

Summary
Transcription factors (TFs) orchestrate the gene expression programs that define each cell’s identity. The canonical TF accomplishes this with two domains, one that binds specific DNA sequences and the other that binds protein coactivators or corepressors. We find that at least half of TFs also bind RNA, doing so through a previously unrecognized domain with sequence and functional features analogous to the arginine-rich motif of the HIV transcriptional activator Tat. RNA binding contributes to TF function by promoting the dynamic association between DNA, RNA and TF on chromatin. TF-RNA interactions are a conserved feature important for vertebrate development and disrupted in disease. We propose that the ability to bind DNA, RNA and protein is a general property of many TFs and is fundamental to their gene regulatory function.
Keywords: Transcription factor, RNA, gene regulation, RNA-binding proteins, arginine-rich motif, single-molecule imaging, development
Graphical Abstract
eTOC (In brief)
Oksuz et al. provide evidence that transcription factors frequently bind RNA at active loci, doing so with a conserved domain resembling the arginine-rich motif of the HIV Tat protein. TF-RNA binding constrains TF mobility in chromatin, contributes to gene regulation, is important for normal development and, when defective, disease pathogenesis.
Main
Transcription factors (TFs), which are encoded by ~1,600 genes in the human genome, comprise the single largest protein family in mammals. Each cell type expresses approximately 150–400 TFs, which together control the gene expression program of the cell1–5. TFs typically contain DNA-binding domains that recognize specific sequences and multiple TFs collectively bind to enhancers and promoter-proximal regions of genes6,7. The DNA-binding domains form stable structures whose conserved features are reliably detected by homology and are therefore used to classify TFs (e.g. C2H2 zinc finger, homeodomain, bHLH, bZIP) (Figure 1A)1,2. TFs also contain effector domains that exhibit less sequence conservation and sample many transient structures that enable multivalent protein interactions8–10. These effector domains recruit coactivator or corepressor proteins, which contribute to gene regulation through mechanisms that include mobilizing nucleosomes, modifying chromatin-associated proteins, influencing genome architecture, recruiting transcription apparatus and controlling aspects of transcription initiation and elongation11,12. This canonical view of TFs that function with two domains, one binding DNA and the other protein, has been foundational for models of gene regulation13,14.
Figure 1. Transcription factor binding to RNA in cells.
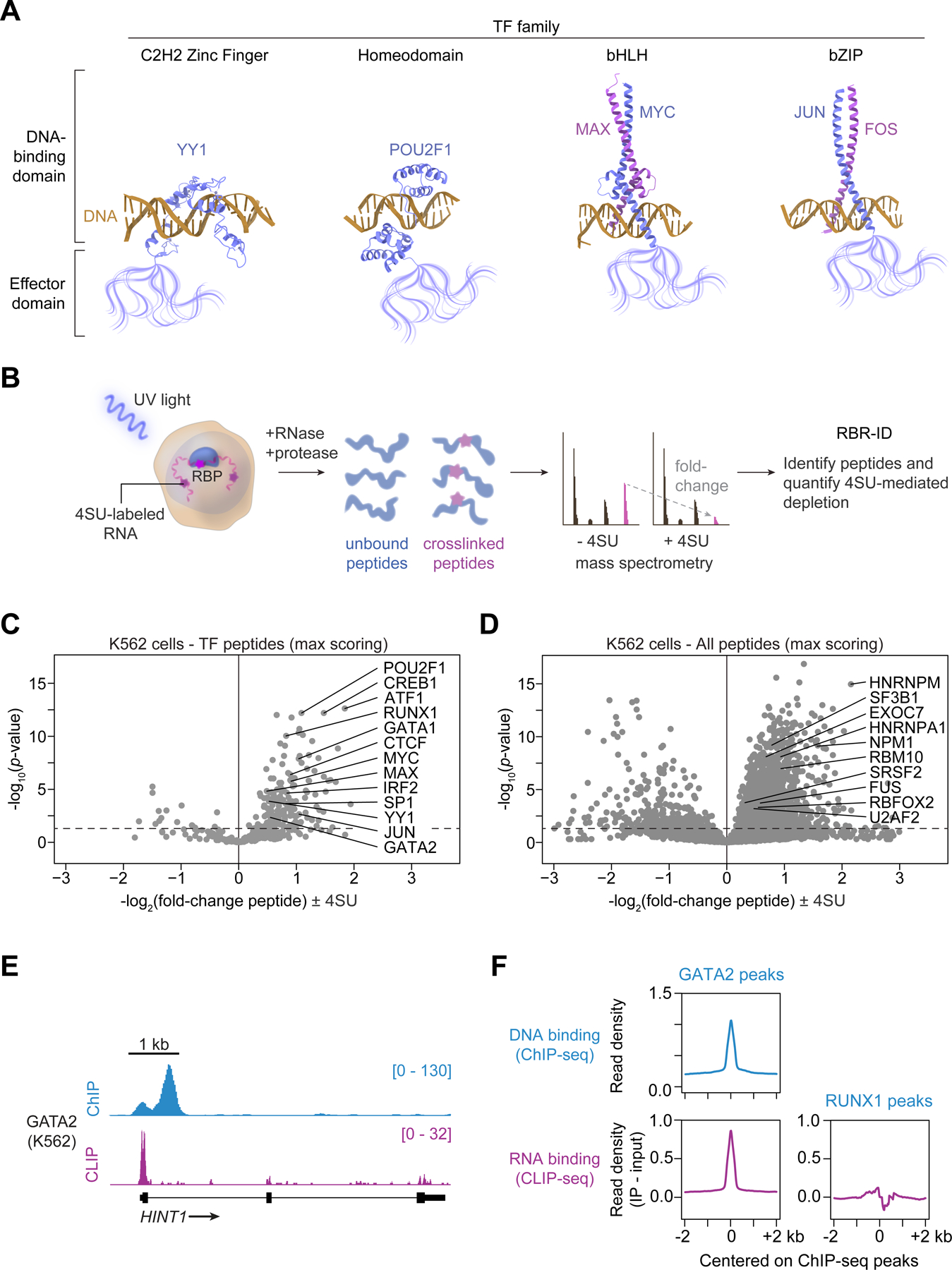
(A) Schematic of DNA-binding and effector domains in transcription factors from different families (PDB accession numbers in Methods).
(B) Experimental scheme for RBR-ID in human K562 cells. 4SU-labeled RNAs are crosslinked to proteins with UV light. RNA-binding peptides are identified by comparing the levels of crosslinked and unbound peptides by mass spectrometry.
(C) Volcano plot of TF peptides in RBR-ID for human K562 cells with select highlighted TFs (dotted line at p=0.05). Each marker represents the peptide with maximum RBR-ID score for each protein.
(D) Volcano plot of all detected peptides in RBR-ID for human K562 cells with select highlighted RBPs (dotted line at p=0.05). Each marker represents the peptide with maximum RBR-ID score for each protein.
(E) ChIP-seq and CLIP signal for GATA2 at the HINT1 locus in K562 cells.
(F) Meta-gene analysis of input-subtracted CLIP signal centered on GATA2 or RUNX1 ChIP-seq peaks in K562 cells.
RNA molecules are produced at loci where TFs are bound, but their roles in gene regulation are not well-understood15,16. A few TFs and cofactors have been reported to bind RNA17–28, but TFs do not harbor domains characteristic of well-studied RNA binding proteins29. We wondered whether TFs might have evolved to interact with RNA molecules that are pervasively present at gene regulatory regions but harbor a heretofore unrecognized RNA-binding domain. Here we present evidence that a broad spectrum of TFs do bind RNA molecules, that TFs accomplish this with a domain analogous to the RNA-binding arginine-rich motif of the HIV Tat transactivator, and that this domain promotes TF occupancy at regulatory loci. These domains are a conserved feature important for vertebrate development, and they are disrupted in cancer and developmental disorders.
Transcription factor binding to RNA in cells
Using nuclei isolated from human K562 cells, we performed a high throughput RNA-protein crosslinking assay (RNA-binding region identification - RBR-ID), which uses UV crosslinking and mass spectrometry to detect angstrom-scale crosslinks, typically thought to reflect direct interactions30, between protein and RNA molecules in cells31 (Figure 1B). The results included the expected distribution of peptides from known RNA-binding proteins (RBPs) and revealed that a broad distribution of TFs had peptides crosslinked to RNA in this assay independent of their cellular abundance (Figures 1C, 1D, and S1A). Nearly half (48%) of TFs identified in the RBR-ID dataset showed evidence of RNA binding in K562 cells (Figure S1B) when the analysis was conducted using thresholds that retain RBPs verified by independent methods31 (Table S1). These results prompted a re-examination of previously published RBR-ID data for murine embryonic stem cells (ESCs)31 which confirmed that a substantial fraction of TFs (41%) in those cells also bind RNA (Figures S1C-1E, and Table S2). A meta-analysis of data from multiple studies using proteomics to identify RNA-binding proteins, including data collected in this study, provides an extensive list of RNA-binding TFs (Table S3).
Specific TFs are notable for their roles in control of cell identity and have been subjected to more extensive study than others. Many well-studied TFs that contribute to the control of cell identity were observed among the TFs that showed evidence of RNA binding. In K562 hematopoietic cells, these included GATA1, GATA2, and RUNX1, which play major roles in regulation of hematopoietic cell genes32, as well as MYC and MAX, oncogenic regulators of these tumor cells33 (Figure 1C). In the ESCs, these included the master pluripotency regulators Oct4, Klf4, and Nanog, as well as the MYC family member that is key to proliferation of these cells, Mycn34 (Figure S1D). The RNA-binding TFs also included those involved in other important cellular processes, including regulation of chromatin structure (CTCF, YY1) and response to signaling (CREB1, IRF2, ATF1) (Figure 1C). It was notable that RNA binding was a property of TFs that span many TF families (Figures S1F and S1G). These results suggest that RNA binding is a property shared by TFs that participate in diverse cellular processes and that possess diverse DNA-binding domains.
We next sought to identify the RNAs that interact with specific TFs. We conducted CLIP for the TF GATA2, a major regulator of hematopoietic genes in K562 cells that showed evidence of RNA binding in our RBR-ID data (Figure 1C). Immunoprecipitation of HA- and FLAG-tagged GATA2 in K562 cells subjected to UV cross-linking showed that GATA2 interacts with RNA in cells in a 4SU-dependent manner (Figure S2A). Interacting RNAs were then sequenced and cross-linked sites were identified with nucleotide resolution (Figure S2B, Table S4, STAR Methods). A diversity of RNA species were bound by GATA2, including many enhancer- and promoter-derived RNAs (Table S4). We reasoned that GATA2 may interact with RNAs transcribed in proximity to regions where GATA2 binds chromatin to regulate genes. Indeed, as illustrated for a specific locus, GATA2 binds chromatin at the HINT1 gene measured by ChIP-seq, and GATA2 interacts with RNA transcribed from the HINT1 gene measured by CLIP-seq (Figure 1E). A metagene analysis revealed that GATA2 CLIP signal was enriched at GATA2 ChIP-seq peaks (Figure 1F). Enrichment of GATA2 CLIP signal was not evident at ChIP-seq peaks of RUNX1, another major regulator of hematopoietic genes (Figure 1F). These results prompted a re-examination of previously published CLIP/ChIP data for RBR-ID+ YY1 and CTCF21,35,36, which also showed that these TFs interact with RNAs transcribed from loci near their chromatin-binding sites (Figures S2C and S2D). These results suggest that TFs bind to RNAs produced in the vicinity of their DNA-binding sites.
Transcription factor binding to RNA in vitro
To corroborate evidence that TFs can bind RNA molecules in cells, we sought to confirm that purified TFs bind RNA molecules in vitro using a fluorescence polarization assay (Figure 2A, STAR Methods). The assay was validated with multiple control proteins with an RNA of random sequence, including three well-studied RNA-binding proteins (U2AF2, HNRNPA1, and SRSF2) and proteins that were not expected to have substantial affinity for RNA (GFP and the DNA-binding restriction enzyme BamHI). The RBPs bound RNA with nanomolar affinities, consistent with previous studies37–40, whereas GFP and BamHI showed little affinity for RNA (Figure 2B). We then selected 13 TFs that showed evidence of crosslinking to RNA in cells, are well-studied for their diverse cellular functions and are members of different TF families, purified them from human cells and measured their RNA-binding affinities. These TFs exhibited a range of binding affinities for the RNA, ranging from 41 to 505 nM, which is remarkably similar to the range of affinities measured for known RBPs (42 to 572 nM) (Figure 2C). Thus, a diverse set of TFs can bind RNA with affinities similar to proteins with known physiological roles in RNA processing. The thousands of enhancers and promoter-proximal regions where TFs bind have diverse sequences, and thus RNA molecules produced from these sites differ in sequence, so we investigated whether TFs bind diverse RNA sequences. Six TFs were investigated, and the results indicate that these TFs do bind various RNA sequences with similar affinities (Figures S2E and S2F).
Figure 2. Transcription factor binding to RNA in vitro.
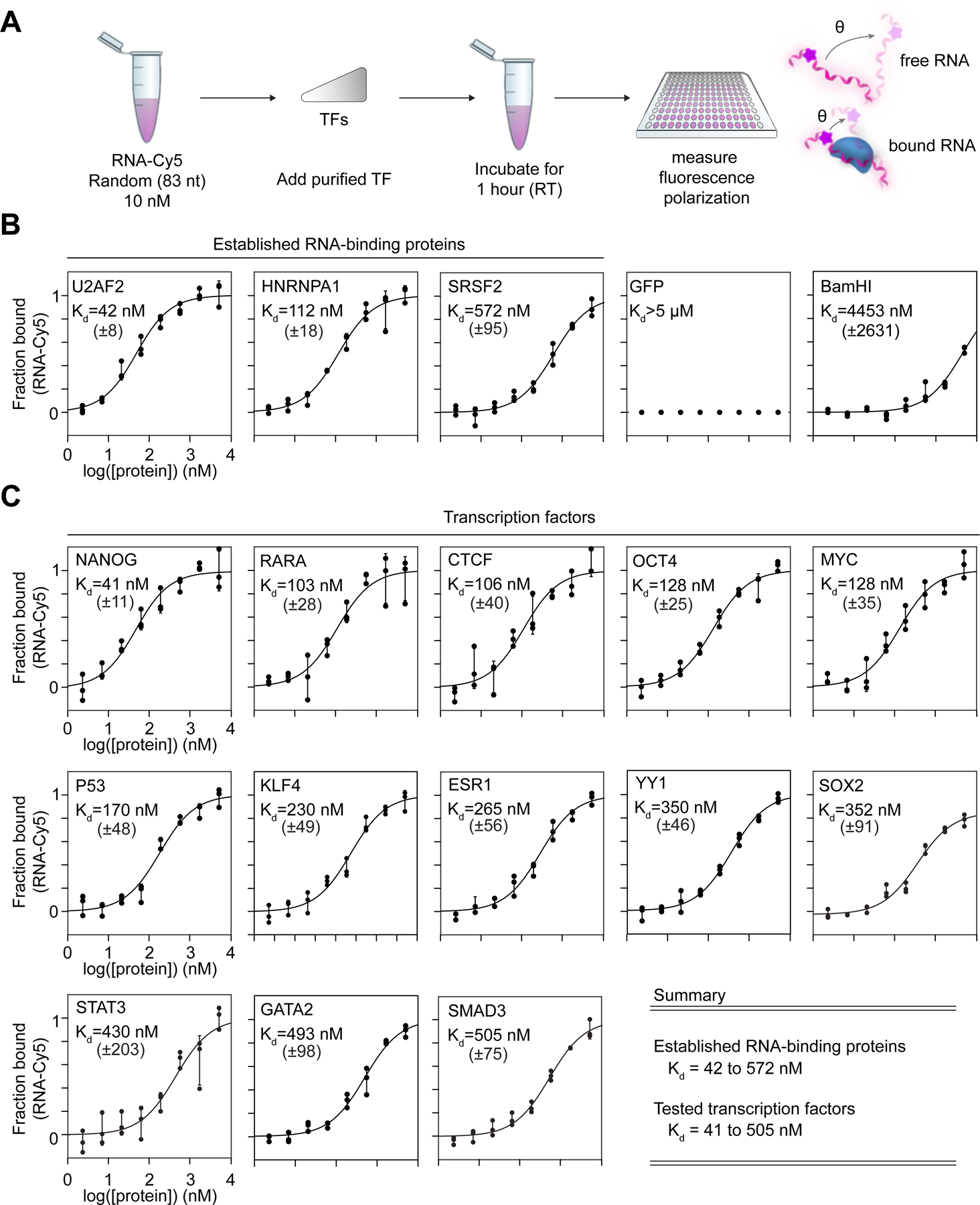
(A) Experimental scheme for measuring the equilibrium dissociation constant for protein-RNA binding. Cy5-labeled RNA and increasing concentrations of purified proteins are incubated and protein-RNA interactions is measured by fluorescence polarization assay.
(B) Fraction bound RNA with increasing protein concentration for established RNA-binding proteins, GFP, and the restriction enzyme BamHI (error bars depict s.d.).
(C) Fraction bound RNA with increasing protein concentration for select transcription factors (error bars depict s.d.). A summary of values for established RNA-binding proteins and TFs are indicated.
An arginine-rich domain in transcription factors
We next sought to identify regions in TFs that contribute to RNA binding. TFs do not contain sequence motifs that resemble those of structured RNA-binding domains29,38 (Figures S3A and S3B), so we searched for local amino acid features that might be common to TFs. Nearly 80% of TFs were found to have a cluster of basic residues (R/K) adjacent to their DNA-binding domain (Figure 3A). Derivation of a position-weight matrix from these “basic patches” revealed that they contain a sequence motif similar to the RNA-binding domain of the HIV Tat transactivator, which has been termed the arginine-rich motif (ARM)41,42 (Figure 3B). These ARM-like domains were enriched in TFs compared to the remainder of the proteome (Figure 3C). Furthermore, the ARM-like domains have sequences that are evolutionarily conserved and appear adjacent to diverse types of DNA-binding domains, as illustrated for KLF4, SOX2, and GATA2 (Figures 3D, S3C, and S3D). This analysis suggests that TFs often contain conserved ARM-like domains, which we will refer to hereafter as TF-ARMs.
Figure 3. An arginine-rich domain in transcription factors.
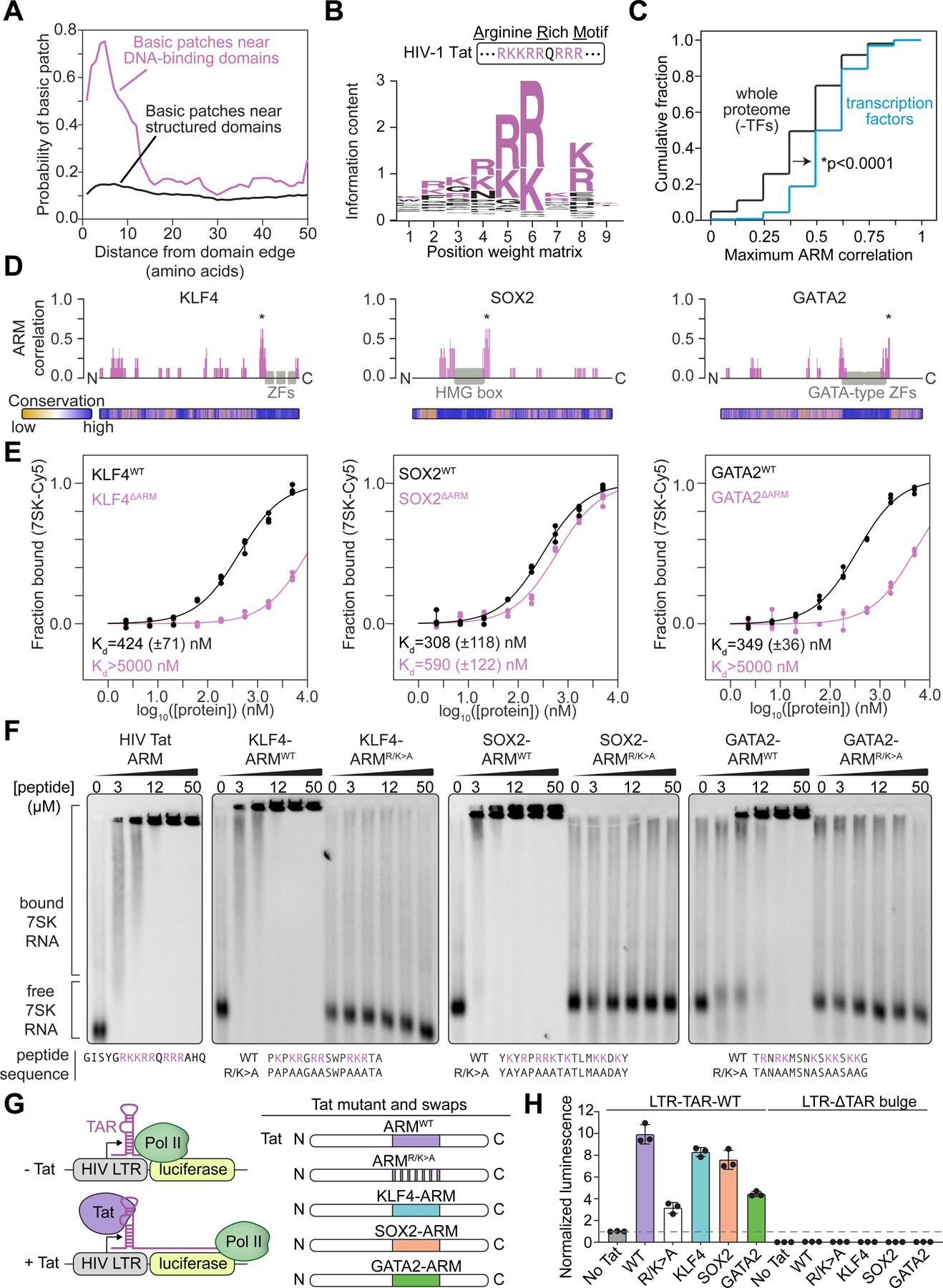
(A) Plot depicting the probability of a basic patch as a function of the distance from either DNA-binding domains (magenta) or all other annotated structured domains (black).
(B) Sequence logo derived from a position-weight matrix generated from the basic patches of TFs.
(C) Cumulative distribution plot of maximum cross-correlation scores between proteins and the Tat ARM (*p < 0.0001, Mann Whitney U test) for the whole proteome excluding TFs (black line) or TFs alone (blue line).
(D) Diagram of select TFs and their cross-correlation to the Tat ARM across a sliding window (*maximum scoring ARM-like region). Evolutionary conservation as calculated by ConSurf (Methods) is provided as a heatmap below the protein diagram.
(E) Fraction bound RNA with increasing protein concentration for wildtype (WT) or deletion (ΔARM) TFs (KLF4 WT vs ΔARM: p=0.017; SOX2 WT vs ΔARM: p=0.0012; GATA2 WT vs ΔARM: p=0.018).
(F) Gel shift assay for 7SK RNA with synthesized peptides encoding wildtype or R/K>A mutations of TF-ARMs.
(G) Experimental scheme for Tat transactivation assay. RNA Pol II transcribes the luciferase gene in the presence of Tat protein and bulge-containing TAR RNA. Indicated TF-ARMs are tested for their ability to replace Tat ARM.
(H) Bar plots depicting the normalized luminescence values for the Tat transactivation assay with or without the TAR RNA bulge with the indicated TF-ARM replacements. Values are normalized to the control condition (padj<0.0001 for Tat RK>A compared to No Tat, WT Tat, KLF4, SOX2, and all conditions with TAR deletion; padj = 0.0086 for Tat RK>A compared to GATA2, Sidak multiple comparison test).
To investigate whether TF-ARMs are necessary for RNA binding, we purified wild-type and deletion mutant versions of KLF4, SOX2 and GATA2 and compared their RNA binding affinities. The 7SK RNA was used in this assay because it is one of a number of RNA species known to be bound by HIV Tat43. RNA binding by the ARM-deleted proteins was substantially reduced (Figure 3E). To determine if the TF-ARMs are sufficient for RNA binding, peptides containing the HIV Tat ARM and TF-ARMs were synthesized and their ability to bind 7SK RNA was investigated using an electrophoretic mobility shift assay (EMSA). The results showed that all the TF-ARM peptides can bind 7SK RNA, as did the control HIV Tat ARM peptide (Figure 3F). This binding was dependent on arginine and lysine residues within the TF-ARMs (Figure 3F), as has been previously demonstrated for the Tat ARM41,43. These results indicate that TF-ARMs are necessary and sufficient for RNA binding.
We considered the possibility that the TF-ARM also contributes to DNA-binding. Synthesized peptides of the SOX2 and KLF4 ARMs were tested for binding to either DNA or RNA. The results show that both ARMs bind RNA with greater affinity compared to DNA (Figures S4A and S4B). Full-length wildtype and ARM-deleted SOX2 and KLF4 were also tested for binding to motif-containing DNA. The results show that deletion of the SOX2 ARM did not affect DNA-binding (Figure S4C). Deletion of the KLF4 ARM did affect DNA-binding (Figure S4D), although not to the extent that it affected RNA binding (Figure 3E). It thus appears possible that some TF-ARMs can contribute to DNA-binding to some extent whereas others do not.
Having found that TF-ARMs bind to RNA in vitro in assays with purified components, we next asked whether TF-ARMs bind RNA in the more complex environment of the cell. To investigate this, we analyzed the RBR-ID data (Figures 1B-D), which can provide spatial information on the regions of proteins that bind RNA in cells. If TF-ARMs were binding to RNA in cells, then we would expect an enrichment of RBR-ID+ peptides overlapping or adjacent to the TF-ARMs. Global analysis of RBR-ID+ peptides in human K562 cells, as well as inspection of RBR-ID+ peptides for individual TFs, confirmed that this was the case (Figure S5). These results provide evidence that ARM-like regions in TFs bind to RNA in cells.
To investigate if TF-ARMs could function similarly to the Tat ARM in cells, we tested whether TF-ARMs could replace the Tat ARM in a classical Tat transactivation assay41. In this assay, the HIV-1 5’ long terminal repeat (LTR) is placed upstream of a luciferase reporter gene. Transcription of the LTR generates an RNA stem loop structure called the Trans-activation Response (TAR), and HIV Tat binds to the TAR RNA to stimulate expression of the reporter gene44 (Figure 3G). We confirmed that expression of full-length Tat stimulates luciferase expression, and that mutation of the lysines and arginines in the Tat ARM reduces this activity (Figure 3H). Replacing the Tat ARM with the TF-ARMs of KLF4, SOX2, or GATA2 rescued the loss of the Tat ARM (Figure 3H). In all cases, activation was dependent on the TAR RNA bulge structure, which is required for Tat binding44 (Figure 3H). These results indicate that the TF-ARMs can perform the functions described for the Tat ARM and activate gene expression in an RNA-dependent manner.
TF-ARMs enhance TF chromatin occupancy and gene expression
TFs bind enhancer and promoter elements in chromatin and regulate transcriptional output, so it is possible that RNA binding, enabled by TF-ARMs, contributes to chromatin occupancy and gene expression. We investigated whether TF-ARMs contributed to TF association with chromatin by measuring the relative levels of TFs in chromatin and nucleoplasmic fractions from ES cells containing HA-tagged TFs with wild-type and mutant ARMs. Genome-wide localization of KLF4 and SOX2 was globally reduced upon deletion of their ARMs (Figure 4A) as determined by CUT&Tag and illustrated for specific genes regulated by KLF4 or SOX2 (Figure 4B). Nuclear fractionation confirmed that deletion of the ARMs reduced the levels of KLF4 and SOX2 in chromatin (Figures S6A and S6B), and treatment of the extracts with RNase reduced TF enrichment in the chromatin fraction (Figures S6C and S6D). These results are consistent with a model whereby TF-RNA interactions enhance the association of TFs with chromatin.
Figure 4. TF-ARMs enhance chromatin occupancy and gene expression.

(A) Meta-gene analysis of CUT&Tag for WT or ΔARM HA-tagged KLF4 or SOX2, centered on called WT peaks in mESCs
(B) Example tracks of CUT&Tag (spike-in normalized) at specific genomic loci.
(C) Diagram of KLF4 and its cross-correlation to the Tat ARM (magenta), predicted disorder (black line), DNA-binding domain (grey boxes) and predicted disordered domain (cyan).
(D) Side and top views of the crystal structure of KLF4 with DNA (PDB: 6VTX) or AlphaFold predicted structure (ID: O43474)
(E) Experimental scheme for TF gene activation assays. KLF4 ZFs are replaced either by GAL4 or TetR DBD. The effect of KLF4-ARM mutation or replacement of KLF4-ARM with Tat-ARM on gene activation is tested by UAS or TetO containing reporter system.
(F) Normalized luminescence of gene activation assays, normalized to the “No TF” condition (error bars depict s.d., GAL4: p<0.0001 for all pairwise comparisons except WT vs. Tat-ARM, p=0.3363; TetR: NoTF vs. WT, p<0.0001, NoTF vs. R/K>A, p=0.5668, NoTF vs. Tat-ARM, p=0.0002, WT vs. R/K>A, p=0.0003, WT vs. Tat-ARM, p=0.7126, Tat-ARM vs. R/K>A, p=0.0008, one-way ANOVA)
We next sought to determine whether TF-ARMs contribute to gene output by using a transcriptional reporter assay that has been used extensively to investigate the functions of domains in TFs that contribute to transcriptional output8. KLF4 was selected for study because previous studies have used this assay to study KLF4 function in various cellular contexts45–47, KLF4 has a single ARM-like domain (Figures 4C and 4D), it has contiguous effector and DNA-binding domains, and our assays show that deletion of the ARM has a strong effect on RNA binding (Figure 3E). In this assay, the KLF4 zinc fingers (DBD) were replaced with the yeast GAL4 DBD, and this fusion was tested for its ability to activate expression of a luciferase reporter downstream of GAL4-binding UAS sites (Figure 4E). GAL4-KLF4WT activated reporter expression, while substitution of arginines and lysines for alanines in the ARM (GAL4-KLF4R/K>A) significantly reduced reporter expression (Figure 4F). Importantly, this reduction was rescued by replacement of the ARM with the HIV Tat ARM (Figure 4F). Similar effects were observed with the replacement of KLF4 DBD with the bacterial TetR DBD, which recognizes TetO elements in the presence of doxycycline (Figures 4E and 4F). The mutation of the KLF4 ARM caused a reduction in reporter expression rather than complete ablation of expression. These results, taken together with previous studies45–47, suggest that while the DNA and protein binding portions of the TF play major roles in gene activation, TF-RNA binding contributes to fine-tune transcriptional output.
A role for TF RNA-binding regions in TF nuclear dynamics
TFs are thought to engage their enhancer and promoter DNA-binding sites through search processes that involve dynamic interactions with diverse components of chromatin. Single molecule image analysis of TF dynamics in cells indicates that TFs conduct a highly dynamic search for their binding sites in chromatin48,49. The tracking data can be fit to a three-state model, where TFs are interpreted to be immobile (potentially DNA-bound), subdiffusive (potentially interacting with chromatin components) and freely diffusing50,51. If TFs interact with chromatin-associated RNA through their ARMs, then we might expect that mutation of their ARMs would reduce the portion of TF molecules in the immobile and sub-diffusive states. To test this, we conducted single-molecule tracking experiments with murine embryonic stem cell (mESC) or human K562 leukemia lines that enable inducible expression of Halo-tagged wildtype or ARM-mutant TFs. For these experiments, we chose the TFs SOX2, KLF4, GATA2, and RUNX1 because of their prominent roles in mES or hematopoietic cells32,34 and our earlier characterization of their RNA-binding regions (Figure 3). As a control, we included the deletion of an ARM-like region from CTCF that overlaps the previously described RNA-binding region (RBR)36, which was shown to reduce both the immobile and subdiffusive fractions of CTCF52. Single-molecule imaging data was fit to a three-state model: immobile, subdiffusive, and freely diffusing (Figures 5A, S7A-S7C, Videos S1-S3, and STAR Methods). Inspection of single-molecule traces for wildtype and ARM-mutant TFs (Figures 5B and S7A), as well as global quantification across replicates (Figures 5C, S7D, and S7E), showed that deletion of the ARM-like domains in TFs reduces the fraction of molecules in the subdiffusive fraction for all factors and immobile fraction for all factors but one (GATA2), while increasing the fraction of freely diffusing molecules. Although diffusive fractions changed with expression level, the behavior of the mutant TF was consistent across expression regimes (Figures S7F). The observed changes in diffusivity upon ARM mutation could reflect changes in binding between TFs and RNA or DNA molecules. The observation that ARM peptides have a preference for RNA binding (Figure S4), and evidence that TF chromatin occupancy is reduced upon RNase treatment or ARM mutation (Figure S6), is consistent with a role for RNA interactions in TF nuclear dynamics. These results suggest that TF-ARMs enhance the timeframe in which TFs are associated with chromatin.
Figure 5. A role for TF RNA-binding regions in TF nuclear dynamics.
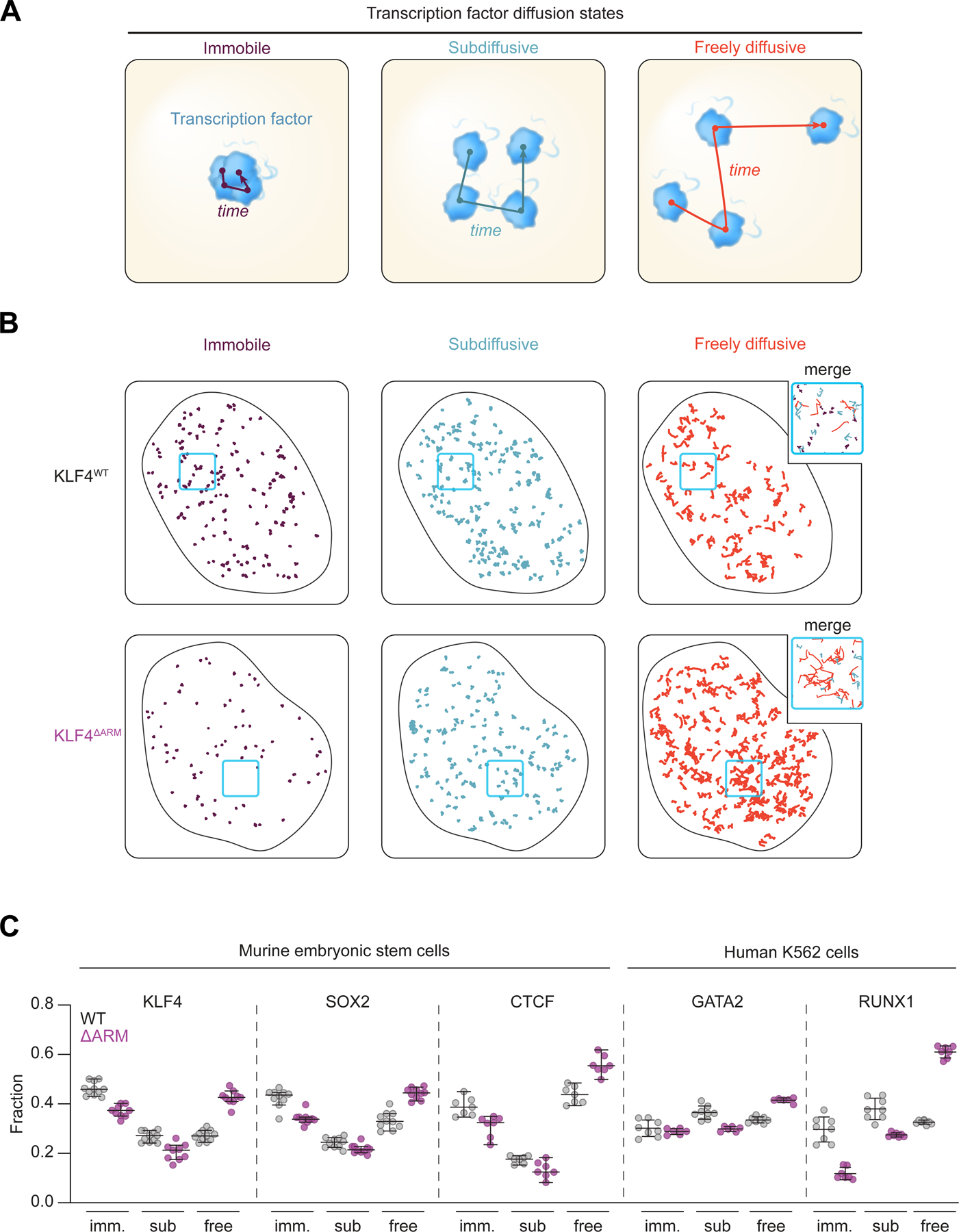
(A) Cartoon depicting a 3-state model of TF diffusion.
(B) Example of single nuclei single-molecule tracking traces for KLF4-WT and KLF4-ARM deletion. The traces are separated by their associated diffusion coefficient (Dimm: <0.04 μm2s™1; Dsub: 0.04–0.2 μm2s™1; Dfree: >0.2 μm2s™1). For each nucleus, 500 randomly sampled traces are shown.
(C) Dot plot depicting the fraction of traces in the immobile, subdiffusive, or freely diffusing states. Each marker represents an independent imaging field (comparing WT and ARM-deletion, p<0.0001 for KLF4free, SOX2free, CTCFfree, GATA2free, RUNX1free, KLF4sub, GATA2sub, RUNX1sub, KLF4imm, SOX2imm, RUNX1imm ; p=0.0094 for SOX2sub; p=0.0101 for CTCFsub, p=0.0034 for CTCFimm, p=0.38 for GATA2imm, two-tailed Student’s t-test; error bars depict 95% C.I.).
TF-ARMs are important for normal development and disrupted in disease
Transcription factors are fundamental controllers of cell-type specific gene expression programs during development, so we next asked whether the TF-ARMs contribute to the factor’s role in normal development in vivo. For this purpose, we turned to the zebrafish, which has served as a valuable model system to study and perturb vertebrate development. Previous study showed that knockdown of zebrafish sox2 by injection of antisense morpholinos at the one-cell stage led to growth defects and embryonic lethality, which could be rescued by co-injection with messenger RNA (mRNA) encoding human SOX253. Using this system, we injected zebrafish with the sox2 morpholino while co-injecting mRNA encoding either wildtype or ARM-mutant human SOX2 (Figures 6A and S7G), which reduced RNA but not DNA binding in vitro (Figures 3E and S4C). Embryos were scored at 48 hours post-fertilization for growth defects by the length of the anterior-posterior axis compared to embryos injected with a non-targeting control morpholino (Figure 6B). Whereas wildtype human SOX2 could partially rescue the growth defect induced by sox2 knockdown, ARM-mutant SOX2 was unable to do so (Figures 6C). These results indicate that TF-ARMs contribute to proper development.
Figure 6. TF-ARMs are important for normal development and disrupted in disease.
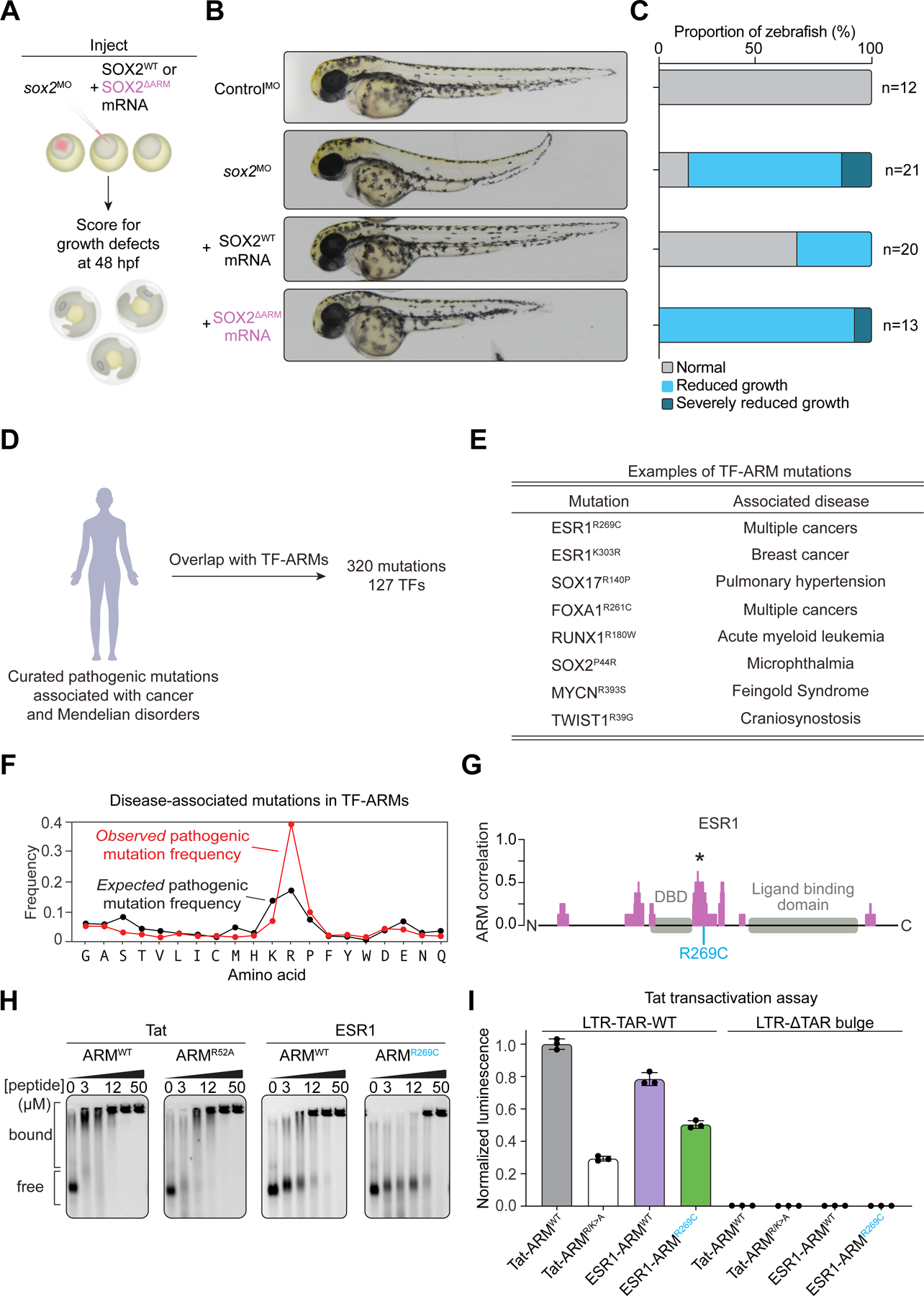
(A) Experimental scheme for injection of zebrafish embryos with morpholinos and rescue by co-injection with the indicated mRNAs (hpf = hours post-fertilization).
(B) Representative images of injected zebrafish embryos at 48 hpf.
(C) Scoring of zebrafish anterior-posterior axis growth.
(D) The landscape of mutations in TF-ARMs associated with human disease
(E) Examples of disease-associated mutations in TF-ARMs.
(F) Line plot of the observed frequency (red) or expected frequency (black) of mutations for amino acids in TF-ARMs (p = 2.7 x 10™74 for enrichment of mutations in arginine, one-side binomial test with Benjamini-Hochberg correction).
(G) Representation of the ESR1 protein and its correlation to the Tat ARM (*Maximum scoring ARM-like region). The selected mutation is provided in blue.
(H) Gel shift assay with 7SK RNA and synthesized peptides for Tat-ARM-WT, Tat-ARM-R52A, ESR1-ARM-WT, and ESR1-ARM-R269C.
(I) Tat transactivation reporter assay with wildtype or mutant versions of Tat and ESR1 ARMs and a version of the reporter without the Tat-binding TAR bulge. Values are normalized to the Tat-ARM-WT condition.
The presence of ARMs in most TFs, and evidence that they can contribute to TF function in a developmental system, prompted us to investigate whether pathological mutations occur in these sequences in human disease. Analysis of curated datasets of pathogenic mutations revealed hundreds of disease-associated missense mutations in TF-ARMs (Figure 6D, Table S5, STAR Methods). These mutations are associated with both germline and somatic disorders, including multiple cancers and developmental syndromes, that affect a range of tissue types (Figure 6E). Variants that mutate arginine residues were the most enriched compared to the other amino acid residues in ARMs (STAR Methods), which is consistent with their importance in RNA binding (Figure 6F) 42. To confirm that such mutations could affect RNA binding, we selected for further study the estrogen receptor (ESR1) R269C mutation (Figure 6G), which is found in multiple cancers and is particularly enriched in a subset of patients with pancreatic cancer54. An EMSA assay showed that RNA binding was reduced with an ESR1 ARM peptide containing the R269C mutation (Figure 6H). Furthermore, when the Tat ARM was replaced with wildtype and mutant versions of the ESR1 ARM in the Tat transactivation assay, the mutation caused reduced reporter expression compared to wildtype (Figure 6I). These results support the hypothesis that disease-associated mutations in TF-ARMs can disrupt TF RNA binding.
Discussion
The canonical view of transcription factors is that they guide the transcription apparatus to genes and control transcriptional output through the concerted function of domains that bind DNA and protein molecules1,3,55,56. The evidence presented here suggests that many transcription factors also harbor RNA-binding domains that contribute to gene regulation (Figure 7A). Given the large portion of TFs that showed evidence of RNA interaction in cells and the presence of an ARM-like sequence in nearly 80% of TFs, it is possible that the majority of TFs engage in RNA binding.
Figure 7. Transcription factors harbor functional RNA-binding domains.
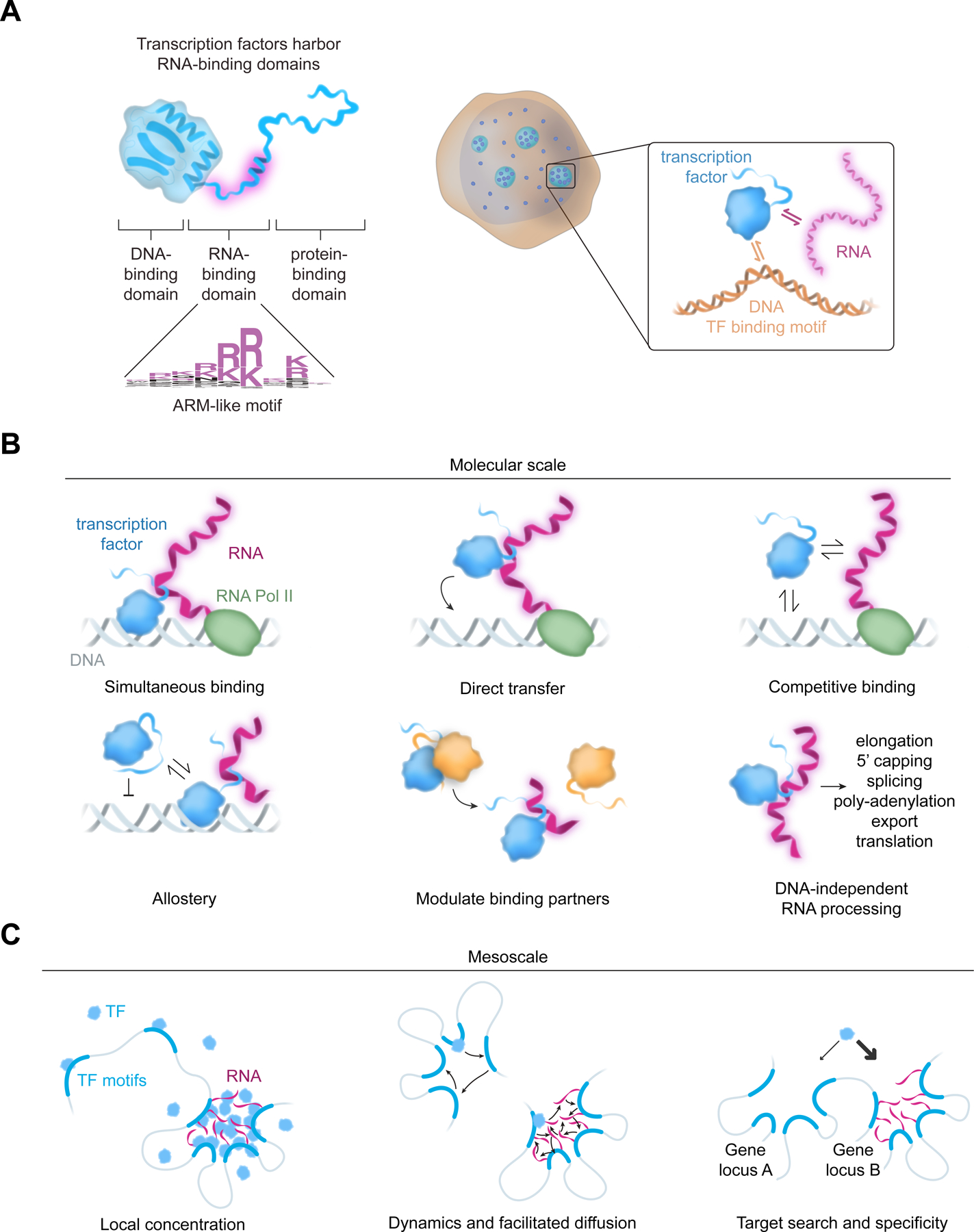
(A) A model depiction of a previously unrecognized RNA-binding domain in a large fraction of transcription factors and its role in TF function.
(B) Various ways by which RNA interactions could impact TF function at the molecular scale
(C) Various ways by which RNA interactions could impact TF function at the mesoscale
RNA molecules are pervasive components of active transcriptional regulatory loci15,16,57–59 and have been implicated in the formation and regulation of spatial compartments60. The noncoding RNAs produced from enhancers and promoters are known to affect gene expression15, and plausible mechanisms by which these RNA species could influence gene regulation have been proposed to include binding to cofactors and chromatin regulators61–64, and electrostatic regulation of condensate compartments58. The evidence that TFs bind RNA suggests additional functions for RNA molecules at enhancers and promoters (Figures 7B and 7C).
Transcription and processing of RNA is a highly localized and dynamic process, producing high local concentrations of RNA at active loci. RNA molecules transcribed by Pol II will typically undergo rapid capping and splicing while tethered to Pol II65. In some cases, RNA molecules accumulate in proximity to the loci where they are transcribed60, but in others they are rapidly exported into the cytoplasm once fully processed. This high local concentration of RNA molecules at sites of transcription would be expected to provide a multivalent interaction network between TFs, DNA and RNA and thereby influence the recruitment and dynamics of TFs at these sites (Figures 7B and 7C). Indeed, previous studies have shown that tethering of RNA molecules to modestly active sites in the genome will enhance the concentration of certain TFs at those loci21.
The observation that many TFs can bind DNA, RNA and protein molecules offers new opportunities to further advance our understanding of gene regulation and its dysregulation in disease. Knowledge that TFs can interact with both DNA and RNA molecules may help with efforts to decipher the “code” by which multiple TFs collectively bind to specific regulatory regions of the genome66–68 and inspire novel hypotheses that may provide additional insight into gene regulatory mechanisms. It might also provide new clues to the pathogenic mechanisms that accompany GWAS variants in enhancers, where those variations occur in both DNA and RNA.
Limitations of the study
This study shows that many transcription factors bind RNA and harbor RNA-binding domains that resemble the HIV Tat ARM. Our results demonstrate for a few tested examples that these domains contribute to the dynamic association of TFs with chromatin, which may provide a mechanism by which TF-RNA interactions contribute to gene control. Although the observed changes in diffusivity of TFs upon ARM mutation was consistent across expression regimes, we cannot exclude the possibility that expression level itself affects TF diffusivity and could explain some of these changes. There are several ways in which the binding of TFs to RNA could affect their function (Figures 7B and 7C), and these mechanisms could result in positive or negative effects on transcriptional output. It is also possible that these domains have additional RNA-dependent functions, some of which may be general and some TF-specific69. Another limitation of the study is the extent to which cellular and organismal phenotypes observed upon deletion of ARM-like domains can be attributed to RNA binding. We believe that characterization of these domains in TFs, including systematic identification of the precise residues required for RNA binding and RNA sequence preferences, will inspire investigation of their roles in many aspects of TF function, including but not limited to locus-specific chromatin association, chromatin architecture, transcriptional output, splicing, translational control, and RNA polymerase II pausing. A key challenge will be to delineate these functions in cells and explore how these functions are related to cooperative or competitive interactions of these domains with RNA, DNA or proteins.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Correspondence and material requests should be addressed to Richard A. Young, young@mit.edu.
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact upon reasonable request with a completed Materials Transfer Agreement.
Data and Code Availability
The RBR-ID mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD035484. CUT&Tag sequencing and CLIP sequencing data have been deposited to GEO with identifier GSE232181. Original images for EMSAs and Western blots as well as single molecule trace data are available through Mendeley Data (DOI: 10.17632/dkx9gsh42h.2). These data are publicly available as of the date of publication.
Code generated during this study is available through Zenodo (DOI: 10.5281/zenodo.7974933; Link: https://zenodo.org/record/7974933)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact by request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
The V6.5 murine embryonic stem cells were a gift from the Jaenisch laboratory of the Whitehead Institute, and these cells are derived from a cross of C57BL/6(F) x 129/sv(M). The human K562 and HEK293 cell lines were purchased from ATCC, and the HEK293F cells for protein purification were a gift from the Sabatini lab. Cell culture conditions are described below. Zebrafish experiments were conducted using male and female zebrafish from a wildtype Tübingen strain. Zebrafish embryos were scored at 48 hours post-fertilization, prior to sex determination, so the influence of sex on the results could not be determined. All animals were housed at Boston Children’s Hospital following standard protocols (water temperature at 28.5 °C and a 14/10-hour light/dark cycle), and handled according to approved Institutional Animal Care and Use Committee (IACUC) of Boston Children’s Hospital protocol 20–10-4254R.
METHOD DETAILS
Structures of known DNA-binding domains in TFs
TF-DNA X-ray structures were obtained from the RCSB Protein Data Bank (Accession numbers: YY1 = 1UBD, MYC/MAX = 1NKP, POU2F1 = 1CQT, JUN/FOS = 1FOS). These entries were modified using ChimeraX70,71, and the effector domains, which are not included in the X-ray structures, are depicted as cartoons highlighting their dynamic and transient structure.
RNA binding region identification (RBR-ID)
K562 cells were cultured in suspension flasks containing culture medium [RPMI-1640 medium with GlutaMAX™ (ThermoFisher Cat. 72400047) supplemented with 10% FBS (ThermoFisher Cat. 10437028), 2 mM L-glutamine (Sigma-Aldrich Cat. G7513), 50 U/mL penicillin and 50 μg/mL streptomycin]. For each biological replicate of RBR-ID, 4 million K562 cells from actively proliferating cultures were aliquoted into 2x T25 flasks. 4-thiouridine (4SU) was added to one of the two flasks for each replicate at a final concentration of 500 μM and incubated for 2 hrs at 37°C with 5% CO2. Cells from each flask were collected and resuspended in 600 μL 1x PBS [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4] and transferred to 6-well plates. Plates were placed on ice with their lids removed and protein–RNA complexes were crosslinked with 1 J/cm2 UVB (312 nm) light. Cells were lysed in Buffer A (10 mM Tris pH 7.94°C, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.2 mM PMSF) with 0.2% IGEPAL CA-630 for 5 min at 4°C, then centrifuged at 2,500 g for 5 min at 4°C to pellet nuclei. Nuclei were washed 3x with 1 mL cold Buffer A (without IGEPAL) and lysed at room temperature in 100 μL denaturing lysis buffer [9 M urea, 100 mM Tris pH 8RT, 1x complete protease inhibitor, EDTA free (Roche Cat. 4693132001)]. Lysates were sonicated using a BioRuptor instrument (Diagenode) as follows: (energy: high, cycle: 15 sec ON, 15 sec OFF, duration: 5 min), centrifuged at 12,000 g for 10 min and supernatant was collected. Extracts were quantified using Pierce BCA assay kit (ThermoFisher Cat. 23225). 5 mM DTT was added to extracts and incubated at room temperature for one hr to reduce proteins, and then alkylated with 10 mM iodoacetamide in the dark for one hr. Samples were then diluted to 1.5 M urea with 50 mM ammonium bicarbonate and treated with 1 μL of 10,000U/μL molecular grade benzonase (Millipore Sigma Cat. E8263) and incubated at room temperature for 30 min. Sequencing grade trypsin (Promega Cat. V5117) was then added to samples at a ratio of 1:50 (trypsin:protein) by mass and incubated at room temperature for 16 hrs. The digested samples were loaded onto Hamilton C18 spin columns, washed twice with 0.1% formic acid, and eluted in 60% acetonitrile in 0.1% formic acid. Samples were dried using a speed vacuum apparatus and reconstituted in 0.1% formic acid, then measured via A205 quantification and diluted to 0.333 μg/μL.
For the proximity analysis in Figure S5, the nearest distance was calculated for each detected protein between RBR-ID+ peptides (p-val<0.05, log2FC<0) and either (1) TF-ARMs (cross-correlation to Tat ARM > 0.5, described below), (2) Known RNA-binding domains (RRM: IPR000504, KH: IPR004087, dsRBD: IPR014720). We required that at least 3 peptides were detected for each protein considered. As a control for the TF-ARM nearest distance analysis, the label (RBR-ID+ or RBR-ID-) of each peptide was randomly shuffled 100 times for all detected RBR-ID peptides for each protein, which provides the null distribution of the dataset. The RBR-ID mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD035484.
LC-MS/MS
Peptide samples were batch randomized and separated using a Thermo Fisher Dionex 3000 nanoLC with a binary gradient consisting of 0.1% formic acid aqueous for mobile phase A and 80% acetonitrile with 0.1% formic acid for mobile phase B. 3 μL of each sample were injected onto a Pepmax C18 trap column and washed with a 0.05% trifluoroacetic acid 2% acetonitrile loading buffer. The linear gradient was 3 minutes until switching the valve at 2% mobile phase B and increasing to 25% by 90 minutes and 45% by 120 minutes at a flow rate of 300 nL/minute. Peptides were separated on a laser-pulled 75 μm ID and 30 cm length analytical column packed with 2.4 μm C18 resin. Peptides were analyzed on a Thermo Fisher QE HF using a DIA method. The precursor scan range was a 385 to 1015 m/z window at a resolution of 60k with an automatic gain control (AGC) target of 106 and a maximum inject time (MIT) of 60 ms. The subsequent product ion scans were 25 windows of 24 m/z at 30k resolution with an AGC target of 106 and MIT of 60 ms and fragmentation of 27 normalized collision energy (NCE). All samples were acquired by LC-MS/MS in three technical replicates. Thermo .raw files were converted to indexed mzML format using ThermoRawFileParser utility (https://github.com/compomics/ThermoRawFileParser). To detect and quantify peptides, indexed mzML files from each set of technical replicates were searched together using Dia-NN v1.8.172 against a FASTA file of the Homo sapiens UniProtKB database (release 2022_02, containing Swiss-Prot + TrEMBL and alternative isoforms). Precursor and fragment m/z ranges of 300–1800 and 200–3000 were considered, respectively with peptides lengths from 6–40. Fixed and variable modifications included carbamidomethyl, N-term acetylation and methionine oxidation. A 0.01 q value cutoff was applied, and the options --peak-translation and --peak-center were enabled, while all other Dia-NN parameters were left as default.
Bioinformatic analysis of the RBR-ID data
After removal of suspected contaminants, identified peptides were re-mapped to an updated human proteome reference (UniProtKB release 2022_02, Swiss-Prot + TrEMBL + isoforms) to reannotate matching proteins. Where multiple protein matches were identified, peptides were assigned to a single protein annotation by first defaulting to Swiss-Prot accessions, where available, then by the accession with the most matching peptides in the dataset and therefore the most likely protein group73. Abundances of the different charge states of the same peptide were summed, and all abundances were normalized by the median peptide intensity in each run. To assess depletion mediated by RNA crosslinking, normalized abundances for each peptide in cells treated or not with 4SU were analyzed by unpaired, two-sided Student’s t tests. For peptides that were missing across all 5 x 3 technical replicates in one of the treatments, Fisher’s exact tests were used comparing the frequency of peptide detection between cells treated with or without 4SU. Statistical significance was determined by adjusting p values from both tests using the Benjamini-Hochberg method74. For mESC RBR-ID data from previous study31, all peptides were re-mapped to an updated mouse reference proteome (UniProtKBrelease 2021_04) as described above while keeping original quantification and P-values. A relaxed p-value threshold (0.10) was used in the original study because it was validated to include additional RBPs31. Peptides were annotated using the InterPro database (release 87, accessed 28 Feb 2022) to identify functional domains. For volcano plots, outliers were removed and each marker represents the peptide with maximum RBR-ID score31 for each protein. Transcription factors annotated in this dataset are from a previous census study1.
Generating list of RNA-binding TFs
RNA-binding proteins identified in the current and previous studies using various methods were collected18,23,31,75–81. The list of RNA-binding proteins from these studies was overlapped with the list of transcription factors from a previous census study1 using merge function in R. Transcription factors that are found at least in one dataset were reported in Table S3.
CLIP
CLIP experiments were performed as previously described82 with minor modifications (see below for details). The protocol is a modified seCLIP protocol with the addition of 4SU incorporation (adapted from PAR-CLIP) and an IR800-conjugated 3’ adapter. CLIP sequencing data have been deposited to GEO with identifier GSE232181.
Protein–RNA crosslinking
K562 cells stably expressing human GATA2 with N-terminal HA-FLAG-Halo tags under dox-inducible promoter were treated for 5 hours with 1 μM doxycycline (Sigma), and 24 hours with 100 μM of 4-Thiouridine (4SU) (Sigma-Aldrich T4509) prior to cell collection. Cells were resuspended in 1X PBS and transferred to a 6-well plate for crosslinking. Plates were placed on ice with lids removed and crosslinked at 365 nm at 0.3 J/cm2. Cell suspension was transferred to microcentrifuge tubes and plates were washed with 1X PBS.
Lysate preparation
Cells were washed in 1X PBS and cell pellets were lysed in eCLIP lysis buffer [20 mM HEPES-NaOH pH 7.4, 1 mM EDTA, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1x cOmplete™ EDTA-free protease inhibitor cocktail (Roche 4693132001)]. Samples were sonicated in a Diagenode Bioruptor (30 s ON/OFF) on medium for 5 minutes. RNase I (ThermoFisher AM2294) was added to lysates for a final concentration of 0.4 U/μL and incubated at 37 °C at 1200 rpm for 5 min. EDTA was immediately added at a final concentration of 21 mM. Lysates were clarified at 15,000g for 10 minutes at 4°C and supernatant was transferred to fresh tubes. Protein concentration was measured using Protein Assay Dye Reagent (Bio-Rad 5000006).
Labeling of crosslinked protein–RNA complexes
Dynabeads™ were washed in eCLIP binding buffer (20 mM HEPES-NaOH pH 7.4, 20 mM EDTA, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate). Antibody was added to bead mixture and incubated, rotating at room temperature for 45 min. Antibody-bead mixture was washed in eCLIP binding buffer and mixed with calculated amount of lysate. Tubes were incubated overnight rotating at 4°C. 2% of lysate-bead mixture was transferred to a new tube to serve as input sample. IP samples were washed with CLIP wash buffer (20 mM HEPES-NaOH pH 7.4, 20 mM EDTA, 5 mM NaCl, 0.2% Tween-20) and IP50 (20 mM Tris pH 7.3RT, 0.2 mM EDTA, 50 mM KCl, 0.05% NP-40). Samples were treated with TURBO™ DNase (ThermoFisher AM2238) and 0.1 U/μL final concentration of RNase I (in some cases, 1 U/μL final concentration was used for better visualization of bands, e.g. Fig. S2A). IP samples were washed in CLIP wash buffer and FastAP buffer (10 mM Tris-Cl pH 7.5RT, 5 mM MgCl2, 100 mM KCl, 0.02% Triton X-100). IP RNA was dephosphorylated using FastAP phosphatase reaction FastAP Thermosensitive Alkaline Phosphotase (ThermoFisher EF0652), and T4 PNK (NEB M0201S).
IP samples were washed in CLIP wash buffer and 1X RNA Ligase buffer (50 mM Tris-Cl pH 7.5RT, 10 mM MgCl2]. A 3’ IR-800 fluorescent adaptor was ligated using T4 RNA Ligase 1 high concentration (NEB M0437M). Samples were washed in eCLIP high-salt wash buffer (50 mM Tris-HCl pH 7.4RT, 1M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) and CLIP wash buffer. IP and input samples were eluted with 4X LDS Sample Buffer (ThermoFisher NP0007), run on an 8% bis-tris gel, and transferred overnight to a nitrocellulose membrane.
Library preparation and sequencing
The transferred membrane was cut ~0–50 kDa above protein size and incubated with Proteinase K (ThermoFisher AM2548) to isolate crosslinked RNA. Remaining steps were performed as per the seCLIP protocol83, with some modifications. RNA was purified and concentrated with phenol:chloroform:IAA (ThermoFisher AM9732) and ethanol precipitation. 3’ and 5’ adapters were designed to include an IR800 fluorophore and an 8-nt UMI for cDNA ligation, respectively. We did not include 5’ deadenylase enzyme in our 5’ ligation reactions and we used the AffinityScript RT (Agilent 600107) for crosslinking-induced truncation. Libraries were sequenced on an Illumina NextSeq 500 in paired-end mode for 47:8:8:29 cycles (read 1 : index 1 : index2 : read 2).
CLIP Analysis
Generating CLIP-seq peaks
Raw CLIP-seq reads were trimmed using Cutadapt84. The adapter sequence AGATCGGAAGAGCACACGTCTGAA was trimmed from the 5’ end of the reads, AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT adapter sequence from the 3’ end, and a universal four nucleotide UMI from the 3’ end. Prior to mapping, UMIs were extracted from the 5’ end of the reads using UMI-tools version 1.0.0 with the argument --bc-pattern=NNNNNNNN85. Bowtie2 was used to map all trimmed reads to the hg19 human genome using parameters -p 40 –end-to-end –no-discordant86,87. Trimmed, mapped, and unique reads were then sorted using the samtools sort function and indexed using the bedtools index function88,89. Lastly, reads were collapsed to account for PCR duplicates using the extracted UMIs with the UMI-tools dedup function. These trimmed, mapped, and collapsed reads were then used for downstream analysis. To call CLIP-seq peaks, .bed files were generated using MACS with parameters -g hs --keep-dup auto –-nomodel90.
Identifying crosslinked nucleotides
As per the seCLIP protocol, during the reverse transcription step, polymerase terminates at the site of the cross-link83. This yields a cDNA product in which the 3’ nucleotide of the cDNA is the nucleotide before the site of the cross-link on the pulled down RNA. During the paired end sequencing, the position 1 of the 5’ end of read1 will therefore map to the site on the genome that is one nucleotide downstream the cross-linked nucleotide. To extract this site from the mapped CLIP-seq reads and generate Table S4 with sequences containing the site of the cross-link +/- 5bp, the genomic locations for the forward strand reads were first extracted. bedtools fasta was then used to extract the -1 position of the 5’ end of the forward strand mapped reads (see CLIP methods) and +/- 5 bps around this site. This generated 11nt sequences in which the site of the cross-link is at the center of the sequence (nucleotide position 6).
To filter out any sequences in which the polymerase terminated early (i.e. prior to the cross-link) in the reverse transcription step, the sequences containing cross-linked nucleotides were filtered further for only the sequences containing a T (U) in the cross-link site (position 6). As expected, there was an enrichment of T (U) nucleotides as compared to G’s, C’s, and A’s at this position within the sequences. The list was further filtered to only include sequences that overlap with called CLIP-seq peaks (see Generating CLIP-seq peaks)
To annotate the cross-link containing sequences with whether they fell within a gene, an enhancer, or a promoter in Table S4, the chromosomal locations of the cross-link containing sequences were overlapped with RefSeq genes, H3K27Ac ChIP-seq peaks (GSM733656), or RefSeq genes TSS +/- 200, respectively. H3K27Ac peaks were called using MACS with parameters -g hs --keep-dup auto –-nomodel.
Generating CLIP-seq metaplots
Fastq files from GATA2 ChIP-seq91 (GSM467648) and RUNX1 ChIP-seq92 (GSM2423457) experiments in K562 cells were downloaded from Gene Omnibus Expression database (GEO) and aligned to the hg19 human genome using Bowtie2. ChIP-seq peaks were called using MACS with parameters -g hs --keep-dup auto –-nomodel. Regions for metaplot analysis were generated using +/-2000 bases from the center of the called peaks. Normalized CLIP-seq densities within these regions were calculated using bamToGFF93. Input-corrected meta-gene plots were generated by subtracting the mean read density per bin of the input CLIP at ChIP peaks from the HA pull down CLIP at ChIP peaks. R matplot function was used to plot the density values across the 4Kb region.
Protein purification
To purify transcription factors (NANOG: pJH201, RARA: pJH203, CTCF: pJH205, OCT4: pJH199, MYC: pJH200, P53: pJH204, KLF4: pJH278, ESR1: pJH202, YY1: pJH087, SOX2: pJH198, STAT3: pJH227, GATA2: pJH247, SMAD3: pJH226, see Key Resource Table for plasmid information), a mammalian purification system using Freestyle HEK 293F cells (gift from Sabatini lab) were used. HEK cells were grown in FreeStyle 293 Expression Medium (Gibco) on an orbital shaker. Coding sequence of desired genes were synthesized by IDT as gBlock fragments (Table S6) containing proper Gibson overhangs. TF-ARM deletion mutants (pJH279, pJH245, pJH272, Key Resource Table) were generated by removal of a stretch of peptide adjacent to DNA binding domains that contain ARMs. The amino acid sequences that are removed in TF-ARM mutants are shown in parentheses as follows: hsKLF4_ΔARM (aa 355–386), hsSOX2_ΔARM (aa 118–178), hsGATA2_ΔARM (aa 360–395), and hsCTCF_ΔARM (576–611). To reduce sequence complexity for gBlock synthesis, codon optimization using the IDT codon optimization tool was applied when needed. The fragments are then cloned into a mammalian expression vector containing Flag and mEGFP (N- or C- terminal) (modified from Addgene #32104) using NEBuilder HiFi DNA Assembly kit (E2611). These vectors were transiently transfected into 293F cells at a concentration of 1 million/ml with 1 μg of DNA per million cells using branched polyethylenimine (PEI) (Polysciences). 60–72 hours post-transfection, cells were resuspended in 45 ml HMSD50 buffer (20 mM HEPES pH 7.5, 5 mM MgCl2, 250 mM sucrose, 1mM DTT, 50mM NaCl, supplemented with 0.2 mM PMSF and 5 mM sodium butyrate) and incubated for 30 min at 4° C with gentle agitation. After a spin down at 3500 rpm at 4°C for 10 min, the supernatant was discarded and the pellet containing nuclei were resuspended in 35 ml of BD450 buffer (10 mM HEPES pH 7.5, 5% Glycerol, 450 mM NaCl, and protease and phosphatase inhibitors) and incubated for 30 min at 4° C with agitation. The solution was spun down at 3500 rpm at 4°C for 10 min to clear the nuclear extract. The supernatant was transferred into fresh tube and the pellet containing chromatin was passed through 18G ½ syringe 5 times. The chromatin containing lysate was spun down at 8000 rpm at 4° C for 10 min and supernatant is combined with the previously collected supernatant. Then the combined supernatants were spun down again at 8000 rpm at 4°C for 10 min to clear the lysate. 500 ul of Flag-M2 beads (Sigma) were added to the cleared lysates and incubated overnight at 4° C. The Flag-M2 beads were washed 2 times with 45 ml BD450 buffer and they were transferred into a purification column (Biorad). The beads on the column were washed 2 more times with 10 ml BD450 buffer and 5 ml Elution buffer (20 mM HEPES pH 7.5, 10% Glycerol, 300 mM NaCl). Elutions were performed by incubating the beads overnight at 4° C with 800 elution buffer and 200 ul of 5mg/ml flag peptide (Sigma). The buffer exchange (into elution buffer) and concentration of proteins were performed using spin columns (Milipore). Proteins were aliquoted and stored at -80°C. Canonical RNA-binding proteins (U2AF2, HNRNPA1, SRSF2) were purified in a previous study94.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Histone H3 antibody | Abcam | ab1791 |
| HA antibody | Abcam | ab9110 |
| Flag | Sigma | F3165 |
| Flag-M2 beads | Sigma | A2220 |
| Sox2 | R&D Systems | MAB2018 |
| Klf4 | R&D Systems | AF3158 |
| Actin | Sigma | A5441 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma | D9891–5G |
| Poly-L-ornithine | Sigma | P4957–50ML |
| U2AF2 | Guo et al., 2019 | N/A |
| HNRNPA1 | Guo et al., 2019 | N/A |
| SRSF2 | Guo et al., 2019 | N/A |
| NANOG | This study | N/A |
| RARA | This study | N/A |
| CTCF | This study | N/A |
| OCT4 | This study | N/A |
| MYC | This study | N/A |
| P53 | This study | N/A |
| KLF4 | This study | N/A |
| ESR1 | This study | N/A |
| YY1 | This study | N/A |
| SOX2 | This study | N/A |
| STAT3 | This study | N/A |
| GATA2 | This study | N/A |
| SMAD3 | This study | N/A |
| KLF4-ΔARM (aa 355–386) | This study | N/A |
| SOX2-ΔARM (aa 118–178) | This study | N/A |
| GATA2-ΔARM (aa 360–395) | This study | N/A |
| GFP | This study | N/A |
| BamHI | NEB | R0136 |
| Critical Commercial Assays | ||
| Dual Luciferase Assay Kit | Promega | E1960 |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | E2621S |
| Monarch Gel Extraction Kit | NEB | T1020S |
| Phusion polymerase | NEB | M0531S |
| MEGAscript T7 Transcription Kit | Invitrogen | AM1334 |
| MEGAclear Transcription Clean-Up Kit | Invitrogen | AM1908 |
| Cy5-labeled UTP | Enzo LifeSciences | ENZ-42506 |
| Lipofectamine 3000 Transfection Agent | ThermoFisher | L3000001 |
| CUT&Tag-IT Assay Kit | Active Motif | 53160 |
| Deposited Data | ||
| RBR-ID mass spectrometry proteomics data | This study | ProteomeXchange: PXD035484 |
| Original imaging data and single molecule traces | This study | Mendeley Data DOI: 10.17632/dkx9gsh42 h.1 |
| GATA2 K562 CLIP-seq | This study | GEO GSE232181 |
| SOX2 and KLF4 CUT&Tag sequencing | This study | GEO GSE232181 |
| Experimental Models: Cell Lines | ||
| V6.5 murine embryonic stem cells | Jaenisch laboratory | N/A |
| pJH308_PBFH_NtermHalo_hsKLF4_WT mESCs | This study | N/A |
| pJH309_PBFH_NtermHalo_hsKLF4_ΔARM (aa 355–386) mESCs | This study | N/A |
| pJH294_PBFH_NtermHalo_hsSOX2_WT mESCs | This study | N/A |
| pJH295_PBFH_NtermHalo_hsSOX2_ΔARM (aa 118–178) mESCs | This study | N/A |
| pJH290_PBFH_NtermHalo_hsCTCF_WT mESCs | This study | N/A |
| pJH291_PBFH_NtermHalo_hsCTCF_ΔARM (aa 576–611) mESCs | This study | N/A |
| pJH357_PBFH-NtermHalo-hsGATA2-WT K562 cells | This study | N/A |
| pJH358_PBFH-NtermHalo-hsGATA2_ΔARM (aa 360–395) K562 cells | This study | N/A |
| pJH337_PBFH-NtermHalo-RUNX1-WT K562 cells | This study | N/A |
| pJH342_PBFH-NtermHalo-RUNX1-_ΔARM (aa 174–197) K562 cells | This study | N/A |
| DNA, RNA and peptides sequences | ||
| See Table S4 for all peptide, RNA, gblock and oligonucleotide sequences | This study | N/A |
| Recombinant DNA | ||
| pJH201_CBFH_NtermGFP_hsNANOG_WT | This study | N/A |
| pJH203_CBFH_NtermGFP_hsRARA_WT | This study | N/A |
| pJH205_CBFH_NtermGFP_hsCTCF_WT | This study | N/A |
| pJH199_CBFH_NtermGFP_hsOCT4_WT | This study | N/A |
| pJH200_CBFH_NtermGFP_hsMYC_WT | This study | N/A |
| pJH204_CBFH_NtermGFP_hsP53_WT | This study | N/A |
| pJH278_CBFH_CtermGFP_hsKLF4_WT | This study | N/A |
| pJH202_CBFH_NtermGFP_hsESR1_WT | This study | N/A |
| pJH087_CBFH_NtermGFP_hsYY1_WT | This study | N/A |
| pJH198_CBFH_NtermGFP_hsSOX2_WT | This study | N/A |
| pJH227_CBFH_NtermGFP_hsSTAT3_WT | This study | N/A |
| pJH247_CBFH_NtermGFP_hsGATA2_WT | This study | N/A |
| pJH226_CBFH_NtermGFP_hsSMAD3_WT | This study | N/A |
| pJH279_CBFH_CtermGFP_hsKLF4_ΔARM (aa 355–386) | This study | N/A |
| pJH245_CBFH_NtermGFP_hsSOX2_ΔARM (aa 118–178) | This study | N/A |
| pJH272_CBFH_NtermGFP_hsGATA2_ΔARM (aa 360–395) | This study | N/A |
| pJH308_PBFH_NtermHalo_hsKLF4_WT | This study | N/A |
| pJH309_PBFH_NtermHalo_hsKLF4_ΔARM (aa 355–386) | This study | N/A |
| pJH294_PBFH_NtermHalo_hsSOX2_WT | This study | N/A |
| pJH295_PBFH_NtermHalo_hsSOX2_ΔARM (aa 118–178) | This study | N/A |
| pJH290_PBFH_NtermHalo_hsCTCF_WT | This study | N/A |
| pJH291_PBFH_NtermHalo_hsCTCF_ΔARM (aa 576–611) | This study | N/A |
| pJH357_PBFH-NtermHalo-hsGATA2-WT | This study | N/A |
| pJH358_PBFH-NtermHalo-hsGATA2_ΔARM (aa 360–395) | This study | N/A |
| pJH337_PBFH-NtermHalo-RUNX1-WT | This study | N/A |
| pJH342_PBFH-NtermHalo-RUNX1-_ΔARM (aa 174–197) | This study | N/A |
| pJH325_HIV-LTR-LUC | This study | N/A |
| pJH326_HIV-LTR-ΔTAR-LUC | This study | N/A |
| pJH327_pcDNA3-HIV-tat-WT | This study | N/A |
| pJH329_pcDNA3-HIV-tat-ARM>KLF4-BP | This study | N/A |
| pJH330_pcDNA3-HIV-tat-ARM>SOX2-BP | This study | N/A |
| pJH361_pcDNA3-HIV-tat-ARM-R/KtoA | This study | N/A |
| pJH371_pcDNA3-HIV-tat-ARM-to-GATA2-peptide | This study | N/A |
| pJH365_pcDNA3-HIV-tat-ARM-to-ESR1 | This study | N/A |
| pJH366_pcDNA3-HIV-tat-ARM-to-ESR1-R269C | This study | N/A |
| pJH438_pb-hUbiC-Gal4-KLF4-WT | This study | N/A |
| pJH439_pb-hUbiC-Gal4-KLF4-ARM-KRtoA | This study | N/A |
| pJH441_pb-hUbiC-Gal4-KLF4-SWAP-Tat-ARM | This study | N/A |
| pJH375_pb-hUbiC-tetR-KLF4-WT | This study | N/A |
| pJH376_pb-hUbiC-tetR-KLF4-ARM-KRtoA | This study | N/A |
| pJH377_pb-hUbiC-tetR-KLF4-SWAP-Tat-ARM | This study | N/A |
| pJH437_pJP080_UAS_luciferase | Li et al., 2020 | N/A |
| pJH175_4xTet_Luc_reporter | This study | N/A |
| Software and Algorithms | ||
| Fiji image processing package | Schindelin et al., 2012 | https://fiji.sc/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Code generated by the study | This study | Zenodo DOI: 10.5281/zenodo.797 4933 https://zenodo.org/record/7974933 |
| Other | ||
| 35 mm glass-bottom imaging dishes | Mattek Corporation | P35G-1.5–20-C |
In vitro RNA synthesis and purification
To synthesize labeled RNA for fluorescence polarization measurements, in vitro transcription templates were generated from ssDNA oligos (for the random RNA template, Integrated DNA Technologies), gBlocks (for 7SK template, Integrated DNA Technologies), or PCR amplification of genomic DNA from V6.5 murine embryonic stem cells (for Pou5f1 enhancer and promoter RNAs)58 (Table S6). Templates were amplified by PCR with primers containing T7 (sense) or SP6 (antisense) promoters:
Templates were amplified using Phusion polymerase (NEB), and the products were gel-purified using the Monarch Gel Extraction Kit (NEB) following the manufacturer’s instructions and eluted in 40 μL H2O. Each template was transcribed using the MEGAscript T7 kit using 200 ng total template according to the manufacturer’s instructions. Reactions included a Cy5-labeled UTP (Enzo LifeSciences ENZ-42506) at a ratio of 1:10 labeled UTP:unlabeled UTP. The transcription reaction was incubated overnight at 37°C, and then it was incubated with 1 μL TURBO DNase (supplied in kit) for 15 minutes at 37°C. Transcribed RNA was purified by the MEGAclear Transcription Clean-Up Kit (Invitrogen) following the manufacturer’s instructions and eluting in 40 μL H2O. The RNA was diluted to 2 μM and aliquoted to limit freeze/thaw cycles. Transcribed RNA was analyzed by gel electrophoresis to verify a single band of correct size.
Fluorescence polarization assay
To determine the binding affinity of a protein with RNA, we conducted the fluorescence polarization assay as previously described with some minor modifications18 (Holmes et al 2020)., The concentration of protein is serially diluted from 5000 nM down to 2 nM by a 3-fold dilution factor. The series of protein concentrations is then mixed with a buffer containing 10 nM Cy5-labeled RNA, 10 mM Tris pH 7.5, 8% Ficoll PM70 (Sigma F2878), 0.05% NP-40 (Sigma), 150 mM NaCl, 1 mM DTT, 0.1 mg/mL non-acetylated BSA (Invitrogen AM2616), and 10 μM ZnCl2. The reactions were performed in triplicates in a 20 μL reaction volume. After incubating the reactions 1 hr at room temperature, they are transferred into flat bottom black 384 well-plate (Corning 3575). Anisotropy was measured by a Tecan i-control infinite M1000 with the following parameters. Excitation Wavelength: 635 nm; Emission Wavelength: 665; Excitation/ Emission Bandwidth: 5 nm; Gain: Auto; Number of Flashes: 20; Settle Time: 200ms; G-Factor: 1. To account for instrument error, the plate was measured 3 times and the mean of the values are used in the affinity calculations. Reagents used for established RNA-binding proteins were generated previously94 and BamHI was purchased from New England Biolabs.
To determine the binding affinity of a protein with DNA, the same buffer conditions and incubation times were used, as described above. The series of protein concentrations from 0.76–1666 nM (3-fold serial dilution) and 10 nM cy5-labeled DNA were used. The motif containing DNA sequences that have been shown to bind SOX218 and KLF495 were ordered from IDT. To prepare motif-containing DNA sequences, 50 μM of oligos with complementary sequences (one unlabeled and the other labeled with cy5) (Table S6) were annealed in TE+100 mM NaCl buffer by ramping down the temperature from 98°C to 4°C on a thermocycler. Then the annealed DNA fragments were diluted to appropriate concentrations with water for the assay.
Binding curves were fit to fluorescence anisotropy data via nonlinear regression with the Levenberg-Marquardt-based ‘curve_fit’ function in scipy (v. 1.7.3). Curve fitting was performed using a monovalent reversible equilibrium binding model accounting for ligand depletion, given by the equation below:
where is the total protein concentration, is the total ligand (RNA) concentration, and , , and are fit parameters. The measured anisotropy value for each condition was determined by first averaging raw anisotropy measurements across three subsequent reads of the same well, then averaging these values across three technical replicates from separate wells. To calculate the bound fraction of RNA, values were normalized to the range between the upper and lower anisotropy asymptotes and . Error bars were computed from the standard deviation of RNA bound fraction across three technical replicates. The script used to calculate the affinities are available on Zenodo (https://zenodo.org/record/7974933).
Electrophoretic mobility shift assay
To determine the binding affinity of a TF-ARM peptides (synthesized by Genscript) (Table S6) with 7SK RNA, we conducted the electrophoretic mobility shift assay as previously described with some minor modifications19,36. The concentration of peptides was serially diluted from 50000 nM down to 3.125 nM by a 2-fold dilution factor in buffer containing 20 mM HEPES, 300 mM NaCl, and 10% Glycerol. The series of protein concentrations was then mixed 1:1 with a buffer containing an initial concentration of 20 nM Cy5-labeled RNA, 20 mM Tris pH 8.0, 5% glycerol, 0.1% NP40 (Sigma), 0.02 mM ZnCl2, 1 mM MgCl2, 2 mM DTT, and 0.2 mg/mL non-acetylated BSA (Invitrogen AM2616). For DNA-binding assays, 20 nM Cy5-labeled dsDNA or 20 nM Cy5-labeled ssRNA were used (Table S6). The reactions were performed in a 20 μL reaction volume. After incubating the reactions in the dark for 1 hr at room temperature, they were loaded into a 2.5% agarose gel that is pre-run for at least 30 min at 4°C. The samples then ran for 1.5 hr at 150V at 4°C. The gel is imaged using Typhoon FLA95 imager with a Cy5 fluorescence module.
Homology search for RNA-binding domains in TFs
We retrieved hidden Markov model based profiles (HMM-profiles) for RNA-binding domains corresponding to the following Pfam96 entries using hmmfetch from the HMMER package (hmmer.org) – RRM_1, RRM_2, RRM_3, RRM_5, RRM_7, RRM_8, RRM_9, DEAD, zf-CCCH, zf-CCCH_2, zf-CCCH_3, zf-CCCH_4, zf-CCCH_6, zf-CCCH_7, zf-CCCH_8, KH_1, KH_2, KH_4, KH_5, KH_6, KH_7, KH_8, KH_9. These domains represent the largest families of RNA-binding domains. We searched for these profiles using hmmsearch form the HMMER package with ‘-T 0’ as a parameter in fasta files with sequences corresponding to TFs1 or RNA-binding proteins97. The log2-odds ratio score from the hmmsearch output was plotted for RBPs with score > 0 (n=350, to provide scores that one would expect if these domains were in the protein) and for all 1651 TFs1. If a TF was not in the output, it was assigned a score of 0.
Analysis of ARM-like regions in TFs
We used an approach based on analogous functions in localCIDER98 and on a previously applied procedure99 used to map basic patches. For each TF, amino acid compositions of Lys and Arg in sliding 5-residue windows were computed. Basic patches were defined as regions of ≥ 5 consecutive residues that consisted of Lys and Arg occurring at a frequency of >0.5. This threshold was based on optimizing this approach against previously described basic patches in MECP299. All identified basic patches were filtered for those that occurred within predicted IDRs (metapredict), determined as described above. For the adjacency analysis, DNA-binding domains were defined based on domains with annotations of DNA-binding in Interpro100. Probabilities of basic patch occurrence in all TFs were computed starting from the N-terminal edge of the first DNA-binding domain and moving N-terminally, or the C-terminal edge of the last DNA-binding domain and moving C-terminally. These probabilities were summed to arrive at the total probability as a function of distance from the bounds of the DNA-binding regions.
A consensus motif for bioinformatically identified basic patches (Figure 3B) was created using MEME (v. 4.11.4)101. Briefly, 963 basic patches found in TFs were padded by appending the 10 amino acid residues upstream and downstream of each the region. Next, a zero-order Markov model was created from 1,290 full sequences of annotated TFs using the ‘fasta_get_markov’ function to generate a background for the motif search. The TF basic patch sequences were input to the ‘MEME’ function using the TF background model, specifying a constraint to identify exactly one site per sequence, a minimum motif width of 5, a maximum motif width of 13, and defaults for the unspecified parameters.
A charge-based cross-correlation method was employed to identify ARMs in TF disordered regions similar to the HIV Tat ARM. Extensive in vitro and cellular analyses of the Tat ARM have mapped the critical residues responsible for Tat RNA-binding and HIV transactivation41,42. To properly function, the Tat ARM requires an arginine positioned near the motif center flanked by an enrichment of basic residues (R/K). The Tat ARM sequence “RKKRRQRRR” was digitized to the amino acid charge pattern “111110111” to create a 9-mer search kernel. A protein target sequence was created by first digitizing the sequence of the protein of interest to “1” for R/K amino acid residues and “0” otherwise, then refining the sequence by setting residues to “0” if they fell outside of disordered regions assessed through the metapredict package102 (v. 2.2) with a disorder threshold of 0.2. The target sequence was further refined by setting all entries to “0” in 9-mer windows where no R’s were originally present. The cross-correlation between the search kernel and the target sequence was then computed using the ‘correlate’ function in scipy using the “direct” method. Maximum cross-correlations were computed as the maximum of the returned array for each protein tested. This method was applied iteratively to all sequences from the UniProt database to generate distributions for TFs and the proteome.
Evolutionary conservation of TF-ARMs
Evolutionary conservation of specific human TFs was assessed using the ConSurf online server103. TF sequences were downloaded from UniProt and run without specifying a 3D structure or MSA, with automatic detection of homologs from the “NR_PROT_DB” database. Defaults were used for all other running parameters. Amino acid conservation scores from the ConSurf GRADES output were re-normalized between 0 and 1 for each protein, such that a score of 1 corresponded to the of the most conserved amino acid in a given protein.
To evaluate the extent of evolutionary conservation for a larger cohort of TF ARMs, the degree of conservation of TF ARMs was compared to non-ARM regions across vertebrates. The OrthoDB v10 database was used to identify the set of vertebrate orthologs for each protein in a list of annotated human TFs. For each TF, a multiple sequence alignment (MSA) of the retrieved vertebrate orthologs was generated using Clustal Omega (v. 1.2.4) with default parameters. The output ALN format MSA files were converted directly to FASTA format. TFs with an ARM maximum cross-correlation score of 5 or above were retained for further analysis. Each MSA file was parsed via the “prody” package (v. 2.3.1)104 in Python using the ‘parseMSA’ command. Reference coordinates for the MSA were set with respect to the human TF of interest by using the ‘refineMSA’ command and specifying the ID of the human TF. The degree of conservation of each amino acid residue in the human TF was quantified by computing the Shannon entropy (H) for each residue via the ‘calcShannonEntropy’ function. Higher values of H represent more sequence variation at a specific residue position and therefore a lower degree of evolutionary conservation. To define ARM regions for the purpose of Shannon entropy analysis, the union of 9-mer regions with an ARM cross-correlation score of 5 or above was used. For each TF analyzed (N=580), the median value of H in the ARM region and the median value of H in the remainder of the sequence (non-ARM region) were calculated and plotted. Distributions of these paired data were compared via a Wilcoxon signed-rank test.
HIV Tat transactivation assay
To generate the HIV LTR luciferase reporter (pJH325, Key Resource Table), the HIV 5’ LTR from the pNL4-3 isolate (Genbank AF324493) was cloned into pGL3-Basic (Promega) via Gibson assembly (NEB 2X HiFi) with a HindIII-digested pGL3-Basic and a gBlock (Integrated DNA Technologies) containing the HIV 5’ LTR with compatible overhangs (Table S6). A mutant version of this reporter lacking the Tat activation site (TAR RNA bulge structure)44 was also generated in a similar fashion (pJH326, Key Resource Table). Mammalian expression vectors encoding Tat, an R/K>A mutant of Tat, and replacements of the Tat ARM with TF-ARMs from KLF4, SOX2, GATA2, and ESR1 were generated by Gibson assembly with a NotI-XhoI-digested pcDNA3 (Invitrogen) and gBlocks encoding these variants with compatible overhangs (pJH327, pJH329, pJH330, pJH361, pJH371, pJH365, pJH366, Key Resource Table and Table S6).
For transfections, HEK293T cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Sigma F4135), 50 U/mL penicillin and 50 μg/mL streptomycin (Life Technologies 15140163). Transfections were conducted in triplicate. 24-well plastic plates were first coated with poly-L-lysine (Sigma) for 30 minutes at 37°C, washed once with 1X PBS, and then allowed to air dry. Cells were seeded in 500 μL of media in coated wells at a density of 2x105 cells per well. The next day, each well was transfected using Lipofectamine 3000 (Life Technologies) (total reaction 50 μL Optimem, 1.5 μL Lipo-3000, 0.6 μL P3000, and the appropriate volume of DNA) with 100 ng of the HIV 5’ LTR reporter vector, 150 ng of the pcDNA3 expression vector (encoding Tat or the variants), and 50 ng of a renilla luciferase plasmid (pRL-SV40, Promega) to normalize transfection efficiency. As a control, we included a pcDNA3 vector expressing LacI-mCherry (labeled as “No Tat” in Figure 3). After 6 hours of incubation, luciferase activity was quantified by the Dual Luciferase Assay kit (Promega) following the manufacturer’s instructions and a Safire II plate reader. The luminescence values were first normalized to the renilla luciferase luminescence for each well, and then all conditions were normalized to the average value of the “No Tat” control condition.
CUT&Tag experimental procedure
CUT&Tag sequencing was performed using the CUT&Tag-IT Assay Kit (Active Motif 53160) according to manufacturer’s instructions. Stable mESC lines expressing HA-tagged versions of WT and ARM-mutant SOX2 and KLF4 were induced with doxycycline (1 μg/mL) for 6 hours, and 4x105 mESCs were collected. The nuclei of the cells were extracted and incubated with 1μg of HA antibody (Abcam ab9110). After incubation with a rabbit secondary antibody and pA-Tn5 Transposomes, DNA was extracted and amplified with i7/i5 indexed primer combinations. SPRI Bead clean-up of the amplified DNA fragments were performed, and libraries were pooled, subjected to gel-based clean up and sequenced by Novaseq (50x50). CUT&Tag sequencing data have been deposited to GEO with identifier GSE232181.
CUT&Tag analysis
Reads were first trimmed by adapter sequence (CTGTCTCTTATACACATCT) in the forward and reverse directions using Cutadapt with default parameters. Subsequent analysis of the data was conducted according to a published protocol with no modification105. Reads were aligned to the mm10 mouse genome, and samples were spike-in normalized according to the protocol by calculating a scale factor from reads aligning to the E. coli genome. Peak calling for both WT and ARM-mutant samples was conducted using the Seacr algorithm using the “non” (non-normalized) and “stringent” parameters106. For meta-gene plots, raw read density was calculated by centering on called peaks for both WT and ARM-mutant TFs that were merged using bedTools merge with default parameters.
TF reporter assays
For KLF4 reporter assays, constructs were designed that replaced the 3 zinc fingers of KLF4 with either the yeast GAL4 DNA-binding domain or the bacterial TetR DNA-binding domain. Plasmids were cloned via Gibson assembly with gBlocks (IDT) encoding wildtype, mutant, or Tat-ARM-swap versions of KLF4, and expression of the KLF4 fusions were driven by the human UbiC promoter (pJH438, pJH439, pJH441, pJH375, pJH376, pJH377, Key Resource Table). Reporter constructs contained either 6X UAS sites (Key Resource Table pJH437) or 4X TetO sites (Key Resource Table, pJH175) upstream of a minimal CMV promoter driving firefly luciferase. For GAL4 experiments, HEK293 cells were plated at 2x105 cells per well in a 24-well plate in triplicate. Cells were transfected with 100 ng reporter, 166 ng KLF4 expression construct, and 50 ng of a renilla luciferase transfection control (pRL-SV40, Promega) the following day using Lipofectamine 3000 following the manufacturer’s instructions. As a control, we included a pcDNA3 vector expressing LacI-mCherry (labeled as “No TF”). After 4 hours of incubation, luciferase activity was quantified by the Dual Luciferase Assay Kit (Promega) following the manufacturer’s instructions and a Safire II plate reader. The luminescence values were first normalized to the renilla luciferase luminescence for each well, and then all conditions were normalized to the average value of the “No TF” control condition. For TetR assays, HEK293 cells were plated at 1x105 cells per well in a 24-well plate in triplicate in media containing tetracycline-free serum. The following day, cells were transfected with 100 ng reporter, 100 ng KLF4 expression construct, and 50 ng of renilla luciferase. After 2 hours of incubation, the media was removed and replaced with a media containing 1 μg/mL doxycycline. After 4 hours in dox, the cells were processed for luminescence readings in an identical fashion to the GAL4 assays.
Single-molecule tracking
Cell line generation
Murine embryonic stem cells were cultured in 2i/LIF media on tissue culture plates coated with 0.2% gelatin (Sigma, G1890). The 2i/LIF media contained: 960 mL DMEM/F12 (Life Technologies, 11320082), 5 mL N2 supplement (Life Technologies, 17502048; stock 100X), 10 mL B27 supplement (Life Technologies, 17504044; stock 50X), 5 mL additional L-glutamine (GIBCO 25030–081; stock 200 mM), 10 mL MEM nonessential amino acids (GIBCO 11140076; stock 100X), 10 mL penicillin-streptomycin (Life Technologies, 15140163; stock 10^4 U/mL), 333 mL BSA fraction V (GIBCO 15260037; stock 7.50%), 7 mL b-mercaptoethanol (Sigma M6250; stock 14.3 M), 100 mL LIF (Chemico, ESG1107; stock 10^7 U/mL), 100 mL PD0325901 (Stemgent, 04–0006-10; stock 10 mM), and 300 mL CHIR99021 (Stemgent, 04–0004-10; stock 10 mM). Cells were passaged by washing once with 1X PBS (Life Technologies, AM9625) and incubating with TrypLE (Life Technologies, 12604021) for 3–5 minutes, then quenched with serum-containing media made by the following recipe: 500 mL DMEM KO (GIBCO 10829–018), MEM nonessential amino acids (GIBCO 11140076; stock 100X), penicillin-streptomycin (Life Technologies, 15140163; stock 10^4 U/mL), 5 mL L-glutamine (GIBCO 25030–081; stock 100X), 4 mL b-mercaptoethanol (Sigma M6250; stock 14.3 M), 50 mL LIF (Chemico, ESG1107; stock 10^7 U/mL), and 75 mL of fetal bovine serum (Sigma, F4135). Cells were passaged every 2 days.
K562 cells were cultured in suspension flasks containing culture medium [RPMI-1640 medium with GlutaMAX™ (ThermoFisher Cat. 72400047) supplemented with 10% FBS (ThermoFisher Cat. 10437028), 2 mM L-glutamine (Sigma-Aldrich Cat. G7513), 50 U/mL penicillin and 50 μg/mL streptomycin].
A piggyBac compatible base vector was assembled containing two tandem gene cassettes: (1) an insertion site downstream of a doxycycline-inducible promoter allowing for the expression of a Flag-HA-Halo-tagged ORF with SV40 NLS and bGH polyA termination sequence, and (2) the Tet-On 3G rtta element driven by the EF1a promoter that also produces hygromycin resistance via a 2A self-cleaving peptide. This base vector was generated by Gibson assembly. Plasmids encoding Halo-tagged versions of TFs (WT and ARM-deletion) were generated by Gibson assembly with BamHI-digested base vector and gBlocks (Integrated DNA Technologies) encoding the WT and ARM-deletion TFs. See Key Resource Table for plasmid information (pJH294, pJH295, pJH290, pJH291, pJH357, pJH358, pJH337, pJH342, pJH308, pJH309; PBFH vectors stand for “PiggyBac Flag HA”).
To generate cell lines, 5x106 mESCs or K562 cells per well were transfected in 6-well plates with 1 μg of the Halo-TF vector and 1 μg of the piggyBac transposase (Systems Biosciences) in serum-containing media (described above) using Lipofectamine-3000 for at least 4 hours. After transfection, the cells were passaged into 10 cm plates in 2i media or K562 media containing 500 μg/mL Hygromycin-B (Gibco 10687010). After 2–4 days of selection for mESC and 2 weeks of selection for K562, cells were maintained as described above.
Sample preparation
mESCs were plated on glass bottom dishes (Mattek Corporation P35G-1.5–20-C) coated with 5 μg/ml of poly-L-ornithine (Sigma-Aldrich P4957) for 2hrs min at 37°C and with 5μg/ml of Laminin (Corning® 354232) for 2hrs-24hrs at 37°C, growing from 20% confluency in 2i for one day. K562 cells were plated on poly-L-lysine coated glass bottom dishes and allowed to attach for at least 4 hours. Doxycycline=10ng/mL was added to dishes for 1hr, followed by adding 5nM of HaloTag-(PA) JF549 for another 3hrs. Cells were then rinsed once with PBS and washed in fresh 2i for 1hr. Dishes were refilled with 2mL prewarmed Leibovitz's L-15 Medium, no phenol red (ThermoFisher 21083027) and brought for imaging.
Imaging
Cells were imaged on an inverted, widefield setup with a Nikon Eclipse Ti microscope and a 100x oil immersion objective as previously described58. Images were acquired with an EMCCD camera (EM gain 1000, exposure time 10ms, conjugated pixel-size on sample 160nm). A 561nm laser beam of 150mW (attenuated with 50% AOTF) was 2x expanded for a uniform illumination across around 200x200 pixel region. 10,000 frames were recorded for each ROI (including 2–4 cells), and the 405nm activation was kept very low to guarantee the molecule sparsity needed for robust reconnection.
Analyses
Particle trajectories were detected and reconnected with customized MATLAB code from MTT107. Detection settings: false-positive threshold=24, window-size 7x7pixel, and Gaussian width fitting allowed. Reconnection settings: , , and . A collection of trajectories from each ROI were fitted to a 3-state model in Spot-on108. Spot-on settings: detection slice dZ=950nm, 8 delays to consider, and only first 10 jumps to consider for each trajectory. The final outputs include fractions and apparent diffusion coefficients of each state (immobile, sub-diffusive, and free, respectively). For expression dependence testing in Figure S7F, trajectories of the same genotype from different nuclei with similar trajectory density were gathered together first and resampled ten times (2,000 trajectories for each resampling) for ten independent Spot-on fittings, respectively. In this way, the accuracy of each fitting and the distributions across different conditions are comparable.
For dwell time analyses in Figure S7E, sparse detections from slow tracking mode were generated with the same MTT settings as for those in the fast tracking. The detections were then grouped to different spatial clusters by running a Density-based spatial clustering of applications with noise (DBSCAN) with short radius. Within each spatial cluster, the time-correlated detections were further grouped into the same trajectory (two dark frames at maximum). In this manner, only immobile (i.e., bound) trajectories will be collected, whose duration () were the apparent dwelling time. The survival probabilities of apparent dwelling time distributions were fitted to a biexponential model for both fixed and live cell samples, where a short dwelling time scale and a long dwelling time scale were fitted. The stable dwell time of each live cell sample was based on the long dwelling time scale, which was calibrated by the long dwelling time scale of a fixed sample with the exact imaging condition as following:
where is the “apparent” long dwelling time scale of the live sample, is the “apparent” long dwelling time scale of a fixed sample on the same date in the same imaging buffer, and is the calibrated stable dwell time actually reported in final figures.
For curve fitting in Figure S7B, the sum of N constrained Gaussian functions is fitted to the probability distributions of logarithm of diffusion coefficients (logD). The total amplitude of N Gaussians is constrained to 1. The center of each Gaussian is constrained within an interval, where the N intervals are determined based on N equally assigned quantiles of the logD distribution. The sigma of each Gaussian is contained below the half width of the corresponding interval. The diffusion coefficients are fitted from 3,000–20,000 individual single-molecule trajectories with at least 5 jumps in assumption of 2-dimensional Brownian motion. Only the first 16 jumps are used to fit the diffusion coefficient if there are more than 16 jumps for a given trajectory.
For subdiffusion analysis in Figure S7C, single-molecule trajectories for KLF4, SOX2, CTCF, GATA2, and RUNX1 were analyzed by computing the mean squared displacement (MSD) of particles as a function of lag time τ according to a standard method109. MSD was computed for trajectories containing 5 or more time steps and the final lag time was trimmed from each trace prior to fitting. Traces were fit to the 2D anomalous diffusion equation:
where MSD is the mean squared displacement (μm2) for each trajectory, is the radial displacement of the particle (μm), is the generalized diffusion coefficient (μm2/sα), t is the lag time (s), and α is the anomalous diffusion exponent. Two-parameter curve fitting of α and was performed in Python using the ‘curve_fit’ function in scipy (v. 1.10.1). Traces with α between 10–5 and 0.8 and with α > 10*σ2(α), where σ(α) is the standard deviation of the α parameter estimate, were classified as subdiffusive. For visualization purposes, datasets for KLF4, CTCF, and SOX2 were randomly downsampled to display 5% of traces.
Sub-nuclear fractionation
mESCs with exogenous expression for SOX2 and KLF4 wild type and ARM deletion mutations expressing HA tag were used for nuclei sub fractionation. To extract nuclei, cells were resuspended in 10 ml HMSD50 buffer (20 mM HEPES pH 7.5, 5 mM MgCl2, 250 mM sucrose, 1mM DTT, 50mM NaCl, supplemented with 0.2 mM PMSF and 5 mM sodium butyrate) and incubated for 30 min at 4°C with gentle agitation. After a spin down at 3500 rpm at 4°C for 10 min, the supernatant was discarded and the pellet containing nuclei were subjected to subcellular protein fractionation for nucleoplasm and chromatin fractions using the Subcellular Protein Fractionation Kit for Cultured Cells (ThermoScientific, Ref 78840) according to manufacturer’s instructions. For RNase treatment in wild type mESCs, nuclei were treated with RNase A (1:100, Thermo Fisher EN0531) and the initial 30-minute incubation at 4°C was adjusted to 20 minutes at 4°C and 10 minutes at 37°C. The pH of the buffer remained the same (~7.5) after RNase A treatment. SDS Page was run on 12% Bis-Tris gel (Criterion XT, BioRad) and western blotting was performed on the subfractions using anti Histone H3 antibody from Abcam (ab1791) and anti HA antibody from Abcam (ab9110) with secondary antibody against Rabbit (IRDye 800CW Goat anti-rabbit LI-COR 926–32211). For wild type transcription factor detection, antibody for Sox2 (R&D Systems, MAB2018) and Klf4 (R&D Systems, AF3158) with secondary antibody anti-mouse for Sox2 (IRDye 680CW goat anti-mouse LI-COR 926–32211) and anti-goat for Klf4 (IRDye 800CW donkey anti-goat LI-COR 926–32214), were used. Fluorescence was assessed using Odyssey CLX LiCOR and quantified using Fiji/ImageJ111.
Zebrafish knockdown and rescue of sox2
Morpholinos (MO, GeneTools) were resuspended in nuclease free water, heated to 65°C for 5 minutes, and stored at room temperature. Wildtype AB zebrafish embryos were injected into the yolk at the 1-cell stage with 7ng of sox2-MO (TCTTGAAAGTCTACCCCACCAGCCG)53, either alone or in combination with 25 pg of human wildtype or ARM-deletion SOX2 mRNA. Messenger RNA was synthesized using the T7 mMessage mMachine (Invitrogen) kit with templates generated from gBlocks (IDT). The mRNA was purified with the MEGAclear Clean-Up Kit (Invitrogen), run on a TBE agarose gel to confirm purity and size, aliquoted, and stored at - 80°C. Embryos injected with 7ng of Standard Control MO (CCTCTTACCTCAGTTACAATTTATA) were used as controls. At 48 hours post fertilization (hpf), MO injected embryos were dechorionated using forceps, anaesthetized using 0.16 mg/ml Tricaine, then visually assessed for growth impairment using a Nikon SMZ18 stereoscope with DS-Ri2 camera and NIS-Elements software. Embryos were scored based on rescue of growth impairment in the presence of wildtype or mutant sox2 mRNA.
To assure that mutant SOX2 was expressed as protein, we conducted Western blots (Figure S7G). Protein extraction for zebrafish embryos (n = 20 per tube) that were uninjected or injected with mRNA encoding HA-tagged ARM-mutant SOX2 was performed with Urea Chaps lysis buffer. Cells were resuspended in Urea Chaps (1% Chaps, 8M Urea, 50mM Tris-Cl pH 7.5 containing protease inhibitors (Thermo Fisher)) and incubated for 30’ at 4°C with gentle agitation. After a spin down at 14,000 rpm for 10’ at 4°C, the supernatant was used for SDS-Page. SDS-Page was run on a 10% Bis-Tris (Criterion XT, BioRad) and western blotting was performed on uninjected and injected samples using anti HA antibody from Abcam (ab9110) and anti beta actin (Sigma A5441) with secondary antibody against Rabbit (IRDye 800CW Goat anti-rabbit LI-COR 926–32211 and IRDye 680RD Goat anti-mouse 926–68070). Fluorescence was assessed using Odyssey CLX LiCOR.
Overlap of pathogenic mutations in TF-ARMs
Pathogenic nonsynonmous substitution mutations were obtained from a prior dataset of pathogenic mutations that integrated multiple databases of somatic and germline variation associated with cancer and Mendelian disorders, including ClinVar (accessed January 29, 2021) and HGMD v2020.4 in hg38. Cancer variants were obtained from AACR Project GENIE v8.1 (AACR Project GENIE Consortium, 2017) and various TCGA and TARGET studies via cBioPortal110. Mutations were subsetted for those affecting TF-ARMs. For mutation frequency analysis, the expected mutation frequency for each amino acid type within TF-ARMs was estimated using the average nucleotide substitution rates within the entire mutation dataset and the frequency of nucleotide types encoding each amino acid type within TF-ARMs. It is important to note that this analysis does not take into account disease-specific mutational signatures, which could introduce potential biases. Enrichment was defined as a significantly higher pathogenic mutation frequency compared to the aforementioned expected amino acid mutation frequency. Statistical significance of the enrichment was determined using a one-sided binomial test, and p-values were corrected for the multiple tests across the twenty amino acids using the Benjamini-Hochberg method.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of quantification and statistical analysis for each experiment can be found in their respective section, and we provide additional details on sample sizes and statistical parameters here. Statistical tests were conducted using Prism software (GraphPad). Confidence intervals for estimates from fluorescence polarization data were computed by multiplying the standard deviation of the curve fit parameter with the Student’s t-value corresponding to the 95% confidence interval with degrees of freedom equal to the number of data points in the concentration curve minus the number of fit parameters. Statistical comparisons between the ’s of two fluorescence polarization curves (for Figure 3E, Figure S2E, and Figure S4) were assessed using a two-tailed Student’s t-test based on the standard errors of the parameters calculated from the diagonals of the covariance matrix returned by ‘curve_fit’ in scipy, with the degrees of freedom as specified above.
The distributions of ARM correlation scores (Figure 3C) for whole proteome (-TFs) vs TFs were compared using a two-tailed Mann Whitney U test, n1=1287, n2=20238.
The Tat reporter assays were conducted on 3 biological replicates per genotype, and luminescence readings were measured in technical duplicates. Each condition was compared to the Tat R/K>A condition using a Sidak multiple comparisons test (DF = 24, t statistics were as follow: TAR-WT - WT=20.15, KLF4=15.3, SOX2=13.17, GATA2=3.805, NoTat=6.419; ΔTAR-bulge – WT=9.263, KLF4=9.319, SOX2=9.329, GATA2=9.315, Tat R/K>A=9.302, No-Tat=9.364).
For comparison of the diffusive fractions reported in Figure 5C, multiple fields of cells were imaged per genotype (KLF4-WT n=11, KLF4-ΔARM n=9, SOX2-WT n=10, SOX2-ΔARM n=9, CTCF-WT n=7, CTCF-ΔARM n=7). The diffusive fractions were compared by 2-tailed Student t-test. The data was confirmed to have equal variance via F test, and the degrees of freedom and t statistics were as follows: KLF4-free (t=13.47, df=18), SOX2-free (t=8.297, df=18), CTCF-free (t=6.044, df=12), KLF4-sub (t=5.152, df=18), SOX2-sub (2.908, df=18), CTCF-sub (t=3.051, df=12), KLF4-imm (t=7.824, df=18), SOX2-imm (t=6.203, df=18), CTCF-imm (t=3.639, df=12).
Supplementary Material
Table S1. RBR-ID dataset for K562 cells, related to Figure 1
Table S2. RBR-ID dataset for mES cells, related to Figure 1
Table S3. List of RNA-binding TFs, related to Figure 1
Table S4. RNAs crosslinked to GATA2 with nucleotide resolution, related to Figure 1
Table S5. Pathogenic mutations in TF-ARMs, related to Figure 6
Table S6. Sequences of reagents used in this study, related to STAR Methods
Video S1. Single-molecule imaging of KLF4-WT-HALO in mESCs, related to Figure 5
Video S2. Single-molecule imaging of SOX2-WT-HALO in mESCs, related to Figure 5
Video S3. Single-molecule imaging of GATA2-WT-HALO in K562 cells, related to Figure 5
MOLECULAR-CELL-D-23–00413R1.
Highlights
Transcription factors (TFs) bind RNA
TFs harbor RNA-binding domains
TF-RNA binding promotes chromatin occupancy
TF-RNA binding is conserved, important for development, and disrupted in disease
Acknowledgements
We are grateful to Phillip Sharp, Amy Gladfelter, Seychelle Vos, and Ibrahim Cissé for discussions regarding HIV Tat, RNA-binding proteins, RNA interactions, and single-molecule imaging. We thank L.D. Lavis (HHMI, Janelia) for the gift of the Halo Tag-(PA)-JF549 dyes. This work was supported by NIH grants GM144283 (R.A.Y), CA155258 (R.A.Y.), NSF PHY2044895 (R.A.Y.), F32CA254216 (J.E.H.), GM138788 (R.B.), GM127408 (R.B.), R01 HL144780–01 (L.I.Z.), R24 OD017870–01 (L.I.Z), CA251062 (S.F.B.) and NSF Graduate Research Fellowship 1745302 (K.J.O.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
R.A.Y. is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics, Dewpoint Therapeutics and Paratus Sciences. R.A.Y. is a member of Molecular Cell's advisory board. O.O. and J.E.H. are consultants at Camp4 Therapeutics. L.I.Z. is a founder and stockholder of Fate Therapeutics, Camp4 Therapeutics, Amagma Therapeutics, Scholar Rock, and Branch Biosciences. L.I.Z. is a consultant for Celularity and Cellarity. The Whitehead Institute has filed a patent application related to this work.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 2.Vaquerizas JM, Kummerfeld SK, Teichmann SA, and Luscombe NM (2009). A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet 10, 252–263. 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 3.Cramer P (2019). Organization and regulation of gene transcription. Nature 573, 45–54. 10.1038/s41586-019-1517-4. [DOI] [PubMed] [Google Scholar]
- 4.Lee TI, and Young RA (2013). Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251. 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadhouders R, Filion GJ, and Graf T (2019). Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354. 10.1038/s41586-019-1182-7. [DOI] [PubMed] [Google Scholar]
- 6.Panne D, Maniatis T, and Harrison SC (2007). An Atomic Model of the Interferon-β Enhanceosome. Cell 129, 1111–1123. 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avsec Ž, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, et al. (2021). Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet 53, 354–366. 10.1038/s41588-021-00782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold CD, Nemčko F, Woodfin AR, Wienerroither S, Vlasova A, Schleiffer A, Pagani M, Rath M, and Stark A (2018). A high‐throughput method to identify trans‐activation domains within transcription factor sequences. EMBO J 37, e98896. 10.15252/embj.201798896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soto LF, Li Z, Santoso CS, Berenson A, Ho I, Shen VX, Yuan S, and Fuxman Bass JI (2022). Compendium of human transcription factor effector domains. Mol. Cell 82, 514–526. 10.1016/j.molcel.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter WF, Nayak S, Iwasa J, and Taatjes DJ (2022). The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol, 1–18. 10.1038/s41580-022-00498-3. [DOI] [PMC free article] [PubMed]
- 12.Vos SM (2021). Understanding transcription across scales: From base pairs to chromosomes. Mol. Cell 81, 1601–1616. 10.1016/j.molcel.2021.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Lelli KM, Slattery M, and Mann RS (2012). Disentangling the many layers of eukaryotic transcriptional regulation. Annu. Rev. Genet 46, 43–68. 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 15.Kaikkonen MU, and Adelman K (2018). Emerging Roles of Non-Coding RNA Transcription. Trends Biochem. Sci 43, 654–667. 10.1016/j.tibs.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, and Sharp PA (2008). Divergent Transcription from Active Promoters. Science 322, 1849–1851. 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassiday LA, and Maher LJ (2002). Having it both ways: transcription factors that bind DNA and RNA. Nucleic Acids Res 30, 4118–4126. 10.1093/nar/gkf512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmes ZE, Hamilton DJ, Hwang T, Parsonnet NV, Rinn JL, Wuttke DS, and Batey RT (2020). The Sox2 transcription factor binds RNA. Nat. Commun 11, 1805. 10.1038/s41467-020-15571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou L, Wei Y, Lin Y, Wang X, Lai Y, Yin M, Chen Y, Guo X, Wu S, Zhu Y, et al. (2020). Concurrent binding to DNA and RNA facilitates the pluripotency reprogramming activity of Sox2. Nucleic Acids Res 48, 3869–3887. 10.1093/nar/gkaa067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saldaña-Meyer R, Rodriguez-Hernaez J, Escobar T, Nishana M, Jácome-López K, Nora EP, Bruneau BG, Tsirigos A, Furlan-Magaril M, Skok J, et al. (2019). RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol. Cell 76, 412–422.e5. 10.1016/j.molcel.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, Jangi M, Giallourakis CC, Sharp PA, and Young RA (2015). Transcription factor trapping by RNA in gene regulatory elements. Science 350, 978–981. 10.1126/science.aad3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theunissen O, Rudt F, Guddat U, Mentzel H, and Pieler T (1992). RNA and DNA binding zinc fingers in Xenopus TFIIIA. Cell 71, 679–690. 10.1016/0092-8674(92)90601-8. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Huangyang P, Wang Y, Xue L, Devericks E, Nguyen HG, Yu X, Oses-Prieto JA, Burlingame AL, Miglani S, et al. (2021). ERα is an RNA-binding protein sustaining tumor cell survival and drug resistance. Cell 0. 10.1016/j.cell.2021.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeon Y, and Lee JT (2011). YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146, 119–133. 10.1016/j.cell.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Yoshida T, Itoh H, and Kohno K (2004). Binding of RNA to p53 regulates its oligomerization and DNA-binding activity. Oncogene 23, 4371–4379. 10.1038/sj.onc.1207583. [DOI] [PubMed] [Google Scholar]
- 26.Steiner HR, Lammer NC, Batey RT, and Wuttke DS (2022). An Extended DNA Binding Domain of the Estrogen Receptor Alpha Directly Interacts with RNAs in Vitro. Biochemistry 61, 2490–2494. 10.1021/acs.biochem.2c00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niessing D, Driever W, Sprenger F, Taubert H, Jäckle H, and Rivera-Pomar R (2000). Homeodomain Position 54 Specifies Transcriptional versus Translational Control by Bicoid. Mol. Cell 5, 395–401. 10.1016/S1097-2765(00)80434-7. [DOI] [PubMed] [Google Scholar]
- 28.Dvir S, Argoetti A, Lesnik C, Roytblat M, Shriki K, Amit M, Hashimshony T, and Mandel-Gutfreund Y (2021). Uncovering the RNA-binding protein landscape in the pluripotency network of human embryonic stem cells. Cell Rep 35. 10.1016/j.celrep.2021.109198. [DOI] [PubMed] [Google Scholar]
- 29.Lunde BM, Moore C, and Varani G (2007). RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol 8, 479–490. 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wheeler EC, Van Nostrand EL, and Yeo GW (2018). Advances and challenges in the detection of transcriptome-wide protein-RNA interactions. Wiley Interdiscip. Rev. RNA 9, e1436. 10.1002/wrna.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He C, Sidoli S, Warneford-Thomson R, Tatomer DC, Wilusz JE, Garcia BA, and Bonasio R (2016). High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Mol. Cell 64, 416–430. 10.1016/j.molcel.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orkin SH, and Zon LI (2008). Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 132, 631–644. 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delgado MD, Lerga A, Cañelles M, Gómez-Casares MT, and León J (1995). Differential regulation of Max and role of c-Myc during erythroid and myelomonocytic differentiation of K562 cells. Oncogene 10, 1659–1665. [PubMed] [Google Scholar]
- 34.Young RA (2011). Control of the embryonic stem cell state. Cell 144, 940–954. 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibarra A, Benner C, Tyagi S, Cool J, and Hetzer MW (2016). Nucleoporin-mediated regulation of cell identity genes. Genes Dev 30, 2253–2258. 10.1101/gad.287417.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saldaña-Meyer R, González-Buendía E, Guerrero G, Narendra V, Bonasio R, Recillas-Targa F, and Reinberg D (2014). CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev 28, 723–734. 10.1101/gad.236869.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burd CG, and Dreyfuss G (1994). RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J 13, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corley M, Burns MC, and Yeo GW (2020). How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 78, 9–29. 10.1016/j.molcel.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maji D, Glasser E, Henderson S, Galardi J, Pulvino MJ, Jenkins JL, and Kielkopf CL (2020). Representative cancer-associated U2AF2 mutations alter RNA interactions and splicing. J. Biol. Chem 295, 17148–17157. 10.1074/jbc.RA120.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, and Manley JL (2015). Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. U. S. A 112, E4726–E4734. 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calnan BJ, Biancalana S, Hudson D, and Frankel AD (1991). Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes Dev 5, 201–210. 10.1101/gad.5.2.201. [DOI] [PubMed] [Google Scholar]
- 42.Calnan BJ, Tidor B, Biancalana S, Hudson D, and Frankel AD (1991). Arginine-Mediated RNA Recognition: the Arginine Fork. Science 252, 1167–1171. 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 43.Pham VV, Salguero C, Khan SN, Meagher JL, Brown WC, Humbert N, de Rocquigny H, Smith JL, and D’Souza VM (2018). HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry. Nat. Commun 9, 4266. 10.1038/s41467-018-06591-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jakobovits A, Smith DH, Jakobovits EB, and Capon DJ (1988). A discrete element 3’ of human immunodeficiency virus 1 (HIV-1) and HIV-2 mRNA initiation sites mediates transcriptional activation by an HIV trans activator. Mol. Cell. Biol 8, 2555–2561. 10.1128/mcb.8.6.2555-2561.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghaleb AM, and Yang VW (2017). Krüppel-like factor 4 (KLF4): What we currently know. Gene 611, 27–37. 10.1016/j.gene.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geiman DE, Ton-That H, Johnson JM, and Yang VW (2000). Transactivation and growth suppression by the gut-enriched Krüppel-like factor (Krüppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res 28, 1106–1113. 10.1093/nar/28.5.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yet SF, McA’Nulty MM, Folta SC, Yen HW, Yoshizumi M, Hsieh CM, Layne MD, Chin MT, Wang H, Perrella MA, et al. (1998). Human EZF, a Krüppel-like zinc finger protein, is expressed in vascular endothelial cells and contains transcriptional activation and repression domains. J. Biol. Chem 273, 1026–1031. 10.1074/jbc.273.2.1026. [DOI] [PubMed] [Google Scholar]
- 48.Chen J, Zhang Z, Li L, Chen B-C, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. (2014). Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285. 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen VQ, Ranjan A, Liu S, Tang X, Ling YH, Wisniewski J, Mizuguchi G, Li KY, Jou V, Zheng Q, et al. (2021). Spatiotemporal coordination of transcription preinitiation complex assembly in live cells. Mol. Cell, S1097276521005918. 10.1016/j.molcel.2021.07.022. [DOI] [PMC free article] [PubMed]
- 50.Garcia DA, Johnson TA, Presman DM, Fettweis G, Wagh K, Rinaldi L, Stavreva DA, Paakinaho V, Jensen RAM, Mandrup S, et al. (2021). An intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. Mol. Cell 81, 1484–1498.e6. 10.1016/j.molcel.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia DA, Fettweis G, Presman DM, Paakinaho V, Jarzynski C, Upadhyaya A, and Hager GL (2021). Power-law behavior of transcription factor dynamics at the single-molecule level implies a continuum affinity model. Nucleic Acids Res 49, 6605–6620. 10.1093/nar/gkab072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen AS, Amitai A, Cattoglio C, Tjian R, and Darzacq X (2020). Guided nuclear exploration increases CTCF target search efficiency. Nat. Chem. Biol 16, 257–266. 10.1038/s41589-019-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pavlou S, Astell K, Kasioulis I, Gakovic M, Baldock R, Heyningen V. van, and Coutinho P (2014). Pleiotropic Effects of Sox2 during the Development of the Zebrafish Epithalamus. PLOS ONE 9, e87546. 10.1371/journal.pone.0087546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boldes T, Merenbakh-Lamin K, Journo S, Shachar E, Lipson D, Yeheskel A, Pasmanik-Chor M, Rubinek T, and Wolf I (2020). R269C variant of ESR1: high prevalence and differential function in a subset of pancreatic cancers. BMC Cancer 20, 531. 10.1186/s12885-020-07005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keegan L, Gill G, and Ptashne M (1986). Separation of DNA binding from the transcription-activating function of a eukaryotic regulatory protein. Science 231, 699–704. 10.1126/science.3080805. [DOI] [PubMed] [Google Scholar]
- 56.Tjian R, and Maniatis T (1994). Transcriptional activation: a complex puzzle with few easy pieces. Cell 77, 5–8. 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 57.Asimi V, Sampath Kumar A, Niskanen H, Riemenschneider C, Hetzel S, Naderi J, Fasching N, Popitsch N, Du M, Kretzmer H, et al. (2022). Hijacking of transcriptional condensates by endogenous retroviruses. Nat. Genet, 1–10. 10.1038/s41588-022-01132-w. [DOI] [PMC free article] [PubMed]
- 58.Henninger JE, Oksuz O, Shrinivas K, Sagi I, LeRoy G, Zheng MM, Andrews JO, Zamudio AV, Lazaris C, Hannett NM, et al. (2021). RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 184, 207–225.e24. 10.1016/j.cell.2020.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharp PA, Chakraborty AK, Henninger JE, and Young RA (2022). RNA in formation and regulation of transcriptional condensates. RNA N. Y. N 28, 52–57. 10.1261/rna.078997.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinodoz SA, Jachowicz JW, Bhat P, Ollikainen N, Banerjee AK, Goronzy IN, Blanco MR, Chovanec P, Chow A, Markaki Y, et al. (2021). RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790.e30. 10.1016/j.cell.2021.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149.e22. 10.1016/j.cell.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, and Shiekhattar R (2013). Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494, 497–501. 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long Y, Wang X, Youmans DT, and Cech TR (2017). How do lncRNAs regulate transcription? Sci. Adv 3, eaao2110. 10.1126/sciadv.aao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hemphill WO, Voong CK, Fenske R, Goodrich JA, and Cech TR (2022). RNA- and DNA-binding proteins generally exhibit direct transfer of polynucleotides: Implications for target site search 2022.11.30.518605. 10.1101/2022.11.30.518605. [DOI] [PMC free article] [PubMed]
- 65.Reimer KA, Mimoso CA, Adelman K, and Neugebauer KM (2021). Co-transcriptional splicing regulates 3′ end cleavage during mammalian erythropoiesis. Mol. Cell 81, 998–1012.e7. 10.1016/j.molcel.2020.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brodsky S, Jana T, Mittelman K, Chapal M, Kumar DK, Carmi M, and Barkai N (2020). Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol. Cell 79, 459–471.e4. 10.1016/j.molcel.2020.05.032. [DOI] [PubMed] [Google Scholar]
- 67.Inukai S, Kock KH, and Bulyk ML (2017). Transcription factor–DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev 43, 110–119. 10.1016/j.gde.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wasserman WW, and Sandelin A (2004). Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet 5, 276–287. 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 69.Han H, Braunschweig U, Gonatopoulos-Pournatzis T, Weatheritt RJ, Hirsch CL, Ha KCH, Radovani E, Nabeel-Shah S, Sterne-Weiler T, Wang J, et al. (2017). Multilayered Control of Alternative Splicing Regulatory Networks by Transcription Factors. Mol. Cell 65, 539–553.e7. 10.1016/j.molcel.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 70.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. Publ. Protein Soc 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. Publ. Protein Soc 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Demichev V, Messner CB, Vernardis SI, Lilley KS, and Ralser M (2020). DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem 75, 4646–4658. 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 74.Hochberg Y, and Benjamini Y (1990). More powerful procedures for multiple significance testing. Stat. Med 9, 811–818. 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 75.Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. (2012). The mRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Mol. Cell 46, 674–690. 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 76.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. (2012). Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell 149, 1393–1406. 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 77.Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, and Kim VN (2013). The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol 20, 1122–1130. 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- 78.Bao X, Guo X, Yin M, Tariq M, Lai Y, Kanwal S, Zhou J, Li N, Lv Y, Pulido-Quetglas C, et al. (2018). Capturing the interactome of newly transcribed RNA. Nat. Methods 15, 213–220. 10.1038/nmeth.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang R, Han M, Meng L, and Chen X (2018). Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proc. Natl. Acad. Sci 115, E3879–E3887. 10.1073/pnas.1718406115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trendel J, Schwarzl T, Horos R, Prakash A, Bateman A, Hentze MW, and Krijgsveld J (2019). The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 176, 391–403.e19. 10.1016/j.cell.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 81.Queiroz RML, Smith T, Villanueva E, Marti-Solano M, Monti M, Pizzinga M, Mirea D-M, Ramakrishna M, Harvey RF, Dezi V, et al. (2019). Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol 37, 169–178. 10.1038/s41587-018-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He C, Bozler J, Janssen KA, Wilusz JE, Garcia BA, Schorn AJ, and Bonasio R (2021). TET2 chemically modifies tRNAs and regulates tRNA fragment levels. Nat. Struct. Mol. Biol 28, 62–70. 10.1038/s41594-020-00526-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blue SM, Yee BA, Pratt GA, Mueller JR, Park SS, Shishkin AA, Starner AC, Van Nostrand EL, and Yeo GW (2022). Transcriptome-wide identification of RNA-binding protein binding sites using seCLIP-seq. Nat. Protoc 17, 1223–1265. 10.1038/s41596-022-00680-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 85.Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Langmead B, Wilks C, Antonescu V, and Charles R (2019). Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35, 421–432. 10.1093/bioinformatics/bty648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang Y-A, Choi K, Farnham PJ, and Bresnick EH (2009). Discovering Hematopoietic Mechanisms Through Genome-Wide Analysis of GATA Factor Chromatin Occupancy. Mol. Cell 36, 667–681. 10.1016/j.molcel.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille J-H, Afeyan LK, Zamudio AV, Shrinivas K, et al. (2019). Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature, 1–6. 10.1038/s41586-019-1464-0. [DOI] [PMC free article] [PubMed]
- 95.Sharma D, Zagore LL, Brister MM, Ye X, Crespo-Hernández CE, Licatalosi DD, and Jankowsky E (2021). The kinetic landscape of an RNA-binding protein in cells. Nature 591, 152–156. 10.1038/s41586-021-03222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gerstberger S, Hafner M, and Tuschl T (2014). A census of human RNA-binding proteins. Nat. Rev. Genet 15, 829–845. 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Holehouse AS, Das RK, Ahad JN, Richardson MOG, and Pappu RV (2017). CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J 112, 16–21. 10.1016/j.bpj.2016.11.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li CH, Coffey EL, Dall’Agnese A, Hannett NM, Tang X, Henninger JE, Platt JM, Oksuz O, Zamudio AV, Afeyan LK, et al. (2020). MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 10.1038/s41586-020-2574-4. [DOI] [PMC free article] [PubMed]
- 100.Blum M, Chang H-Y, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, et al. (2021). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49, D344–D354. 10.1093/nar/gkaa977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME Suite: tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208. 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Emenecker RJ, Griffith D, and Holehouse AS (2021). Metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys. J 120, 4312–4319. 10.1016/j.bpj.2021.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–W350. 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bakan A, Meireles LM, and Bahar I (2011). ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 27, 1575–1577. 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Henikoff S, Henikoff JG, Kaya-Okur HS, and Ahmad K (2020). Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife 9, e63274. 10.7554/eLife.63274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meers MP, Tenenbaum D, and Henikoff S (2019). Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42. 10.1186/s13072-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sergé A, Bertaux N, Rigneault H, and Marguet D (2008). Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat. Methods 5, 687–694. 10.1038/nmeth.1233. [DOI] [PubMed] [Google Scholar]
- 108.Hansen AS, Woringer M, Grimm JB, Lavis LD, Tjian R, and Darzacq X (2018). Robust model-based analysis of single-particle tracking experiments with Spot-On. eLife 7, e33125. 10.7554/eLife.33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Saxton MJ (1997). Single-particle tracking: the distribution of diffusion coefficients. Biophys. J 72, 1744–1753. 10.1016/S0006-3495(97)78820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Banani SF, Afeyan LK, Hawken SW, Henninger JE, Dall’Agnese A, Clark VE, Platt JM, Oksuz O, Hannett NM, Sagi I, et al. (2022). Genetic variation associated with condensate dysregulation in disease. Dev. Cell 10.1016/j.devcel.2022.06.010. [DOI] [PMC free article] [PubMed]
- 111.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RBR-ID dataset for K562 cells, related to Figure 1
Table S2. RBR-ID dataset for mES cells, related to Figure 1
Table S3. List of RNA-binding TFs, related to Figure 1
Table S4. RNAs crosslinked to GATA2 with nucleotide resolution, related to Figure 1
Table S5. Pathogenic mutations in TF-ARMs, related to Figure 6
Table S6. Sequences of reagents used in this study, related to STAR Methods
Video S1. Single-molecule imaging of KLF4-WT-HALO in mESCs, related to Figure 5
Video S2. Single-molecule imaging of SOX2-WT-HALO in mESCs, related to Figure 5
Video S3. Single-molecule imaging of GATA2-WT-HALO in K562 cells, related to Figure 5
Data Availability Statement
The RBR-ID mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD035484. CUT&Tag sequencing and CLIP sequencing data have been deposited to GEO with identifier GSE232181. Original images for EMSAs and Western blots as well as single molecule trace data are available through Mendeley Data (DOI: 10.17632/dkx9gsh42h.2). These data are publicly available as of the date of publication.
Code generated during this study is available through Zenodo (DOI: 10.5281/zenodo.7974933; Link: https://zenodo.org/record/7974933)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact by request.