

Abstract
Background:
Lymphatic vessels are responsible for tissue drainage, and their malfunction is associated with chronic diseases. Lymph uptake occurs via specialized open cell-cell junctions between capillary lymphatic endothelial cells (LECs), while closed junctions in collecting LECs prevent lymph leakage. LEC junctions are known to dynamically remodel in development and disease, but how lymphatic permeability is regulated remains poorly understood.
Methods:
We employed various genetically engineered mouse models in combination with cellular, biochemical, and molecular biology approaches to elucidate the signaling pathways regulating junction morphology and function in lymphatic capillaries.
Results:
By studying the permeability of intestinal lacteal capillaries to lipoprotein particles known as chylomicrons, we show that Rho-associated kinase (ROCK)-dependent cytoskeletal contractility is a fundamental mechanism of LEC permeability regulation. We show that chylomicron-derived lipids trigger neonatal lacteal junction opening via ROCK-dependent contraction of junction-anchored stress fibers. LEC-specific ROCK deletion abolished junction opening and plasma lipid uptake. Chylomicrons additionally inhibited VEGF-A signaling. We show that VEGF-A antagonizes LEC junction opening via VEGFR2 and VEGFR3-dependent PI3K/AKT activation of the small GTPase RAC1, thereby restricting RhoA/ROCK-mediated cytoskeleton contraction.
Conclusions:
Our results reveal that antagonistic inputs into ROCK-dependent cytoskeleton contractions regulate the interconversion of lymphatic junctions in the intestine and in other tissues, providing a tunable mechanism to control the lymphatic barrier.
Keywords: lymphatic endothelial cell junctions, intestinal lipid uptake, lymphatic endothelial permeability, ROCK signaling, VEGF-A/VEGFR2 signaling, Basic Science Research, Cell Signaling/Signal Transduction, Developmental Biology, Lipids and Cholesterol, Vascular Biology
Graphical Abstract
INTRODUCTION
Lymphatic vessels control fluid homeostasis and immune cell trafficking in most tissues of the body, and lipid absorption in the small intestine1–3. They form a unidirectional circulatory system with blind ended capillaries that drain back via collecting vessels and the thoracic duct into the venous circulation. To execute their functions, lymphatic endothelial cells (LECs) lining lymphatic vessels develop two types of cell-cell junctions. Capillary LECs are tied together by discontinuous button-like junctions4. Fluids, lipids, and immune cells enter via openings between the buttons, without disrupting junctional integrity. By contrast, collector LECs have continuous, zipper-like junctions that prevent fluid leak and ensure lymph transport towards the thoracic duct, aided by smooth muscle cells (SMC) and intraluminal valves5.
Both junction types contain the same proteins and can transform into each other via intermediate forms6. A few prior studies show that button junctions appear postnatally and revert back to zippers during inflammation, indicating dynamic remodeling4,6. Infection-induced dermal capillary zippering inhibits dissemination of vaccinia virus7, identifying lymphatic junctions as critical regulators of infection resistance. Junction zippering of intestinal lymphatic capillaries termed lacteals inhibits chylomicron uptake and confers resistance to diet-induced obesity8, demonstrating effects of LEC junction status on whole-body metabolism. Targeting junction remodeling appears as a promising approach to control lymphatic function, but mechanisms regulating this process remain largely unknown.
We have here investigated LEC junction remodeling in the context of lipid uptake in neonatal intestine. Upon ingestion of breastmilk after birth, the newborn mammalian gastrointestinal (GI) tract becomes a lipid-rich environment that supplies energy and essential signaling constituents9. Milk-derived lipids are absorbed by enterocytes and repackaged into chylomicrons, large lipoprotein particles containing triglycerides, cholesterol and apolipoprotein B48 (ApoB48). Newly synthesized chylomicrons exit the abluminal side of enterocytes and are taken up by lacteals10. Lacteal growth inhibition via VEGF-C/VEGFR3 inactivation, loss of lacteal DLL4/Notch activity, or loss of calcitonin receptor–like receptor (Calcrl), resulted in reduced chylomicron absorption11–16. Impaired lacteal extension into the villus, because of abnormal SMC coverage, shifted lipid absorption from the lymphatic system to the portal circulation, leading to hepatic lipidosis17. This indicates that the lacteals are essential for chylomicron uptake and transport.
Herein, we show that newborn lacteal junctions open in response to ingested lipids that trigger their own absorption by activating ROCK-dependent cytoskeletal pull. Chylomicrons also inhibit intestinal VEGF-A signaling by upregulating the VEGF-A decoy receptor VEGFR1 in blood vascular endothelial cells (BECs) via ApoB488,18. We show that VEGF-A antagonizes junction opening via VEGFR2/VEGFR3-dependent PI3K activation of the small GTPase RAC1, which inhibits RhoA/ROCK-mediated cytoskeleton contraction. Our findings thus reveal that ROCK-dependent cytoskeleton remodeling controls the permeability of lymphatic junctions in response to environmental cues.
METHODS
DATA AVAILABILITY
The RNAseq data from HUVECs and HDLECs are available in the NCBI Gene Expression Omnibus (GEO) database (accession numbers GSE209855 and GSE212743). All data are included in the manuscript and the supplementary materials. Additional information can be obtained from the corresponding authors upon reasonable request.
Mice
Vegfr2Y949F 19 and Akt1−/− 20 mice were described previously. Vegfr2flox/flox 21, Vegfr3flox/flox22, PLCγ1flox/flox 23, Erk1−/− (JAX Strain #019113), Erk2flox/flox (JAX Strain #019112), Rock1flox/flox 24 and Rock2flox/flox 25,26 and iMB-Vegfr2 mice27 mice were intercrossed with Prox1CreERT2 mice28. All mice were on a C57BL/6J background and housed in pathogen free animal facilities with a 12 hour (hr)/12 hr light-dark cycle. Mice of both sexes were used. Animal experiments were approved by the Institutional Animal Care and Use Committees (IACUCs) of Yale University and Zhongshan Ophthalmic Center, Sun Yat-sen University.
To induce postnatal gene recombination, we injected mice intraperitoneally (IP) or intragastrically (IG) with tamoxifen (TAM, Sigma; #T5648; 20mg/ml in corn oil). Postnatal mice received 75μg/g TAM daily for three days between P0-P2, P5-P7 or P16-P18 (IP or IG). Adult mice received 2mg TAM on days 1, 2, 3, 5 and 7, starting at age week 5 (IP). All Rock1/2iLKO mice were analyzed within two weeks after the first TAM injection. TAM-injected Cre negative littermates were used as controls.
Statistics
For statistical analysis we used Prism 9-GraphPad software. For comparisons of mouse phenotypes, multiple measurements per mouse were averaged prior to statistical analysis. We analyzed at least 4 mice per condition and mouse numbers per experiment are indicated in Figure legends. Animals were included based on genotypes, proper age, and sex. In some experiments, mice were randomly divided into groups to receive different treatments. For western blot quantifications, protein expression was normalized to endogenous control expression, to correct for loading variation between samples. All timepoints/conditions were then normalized to the average value of baseline/control measurements. Changes were compared using the appropriate statistical test, as indicated in Figure legends for each experiment.
For two group comparisons data were subjected to the non-parametric Mann-Whitney U test. For multiple group comparisons data were analyzed either by One-Way ANOVA (data with equal variances) followed by the Tukey post-hoc test, or by Welch’s ANOVA (data with unequal variances) followed by the Dunnett T3 post-hoc test. For ANOVA, normal distribution was determined using the Shapiro-Wilk test. p < 0.05 was considered statistically significant. Data are expressed as mean ± SEM (standard error of the mean) along with scatter plots of individual data points. Each dot represents one mouse or one experiment, as described in the Figure legends. No outliers were excluded.
RESULTS
Chylomicron-induced cytoskeletal contraction opens lacteal junctions via ROCK
To assess the status of perinatal lacteal junctions, intestinal villi in mouse jejunum were stained for the junctional marker VE-Cadherin (Fig. 1A) and the junctions were categorized as “buttons” or “zippers” based on their morphology (Fig. 1B, see Methods). Lacteals from non-fed prenatal embryonic day (E) 20.5 mice displayed predominantly (>85%) continuous, zipper-like junctions (Fig. 1A–C). By contrast, milk-fed postnatal day (P) 0.5 mice exhibited significantly fewer (<50%) zipper junctions and more discontinuous, button-like junctions (Fig. 1A–C). As the appearance of discontinuous, presumably open LEC junctions, coincided with lacteal exposure to chylomicrons, we asked if the chylomicrons affected junction opening. In vitro, confluent LECs cultured in complete media displayed continuous, straight VE-Cadherin+ adherens junctions, likely due to the presence of VEGF-A in fetal bovine serum8. Upon chylomicron exposure, the junctions transformed into discontinuous, parallel linear segments of VE-cadherin, connected to radial actin stress fibers via focal vinculin-positive patches (Fig. 1D). Treatment with arachidonic acid (AA), the most common long-chain polyunsaturated fatty acid in human milk29, mimicked the effects of chylomicrons on LECs (Fig. 1D). The effect was observed at 30 minutes (min) and disappeared at 120 min (Fig. 1D). The appearance of junction-anchored actin stress fibers suggested increased cytoskeletal contraction, as attested by increased phosphorylation of myosin light chain 2 (MLC2) in response to chylomicrons or AA treatment, compared to untreated cells (Fig. 1E–H). In vivo, wildtype lacteal VE-Cadherin+ discontinuous junctions colocalized with the tension sensor vinculin (Fig. 1I), leading us to hypothesize that chylomicron-derived lipids promote junction opening by triggering cytoskeletal tension changes in lacteal LECs.
Figure 1. Chylomicrons and lipids promote lacteal junction opening via cytoskeletal contractions:

(A) VE-Cadherin and LYVE1 staining of jejunum lacteals of C-sectioned E20.5 wildtype mouse embryos and breastfed postnatal (P) 0.5 neonatal pups.
(B) Example of junction morphology quantification from Figure 1A. The length of zipper and button-like junctions is annotated in green and red respectively and measured using Fiji software (see online Methods).
(C) Quantification of % zipper-like junctions out of total junction length in lacteals shown in A. Each symbol represents one mouse. N=6 mice per group. Error bars, SEM. Mann-Whitney U test.
(D) VE-Cadherin, F-actin and Vinculin staining of confluent HDLECs in complete media with or without treatment with 50μg/mL chylomicrons (CM) or 30μM arachidonic acid (AA) at the indicated timepoints. Arrowheads indicate colocalization of VE-Cadherin, F-actin and Vinculin staining at junctional sites.
(E-H) Western blot and quantification of pMLC2 (Thr18/Ser19), normalized to GAPDH, in HDLECs treated with 50μg/ml CM (E, G) or 30μM AA (F, H) at indicated timepoints. Data are expressed as fold changes compared to the average value of time (T)=0’. N=4 experiments. Error bars, SEM. ns, not significant, One-Way ANOVA with post-hoc Tukey test.
(I) VE-Cadherin, Vinculin and LYVE1 staining of jejunum lacteals of wildtype mice. Arrowheads point to VE-Cadherin and Vinculin colocalization on button-like structures.
Lipids such as AA can activate ROCK, the major regulator of cytoskeletal contraction, in a RhoA-dependent or independent manner30–34, and pharmacological ROCK inhibition zippered lacteal junctions and prevented chylomicron uptake in postnatal mice8. This suggested that lipid-activated ROCK signaling could regulate lacteal permeability. In cultured LECs, serum starvation induced the formation of discontinuous VE-Cadherin+ adherens junctions. Silencing of both mammalian ROCK isoforms, ROCK1 and 235, reverted these junctions back into straighter ones, confirming a LEC-autonomous role for ROCK in zippering lymphatic junctions (Fig. S1A). To determine this effect in vivo, we generated LEC-specific ROCK conditional mutant mice by intercrossing Rock1flox/flox, Rock2flox/flox and Prox1CreERT2 mice (Prox1CreERT2;Rock1flox/flox;Rock2flox/flox: Rock1/2iLKO)24–26,28. Tamoxifen (TAM) administration to these mice reduced ROCK1 and 2 protein expression significantly in initial lymphatics, confirming LEC-specific gene deletion (Fig. S1B–C). Analysis of P6 Rock1/2iLKO mice that had been administered TAM at P0-P2 revealed straight zipper-like junctions in jejunal lacteals, whereas Cre-negative littermate controls (Rock1flox/flox;Rock2flox/flox: Rock1/2f/f) had mainly discontinuous button-like junctions (Fig. 2A–C). Lacteal length or width were not significantly altered in the mutants (Fig. 2D). Functionally, the P6 Rock1/2iLKO pups had significantly less chyle in their mesenteric lymphatic vessels than their littermate controls (Fig. 2E–F), attesting to functional defects in chyle transport. To investigate whether ROCK1/2 loss of function (LOF) affects button maintenance, we analyzed P19–21 Rock1/2iLKO mice after TAM administration between P5-P7 (Fig. 2G). These mice exhibited more lacteal zipper junctions than control littermates (Fig. 2H–I), plus increased length and unaltered width of lacteals (Fig. 2J), and lethality by P35 due to as yet undetermined causes (Fig. 2K). The increase of blood triglyceride levels was also suppressed in adult mutant mice that received olive oil gavage 10 days after completion of TAM administration (Fig. 2L–M). Taken together, our results show that ROCK activity in LECs is required for the formation and maintenance of lacteal button junctions, and for fat uptake in the GI tract.
Figure 2. ROCK is required for lacteal junction opening and lipid uptake:

(A) Experimental strategy to induce Rock1/2 deletion in B-F. Arrowheads represent 3X75μg/g TAM injections, followed by analysis at P6.
(B) VE-Cadherin and LYVE1 staining of jejunum lacteals from P6 Rock1/2f/f and Rock1/2iLKO mice.
(C-D) Quantification of % lacteal zipper-like junctions normalized to total junction length, and lacteal length and width of P6 Rock1/2f/f and Rock1/2iLKO mice. Each dot represents one mouse. N=4 or 5 mice per group. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(E-F) Images of mesenteries and quantification of chyle-filled lymphatic collectors of P6 Rock1/2f/f and Rock1/2iLKO mice. Each dot represents one mouse. N=4 or 5 mice per group. Mann-Whitney U test.
(G) Experimental strategy to induce Rock1/2 deletion in H-J. Arrowheads represent 3X75μg/g TAM injections, followed by analysis at P19–21.
(H) VE-Cadherin and LYVE1 staining of jejunum lacteals from P19–21 Rock1/2f/f and Rock1/2iLKO mice.
(I-J) Quantification of % lacteal zipper-like junctions, normalized to total junction length, and lacteal length and width from P19–21 Rock1/2f/f and Rock1/2iLKO mice. Each dot represents one mouse. N=4 or 5 mice per group. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(K) Survival curves of Rock1/2f/f and Rock1/2iLKO mice after 3X75μg/g TAM injections at P5, P6 and P7. N=7 or 14 mice per group.
(L-M) Timeline of TAM administration (2mg/day for 5 days) and plasma triglyceride measurements of adult Rock1/2f/f and Rock1/2iLKO mice. Mice were fasted for 6 hours, gavaged with 200μl of olive oil, and plasma triglycerides were measured. N=9 or 15 mice per group. Error bars, SEM. Mann-Whitney U test was performed to compare Rock1/2f/f and Rock1/2iLKO groups at the same timepoints.
LEC VEGFR2 activation zippers lymphatic capillary junctions
An increase of VEGF-A bioavailability or exogenous VEGF-A administration can zipper initial lymphatics, and chylomicrons are known to suppress VEGF-A signaling by upregulating the VEGF-A decoy receptor VEGFR1 in BECs via ApoB488,18. In line with this, VEGFR2 phosphorylation was significantly reduced in jejunum from milk-fed P0.5 mice, compared to non-fed prenatal E20.5 mice (Fig. 3A–C). To determine if this process required LEC-autonomous VEGFR2 signaling, we produced LEC-specific inducible Vegfr2 knock-out mice (Prox1CreERT2;Vegfr2flox/flox: Vegfr2iLKO)21. TAM administration between P16–18 abolished VEGFR2 protein expression in Vegfr2iLKO lacteals (Fig. S2A–C). Loss of Vegfr2 from the lymphatics did not affect lacteal junction morphology, or lacteal length or width at baseline conditions (Fig. 3D–F). However, intravenous (IV) VEGF-A injection increased the ratio of lacteal zipper- vs. button-like junctions in control mice, but not in Vegfr2iLKO mice (Fig. 3E, 3G).
Figure 3. VEGFR2 activation zippers lymphatic capillary junctions:

(A-C) Western blot and quantifications of phospho-VEGFR2 (pY949 and pY1173), normalized to total VEGFR2, in jejunum tissue lysates from non-fed prenatal E20.5 and milk-fed P0.5 mice. Data are expressed as fold changes compared to the average value of E20.5. Each dot represents one mouse. N=8 mice per group. Error bars, SEM. Mann-Whitney U test.
(D) Experimental strategy to induce Vegfr2 deletion or overexpression, shown in E-K. Arrowheads represent 3X75μg/g TAM injections, followed by analysis at P21.
(E) VE-Cadherin and LYVE1 staining of jejunum lacteals from P21 Vegfr2f/f and Vegfr2iLKO mice 30 min after IV injection of PBS or 250ng/g VEGF-A.
(F) Quantifications of jejunum lacteal length and width in P21 Vegfr2f/f and Vegfr2iLKO mice. Error bars, SEM. Each dot represents one mouse. N=4 mice per group. ns, not significant. Mann-Whitney U test.
(G) Quantification of % zipper-like junctions out of total junction length in lacteals shown in E. Each symbol represents one mouse. N=4 or 5 mice per group. Error bars, SEM. One-Way ANOVA with post-hoc Tukey test.
(H) pERK and LYVE1 staining of VEGFR2CA-tdTomato (+) and (−) ear dermal lymphatic capillaries of P21 iMB-Vegfr2;Prox1CreERT2 mice. CA: constitutive active. LV: lymphatic vessel.
(I) Quantification of pERK intensity in VEGFR2CA-tdTomato (+) and (−) lymphatic vessels outlined in H. Each dot represents one mouse. N=4 mice per group. Error bars, SEM. Mann-Whitney U test.
(J) VE-Cadherin, tdTomato and LYVE1 staining of ear dermal lymphatic capillaries from P21 iMB-Vegfr2;Prox1CreERT2 mice.
(K) Quantification of zipper-like lymphatic junctions out of total lymphatic junction length in VEGFR2CA-tdTomato (+) and (−) lymphatic vessels in J. Each dot represents one mouse. N=6 mice per group. Error bars, SEM. Mann-Whitney U test.
In a complementary VEGFR2 gain-of-function (GOF) approach, we investigated the effects of VEGFR2 activation on junction morphology by using Prox1CreERT2-iMb-Vegfr2. After TAM treatment, these mice express either a tdTomato-tagged VEGFR2 with constitutive ligand-independent kinase activity, or a YFP fused-dominant negative VEGFR2, or a far-red fluorescent mKate227 in stochastically recombined LECs. Since tissue preparation diminished strong tdTomato signals in the lacteals, we analyzed initial lymphatics in the dermis. As predicted, TAM administration led to LEC-specific expression of red tdTomato fluorescence in a subset of dermal initial lymphatics, along with significantly increased ERK phosphorylation, confirming VEGFR2 overactivation (Fig. 3H–I). We found more zippered junctions in tdTomato-positive than tdTomato-negative LECs (Fig. 3J–K). Taken together, our data show that LEC VEGFR2 signaling is dispensable for junction opening at baseline, whereas its activation is sufficient to zipper junctions.
VEGFR2 Y949 phosphorylation is dispensable for modulation of lacteal junctions
Next, we addressed the mechanisms of VEGF-A/VEGFR2 induced LEC junction zippering. VEGF-A-induced permeability in BECs is regulated by phosphorylation of VEGFR2 at tyrosine (Y) 949, which mediates the binding of VEGFR2 to the Src homology 2-domain of T cell–specific adaptor (TSAd), leading to c-Src activation, and increased VE-Cadherin phosphorylation. This mechanism disrupts adherens junctions and increases vascular permeability19,36. We tested whether Y949 is involved in LEC junction remodeling by analyzing junction morphology in Vegfr2Y949F/Y949F mice that carry a Y to F (Phenylalanine) substitution at position 949, which blocks VEGF-A induced permeability19. Blood vascular leakage and extravasation of fluorescent dextran after intravenous injection of VEGF-A was prevented in the Vegfr2Y949F/Y949F mutant intestinal vasculature (Fig. 4A–B), confirming BEC permeability regulation via Y949. However, LEC junctions were not affected, as VEGF-A injection increased the number of zipper-like junctions to the same extent in control and Vegfr2Y949F/Y949F mice (Fig. 4C–D). Thus, Y949 signaling does not regulate LEC junction morphology downstream of VEGFR2.
Figure 4. VEGFR2 Y949 regulates permeability in blood but not in lymphatic vessels:
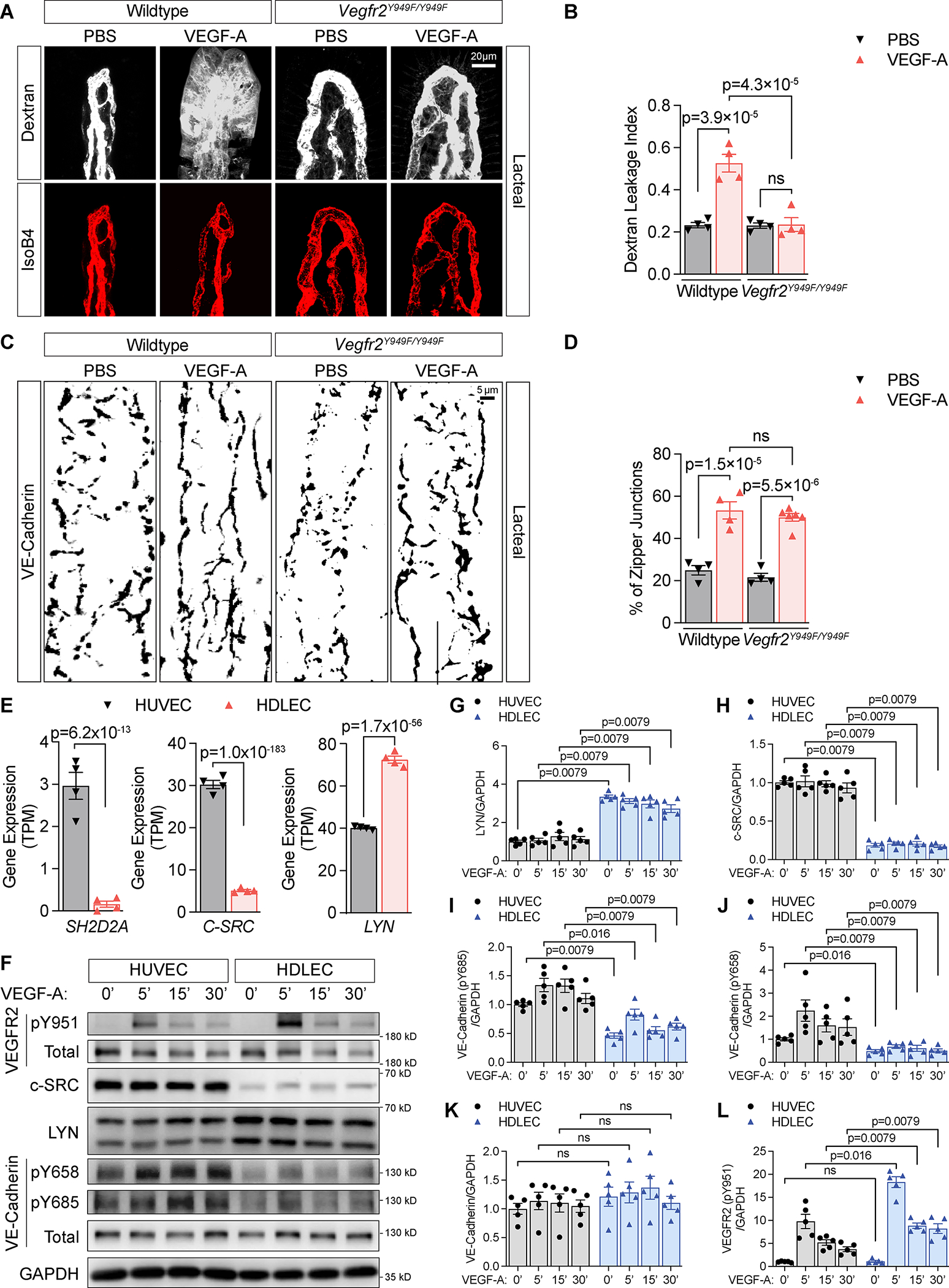
(A) Dextran leakage of jejunum villus blood vessels in P11-P13 Vegfr2Y949F/Y949F mice and wildtype littermates, 10 min after retro-orbital injection of fluorescent labeled IsoB4/dextran (70kDa) with 250ng/g VEGF-A or PBS.
(B) Quantification of dextran leakage shown in A. Each symbol represents one mouse. N=4 mice per group. Error bars, SEM. ns, not significant. One-Way ANOVA with post-hoc Tukey test.
(C) VE-Cadherin staining of jejunum lacteals from P21 wildtype and Vegfr2Y949F/Y949F mice 30 min after IV injection of PBS or 250ng/g VEGF-A.
(D) Quantification of % zipper-like junctions out of total junction length in lacteals shown in C. Each symbol represents one mouse. N=4 or 6 mice per group. Error bars, SEM. ns, not significant. One-Way ANOVA with post-hoc Tukey test.
(E) RNAseq analysis of SH2D2A (encoding TSAd), C-SRC and LYN differential expression in cultured HDLECs vs. HUVECs. Shown are transcript per million (TPM) values of the indicated genes. N=4 experiments. Error bars, SEM. Benjamini-Hochberg test.
(F) Western blot analysis for the indicated proteins in HDLECs vs. HUVECs treated with 50ng/ml VEGF-A for 0, 5, 15 and 30 min.
(G-L) Quantifications of the protein blots, normalized to GAPDH, shown in F. Data are expressed as fold changes compared to the average value of T=0’ of HUVEC group. Error bars, SEM. N=5 experiments. ns, not significant. Mann-Whitney U test was performed to compare HDLECs vs. HUVECs at the same timepoints.
Bulk RNA sequencing revealed very low SH2D2A (encoding TSAd) expression in HDLECs vs. HUVECs (Fig. 4E). Interestingly, we observed a significant decrease in C-SRC transcripts and protein in HDLECs vs. HUVECs, while another Src family kinase (SFK) LYN was significantly increased (Fig. 4E–H). The YES1 and FYN SFK protein levels were not changed despite mild to moderate increase of their transcripts in HDLECs vs. HUVECs (Fig. S3A–D). In BECs in vivo, phosphorylation of VE-Cadherin at Y658 and Y685, mediates junction opening downstream of SFKs37,38. Signaling studies revealed that the VEGF-A-induced phosphorylation of VE-Cadherin at Y685 and Y658 was profoundly reduced in HDLECs vs. HUVECs, despite robust phosphorylation of VEGFR2 Y951 (the human Y949 homologue) (Fig. 4F–L). Thus, intrinsic differences in TSAd/SFKs expression between BECs and LECs could account for the differential responses to VEGF-A activation and VE-Cadherin phosphorylation in BECs vs. LECs, and explain why LECs do not remodel their junctions in response to Y949 phosphorylation.
VEGFR3 is required for VEGF-A induced lacteal junction zippering
VEGFR3 is the main receptor involved in lymphangiogenesis and lymphatic maintenance39. VEGFR3 binds VEGF-C and VEGF-D, but not VEGF-A40. To determine if VEGFR3 regulates LEC junctions, we generated lymphatic specific Vegfr3 mutants by crossbreeding Vegfr3 floxed mice22 with Prox1CreERT2 mice (Prox1CreERT2;Vegfr3flox/flox: Vegfr3iLKO). Deletion of Vegfr3 dramatically decreased lacteal length (Fig. S4A–B), in line with previous reports41. We therefore opted for a short deletion window that did not affect the ratio of lacteal zipper/button junctions at baseline conditions or lacteal length, although it minimally decreased lacteal width (Fig. 5A–E). Intriguingly, while VEGF-A injection increased the number of lacteal zipper-like junctions in control mice, Vegf3iLKO mice maintained mostly buttoned junctions (Fig. 5B–C). As VEGFR3 does not bind VEGF-A directly, we hypothesized that VEGFR3 either modulates VEGFR2 expression levels, or VEGFR2 signaling output via the formation of VEGFR2/VEGFR3 heterodimers42,43. We did not observe any changes in VEGFR2 protein levels in lacteals of Vegfr3iLKO mice (Fig. 5F–G), or any changes to VEGFR2 mRNA levels in cultured LECs after silencing VEGFR3 (Fig. 5H). On the other hand, silencing of VEGFR3 in HDLECs reduced AKT phosphorylation in response to VEGF-A, while phosphorylation of Y951, Y1175, Y1214 and ERK remained unchanged (Fig. 5I–J). These results suggested that VEGFR3 contributes to LEC junction zippering by reinforcing PI3K/AKT signaling downstream of VEGF-A/VEGFR2, presumably via the formation of VEGFR2/VEGFR3 heterodimers.
Figure 5. VEGFR3 is required for VEGF-A/VEGFR2 induced lymphatic junction zippering:
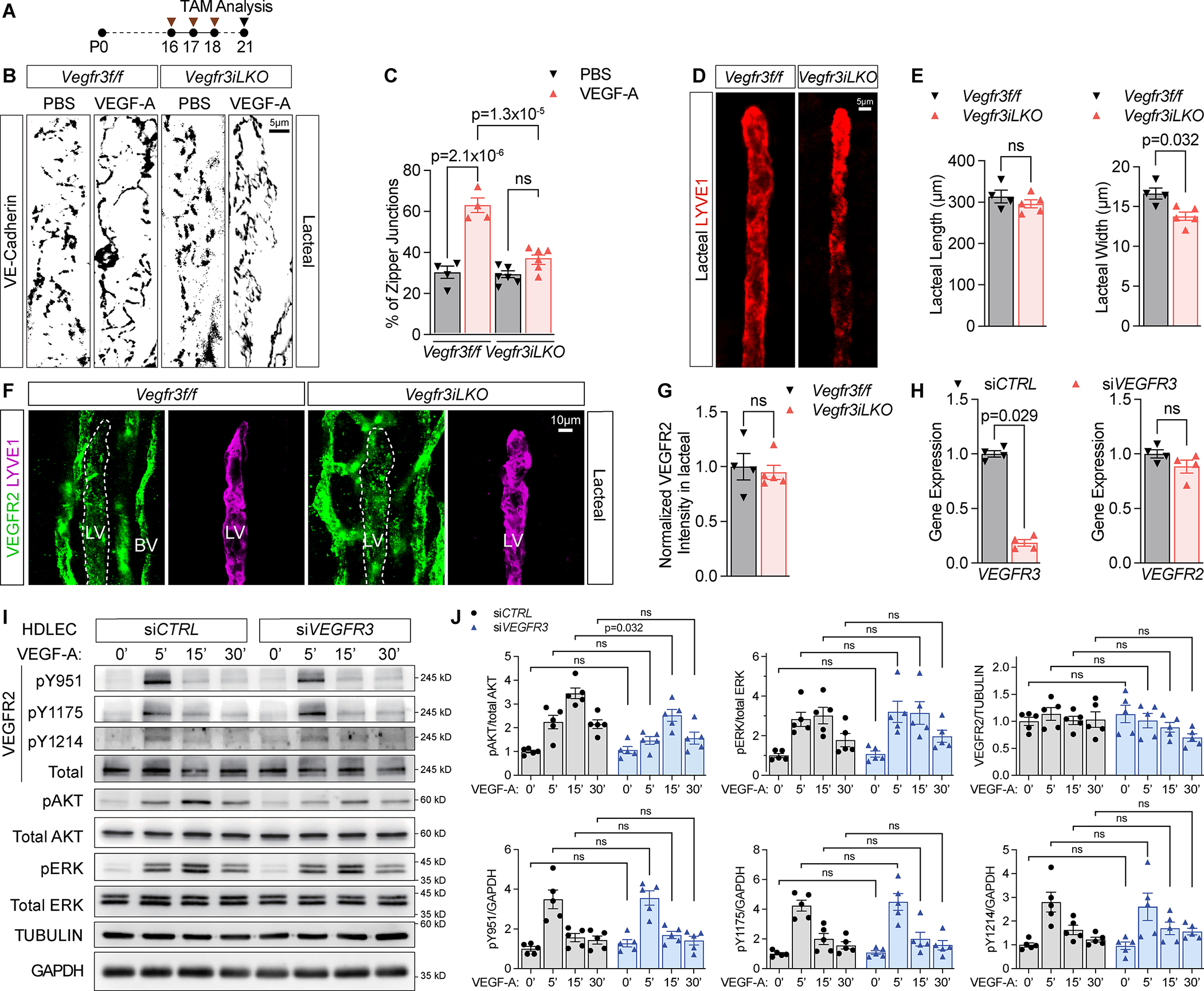
(A) Timeline of experiments shown in B-G. Arrowheads represent 3X75 μg/g TAM injections and analysis at P21.
(B) VE-Cadherin staining of jejunum lacteals from P21 Vegfr3f/f and Vegfr3iLKO littermates, 30 min after PBS or 250ng/g VEGF-A IV injection.
(C) Quantification of % zipper-like junctions out of total junction length in lacteals shown in B. Each symbol represents one mouse. N=4 or 6 mice per group. Error bars, SEM. One-Way ANOVA with post-hoc Tukey test.
(D) LYVE1 staining of jejunum lacteals of P21 Vegfr3f/f and Vegfr3iLKO mice.
(E) Quantifications of lacteal width and length from D. Each dot represents one mouse. N=4 or 5 mice per group. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(F) VEGFR2 and LYVE1 staining of jejunum villi from P21 Vegfr3f/f and Vegfr3iLKO mice. LV: lymphatic vessel. BV: blood vessel.
(G) Quantification of VEGFR2 intensity in the lacteals outlined in F. Data were normalized to the average VEGFR2 intensity of Vegfr3f/f lacteals. Each dot represents one mouse. N=4 or 5 mice per group. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(H) qPCR analysis of VEGFR2 and VEGFR3 expression in HDLECs 2 days after transfection with control (CTRL) or VEGFR3 siRNA. N=4 experiments. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(I-J) Western blots and quantifications of VEGFR2, ERK and AKT phosphorylation, normalized to the indicated loading controls, in HDLECs transfected with siCTRL or siVEGFR3 and treated with 50ng/ml VEGF-A for 0, 5, 15 and 30 min. Data are expressed as fold changes compared to the average value of T=0’ of siCTRL group. N=5 experiments. Error bars, SEM. ns, not significant. Mann-Whitney U test was performed to compare siCTRL vs. siVEGFR3-treated groups at the same timepoints.
VEGF-A zippers lacteal junctions via PI3K/AKT signaling
To determine PI3K/AKT signaling requirement for lacteal junction zippering in vivo, we analyzed Akt1 mouse mutants (Akt1−/−: Akt1KO)20. Global loss of Akt1 did not affect junction morphology or lacteal length or width (Fig. 6A–D). Intravenous VEGF-A injection increased the number of zipper-like junctions in control mice, whereas Akt1KO mice were resistant to VEGF-A induced zippering (Fig. 6A–B). Similar results were observed when we treated the mice with the PI3K inhibitor Wortmannin that blocks VEGF-A induced phosphorylation of AKT (Fig. 6A–B, S5A–B). In parallel, we investigated the role of PLCγ-ERK pathway downstream of VEGFR2 Y1175 phosphorylation44. We analyzed lymphatic specific mutants of Plcγ1 (Prox1CreERT2;Plcγ1flox/flox: Plcγ1iLKO)23, as well as compound mutants of Erk1/2 (Erk1-/;Prox1CreERT2;Erk2flox/flox: Erk1KO;Erk2iLKO). Plcγ1iLKO and Erk1KO;Erk2iLKO mice showed unaltered junction morphology at baseline, and zippered their junctions in response to intravenous VEGF-A (Fig. 6E–G), demonstrating that PI3K/AKT but not PLCγ1/ERK1/2 signaling mediates lacteal junction zippering in response to VEGF-A.
Figure 6. PI3K/AKT signaling mediates VEGF-A-induced lymphatic junction zippering:

(A) VE-Cadherin staining of jejunum lacteals from P21 wildtype, Akt1KO and Wortmannin-treated mice, 30 min after IV administration of PBS or 250ng/g VEGF-A.
(B) Quantification of % zipper-like junctions out of total junction length in lacteals shown in A. Each symbol represents one mouse. N=4 or 6 mice per group. Error bars, SEM. ns, not significant. One-Way ANOVA with post-hoc Tukey test.
(C) LYVE-1 staining and (D) quantifications of length and width of jejunum lacteals from P21 wildtype and Akt1KO mice. Each dot represents one mouse. N=4 mice per group. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(E) Experimental strategy to induce Plcγ or Erk2 deletion in F-G. Arrowheads indicate 3X75μg/g TAM injections and analysis at P21.
(F) VE-Cadherin staining of jejunum lacteals from P21 PlcγiLKO and Erk1KO;Erk2iLKO mice 30 min after IV injection of PBS or 250ng/g VEGF-A.
(G) Quantification of % zipper-like junctions out of total junction length in lacteals shown in F. Each symbol represents one mouse. N=4 to 5 mice per group. Error bars, SEM. Mann-Whitney U test.
VEGF-A/VEGFR2 signaling suppresses RhoA in LECs via RAC1 activation
Next, we investigated how VEGF-A/VEGFR2 interacts with RhoA/ROCK during LEC junction remodeling. Stimulation of serum-starved HDLEC monolayers with VEGF-A for 30 min transformed the discontinuous adherens junctions into straighter zipper junctions and reduced cytoskeletal stress fibers8,45 (Fig. 7A). Consistently, phosphorylated MLC2 was significantly decreased in HDLECs 30 min after VEGF-A stimulation (Fig. 7B). In contrast, serum-starved HUVECs showed increased stress fiber formation and no difference in MLC2 phosphorylation 30 min after VEGF-A stimulation (Fig. 7A–B). In line with the reduced pMLC2 levels in HDLECs, we found a decrease of activated, GTP-bound RhoA following VEGF-A treatment (Fig. 7C). This depended on VEGFR2, since silencing of VEGFR2 sustained RhoA activity (Fig. 7C). VEGF-A stimulation of HDLECs induced GTP-bound RAC1, another small GTPase of the Rho family, in a VEGFR2-dependent manner (Fig. 7D). As RAC1 inactivates RhoA/ROCK, this could explain the reduced RhoA activity and actin depolymerization observed in VEGF-A stimulated HDLECs46. VEGFR2-mediated RAC1 activation depended on PI3K, as pretreatment with the PI3K inhibitor Wortmannin completely abolished the elevation in RAC1-GTP levels after VEGF-A stimulation (Fig. 7E). Thus, VEGF-A/VEGFR2 signaling in LECs reduces stress fiber formation by inhibiting RhoA/ROCK/MLC2 via activation of RAC1, in a PI3K-dependent manner.
Figure 7. VEGF-A/VEGFR2 inhibits stress fiber formation in HDLECs:

(A) VE-Cadherin and phalloidin staining of confluent HUVECs and HDLECs starved in 0.2% serum for 6 hours and treated with 50ng/ml VEGF-A or PBS for 30 min.
(B) Western blot and quantification of pMLC2 (Thr18/Ser19), normalized to TUBULIN, in confluent HUVECs and HDLECs starved for 6 hours and stimulated with 50ng/ml VEGF-A for 0, 5, 15 and 30 min. Data are expressed as fold changes compared to the average value of T=0’ of each cell group. N=6 experiments. Error bars, SEM. ns, not significant. One-Way ANOVA with post-hoc Tukey test (for HDLEC), or Welch’s ANOVA with post-hoc Dunnett T3 test (for HUVEC).
(C-D) RhoA and RAC1 pulldown assays in HDLECs transfected with CTRL or VEGFR2 siRNA and treated with 50ng/ml VEGF-A for 30 min. Top panel: Western blots of GTP-bound RhoA/RAC1 from the pulldown assay and total RhoA/RAC1 from total cell lysates. Bottom panel: Quantifications of the ratios of GTP bound RhoA/RAC1 to total RhoA/RAC1. Data are expressed as fold changes compared to the average value of T=0’ of each siRNA-treated group. N=4–5 experiments. Error bars, SEM. ns, not significant. Mann-Whitney U test.
(E) RAC1 pulldown assay in HDLECs after 5 hours Wortmannin (1μM) or DMSO treatment and 30 min VEGF-A (50ng/ml) stimulation. Shown are western blots (top) and quantification (bottom) of the ratios of GTP bound RAC1 to total RAC1 from cell lysates. Data are expressed as fold changes compared to the average value of T=0’ of each treatment group. N=5 experiments. Error bars, SEM. ns not significant. Mann-Whitney U test.
ROCK regulates lymphatic junction morphology in dermal initial lymphatics
To address whether VEGF-A and ROCK signaling control junction remodeling outside the GI tract, we analyzed junction morphology in dermal lymphatic capillaries. Intradermal injection of VEGF-A induced junction zippering in the dermal initial lymphatics of the ear8 (Fig. 8A–B), while other vascular permeability inducers, such as histamine or bradykinin, did not induce junction zippering (Fig. 8A–B). ROCK signaling was also required for the maintenance of button junctions in dermal initial lymphatics, as Rock1/2iLKO mutants displayed predominantly zipper junctions at baseline (Fig. 8C–E). To examine whether button morphology is necessary for fluid uptake, we injected Evans Blue into the footpads of adult mice. The dye was quickly absorbed by lymphatic capillaries and transported to the draining lymph node in control mice, while VEGF-A injection resulted in significantly reduced dye uptake (Fig. 8F–G). We also observed a significant reduction in the intensity of Evans Blue-filled lymphatics connected to popliteal lymph nodes in the Rock1/2iLKOs (Fig. 8H–J). These results show that ROCK regulates lymphatic junction transformation and solute uptake function across different lymphatic capillary beds.
Figure 8. VEGF-A and ROCK signaling regulate dermal initial lymphatic drainage:

(A) VE-Cadherin staining of ear dermal initial lymphatics from P21 wildtype mice 30 min after intradermal injection of VEGF-A (50ng in 20μl PBS), histamine (500ng in 20μl PBS), bradykinin (20μg in 20μl PBS) or PBS alone.
(B) Quantification of % zipper-like LEC junctions out of total lymphatic junction length in dermal initial lymphatics in A. Each symbol represents one mouse. N=6 or 7 mice per group. Error bars, SEM. ns, not significant. One-Way ANOVA with post-hoc Tukey test.
(C) Experiment strategy to induce Rock1/2 deletion shown in panels D-E. Arrowheads represent 3X75μg/g TAM injections and analysis at P19–21.
(D-E) VE-Cadherin staining and quantification of % zipper-like junctions out of total junction length in ear dermal initial lymphatics from P19–21 Rock1/2f/f and Rock1/2iLKO mice. Each symbol represents one mouse. N=4 mice per group. Error bars, SEM. Mann-Whitney U test.
(F) Evans Blue absorption in lower leg lymphatic vessels of adult wildtype mice at the indicated time points after footpad injection of VEGF-A (20ng in 5μl PBS) or PBS alone, and 5μl of 1.5% Evans Blue solution.
(G) Quantification of dye intensity in Evans Blue-filled lymphatics in F. N=7 or 9 mice per group. Error bars, SEM. Mann-Whitney U test was performed to compare PBS and VEGF-A-treated groups at the same timepoints.
(H) Timeline of TAM administration (2mg per day for 5 days) of adult Rock1/2f/f and Rock1/2iLKO mice. All mice were analyzed 10 days after the last TAM injection.
(I) Evans Blue uptake in lower leg lymphatic vessels of Rock1/2f/f and Rock1/2iLKO mice at the indicated time points after intradermal footpad injection of 9μl 1.5% Evans Blue solution.
(J) Quantification of dye intensity in Evans Blue-filled lymphatics in I. N=7–8 mice per group. Error bars, SEM. ns, not significant. Mann-Whitney U test was performed to compare Rock1/2f/f and Rock1/2iLKO mouse groups at the same timepoints.
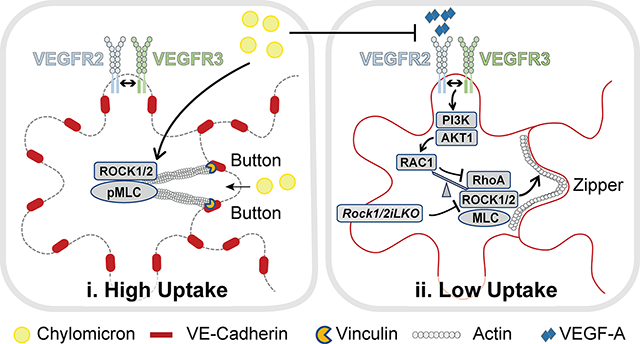
(K) Summary model of LEC junction remodeling in intestinal lacteals. i. High lipid uptake: Chylomicrons derived from ingested lipids activate ROCK (1), which phosphorylates MLC2 and induces actomyosin stress fiber assembly and junction opening, thereby enabling lacteal chylomicron uptake. VEGFR2 is inhibited by chylomicron upregulation of VEGFR1 and NRP1 in BECs, which reduce VEGF-A signals to LEC VEGFR2 (2). ii. Low lipid uptake: VEGF-A signals via VEGFR2/VEGFR3 heterodimers to antagonize ROCK activation and junction opening. VEGFR2/VEGFR3 activate RAC1 via PI3K/AKT, thereby reducing RhoA and ROCK-dependent MLC2 phosphorylation, resulting in actin stress fiber relaxation, cortical actin formation and junction zippering, which prevents chylomicron uptake by lacteals.
DISCUSSION
Our work identifies a two-pronged homeostatic mechanism that triggers lacteal junction opening in intestinal lacteals. Chylomicrons rapidly activate ROCK in LECs, which initiates the formation of junction-anchored stress fibers and generates the tension required to pull junctions apart (Fig. 8Ki). A similar mechanism has been described in BECs during the formation of focal adherens junctions (FAJ), which contain vinculin and partially unzip to increase blood vascular permeability. FAJ formation requires actomyosin contractility and depends on RhoA GTPase activity47. Thus, RhoA and ROCK are key regulators of junctional morphology in both LECs and BECs.
We speculate that lipid activation of ROCK is a driving force that triggers cytoskeletal contraction and junction opening in the intestinal lacteal microenvironment. Lipids, such as lysophospholipids48,49, sphingolipids50 and fatty acids31–34 can activate guanine nucleotide exchange factors (GEFs) for RhoA through G-protein coupled receptors and p38/MAPK. Arachidonic acids can also directly activate ROCK by binding to the autoinhibitory region of ROCK51. The mechanisms of ROCK activation in response to chylomicrons remains to be determined.
Following postnatal fat ingestion, VEGFR2 signaling is inhibited in the intestine. This is likely due to the chylomicron apolipoprotein ApoB48 that could induce postnatal transcriptional upregulation of the VEGF-A decoy receptor VEGFR1 in BECs of the intestinal villi8,18. VEGFR1 sequesters peri-lacteal VEGF-A, and together with Neuropilin 1 (NRP1) inhibits VEGFR2 signaling on LECs and junction zippering12. Therefore, chylomicrons take dual action to ensure perinatal lymphatic junction opening and their own intestinal absorption, which in mammals coincides with milk consumption immediately after birth31.
VEGF-A expression is tightly regulated in the microenvironment of the intestinal villi to maintain the fenestrated blood vasculature52. Increased VEGF-A bioavailability leads to vascular leakage in the intestinal villus8, which could indirectly affect LEC junctions. Here we show that the effects of VEGF-A on LEC junctions are neither a byproduct of concurrent BEC junction opening, nor via effects on other cell types, such as mural cells53. Using VEGFR2 LEC-specific LOF and GOF mice, we show that VEGF-A antagonizes junction opening in a LEC-autonomous manner. VEGFR2 LOF does not alter baseline junction morphology, in contrast to ROCK deletion, revealing antagonistic effects by VEGF-A and ROCK that are both controlled by chylomicrons.
We also show that VEGF-A-mediated inhibition of lacteal permeability is independent of Y949 but abolished by VEGFR3 deletion. Since VEGFR3 does not bind VEGF-A directly, we conclude that VEGFR3 modulates VEGFR2 signaling output via VEGFR2/VEGFR3 heterodimers. Such heterodimers are formed in cultured LECs in response to VEGF-C42, as well as in BECs in angiogenic sprouts in response to VEGF-A and VEGF-C43. As VEGF-A-induced VEGFR2 Y951, Y1175 and Y1214 phosphorylation is unaffected by VEGFR3 knockdown, the formation of such heterodimers may affect the sites of VEGFR3 tyrosine phosphorylation42,43.
VEGF-C-156S, a mutated form of VEGF-C that only binds VEGFR354, was unable to zipper the lacteal junctions of control mice8, and VEGFR3 inhibition with monoclonal antibodies failed to reverse zippered junctions in the inflamed mouse trachea6. On the other hand, VEGF-C produced by lacteal macrophages in response to microbial stimuli is required for button maintenance12. We suggest that these effects are mediated by VEGFR2, or via VEGFR2/VEGFR3 heterodimers, since proteolytically processed VEGF-C also binds to and activates VEGFR255. VEGFR3 knockdown abolished AKT activation downstream of VEGF-A in HDLECs, and AKT deletion prevented VEGF-A induced lacteal junction zippering in vivo. Conversely, VEGFR2 knockdown in LECs prevents VEGF-C induced AKT phosphorylation, but not ERK activation56. Taken together, the data are consistent with a model where VEGF-A or VEGF-C induced VEGFR2/VEGFR3 heterodimer signaling to AKT accounts for LEC permeability inhibition.
VEGF-A-mediated activation of VEGFR2 zippered lacteal junctions by suppressing RhoA via increased RAC1 activation (Fig. 8Kii). RAC1 antagonizes RhoA to remodel apical junctions as described in a variety of cellular contexts57. In this case, RAC1 inhibits RhoA, while RhoA can in turn restrain RAC1 activity. Interestingly, in BECs, signaling via VEGFR2 leads to activation of both RAC158–60 and RhoA61, suggesting that the dynamics of pathway activation are different in BECs vs. LECs. PI3K/AKT signaling can activate RAC1 in many cell types62–64, but how exactly PI3K activation downstream of VEGF-A/VEGFR2 results in activation of RAC1 in LECs remains to be determined.
ROCK deletion resulted ultimately in lethality in the Rock1/2iLKO mice. Determining the cause of death requires further investigation. Prox1CreERT2 is expressed also in other cell types, such as hepatocytes, cardiomyocytes and some neuronal cells, and therefore we cannot exclude off-target effects of ROCK LOF that could underlie the increased mortality of the mutants.
Finally, we show that ROCK activity is essential for button formation also outside of the GI tract. The upstream signaling pathways activating ROCK in skin lymphatics remain to be determined. Dexamethasone activation of LEC glucocorticoid receptors, which increase RhoA-mediated contractility in various settings65,66 promotes button formation and decreases edema in inflamed tracheal lymphatics6. It is therefore likely that signaling through glucocorticoid receptor or other surface receptors such as Calcrl, transforming growth factor β-receptor (TGFβR), platelet derived growth factor receptor (PDFGR) and epidermal growth factor receptor (EGFR) that also activate RhoA, may contribute to button formation in different tissues67–69. Other environmental inputs, including enzymes such as thrombin70, flow shear71 and mechanical forces72 may also be at play. Understanding the inputs into the cytoskeletal mechanism that we here uncover could allow manipulation of lymphatic permeability to control tissue drainage and immune cell trafficking in preclinical disease models.
Supplementary Material
Novelty and Significance.
What is known?
Initial lymphatics at the intestine (termed lacteals) are responsible for absorption of chylomicrons, lipoprotein particles produced by enterocytes.
Chylomicrons enter lacteals through open, ‘button’-like junctions between lymphatic endothelial cells.
Closing (or ‘zippering’) of lymphatic endothelial cell junctions, prevents dietary lipid absorption in mice.
What new information does this article contribute?
In newborn mice, chylomicrons trigger lacteal junction opening, following milk injection right after birth.
Chylomicrons activate ROCK signaling in lymphatic endothelial cells, remodeling junctions and allowing for lipid absorption.
VEGF-A signals via VEGFR2/VEGFR3 to antagonize junction opening by suppressing ROCK.
ROCK suppression is mediated via PI3K-dependent activation or RAC1, which inhibits RhoA.
Lymphatic vessels are essential for intestinal lipid absorption, yet the mechanisms regulating lymphatic permeability are poorly understood. Here we show that chylomicrons, lipoprotein particles produced by enterocytes, regulate their own absorption by triggering the formation of ‘button’-like junctions in the intestinal villus, via activation of ROCK signaling and cytoskeletal remodeling. At the same time chylomicrons suppress VEGFR2 signaling, which limits ROCK activity by inhibiting RhoA. We show that these effects are mediated by VEGFR2/VEGFR3 heterodimer formation in response to VEGF-A, which activate the RhoA suppressor RAC1, via PI3K/AKT. This two-pronged homeostatic mechanism ensures fat ingestion after postnatal milk consumption. In addition, we show that ROCK-mediated cytoskeletal contractions regulate lymphatic junction morphology and permeability also outside the intestine, in the mouse dermis. This study identifies lymphatic junctions as an important regulator of lymphatic vessel function, suggesting a novel target to control lipid absorption and tissue drainage in preclinical models of disease.
ACKNOWLEDGMENTS
We thank Drs J.-L. Thomas, and A. Dubrac for discussion and J. Luo, S. Yi and M. Zhang for technical assistance.
SOURCES OF FUNDING
This project was supported by grants from National Key R&D Program of China (2021YFA1101200), National Natural Science Foundation of China (82070500, 82241009), Natural Science Foundation of Guangdong Province (2022A1515012210), and the Science and Technology Program of Guangzhou City (202102010183) to FZ, and from NIH (1R01DK120373-01A1) and the Leducq Foundation (TNE ATTRACT) to AE.
Non-standard abbreviations and acronyms
- LECs
lymphatic endothelial cells
- BECs
blood vascular endothelial cells
- ROCK
Rho-associated kinase
- ApoB48
apolipoprotein B48
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- SMC
smooth muscle cell
- TAM
tamoxifen
- HDLECs
human dermal lymphatic endothelial cells
- HUVECs
human umbilical vein endothelial cells
- AA
arachidonic acid
- MLC2
myosin light chain 2
- SFK
Src family kinase
Footnotes
DISCLOSURES
The authors declare no competing interests.
REFERENCES
- 1.Jackson DG. Leucocyte Trafficking via the Lymphatic Vasculature- Mechanisms and Consequences. Front Immunol. 2019;10:471. doi: 10.3389/fimmu.2019.00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cifarelli V, Eichmann A. The Intestinal Lymphatic System: Functions and Metabolic Implications. Cell Mol Gastroenterol Hepatol. 2019;7:503–513. doi: 10.1016/j.jcmgh.2018.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrova TV, Koh GY. Organ-specific lymphatic vasculature: From development to pathophysiology. J Exp Med. 2018;215:35–49. doi: 10.1084/jem.20171868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204:2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Zarkada G, Yi S, Eichmann A. Lymphatic Endothelial Cell Junctions: Molecular Regulation in Physiology and Diseases. Front Physiol. 2020;11:509. doi: 10.3389/fphys.2020.00509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol. 2012;180:2561–2575. doi: 10.1016/j.ajpath.2012.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Churchill MJ, du Bois H, Heim TA, Mudianto T, Steele MM, Nolz JC, Lund AW. Infection-induced lymphatic zippering restricts fluid transport and viral dissemination from skin. J Exp Med. 2022;219. doi: 10.1084/jem.20211830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, Zarkada G, Han J, Li J, Dubrac A, Ola R, Genet G, Boye K, Michon P, Kunzel SE, et al. Lacteal junction zippering protects against diet-induced obesity. Science. 2018;361:599–603. doi: 10.1126/science.aap9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koletzko B Human Milk Lipids. Ann Nutr Metab. 2016;69 Suppl 2:28–40. doi: 10.1159/000452819 [DOI] [PubMed] [Google Scholar]
- 10.Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest. 2014;124:929–935. doi: 10.1172/JCI71610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nurmi H, Saharinen P, Zarkada G, Zheng W, Robciuc MR, Alitalo K. VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol Med. 2015;7:1418–1425. doi: 10.15252/emmm.201505731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suh SH, Choe K, Hong SP, Jeong SH, Makinen T, Kim KS, Alitalo K, Surh CD, Koh GY, Song JH. Gut microbiota regulates lacteal integrity by inducing VEGF-C in intestinal villus macrophages. EMBO Rep. 2019;20. doi: 10.15252/embr.201846927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong SP, Yang MJ, Cho H, Park I, Bae H, Choe K, Suh SH, Adams RH, Alitalo K, Lim D, et al. Distinct fibroblast subsets regulate lacteal integrity through YAP/TAZ-induced VEGF-C in intestinal villi. Nat Commun. 2020;11:4102. doi: 10.1038/s41467-020-17886-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shew T, Wolins NE, Cifarelli V. VEGFR-3 Signaling Regulates Triglyceride Retention and Absorption in the Intestine. Front Physiol. 2018;9:1783. doi: 10.3389/fphys.2018.01783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernier-Latmani J, Cisarovsky C, Demir CS, Bruand M, Jaquet M, Davanture S, Ragusa S, Siegert S, Dormond O, Benedito R, et al. DLL4 promotes continuous adult intestinal lacteal regeneration and dietary fat transport. J Clin Invest. 2015;125:4572–4586. doi: 10.1172/JCI82045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis RB, Kechele DO, Blakeney ES, Pawlak JB, Caron KM. Lymphatic deletion of calcitonin receptor-like receptor exacerbates intestinal inflammation. JCI Insight. 2017;2:e92465. doi: 10.1172/jci.insight.92465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu S, Mahadevan A, Elysee IF, Choi J, Souchet NR, Bae GH, Taboada AK, Sanketi B, Duhamel GE, Sevier CS, et al. The asymmetric Pitx2 gene regulates gut muscular-lacteal development and protects against fatty liver disease. Cell Rep. 2021;37:110030. doi: 10.1016/j.celrep.2021.110030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avraham-Davidi I, Ely Y, Pham VN, Castranova D, Grunspan M, Malkinson G, Gibbs-Bar L, Mayseless O, Allmog G, Lo B, et al. ApoB-containing lipoproteins regulate angiogenesis by modulating expression of VEGF receptor 1. Nat Med. 2012;18:967–973. doi: 10.1038/nm.2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Padhan N, Sjostrom EO, Roche FP, Testini C, Honkura N, Sainz-Jaspeado M, Gordon E, Bentley K, Philippides A, et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun. 2016;7:11017. doi: 10.1038/ncomms11017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200 [DOI] [PubMed] [Google Scholar]
- 21.Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, Kopp HG, Shido K, Petit I, Yanger K, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;4:263–274. doi: 10.1016/j.stem.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG, Alitalo K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28:4843–4850. doi: 10.1128/MCB.02214-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu G, Chen Y, Yu M, Podd A, Schuman J, He Y, Di L, Yassai M, Haribhai D, North PE, et al. Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J Exp Med. 2010;207:309–318. doi: 10.1084/jem.20090880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Kong D, Byun KH, Ye C, Koda S, Lee DH, Oh BC, Lee SW, Lee B, Zabolotny JM, et al. Rho-kinase regulates energy balance by targeting hypothalamic leptin receptor signaling. Nat Neurosci. 2012;15:1391–1398. doi: 10.1038/nn.3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto R, Li Y, Noma K, Hiroi Y, Liu PY, Taniguchi M, Ito M, Liao JK. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013;27:1439–1449. doi: 10.1096/fj.12-217018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumper S, Mardakheh FK, McCarthy A, Yeo M, Stamp GW, Paul A, Worboys J, Sadok A, Jorgensen C, Guichard S, et al. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. Elife. 2016;5:e12994. doi: 10.7554/eLife.12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pontes-Quero S, Heredia L, Casquero-Garcia V, Fernandez-Chacon M, Luo W, Hermoso A, Bansal M, Garcia-Gonzalez I, Sanchez-Munoz MS, Perea JR, et al. Dual ifgMosaic: A Versatile Method for Multispectral and Combinatorial Mosaic Gene-Function Analysis. Cell. 2017;170:800–814 e818. doi: 10.1016/j.cell.2017.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bazigou E, Lyons OT, Smith A, Venn GE, Cope C, Brown NA, Makinen T. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J Clin Invest. 2011;121:2984–2992. doi: 58050 [pii] 10.1172/JCI58050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salem N Jr., Van Dael P. Arachidonic Acid in Human Milk. Nutrients. 2020;12. doi: 10.3390/nu12030626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia MC, Ray DM, Lackford B, Rubino M, Olden K, Roberts JD. Arachidonic acid stimulates cell adhesion through a novel p38 MAPK-RhoA signaling pathway that involves heat shock protein 27. J Biol Chem. 2009;284:20936–20945. doi: 10.1074/jbc.M109.020271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, Okawa K, Iwamatsu A, Kaibuchi K, Hartshorne DJ, et al. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem. 1999;274:3744–3752. doi: 10.1074/jbc.274.6.3744 [DOI] [PubMed] [Google Scholar]
- 32.Dietze R, Hammoud MK, Gomez-Serrano M, Unger A, Bieringer T, Finkernagel F, Sokol AM, Nist A, Stiewe T, Reinartz S, et al. Phosphoproteomics identify arachidonic-acid-regulated signal transduction pathways modulating macrophage functions with implications for ovarian cancer. Theranostics. 2021;11:1377–1395. doi: 10.7150/thno.52442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.George MD, Wine RN, Lackford B, Kissling GE, Akiyama SK, Olden K, Roberts JD. p38 mitogen-activated protein kinase interacts with vinculin at focal adhesions during fatty acid-stimulated cell adhesion. Biochem Cell Biol. 2013;91:404–418. doi: 10.1139/bcb-2013-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia MC, Williams J, Johnson K, Olden K, Roberts JD. Arachidonic acid stimulates formation of a novel complex containing nucleolin and RhoA. FEBS Lett. 2011;585:618–622. doi: 10.1016/j.febslet.2011.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loirand G Rho Kinases in Health and Disease: From Basic Science to Translational Research. Pharmacol Rev. 2015;67:1074–1095. doi: 10.1124/pr.115.010595 [DOI] [PubMed] [Google Scholar]
- 36.Sun Z, Li X, Massena S, Kutschera S, Padhan N, Gualandi L, Sundvold-Gjerstad V, Gustafsson K, Choy WW, Zang G, et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med. 2012;209:1363–1377. doi: 10.1084/jem.20111343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208. doi: 10.1038/ncomms2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi Jin YD, Mark Richards, Mika Kaakinen, Wolfgang Giese, Elisabeth Baumann, Anna Szymborska, André Rosa, Sofia Nordling, Lilian Schimmel, Emir Bora Akmeriç, Andreia Pena, Emmanuel Nwadozi, Maria Jamalpour, Katrin Holstein, Miguel Sáinz-Jaspeado, Miguel O. Bernabeu, Michael Welsh, Emma Gordon, Claudio A. Franco, Dietmar Vestweber, Lauri Eklund, Holger Gerhardt & Lena Claesson-Welsh. Tyrosine-protein kinase Yes controls endothelial junctional plasticity and barrier integrity by regulating VE-cadherin phosphorylation and endocytosis. Nature Cardiovascular Research. 2022;1:1156–1173. doi: 10.1038/s44161-022-00172-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaahtomeri K, Karaman S, Makinen T, Alitalo K. Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev. 2017;31:1615–1634. doi: 10.1101/gad.303776.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301 [DOI] [PubMed] [Google Scholar]
- 41.Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K. VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A. 2015;112:761–766. doi: 10.1073/pnas.1423278112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, Claesson-Welsh L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem. 2003;278:40973–40979. doi: 10.1074/jbc.M304499200 [DOI] [PubMed] [Google Scholar]
- 43.Nilsson I, Bahram F, Li X, Gualandi L, Koch S, Jarvius M, Soderberg O, Anisimov A, Kholova I, Pytowski B, et al. VEGF receptor 2/−3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 2010;29:1377–1388. doi: 10.1038/emboj.2010.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen D, Simons M. Emerging roles of PLCgamma1 in endothelial biology. Sci Signal. 2021;14. doi: 10.1126/scisignal.abc6612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cifarelli V, Appak-Baskoy S, Peche VS, Kluzak A, Shew T, Narendran R, Pietka KM, Cella M, Walls CW, Czepielewski R, et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat Commun. 2021;12:3350. doi: 10.1038/s41467-021-23808-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011;21:718–726. doi: 10.1016/j.tcb.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol. 2012;196:641–652. doi: 10.1083/jcb.201108120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skoura A, Hla T. Regulation of vascular physiology and pathology by the S1P2 receptor subtype. Cardiovasc Res. 2009;82:221–228. doi: 10.1093/cvr/cvp088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiang SY, Dusaban SS, Brown JH. Lysophospholipid receptor activation of RhoA and lipid signaling pathways. Biochim Biophys Acta. 2013;1831:213–222. doi: 10.1016/j.bbalip.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh RK, Haka AS, Brumfield A, Grosheva I, Bhardwaj P, Chin HF, Xiong Y, Hla T, Maxfield FR. Ceramide activation of RhoA/Rho kinase impairs actin polymerization during aggregated LDL catabolism. J Lipid Res. 2017;58:1977–1987. doi: 10.1194/jlr.M076398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Araki S, Ito M, Kureishi Y, Feng J, Machida H, Isaka N, Amano M, Kaibuchi K, Hartshorne DJ, Nakano T. Arachidonic acid-induced Ca2+ sensitization of smooth muscle contraction through activation of Rho-kinase. Pflugers Arch. 2001;441:596–603. doi: 10.1007/s004240000462 [DOI] [PubMed] [Google Scholar]
- 52.Bernier-Latmani J, Mauri C, Marcone R, Renevey F, Durot S, He L, Vanlandewijck M, Maclachlan C, Davanture S, Zamboni N, et al. ADAMTS18(+) villus tip telocytes maintain a polarized VEGFA signaling domain and fenestrations in nutrient-absorbing intestinal blood vessels. Nat Commun. 2022;13:3983. doi: 10.1038/s41467-022-31571-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M, Wijelath ES. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol. 2001;188:359–368. doi: 10.1002/jcp.1121 [DOI] [PubMed] [Google Scholar]
- 54.Joukov V, Kumar V, Sorsa T, Arighi E, Weich H, Saksela O, Alitalo K. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem. 1998;273:6599–6602. [DOI] [PubMed] [Google Scholar]
- 55.Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, Saksela O, Kalkkinen N, Alitalo K. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997;16:3898–3911. doi: 10.1093/emboj/16.13.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng Y, Zhang X, Simons M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler Thromb Vasc Biol. 2015;35:421–429. doi: 10.1161/ATVBAHA.114.304881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nguyen LK, Kholodenko BN, von Kriegsheim A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases. 2018;9:316–321. doi: 10.1080/21541248.2016.1224399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oshikawa J, Kim SJ, Furuta E, Caliceti C, Chen GF, McKinney RD, Kuhr F, Levitan I, Fukai T, Ushio-Fukai M. Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2012;302:H724–732. doi: 10.1152/ajpheart.00739.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H, Ramshekar A, Kunz E, Sacks DB, Hartnett ME. IQGAP1 causes choroidal neovascularization by sustaining VEGFR2-mediated Rac1 activation. Angiogenesis. 2020;23:685–698. doi: 10.1007/s10456-020-09740-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Monaghan-Benson E, Hartmann J, Vendrov AE, Budd S, Byfield G, Parker A, Ahmad F, Huang W, Runge M, Burridge K, et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am J Pathol. 2010;177:2091–2102. doi: 10.2353/ajpath.2010.090878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–666. doi: 10.1038/nature06892 [DOI] [PubMed] [Google Scholar]
- 62.Han J, Luby-Phelps K, Das B, Shu X, Xia Y, Mosteller RD, Krishna UM, Falck JR, White MA, Broek D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–560. [DOI] [PubMed] [Google Scholar]
- 63.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3 [DOI] [PubMed] [Google Scholar]
- 64.Zhu G, Fan Z, Ding M, Zhang H, Mu L, Ding Y, Zhang Y, Jia B, Chen L, Chang Z, et al. An EGFR/PI3K/AKT axis promotes accumulation of the Rac1-GEF Tiam1 that is critical in EGFR-driven tumorigenesis. Oncogene. 2015;34:5971–5982. doi: 10.1038/onc.2015.45 [DOI] [PubMed] [Google Scholar]
- 65.Wang HL, Yang CH, Lee HH, Kuo JC, Hur SS, Chien S, Lee OK, Hung SC, Chang ZF. Dexamethasone-induced cellular tension requires a SGK1-stimulated Sec5-GEF-H1 interaction. J Cell Sci. 2015;128:3757–3768. doi: 10.1242/jcs.169961 [DOI] [PubMed] [Google Scholar]
- 66.Fujimoto T, Inoue T, Kameda T, Kasaoka N, Inoue-Mochita M, Tsuboi N, Tanihara H. Involvement of RhoA/Rho-associated kinase signal transduction pathway in dexamethasone-induced alterations in aqueous outflow. Invest Ophthalmol Vis Sci. 2012;53:7097–7108. doi: 10.1167/iovs.12-9989 [DOI] [PubMed] [Google Scholar]
- 67.Tripathi BK, Grant T, Qian X, Zhou M, Mertins P, Wang D, Papageorge AG, Tarasov SG, Hunter KW, Carr SA, et al. Receptor tyrosine kinase activation of RhoA is mediated by AKT phosphorylation of DLC1. J Cell Biol. 2017;216:4255–4270. doi: 10.1083/jcb.201703105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng F, Zhang B, Wu D, Ingram AJ, Gao B, Krepinsky JC. TGFbeta-induced RhoA activation and fibronectin production in mesangial cells require caveolae. Am J Physiol Renal Physiol. 2008;295:F153–164. doi: 10.1152/ajprenal.00419.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang L, Dai F, Liu Y, Yu X, Huang C, Wang Y, Yao W. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol Res. 2018;133:201–212. doi: 10.1016/j.phrs.2018.05.011 [DOI] [PubMed] [Google Scholar]
- 70.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335 [DOI] [PubMed] [Google Scholar]
- 71.Norden PR, Sabine A, Wang Y, Demir CS, Liu T, Petrova TV, Kume T. Shear stimulation of FOXC1 and FOXC2 differentially regulates cytoskeletal activity during lymphatic valve maturation. Elife. 2020;9. doi: 10.7554/eLife.53814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012;51:7420–7432. doi: 10.1021/bi300758e [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data from HUVECs and HDLECs are available in the NCBI Gene Expression Omnibus (GEO) database (accession numbers GSE209855 and GSE212743). All data are included in the manuscript and the supplementary materials. Additional information can be obtained from the corresponding authors upon reasonable request.