPs in the context of cancer. NO has been found to downregulate MMP-9 by destabilizing MMP-9 mRNA in rat glomerular mesangial cells (Eberhardt, Akool et al. 2002); this effect was further demonstrated to arise by inhibiting the expression of the mRNA-stabilizing factor HuR (Akool, Kleinert et al. 2003). The downregulation of MMP-9 by NO was also found to involve the inhibition of PKCδ in a breast cancer cell line in which TPA was used to induce MMP-9 (Jespersen, Doller et al. 2009). On the other hand, iNOS upregulation in tumor-associated macrophages was associated with decreased MMP-2 and MMP-9 expression, and was credited with the synergistic anti-metastatic effects of combined IL-2/anti-CD40 immunotherapy; exogenous NO administration confirmed that NO was responsible for these actions (Weiss, Ridnour et al. 2010). These results highlight the possibility of exogenous NO administration to reduce tumor invasion by thwarting ECM remodeling and emphasize the importance of an NO-producing M1 macrophage phenotype to the host defense against cancer. Thus, considering the finding that cancers are able to induce phenotypic switching of macrophages to the anti-inflammatory M2 phenotype (Sousa, Brion et al. 2015), augmenting NO either by exogenous administration of modulating endogenous signaling may function to restore the host defense against cancer invasion.
Despite the large amount of evidence suggesting a beneficial, anti-metastatic effect of NO on cancer cells, select studies have reported that NO may also be able to increase cancer migration (Figure 2). In one study, Rhus Coriara extract, an agent that was found to reduce breast cancer cell migration and invasiveness, was also found to downregulate iNOS and NO production (El Hasasna, Saleh et al. 2016), raising the possibility that reducing NO levels is a part of the anticancer effects. Correspondingly, iNOS expression has been reported as having a strong association with lymph node metastasis and downregulation of the non-metastatic protein 23 (nm23) protein (Dueñas-Gonzalez, Isales et al. 1997); in fact, NO administration was even found to induce EMT in breast cancer cells, as seen with the reduced cell-to-cell adhesion, decreased E-cadherin, COX-2 overexpression, which has been associated with EMT induction (Neil, Johnson et al. 2008, Ogunwobi and Liu 2011), and increased vimentin (Switzer, Glynn et al. 2012). Correspondingly, in a mammary-adenocarcinoma cell line, NOS inhibitors NMMA and L-NAME significantly reduced invasiveness whereas stimulation of iNOS-derived NO production using LPS and IFN- γ treatment stimulated invasion (Orucevic, Bechberger et al. 1999). A separate study confirmed the role of iNOS, in particular, in inhibiting cancer invasion, as selective inhibition of this enzyme reduced the invasion of human colorectal adenocarcinoma cell line by almost 50% (Siegert, Rosenberg et al. 2002).
One explanation for the differing effects of NO on the invasive ability of cancers is that the gasotransmitter may differentially modulate tumor metastasis based on the identity of the tumor. This is illustrated in one study in which NO was associated with strikingly opposing effects on melanoma and ovarian sarcoma cells lines; whereas the ovarian sarcoma cells produced larger and more metastases in NOS 2-null mice than mice expressing NOS 2, melanoma cells did the opposite (Shi, Xiong et al. 2000). In the same study, NOS 2-expressing macrophages activated to produce NO had cytotoxic effects on ovarian sarcoma, but not melanoma cells, and the cytotoxic effects were reversed with the inhibition of NOS 2 (Shi, Xiong et al. 2000). Additionally, the previous described concentration-dependent effects may also be a factor.
3.2.2. Carbon monoxide in cancer cell invasion and metastasis
Both in vitro and in vivo, CO has demonstrated the ability to inhibit the migration, invasion, and metastasis of several cancers (Figure 3), including breast (Lin, Shen et al. 2008), colorectal (Lv, Su et al. 2019), prostate (Yan, Du et al. 2018), and non-small cell lung cancers (Shao, Gu et al. 2018). Mechanistically, one of the ways in which CO may inhibit cancer invasion and metastasis is through inhibition of EMT, as was demonstrated in models of pancreatic (Yan, Du et al. 2018) and tongue squamous cell carcinoma (Dai, Chen et al. 2022). Moreover, low-dose CO was found to inhibit the migration of breast, pancreatic, colon, prostate, liver, and lung cancer cell lines, and inhibit the lung metastasis of breast cancer and liver metastasis of pancreatic cancer in mice; mechanistically, the effects involved blocking the transcription of heme transporters and reducing the level of cytochrome P4501B1 (Zhang, Zhang et al. 2022), the latter of which has been implicated in cancer metastasis via induction of EMT (Kwon, Baek et al. 2016). In non-cancer models as well, CO may be important in preventing EMT, illustrated by the increase in EMT seen with HO-1 deficiency in the kidneys (Kie, Kapturczak et al. 2008); in this case, however, it is possible that other products of HO-1 played a role.
In addition to its impact on reducing EMT, CO can also reduce ECM remodeling by downregulating MMP-2, MMP-7, and MMP-9 (Megías, Busserolles et al. 2007, Lin, Shen et al. 2008, Lv, Su et al. 2019). As previously mentioned, the ECM-degrading actions of these enzymes are exploited by cancer cells to facilitate invasion through the ECM, and it follows that downregulating MMPs will reduce invasiveness and improve prognosis by allowing for localized therapies such as surgical resection of the tumor tissue prior to spread. Furthermore, in an in vivo study with prostate cancer cells, transfection with HO-1 created tumors with reduced MMP-9 positive staining (Ferrando, Gueron et al. 2011), suggesting that this effect can be seen with both exogenously administered and endogenous increases in CO. Interestingly, the ability of CO to downregulate MMPs has also been seen in non-cancerous, alveolar epithelial cells (Desmard, Amara et al. 2005), and Staphylococcus aureus (S. aureus)-stimulated aortic endothelial cells (Tsai, Yang et al. 2018). In human breast carcinoma cells, through a mechanism involving the upregulation of HO-1, suppression of ERK phosphorylation and decreased c-Jun protein expression, and inhibition of activator protein 1 (AP-1) transcription factor activation by 12-O-tetradecanoylphorbol-13-acetate (TPA, a compound which increases MMP-9 activity), CO was also able to inhibit TPA-induced invasion of breast cancer cells (Lin, Shen et al. 2008). Thus, the modulation of MMP activity may arise from effects on these upstream signaling pathways. IL-6 was also suggested to play a role in another study in which CORM-2 treatment downregulated MMP-7 expression (Megías, Busserolles et al. 2007). Further investigation into the precise mechanisms of the impact of CO on MMPs and EMT induction is warranted, as it seems a promising avenue for thwarting cancer migration and metastasis.
3.2.3. Hydrogen sulfide in cancer cell invasion and metastasis
H2S administration using a variety of H2S-releasing molecules, including SPRC, NaHS, acetyl deacylasadisulfide, the H2S-releasing isothiocyanate erucin, and HA-ADT—a compound made from the combination of hyaluronic acid and 5-(4-hydroxyphenyl)-3H-1,2-dithiol-3-thione (ADT-OH)—has demonstrated the ability to suppress cancer cell migration, and invasion in vitro in gastric cancer cells (Ma, Liu et al. 2011, Zhang, Qi et al. 2015), colon cancer cells (Wu, Wang et al. 2012), breast cancer cells (Dong, Yang et al. 2019), pancreatic adenocarcinoma (Citi, Piragine et al. 2019), metastatic melanoma cells (De Cicco, Panza et al. 2017) and in vivo reduce lung melanoma metastases (De Cicco, Panza et al. 2017).
Mechanistically, there is evidence that H2S can hinder invasion and metastasis through the inhibition of EMT (Figure 4), like the other two gasotransmitters. In A549 human alveolar epithelial cells, the inhibition of CSE using propargylglycine resulted in spontaneous EMT, with the corresponding downregulation in epithelial marker E-cadherin, upregulation in mesenchymal markers N-cadherin and vimentin, and increase in fibroblast-like morphologic features (Fang, Lin et al. 2010). Similar findings were reported in a separate study using human urothelial bladder cancer cells, in which H2S treatment reduced epithelial markers Slug, zinc finger E-box binding homeobox 1 (ZEB-1), caveolin-1, and vimentin, alongside increased expression of E-Cadherin upon exogenous H2S treatment (Panza, Bello et al. 2022). These findings suggest an inhibitory role of endogenous H2S production against EMT, both in cancer as well as non-cancer cells. Interestingly, the former study also demonstrated that the treatment of the A549 cells with TGF-β1, a known inducer of EMT, was seen to suppress CSE expression, while exogenous H2S administration to the TGF-β1-treated cells reversed the EMT-suggestive changes through a mechanism dependent on decreasing Smad2/3 phosphorylation (Fang, Lin et al. 2010). These findings were confirmed in separate studies using the same A549 cell model, in which NaHS was seen to reverse paraquat-induced (Bai, Ye et al. 2019) and NiCl2-induced migration and EMT through suppression of TGF-β1/Smad2/Smad3 signaling (Ye, Yu et al. 2020); the latter study also reported that there was no role of the MAPK signaling pathway in these effects (Ye, Yu et al. 2020). In contrast, in a study with breast cancer cells, the anti-EMT effects of H2S—evidenced by decreased Snail expression—were seen alongside decreased phosphorylation of p38 MAPK (Lv, Li et al. 2014), which has been reported to at least partially account for TGF-β1-induced EMT in alveolar epithelial cells (Chen, Zhou et al. 2013), suggesting a p38 as a potential target of H2S in inhibiting EMT. In addition to these pathways, in H2S-releasing agent diallyl disulfide (DADS) was found to inhibit the β-catenin signaling pathway in breast cancer cells, which was linked to the EMT-suppressing effects of the gasotransmitter (Huang, Yang et al. 2015). The role of both ERK and β-catenin signaling in H2S-mediated EMT inhibition has been confirmed in a non-cancer model using renal tubular epithelial cells (Guo, Peng et al. 2016).
Furthermore, H2S has been found to hinder ECM remodeling through downregulation of MMPs (Figure 4), a crucial step in cancer invasion. The downregulation of MMP-9 by exogenous H2S administration using H2S releasing molecules DADS and DATS has been noted in several models of breast cancer in vitro and in vivo (Huang, Yang et al. 2015, Xiong, Liu et al. 2018), and in one study, the downregulation in MMP-9 was observed alongside MMP-2 downregulation (Liu, Zhu et al. 2015). Interestingly, in a separate study with breast cancer cells, NaHS administration led to the downregulation of MMP-2 expression but had no effect on MMP-9 (Ma, Yan et al. 2018). The downregulation of MMPs by H2S has been attributed to several mechanisms, including suppression of the β-catenin pathway (Huang, Yang et al. 2015), suppression of NF-κB (Liu, Zhu et al. 2015), upregulation of tristetraprolin (Xiong, Liu et al. 2018)—an RNA-binding protein that has been found to induce MMP-2 and MMP-9 mRNA degradation (Van Tubergen, Banerjee et al. 2013)—and the suppression of EGFR and the MAPK/ERK pathway (Liu, Zhu et al. 2015, Wu, Li et al. 2017). Importantly, like with proliferation, there may be a dual effect of H2S on cancer cell migration, with migration being promoted at low concentrations and being significantly inhibited at higher concentrations (Wu, Li et al. 2017). These dual effects coincided with the increase in EGFR and ERK phosphorylation at lower concentrations and reduction in their phosphorylation at higher concentrations (Wu, Li et al. 2017).
Alternatively, H2S may also play a role in increasing tumor migration, invasion, and metastasis. (Figure 4) Both, endogenous CBS-derived H2S and exogenous H2S through NaHS have been found to promote colon cancer cell migration in vitro, whereas inhibition of the enzyme suppressed these effects (Szabo, Coletta et al. 2013). A similar finding was seen with non-small cell lung adenocarcinoma cells and patient specimens, which were found to have upregulated CBS, CSE, and MPST in comparison to non-cancerous tissue whereas inhibition of these enzymes reduced metastatic potential in vitro (Wang, Yan et al. 2020). These findings suggest upregulation of endogenous H2S production as a mechanism used by cancer cells to promote their invasiveness, rendering it as a potential target for combatting cancer. In fact, the CBS-suppressing ribosomal protein L3 was identified to potentiate the efficacy of 5-fluorouracil therapy in lung cancer by inhibiting cell invasion and migration (Russo, Saide et al. 2016). The invasion-promoting role of H2S may, in contrast to the previously discussed studies, be the result of EMT induction. Supporting this possibility, NaHS administration in non-small cell lung adenocarcinoma cells reduced E-cadherin and increased vimentin, indicating induction of EMT, and increased MMP-2 and MMP-9 expression, which allows for enhanced cell invasion (Wang, Yan et al. 2020). Further investigation highlighted a role of hypoxia-inducible factor (HIF)-1α in these effects, as knockdown of HIF-1α reversed the H2S-induced EMT phenotype (Wang, Yan et al. 2020). A second study reported that the H2S-induced increase in the migration of hepatocellular carcinoma cells involves the activation of NF-κB, as inhibition of this transcription factor reversed MMP-2 upregulation seen with NaHS exposure (Zhen, Pan et al. 2015). The role of the NF-κB activation by endogenous H2S in cancer metastasis was also seen in pancreatic cancer cells; this activation led to increased IL-1β signaling and enhanced cancer invasiveness (Wang, Huang et al. 2019). Interestingly, as described above, H2S has also been found to inhibit invasion by suppressing NF-κB (Liu, Zhu et al. 2015), entirely contrary to the results seen in (Wang, Huang et al. 2019). Combined with the finding of the dual, concentration-dependent effects of NaHS in cancer invasion (Wu, Li et al. 2017), there is a possibility that these seemingly contradictory effects may be two sides of the same coin, and therapeutic use of these agents is possible with careful tailoring of delivery concentrations and release kinetics.
3.3. Impact on angiogenesis
Angiogenesis involves the sprouting of new blood vessels from the existing vascular network and is considered a hallmark of cancer (Hanahan and Weinberg 2011). Cancer cells, like normal cells, have requirements for oxygen, nutrients, and a mode to eliminate wastes, which a rich vascular supply provides. However, unlike normal cells in which angiogenesis is a tightly controlled and self-limiting process, in cancer there is a switch to an uncontrolled pro-angiogenic state which not only fulfills the nutritional and oxygen requirements of the cancer, but also provides a route of metastasis to other organs (Baeriswyl and Christofori 2009). Interestingly, conditioned medium from cancer cells has been seen to induce endothelial cell growth (Lian, Xia et al. 2016), indicating that cancer cell secretions are responsible for creating a pro-angiogenic environment.
One of the main triggers for angiogenesis is hypoxia, which is a characteristic of cancer cells. Endothelial cells can sense hypoxia, which leads to the activation of the HIF family proteins that are responsible for various cell survival and angiogenic pathways (Fraisl, Mazzone et al. 2009). Angiogenic activators include, among others, vascular endothelial growth factor (VEGF), angiogenin, TGF-β, TNF-α, platelet-derived endothelial growth factor, and epidermal growth factor (Nishida, Yano et al. 2006). Of these, VEGF has received much interest and is a target of many anti-angiogenic cancer therapies (Rajabi and Mousa 2017). Interestingly, each of the three gasotransmitters has demonstrated either or both pro- and anti- angiogenic properties that must be considered in the context of cancer.
3.3.1. Nitric oxide and cancer angiogenesis
NO has a dual anti-angiogenic and pro-angiogenic role as demonstrated in non-cancer as well as cancer models (Figure 2). In the basal state, eNOS-derived NO is crucial for the maintenance of the endothelium, and low levels of NO from this enzyme can have a pro-angiogenic effect. In fact, the pro-angiogenic effect of prostaglandin E2 (PGE2) in human umbilical vein endothelial cells (HUVECs) was attributed to increased eNOS activity via the PKA/PI3-K/Akt pathway (Namkoong, Lee et al. 2005). These pro-angiogenic actions of NO align with the findings of eNOS recruitment under hypoxic conditions PI3/Akt activation and Hsp90-eNOS binding (Chen and Meyrick 2004), as well as eNOS induction by the pro-angiogenic signal protein VEGF through hsp90-dependent Akt-mediated activation of the enzyme (Brouet, Sonveaux et al. 2001). On the other hand, both sodium nitroprusside (SNP) and L-arginine treatment has demonstrated the ability to curtail angiogenesis in an in vivo model of the chick embryo chorioallantoic membrane, whereas NOS inhibition had the opposite effect (Pipili-Synetos, Sakkoula et al. 1993).
These opposing effects seem to be concentration and flux dependent. The biphasic effects of NO have been illustrated in three different endothelial cell lines, where cGMP-dependent stimulatory effects of NO on cell proliferation, adhesion, and chemotaxis were observed at lower levels of DETA-NO treatment, whereas inhibitory effects were observed at higher levels (Isenberg, Ridnour et al. 2005). There is evidence to suggest that the dual pro- and anti-angiogenic activity of NO is associated with its biphasic effect on the activation of PKC, ERK, and the downstream transcription factor AP-1, which has known pro-angiogenic effects mediated through VEGF (Stanley, Harvey et al. 2005, Jia, Ye et al. 2016) and MMPs (Singh, Quyen et al. 2010, Jin, Park et al. 2011); corresponding to its effects on angiogenesis, NO activated these proteins at low concentrations and inhibited them at higher concentrations (Jones, Tsugawa et al. 2004). In addition to these pathways, the concentration-dependent regulation of angiogenesis by NO may also involve its effects on the expression of the anti-angiogenic protein thrombospondin-1 (TSP-1). The regulation of this protein by NO has been seen to be triphasic, with TSP-1 expression decreasing at 0.1 μM DETA-NO, rebounding at 100 μM, and decreasing at 1,000 μM (Ridnour, Isenberg et al. 2005). These effects were associated with increased phosphorylated ERK (p-ERK), the inhibition of which abolished the pro-angiogenic activity of DETA-NO (Ridnour, Isenberg et al. 2005). Interestingly, exogenous TSP-1 has been found to reduce NO-induced ERK phosphorylation, suggesting a feedback regulation cycle between these two signaling molecules (Isenberg, Ridnour et al. 2005, Ridnour, Isenberg et al. 2005).
Many of the studies conducted with cancer models have demonstrated pro-angiogenic effects of this gasotransmitter, which is a potential limitation in its use in cancer. Exogenous administration of NO has been found to increase VEGF expression in glioblastoma and hepatocellular carcinoma cells (Chin, Kurashima et al. 1997), and furthermore, iNOS expression in lung cells was linked to increased vascularization and VEGF expression compared to iNOS-deficient lines (Ambs, Merriam et al. 1998). These findings suggest that both endogenous and exogenous NO sources exert pro-angiogenic effects. Similarly, iNOS upregulation, which was associated with increased VEGF and tumor vascularity, has been observed in cancerous tissue compared to normal mucosa, and in metastatic tumors compared to non-metastatic ones (Gallo, Fini-Storchi et al. 1998, Cianchi, Cortesini et al. 2003). Thus, iNOS upregulation may be a characteristic of cancer aggressiveness as a result of angiogenesis, and inhibition of endogenous NOS production may counter cancer angiogenesis and therefore metastasis as well. In fact, studies using non-specific NOS inhibitors, iNOS and eNOS short hairpin RNA (shRNA), and an iNOS-selective inhibitor have reported downregulation of pro-angiogenic genes and proteins, reduced tumor neovascularization, and correspondingly, increased necrotic tumor tissue (Jadeski and Lala 1999, Gao, Zhou et al. 2019). Interestingly, however, while iNOS gene transfer was seen to upregulate pro-angiogenic molecules including VEGF and IL-8 in fibrosarcoma and colorectal adenocarcinoma models, the tumor cells were still unable to form metastases, and this dramatic loss of malignancy was attributed to the apoptotic effects of NO (Le, Wei et al. 2005). This suggests that the pro-angiogenic effect of NO might not sufficiently counteract its other anticancer effects.
Alternatively, by increasing tumor perfusion NO may enhance the ability of other cytotoxic therapies to reach the tumor and exert their damage; in one study, the enhanced blood perfusion of tumor tissue allowed increased tumoral accumulation of NO- and O2- delivering ultrasound-responsive nanoparticles, which were used to enhance the anti-cancer effects of sonodynamic therapy and increase the immune response to the tumor (Ji, Si et al. 2021). Still, the balance between the beneficial effects of increasing vs depleting tumor blood supply needs to be investigated further with respect to optimal concentration, to ensure the therapeutic effects of this gasotransmitter in cancer.
3.3.2. Carbon monoxide and cancer angiogenesis
Like NO, CO seems to have a dual role in stimulating or inhibiting angiogenesis (Figure 3), which may depend on the physiological or pathological context in which it is acting. In astrocytes, CORM-2 administration was found to induce angiogenesis by increasing pro-angiogenic VEGF levels through upregulation of the HIF-α protein, which was done using two pathways: promoting HIF-α translation and stabilizing the protein against degradation (Choi, Kim et al. 2010). Endothelial cells exposed to another CORM, CORM-401, also upregulated VEGF levels in vitro, as well as levels of IL-8 (Fayad-Kobeissi, Ratovonantenaina et al. 2016), a chemokine known to induce angiogenesis through endothelial cell proliferation (Li, Dubey et al. 2003).
On the other hand, CO has in numerous studies displayed anti-angiogenic effects against cancer cells. Whereas CO increased IL-8 levels in endothelial cells in the study discussed previously (Li, Dubey et al. 2003), in human gastric cancer cells, CORM-2 treatment decreased IL-1β-induced expression of IL-8, and inhibiting IL-8 using a neutralizing antibody reversed the stimulatory effect of the cancer conditioned medium on endothelial cell growth (Lian, Xia et al. 2016). This effect was the result of modulation of several pathways, including the inhibition of ROS-mediated NF-κB activation, inhibition of ERK1/2 activation, and suppression of MAPK-mediated AP-1 activation (Lian, Xia et al. 2016). These opposing effects in normal compared to cancerous tissue may suggest CO as a useful molecule in depriving neoplastic tissue of blood supply without negatively affecting the vascularization of normal tissue. This selectivity may increase the utility of CO compared with many current anti-angiogenic cancer therapies whose side effects, including impaired wound healing, endothelial dysfunction, hypertension, and increased clotting risk, result from their lack of such selectivity (Rajabi and Mousa 2017). Other studies with CO in both in vitro and in vivo cancer models have shown its ability to reduce de novo angiogenesis by inhibiting phosphorylation of p-Akt—of which angiogenesis is one downstream effect—reducing VEGF levels and inhibiting VEGFR2 phosphorylation, and decreasing the migration and tube-forming ability of endothelial cells; these effects were seen with pancreatic, colorectal, triple-negative breast cancers, and, interestingly, in HUVECs (Vítek, Gbelcová et al. 2014, Ahmad, Hewett et al. 2015, Kourti, Westwell et al. 2019, Lv, Su et al. 2019, Kourti, Cai et al. 2021). The anti-angiogenic effects of CO on non-cancerous HUVECs suggests that the selectivity of CO for cancer may not be absolute.
Yet CO may not have anti-angiogenic effects against all cancers. Increased HO-1 expression has been seen to induce VEGF secretion and angiogenesis and correlate to increased vascular density in models of prostate cancer (Birrane, Li et al. 2013), melanoma (Was, Cichon et al. 2006), glioma (Nishie, Ono et al. 1999), and non-small cell lung cancer (Skrzypek, Tertil et al. 2013), whereas it decreased neovascularization and VEGFR2 expression in PC3 cancer cells (Ferrando, Gueron et al. 2011). It is worth noting here that although HO-1 is responsible for producing CO, it also yields other products such as biliverdin and ferrous iron; therefore, it is possible that CO is not the mediator behind these effects. Still, a dose-dependent increase in vascular permeability and tumor blood supply was observed with treatment using both CORM-2 and HO-1 induction in S180 solid tumors (Fang, Qin et al. 2012), thus, the possible role of CO in increasing tumor blood supply cannot be ignored, albeit this effect was through increasing vascular permeability rather than angiogenesis. Further studies on the precise role of CO on angiogenesis in the context of different cancers would be worthwhile.
3.3.3. Hydrogen sulfide and cancer angiogenesis
Based on the available evidence, H2S seems mainly to be a pro-angiogenic signaling molecule (Figure 4), which may contraindicate its use in cancer. Both in vitro and in vivo, exogenous administration of H2S and its precursors, L-cysteine and 3-MP, has been found to increase endothelial cell growth, proliferation, migration, microvessel formation, and accelerate wound healing through increased neovascularization (Cai, Wang et al. 2007, Papapetropoulos, Pyriochou et al. 2009, Coletta, Papapetropoulos et al. 2012, Coletta, Módis et al. 2015). Likewise, the endogenous production of H2S has been demonstrated to have the same pro-angiogenic effects, whereas inhibition of H2S-producing enzymes suppresses angiogenesis (Papapetropoulos, Pyriochou et al. 2009). All three H2S-producing enzymes have been identified as having a role in this regard. The pro-angiogenic role of CSE is demonstrated by the marked reduction of the pro-angiogenic effects of VEGF upon CSE silencing, with a corresponding enhancement in the outgrowth of branching microvessels with CSE upregulation; this suggests endothelial H2S synthesis to be crucial for VEGF signaling (Coletta, Papapetropoulos et al. 2012). VEGF stimulation of endothelial cells also increases H2S production (Papapetropoulos, Pyriochou et al. 2009), suggesting a self-reinforcing cycle of events to ensure angiogenesis in conditions where it is needed. Correspondingly, CSE expression is upregulated in hypoxic conditions, further supporting its role in angiogenesis and protection against insufficient oxygen delivery (Wang, Guo et al. 2014). Because hypoxia is a major feature of solid tumors and is associated with tumor progression and increased treatment resistance (Brahimi-Horn, Chiche et al. 2007), it is possible that H2S production by tumor tissue promotes tumor survival through its effects on vascularity. CBS is also involved in angiogenesis, and silencing of this enzyme was seen to significantly decrease HUVEC proliferation, diminish the pro-angiogenic effects of VEGF, and reduce transcription of the VEGF receptor VEGF receptor 2 (VEGFR2) and neuropilin 1—both essential for endothelial function—whereas exogenous H2S administration using NaHS and GYY4137 reversed these effects by stabilizing the transcription factor specificity protein 1 through protein S-sulfhydration (Saha, Chakraborty et al. 2016). Lastly, the 3-MP/3-MST/H2S pathway has also been seen to promote angiogenesis both in vitro and in vivo (Coletta, Módis et al. 2015).
Mechanistically, the pro-angiogenic effects of H2S involve several mechanisms. There is evidence to indicate a role of the PI3-K/Akt pathway—which is downstream of VEGFR2 activation—in H2S-induced angiogenesis, and in one study blocking PI3-K was seen to suppress the pro-angiogenic effects of H2S in vitro (Cai, Wang et al. 2007). Additionally, exogenous administration of H2S and NaHS has been found to induce activating phosphorylation of the downstream signaling molecule Akt (Cai, Wang et al. 2007, Wang, Cai et al. 2009, Coletta, Papapetropoulos et al. 2012). Interestingly, one study that reported involvement of the PI3-K/Akt pathway also ruled out any effects of H2S on the phosphorylation of p38 and ERK (Cai, Wang et al. 2007), while a separate study reported the opposite: that H2S increased the phosphorylation of both of these molecules to induce angiogenesis, and that inhibition of the PI3-K/Akt pathway did not affect the migration of H2S-exposed endothelial cells (Papapetropoulos, Pyriochou et al. 2009). The latter demonstrated that inhibition of p38 abolished H2S-induced motility of endothelial cells, further supporting its role in angiogenesis, and highlighted the role of KATP channels in p38 phosphorylation, where blocking these channels inhibited p38 phosphorylation while enhancing their opening increased p38 phosphorylation (Papapetropoulos, Pyriochou et al. 2009). Whether this pathway or the PI3-K/Akt pathway, or both, are involved in H2S-induced angiogenesis is not clear.
As previously mentioned, the signaling and actions of VEGF are intertwined with H2S production and release, illustrated by the finding that CSE silencing reduces the pro-angiogenic effect of VEGF (Coletta, Papapetropoulos et al. 2012). The increased vessel growth and capillary density upon NaHS treatment in a rat model of hind limb ischemia was found to be associated with increased VEGF expression and VEGFR2 phosphorylation (Wang, Cai et al. 2009), and indeed, H2S has been found to upregulate HIF-1α and VEGF mRNA and protein levels and increase HIF-1α binding activity under hypoxic conditions (Liu, Pan et al. 2010). HIF-α, the upstream regulator of VEGF expression, is necessary for the pro-angiogenic effects of H2S, as HIF-α silencing suppressed these effects in vitro (Zhou, Li et al. 2015). Similarly in ex vivo renal artery culture, triple gene therapy with CBS, CSE, and 3-MST, as well as H2S treatment, led to increased VEGF and reduced antiangiogenic factor endostatin (Sen, Sathnur et al. 2012). On the other hand, no change in VEGF levels were seen in NaHS-treated endothelial cells in other studies (Cai, Wang et al. 2007, Tao, Liu et al. 2013), raising the question of whether the upregulation of VEGF and HIF-1α is a direct effect of H2S or a result of other influences such as hypoxia. There is also evidence that H2S can directly activate VEGFR2 through cleavage of an inhibitory disulfide bond of the receptor, allowing VEGFR2 phosphorylation, accompanied by downstream phosphorylation of PI3-K and Akt; interestingly, Akt was not a direct target of H2S, rather its phosphorylation was a downstream effect of VEGFR2 activation (Tao, Liu et al. 2013). This suggests the possibility that the previous studies indicating Akt phosphorylation by H2S as a mechanism for angiogenesis were describing a downstream effect of H2S on VEGFR2 (Tao, Liu et al. 2013).
H2S-induced angiogenesis also seems to involve an NO-dependent mechanism. NOS inhibition using L-NAME has been seen to abolish the proliferation of endothelial cells induced by H2S exposure in vitro, and in vivo, eNOS-deficient mice completely lack the microvessel outgrowthpromoting effect of H2S (Coletta, Papapetropoulos et al. 2012). The opposite was also true: H2S was required for the pro-angiogenic effects of NO donor therapy (Coletta, Papapetropoulos et al. 2012). Thus, reducing the bioavailability of either one of these gasotransmitters while administering the other may allow therapeutic use of one of these gasotransmitters while reducing its potential to induce a harmful pro-angiogenic effect. Further investigation revealed that the angiogenesis-stimulating effects of these gasotransmitters converges on the cGMP/PKG pathway, as inhibiting sGC abolished the pro-angiogenic effects of both (Coletta, Papapetropoulos et al. 2012). This is plausible, given that NO is a known activator of sGC which leads to cGMP production (Bryan, Bian et al. 2009), while H2S is a known inhibitor of PDE5A (Bucci, Papapetropoulos et al. 2010), leading to reduced breakdown and increased accumulation of cGMP. Furthermore, H2S also exerts redox regulation on sGC and thereby enhances its response to NO, allowing more efficient cGMP production (Zhou, Martin et al. 2016). Additionally, H2S stimulates phosphorylation of Akt, which led to activating phosphorylation of eNOS, demonstrating further cooperation between the two gasotransmitter (Coletta, Papapetropoulos et al. 2012). Supporting the role of NO in H2S-induced angiogenesis, H2S has been found to promote ischemic vascular remodeling with increased HIF-1α and VEGF activity in an NO‐dependent manner (Bir, Kolluru et al.), as well as NO-dependent monocyte recruitment and arteriogenesis (Kolluru, Bir et al. 2015).
The combination of these findings gives a strong indication that H2S facilitates tumor progression and spread through the formation of new vascular networks. The overexpression of H2S-producing enzymes and increased H2S levels have been reported in various tumors, including colon cancer (Szabo, Coletta et al. 2013), non-small cell lung adenocarcinoma (Wang, Yan et al. 2020), hepatocellular carcinoma (Zhen, Pan et al. 2015), and Von Hippel Lindau clear cell renal cell carcinoma (Sonke, Verrydt et al. 2015). Aligning with the previous discussion, in these cancer models H2S has been seen to increase tumor neovascularization alongside HIF-1α activation and VEGF upregulation, whereas inhibition of H2S production reverses these pro-angiogenic effects (Szabo, Coletta et al. 2013, Sonke, Verrydt et al. 2015, Zhen, Pan et al. 2015, Wang, Yan et al. 2020). Interestingly, one study demonstrated a difference in the pro-angiogenic effects of H2S on breast cancer endothelial cells vs non-cancerous endothelial cells; whereas NaHS treatment induced cytosolic Ca2+ release in both cell types, at low concentrations (1–10 μM) it promoted VEGF-induced migration and invasion of the cancer endothelial cells while failing to do so in the non-cancerous endothelial cells (Pupo, Fiorio Pla et al. 2011). This indicates that H2S may have a harmful level of selectivity, preferentially promoting cancer angiogenesis and invasion. Overall, the evidence seems to indicate that the pro-angiogenic effects are a drawback to its use in the context of cancer and suggest the need to combine any potential H2S therapy with known antiangiogenic agents or other cytotoxic agents that can take advantage of the increased tumor blood supply induced by H2S exposure.
3.4. Impact on the immune system
Evasion of the host immune response is a hallmark of cancer (Mortezaee 2020), which proves as a major obstacle to the development of effective anticancer therapeutic strategies. There is an entire branch of cancer therapies—termed immunotherapy—that focuses on restoring the host immune response to cancer, an approach that has already demonstrated great promise (Farkona, Diamandis et al. 2016). However, the dysfunction and exclusion of T cells is a major limitation to the efficacy of such therapy (Jiang, Gu et al. 2018), indicating the need for additional strategies that further boost antitumor immune function.
The cytotoxic CD8+ T cell population is one of the main cell types involved in the cytotoxic anticancer immune response, and intratumoral infiltration of these cells has been associated with improved prognosis and increased survival in several cancers (Naito, Saito et al. 1998, Hiraoka, Miyamoto et al. 2006, Ali, Provenzano et al. 2014, Shimizu, Hiratsuka et al. 2019). Unfortunately, cancer cells are able to acquire several adaptations that suppress both CD8+ T cell infiltration into the cell and the cytotoxic effects of this cell, including through recruitment of immunosuppressive myeloid-derived suppressor cells (MDSCs) (Lesokhin, Hohl et al. 2012), increased levels of which has been associated with poorer survival (Jordan, Amaria et al. 2013), induction of immunosuppressive Treg cells (Chen, Pittet et al. 2005, Chen, Pittet et al. 2005), and anti-inflammatory M2 polarization of tumor-associated macrophages (Lepique, Daghastanli et al. 2009, Pu, Xu et al. 2021). Targeting any of these immunosuppressive steps may significantly augment the antitumor immune response. In addition to their other actions, each of the gasotransmitters has been found to play a role in immunomodulation to some degree, with some promising effects and some potential drawbacks to their use.
3.4.1. Nitric oxide immunomodulation in cancer
A large proportion of the studies investigating the immunomodulating actions of NO have described the gasotransmitter to have an immunosuppressive role (Figure 2), which may contraindicate its use in the context of cancer. In fact, NO is involved in the suppression of T cell proliferation by mesenchymal stem cells (Sato, Ozaki et al. 2006), and there is evidence to suggest that cancer cells may take advantage of the immunosuppressive activity of NO and use it to escape immune surveillance. In one study, the coculture of melanoma cells with peripheral blood mononuclear cells (PBMCs) from healthy volunteers demonstrated the former’s ability to suppress the IFN response in PBMCs, an effect that was negatively correlated with NOS1 expression (Liu, Tomei et al. 2014); interestingly, increased NOS1 expression was also associated with reduced responsiveness of melanoma metastases to adoptive T cell transfer immunotherapy (Liu, Tomei et al. 2014). Not only NOS1, but the other NOS enzymes also play a role in this regard. Tumorrecruited myeloid-derived suppressor cells (MDSCs)—which have a purpose of inhibiting cytotoxic T cells and thus allowing tumor growth and metastasis—have been found to be dependent upon tumor-expressed iNOS to induce T cell suppression, whereas iNOS inhibition reduces MDSC accumulation and immunosuppressive activity, and increases intratumoral CD8+ T cell infiltration and cytotoxic activity both in vitro and in vivo (Jayaraman, Parikh et al. 2012, Dufait, Schwarze et al. 2015). Therapeutic targeting of MDSC-mediated immunosuppression is of great interest in combatting cancer, as there is evidence that MDSC accumulation is a limitation of adoptive immunotherapy with cytotoxic T cells, as seen in a murine model of melanoma, where the MDSCs then resulted in NO-mediated immunosuppression that was reversed using the NO scavenger carboxy-PTIO (Hirano, Hosoi et al. 2015). Further study has revealed two different subpopulations of MDSCs—granulocytic and monocytic MDSCs—both of which inhibit T cell function through NO-dependent pathways; however, while granulocytic MDSCs exert their effects through eNOS-derived NO, monocytic MDSCs do so through iNOS-derived NO (Raber, Thevenot et al. 2014). Thus, all three NOS isoforms seem to be implicated in cancer-induced T cell suppression.
There are several pathways through which NOS activity can suppress T cell function. One of these involves the nitration of tyrosine residues necessary for T lymphocyte activity. A study with organ cultures of human prostate adenocarcinoma revealed increased nitrotyrosine in the immunosuppressed tumor-infiltrating lymphocytes, whereas inhibition of iNOS and arginase, another enzyme involved in L-arginine metabolism, was seen to reduce tyrosine nitration and reinstate T cell antitumor activity (Bronte, Kasic et al. 2005). It is possible that NO itself may not be to blame for this, as increased arginase activity can lead to L-arginine depletion leading to increased NOS production of superoxide anions; these then react with NO to form peroxynitrite, a damaging radical known to cause tyrosine nitration, inhibiting T cell activation and inducing T cell apoptosis in vitro (Brito, Naviliat et al. 1999). There is support for these findings from an in vivo murine model, in which the hyperproduction of peroxynitrite and ROS by MDSCs induced nitration of the T cell receptor (TCR)/CD8 complex, thwarting T cell binding to peptide-specific major histocompatibility complex (MHC) molecules, leading to T cell tolerance (Nagaraj, Gupta et al. 2007); interestingly, the ability of these cells to induce T cell tolerance remained even in iNOS-deficient mice, suggesting the upregulation of ROS rather than NO to be the culprit behind the immunosuppressive effects, further supporting that it is peroxynitrite, and not specifically NO, that is responsible for this effect (Nagaraj, Gupta et al. 2007). Other studies have highlighted additional mechanisms for T cell suppression, including iNOS-derived NO-mediated inhibition by MDSCs of IL-2R signaling pathways required for T cell activation (Mazzoni, Bronte et al. 2002), peroxynitrite-mediated nitration of the CCL2 chemokine, which hinders T cell infiltration and sequesters these cells in the stroma surrounding the tumor (Molon, Ugel et al. 2011), and inhibition of vascular E-selectin expression, an adhesion molecule whose expression was positively correlated with the intratumoral infiltration of CLA+ memory T cells (Gehad, Lichtman et al. 2012). The immunosuppressive effects on T cells in these studies were reversed with the inhibition of NOS enzymes (Mazzoni, Bronte et al. 2002) or suppression of peroxynitrite production (Molon, Ugel et al. 2011).
While anti-tumor T cell activity is inhibited by these agents, immune-suppressing T cell phenotypes may be differentially impacted. In one study, iNOS-expressing mice were found to have increased an pro-tumor, IL-17-secreting phenotype of γδ T cells compared to their iNOS-deficient counterparts, leading to the recruitment of MDSCs and impaired cytotoxicity of γδ T cells towards melanoma cells (Douguet, Bod et al. 2016). On the other hand, in a study with melanoma tumor-bearing mice, iNOS inhibition was associated with suppression of intratumoral MDSCs, pointing again to an immunosuppressive role of iNOS (Jayaraman, Alfarano et al. 2014). iNOS deficiency was also associated with an upregulation of immunosuppressive Fox3p+ Treg cells, suggesting the enzyme to protect against Treg accumulation while simultaneously increasing immunosuppression-inducing MDSCs (Jayaraman, Alfarano et al. 2014). In this same study, host iNOS inhibition was unsuccessful in impacting tumor growth by itself, but in combination with Treg depletion using low-dose cyclophosphamide, CD8+ T cell infiltration was boosted, and tumor growth was significantly arrested (Jayaraman, Alfarano et al. 2014).
Immune cell populations other than T cells are also seen to be impacted. MSDCs from patients with cancers of different origins inhibited natural killer (NK) cell antibody-dependent cellular cytotoxicity, an effect involving NO production by MDSCs and subsequent NK cell protein nitration (Stiff, Trikha et al. 2018). The results were confirmed in vivo, as inhibition of iNOS in mice bearing breast cancer tumors enhanced NK cell antibody-dependent cytotoxicity, and inhibiting NO production by MDSCs enhanced the efficacy of anti-cancer monoclonal antibody (mAb) therapy with trastuzumab (Stiff, Trikha et al. 2018). Antigen presentation by dendritic cells to CD4+ is another immune function inhibited by MDSCs via NO production, and correspondingly, NOS inhibition has been found to restore T cell proliferation in this context (Markowitz, Wang et al. 2017). By way of reversing NO/NOS/peroxynitrite-mediated immunosuppression, the anticancer activity of several potential cancer therapies, including the alpha-garactosylceramide (Ito,Ando et al. 2015) and toll-like receptor agonists (Ito,Ando et al. 2015), has been reported to be increased by the inhibition of NOS enzymes in murine cancer models. A phase 1/2 trial of NOS inhibitor L-NMMA with the anti-microtubule agent taxane also demonstrated promising results in treating patients with chemorefractory breast cancer, warranting further investigation in clinical trials (Chung, Anand et al.). Overall, NOS inhibition has earned much interest in the context of cancer therapy.
While these studies point to a detrimental role of NO in the setting of cancer, there is evidence to suggest that exogenous NO administration may be beneficial in this context, though counterintuitive. Illustrating this point, the NO donor GSNO, when administered to ovarian tumor-bearing mice, slowed tumor growth in association with decreased levels of MDSCs, reversed the suppression of T cell proliferation, and increased pro-inflammatory, IFN-γ-producing CD4+ and CD8+ T cell populations (Rattan, Sakr et al. 2018). Within the CD4+ T cell population, a higher ratio of Th1/Th2 cells is favorable for antitumor immunity. Importantly, at low concentrations, NO has been found to selectively induce Th1 but not Th2 differentiation (Niedbala, Wei et al. 1999) by cGMP-dependent induction of IL-12 receptor used by the former cell population without any effect on the IL-4 receptor used by the latter (Niedbala, Wei et al. 2002). Exogenous NO may also exert its immunoprotective effects by downregulating NOS activity. In one study, the furoxan-based NO donor AT38 was seen to block the production of peroxynitrite and downregulate intratumoral iNOS in several murine cancer models, leading to a significant increase in the intratumoral T cell infiltration (Molon, Ugel et al. 2011). Mechanistically, these effects were linked to reduced CCL-2 nitration, which enhanced tumor eradication by adoptive cell therapy and caused sequestration of the immunosuppressive Foxp3+/CD4+ Treg cells at the periphery of the tumor, in contrast to the effect observed on cytotoxic T cells (Molon, Ugel et al. 2011). From these findings, it seems that iNOS overactivity can interfere with antitumor T cell function through the production of reactive nitrogen species and subsequent protein modifications, whereas NO administration can be beneficial in curtailing this enzyme. Thus, the previously described detrimental effects of iNOS in cancer may, rather than a contraindication to NO administration, be mitigated by exogenous administration of NO. The beneficial effects of exogenous NO administration on T cell activity are supported by the results of another study in which NO-releasing aspirin reversed the immunosuppression exerted by MDSCs, restoring T cell proliferation in response to alloantigens in vitro (De Santo, Serafini et al. 2005). The same study demonstrated that in tumor-bearing mice, NO-aspirin reduced the activity of the NOS and arginase enzymes, inhibited intratumoral peroxynitrite formation, and enhanced the efficacy of DNA cancer vaccination (De Santo, Serafini et al. 2005). The increase in T cell proliferation may arise from the impact of NO on the required step of antigen presentation; in a previously discussed study in which NO was implicated in suppression of dendritic cell antigen presentation, pre-treatment with the NO-releasing aspirin derivative NCX-4016 was used to inhibit NO production by MDSCs and found to increase T cell proliferation in the context of dendritic cell antigen presentation (Markowitz, Wang et al. 2017). In each of these studies, NO donors were used to block the activity of iNOS to achieve their beneficial immune-boosting effects. Administering exogenous NO to inhibit NOS activity, and therefore peroxynitrite production, is a strategy that likely takes advantage of the extensive feedback regulation of NO on NOS activity; NO has been seen to inhibit the activity and expression eNOS and iNOS through inhibitory S-nitrosylation of the enzyme and inhibition of NF-κB (Katsuyama, Shichiri et al. 1998, Grumbach, Chen et al. 2005, Altaany, Ju et al. 2014). Thus, the extensive preceding discussion of the evidence that iNOS plays a role in cancer-mediated immunosuppression does not preclude the use of exogenous NO—in fact, NO itself can be used to offset the deleterious immune effects of excessive iNOS activity.
NO may also boost the anti-tumor activity of immune cells besides T cells. In one study, inflammatory M1 macrophages were seen to prevent repolarization into M2 macrophages by inhibiting mitochondrial oxidative phosphorylation, likely by NO production, as inhibition of NO production reversed these effects and allowed the phenotypic switch to anti-inflammatory M2 macrophages (Van den Bossche, Baardman et al. 2016). In this way, NO seems to be key to maintaining a pro-inflammatory macrophage phenotype, which is especially important in cancer states where cancer cells induce macrophage polarization to M2 phenotype to escape immune surveillance. Indeed, NO- and O2 co-delivering ultrasound-responsive nanoparticles enhanced sonodynamic therapy and promoted polarization of macrophages from an M2 to an M1 phenotype after ultrasound radiation (Ji, Si et al. 2021). Additionally, this therapy induced mitochondrial dysfunction by peroxynitrite production, promoted dendritic cell maturation, and depleted MDSCs (Ji, Si et al. 2021).
Because these immune cells work in conjunction with other immune cells rather than in isolation, NO-mediated induction of macrophage switching may also boost the anti-tumor activity of other immune cells. In fact, in murine models of pancreatic carcinomas, the polarization of tumor-associated macrophages into an iNOS-expressing pro-inflammatory M1 phenotype by local low dose gamma-radiation was responsible for increased recruitment of CD8+ T cells into tumor tissue by causing endothelial activation and subsequent upregulation of adhesion molecules, inducing the expression of Th1 chemokines, and suppressing angiogenic, immunosuppressive, and tumor growth factors (Klug, Prakash et al. 2013). The upregulation of adhesion molecules, in particular vascular cell adhesion molecule 1 (VCAM-1), by iNOS-expressing macrophages was also seen in a murine model of pancreatic neuroendocrine tumors, promoting T cell infiltration into the tumor tissue (Sektioglu, Carretero et al. 2016); furthermore, co-transfer of CD8+ T cells with iNOS-expressing macrophages, but not iNOS-deficient macrophages, induced a markedly pro-inflammatory TME, with simultaneous downregulation of angiogenesis-associated genes (Sektioglu, Carretero et al. 2016). Interestingly, the effects of NO were demonstrated to be biphasic in vitro, where low levels of NO donor glyceryl trinitrate (10 μM and 50 μM) induced the expression of adhesion molecules in HUVECs in an NF-κB-dependent manner, whereas higher levels (250 μM and 500 μM) were suppressive (Sektioglu, Carretero et al. 2016), thus emphasizing the importance of considering dosage when using this gasotransmitter for therapeutic purposes.
3.4.2. Carbon monoxide immunomodulation in cancer
CO has demonstrated several immunomodulatory effects, with evidence of both immunosuppressive and immune boosting effects (Figure 3). In the context of LPS stimulation of macrophages in vitro and in vivo LPS challenge of mice, CO was seen to exhibit potent anti-inflammatory effects including downregulation of pro-inflammatory cytokines TNF-α, IL-1β, and MIP-1β, and upregulation of the anti-inflammatory cytokine IL-10 through a mechanism involving the MAPK pathway (Otterbein, Bach et al. 2000). A separate study identified the nuclear hormone peroxisome proliferator-activated receptor-γ (PPARγ) as mediating the anti-inflammatory effects of CO on LPS-stimulated macrophages (Bilban, Bach et al. 2006). In inflammatory conditions, such as LPS stimulation in vitro (Mu, Li et al. 2022) and acute pancreatitis in vivo (Taguchi, Nagao et al. 2018), CO release from different CO donors has also been reported to induce anti-inflammatory M2 macrophage polarization, suggesting that the gasotransmitter has an anti-inflammatory role in setting of increased inflammation. Interestingly, another in vitro study demonstrated that while CORM-3 treatment promoted the M2 phenotype and upregulated iNOS expression in unstimulated alveolar macrophages, it reduced iNOS expression in macrophages stimulated by LPS/IFN-γ (Yamamoto-Oka, Mizuguchi et al. 2018), suggesting that the effects of CO differ based on the level of inflammation present within the system tested.
In addition to inducing anti-inflammatory macrophage polarization, which is beneficial in many disease models but may protect cancer cells from cell death, CO has also demonstrated suppressive effects on T lymphocytes. CO has been found to suppress T cell proliferation and cell cycle entry by inhibiting the secretion of IL-2, possibly through inhibition of ERK (Pae, Oh et al. 2004). There is also evidence that CO may induce CD4+ T cell apoptosis through Fas/CD95-induced apoptosis, in part attributable in part to the inhibition of ERK and MAPK, as seen upon exposure of Jurkat T cells to CO (Song, Zhou et al. 2004). Furthermore, this gasotransmitter has been found to decrease B220+ CD4− CD8− T cells in a murine model of systemic lupus erythematosus (Mackern-Oberti, Obreque et al. 2015), and to be protective against intestinal inflammation by suppressing Th17 cell differentiation (Takagi, Naito et al. 2018). Thus, CO has been found to suppress the immune activity of several different T cell populations, which is a possible drawback to its use in cancer, considering the importance of these cells in anti-tumor host immunity. In fact, increased HO-1 expression has been associated with reduced sensitivity to chemotherapeutic agent pirarubicin in patient colorectal tumor tissues (Yin, Fang et al. 2014), which may be linked to the immunosuppressive effects of CO as discussed. Overall, these studies seem to suggest a harmful immunosuppressive effect of CO in the context of cancer.
There is also evidence to suggest an immunoprotective effect of CO. In one study, CORM treatment was found to reduce the immunosuppressive effects of solar-simulated ultraviolet (UV) radiation and the epidermal UVB byproduct cis-UCA through a mechanism involving guanylyl cyclase (Allanson and Reeve 2005). Further study in a murine model of solar-simulated UV radiation-induced photocarcinogenesis revealed that topical CORM-2 treatment inhibited early tumor appearance, reduced tumor multiplicity, induced the regression of established tumors, and delayed malignant progression; correspondingly, CO reduced expression of immunosuppressive IL-10 and increased immune boosting IL-12 (Allanson and Reeve 2007). These findings suggest an immunopotentiating ability of CO in the context of cancer.
There are several different effects through which CO may yield its immunoprotective effects against cancer, one of which is through the induction of immunogenic cell death. Immunogenic cell death, which results in the release of DAMPs that stimulate the immune system towards recognizing and combatting cancer cells, is one avenue that is currently being investigated for cancer immunotherapy. Interestingly, exogenous CO administration was seen to induce immunogenic cell death in both in vitro and in vivo models of 4T1 breast cancer (Xiao, Liang et al. 2021) and in a murine model of melanoma, in which CO administration suppressed lung metastasis (Zhai, Zhong et al. 2021). Based on these studies, CO-induced immunogenic cell death is successful in stimulating the anti-tumor immune response in vitro and in vivo, with increased dendritic cell maturation, increased CD4+ and CD8+ T cells, and improved ratio of these cell types to Treg cells; furthermore, CO therapy in the former study enhanced the anti-tumor effect of anti-PDL1 immune checkpoint therapy (Xiao, Liang et al. 2021). The immunoprotective role of CO in cancer has been implemented in a study developing a cancer nanovaccine using soft X-ray-triggered CO releasing lanthanide scintillator nanoparticles (ScNPs: NaLuF4:Gd,Tb@NaLuF4) combination with a PhotoCORM, which has demonstrated promise in reversing the immunosuppressive TME, activating the adaptive anti-tumor immunity, and therefore causing systemic tumor growth inhibition through ROS generation and the induction of immunogenic cell death (Li, Jiang et al. 2021). The potential enhancement of the anti-tumor immune response by CO can also increase the efficacy of other currently used cancer chemotherapies. For instance, enhanced effects of doxorubicin therapy has been seen with the use of different CO -generating or -releasing compounds (Wang, Zhang et al. 2019, Zhao, Wang et al. 2022); in one study, the therapeutic effect was increased from 29% to 82.4% in vitro (Zhao, Wang et al. 2022). Thus, combination therapy with CO and immunotherapies or other chemotherapies that ultimately require cancer clearance by the immune system is an avenue to augmenting current therapeutic approaches to cancer. In addition to boosting antitumoral T cell activity through induction of immunogenic cell death, evidence from melanoma antigen-specific T cells has demonstrated that CO boosts T cell antitumor activity in vivo by inducing moderate endoplasmic reticulum stress; mechanistically, this leads to transient activation of ERS sensor protein kinase R-like endoplasmic reticulum kinase (PERK) which induces protective autophagy and epigenetic reprogramming of T cells into a stronger anti-tumoral phenotype (Chakraborty, Parikh et al. 2022). Considering the promising findings of T cell activity enhancement, further investigation of the effects of CO on the T cell responses to cancer may be warranted.
Moreover, in direct contrast to the M2-polarizing activity of CO described previously in inflammatory models, CO has been found to induce anti-tumor macrophage activity in the cancer setting. Exogenous CO administration in a murine model of lung cancer led to inhibition of tumor growth by increasing cancer cell apoptosis; this effect was associated with increased infiltration of CD169+ macrophages into the TME (Nemeth, Csizmadia et al. 2016). This macrophage subtype has been recognized to have a critical role in anti-tumor immunity through cross-presentation of tumor antigens to CD8+ T cells (Asano, Nabeyama et al. 2011). Additionally, CO treatment was associated with skewing of the macrophages into CD86/CD197-positive M1 phenotype alongside suppression of the MMR-expressing M2 phenotype in the TME; mechanistically, the reprogramming of macrophages by CO into an anti-tumoral phenotype was found to be mediated by ROS-dependent activation of MAPK/ERK1/2-c-myc signaling and Notch 1-dependent negative feedback on HO-1 (Nemeth, Csizmadia et al. 2016). Thus, in addition to boosting anti-tumoral immunity through immunogenic cell death-mediated immune activation, CO treatment may also result in enhanced anti-tumoral effects through macrophage activity.
3.4.3. Hydrogen sulfide immunomodulation in cancer
While there has been a large amount of investigation into the immune modulation by H2S, as reviewed in (Dilek, Papapetropoulos et al. 2020), unlike with the other two gasotransmitters, few studies have examined the cancer-specific immunomodulatory role of H 2S. Thus, the following discussion presents evidence from several non-cancer models that may perhaps carry over to the cancer setting.
There is evidence to suggest that H2S plays a role in modulating lymphocyte activity (Figure 4), in particular T cells, which are especially important in the antitumor response. In one study, physiological levels of H2S were found to enhance TCR-stimulated polyclonal and antigen-specific T cell activation, as demonstrated by increases in IL-2, CD69, and CD25 (Miller, Wang et al. 2012). Furthermore, T cell activation also led to increased CSE and CBS, whereas silencing of the latter impaired T cell activation that was reversed upon exogenous H2S supplementation, suggesting a self-reinforcing cycle of T cell activation through H2S (Miller, Wang et al. 2012). Further investigation revealed that H2S-induced T cell activation occurs through ERK1/2 phosphorylation, and inhibition of mitogen-activated protein kinase kinase (MEK)-dependent ERK phosphorylation can suppress T cell activation; in fact, the inhibitory effect of TSP-1 through CD47 was suggested to involve inhibition of ERK phosphorylation, and furthermore, TSP-1 also suppressed activation-dependent CBS and CSE expression in the T cells (Miller, Kaur et al. 2013). From these studies, physiological levels of H2S seem to promote T cell activation.
On the other hand, there is also evidence that H2S can impair the anti-tumor lymphocytic response (Figure 4). Supraphysiological levels of H2S using NaHS have been seen in vitro to impair lymphocyte proliferation and induce lymphocyte cell death by necrosis—not apoptosis—through the loss of mitochondrial membrane potential (Mirandola, Gobbi et al. 2007). The toxicity of H2S towards T cells in this study was specific to CD8+ and NK cells, sparing CD4+ T cells, and interestingly, glutathione was protective against the cytotoxicity of H2S (Mirandola, Gobbi et al. 2007). Similarly, H2S was found to be necessary for gingiva-derived mesenchymal stem cell-induced Fas/FasL-mediated T cell apoptosis in vitro (Yang, Yu et al. 2018). In cancer models, CBS and CSE knockdown in breast cancer cells enhanced their susceptibility to NK and T cell cytotoxicity by allowing increased expression of co-stimulatory ligands on the cancer cells (Youness, Gad et al. 2021). Interestingly, CO was found to increase the sensitivity of breast cancer cells to doxorubicin therapy by inhibiting CBS and thus reducing antioxidant capacity (Kawahara, Moller et al. 2017), aligning with the pro-cancer role of CBS and further supporting the previously discussed anti-tumor potential of CO treatment. Additionally, H2S has been found to be essential for Foxp3+ Treg cell differentiation and function (Yang, Qu et al. 2015), and H2S depletion in colorectal cancer-bearing mice correspondingly decreased CD4+CD25+Foxp3+ Treg cells, while increasing the CD8+ T-cell/Treg ratio, along with enhanced efficacy of anti–programmed cell death ligand 1 (PDL1) and anti–cytotoxic T-lymphocyte–associated antigen 4 (CTLA4) immunotherapies (Yue, Li et al. 2023). Taken together, these studies suggest that the effect of H2S on T cell function are biphasic, as with many of its other effects. This is supported by the results of another study, in which both fast- and slow- donors NaHS and GYY4137, respectively, stimulated proliferation in of lymphocytes healthy and systemic lupus erythematosus patients at lower concentrations and were inhibitory at higher concentrations (Han, Zeng et al. 2013). In this study, the inhibitory effects of H2S were further demonstrated to arise from modulation of the Akt/glycogen synthase kinase-3 beta (GSK3β) (Han, Zeng et al. 2013).
Some of the effects of H2S on immune cells are mediated through its effects on MDSCs, an immunosuppressive cell type recruited by cancer cells. In melanoma-bearing mice, DATS was found to inhibit tumor growth through downregulation of MDSCs in the spleen, blood, and TME, alongside an increase in dendritic cells and CD8+ T cells within the spleen, although no increase in either of the immune cells was seen in the tumor tissue (De Cicco, Ercolano et al. 2020). These effects were at least partially attributable to suppression of immunosuppressive genes in MDSCs, which was linked to restoration of T cell proliferation (De Cicco, Ercolano et al. 2020). In an alternative murine model of H. pylori-induced colitis, oral H2S administration using DATS was able to suppress the recruitment of G-MDSCs to the colon (De Cicco, Sanders et al. 2018).
H2S also has varying impacts on macrophages, another immune cell type that can either facilitate an increased immune response to the cancer or exert an immunosuppressive role allowing cancer cells to escape immune detection. In inflammatory conditions such as LPS stimulation, H2S seems to exert an anti-inflammatory effect on macrophages, reducing the pro-inflammatory mediators TNFα, IL-6, PGE2, and suppressing the expression of IL-1β, COX-2 and iNOS through NF-κB inhibition; these anti-inflammatory effects were further confirmed in vivo in an LPS sepsis mouse model (Huang, Feng et al. 2016). In fact, LPS has been seen to induce increased CSE expression and H2S in macrophages (Zhu, Liu et al. 2010). Similarly, pre-treatment of RAW 264.7 macrophages in vitro with H2S donor JK1 reversed M1 polarization induced by LPS stimulation, yet in this study, H2S also induced M2 polarization in the absence of a pro-inflammatory stimulus (Wu, Chen et al. 2019). On the other hand, NaHS administration has been seen to induce pro-inflammatory cytokine release in monocytes through ERK-dependent NF-κB activation (Zhi, Ang et al. 2007), and to increase early macrophage infiltration of infarcted myocardium in vivo, an effect associated with the internalization of β1 integrin and activation of downstream Src-focal adhesion kinase (FAK)/Pyk2-Rac signaling (Miao, Xin et al. 2016). CSE silencing produced the opposite effects, supporting the role of endogenous H2S production in macrophage infiltration (Miao, Xin et al. 2016). Further support for a pro-inflammatory effect of H2S involving macrophages comes from a murine model of mechanical load-induced tooth movement, in which exogenous H2S administration increased M1 macrophage levels whereas CBS inhibition did the opposite; the pro-M1 polarization effects of H2S were attributed to activation of the STAT1 pathway (He, Liu et al. 2020).
There is evidence for dual effects of H2S on other aspects of the immune response and inflammation (Figure 4). H2S was seen to promote the survival of neutrophils by inhibiting their apoptosis through the inhibition of p38 and caspase 3; intriguingly, H2S also reduced lymphocyte survival in this study (Rinaldi, Gobbi et al. 2006). The gasotransmitter has also been seen in separate studies to decrease leukocyte adherence to the endothelium through the activation of KATP channels (Zanardo, Brancaleone et al. 2006) vs increase neutrophil adhesion and locomotion through the same KATP-dependent mechanism (Dal-Secco, Cunha et al. 2008). Given the role of H2S in protecting the endothelium in the basal state, it is likely that in physiological conditions the gasotransmitter has antiadhesive activity but promotes endothelial activation and inflammation in pathological conditions. In addition to these effects, H2S deficiency can also accelerate cellular senescence in response to oxidative stress, whereas H2S is protective via the activation of antioxidant regulator nuclear factor erythroid 2-related factor 2 (Nrf2) (Yang, Zhao et al. 2012); while beneficial in reducing inflammation in hyperinflammatory states, this may be detrimental when trying to combat, as it may defend these cells against oxidative stress-induced death.
Yet in one study, both the anti-inflammatory and antitumor effects of H2S were emphasized as being important for combatting cancer. The combination of H2S with photothermal therapy (PTT) was found to increase immunogenicity of the latter, as demonstrated by increased tumor-infiltrated CD8+ T cells accompanied by lowered CD4+ T cells (Li, Xie et al. 2021). At the same time, anti-inflammatory effects of H2S were seen through reduction of the PTT-induced proinflammatory cytokines TNF-α, IL-6, and IL-1β; this was an added benefit of H2S therapy, as the pro-inflammatory effect of PTT can reduce its ability to trigger immunogenicity, cause permanent tissue injury with possible tumor regeneration (Li, Xie et al. 2021). In fact, different approaches to curtail the pro-inflammatory response caused by PTT are being investigated (Dong, Wang et al. 2018), indicating the importance of the anti-inflammatory effects seen with H2S. Thus, the seemingly conflicting effects of H2S on the immune/inflammatory response may work to synergistically promote antitumor effects of other therapies.
3.5. Impact on cancer-associated infectious complications
Immunosuppression is an adverse effect of a wide range anticancer therapies, including cytotoxic chemotherapies, radiotherapy, and hematopoietic stem cell transplants, which leave cancer patients vulnerable to infections (Bodey 1986). Bacterial pathogens are a notable cause of infection among cancer patients, particularly gram-negative bacteria that are often antibiotic-resistant (Marin, Gudiol et al. 2014, Perez, Adachi et al. 2014, Vahedian-Ardakani, Moghimi et al. 2019). Those with hematological malignancies are also especially at risk of opportunistic fungal infections (Segal, Almyroudis et al. 2007, Pagano, Busca et al. 2017, Shariati, Moradabadi et al. 2020). Additionally, as illustrated recently by the increased susceptibility to COVID-19 infection and poor outcomes in the recent pandemic (Wang, Sun et al. 2020, Zhang, Zhu et al. 2020), cancer-related immunosuppression can predispose patients to increased severity of viral infections as well. With the increased susceptibility to such a wide range of pathogens, it follows that infectious disease is a leading non-cancer cause of death among cancer patients (Zaorsky, Churilla et al. 2017). This issue is present even among young adult cancer populations, and in one study, an alarming standardized mortality ratio of 5.13 was seen in adolescent and young adult cancer patients for infectious disease (Anderson, Lund et al. 2019). Cancer patients have also been found to have an almost 10-fold increased risk of sepsis in comparison to non-cancer patients (Danai, Moss et al. 2006), accounting for more than 20% of all sepsis hospitalizations in the US (Hensley, Donnelly et al. 2019), and having higher rates of mortality from sepsis compared to non-cancer patients (Abou Dagher, El Khuri et al. 2017). Curtailing infections from a wide range of pathogens is thus of great interest in the context of cancer; all three gasotransmitters have roles in this regard, and thus are worth investigating specifically in the setting of cancer.
3.5.1. Nitric oxide and infection
Increased levels of NO and its derivatives during infection and their association with reduced disease severity, as seen with various viral (Kharitonov, Yates et al. 1995, Sanders, Proud et al. 2004), parasitic (Evans, Thai et al. 1993, Anstey, Weinberg et al. 1996), and bacterial pathogens (Wheeler, Smith et al. 1997), suggests a role of NO in innate host defense. Correspondingly, reduced NO levels can be found in individuals with cystic fibrosis (Lundberg, Nordvall et al. 1996) and Kartagener syndrome (Lundberg, Weitzberg et al. 1994), both of which are known for a predisposition to recurring infections, further highlighting the importance of NO for thwarting infection. NO demonstrates a very broad range of direct antimicrobial activity against different viruses (Sanders, Siekierski et al. 1998, Rimmelzwaan, Baars et al. 1999, Keyaerts, Vijgen et al. 2004, Regev-Shoshani, Vimalanathan et al. 2013, Akaberi, Krambrich et al. 2020), both gram-positive and gram-negative bacteria (Miller, McMullin et al. 2009, Ormerod, Shah et al. 2011), fungi (Ahmadi, Lee et al. 2016, Deppisch, Herrmann et al. 2016, Stasko, McHale et al. 2018), and protozoa (Evans, Thai et al. 1993, Anstey, Weinberg et al. 1996) (Figure 2). Consistent with its ability to combat a wide range of pathogens, the antimicrobial actions of the gasotransmitter involve multiple mechanisms, making it unique compared to current antimicrobial therapies, and reducing the likelihood of antimicrobial resistance; indeed, NO resistance among pathogens has not been reported to our knowledge (Privett, Broadnax et al. 2012, Rouillard, Novak et al. 2021). The activation of mucociliary clearance, which is especially important for the removal of pathogens from the respiratory tract (Nagaki, Shimura et al. 1995, Jiao, Wang et al. 2011), is the first line of defense against pathogens provided by NO. The gasotransmitter also has more specific antimicrobial actions, including protein modifications and induction of DNA damage through oxidative and nitrosative stress (Fang 1997). With viruses, NO can inhibit replication and entry into host cells by nitration, S-nitrosylation, and oxidation of proteins essential for these functions (Colasanti, Persichini et al. 1999, Xu, Zheng et al. 2006). The specific protein target varies based on the identity of the virus; for instance, while NO inhibited HIV-1 reverse transcriptase by oxidation of a cysteine residue (Persichini, Colasanti et al. 1999), in dengue virus type-2 it inhibited the RNA-dependent RNA polymerase (Takhampunya, Padmanabhan et al. 2006). Other reported antiviral mechanisms include inhibition of ribonucleotide reductase and transcription factors,disruptions in post-translational modifications, etc, as described in(Colasanti, Persichini et al. 1999).
With respect to bacteria, while NO alone may not directly exert antibacterial effects, its reaction products and combination with other agents may still be promising to combat bacterial infections. For instance, whereas NO had minimal direct antibacterial effects on E. coli (Pacelli, Wink et al. 1995) and S. typhimurium (De Groote, Granger et al. 1995), it was found to increase the DNA-damaging effects of hydrogen peroxide in the case of the former, and in the latter, exert bacteriostatic and bactericidal effects through its products S-nitrosoglutathione (GSNO) and peroxynitrite, respectively. Interestingly, more recent studies have also found NO treatment to significantly enhance the efficacy of tetracycline therapy against both gram -positive and -negative bacteria (Reger, Meng et al. 2017, Reger, Meng et al. 2017). Interestingly, NO has been found to increase susceptibility bacterial to conventional antibiotics and slow the development of antibiotic resistance (Rouillard, Novak et al. 2021), which is especially important for cancer patients who are susceptible to multi-drug resistant bacterial infections. Additionally, the co-release of NO and CO has been found to be superior in treating methicillin-resistant S. aureus (MRSA) infection in a murine skin wound model vancomycin, suggesting the combined use of these gasotransmitters may be especially beneficial in the context of bacterial infection (Gao, Cheng et al. 2022).
NO has demonstrated fungicidal activity as well (Stasko, McHale et al. 2018); in fact, the antifungal activity of amphotericin B against C. neoformans has been linked to the upregulation of the NO pathway in macrophages (Tohyama, Kawakami et al. 1996, Mozaffarian, Berman et al. 1997). Overall, this broad range antimicrobial activity is likely to be beneficial in the context of cancer, considering the immunosuppressed state induced by cancer therapeutics and susceptibility of these patients to potentially life-threatening infections.
It should be noted that there is also evidence that NO can sometimes perpetuate infection rather than curtail it. The gasotransmitter has been identified as one defense utilized by bacteria against immune oxidative attack (Gusarov and Nudler 2005, Shatalin, Gusarov et al. 2008) and has been associated with resistance of the bacterial pathogens to a range of antibiotics (Gusarov, Shatalin et al. 2009), directly contrary to the increased antibiotic susceptibility and delayed development of antibiotic resistance described more in a more recent study (Rouillard, Novak et al. 2021). With viral infections as well, inhaled NO failed to improve the outcome in mice infected with severe influenza (Darwish, Miller et al. 2012), and in fact, NOS inhibition using non-specific inhibitor L-NMMA improved survival and pulmonary compliance in a murine model of HSV-1 pneumonitis despite a higher viral load, suggesting that the inflammatory response rather than the virus itself was the reason for the damage (Adler, Beland et al. 1997). The reasons for these contradictory findings are not entirely clear; perhaps the effects of NO depend on the identity of the pathogen, and/or the methodological differences such as dosage and release kinetics of NO administration strategies play a role. In any case, while NO has demonstrated great promise in preventing and combatting a wide range of infectious pathogens, understanding the settings in which its use would be beneficial vs harmful is critical, especially when dealing with immunosuppressed cancer patients.
3.5.2. Carbon monoxide and infection
CO has demonstrated a broad range of antimicrobial activity that may be promising for cancer patients predisposed to infection (Figure 3).
A large number of studies have indicated direct antibacterial actions of CO, covering gram-positive, gram-negative, aerobic, and anaerobic bacteria (Nobre Lígia, Seixas João et al. 2007, Desmard, Davidge et al. 2009), and CO treatments can be tailored to preferentially target the bacteria while having no effect on host cells, as seen with a visible-light-induced CORM (Ward, Lynam et al. 2014). The bactericidal activity of macrophages has been attributed to CO production, which leads to bacterial ATP depletion and NALP3 inflammasome activation, promoting bacterial phagocytosis (Wegiel, Larsen et al. 2014). Thus, the exogenous supplementation of CO seems to be one potential avenue in combatting bacterial infections, and indeed, in a murine model of antibiotic-resistant P. aeruginosa, CORM-3 treatment reduced bacterial counts and improved survival in both immunocompetent as well as immunosuppressed hosts (Desmard, Davidge et al. 2009).
In one study, CO administration was found to inhibit the growth of S. aureus and E. coli in culture most potently in near-anaerobic conditions, suggesting that the bactericidal effects of CO are not entirely dependent on its disruption of the electron transport chain through cytochrome c inhibition (Nobre Lígia, Seixas João et al. 2007). Further investigation has revealed several mechanisms by which CO may exert its antibacterial effects, including through the induction of significant transcriptional-level genome-wide changes (Davidge, Sanguinetti et al. 2009, McLean, Begg et al. 2013), direct DNA damage through ROS induction (Tavares, Teixeira et al. 2011), suppression of the bacterial respiration (Desmard, Davidge et al. 2009, Wilson, Jesse et al. 2012), and inhibition of bacterial biofilm formation (Murray, Okegbe et al. 2012, Nguyen, Nguyen et al. 2015). Importantly, CO treatment has also demonstrated promise against drug-resistant bacteria, such as multi-drug resistant uropathogenic E. coli (Sahlberg Bang, Kruse et al. 2016) and methicillin-resistant S. aureus (Cheng, Gan et al. 2021), to which an increased susceptibility among cancer patients has been described (El-Gendy, El-Bondkly et al. 2018, Mahmoud, Salim et al. 2020).
CO may also have direct antiviral activity, although studies focusing on CO itself rather than the CO-producing enzyme HO-1 are relatively limited. Studies with HO-1 have demonstrated its antiviral actions against several viruses, including hepatitis B (Protzer, Seyfried et al. 2007), hepatitis C (Zhu, Wilson et al. 2008), HIV-1 (Devadas and Dhawan 2006), influenza A (Ma, Wang et al. 2016), and dengue virus (Tseng, Lin et al. 2016). As HO-1 is responsible not only for CO production, but also other products such as biliverdin and iron, it is not clear how much of these effects are attributable to CO. There are a few studies that have shown CO specifically to have antiviral actions, seen against the human pathogen enterovirus 71 through inhibition of its replication (Tung, Hsieh et al. 2011), and in several animal viruses, including porcine reproductive and respiratory syndrome virus (PRRSV) (Zhang, Zhao et al. 2016), bovine viral diarrhea virus (BVDV) (Ma, Pu et al. 2017), and spring viremia of carp virus (SVCV) (Li, Li et al. 2018); the antiviral mechanisms in these studies were elucidated to involve cGMP/PKG and NF-κB pathways. Considering these findings, it is possible that CO has yet unexplored antiviral actions that may prove useful in the context of cancer, and further investigation in this area is warranted.
3.5.3. Hydrogen sulfide and infection
Like NO, H2S has a range of antimicrobial actions (Figure 4), although these are not as vast as those demonstrated by NO. As its first line of defense, H2S increases mucociliary clearance of offending particles by reducing transepithelial Na+ transport in lung tissue, reducing mucus viscosity (Althaus, Urness et al. 2012, Agné, Baldin et al. 2015). H2S also has direct antiviral actions against a range of enveloped RNA viruses, including through the inhibition of viral replication (Li, Ma et al. 2015, Bazhanov, Escaffre et al. 2017), entry (Pacheco 2017), and syncytium formation, which is hypothesized to affect viral assembly/release (Li, Ma et al. 2015). A separate, indirect antiviral mechanism of H2S may involve the upregulation of GSH levels (Kimura, Goto et al. 2009), which acts both as an antioxidant as well as an antiviral agent independent of its ROS scavenging activity (Diotallevi, Checconi et al. 2017).
With regards to bacterial infection, there are conflicting reports of the role of H2S. Separate studies have found that suppression of bacterial H2S production rendered the microbes more susceptible to a range of antibiotics by protecting against oxidative stress (Shatalin, Shatalina et al. 2011, Seregina, Lobanov et al. 2022), suggesting a deleterious involvement of the gasotransmitter in the development of antibiotic resistance. On the other hand, H2S has been found to inhibit the growth of several gram-positive and gam-negative bacteria, including S. aureus, S. typhimurium, L. monocytogenes, B. subtilis, B. thuringiensis, E. coli, S. mitis, S. oralis, and E. aerogenes (Fu, Hu et al. 2014, Ooi and Tan 2016, Fu, Wei et al. 2018). Mechanistically, these studies revealed increased ROS and decreased GSH as the mechanisms by which H2S exerted its antibacterial effects (Ooi and Tan 2016, Fu, Wei et al. 2018), directly opposing the observation of H2S protecting against oxidative stress in the previous studies (Shatalin, Shatalina et al. 2011, Seregina, Lobanov et al. 2022). Additionally, a bactericidal role of H2S-containing thermal spring waters has been reported, leading to a suggestion that H2S may have a role in new perspectives for pool treatment (Giampaoli, Valeriani et al. 2013). Interestingly, both toxic and protective effects of H2S were seen in one study with the bacterium Shewanella oneidensis, where the simultaneous addition of H2S with H2O2 enhanced the toxicity of the latter by increasing oxidative stress, while H2S pre-treatment rendered the bacteria resistant to the H2O2 cytotoxicity by activating oxidative-stress-responding regulator OxyR (Wu, Wan et al. 2015); thus, timing seems to be a critical component in the effects of H2S on bacterial pathogens.
Overall, the evidence suggests a benefit to H2S therapy in cancer patients particularly in the context of viral infection.
4. Summary and Perspectives
Overall, the potential for therapeutic application of NO, CO, and H2S in the context of cancer remains unclear. A large body of evidence from a range of experimental cancer models indicates favorable anti-cancer effects of each of these gases. All three have demonstrated an ability to inhibit cancer cell proliferation by inhibiting growth and survival signaling, induce cell cycle arrest and apoptosis, thwart cancer invasion and metastasis by reducing ECM remodeling and EMT induction, and induce anti-tumor immune defense. Additionally, all gasotransmitters have demonstrated tremendous potential in the defense against pathogens, which may be especially important in the context of cancer, considering the burden of immunosuppression that often accompanies this disease and its treatments. NO and CO have also demonstrated the ability to reduce cancer-related angiogenesis, although, in this regard, H2S has demonstrated only pro-angiogenic effects. Despite these promising findings, there is also evidence to suggest NO, CO and H2S have cancer-promoting activity, including through increased growth/survival signaling, increasing angiogenesis, and interfering with tumor-targeting immune defense, which is entirely contradictory to the anti-tumor effects that targeted the same aspects of cancer biology.
The reasons for these contradictory results largely remain to be elucidated. The apparent discrepancies between various studies stay even with a lens that focuses on each type of cancer separately, suggesting other players are involved in these baffling findings. Notably, some studies have emphasized the role of gasotransmitter donor dosage and release kinetics (Wu, Li et al. 2017, Oláh, Módis et al. 2018), demonstrating that the same experimental system can yield opposite findings when these parameters are adjusted. However, it is challenging to establish dose-response curves for these gaseous molecules, leading to difficulties in defining quantitative relationships; this likely plays a significant role in the lack of consistency across studies, which makes it challenging to compare results between different studies. Additionally, the wide range of molecular targets with which these gasotransmitters can interact to elicit effects introduces another variable in interpreting study findings, necessitating affinity and dose-response considerations. As an example, CO itself has over 25 molecular targets, ranging from hemoglobin and cytochrome c oxidase to sGC and several ion channels, binding to each with different affinities (for more information, see (Zhengnan, Ladie Kimberly De La et al. 2022). Furthermore, when using exogenous donors, it is important to consider that some of these compounds have extensive activity independent of the gasotransmitter they release. Once again, using CO as an example, several CO-independent effects of different CORMs have been elucidated, ranging from ion channel modification to antimicrobial activity (Gessner, Sahoo et al. 2017, Juszczak, Kluska et al. 2020, Southam, Williamson et al. 2021), which introduces yet another element that can impact study results. Evidently, there are many issues to be further explored before any conclusions about NO, H2S, and CO in the context of cancer can be made. Still, the preponderous weight of the data strongly suggests a promising anti-cancer potential for NO, CO, and H2S, which merits further serious consideration and efforts to develop as therapeutics.
Decades of research have highlighted the enormous potential of NO, CO, and H 2S in various aspects of medicine, yet few gasotransmitter-releasing agents have reached the marketplace. In the context of cancer, this may be due to the scientific community’s focus on chemoprevention rather than chemotherapy; for more details, see (Kashfi 2018).
An area in which significant progress can be expected is the field of multifunctional compounds, that is, using clinically approved drugs with added NO-, CO-, or H2S-donating moieties, either alone or in combination. In this regard, Otenaproxesul (ATB-346), an H2S-releasing analog of naproxen, has completed phase 2 clinical trials to manage chronic pain, gastric ulcer, and osteoarthritis (NCT03978208, 2019) but not cancer. In addition, there are other H2S-releasing compounds under evaluation for indications other than cancer; for example, a Phase 2a clinical study on the analgesic effect of GIC-1001 and GIC-1002 on visceral pain (NCT02276768; 2016) (NCT01926444; 2019).
With regards to NO, there are several ongoing clinical trials addressing its potential efficacy and application in several indications other than cancer. These include inhaled NO in cystic fibrosis (CF) patients (NCT02498535; 2021), a Phase 2 open-label study of a nebulized NO-generating solution in patients with CF (NCT05101915; 2022), and a Phase 1/2 study of inhaled sodium nitrite as an antimicrobial for Pseudomonas infection in CF (NCT02694393, 2022). The only NO-releasing agent clinically evaluated in managing cancer is an NO derivative of aspirin. A Phase 1 clinical trial of NO-releasing aspirin in preventing colorectal cancer in high-risk patients (NCT00331786; 2007) was terminated due to the possible genotoxicity of a potential metabolite (NicOx 2007). Another NO-releasing NSAID, NO-naproxen (naproxcinod) that, was being evaluated for osteoarthritis of the knee and hip (NCT00504127; 2009) (Lohmander, McKeith et al. 2005, Schnitzer, Kivitz et al. 2005, Karlsson, Pivodic et al. 2009, Schnitzer, Kivitz et al. 2010, Schnitzer, Hochberg et al. 2011) has completed Phase-III clinical trials. However, approval for NDA application was denied due to concerns by the FDA about this agent’s gastrointestinal and cardiovascular effects over long-term use (NicOx 2010).
Regarding CO, translation to the clinical setting is the current focus of an investigation. Several methods of exogenous CO delivery exist, including inhaled CO, light-activated CORMs, liquid-dissolved CO, and metal-based CORMs. A relatively new direction that has recently emerged is the synthesis of organic (non-metal-based, non-photo-activated) CO-releasing prodrugs, which are intended to release CO under physiological conditions in response to a chemical or biological triggers; for details, see (Ji and Wang 2018). This new class of drugs may offer exciting new possibilities for organelle or biomarker-based targeting and enable a more accurate assessment of the biological effect of this gasotransmitter both in vitro and in vivo (Ji and Wang 2018).
Phase II clinical trials using inhaled delivery and PEGylated hemoglobin-carrier delivery of CO for indications such as chronic obstructive pulmonary disease (COPD) (NCT00122694; 2006), idiopathic pulmonary fibrosis (NCT01214187; 2015) (Rosas, Goldberg et al. 2018), vaso-occlusive crises (NCT02672540, NCT02411708; 2017) and ulcerations (NCT02600390; 2016) of sickle cell disease, subarachnoid hemorrhage (NCT02323685; 2016) and kidney transplant (NCT02490202; 2016) (Siracusa, Schaufler et al. 2021) have been conducted. There is also an ongoing phase II clinical trial (May 2022) investigating the safety and efficacy of inhaled CO in the treatment of acute respiratory distress syndrome (ARDS) (NCT03799874). However, no trials are being conducted for treating cancer. As far as we know, CO-donating multifunctional donors have not yet been characterized.
To reiterate, one must also remember that NO, CO, and H2S do not act in isolation but in concert, sometimes through overlapping signaling pathways. These interactions need to be further explicitly elucidated in the context of cancer, as they may potentially enhance or complement the efficacy of other therapeutics. Such efforts have demonstrated the utility of NOSH-aspirin (Chattopadhyay, Kodela et al. 2012, Lee, McGeer et al. 2013, Fonseca, Cunha et al. 2015, Kodela, Chattopadhyay et al. 2015, Chattopadhyay, Kodela et al. 2020), NOSH-naproxen (Kodela, Chattopadhyay et al. 2013, Chattopadhyay, Kodela et al. 2016), and NOSH-sulindac (Kashfi, Chattopadhyay et al. 2015), as dual NO- and H2S-donating hybrids against cancer and other inflammatory-based diseases in preclinical studies.
Finally, selective inducers/inhibitors of the gasotransmitter-generating enzymes may serve as another possibility for therapeutic applications. Importantly, however, each of these scenarios has both benefits and drawbacks. For instance, selective inhibitors of CBS could potentially increase homocysteine levels and thus increase cardiovascular risks, and iNOS-specific inhibitors could induce immunosuppression.
Many scientific discoveries have had an element of serendipity, curiosity, and a prepared mind; the NO, CO, and H2S arenas are no exception.
Acknowledgments
Supported in part by the National Institutes of Health [R24 DA018055; R01GM123508] and the Professional Staff Congress-City University of New York (PSC-CUNY) [TRADB-49–271].
Abbreviations:
- 3-MP
3-mercaptopyruvate
- 3-MST
3-mercaptopyruvate sulfurtransferase
- AP-1
activator protein
- Apaf-1
apoptosis protease activating factor 1
- ARDS
acute respiratory distress syndrome
- Bak
Bcl-2 homologous antagonist/killer
- Bax
Bcl-2-associated X protein
- Bcl-2
B-cell lymphoma 2
- Ca2+
calcium ions
- CAT
cysteine aminotransferas
- CBS
cystathionine β-synthase
- CDK
cyclin-dependent kinase
- cGMP
cyclic guanosine monophosphate
- cIAP1
cellular inhibitor of apoptosis protein 1
- c-Myc
cellular myelocytomatosis
- CO
carbon monoxide
- CORM
carbon monoxide-releasing molecule
- COX
cyclooxygenase
- CSE
cystathionine γ-lyase
- CTLA4
cytotoxic T-lymphocyte–associated antigen
- DADS
diallyl disulfide
- DAO
D-amino acid oxidase
- DATS
diallyl trisulfide
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transitio
- eNOS
endothelial nitric oxide synthase
- ER
endoplasmic reticulum
- ERα
estrogen receptor alpha
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- FasL
Fas-ligand
- Fox
Forkhead box
- GSK3β
glycogen synthase kinase-3 beta
- GSNO
S-nitrosoglutathione
- H2S
hydrogen sulfide
- H2Sn
polysulfide
- Hb
hemoglobin
- HIF
hypoxia-inducible factor
- HNO
nitroxyl
- HO
heme oxygenase
- HSNO
thionitrous acid
- Hsp
heat-shock protein
- hTERT
human telomerase reverse transcriptase
- HUVEC
human umbilical vein endothelial cell
- IAP
inhibitor of apoptosis protein
- IFN
interferon
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IP3
inositol triphosphate
- JNK
c-Jun N-terminal kinase
- K+
potassium ion
- KATP
ATP-dependent potassium channel
- L-NAME
L-NG-Nitro arginine methyl este
- LPS
lipopolysaccharide
- mAb
monoclonal antibody
- MAPK
mitogen-activated protein kinase
- MDSC
myeloid-derived suppressor cells
- MEK
mitogen-activated protein kinase kinase
- MHC
major histocompatibility complex
- miR
micro-RNA
- MMP
matrix metalloproteinase
- MPO
myeloperoxidase
- MRSA
methicillin-resistant Staphylococcus aureus
- mTOR
mammalian target of rapamycin
- NADPH
nicotinamide adenine dinucleotide phosphate
- NaHS
sodium hydrosulfide
- NDRG-1
N-Myc-downstream-regulated gene 1
- NF-κB
nuclear factor kappa light chain enhancer of activated B cells
- nm23
non-metastatic protein 23
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NOS
nitric oxide synthase
- Notch-1
neurogenic locus notch homolog protein 1
- Nrf2
nuclear factor erythroid 2-related factor 2
- ONSS−
nitropersulfide
- NSAID
non-steroidal anti-inflammatory drugs
- PBMC
peripheral blood mononuclear cells
- PCNA
proliferating cell nuclear antigen
- PDE5A
phosphodiesterase 5A
- PDL1
programmed cell death ligand 1
- PEITC
β-phenylethyl isothiocyanate
- PERK
protein kinase R-like endoplasmic reticulum kinase
- PGE2
prostaglandin E2
- PI3-K
phosphoinositide 3-kinase
- PK
protein kinase
- PLP
pyridoxal 5´-phosphate
- PP2A
protein phosphatase 2A
- PPAR
peroxisome proliferator-activated receptor
- PTT
photothermal therapy
- Rb
retinoblastoma
- RKIP
Raf-1 kinase inhibitor protein
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RSNO
nitrosothiol
- sGC
soluble guanylate cyclas
- shRNA
short hairpin RNA
- SNAP
S-nitroso-N-acetylpenicillamine
- SNP
sodium nitroprusside
- SPRC
S-propargyl-cysteine
- Src
steroid receptor coactivator
- STAT
signal transducer and activator of transcription
- Tcf
T-cell factor
- TCR
T cell receptor
- TGF
transforming growth factor
- TME
tumor microenvironment
- TNF-α
tumor necrosis factor-α
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- TSP-1
thrombospondin-1
- UV
ultraviolet
- VCAM-1
vascular cell adhesion molecule 1
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor 2
- Wnt
Wingless/Integrated
- XIAP
X-linked inhibitor of apoptosis protein
- ZEB-1
zinc finger E-box binding homeobox 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- Abe K and Kimura H (1996). “The possible role of hydrogen sulfide as an endogenous neuromodulator.” J Neurosci 16(3): 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou Dagher G, El Khuri C, Chehadeh AA-H, Chami A, Bachir R, Zebian D and Bou Chebl R (2017). “Are patients with cancer with sepsis and bacteraemia at a higher risk of mortality? A retrospective chart review of patients presenting to a tertiary care centre in Lebanon.” BMJ Open 7(3): e013502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams C, McCarthy HO, Coulter JA, Worthington J, Murphy C, Robson T and Hirst DG (2009). “Nitric oxide synthase gene therapy enhances the toxicity of cisplatin in cancer cells.” The Journal of Gene Medicine 11(2): 160–168. [DOI] [PubMed] [Google Scholar]
- Adler H, Beland JL, Del-Pan NC, Kobzik L, Brewer JP, Martin TR and Rimm IJ (1997). “Suppression of herpes simplex virus type 1 (HSV-1)-induced pneumonia in mice by inhibition of inducible nitric oxide synthase (iNOS, NOS2).” J Exp Med 185(9): 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agné AM, Baldin J-P, Benjamin AR, Orogo-Wenn MC, Wichmann L, Olson KR, Walters DV and Althaus M (2015). “Hydrogen sulfide decreases β-adrenergic agonist-stimulated lung liquid clearance by inhibiting ENaC-mediated transepithelial sodium absorption.” American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 308(7): R636–R649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Hewett PW, Fujisawa T, Sissaoui S, Cai M, Gueron G, Al-Ani B, Cudmore M, Ahmed SF, Wong MK, Wegiel B, Otterbein LE, Vítek L, Ramma W, Wang K and Ahmed A (2015). “Carbon monoxide inhibits sprouting angiogenesis and vascular endothelial growth factor receptor-2 phosphorylation.” Thromb Haemost 113(2): 329–337. [DOI] [PubMed] [Google Scholar]
- Ahmadi MS, Lee HH, Sanchez DA, Friedman AJ, Tar MT, Davies KP, Nosanchuk JD and Martinez LR (2016). “Sustained Nitric Oxide-Releasing Nanoparticles Induce Cell Death in Candida albicans Yeast and Hyphal Cells, Preventing Biofilm Formation In Vitro and in a Rodent Central Venous Catheter Model.” Antimicrobial Agents and Chemotherapy 60(4): 2185–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaberi D, Krambrich J, Ling J, Luni C, Hedenstierna G, Järhult JD, Lennerstrand J and Lundkvist Å (2020). “Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro.” Redox Biology 37: 101734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, Ihara H, Sawa T, Matsunaga T, Kasamatsu S, Nishimura A, Morita M, Tomizawa K, Nishimura A, Watanabe S, Inaba K, Shima H, Tanuma N, Jung M, Fujii S, Watanabe Y, Ohmuraya M, Nagy P, Feelisch M, Fukuto JM and Motohashi H (2017). “Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics.” Nat Commun 8(1): 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akool E-S, Kleinert H, M. A. HF, Abdelwahab H, Förstermann U, Pfeilschifter Jand Eberhardt W(2003). “Nitric Oxide Increases the Decay of Matrix Metalloproteinase 9 mRNA by Inhibiting the Expression of mRNA-Stabilizing Factor HuR.” Molecular and Cellular Biology 23(14): 4901–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Magableh MR, Kemp-Harper BK and Hart JL (2015). “Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice.” Hypertension Research 38(1): 13–20. [DOI] [PubMed] [Google Scholar]
- Al-Owais MMA, Scragg JL, Dallas ML, Boycott HE, Warburton P, Chakrabarty A, Boyle JP and Peers C (2012). “Carbon Monoxide Mediates the Anti-apoptotic Effects of Heme Oxygenase-1 in Medulloblastoma DAOY Cells via K+ Channel Inhibition *.” Journal of Biological Chemistry 287(29): 24754–24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE and Knowles RG (2001). “Nitric oxide synthases: structure, function and inhibition.” Biochem J 357(Pt 3): 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali HR, Provenzano E, Dawson SJ, Blows FM, Liu B, Shah M, Earl HM, Poole CJ, Hiller L, Dunn JA, Bowden SJ, Twelves C, Bartlett JMS, Mahmoud SMA, Rakha E, Ellis IO, Liu S, Gao D, Nielsen TO, Pharoah PDP and Caldas C (2014). “Association between CD8+ T-cell infiltration and breast cancer survival in 12 439 patients.” Annals of Oncology 25(8): 1536–1543. [DOI] [PubMed] [Google Scholar]
- Allanson M and Reeve VE (2005). “Ultraviolet A (320–400 nm) Modulation of Ultraviolet B (290–320 nm)-Induced Immune Suppression Is Mediated by Carbon Monoxide.” Journal of Investigative Dermatology 124(3): 644–650. [DOI] [PubMed] [Google Scholar]
- Allanson M and Reeve VE (2007). “Carbon monoxide signalling reduces photocarcinogenesis in the hairless mouse.” Cancer Immunology, Immunotherapy 56(11): 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaany Z, Ju Y, Yang G and Wang R (2014). “The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide.” Sci Signal 7(342): ra87. [DOI] [PubMed] [Google Scholar]
- Althaus M, Urness KD, Clauss WG, Baines DL and Fronius M (2012). “The gasotransmitter hydrogen sulphide decreases Na+ transport across pulmonary epithelial cells.” British Journal of Pharmacology 166(6): 1946–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambs S, Merriam WG, Ogunfusika MO, Bennett WP, Ishibe N, Hussain SP, Tzeng EE, Geller DA, Billiar TR and Harris CC (1998). “p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells.” Nature Medicine 4(12): 1371–1376. [DOI] [PubMed] [Google Scholar]
- Anderson C, Lund JL, Weaver MA, Wood WA, Olshan AF and Nichols HB (2019). “Noncancer mortality among adolescents and young adults with cancer.” Cancer 125(12): 2107–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstey NM, Weinberg JB, Hassanali MY, Mwaikambo ED, Manyenga D, Misukonis MA, Arnelle DR, Hollis D, McDonald MI and Granger DL (1996). “Nitric oxide in Tanzanian children with malaria: inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression.” J Exp Med 184(2): 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anuar F, Whiteman M, Siau JL, Kwong SE, Bhatia M and Moore PK (2006). “Nitric oxide-releasing flurbiprofen reduces formation of proinflammatory hydrogen sulfide in lipopolysaccharide-treated rat.” Br J Pharmacol 147(8): 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate LA, Luscher P and Tyrrell RM (1991). “Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells.” Cancer Res 51(3): 974–978. [PubMed] [Google Scholar]
- Arp AJ, Childress JJ and Vetter RD (1987). “The sulphide-binding protein in the blood of the vestimentiferan tubeworm, Riftia pachyptila, is the extracellular hemoglobin.” J. Exp. Biol 128: 139–158. [Google Scholar]
- Asano K, Nabeyama A, Miyake Y, Qiu C-H, Kurita A, Tomura M, Kanagawa O, Fujii S.-i. and Tanaka M(2011). “CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens.” Immunity 34(1): 85–95. [DOI] [PubMed] [Google Scholar]
- Axelsson S, Magnuson A, Lange A, Alshamari A, Hörnquist EH and Hultgren O (2020). “A combination of the activation marker CD86 and the immune checkpoint marker B and T lymphocyte attenuator (BTLA) indicates a putative permissive activation state of B cell subtypes in healthy blood donors independent of age and sex.” BMC Immunol 21(1): 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeriswyl V and Christofori G (2009). “The angiogenic switch in carcinogenesis.” Seminars in Cancer Biology 19(5): 329–337. [DOI] [PubMed] [Google Scholar]
- Bahnassy AA, Zekri A-RN, El-Houssini S, El-Shehaby AMR, Mahmoud MR, Abdallah S and El-Serafi M (2004). “Cyclin A and cyclin D1 as significant prognostic markers in colorectal cancer patients.” BMC Gastroenterology 4(1): 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai YW, Ye MJ, Yang DL, Yu MP, Zhou CF and Shen T (2019). “Hydrogen sulfide attenuates paraquat-induced epithelial-mesenchymal transition of human alveolar epithelial cells through regulating transforming growth factor-β1/Smad2/3 signaling pathway.” J Appl Toxicol 39(3): 432–440. [DOI] [PubMed] [Google Scholar]
- Banerjee K, Ganguly A, Chakraborty P, Sarkar A, Singh S, Chatterjee M, Bhattacharya S and Choudhuri SK (2014). “ROS and RNS induced apoptosis through p53 and iNOS mediated pathway by a dibasic hydroxamic acid molecule in leukemia cells.” European Journal of Pharmaceutical Sciences 52: 146–164. [DOI] [PubMed] [Google Scholar]
- Baritaki S, Huerta-Yepez S, Sahakyan A, Karagiannides I, Bakirtzi K, Jazirehi A and Bonavida B (2010). “Mechanisms of nitric oxide-mediated inhibition of EMT in cancer.” Cell Cycle 9(24): 4931–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhanov N, Escaffre O, Freiberg AN, Garofalo RP and Casola A (2017). “Broad-Range Antiviral Activity of Hydrogen Sulfide Against Highly Pathogenic RNA Viruses.” Scientific reports 7: 41029–41029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenyiova A, Grman M, Mijuskovic A, Stasko A, Misak A, Nagy P, Ondriasova E, Cacanyiova S, Brezova V, Feelisch M and Ondrias K (2015). “The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants.” Nitric Oxide 46: 123–130. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Sidhapuriwala JN, Sparatore A and Moore PK (2008). “Treatment with H2S-releasing diclofenac protects mice against acute pancreatitis-associated lung injury.” Shock 29(1): 84–88. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Wong FL, Fu D, Lau HY, Moochhala SM and Moore PK (2005). “Role of hydrogen sulfide in acute pancreatitis and associated lung injury.” Faseb J 19(6): 623–625. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, RodriguezAguayo C, Lopez-Berestein G, Basal E, Weaver AL, Visscher DW, Cliby W, Sood AK, Bhattacharya R and Mukherjee P (2013). “Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance.” PLoS One 8(11): e79167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco CL, Akaike T, Ida T, Nagy P, Bogdandi V, Toscano JP, Kumagai Y, Henderson CF, Goddu RN, Lin J and Fukuto JM (2019). “The reaction of hydrogen sulfide with disulfides: formation of a stable trisulfide and implications for biological systems.” Br J Pharmacol 176(4): 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilban M, Bach FH, Otterbein SL, Ifedigbo E, de Costa d’Avila J, Esterbauer H, Chin BY, Usheva A, Robson SC, Wagner O and Otterbein LE (2006). “Carbon Monoxide Orchestrates a Protective Response through PPARγ.” Immunity 24(5): 601–610. [DOI] [PubMed] [Google Scholar]
- Bir SC, Kolluru GK, McCarthy P, Shen X, Pardue S, Pattillo CB and Kevil CG “Hydrogen Sulfide Stimulates Ischemic Vascular Remodeling Through Nitric Oxide Synthase and Nitrite Reduction Activity Regulating Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor–Dependent Angiogenesis.” Journal of the American Heart Association 1(5): e004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrane G, Li H, Yang S, Tachado SD and Seng S (2013). “Cigarette smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in prostate cancer cells: Nuclear HO-1 promotes vascular endothelial growth factor secretion.” Int J Oncol 42(6): 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodey GP (1986). “Infection in cancer patients. A continuing association.” Am J Med 81(1a): 11–26. [DOI] [PubMed] [Google Scholar]
- Bouton C and Demple B (2000). “Nitric Oxide-inducible Expression of Heme Oxygenase-1 in Human Cells: TRANSLATION-INDEPENDENT STABILIZATION OF THE mRNA AND EVIDENCE FOR DIRECT ACTION OF NITRIC OXIDE *.” Journal of Biological Chemistry 275(42): 32688–32693. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn MC, Chiche J and Pouysségur J (2007). “Hypoxia and cancer.” Journal of Molecular Medicine 85(12): 1301–1307. [DOI] [PubMed] [Google Scholar]
- Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, Rusu A, Irimie A, Atanasov AG, Slaby O, Ionescu C and Berindan-Neagoe I (2019). “A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer.” Cancers (Basel) 11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PA, Thomas GJ and Langdon JD (2003). “The role of nitric oxide in oral diseases.” Arch Oral Biol 48(2): 93–100. [DOI] [PubMed] [Google Scholar]
- Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, Radi R and Cayota AM (1999). “Peroxynitrite Inhibits T Lymphocyte Activation and Proliferation by Promoting Impairment of Tyrosine Phosphorylation and Peroxynitrite-Driven Apoptotic Death1.” The Journal of Immunology 162(6): 3356–3366. [PubMed] [Google Scholar]
- Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, Battistini L, Iafrate M, Prayer-Galetti T, Pagano F and Viola A (2005). “Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers.” Journal of Experimental Medicine 201(8): 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouet A, Sonveaux P, Dessy C, Balligand J-L and Feron O (2001). “Hsp90 Ensures the Transition from the Early Ca2+-dependent to the Late Phosphorylation-dependent Activation of the Endothelial Nitric-oxide Synthase in Vascular Endothelial Growth Factor-exposed Endothelial Cells *.” Journal of Biological Chemistry 276(35): 32663–32669. [DOI] [PubMed] [Google Scholar]
- Bryan NS, Bian K and Murad F (2009). “Discovery of the nitric oxide signaling pathway and targets for drug development.” Front Biosci (Landmark Ed) 14(1): 1–18. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V and Cirino G (2010). “Hydrogen Sulfide Is an Endogenous Inhibitor of Phosphodiesterase Activity.” Arteriosclerosis, Thrombosis, and Vascular Biology 30(10): 1998–2004. [DOI] [PubMed] [Google Scholar]
- Bukovska G, Kery V and Kraus JP (1994). “Expression of human cystathionine beta-synthase in Escherichia coli: purification and characterization.” Protein Expr Purif 5(5): 442–448. [DOI] [PubMed] [Google Scholar]
- Butler AR and Nicholson R (2003). Life, death and nitric oxide. Cambridge, UK, Royal Society of Chemistry. [Google Scholar]
- Cai W-J, Wang M-J, Ju L-H, Wang C and Zhu Y-C (2010). “Hydrogen sulfide induces human colon cancer cell proliferation: Role of Akt, ERK and p21.” Cell Biology International 34(6): 565–572. [DOI] [PubMed] [Google Scholar]
- Cai W-J, Wang M-J, Moore PK, Jin H-M, Yao T and Zhu Y-C (2007). “The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation.” Cardiovascular Research 76(1): 29–40. [DOI] [PubMed] [Google Scholar]
- Campana D, Coustan-Smith E, Manabe A, Buschle M, Raimondi SC, Behm FG, Ashmun R, Arico M, Biondi A and Pui CH (1993). “Prolonged survival of B-lineage acute lymphoblastic leukemia cells is accompanied by overexpression of bcl-2 protein.” Blood 81(4): 1025–1031. [PubMed] [Google Scholar]
- Cardounel A, Julian D and Predmore B (2011). “Hydrogen Sulfide Increases Nitric Oxide Production from Endothelial Cells by an Akt-Dependent Mechanism.” Frontiers in Physiology 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington SJ, Chakraborty I, Bernard JML and Mascharak PK (2014). “Synthesis and Characterization of a “Turn-On” photoCORM for Trackable CO Delivery to Biological Targets.” ACS Medicinal Chemistry Letters 5(12): 1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington SJ, Chakraborty I and Mascharak PK (2013). “Rapid CO release from a Mn(i) carbonyl complex derived from azopyridine upon exposure to visible light and its phototoxicity toward malignant cells.” Chemical Communications 49(96): 11254–11256. [DOI] [PubMed] [Google Scholar]
- Chakraborty I, Carrington SJ, Hauser J, Oliver SRJ and Mascharak PK (2015). “Rapid Eradication of Human Breast Cancer Cells through Trackable Light-Triggered CO Delivery by Mesoporous Silica Nanoparticles Packed with a Designed photoCORM.” Chemistry of Materials 27(24): 8387–8397. [Google Scholar]
- Chakraborty P, Parikh RY, Choi S, Tran D, Gooz M, Hedley ZT, Kim D-S, Pytel D, Kang I, Nadig SN, Beeson GC, Ball L, Mehrotra M, Wang H, Berto S, Palanisamy V, Li H, Chatterjee S, Rodriguez PC, Maldonado EN, Diehl JA, Gangaraju VK and Mehrotra S (2022). “Carbon Monoxide Activates PERK-Regulated Autophagy to Induce Immunometabolic Reprogramming and Boost Antitumor T-cell Function.” Cancer Research 82(10): 1969–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Duvalsaint PL and Kashfi K (2016). “Gastrointestinal safety, chemotherapeutic potential, and classic pharmacological profile of NOSH-naproxen (AVT-219) a dual NO- and H2S-releasing hybrid.” Pharmacol Res Perspect 4(2): e00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D and Kashfi K (2012). “Hydrogen sulfide-releasing aspirin suppresses NF-κB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo.” Biochemical Pharmacology 83(6): 723–732. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Nath N, Dastagirzada YM, Velázquez-Martínez CA, Boring D and Kashfi K (2012). “Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: A general property and evidence of a tissue type-independent effect.” Biochemical Pharmacology 83(6): 715–722. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Olson KR and Kashfi K (2012). “NOSH–aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model.” Biochemical and Biophysical Research Communications 419(3): 523–528. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Santiago G, Le TTC, Nath N and Kashfi K (2020). “NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS.” Biochem Pharmacol 176: 113857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HH, Zhou XL, Shi YL and Yang J (2013). “Roles of p38 MAPK and JNK in TGF-β1-induced human alveolar epithelial to mesenchymal transition.” Arch Med Res 44(2): 93–98. [DOI] [PubMed] [Google Scholar]
- Chen J. x. and Meyrick B (2004). “Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium.” Laboratory Investigation 84(2): 182–190. [DOI] [PubMed] [Google Scholar]
- Chen M-L, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H and Khazaie K (2005). “Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo.” Proceedings of the National Academy of Sciences 102(2): 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H and Khazaie K (2005). “Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo.” Proc Natl Acad Sci U S A 102(2): 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Gan G, Shen Z, Gao L, Zhang G and Hu J (2021). “Red Light-Triggered Intracellular Carbon Monoxide Release Enables Selective Eradication of MRSA Infection.” Angewandte Chemie International Edition 60(24): 13513–13520. [DOI] [PubMed] [Google Scholar]
- Chiesa JJ, Baidanoff FM and Golombek DA (2018). “Don’t just say no: Differential pathways and pharmacological responses to diverse nitric oxide donors.” Biochemical Pharmacology 156: 1–9. [DOI] [PubMed] [Google Scholar]
- Chin K, Kurashima Y, Ogura T, Tajiri H, Yoshida S and Esumi H (1997). “Induction of vascular endothelial growth factor by nitric oxide in human glioblastoma and hepatocellular carcinoma cells.” Oncogene 15(4): 437–442. [DOI] [PubMed] [Google Scholar]
- Choi S, Kim J, Kim J-H, Lee D-K, Park W, Park M, Kim S, Hwang JY, Won M-H, Choi YK, Ryoo S, Ha K-S, Kwon Y-G and Kim Y-M (2017). “Carbon monoxide prevents TNF-α-induced eNOS downregulation by inhibiting NF-κB-responsive miR-155–5p biogenesis.” Experimental & Molecular Medicine 49(11): e403–e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Kim CK, Lee H, Jeoung D, Ha KS, Kwon YG, Kim KW and Kim YM (2010). “Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein.” J Biol Chem 285(42): 32116–32125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Anand K, Anselme AC, Chan AA, Gupta N, Venta LA, Schwartz MR, Qian W, Xu Y, Zhang L, Kuhn J, Patel T, Rodriguez AA, Belcheva A, Darcourt J, Ensor J, Bernicker E, Pan P-Y, Chen SH, Lee DJ, Niravath PA and Chang JC “A phase 1/2 clinical trial of the nitric oxide synthase inhibitor L-NMMA and taxane for treating chemoresistant triple-negative breast cancer.” Science Translational Medicine 13(624): eabj5070. [DOI] [PubMed] [Google Scholar]
- Chung P, Cook T, Liu K, Vodovotz Y, Zamora R, Finkelstein S, Billiar T and Blumberg D (2003). “Overexpression of the human inducible nitric oxide synthase gene enhances radiation-induced apoptosis in colorectal cancer cells via a caspase-dependent mechanism.” Nitric Oxide 8(2): 119–126. [DOI] [PubMed] [Google Scholar]
- Cianchi F, Cortesini C, Fantappiè O, Messerini L, Schiavone N, Vannacci A, Nistri S, Sardi I, Baroni G, Marzocca C, Perna F, Mazzanti R, Bechi P and Masini E (2003). “Inducible Nitric Oxide Synthase Expression in Human Colorectal Cancer: Correlation with Tumor Angiogenesis.” The American Journal of Pathology 162(3): 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citi V, Piragine E, Pagnotta E, Ugolini L, Di Cesare Mannelli L, Testai L, Ghelardini C, Lazzeri L, Calderone V and Martelli A (2019). “Anticancer properties of erucin, an H2S-releasing isothiocyanate, on human pancreatic adenocarcinoma cells (AsPC-1).” Phytotherapy Research 33(3): 845–855. [DOI] [PubMed] [Google Scholar]
- Colasanti M, Persichini T, Venturini G and Ascenzi P (1999). “S-nitrosylation of viral proteins: molecular bases for antiviral effect of nitric oxide.” IUBMB Life 48(1): 25–31. [DOI] [PubMed] [Google Scholar]
- Coletta C, Módis K, Szczesny B, Brunyánszki A, Oláh G, Rios EC, Yanagi K, Ahmad A, Papapetropoulos A and Szabo C (2015). “Regulation of Vascular Tone, Angiogenesis and Cellular Bioenergetics by the 3-Mercaptopyruvate Sulfurtransferase/H2S Pathway: Functional Impairment by Hyperglycemia and Restoration by DL-α-Lipoic Acid.” Mol Med 21(1): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E and Szabo C (2012). “Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation.” Proc Natl Acad Sci U S A 109(23): 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T, Wang Z, Alber S, Liu K, Watkins SC, Vodovotz Y, Billiar TR and Blumberg D (2004). “Nitric Oxide and Ionizing Radiation Synergistically Promote Apoptosis and Growth Inhibition of Cancer by Activating p53.” Cancer Research 64(21): 8015–8021. [DOI] [PubMed] [Google Scholar]
- Cortese-Krott MM, Fernandez BO, Santos JLT, Mergia E, Grman M, Nagy P, Kelm M, Butler A and Feelisch M (2014). “Nitrosopersulfide (SSNO−) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide.” Redox Biology 2: 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese-Krott MM, Kuhnle GGC, Dyson A, Fernandez BO, Grman M, DuMond JF, Barrow MP, McLeod G, Nakagawa H, Ondrias K, Nagy P, King SB, Saavedra JE, Keefer LK, Singer M, Kelm M, Butler AR and Feelisch M (2015). “Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl.” Proceedings of the National Academy of Sciences 112(34): E4651–E4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotta E and Koshland DE Jr. (1992). “NO news is good news.” Science 258(5090): 1862–1865. [DOI] [PubMed] [Google Scholar]
- Czikora Á, Erdélyi K, Ditrói T, Szántó N, Jurányi EP, Szanyi S, Tóvári J, Strausz T and Nagy P (2022). “Cystathionine β-synthase overexpression drives metastatic dissemination in pancreatic ductal adenocarcinoma via inducing epithelial-to-mesenchymal transformation of cancer cells.” Redox Biol 57: 102505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Chen H, Pan Y and Song H (2022). “Carbon Monoxide-Releasing Molecule-3 Suppresses the Malignant Biological Behavior of Tongue Squamous Cell Carcinoma via Regulating Keap1/Nrf2/HO-1 Signaling Pathway.” Biomed Res Int 2022: 9418332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal-Secco D, Cunha TM, Freitas A, Alves-Filho JC, Souto F. c. O., Fukada SY, Grespan R, Alencar NMN, Neto AF, Rossi MA, Ferreira S. r. H., Hothersall JSand Cunha FQ(2008). “Hydrogen Sulfide Augments Neutrophil Migration through Enhancement of Adhesion Molecule Expression and Prevention of CXCR2 Internalization: Role of ATP-Sensitive Potassium Channels1.” The Journal of Immunology 181(6): 4287–4298. [DOI] [PubMed] [Google Scholar]
- Danai PA, Moss M, Mannino DM and Martin GS (2006). “The epidemiology of sepsis in patients with malignancy.” Chest 129(6): 1432–1440. [DOI] [PubMed] [Google Scholar]
- Dangel O, Mergia E, Karlisch K, Groneberg D, Koesling D and Friebe A (2010). “Nitric oxide-sensitive guanylyl cyclase is the only nitric oxide receptor mediating platelet inhibition.” J Thromb Haemost 8(6): 1343–1352. [DOI] [PubMed] [Google Scholar]
- Darwish I, Miller C, Kain KC and Liles WC (2012). “Inhaled nitric oxide therapy fails to improve outcome in experimental severe influenza.” Int J Med Sci 9(2): 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidge KS, Sanguinetti G, Yee CH, Cox AG, McLeod CW, Monk CE, Mann BE, Motterlini R and Poole RK (2009). “Carbon Monoxide-releasing Antibacterial Molecules Target Respiration and Global Transcriptional Regulators * .” Journal of Biological Chemistry 284(7): 4516–4524. [DOI] [PubMed] [Google Scholar]
- De Cicco P, Ercolano G, Rubino V, Terrazzano G, Ruggiero G, Cirino G and Ianaro A (2020). “Modulation of the functions of myeloid-derived suppressor cells : a new strategy of hydrogen sulfide anti-cancer effects.” British Journal of Pharmacology 177(4): 884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cicco P, Panza E, Armogida C, Ercolano G, Taglialatela-Scafati O, Shokoohinia Y, Camerlingo R, Pirozzi G, Calderone V, Cirino G and Ianaro A (2017). “The Hydrogen Sulfide Releasing Molecule Acetyl Deacylasadisulfide Inhibits Metastatic Melanoma.” Frontiers in Pharmacology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cicco P, Sanders T, Cirino G, Maloy KJ and Ianaro A (2018). “Hydrogen Sulfide Reduces Myeloid-Derived Suppressor Cell-Mediated Inflammatory Response in a Model of Helicobacter hepaticus-Induced Colitis.” Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groote MA, Granger D, Xu Y, Campbell G, Prince R and Fang FC (1995). “Genetic and redox determinants of nitric oxide cytotoxicity in a Salmonella typhimurium model.” Proc Natl Acad Sci U S A 92(14): 6399–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Preter G, Deriemaeker C, Danhier P, Brisson L, Cao Pham TT, Grégoire V, Jordan BF, Sonveaux P and Gallez B (2016). “A Fast Hydrogen Sulfide–Releasing Donor Increases the Tumor Response to Radiotherapy.” Molecular Cancer Therapeutics 15(1): 154–161. [DOI] [PubMed] [Google Scholar]
- De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, Melani C, Guiducci C, Colombo MP, Iezzi M, Musiani P, Zanovello P and Bronte V (2005). “Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination.” Proc Natl Acad Sci U S A 102(11): 4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos J, Thykjaer T, Tarte K, Ensslen M, Raynaud P, Requirand G, Pellet F, Pantesco V, Reme T, Jourdan M, Rossi JF, Orntoft T and Klein B (2002). “Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays.” Oncogene 21(44): 6848–6857. [DOI] [PubMed] [Google Scholar]
- Deppisch C, Herrmann G, Graepler-Mainka U, Wirtz H, Heyder S, Engel C, Marschal M, Miller CC and Riethmüller J (2016). “Gaseous nitric oxide to treat antibiotic resistant bacterial and fungal lung infections in patients with cystic fibrosis: a phase I clinical study.” Infection 44(4): 513–520. [DOI] [PubMed] [Google Scholar]
- Desmard M, Amara N, Lanone S, Motterlini R and Boczkowski J (2005). “Carbon monoxide reduces the expression and activity of matrix metalloproteinases 1 and 2 in alveolar epithelial cells.” Cell Mol Biol (Noisy-le-grand) 51(4): 403–408. [PubMed] [Google Scholar]
- Desmard M, Davidge KS, Bouvet O, Morin D, Roux D, Foresti R, Ricard JD, Denamur E, Poole RK, Montravers P, Morterlini R and Boczkowski J (2009). “A carbon monoxide-releasing molecule (CORM-3) exerts bactericidal activity against Pseudomonas aeruginosa and improves survival in an animal model of bacteraemia.” The FASEB Journal 23(4): 1023–1031. [DOI] [PubMed] [Google Scholar]
- Devadas K and Dhawan S (2006). “Hemin Activation Ameliorates HIV-1 Infection via Heme Oxygenase-1 Induction.” The Journal of Immunology 176(7): 4252–4257. [DOI] [PubMed] [Google Scholar]
- Dilek N, Papapetropoulos A, Toliver-Kinsky T and Szabo C (2020). “Hydrogen sulfide: An endogenous regulator of the immune system.” Pharmacological Research 161: 105119. [DOI] [PubMed] [Google Scholar]
- Dillekås H, Rogers MS and Straume O (2019). “Are 90% of deaths from cancer caused by metastases?” Cancer Med 8(12): 5574–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R and Zeiher AM (1999). “Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation.” Nature 399(6736): 601–605. [DOI] [PubMed] [Google Scholar]
- Diotallevi M, Checconi P, Palamara AT, Celestino I, Coppo L, Holmgren A, Abbas K, Peyrot F, Mengozzi M and Ghezzi P (2017). “Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties.” Frontiers in Immunology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Q, Wang X, Hu X, Xiao L, Zhang L, Song L, Xu M, Zou Y, Chen L, Chen Z and Tan W (2018). “Simultaneous Application of Photothermal Therapy and an Anti-inflammatory Prodrug using Pyrene-Aspirin-Loaded Gold Nanorod Graphitic Nanocapsules.” Angew Chem Int Ed Engl 57(1): 177–181. [DOI] [PubMed] [Google Scholar]
- Dong Q, Yang B, Han J-G, Zhang M-M, Liu W, Zhang X, Yu H-L, Liu Z-G, Zhang S-H, Li T, Wu D-D, Ji X-Y and Duan S-F (2019). “A novel hydrogen sulfide-releasing donor, HA-ADT, suppresses the growth of human breast cancer cells through inhibiting the PI3K/AKT/mTOR and Ras/Raf/MEK/ERK signaling pathways.” Cancer Letters 455: 60–72. [DOI] [PubMed] [Google Scholar]
- Dong Y, Sui L, Sugimoto K, Tai Y and Tokuda M (2001). “Cyclin D1-CDK4 complex, a possible critical factor for cell proliferation and prognosis in laryngeal squamous cell carcinomas.” International Journal of Cancer 95(4): 209–215. [DOI] [PubMed] [Google Scholar]
- Dong Z, Staroselsky AH, Qi X, Xie K and Fidler IJ (1994). “Inverse Correlation between Expression of Inducible Nitric Oxide Synthase Activity and Production of Metastasis in K-1735 Murine Melanoma Cells1.” Cancer Research 54(3): 789–793. [PubMed] [Google Scholar]
- Douguet L, Bod L, Lengagne R, Labarthe L, Kato M, Avril M-F and Prévost-Blondel A (2016). “Nitric oxide synthase 2 is involved in the pro-tumorigenic potential of γδ17 T cells in melanoma.” OncoImmunology 5(8): e1208878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachuk KO, Kotsjuruba AV and Sagach VF (2015). “[HYDROGEN SULFIDE DONOR, NAHS, RECOVERS CONSTITUTIVE NO SYNTHESIS AND ENDOTHELIUM-DEPENDENT RELAXATION OF ISOLATED AORTA IN OLD RATS].” Fiziol Zh 61(6): 3–10. [DOI] [PubMed] [Google Scholar]
- Dueñas-Gonzalez A, Isales CM, del Mar Abad-Hernandez M, Gonzalez-Sarmiento R, Sangueza O and Rodriguez-Commes J (1997). “Expression of inducible nitric oxide synthase in breast cancer correlates with metastatic disease.” Mod Pathol 10(7): 645–649. [PubMed] [Google Scholar]
- Dufait I, Schwarze JK, Liechtenstein T, Leonard W, Jiang H, Escors D, De Ridder M and Breckpot K (2015). “Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer.” Oncotarget 6(14): 12369–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt W, Akool ELS, Rebhan J. r., Frank S, Beck K-F, Franzen R, Hamada FMA and Pfeilschifter J(2002). “Inhibition of Cytokine-induced Matrix Metalloproteinase 9 Expression by Peroxisome Proliferator-activated Receptor α Agonists Is Indirect and Due to a NO-mediated Reduction of mRNA Stability*.” Journal of Biological Chemistry 277(36): 33518–33528. [DOI] [PubMed] [Google Scholar]
- El Hasasna H, Saleh A, Samri HA, Athamneh K, Attoub S, Arafat K, Benhalilou N, Alyan S, Viallet J, Dhaheri YA, Eid A and Iratni R (2016). “Rhus coriaria suppresses angiogenesis, metastasis and tumor growth of breast cancer through inhibition of STAT3, NFκB and nitric oxide pathways.” Scientific Reports 6(1): 21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gendy MMAA, El-Bondkly AMA, Keera AA and Ali AM (2018). “Incidence of Methicillin-Resistant Staphylococcus aureus (MRSA) in Microbial Community of Cancer Patients and Evaluation of Their Resistant Pattern.” Arabian Journal for Science and Engineering 43(1): 83–92. [Google Scholar]
- Erickson PF, Maxwell IH, Su LJ, Baumann M and Glode LM (1990). “Sequence of cDNA for rat cystathionine gamma-lyase and comparison of deduced amino acid sequence with related Escherichia coli enzymes.” Biochem J 269(2): 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito I, Kleeff J, Abiatari I, Shi X, Giese N, Bergmann F, Roth W, Friess H and Schirmacher P (2007). “Overexpression of cellular inhibitor of apoptosis protein 2 is an early event in the progression of pancreatic cancer.” Journal of Clinical Pathology 60(8): 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TG, Thai L, Granger DL and Hibbs JB Jr. (1993). “Effect of in vivo inhibition of nitric oxide production in murine leishmaniasis.” J Immunol 151(2): 907–915. [PubMed] [Google Scholar]
- Fabbri F, Brigliadori G, Ulivi P, Tesei A, Vannini I, Rosetti M, Bravaccini S, Amadori D, Bolla M and Zoli W (2005). “Pro-apoptotic effect of a nitric oxide-donating NSAID, NCX 4040, on bladder carcinoma cells.” Apoptosis 10(5): 1095–1103. [DOI] [PubMed] [Google Scholar]
- Fang FC (1997). “Perspectives series: host/pathogen interactions. Mechanisms of nitric oxiderelated antimicrobial activity.” J Clin Invest 99(12): 2818–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Qin H, Nakamura H, Tsukigawa K, Shin T and Maeda H (2012). “Carbon monoxide, generated by heme oxygenase-1, mediates the enhanced permeability and retention effect in solid tumors.” Cancer Sci 103(3): 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L-P, Lin Q, Tang C-S and Liu X-M (2010). “Hydrogen sulfide attenuates epithelial- mesenchymal transition of human alveolar epithelial cells.” Pharmacological Research 61(4): 298–305. [DOI] [PubMed] [Google Scholar]
- Farkona S, Diamandis EP and Blasutig IM (2016). “Cancer immunotherapy: the beginning of the end of cancer?” BMC Medicine 14(1): 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayad-Kobeissi S, Ratovonantenaina J, Dabiré H, Wilson JL, Rodriguez AM, Berdeaux A, Dubois-Randé JL, Mann BE, Motterlini R and Foresti R (2016). “Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule.” Biochem Pharmacol 102: 64–77. [DOI] [PubMed] [Google Scholar]
- Fernald K and Kurokawa M (2013). “Evading apoptosis in cancer.” Trends in Cell Biology 23(12): 620–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando M, Gueron G, Elguero B, Giudice J, Salles A, Leskow FC, Jares-Erijman EA, Colombo L, Meiss R, Navone N, De Siervi A and Vazquez E (2011). “Heme oxygenase 1 (HO-1) challenges the angiogenic switch in prostate cancer.” Angiogenesis 14(4): 467–479. [DOI] [PubMed] [Google Scholar]
- Fonseca MD, Cunha FQ, Kashfi K and Cunha TM (2015). “NOSH-aspirin (NBS-1120), a dual nitric oxide and hydrogen sulfide-releasing hybrid, reduces inflammatory pain.” Pharmacol Res Perspect 3(3): e00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresti R, Hammad J, Clark JE, Johnson TR, Mann BE, Friebe A, Green CJ and Motterlini R (2004). “Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule.” British Journal of Pharmacology 142(3): 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresti R and Motterlini R (1999). “The heme oxygenase pathway and its interaction with nitric oxide in the control of cellular homeostasis.” Free Radic Res 31(6): 459–475. [DOI] [PubMed] [Google Scholar]
- Foresti R, Sarathchandra P, Clark JE, Green CJ and Motterlini R (1999). “Peroxynitrite induces haem oxygenase-1 in vascular endothelial cells: a link to apoptosis.” Biochemical Journal 339(3): 729–736. [PMC free article] [PubMed] [Google Scholar]
- Fouad YA and Aanei C (2017). “Revisiting the hallmarks of cancer.” Am J Cancer Res 7(5): 1016–1036. [PMC free article] [PubMed] [Google Scholar]
- Fraisl P, Mazzone M, Schmidt T and Carmeliet P (2009). “Regulation of Angiogenesis by Oxygen and Metabolism.” Developmental Cell 16(2): 167–179. [DOI] [PubMed] [Google Scholar]
- Friebe A and Koesling D (2003). “Regulation of nitric oxide-sensitive guanylyl cyclase.” Circ Res 93(2): 96–105. [DOI] [PubMed] [Google Scholar]
- Fu L-H, Wei Z-Z, Hu K-D, Hu L-Y, Li Y-H, Chen X-Y, Han Z, Yao G-F and Zhang H (2018). “Hydrogen sulfide inhibits the growth of Escherichia coli through oxidative damage.” Journal of Microbiology 56(4): 238–245. [DOI] [PubMed] [Google Scholar]
- Fu LH, Hu KD, Hu LY, Li YH, Hu LB, Yan H, Liu YS and Zhang H (2014). “An antifungal role of hydrogen sulfide on the postharvest pathogens Aspergillus niger and Penicillium italicum.” PLoS One 9(8): e104206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K.-i. and Nagai R (2004). “Carbon Monoxide Protects Against Cardiac Ischemia—Reperfusion Injury In Vivo via MAPK and Akt—eNOS Pathways.” Arteriosclerosis, Thrombosis, and Vascular Biology 24(10): 1848–1853. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Ignarro LJ, Nagy P, Wink DA, Kevil CG, Feelisch M, CorteseKrott MM, Bianco CL, Kumagai Y, Hobbs AJ, Lin J, Ida T and Akaike T (2018). “Biological hydropersulfides and related polysulfides – a new concept and perspective in redox biology.” FEBS Letters 592(12): 2140–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo O, Fini-Storchi I, Vergari WA, Masini E, Morbidelli L, Ziche M and Franchi A (1998). “Role of Nitric Oxide in Angiogenesis and Tumor Progression in Head and Neck Cancer.” JNCI: Journal of the National Cancer Institute 90(8): 587–596. [DOI] [PubMed] [Google Scholar]
- Gao J, Liu X and Rigas B (2005). “Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress.” Proc Natl Acad Sci U S A 102(47): 17207–17212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Cheng J, Shen Z, Zhang G, Liu S and Hu J (2022). “Orchestrating Nitric Oxide and Carbon Monoxide Signaling Molecules for Synergistic Treatment of MRSA Infections.” Angewandte Chemie International Edition 61(3): e202112782. [DOI] [PubMed] [Google Scholar]
- Gao L and Williams JL (2012). “Nitric oxide-donating aspirin induces G2/M phase cell cycle arrest in human cancer cells by regulating phase transition proteins.” Int J Oncol 41(1): 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhou S, Xu Y, Sheng S, Qian SY and Huo X (2019). “Nitric oxide synthase inhibitors 1400W and L-NIO inhibit angiogenesis pathway of colorectal cancer.” Nitric Oxide 83: 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehad AE, Lichtman MK, Schmults CD, Teague JE, Calarese AW, Jiang Y, Watanabe R and Clark RA (2012). “Nitric Oxide–Producing Myeloid-Derived Suppressor Cells Inhibit Vascular E-Selectin Expression in Human Squamous Cell Carcinomas.” Journal of Investigative Dermatology 132(11): 2642–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller DA and Billiar TR (1998). “Molecular biology of nitric oxide synthases.” Cancer Metastasis Rev 17(1): 7–23. [DOI] [PubMed] [Google Scholar]
- Gessner G, Sahoo N, Swain SM, Hirth G, Schönherr R, Mede R, Westerhausen M, Brewitz HH, Heimer P, Imhof D, Hoshi T and Heinemann SH (2017). “CO-independent modification of K(+) channels by tricarbonyldichlororuthenium(II) dimer (CORM-2).” Eur J Pharmacol 815: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemi A and Kashfi K (2022). Nitric oxide: A brief history of discovery and timeline of its research. The role of nitric oxide in type 2 diabetes. Ghasemi BZ A.Kashfi K Singapore, Bentham Science Publishers Ltd: 27–38. [Google Scholar]
- Giampaoli S, Valeriani F, Gianfranceschi G, Vitali M, Delfini M, Festa MR, Bottari E and Romano Spica V (2013). “Hydrogen sulfide in thermal spring waters and its action on bacteria of human origin.” Microchemical Journal 108: 210–214. [Google Scholar]
- Giri S, Rattan R, Deshpande M, Maguire JL, Johnson Z, Graham RP and Shridhar V (2014). “Preclinical Therapeutic Potential of a Nitrosylating Agent in the Treatment of Ovarian Cancer.” PLOS ONE 9(6): e97897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globus O, Sagie S, Lavine N, Barchana DI and Urban D (2021). “Early mortality in metastatic cancer: A SEER population data analysis.” Journal of Clinical Oncology 39(15_suppl): e18789–e18789. [Google Scholar]
- Goligorsky MS, Brodsky SV and Noiri E (2004). “NO bioavailability, endothelial dysfunction, and acute renal failure: new insights into pathophysiology.” Semin Nephrol 24(4): 316–323. [DOI] [PubMed] [Google Scholar]
- Gould SJ, Keller GA and Subramani S (1988). “Identification of peroxisomal targeting signals located at the carboxy terminus of four peroxisomal proteins.” J Cell Biol 107(3): 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granados-Principal S, Liu Y, Guevara ML, Blanco E, Choi DS, Qian W, Patel T, Rodriguez AA, Cusimano J, Weiss HL, Zhao H, Landis MD, Dave B, Gross SS and Chang JC (2015). “Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer.” Breast Cancer Res 17: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumbach IM, Chen W, Mertens SA and Harrison DG (2005). “A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription.” J Mol Cell Cardiol 39(4): 595–603. [DOI] [PubMed] [Google Scholar]
- Guan Q, Zhou L-L, Li Y-A and Dong Y-B (2019). “A nanoscale metal–organic framework for combined photodynamic and starvation therapy in treating breast tumors.” Chemical Communications 55(99): 14898–14901. [DOI] [PubMed] [Google Scholar]
- Guo H, Gai JW, Wang Y, Jin HF, Du JB and Jin J (2012). “Characterization of hydrogen sulfide and its synthases, cystathionine beta-synthase and cystathionine gamma-lyase, in human prostatic tissue and cells.” Urology 79(2): 483 e481–485. [DOI] [PubMed] [Google Scholar]
- Guo L, Peng W, Tao J, Lan Z, Hei H, Tian L, Pan W, Wang L and Zhang X (2016). “Hydrogen Sulfide Inhibits Transforming Growth Factor-β1-Induced EMT via Wnt/Catenin Pathway.” PLOS ONE 11(1): e0147018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I and Nudler E (2005). “NO-mediated cytoprotection: Instant adaptation to oxidative stress in bacteria.” Proceedings of the National Academy of Sciences 102(39): 13855–13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I, Shatalin K, Starodubtseva M and Nudler E (2009). “Endogenous Nitric Oxide Protects Bacteria Against a Wide Spectrum of Antibiotics.” Science 325(5946): 1380–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, West NEJ, Pillai R, Taggart DP and Channon KM (2002). “Nitric Oxide Modulates Superoxide Release and Peroxynitrite Formation in Human Blood Vessels.” Hypertension 39(6): 1088–1094. [DOI] [PubMed] [Google Scholar]
- Hadler-Olsen E, Winberg J-O and Uhlin-Hansen L (2013). “Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets.” Tumor Biology 34(4): 2041–2051. [DOI] [PubMed] [Google Scholar]
- Hamidizad Z, Kadkhodaee M, Karimian SM, Ranjbaran M, Heidari F, Bakhshi E, Kianian F, Zahedi E and Seifi B (2022). “Therapeutic effects of CORM3 and NaHS in chronic kidney disease induced cognitive impairment via the interaction between carbon monoxide and hydrogen sulfide on Nrf2/HO-1 signaling pathway in rats.” Chemico-Biological Interactions 368: 110217. [DOI] [PubMed] [Google Scholar]
- Han Y, Zeng F, Tan G, Yang C, Tang H, Luo Y, Feng J, Xiong H and Guo Q (2013). “Hydrogen Sulfide Inhibits Abnormal Proliferation of Lymphocytes via AKT/GSK3β Signal Pathway in Systemic Lupus Erythematosus Patients.” Cellular Physiology and Biochemistry 31(6): 795–804. [DOI] [PubMed] [Google Scholar]
- Hanahan D and Weinberg Robert A. (2011). “Hallmarks of Cancer: The Next Generation.” Cell 144(5): 646–674. [DOI] [PubMed] [Google Scholar]
- Hannen R and Bartsch JW (2018). “Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis.” FEBS Letters 592(12): 2023–2031. [DOI] [PubMed] [Google Scholar]
- Harada K, Supriatno, Kawaguchi S-I, Tomitaro O, Yoshida H and Sato M (2004). “Overexpression of iNOS Gene Suppresses the Tumorigenicity and Metastasis of Oral Cancer Cells.” In Vivo 18(4): 449. [PubMed] [Google Scholar]
- Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, Davey S, Squire J, Park PC and Feilotter H (2012). “EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer.” BMC Cancer 12: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Kasai K and Gross SS (2004). “NO suppresses while peroxynitrite sustains NF-kappaB: a paradigm to rationalize cytoprotective and cytotoxic actions attributed to NO.” Cardiovasc Res 63(1): 31–40. [DOI] [PubMed] [Google Scholar]
- He D, Liu F, Cui S, Jiang N, Yu H, Zhou Y, Liu Y and Kou X (2020). “Mechanical load-induced H2S production by periodontal ligament stem cells activates M1 macrophages to promote bone remodeling and tooth movement via STAT1.” Stem Cell Research & Therapy 11(1): 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke JL, Ridnour LA, Cheng RY, Switzer CH, Lizardo MM, Khanna C, Glynn SA, Hussain SP, Young HA, Ambs S and Wink DA (2014). “Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression.” Proc Natl Acad Sci U S A 111(17): 6323–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich MR, Coletta C, Chao C and Szabo C (2015). “The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer.” Antioxid Redox Signal 22(5): 424–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich MR and Szabo C (2015). “Hydrogen Sulfide and Cancer.” Handb Exp Pharmacol 230: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley MK, Donnelly JP, Carlton EF and Prescott HC (2019). “Epidemiology and Outcomes of Cancer-Related Versus Non-Cancer-Related Sepsis Hospitalizations.” Crit Care Med 47(10): 1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez JM, Farma JM, Coppola D, Hakam A, Fulp WJ, Chen D-T, Siegel EM, Yeatman TJ and Shibata D (2011). “Expression of the Antiapoptotic Protein Survivin in Colon Cancer.” Clinical Colorectal Cancer 10(3): 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickok JR, Sahni S, Mikhed Y, Bonini MG and Thomas DD (2011). “Nitric Oxide Suppresses Tumor Cell Migration through N-Myc Downstream-regulated Gene-1 (NDRG1) Expression: ROLE OF CHELATABLE IRON *.” Journal of Biological Chemistry 286(48): 41413–41424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hientz K, Mohr A, Bhakta-Guha D and Efferth T (2017). “The role of p53 in cancer drug resistance and targeted chemotherapy.” Oncotarget 8(5): 8921–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Trevino-Villarreal JH, Mejia P, Ozaki CK, Wang R, Gladyshev VN, Madeo F, Mair WB and Mitchell JR (2015). “Endogenous hydrogen sulfide production is essential for dietary restriction benefits.” Cell 160(1–2): 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Hosoi A, Matsushita H, Iino T, Ueha S, Matsushima K, Seto Y and Kakimi K (2015). “The nitric oxide radical scavenger carboxy-PTIO reduces the immunosuppressive activity of myeloid-derived suppressor cells and potentiates the antitumor activity of adoptive cytotoxic T lymphocyte immunotherapy.” OncoImmunology 4(8): e1019195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka K, Miyamoto M, Cho Y, Suzuoki M, Oshikiri T, Nakakubo Y, Itoh T, Ohbuchi T, Kondo S and Katoh H (2006). “Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma.” British Journal of Cancer 94(2): 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogaboam CM, Befus AD and Wallace JL (1993). “Modulation of rat mast cell reactivity by IL-1 beta. Divergent effects on nitric oxide and platelet-activating factor release.” J Immunol 151(7): 3767–3774. [PubMed] [Google Scholar]
- Hopper CP, Zambrana PN, Goebel U and Wollborn J (2021). “A brief history of carbon monoxide and its therapeutic origins.” Nitric Oxide 111–112: 45–63. [DOI] [PubMed] [Google Scholar]
- Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, Shen J, Cai L, Cai X and Chen M (2021). “Targeting mutant p53 for cancer therapy: direct and indirect strategies.” Journal of Hematology & Oncology 14(1): 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CW, Feng W, Peh MT, Peh K, Dymock BW and Moore PK (2016). “A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo.” Pharmacological Research 113: 533–546. [DOI] [PubMed] [Google Scholar]
- Huang J, Yang B, Xiang T, Peng W, Qiu Z, Wan J, Zhang L, Li H, Li H and Ren G (2015). “Diallyl disulfide inhibits growth and metastatic potential of human triple-negative breast cancer cells through inactivation of the β-catenin signaling pathway.” Molecular Nutrition & Food Research 59(6): 1063–1075. [DOI] [PubMed] [Google Scholar]
- Huang JQ, Sridhar S, Chen Y and Hunt RH (1998). “Meta-analysis of the relationship between helicobacter pylori seropositivity and gastric cancer.” Gastroenterology 114(6): 1169–1179. [DOI] [PubMed] [Google Scholar]
- Huerta-Yepez S, Vega M, Jazirehi A, Garban H, Hongo F, Cheng G and Bonavida B (2004). “Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-κB and inhibition of Bcl-xL expression.” Oncogene 23(29): 4993–5003. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ (1989). “Endothelium-derived nitric oxide: actions and properties.” FASEB J 3(1): 31–36. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE and Chaudhuri G (1987). “Endotheliumderived relaxing factor produced and released from artery and vein is nitric oxide.” Proc Natl Acad Sci U S A 84(24): 9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingi T, Cheng J and Ronnett GV (1996). “Carbon Monoxide: An Endogenous Modulator of the Nitric Oxide–Cyclic GMP Signaling System.” Neuron 16(4): 835–842. [DOI] [PubMed] [Google Scholar]
- Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA and Roberts DD (2005). “Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner.” Proceedings of the National Academy of Sciences 102(37): 13141–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Ando T, Ogiso H, Arioka Y and Seishima M (2015). “Inhibition of induced nitric oxide synthase enhances the anti-tumor effects on cancer immunotherapy using TLR7 agonist in mice.” Cancer Immunology, Immunotherapy 64(4): 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Ando T and Seishima M (2015). “Inhibition of iNOS activity enhances the anti-tumor effects of alpha-galactosylceramide in established murine cancer model.” Oncotarget 6(39): 41863–41874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, Hodder MC and Sansom OJ (2020). “WNT and β-Catenin in Cancer: Genes and Therapy.” Annual Review of Cancer Biology 4(1): 177–196. [Google Scholar]
- Jadeski LC and Lala PK (1999). “Nitric Oxide Synthase Inhibition by NG-Nitro-l-Arginine Methyl Ester Inhibits Tumor-Induced Angiogenesis in Mammary Tumors.” The American Journal of Pathology 155(4): 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani-Asl A and Bonni A (2013). “iNOS: a potential therapeutic target for malignant glioma.” Curr Mol Med 13(8): 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman P, Alfarano MG, Svider PF, Parikh F, Lu G, Kidwai S, Xiong H and Sikora AG (2014). “iNOS Expression in CD4+ T Cells Limits Treg Induction by Repressing TGFβ1: Combined iNOS Inhibition and Treg Depletion Unmask Endogenous Antitumor Immunity.” Clinical Cancer Research 20(24): 6439–6451. [DOI] [PubMed] [Google Scholar]
- Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, Chen S-H, Umansky VY and Sikora AG (2012). “Tumor-Expressed Inducible Nitric Oxide Synthase Controls Induction of Functional Myeloid-Derived Suppressor Cells through Modulation of Vascular Endothelial Growth Factor Release.” The Journal of Immunology 188(11): 5365–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC and Moncada S (1995). “Roles of nitric oxide in tumor growth.” Proc Natl Acad Sci U S A 92(10): 4392–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S-O, Pae H-O, Oh G-S, Jeong G-S, Lee B-S, Lee S, Kim DY, Rhew HY, Lee K-M and Chung H-T (2006). “Hydrogen sulfide potentiates interleukin-1β-induced nitric oxide production via enhancement of extracellular signal-regulated kinase activation in rat vascular smooth muscle cells.” Biochemical and Biophysical Research Communications 345(3): 938–944. [DOI] [PubMed] [Google Scholar]
- Jespersen C, Doller A, Akool E-S, Bachmann M, Müller R, Gutwein P, Mühl H, Pfeilschifter J and Eberhardt W (2009). “Molecular mechanisms of nitric oxide-dependent inhibition of TPA-induced matrix metalloproteinase-9 (MMP-9) in MCF-7 cells.” Journal of Cellular Physiology 219(2): 276–287. [DOI] [PubMed] [Google Scholar]
- Ji C, Si J, Xu Y, Zhang W, Yang Y, He X, Xu H, Mou X, Ren H and Guo H (2021). “Mitochondria-targeted and ultrasound-responsive nanoparticles for oxygen and nitric oxide codelivery to reverse immunosuppression and enhance sonodynamic therapy for immune activation.” Theranostics 11(17): 8587–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X and Wang B (2018). “Strategies toward Organic Carbon Monoxide Prodrugs.” Accounts of Chemical Research 51(6): 1377–1385. [DOI] [PubMed] [Google Scholar]
- Jia J, Ye T, Cui P, Hua Q, Zeng H and Zhao D (2016). “AP-1 transcription factor mediates VEGF-induced endothelial cell migration and proliferation.” Microvascular Research 105: 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Chan A, Ali S, Saha A, Haushalter KJ, Lam WL, Glasheen M, Parker J, Brenner M, Mahon SB, Patel HH, Ambasudhan R, Lipton SA, Pilz RB and Boss GR (2016). “Hydrogen Sulfide--Mechanisms of Toxicity and Development of an Antidote.” Sci Rep 6: 20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J, Freeman GJ, Brown MA, Wucherpfennig KW and Liu XS (2018). “Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response.” Nature Medicine 24(10): 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Wang H, Lou W, Jin S, Fan E, Li Y, Han D and Zhang L (2011). “Regulation of ciliary beat frequency by the nitric oxide signaling pathway in mouse nasal and tracheal epithelial cells.” Experimental Cell Research 317(17): 2548–2553. [DOI] [PubMed] [Google Scholar]
- Jin H. f., Du J.-b., Li X.-h., Wang Y.-f., Liang Y.-f.and Tang C.-s.(2006). “Interaction between hydrogen sulfide/cystathionine γ-lyase and carbon monoxide/heme oxygenase pathways in aortic smooth muscle cells.” Acta Pharmacologica Sinica 27(12): 1561–1566. [DOI] [PubMed] [Google Scholar]
- Jin Y-J, Park I, Hong I-K, Byun H-J, Choi J, Kim Y-M and Lee H (2011). “Fibronectin and vitronectin induce AP-1-mediated matrix metalloproteinase-9 expression through integrin α5β1/αvβ3-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells.” Cellular Signalling 23(1): 125–134. [DOI] [PubMed] [Google Scholar]
- Jones MK, Tsugawa K, Tarnawski AS and Baatar D (2004). “Dual actions of nitric oxide on angiogenesis: possible roles of PKC, ERK, and AP-1.” Biochemical and Biophysical Research Communications 318(2): 520–528. [DOI] [PubMed] [Google Scholar]
- Jordan KR, Amaria RN, Ramirez O, Callihan EB, Gao D, Borakove M, Manthey E, Borges VF and McCarter MD (2013). “Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients.” Cancer Immunology, Immunotherapy 62(11): 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowska H, Placha W, Nagahara N and Wrobel M (2011). “The expression and activity of cystathionine-gamma-lyase and 3-mercaptopyruvate sulfurtransferase in human neoplastic cell lines.” Amino Acids 41(1): 151–158. [DOI] [PubMed] [Google Scholar]
- Juszczak M, Kluska M, Wysokiński D and Woźniak K (2020). “DNA damage and antioxidant properties of CORM-2 in normal and cancer cells.” Sci Rep 10(1): 12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Yadav V and Banerjee R (2016). “Heme-dependent Metabolite Switching Regulates H2S Synthesis in Response to Endoplasmic Reticulum (ER) Stress * ♦.” Journal of Biological Chemistry 291(32): 16418–16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R and Weinberg RA (2009). “The basics of epithelial-mesenchymal transition.” The Journal of Clinical Investigation 119(6): 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson J, Pivodic A, Aguirre D and Schnitzer TJ (2009). “Efficacy, safety, and tolerability of the cyclooxygenase-inhibiting nitric oxide donator naproxcinod in treating osteoarthritis of the hip or knee.” J Rheumatol 36(6): 1290–1297. [DOI] [PubMed] [Google Scholar]
- Kashfi K (2018). “The dichotomous role of H(2)S in cancer cell biology? Déjà vu all over again.” Biochem Pharmacol 149: 205–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashfi K, Chattopadhyay M and Kodela R (2015). “NOSH-sulindac (AVT-18A) is a novel nitric oxide- and hydrogen sulfide-releasing hybrid that is gastrointestinal safe and has potent anti-inflammatory, analgesic, antipyretic, anti-platelet, and anti-cancer properties.” Redox Biol 6: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashfi K and Duvalsaint PL (2017). Nitric oxide donors and therapeutic applications in cancer. Nitric Oxide Donors: Novel Biomedical Applications. Seabra AB. Oxford, United Kingdom, Elsevier/Academic Press: 75–119. [Google Scholar]
- Kashfi K and Esmaili M (2017). NO-H2S-Releasing Chimeras as a Multifaceted Approach to Cancer Therapy. Cancer Sensitizing Agents for Chemotherapy; Nitric Oxide (Donor/Induced) in Chemosensitization. Bonavida B, Elsevier/Academic Press. 1: 105–142. [Google Scholar]
- Kashfi K and Olson KR (2012). “Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras.” Biochem Pharmacol 24(12): 019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashfi K and Patel KK (2022). “Carbon monoxide and its role in human physiology: A brief historical perspective.” Biochem Pharmacol 204: 115230. [DOI] [PubMed] [Google Scholar]
- Kashfi K, Rayyan Y, Qiao LL, Williams JL, Chen J, del Soldato P, Traganos F and Rigas B (2002). “Nitric Oxide-Donating Nonsteroidal Anti-Inflammatory Drugs Inhibit the Growth of Various Cultured Human Cancer Cells: Evidence of a Tissue Type-Independent Effect.” Journal of Pharmacology and Experimental Therapeutics 303(3): 1273. [DOI] [PubMed] [Google Scholar]
- Katsuyama K, Shichiri M, Marumo F and Hirata Y (1998). “NO inhibits cytokine-induced iNOS expression and NF-kappaB activation by interfering with phosphorylation and degradation of IkappaB-alpha.” Arterioscler Thromb Vasc Biol 18(11): 1796–1802. [DOI] [PubMed] [Google Scholar]
- Katsuyama K, Shichiri M, Marumo F and Hirata Y (1998). “NO Inhibits Cytokine-Induced iNOS Expression and NF-κB Activation by Interfering With Phosphorylation and Degradation of IκB-α.” Arteriosclerosis, Thrombosis, and Vascular Biology 18(11): 1796–1802. [DOI] [PubMed] [Google Scholar]
- Kawahara B, Faull KF, Janzen C and Mascharak PK (2021). “Carbon Monoxide Inhibits Cytochrome P450 Enzymes CYP3A4/2C8 in Human Breast Cancer Cells, Increasing Sensitivity to Paclitaxel.” Journal of Medicinal Chemistry 64(12): 8437–8446. [DOI] [PubMed] [Google Scholar]
- Kawahara B, Gao L, Cohn W, Whitelegge JP, Sen S, Janzen C and Mascharak PK (2020). “Diminished viability of human ovarian cancer cells by antigen-specific delivery of carbon monoxide with a family of photoactivatable antibody-photoCORM conjugates.” Chemical Science 11(2): 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara B, Moller T, Hu-Moore K, Carrington S, Faull KF, Sen S and Mascharak PK (2017). “Attenuation of Antioxidant Capacity in Human Breast Cancer Cells by Carbon Monoxide through Inhibition of Cystathionine β-Synthase Activity: Implications in Chemotherapeutic Drug Sensitivity.” Journal of Medicinal Chemistry 60(19): 8000–8010. [DOI] [PubMed] [Google Scholar]
- Kawahara B, Ramadoss S, Chaudhuri G, Janzen C, Sen S and Mascharak PK (2019). “Carbon monoxide sensitizes cisplatin-resistant ovarian cancer cell lines toward cisplatin via attenuation of levels of glutathione and nuclear metallothionein.” Journal of Inorganic Biochemistry 191: 29–39. [DOI] [PubMed] [Google Scholar]
- Kenny FS, Hui R, Musgrove EA, Gee JMW, Blamey RW, Nicholson RI, Sutherland RL and Robertson JFR (1999). “Overexpression of Cyclin D1 Messenger RNA Predicts for Poor Prognosis in Estrogen Receptor-positive Breast Cancer1.” Clinical Cancer Research 5(8): 2069–2076. [PubMed] [Google Scholar]
- Kevil C, Cortese-Krott MM, Nagy P, Papapetropoulos A, Feelisch M and Szabo C (2017). Chapter 5 - Cooperative Interactions Between NO and H2S: Chemistry, Biology, Physiology, Pathophysiology. Nitric Oxide (Third Edition). Ignarro LJand Freeman BA, Academic Press: 57–83. [Google Scholar]
- Keyaerts E, Vijgen L, Chen L, Maes P, Hedenstierna G and Van Ranst M (2004). “Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound.” International journal of infectious diseases : IJID : official publication of the International Society for Infectious Diseases 8(4): 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NH, Wang D, Wang W, Shahid M, Khattak S, Ngowi EE, Sarfraz M, Ji X-Y, Zhang C-Y and Wu D-D (2022) “Pharmacological Inhibition of Endogenous Hydrogen Sulfide Attenuates Breast Cancer Progression.” Molecules 27 DOI: 10.3390/molecules27134049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NI, Bradstock KF and Bendall LJ (2007). “Activation of Wnt/beta-catenin pathway mediates growth and survival in B-cell progenitor acute lymphoblastic leukaemia.” Br J Haematol 138(3): 338–348. [DOI] [PubMed] [Google Scholar]
- Khan NI, Cisterne A, Baraz R, Bradstock KF and Bendall LJ (2012). “ParaNO-aspirin inhibits NF-<strong>κ</strong>B and induces apoptosis in B-cell progenitor acute lymphoblastic leukemia.” Experimental Hematology 40(3): 207–215.e201. [DOI] [PubMed] [Google Scholar]
- Khan S, Lopez-Dee Z, Kumar R and Ling J (2013). “Activation of NFkB is a novel mechanism of pro-survival activity of glucocorticoids in breast cancer cells.” Cancer Letters 337(1): 90–95. [DOI] [PubMed] [Google Scholar]
- Kharitonov SA, Yates D and Barnes PJ (1995). “Increased nitric oxide in exhaled air of normal human subjects with upper respiratory tract infections.” Eur Respir J 8(2): 295–297. [DOI] [PubMed] [Google Scholar]
- Kida M, Sugiyama T, Yoshimoto T and Ogawa Y (2013). “Hydrogen sulfide increases nitric oxide production with calcium-dependent activation of endothelial nitric oxide synthase in endothelial cells.” European Journal of Pharmaceutical Sciences 48(1): 211–215. [DOI] [PubMed] [Google Scholar]
- Kie J-H, Kapturczak MH, Traylor A, Agarwal A and Hill-Kapturczak N (2008). “Heme Oxygenase-1 Deficiency Promotes Epithelial-Mesenchymal Transition and Renal Fibrosis.” Journal of the American Society of Nephrology 19(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D-H, Yoon H-J, Cha Y-N and Surh Y-J (2018). “Role of heme oxygenase-1 and its reaction product, carbon monoxide, in manifestation of breast cancer stem cell-like properties: Notch-1 as a putative target.” Free Radical Research 52(11–12): 1336–1347. [DOI] [PubMed] [Google Scholar]
- Kimura H (2015). “Signaling of hydrogen sulfide and polysulfides.” Antioxid Redox Signal 22(5): 347–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Goto Y-I and Kimura H (2009). “Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria.” Antioxidants & Redox Signaling 12(1): 1–13. [DOI] [PubMed] [Google Scholar]
- King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao Y-X, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R and Lefer DJ (2014). “Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent.” Proceedings of the National Academy of Sciences 111(8): 3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li C-Q, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ and Anderson KC (2007). “JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells.” Blood 110(2): 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert H, Schwarz PM and Forstermann U (2003). “Regulation of the expression of inducible nitric oxide synthase.” Biol Chem 384(10–11): 1343–1364. [DOI] [PubMed] [Google Scholar]
- Klug F, Prakash H, Huber Peter E., Seibel T, Bender N, Halama N, Pfirschke C, Voss Ralf H., Timke C, Umansky L, Klapproth K, Schäkel K, Garbi N, Jäger D, Weitz J, Schmitz-Winnenthal H, Hämmerling Günter J. and Beckhove P (2013). “Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy.” Cancer Cell 24(5): 589–602. [DOI] [PubMed] [Google Scholar]
- Kodela R, Chattopadhyay M and Kashfi K (2013). “Synthesis and biological activity of NOSH-naproxen (AVT-219) and NOSH-sulindac (AVT-18A) as potent anti-inflammatory agents with chemotherapeutic potential.” Medchemcomm 4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodela R, Chattopadhyay M, Velázquez-Martínez CA and Kashfi K (2015). “NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid has enhanced chemopreventive properties compared to aspirin, is gastrointestinal safe with all the classic therapeutic indications.” Biochem Pharmacol 98(4): 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolios G, Valatas V and Ward SG (2004). “Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle.” Immunology 113(4): 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru GK, Bir SC, Yuan S, Shen X, Pardue S, Wang R and Kevil CG (2015). “Cystathionine γ-lyase regulates arteriogenesis through NO-dependent monocyte recruitment.” Cardiovasc Res 107(4): 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru GK, Shen X and Kevil CG (2013). “A tale of two gases: NO and H2S, foes or friends for life?” Redox Biol 1: 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru GK, Shen X and Kevil CG (2020). “Reactive Sulfur Species: A New Redox Player in Cardiovascular Pathophysiology.” Arterioscler Thromb Vasc Biol 40(4): 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourti M, Cai J, Jiang W and Westwell AD (2021). “Structural Modifications on CORM-3 Lead to Enhanced Anti-angiogenic Properties Against Triple-negative Breast Cancer Cells.” Med Chem 17(1): 40–59. [DOI] [PubMed] [Google Scholar]
- Kourti M, Westwell A, Jiang W and Cai J (2019). “Repurposing old carbon monoxide-releasing molecules towards the anti-angiogenic therapy of triple-negative breast cancer.” Oncotarget 10(10): 1132–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ and Tonks NK (2011). “H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response.” Sci Signal 4(203): ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Król M and Kepinska M (2020). “Human Nitric Oxide Synthase-Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases.” Int J Mol Sci 22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y-J, Baek H-S, Ye D-J, Shin S, Kim D and Chun Y-J (2016). “CYP1B1 Enhances Cell Proliferation and Metastasis through Induction of EMT and Activation of Wnt/β-Catenin Signaling via Sp1 Upregulation.” PLOS ONE 11(3): e0151598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster JR Jr. (2020). “Historical origins of the discovery of mammalian nitric oxide (nitrogen monoxide) production/physiology/pathophysiology.” Biochem Pharmacol: 113793. [DOI] [PubMed] [Google Scholar]
- Lancaster JR Jr. (2020). “Historical origins of the discovery of mammalian nitric oxide (nitrogen monoxide) production/physiology/pathophysiology.” Biochem Pharmacol 176: 113793. [DOI] [PubMed] [Google Scholar]
- Le X, Wei D, Huang S, Lancaster JR and Xie K (2005). “Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors.” Proceedings of the National Academy of Sciences 102(24): 8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, McGeer E, Kodela R, Kashfi K and McGeer PL (2013). “NOSH-aspirin (NBS-1120), a novel nitric oxide and hydrogen sulfide releasing hybrid, attenuates neuroinflammation induced by microglial and astrocytic activation: a new candidate for treatment of neurodegenerative disorders.” Glia 61(10): 1724–1734. [DOI] [PubMed] [Google Scholar]
- Lee W-Y, Chen Y-C, Shih C-M, Lin C-M, Cheng C-H, Chen K-C and Lin C-W (2014). “The induction of heme oxygenase-1 suppresses heat shock protein 90 and the proliferation of human breast cancer cells through its byproduct carbon monoxide.” Toxicology and Applied Pharmacology 274(1): 55–62. [DOI] [PubMed] [Google Scholar]
- Lee ZW, Teo XY, Tay EYW, Tan CH, Hagen T, Moore PK and Deng LW (2014). “Utilizing hydrogen sulfide as a novel anti-cancer agent by targeting cancer glycolysis and pH imbalance.” British Journal of Pharmacology 171(18): 4322–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ZW, Zhou J, Chen C-S, Zhao Y, Tan C-H, Li L, Moore PK and Deng L-W (2011). “The Slow-Releasing Hydrogen Sulfide Donor, GYY4137, Exhibits Novel Anti-Cancer Effects In Vitro and In Vivo.” PLOS ONE 6(6): e21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepique AP, Daghastanli KRP, Cuccovia IM and Villa LL (2009). “HPV16 Tumor Associated Macrophages Suppress Antitumor T Cell Responses.” Clinical Cancer Research 15(13): 4391–4400. [DOI] [PubMed] [Google Scholar]
- Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, Merghoub T and Wolchok JD (2012). “Monocytic CCR2+ Myeloid-Derived Suppressor Cells Promote Immune Escape by Limiting Activated CD8 T-cell Infiltration into the Tumor Microenvironment.” Cancer Research 72(4): 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Dubey S, Varney ML, Dave BJ and Singh RK (2003). “IL-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis1.” The Journal of Immunology 170(6): 3369–3376. [DOI] [PubMed] [Google Scholar]
- Li C, Li L, Jin L and Yuan J (2018). “Heme Oxygenase-1 inhibits spring viremia of carp virus replication through carbon monoxide mediated cyclic GMP/Protein kinase G signaling pathway.” Fish & Shellfish Immunology 79: 65–72. [DOI] [PubMed] [Google Scholar]
- Li H, Ma Y, Escaffre O, Ivanciuc T, Komaravelli N, Kelley JP, Coletta C, Szabo C, Rockx B, Garofalo RP and Casola A (2015). “Role of hydrogen sulfide in paramyxovirus infections.” Journal of virology 89(10): 5557–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Witte K, August M, Brausch I, Gödtel-Armbrust U, Habermeier A, Closs EI, Oelze M, Münzel T and Förstermann U (2006). “Reversal of Endothelial Nitric Oxide Synthase Uncoupling and Up-Regulation of Endothelial Nitric Oxide Synthase Expression Lowers Blood Pressure in Hypertensive Rats.” Journal of the American College of Cardiology 47(12): 2536–2544. [DOI] [PubMed] [Google Scholar]
- Li J, Xie L, Li B, Yin C, Wang G, Sang W, Li W, Tian H, Zhang Z, Zhang X, Fan Q and Dai Y (2021). “Engineering a Hydrogen-Sulfide-Based Nanomodulator to Normalize Hyperactive Photothermal Immunogenicity for Combination Cancer Therapy.” Advanced Materials 33(22): 2008481. [DOI] [PubMed] [Google Scholar]
- Li L, Bhatia M and Moore PK (2006). “Hydrogen sulphide--a novel mediator of inflammation?” Curr Opin Pharmacol 6(2): 125–129. [DOI] [PubMed] [Google Scholar]
- Li L, Hsu A and Moore PK (2009). “Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation--a tale of three gases!” Pharmacol Ther 123(3): 386–400. [DOI] [PubMed] [Google Scholar]
- Li Y, Dang J, Liang Q and Yin L (2019). “Carbon monoxide (CO)-Strengthened cooperative bioreductive anti-tumor therapy via mitochondrial exhaustion and hypoxia induction.” Biomaterials 209: 138–151. [DOI] [PubMed] [Google Scholar]
- Li Y, Dang J, Liang Q and Yin L (2019). “Thermal-Responsive Carbon Monoxide (CO) Delivery Expedites Metabolic Exhaustion of Cancer Cells toward Reversal of Chemotherapy Resistance.” ACS Central Science 5(6): 1044–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jiang M, Deng Z, Zeng S and Hao J (2021). “Low Dose Soft X-Ray Remotely Triggered Lanthanide Nanovaccine for Deep Tissue CO Gas Release and Activation of Systemic Anti-Tumor Immunoresponse.” Advanced Science 8(12): 2004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian S, Xia Y, Ung TT, Khoi PN, Yoon HJ, Kim NH, Kim KK and Jung YD (2016). “Carbon monoxide releasing molecule-2 ameliorates IL-1β-induced IL-8 in human gastric cancer cells.” Toxicology 361–362: 24–38. [DOI] [PubMed] [Google Scholar]
- Lin C-W, Shen S-C, Hou W-C, Yang L-Y and Chen Y-C (2008). “Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9.” Molecular Cancer Therapeutics 7(5): 1195–1206. [DOI] [PubMed] [Google Scholar]
- Lin K, Rockliffe N, Johnson GG, Sherrington PD and Pettitt AR (2008). “Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells.” Oncogene 27(17): 2445–2455. [DOI] [PubMed] [Google Scholar]
- Lincoln T, Cornwell T, Komalavilas P, MacMillan-Crow LA and Boerth NJ (1996). The nitric oxide-cyclic GMP signaling system. . Biochemistry of smooth muscle contraction. San Diego BM, Academic Press: 257–268. [Google Scholar]
- Liu Q, Tomei S, Ascierto ML, De Giorgi V, Bedognetti D, Dai C, Uccellini L, Spivey T, Pos Z, Thomas J, Reinboth J, Murtas D, Zhang Q, Chouchane L, Weiss GR, Slingluff CL Jr., Lee PP, Rosenberg SA, Alter H, Yao K, Wang E and Marincola FM (2014). “Melanoma NOS1 expression promotes dysfunctional IFN signaling.” The Journal of Clinical Investigation 124(5): 2147–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Mukosera GT and Blood AB (2020). “The role of gasotransmitters in neonatal physiology.” Nitric Oxide 95: 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Pan L, Zhuo Y, Gong Q, Rose P and Zhu Y (2010). “Hypoxia-Inducible Factor-1α Is Involved in the Pro-angiogenic Effect of Hydrogen Sulfide under Hypoxic Stress.” Biological and Pharmaceutical Bulletin 33(9): 1550–1554. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhu P, Wang Y, Wei Z, Tao L, Zhu Z, Sheng X, Wang S, Ruan J, Liu Z, Cao Y, Shan Y, Sun L, Wang A, Chen W and Lu Y (2015). “Antimetastatic Therapies of the Polysulfide Diallyl Trisulfide against Triple-Negative Breast Cancer (TNBC) via Suppressing MMP2/9 by Blocking NF-κB and ERK/MAPK Signaling Pathways.” PLOS ONE 10(4): e0123781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh C-Y, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP and Looi CY (2019) “The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges.” Cells 8 DOI: 10.3390/cells8101118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmander LS, McKeith D, Svensson O, Malmenas M, Bolin L, Kalla A, Genti G, Szechinski J and Ramos-Remus C (2005). “A randomised, placebo controlled, comparative trial of the gastrointestinal safety and efficacy of AZD3582 versus naproxen in osteoarthritis.” Ann Rheum Dis 64(3): 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Gao Y, Huang X and Wang X (2014). “GYY4137, a hydrogen sulfide (H2S) donor, shows potent anti-hepatocellular carcinoma activity through blocking the STAT3 pathway.” Int J Oncol 44(4): 1259–1267. [DOI] [PubMed] [Google Scholar]
- Lucetti LT, Silva RO, Santana APM, de Melo Tavares B, Vale ML, Soares PMG, de Lima Júnior FJB, Magalhães PJC, de Queiroz Cunha F, de Albuquerque Ribeiro R, Medeiros J-VR and Souza MHLP (2017). “Nitric Oxide and Hydrogen Sulfide Interact When Modulating Gastric Physiological Functions in Rodents.” Digestive Diseases and Sciences 62(1): 93–104. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Nordvall SL, Weitzberg E, Kollberg H and Alving K (1996). “Exhaled nitric oxide in paediatric asthma and cystic fibrosis.” Archives of Disease in Childhood 75(4): 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Nordvall SL, Kuylenstierna R, Lundberg JM and Alving K (1994). “Primarily nasal origin of exhaled nitric oxide and absence in Kartagener’s syndrome.” Eur Respir J 7(8): 1501–1504. [DOI] [PubMed] [Google Scholar]
- Lv C, Su Q, Fang J and Yin H (2019). “Styrene-maleic acid copolymer-encapsulated carbon monoxide releasing molecule-2 (SMA/CORM-2) suppresses proliferation, migration and invasion of colorectal cancer cells in vitro and in vivo.” Biochemical and Biophysical Research Communications 520(2): 320–326. [DOI] [PubMed] [Google Scholar]
- Lv M, Li Y, Ji M-H, Zhuang M and Tang J-H (2014). “Inhibition of invasion and epithelial-mesenchymal transition of human breast cancer cells by hydrogen sulfide through decreased phospho-p38 expression.” Mol Med Rep 10(1): 341–346. [DOI] [PubMed] [Google Scholar]
- Ma K, Liu Y, Zhu Q, Liu C.-h., Duan J-L, Tan BKHand Zhu YZ(2011). “H2S Donor, S-Propargyl-Cysteine, Increases CSE in SGC-7901 and Cancer-Induced Mice: Evidence for a Novel Anti-Cancer Effect of Endogenous H2S?” PLOS ONE 6(6): e20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-L, Wang H-Q, Wu P, Hu J, Yin J-Q, Wu S, Ge M, Sun W-F, Zhao J-Y, Aisa HA, Li Y-H and Jiang J-D (2016). “Rupestonic acid derivative YZH-106 suppresses influenza virus replication by activation of heme oxygenase-1-mediated interferon response.” Free Radical Biology and Medicine 96: 347–361. [DOI] [PubMed] [Google Scholar]
- Ma Y, Yan Z, Deng X, Guo J, Hu J, Yu Y and Jiao F (2018). “Anticancer effect of exogenous hydrogen sulfide in cisplatin‑resistant A549/DDP cells.” Oncol Rep 39(6): 2969–2977. [DOI] [PubMed] [Google Scholar]
- Ma Z, Bi Q and Wang Y (2015). “Hydrogen sulfide accelerates cell cycle progression in oral squamous cell carcinoma cell lines.” Oral Diseases 21(2): 156–162. [DOI] [PubMed] [Google Scholar]
- Ma Z, Pu F, Zhang X, Yan Y, Zhao L, Zhang A, Li N, Zhou E-M and Xiao S (2017). “Carbon monoxide and biliverdin suppress bovine viral diarrhoea virus replication.” Journal of General Virology 98(12): 2982–2992. [DOI] [PubMed] [Google Scholar]
- Mackern-Oberti JP, Obreque J, Méndez GP, Llanos C and Kalergis AM (2015). “Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus.” Clinical and Experimental Immunology 182(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magierowski M, Magierowska K, Hubalewska-Mazgaj M, Adamski J, Bakalarz D, Sliwowski Z, Pajdo R, Kwiecien S and Brzozowski T (2016). “Interaction between endogenous carbon monoxide and hydrogen sulfide in the mechanism of gastroprotection against acute aspirin-induced gastric damage.” Pharmacological Research 114: 235–250. [DOI] [PubMed] [Google Scholar]
- Magierowski M, Magierowska K, Hubalewska-Mazgaj M, Surmiak M, Sliwowski Z, Wierdak M, Kwiecien S, Chmura A and Brzozowski T (2018). “Cross-talk between hydrogen sulfide and carbon monoxide in the mechanism of experimental gastric ulcers healing, regulation of gastric blood flow and accompanying inflammation.” Biochemical Pharmacology 149: 131–142. [DOI] [PubMed] [Google Scholar]
- Mahmoud AT, Salim MT, Ibrahem RA, Gabr A and Halby HM (2020) “Multiple Drug Resistance Patterns in Various Phylogenetic Groups of Hospital-Acquired Uropathogenic E. coli Isolated from Cancer Patients.” Antibiotics9 DOI: 10.3390/antibiotics9030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines MD (1997). “The heme oxygenase system: a regulator of second messenger gases.” Annu Rev Pharmacol Toxicol 37: 517–554. [DOI] [PubMed] [Google Scholar]
- Mann BE and Motterlini R (2007). “CO and NO in medicine.” Chem Commun (Camb)(41): 4197–4208. [DOI] [PubMed] [Google Scholar]
- Marin M, Gudiol C, Ardanuy C, Garcia-Vidal C, Calvo M, Arnan M and Carratalà J (2014). “Bloodstream infections in neutropenic patients with cancer: Differences between patients with haematological malignancies and solid tumours.” Journal of Infection 69(5): 417–423. [DOI] [PubMed] [Google Scholar]
- Markowitz J, Wang J, Vangundy Z, You J, Yildiz V, Yu L, Foote IP, Branson OE, Stiff AR, Brooks TR, Biesiadecki B, Olencki T, Tridandapani S, Freitas MA, Papenfuss T, Phelps MA and Carson WE (2017). “Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration.” Scientific Reports 7(1): 15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ruiz A, Cadenas S and Lamas S (2011). “Nitric oxide signaling: Classical, less classical, and nonclassical mechanisms.” Free Radical Biology and Medicine 51(1): 17–29. [DOI] [PubMed] [Google Scholar]
- Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P and Segal DM (2002). “Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism1.” The Journal of Immunology 168(2): 689–695. [DOI] [PubMed] [Google Scholar]
- McEvoy JG (2015). “Gases, God and the balance of nature: a commentary on Priestley (1772) ‘Observations on different kinds of air’.” Philos Trans A Math Phys Eng Sci 373(2039). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean S, Begg R, Jesse HE, Mann BE, Sanguinetti G and Poole RK (2013). “Analysis of the Bacterial Response to Ru(CO)3Cl(Glycinate) (CORM-3) and the Inactivated Compound Identifies the Role Played by the Ruthenium Compound and Reveals Sulfur-Containing Species as a Major Target of CORM-3 Action.” Antioxidants & Redox Signaling 19(17): 1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megías J, Busserolles J and Alcaraz MJ (2007). “The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells.” Br J Pharmacol 150(8): 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Zhao S, Xie L, Han Y and Ji Y (2018). “Protein S-sulfhydration by hydrogen sulfide in cardiovascular system.” Br J Pharmacol 175(8): 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Martinez MA, Raymond-Stintz MA, Winter SS and Wilson BS (2010). “IKK inhibitor bay 11–7082 induces necroptotic cell death in precursor-B acute lymphoblastic leukaemic blasts.” British Journal of Haematology 148(3): 487–490. [DOI] [PubMed] [Google Scholar]
- Miao L, Xin X, Xin H, Shen X and Zhu Y-Z (2016). “Hydrogen Sulfide Recruits Macrophage Migration by Integrin β1-Src-FAK/Pyk2-Rac Pathway in Myocardial Infarction.” Scientific Reports 6(1): 22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel T and Feron O (1997). “Nitric oxide synthases: which, where, how, and why?” J Clin Invest 100(9): 2146–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, McMullin B, Ghaffari A, Stenzler A, Pick N, Roscoe D, Ghahary A, Road J and Av-Gay Y (2009). “Gaseous nitric oxide bactericidal activity retained during intermittent highdose short duration exposure.” Nitric Oxide 20(1): 16–23. [DOI] [PubMed] [Google Scholar]
- Miller TW, Kaur S, Ivins-O’Keefe K and Roberts DD (2013). “Thrombospondin-1 is a CD47-dependent endogenous inhibitor of hydrogen sulfide signaling in T cell activation.” Matrix Biology 32(6): 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TW, Wang EA, Gould S, Stein EV, Kaur S, Lim L, Amarnath S, Fowler DH and Roberts DD (2012). “Hydrogen Sulfide Is an Endogenous Potentiator of T Cell Activation *.” Journal of Biological Chemistry 287(6): 4211–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirandola P, Gobbi G, Sponzilli I, Pambianco M, Malinverno C, Cacchioli A, De Panfilis G and Vitale M (2007). “Exogenous hydrogen sulfide induces functional inhibition and cell death of cytotoxic lymphocytes subsets.” Journal of Cellular Physiology 213(3): 826–833. [DOI] [PubMed] [Google Scholar]
- Mitchell CW and Davenport SJ (1924). “Hydrogen Sulphide Literature.” Public Health Reports (1896–1970) 39(1): 1–13. [Google Scholar]
- Miyamoto R, Koike S, Takano Y, Shibuya N, Kimura Y, Hanaoka K, Urano Y, Ogasawara Y and Kimura H (2017). “Polysulfides (H2Sn) produced from the interaction of hydrogen sulfide (H2S) and nitric oxide (NO) activate TRPA1 channels.” Scientific Reports 7(1): 45995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, Savino B, Colombo P, Jonjic N, Pecanic S, Lazzarato L, Fruttero R, Gasco A, Bronte V and Viola A (2011). “Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells.” Journal of Experimental Medicine 208(10): 1949–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S and Higgs A (1993). “The L-arginine-nitric oxide pathway.” N Engl J Med 329(27): 2002–2012. [DOI] [PubMed] [Google Scholar]
- Monti M, Hyseni I, Pacini A, Monzani E, Casella L and Morbidelli L (2018). “Cross-talk between endogenous H2S and NO accounts for vascular protective activity of the metal-nonoate Zn(PipNONO)Cl.” Biochemical Pharmacology 152: 143–152. [DOI] [PubMed] [Google Scholar]
- Moody SE, Perez D, Pan T.-c., Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RDand Chodosh LA(2005). “The transcriptional repressor Snail promotes mammary tumor recurrence.” Cancer Cell 8(3): 197–209. [DOI] [PubMed] [Google Scholar]
- Mortezaee K (2020). “Immune escape: A critical hallmark in solid tumors.” Life Sciences 258: 118110. [DOI] [PubMed] [Google Scholar]
- Motterlini R and Otterbein LE (2010). “The therapeutic potential of carbon monoxide.” Nat Rev Drug Discov 9(9): 728–743. [DOI] [PubMed] [Google Scholar]
- Mozaffarian N, Berman JW and Casadevall A (1997). “Enhancement of nitric oxide synthesis by macrophages represents an additional mechanism of action for amphotericin B.” Antimicrobial Agents and Chemotherapy 41(8): 1825–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Li W, Yang X, Chen J and Weng Y (2022). “Partially Reduced MIL-100(Fe) as a CO Carrier for Sustained CO Release and Regulation of Macrophage Phenotypic Polarization.” ACS Biomater Sci Eng 8(11): 4777–4788. [DOI] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Ullrich V and Mülsch A (2005). “Vascular Consequences of Endothelial Nitric Oxide Synthase Uncoupling for the Activity and Expression of the Soluble Guanylyl Cyclase and the cGMP-Dependent Protein Kinase.” Arteriosclerosis, Thrombosis, and Vascular Biology 25(8): 1551–1557. [DOI] [PubMed] [Google Scholar]
- Murray TS, Okegbe C, Gao Y, Kazmierczak BI, Motterlini R, Dietrich LEP and Bruscia EM (2012). “The Carbon Monoxide Releasing Molecule CORM-2 Attenuates Pseudomonas aeruginosa Biofilm Formation.” PLOS ONE 7(4): e35499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki M, Shimura S, Irokawa T, Sasaki T and Shirato K (1995). “Nitric oxide regulation of glycoconjugate secretion from feline and human airways in vitro.” Respiration Physiology 102(1): 89–95. [DOI] [PubMed] [Google Scholar]
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J and Gabrilovich DI (2007). “Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer.” Nature Medicine 13(7): 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H and Ohtani H (1998). “CD8+ T Cells Infiltrated within Cancer Cell Nests as a Prognostic Factor in Human Colorectal Cancer.” Cancer Research 58(16): 3491–3494. [PubMed] [Google Scholar]
- Namkoong S, Lee S-J, Kim C-K, Kim Y-M, Chung H-T, Lee H, Han J-A, Ha K-S, Kwon YG and Kim Y-M (2005). “Prostaglandin E2 stimulates angiogenesis by activating the nitric oxide/cGMP pathway in human umbilical vein endothelial cells.” Experimental & Molecular Medicine 37(6): 588–600. [DOI] [PubMed] [Google Scholar]
- Naseem KM (2005). “The role of nitric oxide in cardiovascular diseases.” Mol Aspects Med 26(1–2): 33–65. [DOI] [PubMed] [Google Scholar]
- Nath N, Vassell R, Chattopadhyay M, Kogan M and Kashfi K (2009). “Nitro-aspirin inhibits MCF-7 breast cancer cell growth: Effects on COX-2 expression and Wnt/β-catenin/TCF-4 signaling.” Biochemical Pharmacology 78(10): 1298–1304. [DOI] [PubMed] [Google Scholar]
- Nathan C and Xie QW (1994). “Nitric oxide synthases: roles, tolls, and controls.” Cell 78(6): 915–918. [DOI] [PubMed] [Google Scholar]
- Neil JR, Johnson KM, Nemenoff RA and Schiemann WP (2008). “Cox-2 inactivates Smad signaling and enhances EMT stimulated by TGF-β through a PGE 2 -dependent mechanisms.” Carcinogenesis 29(11): 2227–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth Z, Csizmadia E, Vikstrom L, Li M, Bisht K, Feizi A, Otterbein S, Zuckerbraun B, Costa DB, Pandolfi PP, Fillinger J, Döme B, Otterbein LE and Wegiel B (2016). “Alterations of tumor microenvironment by carbon monoxide impedes lung cancer growth.” Oncotarget 7(17): 23919–23932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Nguyen T-K, Rice SA and Boyer C (2015). “CO-Releasing Polymers Exert Antimicrobial Activity.” Biomacromolecules 16(9): 2776–2786. [DOI] [PubMed] [Google Scholar]
- Nicholls P and Kim JK (1981). “Oxidation of sulphide by cytochrome aa3.” Biochim Biophys Acta 637(2): 312–320. [DOI] [PubMed] [Google Scholar]
- NicOx SA (2007). “NicOx provides an update on NCX-4016. Sophia Antipolis, France” Updated June 18, 2007; cited August 12, 2016. [Google Scholar]
- NicOx SA (2010). “FDA provides Complete Response Letter to NicOx’s New Drug Application for naproxcinod.”, Updated July 22, 2010; cited August 12, 2016. [Google Scholar]
- Niedbala W, Wei X-Q, Piedrafita D, Xu D and Liew FY (1999). “Effects of nitric oxide on the induction and differentiation of Th1 cells.” European Journal of Immunology 29(8): 2498–2505. [DOI] [PubMed] [Google Scholar]
- Niedbala W, Wei XQ, Campbell C, Thomson D, Komai-Koma M and Liew FY (2002). “Nitric oxide preferentially induces type 1 T cell differentiation by selectively up-regulating IL-12 receptor beta 2 expression via cGMP.” Proc Natl Acad Sci U S A 99(25): 16186–16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Yano H, Nishida T, Kamura T and Kojiro M (2006). “Angiogenesis in cancer.” Vasc Health Risk Manag 2(3): 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, Ito Y, Inamura T, Ikezaki K, Fukui M, Iwaki T and Kuwano M (1999). “Macrophage Infiltration and Heme Oxygenase-1 Expression Correlate with Angiogenesis in Human Gliomas1.” Clinical Cancer Research 5(5): 1107–1113. [PubMed] [Google Scholar]
- Nobre Lígia S, Seixas João D, Romão Carlos C and Saraiva Lígia M (2007). “Antimicrobial Action of Carbon Monoxide-Releasing Compounds.” Antimicrobial Agents and Chemotherapy 51(12): 4303–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorolyai S, Shajari N, Baghbani E, Sadreddini S and Baradaran B (2019). “The relation between PI3K/AKT signalling pathway and cancer.” Gene 698: 120–128. [DOI] [PubMed] [Google Scholar]
- Nowaczyk A, Kowalska M, Nowaczyk J and Grześk G (2021). “Carbon Monoxide and Nitric Oxide as Examples of the Youngest Class of Transmitters.” International journal of molecular sciences 22(11): 6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunwobi OO and Liu C (2011). “Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways.” Clinical & Experimental Metastasis 28(8): 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh G-S, Pae H-O, Lee B-S, Kim B-N, Kim J-M, Kim H-R, Jeon SB, Jeon WK, Chae H-J and Chung H-T (2006). “Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide.” Free Radical Biology and Medicine 41(1): 106–119. [DOI] [PubMed] [Google Scholar]
- Oláh G, Módis K, Törö G, Hellmich MR, Szczesny B and Szabo C (2018). “Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation.” Biochemical Pharmacology 149: 186–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR (2017). “H2S and polysulfide metabolism: Conventional and unconventional pathways.” Biochem Pharmacol. [DOI] [PubMed] [Google Scholar]
- Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J and Fukuto JM (2014). “Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications of their possible biological activity and utility.” Free Radic Biol Med 77: 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi XJ and Tan KS (2016). “Reduced Glutathione Mediates Resistance to H2S Toxicity in Oral Streptococci.” Appl Environ Microbiol 82(7): 2078–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod AD, Shah AAJ, Li H, Benjamin NB, Ferguson GP and Leifert C (2011). “An observational prospective study of topical acidified nitrite for killing methicillin-resistant Staphylococcus aureus (MRSA) in contaminated wounds.” BMC Research Notes 4(1): 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orucevic A, Bechberger J, Green AM, Shapiro RA, Billiar TR and Lala PK (1999). “Nitric-oxide production by murine mammary adenocarcinoma cells promotes tumor-cell invasiveness.” International Journal of Cancer 81(6): 889–896. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA and Choi AMK (2000). “Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway.” Nature Medicine 6(4): 422–428. [DOI] [PubMed] [Google Scholar]
- Otterbein LE and Choi AM (2000). “Heme oxygenase: colors of defense against cellular stress.” Am J Physiol Lung Cell Mol Physiol 279(6): L1029–1037. [DOI] [PubMed] [Google Scholar]
- V. P, D D, Bala Mand N S (2018). “Photoactivated [Mn(CO)3Br(μ-bpcpd)]2 induces apoptosis in cancer cells via intrinsic pathway.” Journal of Photochemistry and Photobiology B: Biology 188: 28–41. [DOI] [PubMed] [Google Scholar]
- Pacelli R, Wink DA, Cook JA, Krishna MC, DeGraff W, Friedman N, Tsokos M, Samuni A and Mitchell JB (1995). “Nitric oxide potentiates hydrogen peroxide-induced killing of Escherichia coli.” J Exp Med 182(5): 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco A (2017). SULFUR-CONTAINING COMPOUNDS AS HYDROGEN SULFIDE AND BROAD-SPECTRUM ANTIVIRAL AGENTS.
- Pae H-O, Oh G-S, Choi B-M, Chae S-C, Kim Y-M, Chung K-R and Chung H-T (2004). “Carbon Monoxide Produced by Heme Oxygenase-1 Suppresses T Cell Proliferation via Inhibition of IL-2 Production1.” The Journal of Immunology 172(8): 4744–4751. [DOI] [PubMed] [Google Scholar]
- Pagano L, Busca A, Candoni A, Cattaneo C, Cesaro S, Fanci R, Nadali G, Potenza L, Russo D, Tumbarello M, Nosari A, Aversa F, Lessi F, Criscuolo M, Farina F, Tisi MC, Turri G, Barone A, Spolzino A, Del Principe MI, Quinto AM, Di Blasi R, Maracci L, Nabergoj M, Cambò B, Pegoraro A, Marchesi F, Pascale S, Passi A, Carlisi M, Polverelli N, Beggia B, Rambaldi B, Prezioso L and Sanna M (2017). “Risk stratification for invasive fungal infections in patients with hematological malignancies: SEIFEM recommendations.” Blood Reviews 31(2): 17–29. [DOI] [PubMed] [Google Scholar]
- Pan Y, Ye S, Yuan D, Zhang J, Bai Y and Shao C (2014). “Hydrogen sulfide (H2S)/cystathionine γ-lyase (CSE) pathway contributes to the proliferation of hepatoma cells.” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 763–764: 10–18. [DOI] [PubMed] [Google Scholar]
- Panza E, Bello I, Smimmo M, Brancaleone V, Mitidieri E, Bucci M, Cirino G, Sorrentino R and R. d′Emmanuele di Villa Bianca (2022). “Endogenous and exogenous hydrogen sulfide modulates urothelial bladder carcinoma development in human cell lines.” Biomedicine & Pharmacotherapy 151: 113137. [DOI] [PubMed] [Google Scholar]
- Panza E, De Cicco P, Armogida C, Scognamiglio G, Gigantino V, Botti G, Germano D, Napolitano M, Papapetropoulos A, Bucci M, Cirino G and Ianaro A (2015). “Role of the cystathionine gamma lyase/hydrogen sulfide pathway in human melanoma progression.” Pigment Cell Melanoma Res 28(1): 61–72. [DOI] [PubMed] [Google Scholar]
- Panza E, De Cicco P, Armogida C, Scognamiglio G, Gigantino V, Botti G, Germano D, Napolitano M, Papapetropoulos A, Bucci M, Cirino G and Ianaro A (2015). “Role of the cystathionine γ lyase/hydrogen sulfide pathway in human melanoma progression.” Pigment Cell & Melanoma Research 28(1): 61–72. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R and Szabó C (2009). “Hydrogen sulfide is an endogenous stimulator of angiogenesis.” Proceedings of the National Academy of Sciences 106(51): 21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee SK, Lim CR, Park HW, Liu F, Kim S-J and Kim B-C (2018). “Heme oxygenase-1/carbon monoxide axis suppresses transforming growth factor-β1-induced growth inhibition by increasing ERK1/2-mediated phosphorylation of Smad3 at Thr-179 in human hepatocellular carcinoma cell lines.” Biochemical and Biophysical Research Communications 498(3): 609–615. [DOI] [PubMed] [Google Scholar]
- Paul BD and Snyder SH (2015). “H2S: A Novel Gasotransmitter that Signals by Sulfhydration.” Trends Biochem Sci 40(11): 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Wu B, Cao Q, Wu L and Yang G (2011). “Hydrogen sulfide mediates the anti-survival effect of sulforaphane on human prostate cancer cells.” Toxicology and Applied Pharmacology 257(3): 420–428. [DOI] [PubMed] [Google Scholar]
- Perez F, Adachi J and Bonomo RA (2014). “Antibiotic-Resistant Gram-Negative Bacterial Infections in Patients With Cancer.” Clinical Infectious Diseases 59(suppl_5): S335–S339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta C, Cervia D, Di Renzo I, Moscheni C, Bassi MT, Campana L, Martelli C, Catalani E, Giovarelli M, Zecchini S, Coazzoli M, Capobianco A, Ottobrini L, Lucignani G, Rosa P, Rovere-Querini P, De Palma C and Clementi E (2018). “Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition.” Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persichini T, Colasanti M, Fraziano M, Colizzi V, Medana C, Polticelli F, Venturini G and Ascenzi P (1999). “Nitric oxide inhibits the HIV-1 reverse transcriptase activity.” Biochem Biophys Res Commun 258(3): 624–627. [DOI] [PubMed] [Google Scholar]
- Phillips CM, Zatarain JR, Nicholls ME, Porter C, Widen SG, Thanki K, Johnson P, Jawad MU, Moyer MP, Randall JW, Hellmich JL, Maskey M, Qiu S, Wood TG, Druzhyna N, Szczesny B, Módis K, Szabo C, Chao C and Hellmich MR (2017). “Upregulation of Cystathionine-β-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis.” Cancer Res 77(21): 5741–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipili-Synetos E, Sakkoula E and Maragoudakis ME (1993). “Nitric oxide is involved in the regulation of angiogenesis.” Br J Pharmacol 108(4): 855–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte T, Abate A, Dennery PA and Schröder H (2000). “Heme Oxygenase-1 Is a cGMP-Inducible Endothelial Protein and Mediates the Cytoprotective Action of Nitric Oxide.” Arteriosclerosis, Thrombosis, and Vascular Biology 20(5): 1209–1215. [DOI] [PubMed] [Google Scholar]
- Priestley J (1772). “Observations on different kinds of air.” Phil. Trans 62: 147–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privett BJ, Broadnax AD, Bauman SJ, Riccio DA and Schoenfisch MH (2012). “Examination of bacterial resistance to exogenous nitric oxide.” Nitric Oxide 26(3): 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protzer U, Seyfried S, Quasdorff M, Sass G, Svorcova M, Webb D, Bohne F, Hösel M, Schirmacher P and Tiegs G (2007). “Antiviral Activity and Hepatoprotection by Heme Oxygenase-1 in Hepatitis B Virus Infection.” Gastroenterology 133(4): 1156–1165. [DOI] [PubMed] [Google Scholar]
- Pu J, Xu Z, Nian J, Fang Q, Yang M, Huang Y, Li W, Ge B, Wang J and Wei H (2021). “M2 macrophage-derived extracellular vesicles facilitate CD8+T cell exhaustion in hepatocellular carcinoma via the miR-21–5p/YOD1/YAP/β-catenin pathway.” Cell Death Discovery 7(1): 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupo E, Fiorio Pla A, Avanzato D, Moccia F, Avelino Cruz J-E, Tanzi F, Merlino A, Mancardi D and Munaron L (2011). “Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells.” Free Radical Biology and Medicine 51(9): 1765–1773. [DOI] [PubMed] [Google Scholar]
- Puranik M, Weeks CL, Lahaye D, Kabil Ö, Taoka S, Nielsen SB, Groves JT, Banerjee R and Spiro TG (2006). “Dynamics of Carbon Monoxide Binding to Cystathionine β-Synthase *.” Journal of Biological Chemistry 281(19): 13433–13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, Ochoa AC, Fletcher M, Velasco C, Wilk A, Reiss K and Rodriguez PC (2014). “Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxiderelated pathways.” International Journal of Cancer 134(12): 2853–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Jenkins DC, Holmes L and Moncada S (1991). “Human colorectal adenocarcinoma cells: differential nitric oxide synthesis determines their ability to aggregate platelets.” Cancer Res 51(22): 6073–6078. [PubMed] [Google Scholar]
- Rajabi M and Mousa SA (2017) “The Role of Angiogenesis in Cancer Treatment.” Biomedicines 5 DOI: 10.3390/biomedicines5020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattan R, Sakr S, Ali-Fehmi R, Abdulfatah E, Hanna RK, Giri S and Munkarah AR (2018). “S-nitrosoglutathione, a physiologic nitric oxide carrier, reduces immunosupression in ovarian cancer.” Gynecologic Oncology 149: 47. [Google Scholar]
- Reger NA, Meng WS and Gawalt ES (2017) “Antimicrobial Activity of Nitric OxideReleasing Ti-6Al-4V Metal Oxide.” Journal of Functional Biomaterials 8 DOI: 10.3390/jfb8020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger NA, Meng WS and Gawalt ES (2017). “Surface modification of PLGA nanoparticles to deliver nitric oxide to inhibit Escherichia coli growth.” Applied Surface Science 401: 162–171. [Google Scholar]
- Regev-Shoshani G, Vimalanathan S, McMullin B, Road J, Av-Gay Y and Miller C (2013). “Gaseous nitric oxide reduces influenza infectivity in vitro.” Nitric Oxide 31: 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revathidevi S and Munirajan AK (2019). “Akt in cancer: Mediator and more.” Seminars in Cancer Biology 59: 80–91. [DOI] [PubMed] [Google Scholar]
- Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD and Wink DA (2005). “Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1.” Proceedings of the National Academy of Sciences 102(37): 13147–13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA and Isenberg JS (2006). “The biphasic nature of nitric oxide responses in tumor biology.” Antioxid Redox Signal 8(7–8): 1329–1337. [DOI] [PubMed] [Google Scholar]
- Rimmelzwaan GF, Baars MM, de Lijster P, Fouchier RA and Osterhaus AD (1999). “Inhibition of influenza virus replication by nitric oxide.” Journal of virology 73(10): 8880–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi L, Gobbi G, Pambianco M, Micheloni C, Mirandola P and Vitale M (2006). “Hydrogen sulfide prevents apoptosis of human PMN via inhibition of p38 and caspase 3.” Laboratory Investigation 86(4): 391–397. [DOI] [PubMed] [Google Scholar]
- Rivlin N, Brosh R, Oren M and Rotter V (2011). “Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis.” Genes Cancer 2(4): 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha M, Krüger A, Van Rooijen N, Schirrmacher V and Umansky V (1995). “Liver endothelial cells participate in T-cell-dependent host resistance to lymphoma metastasis by production of nitric oxide in vivo.” International Journal of Cancer 63(3): 405–411. [DOI] [PubMed] [Google Scholar]
- Rosas IO, Goldberg HJ, Collard HR, El-Chemaly S, Flaherty K, Hunninghake GM, Lasky JA, Lederer DJ, Machado R, Martinez FJ, Maurer R, Teller D, Noth I, Peters E, Raghu G, Garcia JGN and Choi AMK (2018). “A Phase II Clinical Trial of Low-Dose Inhaled Carbon Monoxide in Idiopathic Pulmonary Fibrosis.” Chest 153(1): 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose P, Moore PK, Ming SH, Nam OC, Armstrong JS and Whiteman M (2005). “Hydrogen sulfide protects colon cancer cells from chemopreventative agent beta-phenylethyl isothiocyanate induced apoptosis.” World J Gastroenterol 11(26): 3990–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard KR, Novak OP, Pistiolis AM, Yang L, Ahonen MJR, McDonald RA and Schoenfisch MH (2021). “Exogenous Nitric Oxide Improves Antibiotic Susceptibility in Resistant Bacteria.” ACS Infectious Diseases 7(1): 23–33. [DOI] [PubMed] [Google Scholar]
- Russo A, Saide A, Cagliani R, Cantile M, Botti G and Russo G (2016). “rpL3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating CBS and NFκB upon 5-FU treatment.” Scientific Reports 6(1): 38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Chakraborty PK, Xiong X, Dwivedi SK, Mustafi SB, Leigh NR, Ramchandran R, Mukherjee P and Bhattacharya R (2016). “Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration.” Faseb j 30(1): 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlberg Bang C, Kruse R, Johansson K and Persson K (2016). “Carbon monoxide releasing molecule-2 (CORM-2) inhibits growth of multidrug-resistant uropathogenic Escherichia coli in biofilm and following host cell colonization.” BMC Microbiology 16(1): 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA and Poulikakos PI (2014). “Targeting RAS–ERK signalling in cancer: promises and challenges.” Nature Reviews Drug Discovery 13(12): 928–942. [DOI] [PubMed] [Google Scholar]
- Sanders SP, Proud D, Permutt S, Siekierski ES, Yachechko R and Liu MC (2004). “Role of nasal nitric oxide in the resolution of experimental rhinovirus infection.” Journal of Allergy and Clinical Immunology 113(4): 697–702. [DOI] [PubMed] [Google Scholar]
- Sanders SP, Siekierski ES, Porter JD, Richards SM and Proud D (1998). “Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line.” Journal of virology 72(2): 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhanam AVR, d’Uscio LV, Smith LA and Katusic ZS (2012). “Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice.” Journal of neurochemistry 122(6): 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarady JK, Zuckerbraun BS, Bilban M, Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AMK and Otterbein LE (2004). “Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver.” The FASEB Journal 18(7): 854–856. [DOI] [PubMed] [Google Scholar]
- Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, Muroi K and Ozawa K (2006). “Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells.” Blood 109(1): 228–234. [DOI] [PubMed] [Google Scholar]
- Sawa T, Takata T, Matsunaga T, Ihara H, Motohashi H and Akaike T (2022). “Chemical Biology of Reactive Sulfur Species: Hydrolysis-Driven Equilibrium of Polysulfides as a Determinant of Physiological Functions.” Antioxid Redox Signal 36(4–6): 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer TJ, Hochberg MC, Marrero CE, Duquesroix B, Frayssinet H and Beekman M (2011). “Efficacy and safety of naproxcinod in patients with osteoarthritis of the knee: a 53-week prospective randomized multicenter study.” Semin Arthritis Rheum 40(4): 285–297. [DOI] [PubMed] [Google Scholar]
- Schnitzer TJ, Kivitz A, Frayssinet H and Duquesroix B (2010). “Efficacy and safety of naproxcinod in the treatment of patients with osteoarthritis of the knee: a 13-week prospective, randomized, multicenter study.” Osteoarthritis Cartilage 18(5): 629–639. [DOI] [PubMed] [Google Scholar]
- Schnitzer TJ, Kivitz AJ, Lipetz RS, Sanders N and Hee A (2005). “Comparison of the COX-inhibiting nitric oxide donator AZD3582 and rofecoxib in treating the signs and symptoms of Osteoarthritis of the knee.” Arthritis Rheum 53(6): 827–837. [DOI] [PubMed] [Google Scholar]
- Schofield RE (1967). “Joseph Priestley, natural philosopher.” Ambix 14: 1–15. [DOI] [PubMed] [Google Scholar]
- Searcy DG and Lee SH (1998). “Sulfur reduction by human erythrocytes.” J Exp Zool 282(3): 310–322. [DOI] [PubMed] [Google Scholar]
- Segal BH, Almyroudis NG, Battiwalla M, Herbrecht R, Perfect JR, Walsh TJ and Wingard JR (2007). “Prevention and Early Treatment of Invasive Fungal Infection in Patients with Cancer and Neutropenia and in Stem Cell Transplant Recipients in the Era of Newer Broad-Spectrum Antifungal Agents and Diagnostic Adjuncts.” Clinical Infectious Diseases 44(3): 402–409. [DOI] [PubMed] [Google Scholar]
- Sektioglu IM, Carretero R, Bender N, Bogdan C, Garbi N, Umansky V, Umansky L, Urban K, von Knebel-Döberitz M, Somasundaram V, Wink D, Beckhove P and Hämmerling GJ (2016). “Macrophage-derived nitric oxide initiates T-cell diapedesis and tumor rejection.” OncoImmunology 5(10): e1204506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S and Snyder SH (2012). “Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions.” Mol Cell 45(1): 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul Bindu D., Gadalla Moataz M., Mustafa Asif K., Sen T, Xu R, Kim S and Snyder Solomon H. (2012). “Hydrogen Sulfide-Linked Sulfhydration of NF-κB Mediates Its Antiapoptotic Actions.” Molecular Cell 45(1): 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Kawahara B, Gupta D, Tsai R, Khachatryan M, Roy-Chowdhuri S, Bose S, Yoon A, Faull K, Farias-Eisner R and Chaudhuri G (2015). “Role of cystathionine beta-synthase in human breast Cancer.” Free Radic Biol Med 86: 228–238. [DOI] [PubMed] [Google Scholar]
- Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N and Tyagi SC (2012). “Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia.” Am J Physiol Cell Physiol 303(1): C41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seregina TA, Lobanov KV, Shakulov RS and Mironov AS (2022). “Enhancement of the Bactericidal Effect of Antibiotics by Inhibition of Enzymes Involved in Production of Hydrogen Sulfide in Bacteria.” Molecular Biology 56(5): 638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa WC (2004). “eNOS at a glance.” J Cell Sci 117(Pt 12): 2427–2429. [DOI] [PubMed] [Google Scholar]
- Shao L, Gu YY, Jiang CH, Liu CY, Lv LP, Liu JN and Zou Y (2018). “Carbon monoxide releasing molecule-2 suppresses proliferation, migration, invasion, and promotes apoptosis in non-small cell lung cancer Calu-3 cells.” Eur Rev Med Pharmacol Sci 22(7): 1948–1957. [DOI] [PubMed] [Google Scholar]
- Shariati A, Moradabadi A, Chegini Z, Khoshbayan A and Didehdar M (2020). “An Overview of the Management of the Most Important Invasive Fungal Infections in Patients with Blood Malignancies.” Infect Drug Resist 13: 2329–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatalin K, Gusarov I, Avetissova E, Shatalina Y, McQuade LE, Lippard SJ and Nudler E (2008). “Bacillus anthracis-derived nitric oxide is essential for pathogen virulence and survival in macrophages.” Proceedings of the National Academy of Sciences 105(3): 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatalin K, Shatalina E, Mironov A and Nudler E (2011). “H2S: A Universal Defense Against Antibiotics in Bacteria.” Science 334(6058): 986–990. [DOI] [PubMed] [Google Scholar]
- Shi Q, Xiong Q, Wang B, Le X, Khan NA and Xie K (2000). “Influence of Nitric Oxide Synthase II Gene Disruption on Tumor Growth and Metastasis1.” Cancer Research 60(10): 2579–2583. [PubMed] [Google Scholar]
- Shibuya N and Kimura H (2013). “Production of hydrogen sulfide from d-cysteine and its therapeutic potential.” Front Endocrinol (Lausanne) 4: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N and Kimura H (2013). “A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells.” Nat Commun 4: 1366. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K and Kimura H (2009). “3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain.” Antioxid Redox Signal 11(4): 703–714. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Hiratsuka H, Koike K, Tsuchihashi K, Sonoda T, Ogi K, Miyakawa A, Kobayashi J, Kaneko T, Igarashi T, Hasegawa T and Miyazaki A (2019). “Tumor-infiltrating CD8+ T-cell density is an independent prognostic marker for oral squamous cell carcinoma.” Cancer Medicine 8(1): 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Iwabuchi T, Soga T, Kato Y, Yamamoto T, Takano N, Hishiki T, Ueno Y, Ikeda S, Sakuragawa T, Ishikawa K, Goda N, Kitagawa Y, Kajimura M, Matsumoto K and Suematsu M (2009). “Cystathionine β-synthase as a carbon monoxide–sensitive regulator of bile excretion.” Hepatology 49(1): 141–150. [DOI] [PubMed] [Google Scholar]
- Siegert A, Rosenberg C, Schmitt WD, Denkert C and Hauptmann S (2002). “Nitric oxide of human colorectal adenocarcinoma cell lines promotes tumour cell invasion.” British Journal of Cancer 86(8): 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, Hailemichael Y, Jayaraman P, Myers JN, Grimm EA and Overwijk WW (2010). “Targeted Inhibition of Inducible Nitric Oxide Synthase Inhibits Growth of Human Melanoma In vivo and Synergizes with Chemotherapy.” Clinical Cancer Research 16(6): 1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, Quyen DV, Kundumani-Sridharan V, Brooks PC and Rao GN (2010). “AP-1 (Fra-1/c-Jun)-mediated Induction of Expression of Matrix Metalloproteinase-2 Is Required for 15(S)-Hydroxyeicosatetraenoic Acid-induced Angiogenesis *.” Journal of Biological Chemistry 285(22): 16830–16843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Chaudhary SC, Kapur P, Weng Z, Elmets CA, Kopelovich L and Athar M (2012). “Nitric Oxide Donor Exisulind Is an Effective Inhibitor of Murine Photocarcinogenesis†.” Photochemistry and Photobiology 88(5): 1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siracusa R, Schaufler A, Calabrese V, Fuller PM and Otterbein LE (2021). “Carbon Monoxide: from Poison to Clinical Trials.” Trends in pharmacological sciences 42(5): 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrand T (1951). “Endogenous formation of carbon monoxide; the CO concentration in the inspired and expired air of hospital patients.” Acta Physiol Scand 22(2–3): 137–141. [DOI] [PubMed] [Google Scholar]
- Skrzypek K, Tertil M, Golda S, Ciesla M, Weglarczyk K, Collet G, Guichard A, Kozakowska M, Boczkowski J, Was H, Gil T, Kuzdzal J, Muchova L, Vitek L, Loboda A, Jozkowicz A, Kieda C and Dulak J (2013). “Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis.” Antioxid Redox Signal 19(7): 644–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S, Keerthivasan G, Huang Z, Gurbuxani S and Crispino JD (2010). “Overexpression of survivin initiates hematologic malignancies in vivo.” Leukemia 24(11): 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Zhou Z, Kim PKM, Shapiro RA, Liu F, Ferran C, Choi AMK and Otterbein LE (2004). “Carbon Monoxide Promotes Fas/CD95-induced Apoptosis in Jurkat Cells *.” Journal of Biological Chemistry 279(43): 44327–44334. [DOI] [PubMed] [Google Scholar]
- Sonke E, Verrydt M, Postenka CO, Pardhan S, Willie CJ, Mazzola CR, Hammers MD, Pluth MD, Lobb I, Power NE, Chambers AF, Leong HS and Sener A (2015). “Inhibition of endogenous hydrogen sulfide production in clear-cell renal cell carcinoma cell lines and xenografts restricts their growth, survival and angiogenic potential.” Nitric Oxide 49: 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soung YH, Lee JW, Kim SY, Park WS, Nam SW, Lee JY, Yoo NJ and Lee SH (2004). “Somatic mutations of CASP3 gene in human cancers.” Human Genetics 115(2): 112–115. [DOI] [PubMed] [Google Scholar]
- Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Mönkkönen J, KellokumpuLehtinen P-L, Lauttia S, Tynninen O, Joensuu H, Heymann D and Määttä JA (2015). “Human breast cancer cells educate macrophages toward the M2 activation status.” Breast Cancer Research 17(1): 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southam HM, Williamson MP, Chapman JA, Lyon RL, Trevitt CR, Henderson PJF and Poole RK (2021). “‘Carbon-Monoxide-Releasing Molecule-2 (CORM-2)’ Is a Misnomer: Ruthenium Toxicity, Not CO Release, Accounts for Its Antimicrobial Effects.” Antioxidants (Basel) 10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley G, Harvey K, Slivova V, Jiang J and Sliva D (2005). “Ganoderma lucidum suppresses angiogenesis through the inhibition of secretion of VEGF and TGF-β1 from prostate cancer cells.” Biochemical and Biophysical Research Communications 330(1): 46–52. [DOI] [PubMed] [Google Scholar]
- Stasko N, McHale K, Hollenbach Stanley J, Martin M and Doxey R (2018). “Nitric OxideReleasing Macromolecule Exhibits Broad-Spectrum Antifungal Activity and Utility as a Topical Treatment for Superficial Fungal Infections.” Antimicrobial Agents and Chemotherapy 62(7): e01026–01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, Kendra K, Campbell A, Gautam S, Abood D, Landi I, Hsu V, Duggan M, Wesolowski R, Old M, Howard JH, Yu L, Stasik N, Olencki T, Muthusamy N, Tridandapani S, Byrd JC, Caligiuri M and Carson WE (2018). “Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor–Mediated Natural Killer Cell Function.” Clinical Cancer Research 24(8): 1891–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr DJ (1997). “Structure-function aspects in the nitric oxide synthases.” Annu Rev Pharmacol Toxicol 37: 339–359. [DOI] [PubMed] [Google Scholar]
- Sun T, Gao Y, Tan W, Ma S, Shi Y, Yao J, Guo Y, Yang M, Zhang X, Zhang Q, Zeng C and Lin D (2007). “A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers.” Nature Genetics 39(5): 605–613. [DOI] [PubMed] [Google Scholar]
- Switzer CH, Glynn SA, Cheng RYS, Ridnour LA, Green JE, Ambs S and Wink DA (2012). “S-Nitrosylation of EGFR and Src Activates an Oncogenic Signaling Network in Human Basal-Like Breast Cancer.” Molecular Cancer Research 10(9): 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (2016). “Gasotransmitters in cancer: from pathophysiology to experimental therapy.” Nat Rev Drug Discov 15(3): 185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (2017). “A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator.” Biochem Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (2018). “A timeline of hydrogen sulfide (H(2)S) research: From environmental toxin to biological mediator.” Biochem Pharmacol 149: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A and Hellmich MR (2013). “Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer.” Proc Natl Acad Sci U S A 110(30): 12474–12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A and Hellmich MR (2013). “Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer.” Proceedings of the National Academy of Sciences 110(30): 12474–12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Marcatti M, Zatarain JR, Druzhyna N, Wiktorowicz JE, Nagy P, Hellmich MR and Szabo C (2016). “Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics.” Scientific Reports 6(1): 36125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei ML, Giannoni E, Fiaschi T and Chiarugi P (2012). “Anoikis: an emerging hallmark in health and diseases.” The Journal of Pathology 226(2): 380–393. [DOI] [PubMed] [Google Scholar]
- Taguchi K, Nagao S, Maeda H, Yanagisawa H, Sakai H, Yamasaki K, Wakayama T, Watanabe H, Otagiri M and Maruyama T (2018). “Biomimetic carbon monoxide delivery based on hemoglobin vesicles ameliorates acute pancreatitis in mice via the regulation of macrophage and neutrophil activity.” Drug Deliv 25(1): 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi T, Naito Y, Tanaka M, Mizushima K, Ushiroda C, Toyokawa Y, Uchiyama K, Hamaguchi M, Handa O and Itoh Y (2018). “Carbon monoxide ameliorates murine T-cell-dependent colitis through the inhibition of Th17 differentiation.” Free Radical Research 52(11–12): 1328–1335. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Setoguchi T, Machigashira M, Kanbara K and Izumi Y (2008). “Hydrogen sulfide inhibits cell proliferation and induces cell cycle arrest via an elevated p21Cip1 level in Ca9-22 cells.” Journal of Periodontal Research 43(1): 90–95. [DOI] [PubMed] [Google Scholar]
- Takhampunya R, Padmanabhan R and Ubol S (2006). “Antiviral action of nitric oxide on dengue virus type 2 replication.” J Gen Virol 87(Pt 10): 3003–3011. [DOI] [PubMed] [Google Scholar]
- Tamizhselvi R, Moore PK and Bhatia M (2007). “Hydrogen sulfide acts as a mediator of inflammation in acute pancreatitis: in vitro studies using isolated mouse pancreatic acinar cells.” J Cell Mol Med 11(2): 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Kang L, Kimura T and Niki I (2011). “Hydrogen sulphide protects mouse pancreatic β-cells from cell death induced by oxidative stress, but not by endoplasmic reticulum stress.” British Journal of Pharmacology 162(5): 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao BB, Liu SY, Zhang CC, Fu W, Cai WJ, Wang Y, Shen Q, Wang MJ, Chen Y, Zhang LJ, Zhu YZ and Zhu YC (2013). “VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells.” Antioxid Redox Signal 19(5): 448–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoka S and Banerjee R (2001). “Characterization of NO binding to human cystathionine β-synthase:: Possible implications of the effects of CO and NO binding to the human enzyme.” Journal of Inorganic Biochemistry 87(4): 245–251. [DOI] [PubMed] [Google Scholar]
- Taoka S, West M and Banerjee R (1999). “Characterization of the Heme and Pyridoxal Phosphate Cofactors of Human Cystathionine β-Synthase Reveals Nonequivalent Active Sites.” Biochemistry 38(9): 2738–2744. [DOI] [PubMed] [Google Scholar]
- Tavares AFN, Teixeira M, Romão CC, Seixas JD, Nobre LS and Saraiva LM (2011). “Reactive Oxygen Species Mediate Bactericidal Killing Elicited by Carbon Monoxide-releasing Molecules *.” Journal of Biological Chemistry 286(30): 26708–26717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayem Y, Johnson TR, Mann BE, Green CJ and Motterlini R (2006). “Protection against cisplatin-induced nephrotoxicity by a carbon monoxide-releasing molecule.” American Journal of Physiology-Renal Physiology 290(4): F789–F794. [DOI] [PubMed] [Google Scholar]
- Tenhunen R, Marver HS and Schmid R (1969). “Microsomal heme oxygenase. Characterization of the enzyme.” J Biol Chem 244(23): 6388–6394. [PubMed] [Google Scholar]
- Thom SR, Xu YA and Ischiropoulos H (1997). “Vascular Endothelial Cells Generate Peroxynitrite in Response to Carbon Monoxide Exposure.” Chemical Research in Toxicology 10(9): 1023–1031. [DOI] [PubMed] [Google Scholar]
- Thomsen LL and Miles DW (1998). “Role of nitric oxide in tumour progression: lessons from human tumours.” Cancer Metastasis Rev 17(1): 107–118. [DOI] [PubMed] [Google Scholar]
- Thorup C, Jones CL, Gross SS, Moore LC and Goligorsky MS (1999). “Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS.” American Journal of Physiology-Renal Physiology 277(6): F882–F889. [DOI] [PubMed] [Google Scholar]
- Tiong CX, Lu M and Bian J-S (2010). “Protective effect of hydrogen sulphide against 6-OHDA-induced cell injury in SH-SY5Y cells involves PKC/PI3K/Akt pathway.” British Journal of Pharmacology 161(2): 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama M, Kawakami K and Saito A (1996). “Anticryptococcal effect of amphotericin B is mediated through macrophage production of nitric oxide.” Antimicrobial Agents and Chemotherapy 40(8): 1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M-H, Yang C-M, Chang K-T, Chuang C-C, Lin W-N, Jiang R-S, Wu C-H and Lee IT (2018). “Carbon monoxide ameliorates Staphylococcus aureus-elicited COX-2/IL6/MMP-9-dependent human aortic endothelial cell migration and inflammatory responses.” Immunology Letters 203: 40–49. [DOI] [PubMed] [Google Scholar]
- Tseng CK, Lin CK, Wu YH, Chen YH, Chen WC, Young KC and Lee JC (2016). “Human heme oxygenase 1 is a potential host cell factor against dengue virus replication.” Sci Rep 6: 32176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoyi K, Ha YM, Kim YM, Lee YS, Kim HJ, Kim HJ, Seo HG, Lee JH and Chang KC (2009). “Activation of PPAR-γ by Carbon Monoxide from CORM-2 Leads to the Inhibition of iNOS but not COX-2 Expression in LPS-Stimulated Macrophages.” Inflammation 32(6): 364–371. [DOI] [PubMed] [Google Scholar]
- Tung W-H, Hsieh H-L, Lee IT and Yang C-M (2011). “Enterovirus 71 induces integrin β1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: Role of HO-1/CO in viral replication.” Journal of Cellular Physiology 226(12): 3316–3329. [DOI] [PubMed] [Google Scholar]
- Vahedian-Ardakani HA, Moghimi M, Shayestehpour M, Doosti M and Amid N (2019). “Bacterial Spectrum and Antimicrobial Resistance Pattern in Cancer Patients with Febrile Neutropenia.” Asian Pac J Cancer Prev 20(5): 1471–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J, Baardman J, Otto Natasja A., van der Velden S, Neele Annette E., van den Berg Susan M., Luque-Martin R, Chen H-J, Boshuizen Marieke C. S., Ahmed M, Hoeksema Marten A., de Vos Alex F.and Menno P de Winther J(2016). “Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages.” Cell Reports 17(3): 684–696. [DOI] [PubMed] [Google Scholar]
- Van Tubergen EA, Banerjee R, Liu M, Vander Broek R, Light E, Kuo S, Feinberg SE, Willis AL, Wolf G, Carey T, Bradford C, Prince M, Worden FP, Kirkwood KL and D’Silva NJ (2013). “Inactivation or loss of TTP promotes invasion in head and neck cancer via transcript stabilization and secretion of MMP9, MMP2, and IL-6.” Clin Cancer Res 19(5): 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini F, Kashfi K and Nath N (2015). “The dual role of iNOS in cancer.” Redox Biol 6: 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini F, MacKessack-Leitch AC, Eschbach EK, Chattopadhyay M, Kodela R and Kashfi K (2015). “Synthesis and anti-cancer potential of the positional isomers of NOSH-aspirin (NBS-1120) a dual nitric oxide and hydrogen sulfide releasing hybrid.” Bioorg Med Chem Lett 25(20): 4677–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellecco V, Mancini A, Ianaro A, Calderone V, Attanasio C, Cantalupo A, Andria B, Savoia G, Panza E, Di Martino A, Cirino G and Bucci M (2016). “Cystathionine beta-synthase-derived hydrogen sulfide is involved in human malignant hyperthermia.” Clin Sci (Lond) 130(1): 35–44. [DOI] [PubMed] [Google Scholar]
- Verma A, Hirsch DJ, Glatt CE, Ronnett GV and Snyder SH (1993). “Carbon monoxide: a putative neural messenger.” Science 259(5093): 381–384. [DOI] [PubMed] [Google Scholar]
- Vítek L, Gbelcová H, Muchová L, Váňová K, Zelenka J, Koníčková R, Šuk J, Zadinova M, Knejzlík Z, Ahmad S, Fujisawa T, Ahmed A and Ruml T (2014). “Antiproliferative effects of carbon monoxide on pancreatic cancer.” Digestive and Liver Disease 46(4): 369–375. [DOI] [PubMed] [Google Scholar]
- Vyas-Read S, Shaul PW, Yuhanna IS and Willis BC (2007). “Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells.” American Journal of Physiology-Lung Cellular and Molecular Physiology 293(1): L212–L221. [DOI] [PubMed] [Google Scholar]
- Wagner EF and Nebreda ÁR (2009). “Signal integration by JNK and p38 MAPK pathways in cancer development.” Nature Reviews Cancer 9(8): 537–549. [DOI] [PubMed] [Google Scholar]
- Wallace JL (2007). “Hydrogen sulfide-releasing anti-inflammatory drugs.” Trends Pharmacol Sci 28(10): 501–505. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Viappiani S and Bolla M (2009). “Cyclooxygenase-inhibiting nitric oxide donators for osteoarthritis.” Trends Pharmacol Sci 30(3): 112–117. [DOI] [PubMed] [Google Scholar]
- Wang L, Sun Y, Yuan Y, Mei Q and Yuan X (2020). “Clinical challenges in cancer patients with COVID-19: Aging, immunosuppression, and comorbidities.” Aging (Albany NY) 12(23): 24462–24474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Guo Z and Wang S (2014). “Regulation of Cystathionine γ-Lyase in Mammalian Cells by Hypoxia.” Biochemical Genetics 52(1): 29–37. [DOI] [PubMed] [Google Scholar]
- Wang M, Yan J, Cao X, Hua P and Li Z (2020). “Hydrogen sulfide modulates epithelial-mesenchymal transition and angiogenesis in non-small cell lung cancer via HIF-1α activation.” Biochemical Pharmacology 172: 113775. [DOI] [PubMed] [Google Scholar]
- Wang M-J, Cai W-J, Li N, Ding Y-J, Chen Y and Zhu Y-C (2009). “The Hydrogen Sulfide Donor NaHS Promotes Angiogenesis in a Rat Model of Hind Limb Ischemia.” Antioxidants & Redox Signaling 12(9): 1065–1077. [DOI] [PubMed] [Google Scholar]
- Wang R (1998). “Resurgence of carbon monoxide: an endogenous gaseous vasorelaxing factor.” Can J Physiol Pharmacol 76(1): 1–15. [DOI] [PubMed] [Google Scholar]
- Wang R (2002). “Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter?” Faseb j 16(13): 1792–1798. [DOI] [PubMed] [Google Scholar]
- Wang S-B, Zhang C, Chen Z-X, Ye J-J, Peng S-Y, Rong L, Liu C-J and Zhang X-Z (2019). “A Versatile Carbon Monoxide Nanogenerator for Enhanced Tumor Therapy and Anti-Inflammation.” ACS Nano 13(5): 5523–5532. [DOI] [PubMed] [Google Scholar]
- Wang W-J, Kan C-D, Chen C-Y, Meng Y-Y, Wang J-N, Chen W-L, Chen C-H and Li W-P (2021) “Synthetic Poly(lactic-co-glycolic Acid) Microvesicles as a Feasible Carbon Monoxide-Releasing Platform for Cancer Treatment.” Membranes 11 DOI: 10.3390/membranes11110818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zalcenstein A and Oren M (2003). “Nitric oxide promotes p53 nuclear retention and sensitizes neuroblastoma cells to apoptosis by ionizing radiation.” Cell Death & Differentiation 10(4): 468–476. [DOI] [PubMed] [Google Scholar]
- Wang Y-H, Huang J-T, Chen W-L, Wang R-H, Kao M-C, Pan Y-R, Chan S-H, Tsai K-W, Kung H-J, Lin K-T and Wang L-H (2019). “Dysregulation of cystathionine γ-lyase promotes prostate cancer progression and metastasis.” EMBO reports 20(10): e45986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Cook T, Alber S, Liu K, Kovesdi I, Watkins SK, Vodovotz Y, Billiar TR and Blumberg D (2004). “Adenoviral Gene Transfer of the Human Inducible Nitric Oxide Synthase Gene Enhances the Radiation Response of Human Colorectal Cancer Associated with Alterations in Tumor Vascularity.” Cancer Research 64(4): 1386–1395. [DOI] [PubMed] [Google Scholar]
- Ward JS, Lynam JM, Moir J and Fairlamb IJS (2014). “Visible-Light-Induced CO Release from a Therapeutically Viable Tryptophan-Derived Manganese(I) Carbonyl (TryptoCORM) Exhibiting Potent Inhibition against E. coli.” Chemistry – A European Journal 20(46): 15061–15068. [DOI] [PubMed] [Google Scholar]
- Was H, Cichon T, Smolarczyk R, Rudnicka D, Stopa M, Chevalier C, Leger JJ, Lackowska B, Grochot A, Bojkowska K, Ratajska A, Kieda C, Szala S, Dulak J and Jozkowicz A (2006). “Overexpression of Heme Oxygenase-1 in Murine Melanoma: Increased Proliferation and Viability of Tumor Cells, Decreased Survival of Mice.” The American Journal of Pathology 169(6): 2181–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, Penacho N, Seth P, Sukhatme V, Ahmed A, Pandolfi PP, Helczynski L, Bjartell A, Persson JL and Otterbein LE (2013). “Carbon Monoxide Expedites Metabolic Exhaustion to Inhibit Tumor Growth.” Cancer Research 73(23): 7009–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, Soares MP and Otterbein LE (2014). “Macrophages sense and kill bacteria through carbon monoxide–dependent inflammasome activation.” The Journal of Clinical Investigation 124(11): 4926–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Richardson EL, Zhu K, Wang L, Le X, He Y, Huang S and Xie K (2003). “Direct Demonstration of Negative Regulation of Tumor Growth and Metastasis by Host-inducible Nitric Oxide Synthase1.” Cancer Research 63(14): 3855–3859. [PubMed] [Google Scholar]
- Weiss JM, Ridnour LA, Back T, Hussain SP, He P, Maciag AE, Keefer LK, Murphy WJ, Harris CC, Wink DA and Wiltrout RH (2010). “Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy.” Journal of Experimental Medicine 207(11): 2455–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westley AM and Westley J (1991). “Biological sulfane sulfur.” Anal Biochem 195(1): 63–67. [DOI] [PubMed] [Google Scholar]
- Wheeler MA, Smith SD, García-Cardeña G, Nathan CF, Weiss RM and Sessa WC (1997). “Bacterial infection induces nitric oxide synthase in human neutrophils.” J Clin Invest 99(1): 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M and Moore PK (2006). “Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide.” Biochemical and Biophysical Research Communications 343(1): 303–310. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Li L, Rose P, Tan C-H, Parkinson DB and Moore PK (2009). “The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages.” Antioxidants & Redox Signaling 12(10): 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JL, Nath N, Chen J, Hundley TR, Gao J, Kopelovich L, Kashfi K and Rigas B (2003). “Growth Inhibition of Human Colon Cancer Cells by Nitric Oxide (NO)-Donating Aspirin Is Associated with Cyclooxygenase-2 Induction and β-Catenin/T-Cell Factor Signaling, Nuclear Factor-κB, and NO Synthase 2 Inhibition: Implications for Chemoprevention.” Cancer Research 63(22): 7613–7618. [PubMed] [Google Scholar]
- Wilson JL, Jesse HE, Hughes B, Lund V, Naylor K, Davidge KS, Cook GM, Mann BE and Poole RK (2012). “Ru(CO)3Cl(Glycinate) (CORM-3): A Carbon Monoxide–Releasing Molecule with Broad-Spectrum Antimicrobial and Photosensitive Activities Against Respiration and Cation Transport in Escherichia coli.” Antioxidants & Redox Signaling 19(5): 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Wu B, Guo J, Luo W, Wu D, Yang H, Zhen Y, Yu X, Wang H, Zhou Y, Liu Z, Fang W and Yang Z (2011). “Elevated expression of CDK4 in lung cancer.” Journal of Translational Medicine 9(1): 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Li M, Tian W, Wang S, Cui L, Li H, Wang H, Ji A and Li Y (2017). “Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways.” Scientific Reports 7(1): 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Wan F, Fu H, Li N and Gao H (2015). “A Matter of Timing: Contrasting Effects of Hydrogen Sulfide on Oxidative Stress Response in Shewanella oneidensis.” Journal of Bacteriology 197(22): 3563–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chen A, Zhou Y, Zheng S, Yang Y, An Y, Xu K, He H, Kang J, Luckanagul JA, Xian M, Xiao J and Wang Q (2019). “Novel H2S-Releasing hydrogel for wound repair via in situ polarization of M2 macrophages.” Biomaterials 222: 119398. [DOI] [PubMed] [Google Scholar]
- Wu L and Wang R (2005). “Carbon monoxide: endogenous production, physiological functions, and pharmacological applications.” Pharmacol Rev 57(4): 585–630. [DOI] [PubMed] [Google Scholar]
- Wu YC, Wang XJ, Yu L, Chan FKL, Cheng ASL, Yu J, Sung JJY, Wu WKK and Cho CH (2012). “Hydrogen Sulfide Lowers Proliferation and Induces Protective Autophagy in Colon Epithelial Cells.” PLOS ONE 7(5): e37572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wushou A, Hou J, Zhao Y-J and Shao Z-M (2014) “Twist-1 Up-Regulation in Carcinoma Correlates to Poor Survival.” International Journal of Molecular Sciences 15, 21621–21630 DOI: 10.3390/ijms151221621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Liang S, Zhao Y, Pang M, Ma P. a., Cheng Zand Lin J(2021). “Multifunctional carbon monoxide nanogenerator as immunogenic cell death drugs with enhanced antitumor immunity and antimetastatic effect.” Biomaterials 277: 121120. [DOI] [PubMed] [Google Scholar]
- Xie K, Wang Y, Huang S, Xu L, Bielenberg D, Salas T, McConkey DJ, Jiang W and Fidler IJ (1997). “Nitric oxide-mediated apoptosis of K-1735 melanoma cells is associated with downregulation of Bcl-2.” Oncogene 15(7): 771–779. [DOI] [PubMed] [Google Scholar]
- Xie L, Feng H, Li S, Meng G, Liu S, Tang X, Ma Y, Han Y, Xiao Y, Gu Y, Shao Y, Park CM, Xian M, Huang Y, Ferro A, Wang R, Moore PK, Wang H and Ji Y (2016). “SIRT3 Mediates the Antioxidant Effect of Hydrogen Sulfide in Endothelial Cells.” Antioxid Redox Signal 24(6): 329–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong T, Liu XW, Huang XL, Xu XF, Xie WQ, Zhang SJ and Tu J (2018). “Tristetraprolin: A novel target of diallyl disulfide that inhibits the progression of breast cancer.” Oncol Lett 15(5): 7817–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Zheng S, Dweik RA and Erzurum SC (2006). “Role of epithelial nitric oxide in airway viral infection.” Free Radical Biology and Medicine 41(1): 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Takano N, Ishiwata K and Suematsu M (2010). “Carbon monoxide stimulates global protein methylation via its inhibitory action on cystathionine β-synthase.” Journal of Clinical Biochemistry and Nutrition 48(1): 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Oka H, Mizuguchi S, Toda M, Minamiyama Y, Takemura S, Shibata T, Cepinskas G and Nishiyama N (2018). “Carbon monoxide-releasing molecule, CORM-3, modulates alveolar macrophage M1/M2 phenotype in vitro.” Inflammopharmacology 26(2): 435–445. [DOI] [PubMed] [Google Scholar]
- Yan C and Boyd DD (2007). “Regulation of matrix metalloproteinase gene expression.” Journal of Cellular Physiology 211(1): 19–26. [DOI] [PubMed] [Google Scholar]
- Yan Y, Du C, Li G, Chen L, Yan Y, Chen G, Hu W and Chang L (2018). “CO suppresses prostate cancer cell growth by directly targeting LKB1/AMPK/mTOR pathway in vitro and in vivo.” Urologic Oncology: Seminars and Original Investigations 36(6): 312.e311–312.e318. [DOI] [PubMed] [Google Scholar]
- Yanfei W, Lin S, Junbao D and Chaoshu T (2006). “Impact of L-arginine on hydrogen sulfide/cystathionine-gamma-lyase pathway in rats with high blood flow-induced pulmonary hypertension.” Biochem Biophys Res Commun 345(2): 851–857. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L and Wang R (2012). “Hydrogen Sulfide Protects Against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2.” Antioxidants & Redox Signaling 18(15): 1906–1919. [DOI] [PubMed] [Google Scholar]
- Yang J, Minkler P, Grove D, Wang R, Willard B, Dweik R and Hine C (2019). “Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B(6).” Commun Biol 2: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P-M, Huang Y-T, Zhang Y-Q, Hsieh C-W and Wung B-S (2016). “Carbon monoxide releasing molecule induces endothelial nitric oxide synthase activation through a calcium and phosphatidylinositol 3-kinase/Akt mechanism.” Vascular Pharmacology 87: 209–218. [DOI] [PubMed] [Google Scholar]
- Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, Chen C, Liu S, Liu D, Chen Y, Zandi E, Chen W, Zhou Y and Shi S (2015). “Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis.” Immunity 43(2): 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Yu T, Liu D, Shi S and Zhou Y (2018). “Hydrogen sulfide promotes immunomodulation of gingiva-derived mesenchymal stem cells via the Fas/FasL coupling pathway.” Stem Cell Research & Therapy 9(1): 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Yu M, Yang D, Li J, Wang H, Chen F, Yu H, Shen T, Zhu Q and Zhou C (2020). “Exogenous hydrogen sulfide donor NaHS alleviates nickel-induced epithelial-mesenchymal transition and the migration of A549 cells by regulating TGF-β1/Smad2/Smad3 signaling.” Ecotoxicology and Environmental Safety 195: 110464. [DOI] [PubMed] [Google Scholar]
- Yin H, Fang J, Liao L, Maeda H and Su Q (2014). “Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity.” BMC Cancer 14(1): 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong Q-C, Hu L-F, Wang S, Huang D and Bian J-S (2010). “Hydrogen sulfide interacts with nitric oxide in the heart: possible involvement of nitroxyl.” Cardiovascular Research 88(3): 482–491. [DOI] [PubMed] [Google Scholar]
- Yoo YM, Jung EM, Ahn C and Jeung EB (2018). “Nitric oxide prevents H(2)O(2)-induced apoptosis in SK-N-MC human neuroblastoma cells.” Int J Biol Sci 14(14): 1974–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youness RA, Gad AZ, Sanber K, Ahn YJ, Lee G-J, Khallaf E, Hafez HM, Motaal AA, Ahmed N and Gad MZ (2021). “Targeting hydrogen sulphide signaling in breast cancer.” Journal of Advanced Research 27: 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Jin H, Xu J, Gu J, Li X, Xie Q, Huang H, Li J, Tian Z, Jiang G, Chen C, He F, Wu X-R and Huang C (2018). “XIAP overexpression promotes bladder cancer invasion in vitro and lung metastasis in vivo via enhancing nucleolin-mediated Rho-GDIβ mRNA stability.” International Journal of Cancer 142(10): 2040–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue T, Li J, Zhu J, Zuo S, Wang X, Liu Y, Liu J, Liu X, Wang P and Chen S (2023). “Hydrogen Sulfide Creates a Favorable Immune Microenvironment for Colon Cancer.” Cancer Research 83(4): 595–612. [DOI] [PubMed] [Google Scholar]
- Zanardo RCO, Brancaleone V, Distrutti E, Fiorucci S, Cirino G and Wallace JL (2006). “Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation.” The FASEB Journal 20(12): 2118–2120. [DOI] [PubMed] [Google Scholar]
- Zaorsky NG, Churilla TM, Egleston BL, Fisher SG, Ridge JA, Horwitz EM and Meyer JE (2017). “Causes of death among cancer patients.” Annals of Oncology 28(2): 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai T, Zhong W, Gao Y, Zhou H, Zhou Z, Liu X, Yang S and Yang H (2021). “Tumor Microenvironment-Activated Nanoparticles Loaded with an Iron-Carbonyl Complex for Chemodynamic Immunotherapy of Lung Metastasis of Melanoma In Vivo.” ACS Applied Materials & Interfaces 13(33): 39100–39111. [DOI] [PubMed] [Google Scholar]
- Zhang A, Zhao L, Li N, Duan H, Liu H, Pu F, Zhang G, Zhou E-M and Xiao S (2016). “Carbon Monoxide Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by the Cyclic GMP/Protein Kinase G and NF-κB Signaling Pathway.” Journal of Virology 91(1): e01866–01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Qi Q, Yang J, Sun D, Li C, Xue Y, Jiang Q, Tian Y, Xu C and Wang R (2015). “An Anticancer Role of Hydrogen Sulfide in Human Gastric Cancer Cells.” Oxidative Medicine and Cellular Longevity 2015: 636410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhu F, Xie L, Wang C, Wang J, Chen R, Jia P, Guan HQ, Peng L, Chen Y, Peng P, Zhang P, Chu Q, Shen Q, Wang Y, Xu SY, Zhao JP and Zhou M (2020). “Clinical characteristics of COVID-19-infected cancer patients: a retrospective case study in three hospitals within Wuhan, China.” Annals of Oncology 31(7): 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang G, Chen X, Chen Z, Tan AY, Lin A, Zhang C, Torres LK, Bajrami S, Zhang T, Zhang G, Xiang JZ, Hissong EM, Chen Y-T, Li Y and Du Y-CN (2022). “Low-dose carbon monoxide suppresses metastatic progression of disseminated cancer cells.” Cancer Letters 546: 215831. [DOI] [PubMed] [Google Scholar]
- Zhang W and Liu HT (2002). “MAPK signal pathways in the regulation of cell proliferation in mammalian cells.” Cell Research 12(1): 9–18. [DOI] [PubMed] [Google Scholar]
- Zhao K, Ju Y, Li S, Altaany Z, Wang R and Yang G (2014). “S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair.” EMBO reports 15(7): 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y and Wang R (2001). “The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener.” The EMBO Journal 20(21): 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y and Wang R (2001). “The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener.” Embo j 20(21): 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W-N, Wang M, Zhang C, Sun S and Xu Y (2022). “Cancer cell membrane targeting and red light-triggered carbon monoxide (CO) release for enhanced chemotherapy.” Chemical Communications 58(61): 8512–8515. [DOI] [PubMed] [Google Scholar]
- Zhen Y, Pan W, Hu F, Wu H, Feng J, Zhang Y and Chen J (2015). “Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-κB pathway in PLC/PRF/5 hepatoma cells.” Int J Oncol 46(5): 2194–2204. [DOI] [PubMed] [Google Scholar]
- Zheng D-W, Li B, Li C-X, Xu L, Fan J-X, Lei Q and Zhang X-Z (2017). “Photocatalyzing CO2 to CO for Enhanced Cancer Therapy.” Advanced Materials 29(44): 1703822. [DOI] [PubMed] [Google Scholar]
- Zhengnan Y, Ladie Kimberly De La C, Xiaoxiao Yand Binghe W(2022). “Carbon Monoxide Signaling: Examining Its Engagement with Various Molecular Targets in the Context of Binding Affinity, Concentration, and Biologic Response.” Pharmacological Reviews 74(3): 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi L, Ang AD, Zhang H, Moore PK and Bhatia M (2007). “Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-κB pathway.” Journal of Leukocyte Biology 81(5): 1322–1332. [DOI] [PubMed] [Google Scholar]
- Zhong G, Chen F, Cheng Y, Tang C and Du J (2003). “The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase.” Journal of Hypertension 21(10). [DOI] [PubMed] [Google Scholar]
- Zhou Y, Li X-H, Zhang C-C, Wang M-J, Xue W-L, Wu D-D, Ma F-F, Li W-W, Tao B-B and Zhu Y-C (2015). “Hydrogen sulfide promotes angiogenesis by downregulating miR-640 via the VEGFR2/mTOR pathway.” American Journal of Physiology-Cell Physiology 310(4): C305–C317. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Martin E, Sharina I, Esposito I, Szabo C, Bucci M, Cirino G and Papapetropoulos A (2016). “Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide.” Pharmacological Research 111: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X-Y, Liu S-J, Liu Y-J, Wang S and Ni X (2010). “Glucocorticoids suppress cystathionine gamma-lyase expression and H2S production in lipopolysaccharide-treated macrophages.” Cellular and Molecular Life Sciences 67(7): 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XY, Liu SJ, Liu YJ, Wang S and Ni X (2010). “Glucocorticoids suppress cystathionine gamma-lyase expression and H2S production in lipopolysaccharide-treated macrophages.” Cell Mol Life Sci 67(7): 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Wilson AT, Mathahs MM, Wen F, Brown KE, Luxon BA and Schmidt WN (2008). “Heme oxygenase-1 suppresses hepatitis C virus replication and increases resistance of hepatocytes to oxidant injury.” Hepatology 48(5): 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z, Liu J, Sun Z, Cheng Y-W, Ewing M, Bugge TH, Finkel T, Leppla SH and Liu S (2023). “ERK and c-Myc signaling in host-derived tumor endothelial cells is essential for solid tumor growth.” Proceedings of the National Academy of Sciences 120(1): e2211927120. [DOI] [PMC free article] [PubMed] [Google Scholar]