

Abstract
Open vascular reconstructions such as bypass are common treatments for cardiovascular disease. Unfortunately, neointimal hyperplasia (IH) follows, leading to treatment failure for which there is no approved therapy. Here we combined the strengths of tailoring nanoplatforms for open vascular reconstructions and targeting new epigenetic mechanisms. We produced adhesive nanoparticles (ahNP) that could be pen-brushed and immobilized on the adventitia to sustainably release pinometostat, an inhibitor drug selective to the epigenetic writer DOT1L that catalyzes histone-3 lysine-79 dimethylation (H3K79me2). This treatment not only reduced IH by 76.8% in injured arteries mimicking open reconstructions in obese Zucker rats with human-like diseases but also avoided the shortcoming of endothelial impairment in IH management. In mechanistic studies, chromatin immunoprecipitation (ChIP) sequencing revealed co-enrichment of the histone mark H3K27ac(acetyl) and its reader BRD4 at the gene of aurora kinase B (AURKB), where H3K79me2 was also enriched as indicated by ChIP-qPCR. Accordingly, DOT1L co-immunoprecipitated with H3K27ac. Furthermore, the known IH driver BRD4 governed the expression of DOT1L which controlled AURKB’s protein level, revealing a BRD4->DOT1L->AURKB axis. Consistently, AURKB-selective inhibition reduced IH. Thus, this study presents a prototype nanoformulation suited for open vascular reconstructions, and the new insights into chromatin modulators may aid future translational advances.
Keywords: Open vascular reconstructions, epigenetic targeting, adhesive nanoparticles, adventitia-localized nanoformulations, DOT1L inhibitors, neointima-abating, endothelium-preserving
Introduction
Despite technological advances in angioplasty and stenting, bypass grafting and endarterectomy are still commonly performed (~400,000 cases a year in the US) to resume blood flow in treating cardiovascular diseases1, 2. Moreover, an arteriovenous fistula is required for vascular access in hemodialysis3. These procedures, herein collectively termed open vascular reconstructions as they require open-body surgery, amount to over a million cases a year in the US alone4. Unfortunately, post-operative failure occurs at unacceptably high rates (e.g. bypass initial year 15–70%) largely due to neointimal hyperplasia (IH)1, 5, 6, inflicting tremendous human and financial costs. After numerous studies, there remains the absence of FDA-approved methods to prevent post-surgery failure of open vascular reconstructions1, 7. Drug-eluting stents, primarily designed for endovascular drug delivery following angioplasty, have been tested for vein grafts yet without approval for clinical use8. A recent 12-month clinical trial of external support for saphenous vein grafts did not produce significant IH mitigation9. On the other hand, a peri or epi-vascular external route for drug delivery is advantageous given convenient access to the vein grafts, arteriovenous fistulas, or prosthetic conduits upon open surgery10–12. To this end, clinical trials involved rapamycin-impregnated polymer mesh, vascular endothelial growth factor (VEGF)-expressing adenovirus in collagen collars or rapamycin-eluting collagen wrap, etc.10, 13 But none has become a clinical method. In preclinical studies, drug-releasing gels, wraps, cuffs, and nanoparticles have been tested yet shown various limitations including physical stress on the vessel, toxicity of breakdown products, and short-term efficacy11. This status quo highlights a two-fold problem — not only an insufficient understanding of IH pathogenic mechanisms but also a lack of suitable drug delivery methods. Accordingly, a two-pronged approach of mechanistic and translational studies is warranted to make synergistic progress in these two areas.
Rapidly growing evidence supports that epigenetics plays a key role in health and disease14 and that epigenetic modulatory agents, dubbed epi-drugs, emerge as new medicine15. Indeed, many pathogenic processes can be traced down to aberrant epigenetic remodeling, as exemplified by dysregulated vascular smooth muscle cell (SMC) plasticity16. Normal SMCs constitute the strength and integrity of the vascular wall. However, if pathogenically stimulated, they undergo phenotypic transition, becoming proliferative and migratory thereby forming neointimal lesions that obstruct blood flow16. Histone modifiers have been implicated in SMC phenotypic transition and IH16. These proteins include writers, readers, and erasers. While writers add chemical marks onto histones, readers transmit them into changes in gene expression, and erasers undo the modifications. Histone deacetylases (erasers) have been extensively studied in SMCs and IH16. However, less knowledge is available on writers and readers of histone modifications in this context17, 18.
Disruptor of telomeric silencing 1-Like (DOT1L) is an epigenetic writer that catalyzes histone-3 lysine-79 (H3K79) methylation19. Unlike other methyltransferases that modify the H3 tail, DOT1L targets H3K79 in the H3 globular domain and is less understood20. On the other hand, DOT1Lselective inhibitors have been well developed, including pinometostat (herein abbreviated as Pino) which is in human trials for cancer treatment20, 21. However, the role of DOT1L in IH pathogenesis remains little known. We recently reported that perivascular delivery of Pino in hydrogel effectively reduced IH in the open surgery model of rat carotid artery injury, and DOT1L-specific silencing recapitulated this effect22. Most recently, Farina et al reported DOT1L’s atherogenic role and genetic variations associated with atherosclerosis23. These studies suggested that DOT1L is a potential epigenetic target for abating IH although the underlying regulatory mechanisms remain to be investigated22.
From the translational perspective for open vascular reconstructions, herein we designed adhesive nanoparticles (ahNP) that can be easily brushed onto the reconstructed vessel and thereby attached (i.e. epi-vascular) for localized drug delivery. We call it an “Epi^NanoPaint” method, which is simple and gentle needing only a pen brush, and quick (<1 min). By using Epi^NanoPaint to deliver Pino, we examined its IH-mitigating efficacy and local toxicity, in healthy rats and also in obese Zucker rats bearing human-like disease backgrounds. From the mechanistic perspective, we identified a DOT1L-centered novel epigenetic regulatory axis. Thus, by combining epigenetic targeting and nanoparticle-based local delivery, herein we present a proof of concept for IH mitigation tailored to open vascular reconstructions.
Results
Epi^NanoPaint exhibits prolonged pinometostat (Pino) release in vitro and retention in the vascular vessel adventitia
To produce adhesive nanoparticles (ahNP), we used a unimolecular micelle as a drug carrier. This NP contains a hydrophobic core that can harbor hydrophobic drugs with high capacity (~20 wt%)24 and a PEG shell that is water soluble and also amenable for conjugation, e.g. attaching a Cy5 tag (Figure 1A). To render the NP adhesive, we modified the terminal end of PEG with N-hydroxysulfosuccinimide (sulfo-NHS) ester that can readily interact with primary amines abundant in proteins to form amide bonds.
Figure 1. Schematic of EpîNanoPaint and its application by pen-brushing.
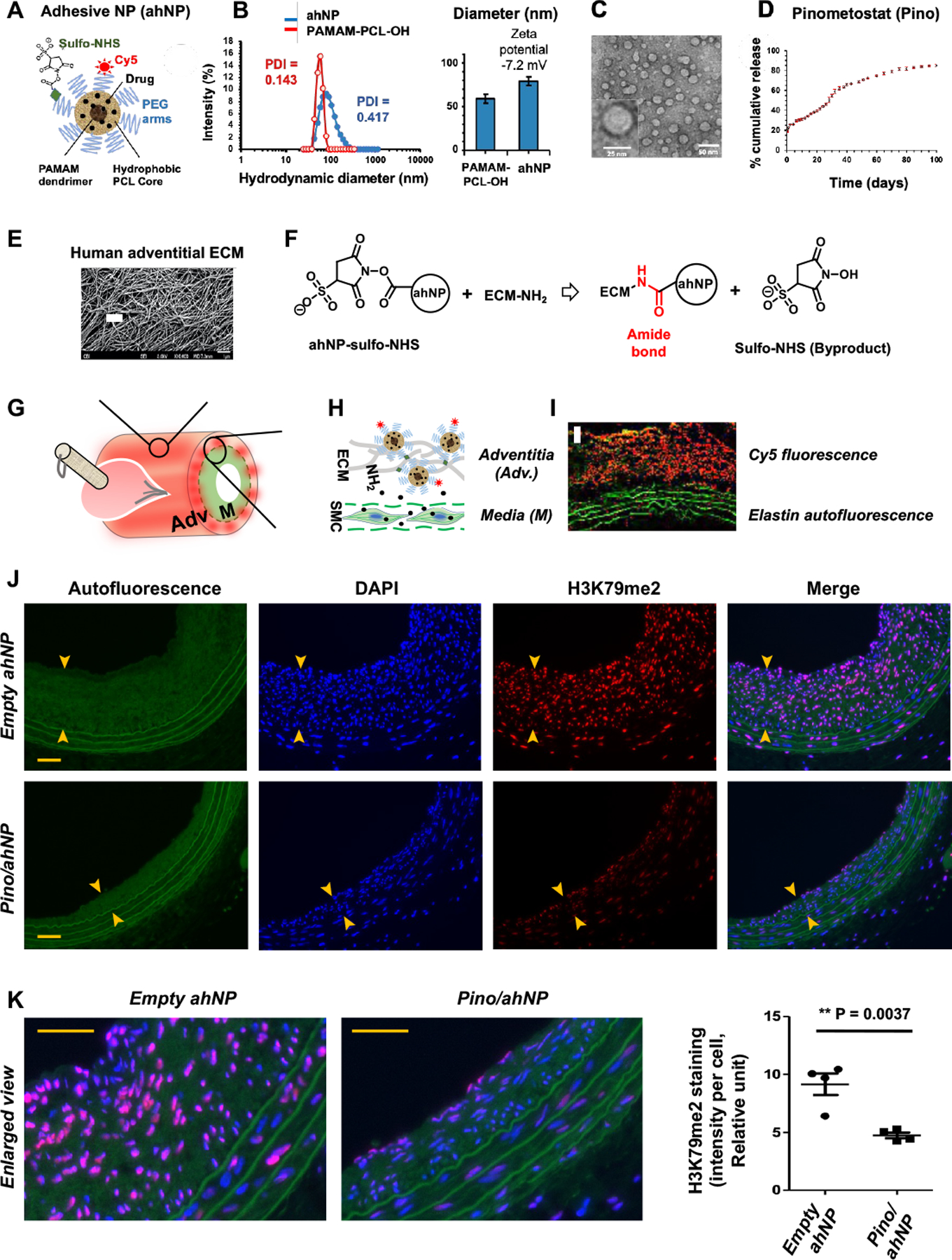
A. Illustration of ahNP structure. The ahNP has a hydrophobic PCL core and is adhesive because of the sulfo-NHS groups on the PEG arms. For ahNP immobilization on extracellular matrix (ECM), sulfo-NHS reacts with NH2 on ECM proteins to form an amide bond. The sulfo-NHS byproduct is highly water-soluble and can be easily removed.
B. Size and zeta potential of PAMAM-PCL-OH and ahNP from DLS. Quantification: Data are presented as mean ± SEM, n= 3 independent repeat experiments.
C. TEM image demonstrating the spherical shape of ahNP.
D. Cumulative release of pinometostat from ahNP at 37°C for 100 days. Quantification: Data are presented as mean ± SEM, n=3 independent repeat experiments.
E. SEM image of human adventitial ECM33. Scale bar: 1 μm.
F. The sulfo-NHS (N-hydroxysulfosuccinimide) ester on ahNP forms an amide bond with NH2 rich on the adventitial ECM.
G. Schematic of epi-vascular application by pen-brushing ahNP onto the vessel adventitia.
H. Schematic of ahNP immobilization on the adventitial ECM.
I. Confocal image (collected 7 days after brushing) showing adventitial Cy5 signal and medial (M) elastin auto-fluorescence (scale bar: 50 μm).
J. Effect of Pino/ahNP on H3K79me2. IH was induced by open-surgery and balloon injury in obese Zucker rat common carotid arteries, and Pino-loaded ahNP or empty ahNP was pen-brushed on the adventitia of the injured arteries. Arteries were collected 28 days later and cross-sections were used for immunofluorescence staining of H3K79me2. Neointima is demarcated by arrow heads. Scale bar: 50 μm.
K. Quantification for J. Shown are enlarged merge images. Scale bar: 50 μm. The values from 4–6 sections were pooled to generate the mean for each animal. The means from 4 animals in each group were then averaged to produce the final mean ± SEM (n = 4 rats). Unpaired Student t-test: **p<0.01
The ahNP was synthesized as indicated by the scheme in Figure S1. The hydrodynamic diameters and zeta potentials of PAMAM-PCL-OH and ahNP were measured via dynamic light scattering (DLS) (Figure 1B). The hydrodynamic diameter of PAMAM-PCL-OH and ahNP was 58.8, and 78.8 nm, respectively, while the zeta potential of ahNP was negative (−7.2 mV) due to the sulfonate group in sulfo-NHS ester after PEGylation25. Transmission electron microscope (TEM) imaging illustrated spherical ahNP with a dynamic diameter of ~30–35 nm (Figure 1C). The chemical structures of PAMAM-PCL-OH, NP-NHBoc, NP-NH2, NP-COOH, and ahNP were characterized by 1H NMR analysis (Figure S2, A–E). The average number of repeating units of the PCL was evaluated to be 28, based on the area ratio of the peaks at (d) δ4.06 ppm and (d′) δ3.65 ppm (Figure S2A).
For ahNP-aided drug delivery, we chose the DOT1L-selective inhibitor pinometostat, which showed a function of reducing H3K79me2/me3 and IH in injured rat arteries in our previous study using a hydrogel approach for perivascular delivery22. We also applied rapamycin for positive control as it is commonly used in preclinical24, 26–29 and clinical30, 31 management of IH. Each drug was loaded into the ahNP through a standard dialysis method32. The loading contents of pinometostat and rapamycin in ahNP were 17.5% and 19.1% by weight, respectively, while the encapsulation efficiency of pinometostat and rapamycin were 58.3% and 72.4%, respectively, measured by HPLC following our previously reported method24. Figure 1D shows in vitro drug (Pino) release profile over 100 days from ahNP. There was a burst release during the first 3 days (26.3 wt%), followed by sustained release (85.0 wt%) until day 100.
The adventitia of a vascular vessel is a porous tissue densely packed with extracellular matrix (ECM) fibers33 (Figure 1E), which provide extensive surface areas for ahNP to attach to. Enabled by sulfo-NHS groups on the surface, ahNP can readily and covalently react with the abundant amine groups on ECM proteins to form amide bonds34 (Figure 1F), and the ahNP can be simply pen-brushed onto the vessel surface and immobilized on the ECM, to release drug locally and durably (schematized in Figure 1, G and H). Because the elastic laminae in the vessel wall are fenestrated sheets35, 36 that allow small molecules to pass through37, it is conceivable that the drug released from the immobilized ahNP should readily diffuse into the neighboring medial layer where SMCs reside. To mimic open vascular reconstruction, we opened the neck of the rat and injured the common carotid artery with a balloon catheter. We then used gauze to dry the vessel surface so that proteins in the body fluid would not scavenge ahNP. To apply ahNP, we dipped a pen brush in the ahNP solution and quickly brushed the outer surface of the injured artery, and used gauze to absorb unreacted ahNP before closing the neck skin of the animal. Confocal imaging confirmed that substantial Cy5-ahNP remained in the adventitia of the injured artery 7 days after the ahNP application (Figure 1I).
Epi^NanoPaint loaded with the DOT1L-selective inhibitor pinometostat abates IH in obese Zucker rats in a one-month treatment period
The prolonged drug release (>3 months) from ahNP (Figure 1D) is advantageous for testing its IH-mitigating efficacy in a relatively long term. Whereas injury-induced IH in healthy rats tends to regress, progressive IH occurs in obese Zucker rats that spontaneously develop human-like disease conditions including hypercholesterolemia and diabetes38. We thus performed therapeutic tests in obese Zucker rats by pen-brushing Pino-loaded ahNP onto balloon-injured carotid arteries. Rapamycin-loaded ahNP was used for positive control because of its established IH-abating efficacy. For each drug, we chose a dose 100-fold lower than that used in a common approach of perivascular hydrogel delivery, based on our previous reports22, 24 and pilot experiments (Figure S3). Animals were euthanized 4 weeks after the ahNP application. To determine whether the drug (Pino) released from the adventitia-immobilized ahNP was able to reach its target protein DOT1L in the vessel wall and inhibit its hyperactivity of catalyzing H3K79me2, we measured H3K79me2 immunofluorescence on the cross-sections of the arteries collected 28 days post injury. Figure 1 (J and K) indicates that H3K79me2 was indeed markedly reduced by Pino/ahNP compared to empty ahNP, mainly in the medial and neointimal layers. We then measured IH as the neointima/media area ratio (N/M). While DOT1L was upregulated in human coronary arteries that developed IH (Figure 2A), and also in injured rat carotid arteries that underwent IH as we previously reported22, treatment of injured rat arteries with rapamycin-loaded ahNP reduced IH by 62.8% (p < 0.0001) as compared with empty ahNP, and Pino-loaded ahNP reduced IH by 76.8% (p < 0.0001) (Figure 2, B and C). Both Rapa/ahNP and Pino/ahNP increased the lumen area, by 43.1% (p < 0.01) and 54.6% (p < 0.01), respectively. Accordingly, they reduced the stenosis rate by 59.3% (p < 0.0001) and 70.7% (p < 0.0001), respectively. There was no adverse effect of vessel shrinkage, as indicated by no change in the EEL perimeter that measures the overall vessel size. We then immunostained the macrophage marker CD68 and the cell death marker caspase-3 (cleaved) on sections to assess the local tissue toxicity of the two nanoformulations. As seen in Figure 3, either Rapa/ahNP or Pino/ahNP markedly reduced these markers as compared to the empty ahNP control. In addition, the drug Pino did not alter the expression of the full-length caspase-3 protein in cultured SMCs (Figure S4). Together, these results indicate that the Epi^NanoPaint loaded with the epigenetic modulatory drug Pino effectively inhibits injury-induced IH and ameliorates local tissue damage in obese Zucker rats after one month of treatment.
Figure 2. Pino-loaded Epi^NanoPaint (Pino/ahNP) abates IH in a 1-month term in obese Zucker rats.
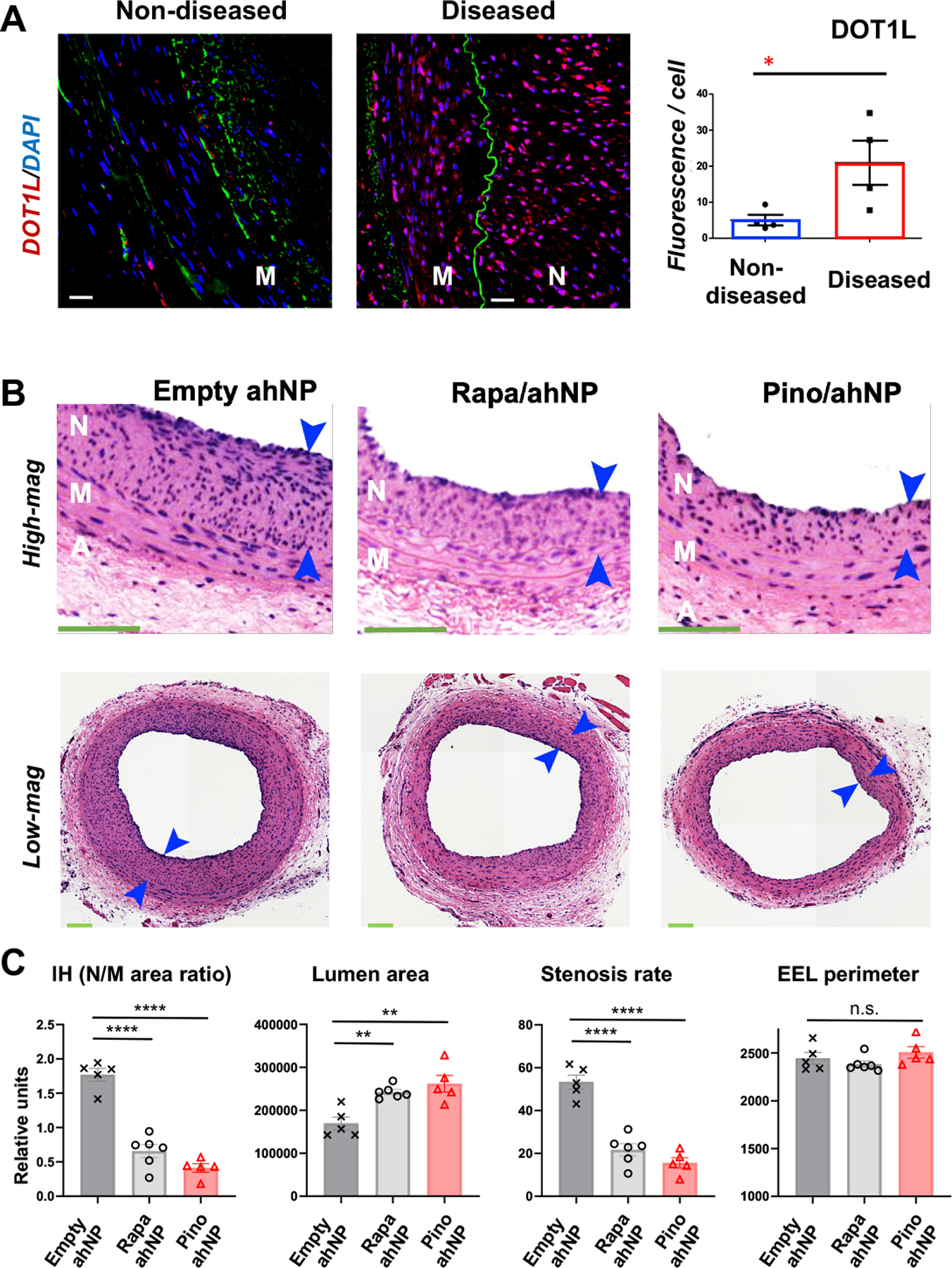
IH was induced via open-surgery and balloon injury in obese Zucker rat common carotid arteries, and drug-loaded ahNP or empty ahNP was pen-brushed on the adventitia of the injured arteries. After 28 days, arteries were collected and animals were euthanized.
A. Immunofluorescence showing DOT1L upregulation in human stenotic coronary arteries. Diseased: Stent-edge neointima; non-diseased: no stent (samples from CVPath Institute, MD). M, media; N, neointima. Green: Elastin autofluorescence. Scale bar: 50 μm. Mean ± SEM, n= 4 patients. Unpaired Student t-test: *p<0.05
B. Representative H&E-stained cross-sections of Zucker rat arteries. To mimic open vascular reconstruction, an incision was made in the neck of the rat, and the common carotid artery was dissected and balloon-injured. Epi^NanoPaint was brushed on the artery adventitia immediately after balloon injury. After 28 days of treatment with empty ahNP (no drug) or ahNP loaded with rapamycin (Rapa, 0.018 mg/kg) or with Pino (0.3 mg/kg), arteries were collected for morphometric analysis. N, neointima (between arrows); M, media; A, adventitia. Scale bar:100 μm.
C. Quantification for B. IH was measured as the N/M area ratio. Mean ± SEM, n = 5 or 6 rats. One-way ANOVA and Tukey test: **P<0.01, ****P<0.0001, n.s., not significant.
Figure 3. Pino/ahNP reduces CD68 and active (cleaved) caspase-3 in injured Zucker rat carotid arteries.
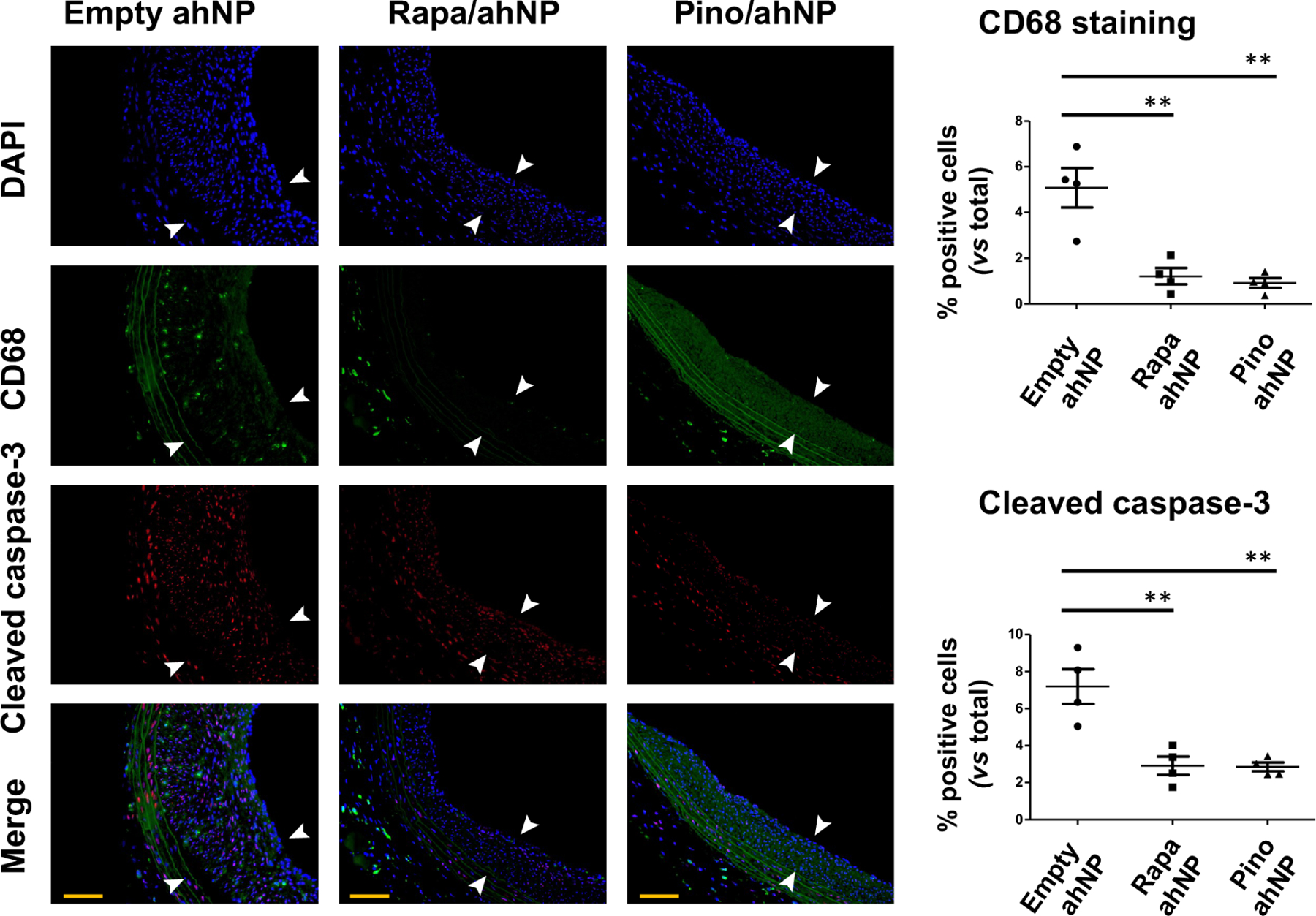
Obese Zucker rat carotid arteries were injured to induce IH, and drug-loaded ahNP or empty ahNP were pen-brushed on the adventitia. Pino: 0.3 mg/kg; Rapa: 0.018 mg/kg. Arteries were collected 28 days later. Green autofluorescence illuminates elastin laminae. Neointima is demarcated between arrows. Scale bar: 100 μm. Quantification: Data are presented as mean ± SEM of percent positively stained cells versus total cells; n = 4 rats. ANOVA and Tukey test: **P<0.01.
Pinometostat delivered via Epi^NanoPaint, but not the free drug, is effective in mitigating IH in injured rat arteries
We next characterized the ahNP in more detail by including additional control groups without ahNP; i.e. injury only and free drug. For this specific purpose, we chose Sprague-Dawley rats which are widely used to study IH, with a 2-week standard endpoint for using the balloon injury model in Sprague-Dawley rats. As seen in Figure 4, Pino/ahNP effectively decreased IH that was measured as the neointima/media ratio (N/M) by 55.7% (p < 0.0001) and 39.6% (p < 0.05) as compared with the injury-only (saline) and empty UM groups, respectively. In contrast, there was no significant difference in IH between the injury-only and free Pino groups, but the same dose of Pino loaded in ahNP resulted in a lower N/M ratio compared with free Pino. There appeared to be a trend of increased lumen area in the Pino/ahNP group relative to the other three groups. Moreover, there was a decrease in stenosis rate in the Pino/ahNP group compared to the injuryonly group. No significant difference was observed in EEL perimeter among the four groups, ruling out vessel shrinkage caused by either ahNP alone or Pino/ahNP. The experiments using rapamycin as a positive control drug produced similar results, with Rapa/ahNP reducing IH by 69.6% (p < 0.0001) and 58.5% (p < 0.01) compared with the injury-only and empty UM groups, respectively. However, the arteries treated with free rapamycin shrank significantly as compared with the injury-only group, and the arteries treated with Rapa/ahNP showed the same trend. The reason for the smaller sizes of the drug-treated arteries is not clear. Fibrosis has been thought to cause vessel shrinkage, but we did not see an increase of collagen deposition in rapamycin-treated arteries compared to the injury-only or empty ahNP group (Figure S5). We also determined the effect of these nanoformulations on local tissue damage (Figure S6). Immunostaining assays indicated that Pino/ahNP and Rapa/ahNP both reduced CD68 and active caspase-3, whether compared to the injury-only control (saline) or empty ahNP, whereas free drugs (Pino and Rapa, without ahNP) did not have this effect.
Figure 4. Pino/Epi^NanoPaint reduces IH in a short term (14 days after painting) in Sprague-Dawley rats.
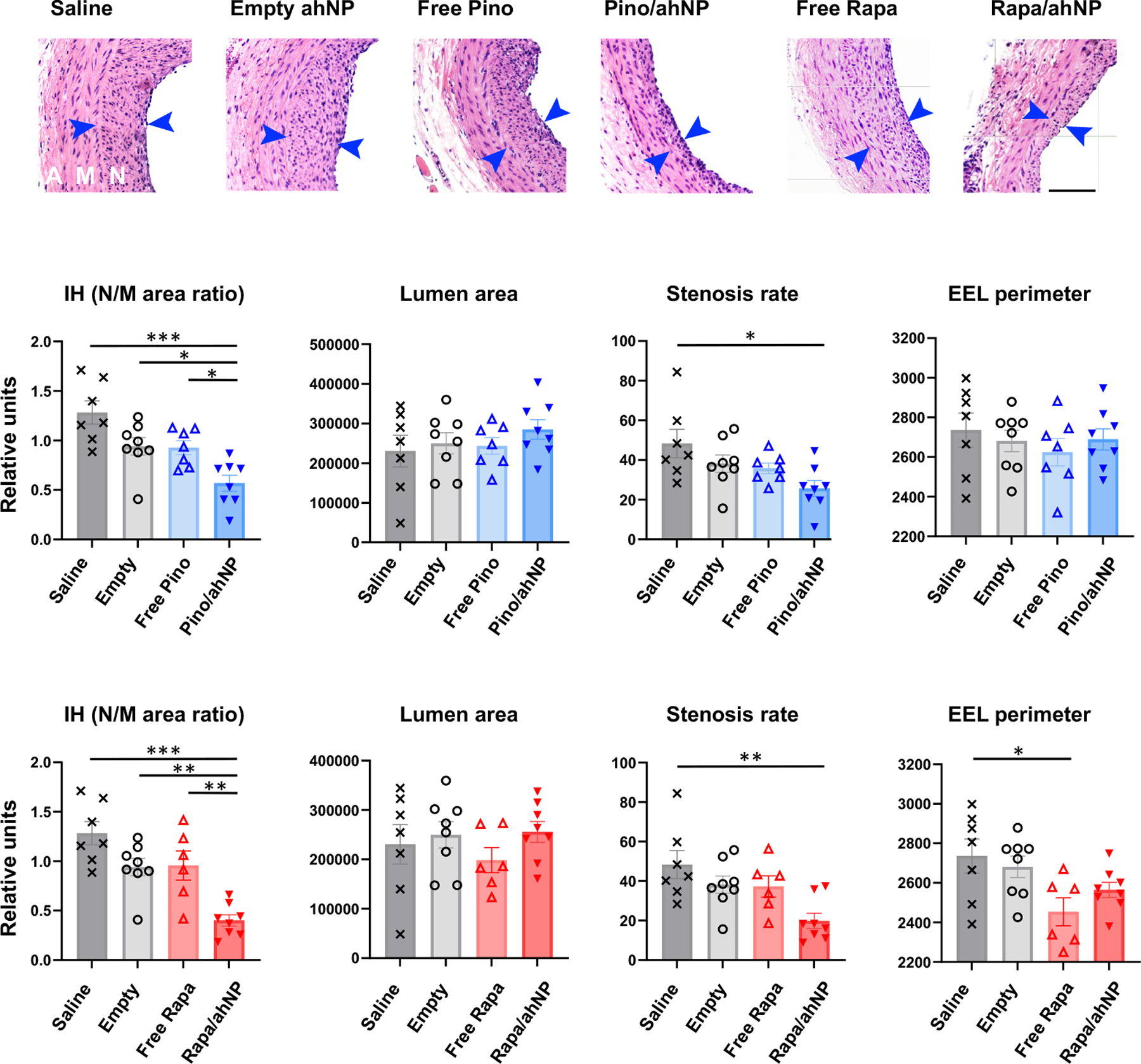
IH was induced via the open-surgery of balloon injury in carotid arteries, and drug-loaded Epi^NanoPaint was pen-brushed on the adventitia of the injured artery. Pino: 0.3 mg/kg; Rapa: 0.018 mg/kg. The control groups include saline (injury only), empty ahNP, and free drug in a PBS solution (no ahNP). A, adventitia; M, media; N, neointima (between arrows). Scale bar:100 μm. IH: N/M area ratio. Quantification: Mean ± SEM, n=7 or 8 rats; ANOVA and Tukey test: *p<0.05, **P<0.01, ***P<0.001.
Taken together, the results presented above demonstrate that the Pino/ahNP formulation of Epi^NanoPaint confers a remarkable inhibitory effect on injury-induced IH and inflammatory/apoptotic local tissue damage in either a long-term (in obese Zucker rats) or short-term treatment (in Sprague-Dawley rats).
Pinometostat, in contrast to rapamycin, does not impede re-endothelialization when delivered via Epi^NanoPaint
It is interesting to note that the Rapa/ahNP formulation produced an IH-abating effect very similar to that of Pino/ahNP. Rapamycin has been clinically used on drug-eluting stents to manage postangioplasty restenosis. Unfortunately, this drug is concerning because of its endothelial toxicity and thrombogenicity, as implicated by increasing evidence39–42. To monitor the toxicity of Rapa/ahNP to the endothelium, we measured rat carotid artery re-endothelialization on post-injury day 7. Indeed, compared to empty ahNP, Rapa/ahNP impeded re-endothelialization of the injured artery (Figure 5A). In contrast, Pino/ahNP did not delay re-endothelialization as compared to the empty ahNP group. Therefore, Rapa/ahNP but not the Pino/ahNP formulation is detrimental to the recovery of damaged endothelium. To further assess the influence of DOT1L inhibition on the growth of endothelial cells (ECs), we pretreated cultured ECs with rapamycin or a DOT1L inhibitor (SGC0946 or Pino; SGC0946 is more cell permeable)43. We then challenged the cells with TNFα — a commonly used in vitro model44. We found that whereas rapamycin substantially reduced EC viability, the DOT1L inhibitor SGC0946 did not, whether in the absence or presence of TNFα (Figure 5B). In accordance, whereas rapamycin markedly reduced the protein levels of eNOS, KLF4, and PCNA, all key factors for EC growth and survival26, 41, SGC0946 did not (except for the +TNFα condition for PCNA) (Figure 5, C–D). In accordance, in a separate set of experiments, we observed a similar pattern of rapamycin impairing eNOS protein expression in contrast to Pino (Figure 5F). Since pretreatment of ECs with 0.5 μM Pino for 5 days (see hashtag) nearly completely blocked the H3K79me2-catalyzing function of DOT1L, we used this condition to determine the effect of Pino on PCNA expression. We found that unlike rapamycin, Pino (vs vehicle) increased PCNA protein in TNFα-challenged ECs (Figure 5G). Thus, in vitro results demonstrate that rapamycin, but not DOT1L-selective inhibitors, is endothelial toxic.
Figure 5. Pinometostat delivered by Epi^NanoPaint is not toxic to the endothelium.
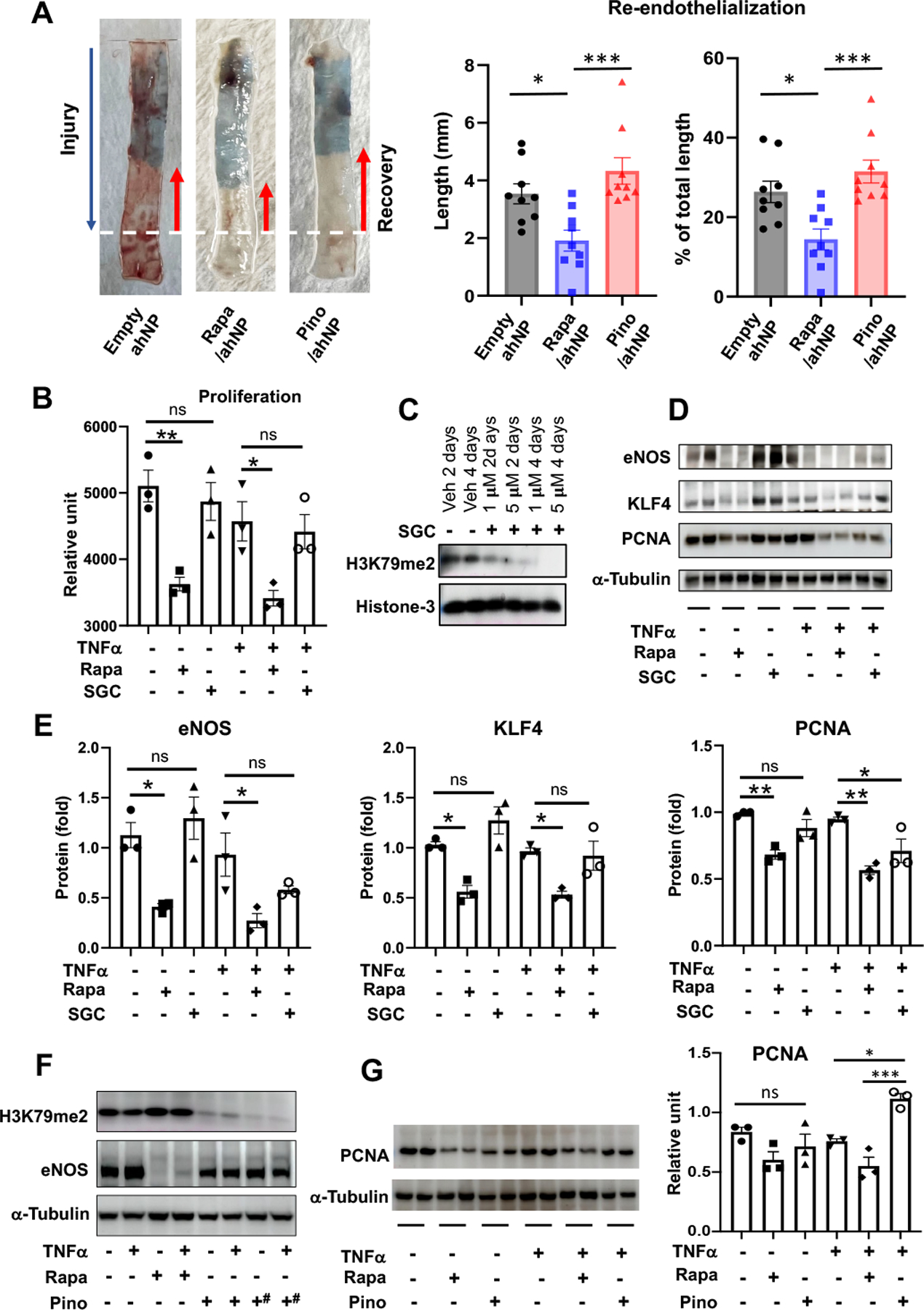
A. Re-endothelialization. The endothelium was damaged as far as the dotted white line in the IH model of rat common carotid artery injury and grew back (after 7 days) as much as the length of red arrow marking the unstained segment. The remainder damaged segment was stained blue. Drug dose: 0.3 mg/kg pinometostat; 0.018 mg/kg rapamycin. Mean ±SEM, n= 8 or 9 Sprague-Dawley rats. One-way ANOVA and Tukey test: *p<0.05, ***P<0.001.
B-E. In vitro assays using the DOT1L-selective inhibitor SGC0946. Human umbilical vein endothelial cells were cultured in Lonza EGM-2 growth medium until ~80% confluency and starved in basal medium overnight. Rapamycin (final 0.5 μM) or SGC0946 (final 5 μM) or vehicle (veh, equal volume of DMSO) was added to the starved cells and incubated for 2 days (note different concentrations of SGC and pretreatment days tested in C). The cells were then treated with TNFα (20 ng/ml) for 4h followed by Cell Titer Glo viability assay and immunoblot assay. In D, duplicate immunoblot bands represent two separate experiments. Quantification: Mean ± SEM, n = 3 independent repeat experiments; oneway ANOVA and Tukey test: *p<0.05, **P<0.01.
F-G. In vitro assays using the DOT1L-selective inhibitor Pino. Human umbilical vein endothelial cells were cultured in Lonza EGM-2 growth medium until ~80% confluency and starved in basal medium overnight. Rapamycin (final 0.5 μM) or vehicle (equal volume of DMSO) was added to the starved cells and incubated for 2 days. Pretreatment with Pino (final 0.5 μM) continued for 3 or 5 days (hashtags in F). The cells were then treated with TNFα (20 ng/ml) for 4h followed by immunoblot assay. In G, cells were pretreated with 0.5 μM Pino for 5 days and 0.5 μM Rapa for 2 days. Duplicate bands on the immunoblot represent two separate experiments. Quantification: Mean ± SEM, n= 3 independent repeat experiments; one-way ANOVA and Tukey test: *p<0.05, ***P<0.001.
The histone code reader BRD4 is an upstream determinant of DOT1L expression in SMCs
We22 and Dr. Elia’s group23 observed marked DOT1L upregulation in human and animal atherosclerotic and stenotic lesions (also see Figure 2A), a result implicating a pathogenic role of its hyperactivity. However, in this context, the epigenetic determinants that control DOT1L expression levels remained elusive. Previous studies highlighted that the family of bromo/extra terminal domain proteins (BETs), especially BRD4 (bromodomain 4), is a key epigenetic driver of gene activation in proliferative disease settings17. BRD4 and DOT1L are seemingly unrelated players, in view of that BRD4 “reads” acetyl marks on histone tails whereas DOT1L “writes” methyl marks at the H3K79 site of the H3 globular domain. Our recent studies demonstrated that both BRD4 and DOT1L promote IH22, 44–47. Moreover, another study identified synergistic activity of BRD4 and DOT1L in regulating leukemogenic transcription programs, although whether BRD4 regulates DOT1L expression was not determined48. We used siRNAs to silence BRD2, BRD3, and BRD4 individually in SMCs and found that the silencing of BRD4, but not the silencing of BRD2 or BRD3, reduced DOT1L mRNA and protein (Figure 6A). These results demonstrate that the epigenetic reader BRD4 governs DOT1L’s expression levels.
Figure 6. BRD4 is an upstream regulator of both DOT1L and AURKB in SMCs.
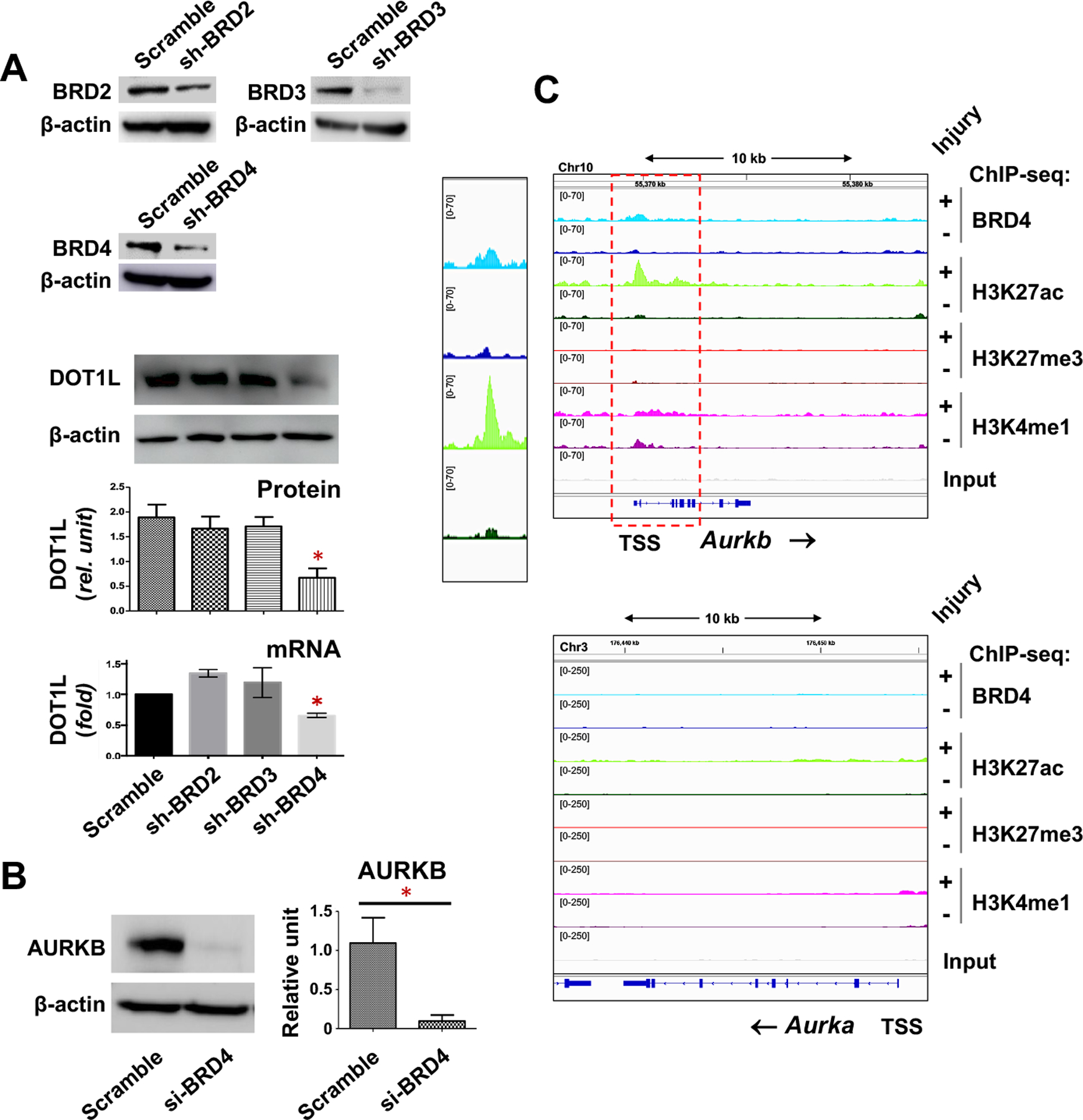
A. BRD4 silencing reduces DOT1L expression. Mouse SMCs (MOVAS) were transduced with lentivirus to express scrambled or gene-specific shRNAs. The transduced cells were allowed to recover in fresh medium for 24h and then starved overnight in basal medium prior to stimulation with PDGF-BB (20ng/ml) for 24 h. Immunoblots: Mean ± SEM, n = 3 independent repeat experiments. qRT-PCR: Mean ± SD, n = 3 replicates. ANOVA and Tukey test: *p<0.05, compared to any of the other 3 bars.
B. BRD4 silencing down-regulates AURKB. Rat primary aortic SMCs were transfected with scrambled or BRD4-specific siRNA, and stimulated with PDGF-BB (20ng/ml, 48h). Mean ± SEM, n= 3 independent repeat experiments. Student’s t-test: *p<0.05.
C. BRD4 and H3K27ac co-occupy the genomic region of Aurkb. The data was retrieved by using accession number GSE194390 (see our recent report45). Integrative genomics viewer (IGV) tracks show the comparison of normalized ChIP-seq peaks between injured (+, light color) and uninjured sham-control (-, dark color) arteries. The box on the left highlights the Aurkb TSS region where the binding of H3K27ac and BRD4 increased after arterial injury. Non-specific input indicates low background noise.
Inhibition of DOT1L reduces the expression of AURKB in SMCs
After identifying BRD4 as an upstream regulator of DOT1L expression, we next pursued a downstream effector that mediates DOT1L’s pro-IH function. While BRD4 is pro-proliferative in many cell types17, aurora kinases A and B (AURKA, AURKB) are pro-proliferative mitotic factors49. It was recently reported that BETs-selective inhibition using JQ1 down-regulated AURKA and AURKB50. This prompted us to perform a pilot experiment, using JQ1 to pretreat SMCs prior to the mitogenic stimulation with PDGF-BB (platelet-derived growth factor). Interestingly, JQ1 reduced AURKB but not AURKA, in either human, rat, or mouse SMCs, without or with PDGF-BB (Figure S7A). We further confirmed that JQ1 inhibited AURKB mRNA expression in either rat or mouse SMCs (Figure S7B). We also performed gene silencing with a BRD4-specific siRNA that proved effective in our previous reports44, 51, and found that BRD4 silencing reduced AURKB protein (Figure 6B). Consistently, based on our ChIP-seq data obtained from injured rat carotid artery tissues (uninjured arteries as controls)45, we found that both BRD4 and H3K27ac were enriched at and after the Aurkb transcription start site (TSS) (Figure 6C). The pattern of H3K4me1 peaks appeared different from that of BRD4 and H3K27ac. It is reasonable considering that unlike BRD4 and H3K27ac, H3K4me1 is known to associate not only with active enhancers but also inactive and poised enhancers45. BRD4 reads/binds the transcription-activating chromatin mark H3K27ac and forms a complex with the central transcription machinery thereby potently promoting gene expression17. In this light, the injury-induced BRD4/H3K27ac enrichment at Aurkb implicated BRD4 regulation of AURKB gene expression. Consistently, H3K4me1, another histone mark associated with gene activation, was also found to be enriched in the genomic region downstream of the Aurkb TSS. There was no obvious peak of H3K27me3, a bona fide gene repression mark45, providing a negative control to the peaks of the gene-activation mark H3K27ac. In stark contrast to the ChIP-seq results of Aurkb, there were essentially no peaks of the aforementioned chromatin marks at or after the Aurkb TSS. Thus, the immunoblot and ChIPseq results together support that BRD4 positively regulates AURKB expression.
We previously observed that BRD4 and DOT1L protein levels were both elevated in the rat carotid arteries that underwent IH22, 44. Here we further show that the mRNA levels of BRD4 and DOT1L were also markedly upregulated in rat carotid arteries with IH, and the mRNA of AURKB was increased as well (Figure 7A). Recent reports from the Elia group23 and ours22, 44 indicated that DOT1L and BRD4 were upregulated in vitro in phenotypically transformed SMCs after PDGFBB stimulation. Moreover, consistent with injury-induced DOT1L activation in rat arteries22, H3K79m2 which is the catalytic product of the DOT1L enzymatic activity, was also increased in response to PDGF-BB (Figure 7B). In line with this observation of PDGF-stimulated hyperactive DOT1L, AURKB expression was also stimulated by PDGF-BB in SMCs (Figure 7C). Given the above results indicating that BRD4 governed the expression of both DOT1L and AURKB, we were motivated to determine whether AURKB is an effector downstream of DOT1L. Interestingly, selective DOT1L inhibition reduced AURKB protein levels in SMCs (Figure 7D). We then explored the chromatin-associated mechanisms pertaining to the DOT1L->AURKB axis. We first performed ChIP-qPCR by using an antibody against H3K79me2 catalyzed by DOT1L. Our data showed that the DNA fragment of an upstream genomic region (~1kb from the Aurkb TSS) coIPed with H3K79me2 (Figure 7E), suggesting that H3K79me2 (hence DOT1L) was enriched at Aurkb. Since H3K27ac is associated with the gene-activation machinery, we also determined whether DOT1L was associated with H3K27ac. Indeed, Figure 7F indicates that endogenous DOT1L co-IPed with anti-H3K27ac. Thus, the results together suggest that DOT1L promotes Aurkb activation (see schematic in Figure 7G).
Figure 7. AURKB is downstream of DOT1L and regulates SMC phenotypic transition.
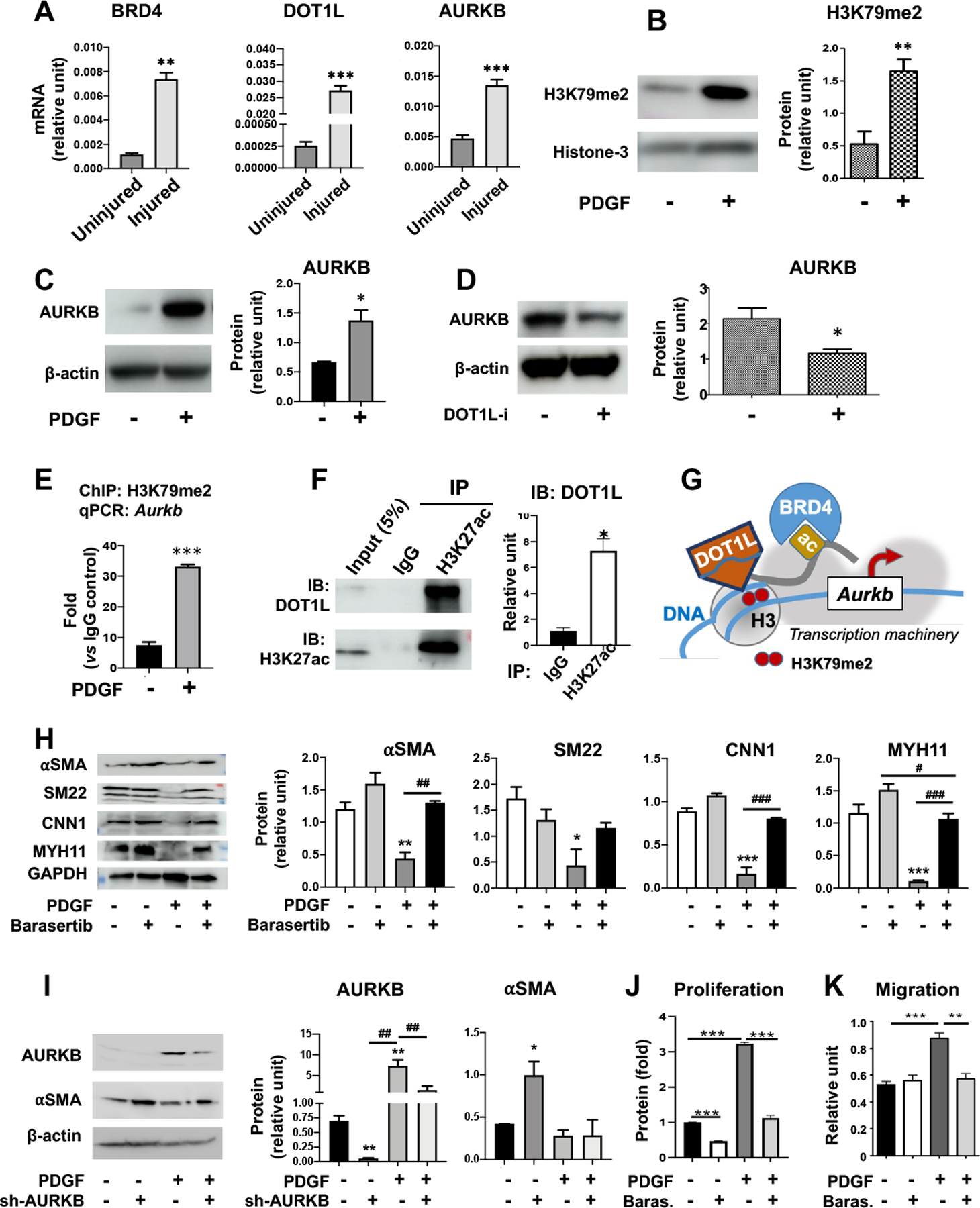
Sub-confluent mouse SMCs (MOVAS) were starved in basal medium overnight and then stimulated with solvent control or PDGF-BB (20 ng/ml, 24h) prior to harvest for immunoblot or co-IP assays. Otherwise, cells or tissues are specified below.
A. BRD4, DOT1L, and AURKB mRNA levels increase in injured arteries. IH-inducing balloon angioplasty was performed in common carotid arteries of Sprague-Dawley rats. The injured arteries and uninjured controls were collected 7 days later for qRT-PCR analysis. Mean ± SD, n = 3 repeats. Unpaired Student’s t-test: **P<0.01, ***P<0.001.
B-C. Stimulation of SMCs (MOVAS) with the mitogen PDGF-BB increases H3K79me2 and AURKB. Mean ± SEM, n = 4 independent repeat experiments for AURKB; mean ± SD, n = 3 replicates for H3K79me2. Student’s t-test: *p<0.05, **P<0.01.
D. DOT1L inhibition reduces AURKB. Starved rat primary SMCs were pretreated with the DOT1L-selective inhibitor SGC0946 (1 μM) for 3 days prior to stimulation with PDGF-BB. While both Pino and SGC0946 are highly DOT1L-selective inhibitors, we chose SGC for in vitro studies because of its better cell permeability. Mean ± SEM, n = 3 independent repeat experiments. Student’s t-test: *P<0.05.
E. H3K79me2 enriches at Aurkb. Chromatin immunoprecipitation (ChIP) was performed using an antibody specific for H3K79me2 and rat primary aortic SMCs without or with PDGF-BB stimulation (20 ng/ml, 10 h). qPCR detects IP’ed DNA corresponding to the promoter region of Aurkb. Data was normalized to IgG control. Mean ± SD, n=3 repeats. Student t-test: ***p<0.001.
F. Co-IP of DOT1L with anti-H3K27ac. Mouse SMCs were incubated with PDGF-BB and then used for the co-IP experiment. Immunoblots detect the endogenous DOT1L protein. Mean ± SEM, n= 3 independent repeat experiments. Student’s t-test: *P<0.05.
G. Schematic of the DOT1L regulation of Aurkb gene expression. While the histone acetylation (ac) reader BRD4 binds H3K27ac on the histone tail, the writer DOT1L adds H3K79me2 to the histone globular domain. Both players act as gene activators by associating with the central transcription machinery (gray patch in the back). Our data show that while H3K79me2 enriches at Aurkb (Figure 7E), DOT1L is associated with the histone protein that has a H3K27ac-marked tail (Figure 7F).
H. AURKB inhibition increases SMC differentiation marker proteins. MOVAS cells were pretreated with barasertib (1μM) for 2h prior to stimulation with PDGF-BB (20ng/ml, 48h). Mean ± SEM, n = 3 independent repeat experiments. ANOVA and Tukey test: *P<0.05, **P<0.01, ***P<0.001 compared to the basal condition (no PDGF, no inhibitor, white bar); #P<0.05, ##P<0.01, ###P<0.001, pairwise comparison indicated by a horizontal bar.
I. AURKB silencing increases αSMA. MOVAS cells were transduced with scrambled or AURKB-specific shRNA. Starved celled were treated without or with PDGF-BB (40 ng/ml, 24h). Mean ± SEM, n = 3 independent repeat experiments. ANOVA and Tukey test: *P<0.05, **P<0.01, compared to the basal condition (scrambled, no PDGF, black bar); ##P<0.01, pairwise comparison indicated by a horizontal bar.
J-K. AURKB inhibition abrogates PDGF-stimulated SMC proliferation and migration. MOVAS cells were pretreated with the AURKB-selective inhibitor barasertib (1μM) for 2h prior to stimulation with PDGF-BB. Cell proliferation was determined by the CellTiterGlo assay. Migration was determined by scratch assay. Mean ± SEM, n = 3 independent repeat experiments. ANOVA and Tukey test: **P<0.01, ***p<0.001.
Inhibition of AURKB in injured rat arteries hampers neointima formation
Aurora kinase B (AURKB) is a kinetochore protein intimately involved in cell division. Its proproliferative/pro-migratory function has been well documented in oncology and pharmacologically targeted for treating cancers in clinical trials50. However, to our knowledge, the role of AURKB in neointima formation has not been previously reported. Herein, we used the in vitro model of PDGF-stimulated, pro-IH SMC phenotypic transition to determine the effect of AURKB inhibition on this process. As seen in Figure 7H, whereas treatment with PDGF-BB remarkably reduced SMC marker proteins, including αSMA, SM22, CNN1, and MYH11, pretreatment of SMCs with the AURKB-selective inhibitor barasertib restored these markers to various degrees. The silencing of AURKB with shRNA also increased αSMA in the absence of PDGF-BB (Figure 7I). In addition, pretreatment with barasertib abrogated PDGF-stimulated SMC proliferation and migration. (Figure 7J/K). Thus, AURKB appeared to regulate SMC’s IHformative behaviors in vitro. Consistently, the silencing of DOT1L in injured rat arteries, which mitigated IH22, also reduced AURKB in vivo (Figure 8A). In view of the upregulation of AURKB in human neointimal lesions (Figure S8) and its regulation by BRD4 and DOT1L, both known to promote IH22, 44, we surmised that AURKB could be pharmacologically targeted for treating IH. Using the artery injury model in Sprague-Dawley rats, we found that after 14 days of treatment with the selective AURKB inhibitor barasertib, IH and the stenosis rate were decreased by 49.5% (p < 0.05) and 47.0% (p < 0.05), respectively, each compared to the vehicle control; lumen area increased by 65.8% (p = 0.07), and no EEL perimeter change was observed (Figure 8, B and C). The treatment with the AURKA-selective inhibitor alisertib also reduced IH and stenosis rate, yet to a lesser extent (Figure S9). Thus, these preclinical tests identify AURKB as a novel target for intervention to impede neointimal progression.
Figure 8. DOT1L silencing reduces AURKB and AURKB inhibition mitigates IH in injured rat carotid arteries.
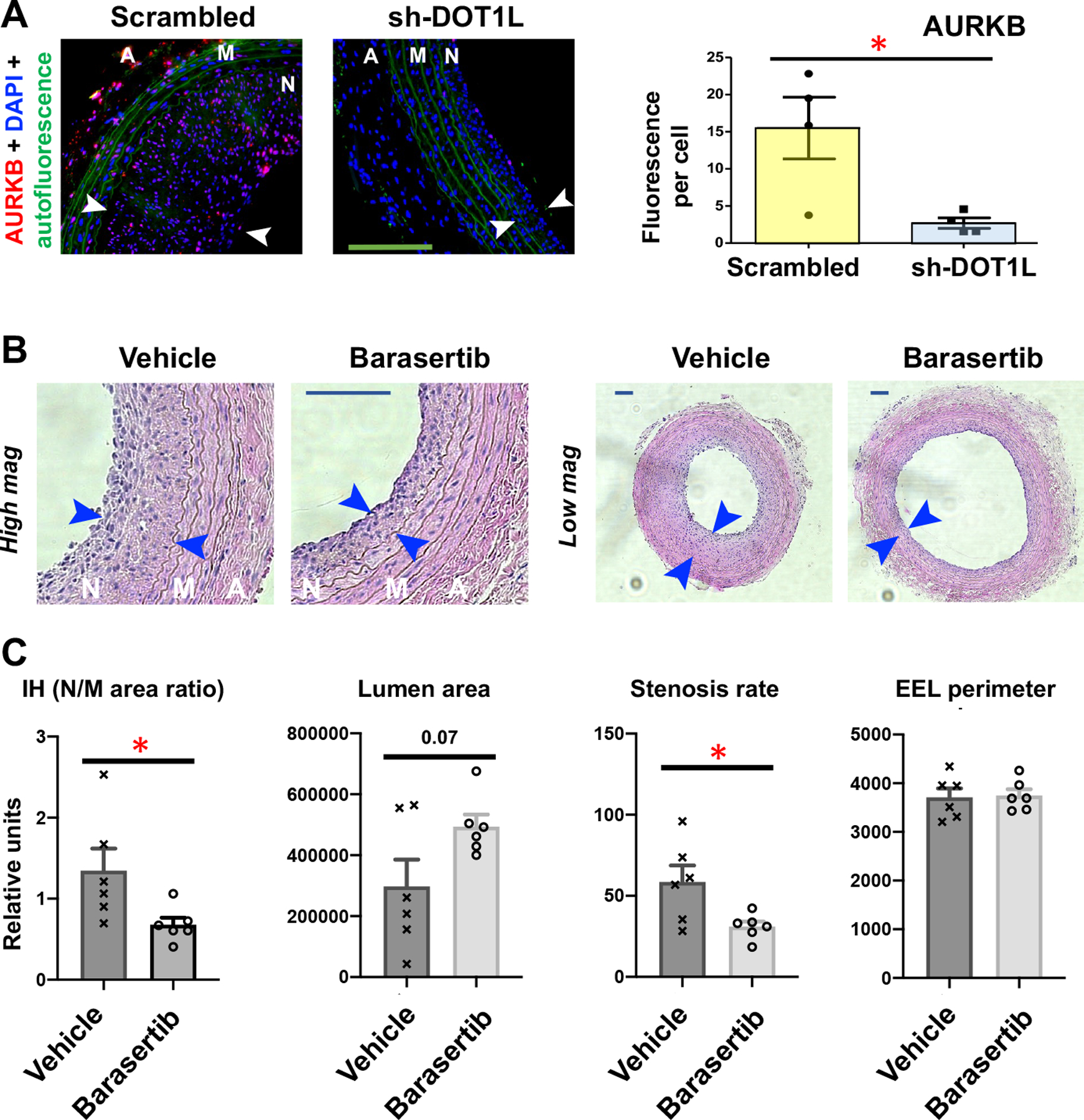
To induce IH, balloon injury of common carotid artery was performed in Sprague-Dawley rats. Immediately after the injury, lentivirus was intra-luminally infused into the injured artery wall to express scrambled or DOT1L-specific shRNA. In a separate experiment, the AURKB-selective inhibitor barasertib (10 mg/kg) was applied in perivascular hydrogel. Arteries were collected 14 days after injury for immunostaining or morphometric analysis. N, neointima (between arrows); M, media; A, adventitia. Scale bar:100 μm.
A. DOT1L silencing reduces AURKB. Quantification: Mean ± SEM, n=4 rats; unpaired Student’s t-test: *p<0.05.
B-C. AURKB inhibition mitigates IH (neointima vs media area ratio). Quantification: Mean ± SEM, n= 6 rats; unpaired Student’s t-test: *p<0.05.
Discussion
In this study, we developed an Epi^NanoPaint method for adventitia-localized delivery of a DOT1L-selective inhibitor, which effectively mitigated IH not only in healthy rats but also in obese Zucker rats that bear human-like disease backgrounds. Mechanistically, we found that the epigenetic reader BRD4 governed the expression of the epigenetic writer DOT1L, and inhibition of DOT1L reduced AURKB – a herein-identified new target for treating IH. Thus, by combining nanoplatforms with epigenetic targeting, we present here an IH-mitigating prototypical nanoformulation tailored to open vascular reconstructions. The molecular insights into a novel BRD4->DOT1L->AURKB epigenetic axis may contribute to precise therapeutic targeting in future translational advances.
Atherosclerosis is the leading cause of mortality and morbidity worldwide. Currently, there are two major treatments for this stenotic cardiovascular disease: angioplasty and bypass surgery1. In either case, post-procedure IH develops which could eventually re-occlude the vessel lumen52. Drug-eluting stents represent a major medical advance in ameliorating post-angioplasty IH52. However, despite decades of investigation, there remains a lack of clinical methods to prevent post-operative failure of bypass and other open vascular reconstructions such as arteriovenous fistula3, 11. This underscores that ultimate success requires synergistic innovations, not only in therapeutic targets but also in suitable drug delivery methods. To this end, the unimolecular micelle nanoparticle, which is minute in mass yet highly amenable to modifications to fulfill diverse functions, has been advanced as a promising drug delivery platform24. On another front, epi-drugs emerge as a new thrust in preclinical and human trials, though mainly in treating cancers at present15. Dysregulated epigenetic homeostasis is increasingly recognized as a root problem for many pathological conditions. Epi-drugs confer an approach to restoring epigenetic homeostasis by dialing down whole sets of overactive genes without altering DNA sequences – one that is reversible and hence relatively safe17. Our current study was designed to combine epigenetic targeting with nanotechnology to tackle the issue of the persistent lack of clinical methods for the prevention of IH and failure of open vascular reconstructions.
As recently reviewed11, 13, the peri- or epi-vascular route of drug delivery is suitable for open vascular reconstructions taking advantage of exposed vascular or prosthetic conduits during surgery. However, given the limited time window of invasive open surgeries such as bypass, a clinically viable method should be not only efficacious, non-invasive, and non-toxic, but also easy and quick to apply. With this in mind, we conceived the Epi^NanoPaint design using ahNP that can be simply pen-brushed on the adventitia of a reconstructed vessel. This method incorporates multiple merits: 1) It is easy, quick (<1 min), and gentle imposing little mechanical stress on the vascular vessel. 2) The covalently bonded unimolecular micelle is resistant to disassembly and hence durable. Though minute in mass, it is highly capable of drug loading (17.5 wt% for pinometostat and 19.1 wt% for rapamycin)24. 3) The hydrophilic sulfo-NHS ester terminal groups render ahNP adhesive by forming stable amide bonds with the NH2 functional groups of adventitial ECM proteins. 4) The byproduct sulfo-NHS is highly soluble34, thus readily removable with absorbents such as gauze. 5) Compared to the conventional perivascular gel method22, Epi^NanoPaint reduced the IH-mitigating effective dose of pinometostat by 100 fold (0.3 mg/kg vs 30 mg/kg)22. A rationale is that the ahNP immobilized on the adventitial ECM may function like “drip irrigation”, releasing the drug locally, slowly, and durably without needing large amounts of the drug. In contrast, the free drug (no ahNP) that “flooded” the vessel adventitia failed to produce an IH-mitigating effect. 6) Importantly, ahNP is amenable to modifications (e.g. size, drug release kinetics, and Cy5 tag, etc.) and hence multifunctionality, providing great potential for further optimization in broad translational applications.
Although the Epi^NanoPaint method appears simple, there has been a lack of reports of using adhesive nanoplatforms for the treatment of IH29. Various tissue-adhesive molecules have been explored for other applications (e.g. skin wound healing), but there are shortcomings with such approaches. For instance, the aldehyde is associated with fibrotic toxicity53. Fibronectin adheres to collagen non-covalently54 thus unstably. Catechol-based adhesives are attractive biomaterials. However, the underlying Dopa chemistry submits to microenvironmental conditions, e.g. redox, pH, metal ion chelating, and surface drying, etc55. We chose the sulfo-NHS ester, a moiety widely applied in the pharmaceutical protein industry56, for its robust reactivity with the NH2 group abundant on the adventitial ECM34, the stability of the resulting amide bond, and sulfo-NHS as the byproduct that is non-toxic and nonetheless easy to remove56, 57. Indeed, we did not detect obvious local tissue toxicity of Epi^NanoPaint. Rather, the Pino/ahNP formulation (vs empty ahNP) reduced pro-inflammatory and pro-apoptotic marker proteins (CD68 and active caspase 3) that are often used to monitor tissue damage29.
Another important consideration of toxicity is the effect on the endothelium. A healthy endothelial lining stakes off pathogenic cells and cytokines from infiltrating into the inner vessel wall. Dysfunctional ECs, however, become non-proliferative/apoptotic and fervent producers of thrombogenic factors and inflammatory cytokines that exacerbate SMC phenotypic instability and neointimal growth30. Therefore, an ideal IH-preventing agent should avert not only SMC phenotypic changes but also EC dysfunction58. In choosing an IH-inhibiting therapeutic agent, rapamycin would appear as the top choice because it is clinically used on drug-eluting stents to mitigate post-angioplasty restenosis. However, despite the many therapeutic benefits of rapamycin observed in various disease models, concerns arise from increasing evidence that rapamycincoated stents can harm the endothelium and heighten thrombotic risks30, 41, 42. Similarly, Paclitaxel, another popular drug clinically used on stents, has been reportedly associated with mortality in some stent recipients59. Consistently, our data showed that rapamycin-loaded ahNP impeded the growth of the endothelium in rats. Compared to the therapeutic dosages of 1–2 mg/kg/day in the literature27, 28, 31, 40, the dose of rapamycin in ahNP applied on the adventitia was very low (0.018 mg/kg, one time), and the actual amount of the drug that reached the endothelium by traversing the vessel wall could be even lower. Yet endothelial toxicity still occurred. Therefore, these lines of evidence underscore the imperative to circumvent the limitations of these indiscriminately antiproliferative drugs, via studies on more specific interventional agents. Along this line, previous reports showed that the BETs-selective inhibitor JQ1 disrupted neointimal formation without harming the endothelium26, 44, 60. Herein, we found yet another epi-drug, pinometostat, was nontoxic to the recovering endothelium in the IH model of arterial injury. More detailed future studies are needed to better understand why the pharmacological intervention using DOT1L is non-toxic to the endothelium.
DOT1L is unique in that it is the only known writer of the methylation at H3K79, and unlike the modification sites densely packed on the flexible histone tail, H3K79 is situated in the wellstructured H3 globular domain21. Overall, the biological function and regulations of DOT1L, especially in cardiovascular disease, remain poorly understood23. In this regard, our data indicating BRD4 as an upstream determinant of DOT1L expression presents new knowledge. In a recent report, BRD4 and DOT1L were found to exist in separate protein complexes while acting in synergy to activate highly transcribed genes in leukemia cells48. However, it was not known whether the BRD4->DOT1L hierarchical regulation was involved. Another novel regulation we identified here is the control of AURKB’s expression by DOT1L. AURKB is involved in numerous cellular events, the most prominent being chromosome segregation in mitosis49. Recently, AURKB was found to phosphorylate S28 and S10 on the histone-3 tail, hence a novel epigenetic writer50. Herein we first observed that AURKB inhibition with barasertib attenuated IH and SMC phenotypic transition. In accordance, DOT1L and AURKB both were upregulated in human and rat arteries that underwent IH, and deactivating DOT1L abated IH. Therefore, the next important question was how DOT1L governed AURKB levels. Our results provided new insight into the DOT1L-associated epigenetic mechanism. That is, while the H3K79me2 mark enriched at Aurkb, as revealed by ChIP-qPCR, the H3K79me2 writer DOT1L associated (co-IPed) with the gene-activating mark H3K27ac that enriched at Aurkb as well, as evidenced by ChIP-seq. This result is in line with the reports of DOT1L in complex with the central transcription machinery48. Moreover, given the recent evidence for DOT1L’s role in maintaining enhancer/promoter interactions61, more intriguing and nuanced epigenetic mechanisms await further exploration. In an overview, interest in epigenetic remodeling factors is surging yet these proteins have been mostly studied separately, and the understanding of their functional interplay has lagged45, 62. Herein we identified a previously uncharacterized functional epigenetic axis, namely, BRD4->DOT1L->AURKB. Because of the complexity of disease mechanisms and the intricate chromatin architecture and regulations, further investigation into this axis could contribute to the precise targeting of the IH pathogenesis for optimal therapeutic outcomes.
Study limitations
Previous reports demonstrated that an IH-mitigating efficacy of drug-loaded nanoplatforms extends much beyond the end of drug release24, 37, likely due to the importance of early-stage pathogenesis in long-term disease progression. It is thus expectable that Pino-containing Epi^NanoPaint could be capable of IH mitigation beyond the drug release period of ~3 months. Therefore, while our study showed one-month efficacy of the test in obese Zucker rats, the full potential of this Pino/ahNP formulation is not known at present. As another limitation of our study, it is not clear whether a long-release formulation alone is sufficient, or whether a mixture of longrelease and short-release ahNP formulations is required for prolonged efficacy. Answering these questions would require future tests in obese Zucker rats over a longer period. In addition, given the advantage that Epi^NanoPaints loaded with different drugs (e.g. inhibitors to DOT1L and AURKB) can be conveniently mixed and applied, the potential of combination therapy using this nanosystem deserves further exploration. Nevertheless, our work presents a prototype for future translational development.
Conclusions
Using pinometostat as a model drug for epigenetic targeting and Epi^NanoPaint as a modular platform for drug delivery, we present here a proof of concept that uniquely caters to open vascular reconstructions. This nanosystem showed IH-abating efficacy in obese Zucker rats that mimic human-like disease backgrounds. Furthermore, from the mechanistic perspective, we identified BRD4 as an upstream determinant of the expression of DOT1L and AURKB as a downstream effector for its pro-IH function. Thus, this study lays the basis for further mechanistic understanding and translational advances to tackle persistent, IH-associated recurrent vascular disease conditions.
Methods
Materials
Various resources, including kits and reagents, are presented in Table S1 or included in the corresponding texts below.
Pinometostat and rapamycin were purchased from LC Laboratories (Woburn, MA). MethoxyPEG-COOH (mPEGNH2, Mn = 5 kDa) and NH2-PEG-COOH (Mn = 5 kDa) were obtained from JenKem Technology USA Inc. (Plano, TX, USA). Cyanine5-N-hydroxysuccinimide ester (Cy5NHS ester) dye was obtained from Lumiprobe Corporation (Hunt Valley, MD, USA). Succinic anhydride, tin(II) 2-ethylhexanoate (Sn(Oct)2), and 4-dimethylamino pyridine (DMAP) were purchased from Thermo Fisher scientific (Waltham, MA, USA). Anhydrous pyridine was acquired from EMD Millipore Corporation (Billerica, MA, USA). Other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Synthesis schemes of Epi^NanoPaint using adhesive nanoparticles (ahNP)
Synthesis of Cy5-PEG-COOH, and Boc-protected amine-PEG-COOH
To prepare Cy5-PEG-COOH, Cy5-NHS ester (9.80 mg, 15.0 μmol) was conjugated with HOOCPEG-NH2 (50 mg, 10.0 μmol) in dimethylformamide (DMF) (10 ml). The reaction was stirred at room temperature for 24 h in the dark. Thereafter, it was purified by dialysis using a RC dialysis bag (MWCO 1 kDa) against DI water for 24 h. The product was obtained after lyophilization under vacuum. To synthesize HOOC-PEG-NHBoc, di-tert-butyl decarbonate ((Boc)2O) (157.14 mg, 0.72 mmol) and HOOC-PEG-NH2 (400 mg, 0.08 mmol) were dissolved in dichloromethane (DCM) (20 ml). The reaction was stirred for 24 h before precipitation in cold ethyl ether and the precipitate was lyophilized for 48 h.
Synthesis of PAMAM-PCL-OH and PAMAM-PCL-PEG-OCH3/Cy5/NH2 (NP-NH2)
PAMAM-PCL-OH was first synthesized through ring-opening polymerization as previously reported24 with some modifications using PAMAM-OH (4th generation, 23.69 mg, 1.67 μmol), εcaprolactone (11.81 mg, 4.76 mmol), and Sn(Oct)2, (1.54 μl, 4.76 μmol). The reaction was stirred at 120°C under N2 atmosphere for 24 h. The crude product was precipitated in cold methanol using a centrifuge (10,000 ×g, 10 min). The precipitation step was repeated 3 times, followed by lyophilization of the obtained product for 48 h.
Next, NP-NHBoc was prepared via esterification24. PAMAM-PCL-OH (50 mg, 0.43 μmol, Mw 116 kDa), mPEG-COOH (91.63 mg, 18.3 μmol), Cy5-PEG-COOH (30.54 mg, 6.11 μmol), HOOC-PEG-NHBoc (122.17 mg, 24.43 μmol), DMAP (6.72 mg, 54.98 μmol), and 1-ethyl-3-(3dimethylaminopropyl) carbodiimide (EDC) (5.27 mg, 27.49 μmol) were first dried under vacuum for 30 min, followed by adding DMF (10 ml) to dissolve all reagents. The reaction was stirred at room temperature for 48 h under N2 atmosphere in the dark and purified via dialysis against DI water using a regenerated cellulose (RC) dialysis bag (molecular weight cut-off (MWCO) 8 kDa) for 24 h. The product was then lyophilized for 48 h.
To deprotect/cleave the Boc protecting groups, NP-NHBoc (200 mg, 18.3 μmol) was dissolved in dichloromethane DCM (15 ml). TFA (15 ml) was added into the solution dropwise and the reaction was stirred at room temperature for 2 h in the dark. To remove TFA, the reaction was connected to a rotary-evaporator to evaporate TFA. The crude product was then purified by precipitation in cold ethyl ether using centrifuge (10,000 g, 10 min) and repeated it 3 times before lyophilization to obtain NP-NH2.
Synthesis of PAMAM-PCL-PEG-OCH3/Cy5/Sulfo-NHS ester (ahNP)
NP-NH2 (250 mg, 1.01 μmol) and succinic anhydride (38.77 mg, 387.44 μmol) were dissolved in anhydrous DCM (20 ml) before adding DMAP (142.00 mg, 1.16 mmol)26. The reaction was stirred at room temperature for 48 h under N2 atmosphere in the dark. To collect the product, the reaction was precipitated in cold diethyl ether via centrifugation (10,000 ×g, 10 min). The precipitation step was repeated 3 times. The precipitate was purified by dialysis against DI water using a RC dialysis bag (MWCO 15 kDa) for 48 h and lyophilized under vacuum to produce NP-COOH. Next, NPCOOH (250 mg, 1.01 μmol), EDC (37.14 mg, 193.72 μmol), and sulfo-NHS (44.59 mg, 387.44 μmol) were dried under vacuum for 30 min. DMF (15 ml) was then added to dissolve all reagents and the reaction was stirred at room temperature for 2 h in the dark. To purify the reaction, it was dialyzed against DI water using a RC dialysis bag (MWCO 15 kDa) for 24 h and lyophilized under vacuum for 48 h to obtain the ahNP (MW 276 kDa).
Preparation of drug-loaded ahNP
ahNP (50 mg) and pinometostat (15 mg) were dissolved in DMSO (10 ml). DI water (30 ml) was added dropwise into the mixture, followed by dialysis against DI water using a RC dialysis bag (MWCO 15 kDa) for 24 h before lyophilization through a standard dialysis method32. Rapamycinloaded ahNP was prepared using the same method.
Characterization of ahNP
The chemical structures and molecular weights were determined (Figure S2, A–F) by the 1H NMR spectroscopy (Bruker Avance-400 MHz), and gel permeation chromatography equipped with triple detectors (GPC, Viscotek, Malvern), respectively26. The NMR samples were prepared in deuterated chloroform (CDCl3) (~5 mg/ml) while the samples for GPC analysis were dissolved in DMF (1.5 to 5.0 mg/ml). Fourier-transform infrared (FTIR) spectroscopy (Tensor 27, Bruker) was used to identify the hydroxyl in PAMAM-PCL-OH, carboxylic acid at the terminal end in NPCOOH, and sulfo-NHS functional groups in ahNP. The hydrodynamic diameter and zeta potential of PAMAM-PCL-OH and UM-NHS were measured using a DLS machine (Zetasizer NanoZS90, Malvern). For diameter quantification, PAMAM-PCL-OH was suspended in chloroform (0.02 mg/ml), while ahNP was dispersed in DI water (0.02 mg/ml). For zeta potential measurement, the ahNP was suspended in DI water (0.02 mg/ml) (Figure 1B). Additionally, the morphology of ahNP was observed under TEM (FEI Tecnai T12) using phosphotungstic acid (PTA) as a negative stain. To prepare the TEM sample, the ahNP solution (0.05 mg/ml in DI water) was dropped onto a TEM grid (Formvar/Carbon coated copper grids, 200 mesh). The grid is air-dried, and a drop of PTA solution (1%, pH 7) was then added to the grid. After 30 seconds, the excessive PTA solution was removed by filter paper, and the grid was air-dried again. Pinometostat and rapamycin loading content/efficiency were quantified by HPLC (Elite LaChrom, Hitachi) at 255 and 278 nm, respectively, using 0.5 mg/ml of drug-loaded ahNP in DMSO26.
Evaluation of pinometostat release from ahNP in vitro
The pinometostat release profiles from ahNP were measured following the reported protocol26 with some modifications. ahNP (5 ml, 5 mg/ml in PBS, pH 7.4) was enclosed in a RC dialysis bag (MWCO 8 kDa). The dialysis bags were then immersed in PBS (20 ml, pH 7.4) with 0.2% tween80 and kept in a horizontal shaker (60 rpm) at 37°C. At the certain time points mentioned in the figure legend, supernatants were collected and replaced with fresh media at the same volume. The amounts of pinometostat in the supernatants were quantified by HPLC at 255 nm.
Animals
We performed all animal experiments according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) with the protocols approved by the Institutional Animal Care and Use Committee of the University of Virginia. Male Sprague-Dawley rats weighing 300–330 g or male Zucker obese rats at the age of 8–9 weeks were purchased from Charles River Laboratories (Wilmington, MA). These rats were used for surgery at the age of 10–11 weeks. They were housed in an air-conditioned room with a 12 h–12 h light-dark cycle in isolation racks, fed with a normal diet and water without any restrictions.
Animal model of neointimal hyperplasia (IH)
An experienced animal handler performed all surgeries as previously reported. In brief, after inhalation of isoflurane for anesthesia, a skin incision was made in the midline of the neck, and left carotid arteries were dissected. Through an arteriotomy on the external carotid artery, a 2Fr. Fogarty catheter was inserted until reaching the root of the common carotid artery (CCA). The CCA wall was injured and the endothelial inner lining was damaged by retrieving the balloon catheter (inflated at 1.5 atm) 4 times. The external carotid artery and its branches were ligated, and blood flow was resumed toward the internal carotid artery, and Epi^NanoPaint was applied immediately. We used a pen brush to dip into the solution of drug-loaded ahNP and brushed it on the adventitia of the injured CCA that was gently wiped with gauze beforehand to remove body fluid. In control groups, the same action was taken to paint empty ahNP and free drug solutions. The application of Epi^NanoPaint took less than a minute. After painting, excessive solutions were removed with gauze. In separate experiments, an AURKB-selective (barasertib) or AURKAselective (alisertib) inhibitor (each 10 mg/kg) or an equal amount of DMSO (vehicle control) was dispersed in mixed thermosensitive hydrogels (200 μl Triblock gel, AK12, Akina Inc., IN, and 200 μl Pluronic gel, Sigma, St. Louis, MO). The mix was applied around the balloon-injured carotid artery. The surgery was then finished, and the neck skin was closed. The rats were observed until full recovery. The analgesic plan included slow-release bupivacaine (up to 2 mg/kg) and carprofen (5 mg/kg). At day 28 or day 14 (for Zucker rats and Sprague-Dawley rats, respectively), rats were euthanized and CCAs were collected after perfusion fixation with 4% paraformaldehyde (PFA). The arterial specimens were fixed in 4% PFA for 12–24 h, paraffin-embedded, and sectioned for histological staining and morphometric analysis.
Morphometric analysis
Two in the rapamycin-loaded UM and one in the free rapamycin group of Sprague-Dawley rats were excluded due to suboptimal histology intractable for quantification. One in the Pino-loaded ahNP group of Zucker rats was excluded because of unsuccessful surgery. Each of the collected CCAs was cut into 3–4 pieces before embedding and slides were cut at 5-μm thick intervals. The slides were stained for Hematoxylin & Eosin staining and Masson’s trichrome staining. We used Image J software (NIH, Bethesda, MD, USA) for morphometric analysis. The Lumen area, lumen perimeter, the area inside the internal elastic lamina (IEL), IEL perimeter, the area inside the external elastic lamina (EEL), and the EEL perimeter were measured. The neointima/media ratio (N/M ratio) was calculated as (IEL area - lumen area) / (EEL area – IEL area) and stenosis rate was (IEL area - lumen area) / IEL area. The average of 3–4 levels was calculated in each animal.
We also quantified collagen density based on Masson’s trichrome staining performed at the same time. The color images were converted into 8-bit scale images, and integrated density below the specific threshold consistent through the analysis was evaluated. Values from 3–4 slides were averaged for each animal.
Immunofluorescence
The assay was performed following our reported protocol22. Briefly, artery sections were incubated with a primary antibody (Table S2) overnight, followed by rinsing and incubating with an antirabbit/mouse secondary antibody conjugated with Alexa Fluor 594 (A-11037/A-21203, Invitrogen, Carlsbad, CA), and/or Alexa Fluor 488 (ab150117, Abcam). DAPI was used to stain the nuclei. Fluorescence microscopy was applied to image the specific staining of the protein of interest. For quantification (using ImageJ), 3–4 immunostained sections from each animal were used. Fluorescence intensity was normalized to the number of DAPI-stained nuclei in the media and neointima layers. The values from all sections were pooled to generate the mean for each animal. The means from all animals in each group were then averaged, and the final mean (± SEM) was calculated.
Re-endothelialization
Endothelial cell recovery (reendothelialization) after arterial injury was evaluated on day 7 as previously described26. In brief, balloon injury was performed 8mm shorter than the general procedure. Evans blue solution (0.5%, 1mL) was injected through the tail vein 30 min prior to euthanasia. Rats were subsequently perfused with saline and the entire CCAs were collected. The area stained for blue was considered not covered with endothelial cells, while the unstained area (white) was evaluated as covered with endothelial cells. The baseline length of the white area was determined immediately after injury (n = 3) in a separate experiment and subtracted from the length measured on day 7 models (n = 9, each group). Percent reendothelialization was calculated and compared between the groups.
Artery tissue ChIP sequencing and data processing
The experiments were described in our recent report45. Briefly, the IH-inducing balloon injury of rat common carotid arteries was performed as described above, and 7 days later the injured (and the contralateral uninjured) arteries were collected and snap-frozen in liquid N2. Artery tissues from 50 rats were pooled for ChIP-seq analysis at Active Motif per company standard procedures45. Genomic DNA was sheared to an average length of 300–500 bp by sonicating the lysates, and the segments of interest were immunoprecipitated using an antibody (4 μg) against BRD4, H3K27ac, H3K27me3, or H3K4me1. Purified ChIP DNA was used in the preparation of Illumina sequencing libraries. Standard steps included end-polishing, dA-addition, adaptor ligation, and PCR amplification. The DNA libraries were quantified and sequenced on Illumina’s NextSeq 500. Sequence reads were aligned to the reference genome Rno5, peak locations were identified using Macs2 algorithm63 and annotated based on UCSC RefSeq. Differential peak locations were called using SICER64. In-house shell and R scripts (https://www.r-project.org) were used for data integration. IGV (http://www.broadinstitute.org/igv/) was used for visualization. Data is available through GEO with accession number GSE194390.
Cell culture
The mouse aortic SMC line (MOVAS), human primary aortic SMCs, and human umbilical vein endothelial cells (HUVEC) were purchased from ATCC (Manassas, VA, USA). SMCs were cultured in DMEM high-glucose full medium (catalog no. [cat.] 11965092; ThermoFisher) supplemented with 10% FBS and 50 μg/mL G418 (antibiotic, cat. 4727878001; Sigma-Aldrich) at 5% CO2 and 37°C. Primary rat aortic SMCs were isolated as recently described65 with minor modifications66. The cells were cultured in smooth muscle complete (full) medium (cat. M2268; Cell Biologics) and used at passage 5.
Proliferation assay
The CellTiter-Glo viability assay was used as we described previously44. SMCs were seeded in 96-well plates at a density of 2000 cells per well and starved for 24 h in DMEM containing 0.5% FBS. Prior to the mitogenic stimulation with PDGF-BB (human recombinant, R&D Systems Inc., MN), cells were pretreated with the AURKB-selective inhibitor barasertib for 2h. At the end of PDGF-BB treatment, plates were decanted, re-filled with 50 μl CellTiter-Glo reagent/50 μl PBS per well, incubated for 10 min, and then read in FlexStation 3 Benchtop Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) using a 250 ms integration.
Migration assay
The scratch wound assay was performed44. Briefly, SMCs were cultured to a 90% confluency in 6-well plates and then starved for 24 h in DMEM medium containing 0.5% FBS. Barasertib was added and incubated for 2h. A sterile pipette tip was used to generate an ~ 1 mm cell-free gap. Dislodged cells were washed away with PBS, and the plates were refilled with fresh medium containing 20 ng/ml PDGF-BB and incubated for 24 h. At the end of the treatment, for illumination of the cells, Calcein AM was added at a final concentration of 2 μM. After a 30-min incubation, the plates were washed with PBS, and images were taken. Cell migration was quantified by Image J based on the change of cell-free area before and after PDGF-BB stimulation.
Gene silencing
We used lentivirus to express shRNAs in MOVAS cells to knock down genes specifically, as we previously described65. Gene silencing in rat primary SMCs was done by using small interfering RNAs (siRNAs). Transfection was kept for 24 h by incubating cells with Lipofectamine RNAiMAX Transfection Reagent (Cat. 13778075; ThermoFisher Scientific). Sequences of shRNAs and siRNAs are listed in Table S3.
Immunoblotting (IB)
Cells were harvested and lysed on ice in the RIPA buffer (cat. 89900; ThermoFisher Scientific) that includes a Halt protease and phosphatase inhibitor cocktail (cat. 78440; ThermoFisher Scientific). Cell lysates were quantified for protein concentrations using the Bio-Rad DC Protein Assay kit (cat. 5000112; Bio-Rad) prior to loading to a 5–12% SDS-PAGE gel. The separated proteins were transferred to a polyvinylidene fluoride (PVDF) membrane, which was incubated with a primary antibody (see list in Table S5) followed by rinses of the membrane and incubation with an HRP-conjugated secondary antibody. The signals from immunoblots were recorded with Amersham ImageQuant™ 800 immunoblot imaging systems (GE Healthcare Life Sciences). Protein band densitometry was quantified using National Institutes of Health (NIH) ImageJ and normalized to loading control for statistical analysis. Antibodies are included in Table S2.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from cultured cells using the TRIzol reagent (cat. 15596018; ThermoFisher Scientific). gDNA Eliminator columns were used to eliminate potential contaminating genomic DNA. RNA was quantified with a NanoDrop NP-1000 spectrometer, and 1 μg was used for the first-strand cDNA synthesis. qRT-PCR was run in Quant Studio 3 (Applied Biosystems, Carlsbad, CA, USA). The housekeeping gene GAPDH was used for normalization using the ΔΔCt method. Each cDNA template was amplified in triplicate reactions using PerfeCTa SYBR Green SuperMix (cat. 95054; Quantabio) with gene-specific primers listed in Table S4.
Chromatin immunoprecipitation qPCR (ChIP-qPCR)
As we recently reported45, cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Following the termination of the reaction, cells were washed with ice-cold PBS and lysed in the buffer containing 10 mM Tris-HCl (pH 7.9), 0.25% Triton X-100, 10 mM EDTA, and protease inhibitors. The lysate was pelleted by centrifugation at 4,000 rpm for 5 min, re-suspended in the nuclei extraction buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 mM EDTA, and protease inhibitors), and then sonicated (forty 30-s pulses at 20 W per 1×107 cells) to disrupt the nuclear membrane. Chromatin extracts containing DNA fragments sheared by sonication were IPed with a specific antibody (or nonspecific IgG) overnight at 4°C. ChIP-grade Protein A/G Magnetic beads were added and incubated for 4h at 4°C. RNA and proteins were digested with RNase A (5 U) and Proteinase K (2 μg/μL), and purified DNA was used for qPCR with primers (Table S5) that detect the Aurkb region from −1154bp to −1028bp.
Co-immunoprecipitation (co-IP)
The procedures were described in our recent reports67, 68. The Pierce Crosslink Immunoprecipitation kit (Thermo Scientific, 26147) was used. In brief, MOVAS cell lysates were prepared (see above) and centrifuged at 13,200 × g for 15 min at 4°C. Nuclei were collected, sonicated, and cleared by centrifugation. The samples were incubated at 4°C for 4 h with Magnetic beads (Dynabeads Protein A or G, Invitrogen) that were preloaded with an antibody specific for H3K27ac. The beads were washed 3× with the binding buffer (50 mM Tris-Cl, 150 mM NaCl, 1 mM EDTA, 10% glycerin), and SDS sample buffer was then added to elute the co-IPed proteins for immunoblot determination of endogenous DOT1L.
Statistical analysis
Normality of data distribution was checked by Shapiro–Wilk test. One-way analysis of variance (ANOVA) was performed for multiple group comparison followed by a post hoc test (specified in figure legends). Two-group comparison was analyzed with Student’s t-test. Data is presented as mean ± standard error of the mean (SEM) or ± standard deviation (SD). A difference with a p value < 0.05 is considered significant.
Supplementary Material
Acknowledgments
We thank Dr. Todd Stukenberg for the discussions on AURKB. We also thank Dr. Ziv Haskal for reviewing the manuscript and providing suggestions.
Funding
This work was supported by NIH R01 grants HL133665 (to L.-W.G.), R01HL143469, R01HL129785 (to K.C.K, S.G., and L.-W.G.), and R01HL162895 (to B.W.). The work was also supported by AHA predoctoral fellowship award#903227/2022 (grant#2021AHA000PRE0000216149, to Y.H.), post-doctoral fellowship award 20POST35210967 (to M.Z.) and Overseas Research Fellowships, the Uehara Memorial Foundation in Japan (to T.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit author statement
Takuro Shirasu: Investigation, Methodology, Writing - Original Draft
Nisakorn Yodsanit: Investigation, Methodology, Writing - Original Draft
Jing Li: Investigation, Methodology
Yitao Huang: Investigation, Methodology
Xiujie Xie: Investigation, Methodology
Runze Tang: Investigation, Methodology
Qingwei Wang: Investigation, Methodology
Mengxue Zhang: Investigation, Methodology
Go Urabe: Investigation, Methodology
Amy Webb: Investigation, Methodology
Yuyuan Wang: Investigation,
Xiuxiu Wang: Investigation,
Ruosen Xie: Conceptualization, Investigation
Bowen Wang: Conceptualization, Investigation
K. Craig Kent: Supervision
Shaoqin Gong: Writing - Review & Editing, Supervision
Lian-Wang Guo: Writing - Review & Editing, Supervision
Declaration of interests
None
References
- 1.de Vries MR, Simons KH, Jukema JW, Braun J and Quax PH. Vein graft failure: from pathophysiology to clinical outcomes. Nat Rev Cardiol. 2016;13:451–70. [DOI] [PubMed] [Google Scholar]
- 2.Head SJ, Milojevic M, Daemen J, Ahn JM, Boersma E, Christiansen EH, Domanski MJ, Farkouh ME, Flather M, Fuster V, Hlatky MA, Holm NR, Hueb WA, Kamalesh M, Kim YH, Makikallio T, Mohr FW, Papageorgiou G, Park SJ, Rodriguez AE, Sabik JF 3rd, Stables RH, Stone GW, Serruys PW and Kappetein AP. Mortality after coronary artery bypass grafting versus percutaneous coronary intervention with stenting for coronary artery disease: a pooled analysis of individual patient data. Lancet. 2018;391:939–948. [DOI] [PubMed] [Google Scholar]
- 3.Lawson JH, Niklason LE and Roy-Chaudhury P. Challenges and novel therapies for vascular access in haemodialysis. Nat Rev Nephrol. 2020;16:586–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodney PP, Beck AW, Nagle J, Welch HG and Zwolak RM. National trends in lower extremity bypass surgery, endovascular interventions, and major amputations. J Vasc Surg. 2009;50:54–60. [DOI] [PubMed] [Google Scholar]
- 5.Wu W, Wang C, Zang H, Qi L, Azhar M, Nagarkatti M, Nagarkatti P, Cai G, Weiser-Evans MCM and Cui T. Mature Vascular Smooth Muscle Cells, but Not Endothelial Cells, Serve as the Major Cellular Source of Intimal Hyperplasia in Vein Grafts. Arterioscler Thromb Vasc Biol. 2020;40:1870–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trocha KM, Kip P, Tao M, MacArthur MR, Trevino-Villarreal JH, Longchamp A, Toussaint W, Lambrecht BN, de Vries MR, Quax PHA, Mitchell JR and Ozaki CK. Shortterm preoperative protein restriction attenuates vein graft disease via induction of cystathionine gamma-lyase. Cardiovasc Res. 2020;116:416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi EA, Kilari S and Misra S. Novel Clinical Therapies and Technologies in Dialysis Vascular Access. Kidney360. 2021;2:1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel NJ, Bavishi C, Atti V, Tripathi A, Nalluri N, Cohen MG, Kini AS, Sharma SK, Dangas G and Bhatt DL. Drug-Eluting Stents Versus Bare-Metal Stents in Saphenous Vein Graft Intervention. Circ Cardiovasc Interv. 2018;11:e007045. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein DJ, Puskas JD, Alexander JH, Chang HL, Gammie JS, Marks ME, Iribarne A, Vengrenyuk Y, Raymond S, Taylor BS, Yarden O, Orion E, Dagenais F, Ailawadi G, Chu MWA, DiMaio JM, Narula J, Moquete EG, O’Sullivan K, Williams JB Jr., Crestanello JA, Jessup M, Rose EA, Scavo V, Acker MA, Gillinov M, Mack MJ, Gelijns AC, O’Gara PT, Moskowitz AJ, Bagiella E and Voisine P. External Support for Saphenous Vein Grafts in Coronary Artery Bypass Surgery: A Randomized Clinical Trial. JAMA Cardiol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conte MS, Bandyk DF, Clowes AW, Moneta GL, Seely L, Lorenz TJ, Namini H, Hamdan AD, Roddy SP, Belkin M, Berceli SA, DeMasi RJ, Samson RH, Berman SS and Investigators PI. Results of PREVENT III: a multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J Vasc Surg. 2006;43:742–751; discussion 751. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhary MA, Guo LW, Shi X, Chen G, Gong S, Liu B and Kent KC. Periadventitial drug delivery for the prevention of intimal hyperplasia following open surgery. J Control Release. 2016;233:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu B, Werlin EC, Chen M, Mottola G, Chatterjee A, Lance KD, Bernards DA, Sansbury BE, Spite M, Desai TA and Conte MS. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rabbit vein graft model. J Vasc Surg. 2018;68:188S–200S e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mylonaki I, Allemann E, Saucy F, Haefliger JA, Delie F and Jordan O. Perivascular medical devices and drug delivery systems: Making the right choices. Biomaterials. 2017;128:56–68. [DOI] [PubMed] [Google Scholar]
- 14.Feinberg AP. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N Engl J Med. 2018;378:1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamdani N, Costantino S, Mugge A, Lebeche D, Tschope C, Thum T and Paneni F. Leveraging clinical epigenetics in heart failure with preserved ejection fraction: a call for individualized therapies. Eur Heart J. 2021;42:1940–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM and Martin KA. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc Sci. 2021;2:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borck PC, Guo LW and Plutzky J. BET Epigenetic Reader Proteins in Cardiovascular Transcriptional Programs. Circ Res. 2020;126:1190–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty R, Ostriker AC, Xie Y, Dave JM, Gamez-Mendez A, Chatterjee P, Abu Y, Valentine J, Lezon-Geyda K, Greif DM, Schulz VP, Gallagher PG, Sessa WC, Hwa J and Martin KA. Histone Acetyltransferases p300 and CBP Coordinate Distinct Chromatin Remodeling Programs in Vascular Smooth Muscle Plasticity. Circulation. 2022;145:1720–1737. [DOI] [PubMed] [Google Scholar]
- 19.Worden EJ, Hoffmann NA, Hicks CW and Wolberger C. Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell. 2019;176:1490–1501 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarno F, Nebbioso A and Altucci L. DOT1L: a key target in normal chromatin remodelling and in mixed-lineage leukaemia treatment. Epigenetics. 2020;15:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood K, Tellier M and Murphy S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Urabe G, Zhang M, Li J, Ozer HG, Wang B, Kent KC and Guo LW. Nullifying epigenetic writer DOT1L attenuates neointimal hyperplasia. Atherosclerosis. 2020;308:2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farina FM, Serio S, Hall IF, Zani S, Cassanmagnago GA, Climent M, Civilini E, Condorelli G, Quintavalle M and Elia L. The epigenetic enzyme DOT1L orchestrates vascular smooth muscle cell-monocyte crosstalk and protects against atherosclerosis via the NF-kappaB pathway. Eur Heart J. 2022. [DOI] [PubMed] [Google Scholar]
- 24.Chen G, Shi X, Wang B, Xie R, Guo LW, Gong S and Kent KC. Unimolecular MicelleBased Hybrid System for Perivascular Drug Delivery Produces Long-Term Efficacy for Neointima Attenuation in Rats. Biomacromolecules. 2017;18:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meschaninova MI, Novopashina DS, Semikolenova OA, Silnikov VN and Venyaminova AG. Novel Convenient Approach to the Solid-Phase Synthesis of Oligonucleotide Conjugates. Molecules. 2019;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang B, Chen G, Urabe G, Xie R, Wang Y, Shi X, Guo LW, Gong S and Kent KC. A paradigm of endothelium-protective and stent-free anti-restenotic therapy using biomimetic nanoclusters. Biomaterials. 2018;178:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parlar A, Can C, Erol A and Ulker S. Posttransplantation therapeutic rapamycin concentration protects nitric oxide-related vascular endothelial function: comparative effects in rat thoracic aorta and coronary endothelial cell culture. Transplant Proc. 2010;42:192–330. [DOI] [PubMed] [Google Scholar]
- 28.Jahnke T, Schafer FK, Bolte H, Heuer G, Karbe U, Brossmann J, Brandt M, Heller M and Muller-Hulsbeck S. Short-term rapamycin for inhibition of neointima formation after balloon-mediated aortic injury in rats: is there a window of opportunity for systemic prophylaxis of restenosis? Journal of endovascular therapy : an official journal of the International Society of Endovascular Specialists. 2005;12:332–42. [DOI] [PubMed] [Google Scholar]
- 29.Shirasu T, Yodsanit N, Xie X, Zhao Y, Wang Y, Xie R, Huang Y, Wang B, Urabe G, Gong S, Guo LW and Kent KC. An adventitial painting modality of local drug delivery to abate intimal hyperplasia. Biomaterials. 2021;275:120968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofma SH, van der Giessen WJ, van Dalen BM, Lemos PA, McFadden EP, Sianos G, Ligthart JM, van Essen D, de Feyter PJ and Serruys PW. Indication of long-term endothelial dysfunction after sirolimus-eluting stent implantation. Eur Heart J. 2006;27:166–70. [DOI] [PubMed] [Google Scholar]
- 31.Tigerstedt NM, Aavik E, Lehti S, Hayry P and Savolainen-Peltonen H. Mechanisms behind the synergistic effect of sirolimus and imatinib in preventing restenosis after intimal injury. Journal of vascular research. 2009;46:240–52. [DOI] [PubMed] [Google Scholar]
- 32.Chen G, Jaskula–Sztul R, Harrison A, Dammalapati A, Xu W, Cheng Y, Chen H and Gong S. KE108-Conjugated Unimolecular Micelles Loaded with a Novel HDAC Inhibitor Thailandepsin-A for Targeted Neuroendocrine Cancer Therapy. Biomaterials. 2016;97:2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fercana GR, Yerneni S, Billaud M, Hill JC, VanRyzin P, Richards TD, Sicari BM, Johnson SA, Badylak SF, Campbell PG, Gleason TG and Phillippi JA. Perivascular extracellular matrix hydrogels mimic native matrix microarchitecture and promote angiogenesis via basic fibroblast growth factor. Biomaterials. 2017;123:142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams MR, Moody CT, Sollinger JL and Brudno Y. Extracellular-Matrix-Anchored Click Motifs for Specific Tissue Targeting. Mol Pharm. 2020;17:392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCreary DD, Skirtich NF, Andraska EA, Tzeng E and McEnaney RM. Survey of the extracellular matrix architecture across the rat arterial tree. JVS Vasc Sci. 2022;3:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirby BS, Bruhl A, Sullivan MN, Francis M, Dinenno FA and Earley S. Robust internal elastic lamina fenestration in skeletal muscle arteries. PloS one. 2013;8:e54849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi X, Chen G, Guo LW, Si Y, Zhu M, Pilla S, Liu B, Gong S and Kent KC. Periadventitial application of rapamycin-loaded nanoparticles produces sustained inhibition of vascular restenosis. PloS one. 2014;9:e89227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonas M, Edelman ER, Groothuis A, Baker AB, Seifert P and Rogers C. Vascular neointimal formation and signaling pathway activation in response to stent injury in insulinresistant and diabetic animals. Circ Res. 2005;97:725–33. [DOI] [PubMed] [Google Scholar]
- 39.Hao D, Fan Y, Xiao W, Liu R, Pivetti C, Walimbe T, Guo F, Zhang X, Farmer DL, Wang F, Panitch A, Lam KS and Wang A. Rapid endothelialization of small diameter vascular grafts by a bioactive integrin-binding ligand specifically targeting endothelial progenitor cells and endothelial cells. Acta Biomater. 2020;108:178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajathurai T, Rizvi SI, Lin H, Angelini GD, Newby AC and Murphy GJ. Periadventitial rapamycin-eluting microbeads promote vein graft disease in long-term pig vein-into-artery interposition grafts. Circ Cardiovasc Interv. 2010;3:157–65. [DOI] [PubMed] [Google Scholar]
- 41.Holy EW, Jakob P, Eickner T, Camici GG, Beer JH, Akhmedov A, Sternberg K, Schmitz KP, Luscher TF and Tanner FC. PI3K/p110alpha inhibition selectively interferes with arterial thrombosis and neointima formation, but not re-endothelialization: potential implications for drug-eluting stent design. Eur Heart J. 2014;35:808–20. [DOI] [PubMed] [Google Scholar]
- 42.Steffel J, Latini RA, Akhmedov A, Zimmermann D, Zimmerling P, Luscher TF and Tanner FC. Rapamycin, but not FK-506, increases endothelial tissue factor expression: implications for drug-eluting stent design. Circulation. 2005;112:2002–11. [DOI] [PubMed] [Google Scholar]
- 43.Scheer S, Ackloo S, Medina TS, Schapira M, Li F, Ward JA, Lewis AM, Northrop JP, Richardson PL, Kaniskan HU, Shen Y, Liu J, Smil D, McLeod D, Zepeda-Velazquez CA, Luo M, Jin J, Barsyte-Lovejoy D, Huber KVM, De Carvalho DD, Vedadi M, Zaph C, Brown PJ and Arrowsmith CH. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat Commun. 2019;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang B, Zhang M, Takayama T, Shi X, Roenneburg DA, Kent KC and Guo LW. BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine. 2015;2:1650–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang M, Urabe G, Ozer HG, Xie X, Webb A, Shirasu T, Li J, Han R, Kent KC, Wang B and Guo LW. Angioplasty induces epigenomic remodeling in injured arteries. Life Sci Alliance. 2022;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang M, Wang B, Urabe G, Huang Y, Plutzky J, Kent KC and Guo LW. The BD2 domain of BRD4 is a determinant in EndoMT and vein graft neointima formation. Cell Signal. 2019;61:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutzmann J, Haertle M, Daniel JM, Kloss F, Musmann RJ, Kalies K, Knopp K, Pilowski C, Sirisko M, Sieweke JT, Bauersachs J, Sedding DG and Gegel S. BET bromodomaincontaining epigenetic reader proteins regulate vascular smooth muscle cell proliferation and neointima formation. Cardiovasc Res. 2021;117:850–862. [DOI] [PubMed] [Google Scholar]
- 48.Gilan O, Lam EY, Becher I, Lugo D, Cannizzaro E, Joberty G, Ward A, Wiese M, Fong CY, Ftouni S, Tyler D, Stanley K, MacPherson L, Weng CF, Chan YC, Ghisi M, Smil D, Carpenter C, Brown P, Garton N, Blewitt ME, Bannister AJ, Kouzarides T, Huntly BJ, Johnstone RW, Drewes G, Dawson SJ, Arrowsmith CH, Grandi P, Prinjha RK and Dawson MA. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat Struct Mol Biol. 2016;23:673–81. [DOI] [PubMed] [Google Scholar]
- 49.Goldenson B and Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34:537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borah NA and Reddy MM. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules. 2021;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Urabe G, Huang Y, Zhang M, Wang B, Marcho L, Shen H, Kent KC and Guo LW. A Role for Polo-Like Kinase 4 in Vascular Fibroblast Cell-Type Transition. JACC Basic Transl Sci. 2021;6:257–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colleran R, Kufner S, Mehilli J, Rosenbeiger C, Schupke S, Hoppmann P, Joner M, Mankerious N, Fusaro M, Cassese S, Abdel-Wahab M, Neumann FJ, Richardt G, Ibrahim T, Schunkert H, Laugwitz KL, Kastrati A, Byrne RA and Investigators I-C. Efficacy Over Time With Drug-Eluting Stents in Saphenous Vein Graft Lesions. J Am Coll Cardiol. 2018;71:1973–1982. [DOI] [PubMed] [Google Scholar]
- 53.LoPachin RM and Gavin T. Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem Res Toxicol. 2014;27:1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain M, Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Cornelissen A, Guo L, Finn AV, Lentz SR and Chauhan AK. Smooth muscle cell-specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J Clin Invest. 2020;130:295–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kord Forooshani P and Lee BP. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J Polym Sci A Polym Chem. 2017;55:9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leichner C, Wulz P, Baus RA, Menzel C, Gotzfried SK, Gust R and Bernkop-Schnurch A. N-Hydroxysulfosuccinimide Esters versus Thiomers: A Comparative Study Regarding Mucoadhesiveness. Mol Pharm. 2019;16:1211–1219. [DOI] [PubMed] [Google Scholar]
- 57.Girardot JM and Girardot MN. Amide cross-linking: an alternative to glutaraldehyde fixation. J Heart Valve Dis. 1996;5:518–25. [PubMed] [Google Scholar]
- 58.Wang B, Zhang M, Urabe G, Huang Y, Chen G, Wheeler D, Dornbos DJ 3rd, Huttinger A, Nimjee SM, Gong S, Guo LW and Kent KC. PERK Inhibition Mitigates Restenosis and Thrombosis: A Potential Low-Thrombogenic Antirestenotic Paradigm. JACC Basic Transl Sci. 2020;5:245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rocha-Singh KJ, Duval S, Jaff MR, Schneider PA, Ansel GM, Lyden SP, Mullin CM, Ioannidis JPA, Misra S, Tzafriri AR, Edelman ER, Granada JF, White CJ, Beckman JA and Viva Physicians I. Mortality and Paclitaxel-Coated Devices: An Individual Patient Data Meta-Analysis. Circulation. 2020;141:1859–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL, Yang T, Wang H, Luscinskas FW, Croce KJ, Bradner JE and Plutzky J. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Godfrey L, Crump NT, Thorne R, Lau IJ, Repapi E, Dimou D, Smith AL, Harman JR, Telenius JM, Oudelaar AM, Downes DJ, Vyas P, Hughes JR and Milne TA. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat Commun. 2019;10:2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang M, Li J, Wang Q, Urabe G, Tang R, Huang Y, Mosquera JV, Kent KC, Wang B, Miller CL and Guo LW. Gene-repressing epigenetic reader EED unexpectedly enhances cyclinD1 gene activation. Mol Ther Nucleic Acids. 2023;31:717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W and Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome biology. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zang C, Schones DE, Zeng C, Cui K, Zhao K and Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J, Shen H, Owens GK and Guo LW. SREBP1 regulates Lgals3 activation in response to cholesterol loading. Mol Ther Nucleic Acids. 2022;28:892–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chi J, Meng L, Pan S, Lin H, Zhai X, Liu L, Zhou C, Jiang C and Guo H. Primary Culture of Rat Aortic Vascular Smooth Muscle Cells: A New Method. Med Sci Monit. 2017;23:4014–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang M, Urabe G, Little C, Wang B, Kent AM, Huang Y, Kent KC and Guo LW. HDAC6 Regulates the MRTF-A/SRF Axis and Vascular Smooth Muscle Cell Plasticity. JACC Basic Transl Sci. 2018;3:782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Q, Ozer HG, Wang B, Zhang M, Urabe G, Huang Y, Kent KC and Guo LW. A hierarchical and collaborative BRD4/CEBPD partnership governs vascular smooth muscle cell inflammation. Mol Ther Methods Clin Dev. 2021;21:54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.