

PURPOSE
Selinexor is the first selective inhibitor of nuclear export to be approved for the treatment of relapsed or refractory multiple myeloma (MM). Currently, there are no known genomic biomarkers or assays to help select MM patients at higher likelihood of response to selinexor. Here, we aimed to characterize the transcriptomic correlates of response to selinexor-based therapy.
METHODS
We performed RNA sequencing on CD138+ cells from the bone marrow of 100 patients with MM who participated in the BOSTON study, followed by differential gene expression and pathway analysis. Using the differentially expressed genes, we used cox proportional hazard models to identify a gene signature predictive of response to selinexor, followed by validation in external cohorts.
RESULTS
The three-gene signature predicts response to selinexor-based therapy in patients with MM in the BOSTON cohort. Then, we validated this gene signature in 64 patients from the STORM cohort of triple-class refractory MM and additionally in an external cohort of 35 patients treated in a real-world setting outside of clinical trials. We found that the signature tracks with both depth and duration of response, and it also validates in a different tumor type using a cohort of pretreatment tumors from patients with recurrent glioblastoma. Furthermore, the genes involved in the signature, WNT10A, DUSP1, and ETV7, reveal a potential mechanism through upregulated interferon-mediated apoptotic signaling that may prime tumors to respond to selinexor-based therapy.
CONCLUSION
In this study, we present a present a novel, three-gene expression signature that predicts selinexor response in MM. This signature has important clinical relevance as it could identify patients with cancer who are most likely to benefit from treatment with selinexor-based therapy.
INTRODUCTION
Selinexor is the first selective inhibitor of nuclear export (SINE) approved for the treatment of relapsed or refractory multiple myeloma (MM).1 This approval is well supported by recent clinical trial data, most notably including the STORM and BOSTON studies. In the STORM phase II clinical trial, oral selinexor and dexamethasone were administered twice weekly in patients with triple-class refractory MM, with 26% of patients exhibiting a partial response (PR) or better and a median progression-free survival (PFS) of 3.7 months.1,2 The phase III BOSTON trial compared the efficacy of once weekly selinexor in combination with once weekly bortezomib and dexamethasone (Vd) with standard twice weekly Vd in patients with previously treated MM and found an overall response rate (ORR) of 76.4% and a median PFS of 13.9 months for patients receiving the novel treatment regimen, compared with 9.5 months for those receiving the standard treatment.1
CONTEXT
Key Objective
Although the mechanism of action for selinexor is well characterized, there are currently no known biomarkers to guide patient selection for selinexor-based therapy. This study aims to characterize transcriptomic correlates of response to selinexor-based therapy in multiple myeloma.
Knowledge Generated
Using RNA-sequencing from 256 patients across multiple cohorts of selinexor-treated tumors, this study identified a novel, three-gene expression signature that predicts response to selinexor in multiple cohorts and tracks with both depth and duration of response.
Relevance
A robustly validated predictive gene signature can help identify patients who may respond favorably to selinexor-based therapy. Future studies could evaluate the implementation of the signature in a clinical setting using prospective trials.
Mechanistically, selinexor binds to the karyopherin exportin 1 (XPO1), which is responsible for shuttling more than 200 oncogenic and tumor suppressor proteins and mRNA transcripts to the cytoplasm.3 This inhibition of nuclear export, the sequestration of tumor suppressor proteins in the nucleus, and the prevention of oncogene mRNA translation into oncoproteins ultimately induces cancer cell death while permitting the survival of nonmalignant cells.4-6 Selinexor has a unique adverse event (AE) profile because of its novel mechanism of action, and AEs are common, with severe AE reported in 52% of patients in the BOSTON trial and 80% of patients in the STORM trial.1,2 Identifying biomarkers that help predict treatment responses and toxicity is essential for targeted selinexor-based therapeutic intervention.
Systematic approaches to biomarker discovery for selinexor response that leverage next-generation sequencing are generally lacking in the literature. Although MM cells tend to overexpress XPO1 compared with normal plasma cells, XPO1 alterations have not correlated significantly with response to treatment with SINE compounds with the exception of a few in-vitro studies.3-5,7 The STORM study presented a brief transcriptomic analysis that identified a potential four-gene signature on the basis of imputed protein activity.2 A few candidate biomarkers identified through bioinformatics analyses have also been reported in conference proceedings.8,9 However, limitations of these studies include small sample sizes and lack of validation with external cohorts of selinexor-treated patients.
Here, we analyzed RNA sequencing data from 256 patients from multiple studies of selinexor-based therapy to characterize the transcriptomic correlates of response to selinexor. We discovered a novel three-gene signature predicting response to selinexor using RNA-seq data from the BOSTON study and validated it on data from the STORM clinical trial and on an additional independent cohort of patients treated with selinexor at the Mount Sinai Hospital in real-world conditions outside of a clinical trial. The three-gene signature is biologically interpretable and opens a path for evaluating a response mechanism in future studies. More importantly, this signature has the potential to identify patients most likely to benefit from selinexor-based therapy, ultimately reducing toxicities and improving outcomes.
METHODS
Patient Selection and RNA Sequencing
BOSTON and STORM.
CD138+ cells were purified from bone marrow aspirates obtained from 100 patients who participated in the BOSTON study and 64 patients who participated in the STORM study. RNA extraction was performed using the Qiagen Allprep RNA mini kit, and library preparation was performed with the TruSeq Stranded mRNA (non–formalin-fixed paraffin-embedded compatible) kit. For samples with low RNA quality, the Smart-Seq V4 Ultra Low Input Nextera XT kit was used. Total RNA sequencing was performed with 100 bp reads using an Illumina HiSeq 2500 instrument.
MSSM.
The 35 patients were physician-referred as part of the MM banking protocol approved by the Mount Sinai Institutional Review Board. Written informed consent was obtained from each patient. CD138+ cells were isolated from bone marrow aspirates obtained before the start of selinexor-based therapy, and RNA isolation and sequencing were performed as previously described.10
MMRF-CoMMpass.
Gene-level counts for 767 RNA-seq samples from the MMRF-CoMMpass data set were downloaded from dbGaP (accession #phs00748).
KING.
Gene-level read counts were obtained from pretreatment tumors from 57 patients with recurrent glioblastoma that were enrolled in the phase II KING study.11
Bioinformatics processing.
Raw reads were aligned to the GRCh38 human reference genome using STAR.12 Gene-level counts, obtained through featureCounts, were filtered to remove immunoglobulin and ribosomal transcripts and to remove genes whose counts across all samples had zero variance.13,14 Counts were converted to Log2CPM, normalized with voom, and corrected for batch effects or covariates identified through variancePartition analysis using the sva ComBat package.14-16
Differential Expression
Differential expression (DE) genes were identified in the BOSTON selinexor, bortezomib, and dexamethasone (XVd) and the Vd arm separately using DESeq2.17 Within each arm, DE genes relative to responders versus nonresponders were generated across a total of nine response cutoffs on the basis of PFS, ORR, or a combination of both. For each response comparison, genes were designated as DE in selinexor responders if they were significantly DE in the XVd arm (PAdj < .05) and were not significantly DE in the corresponding comparison within the Vd arm (P > .05). DESeq2 analysis was performed on unfiltered raw counts per tool requirements.17 Gene set enrichment analysis (GSEA) was applied to the selinexor-specific DESeq2 results in the XVd arm using MSigDB Hallmark pathway definitions using the fGSEA package.18,19
Survival Analysis
Each DE gene set identified through the nine comparisons was split into upregulated and downregulated subsets on the basis of log fold change (logFC) in the XVd Arm, resulting in 17 candidate gene signatures for further analysis (nine upregulated and eight downregulated). Gene Set Variation Analysis (GSVA) scores were calculated for all samples for the 17 candidate gene signatures from the covariate-normalized expression matrix.20 For each candidate gene signature, a univariate Cox proportional hazard model was generated in the BOSTON XVd arm. The gene signature with the best model performance in the discovery cohort was selected by ranking the highest Somer's Dxy after repeated four-fold cross-validation (no.repeats = 1,000). It was also evaluated with a Cox proportional hazards model in the Vd arm as a negative control to ensure that performance was specific to selinexor. A permutation test was performed to evaluate whether the selected gene signature is more significant via log-rank Kaplan-Meier testing than a GSVA score composed of three randomly selected genes.
All validation tests were executed by first performing feature-specific quantile normalization of the expression matrix with the distribution of expression in the BOSTON data set, followed by calculation of a GSVA score for the gene signature.20,21 Survival was tested between groups that had low expression versus high expression, using a cutoff of zero, with a log-rank test and Cox regression performed with the same procedures used for the discovery cohort.
Statistical Analysis
All statistical analyses were performed using R. Regression models, including Cox proportional hazard models and ordinal regressions, were performed using the rms package.22 All other statistical comparisons were performed using a Wilcoxon-Mann-Whitney test unless otherwise noted.
RESULTS
Patient Characteristics and Transcriptomic Profiling of Selinexor-Treated MM Tumors: BOSTON
We performed RNA-seq on CD138+ bone marrow plasma cells from 100 patients who participated in the BOSTON study, and their clinical and demographic characteristics are detailed in Table 1. The ORR in the XVd arm, defined as PR or better, was 75.47%, compared with an ORR of 65.95% in the Vd arm. These observations suggest that the addition of selinexor to a regimen of Vd in MM offers a clinical benefit over Vd alone and are consistent with the findings of previous studies, including the main BOSTON trial.1
TABLE 1.
Cohort Clinical Characteristics

We leveraged RNA sequencing on CD138+-selected cells from two external cohorts: patients with MM treated with selinexor-dexamethasone who participated in the STORM trial (n = 64) and patients with MM treated post-US Food and Drug Administration approval at Mount Sinai Hospital outside of any clinical trial setting (MSSM cohort, n = 35).2 Patients in the STORM cohort had failed at least five prior lines of therapy and had an ORR of 28.1% (18 patients, Table 1).2 Patients in the MSSM cohort had an ORR of 28.5% (10 patients) for selinexor-based therapy, which was often administered in combination with a variety of other agents (Table 1). In this cohort, the most common combination drug strategy in addition to the selinexor backbone was carfilzomib and dexamethasone, which were used in seven (20%) patients. We did not find any significant predictors of response to selinexor from clinical, demographic, or cytogenetic markers.
Differential Expression Analysis Identifies Genes Associated With Selinexor Response
The strategy used for DE analysis is summarized in Figure 1A. To identify genes whose expression is associated with longer PFS or better depth of response to selinexor, we performed a series of DE comparisons across nine unique different PFS or depth of response (defined according to International Myeloma Working Group [IMWG] criteria) thresholds in both the XVd and Vd arms (Appendix Fig A1, Appendix Table A1).23 Across all response thresholds, there were a total of 107 unique significant DE genes between better and worse responders in the XVd arm (27 upregulated and 70 downregulated, P Adj < 0.05) and 560 unique DE genes in the Vd arm (398 upregulated and 162 downregulated, P Adj < 0.05). To identify genes whose association with PFS or IMWG response category was selinexor-specific, we retained genes that were significant in the XVd arm but not in the Vd arm for each corresponding cutoff for further analysis. There was a moderate overlap (up to 31%) across genes identified through the different comparisons, with six of 24 (25.0%) uniquely downregulated genes and 12 of 33 (36.26%) uniquely upregulated genes overlapping across at least two different cutoffs.
FIG 1.

Differentially expressed genes in selinexor responders. (A) Experimental design for identifying DE genes specific to selinexor. (B) Volcano plot of the BOSTON XVd DE comparison with a PFS ≥ 120 days response cutoff showing the three significantly upregulated genes used to generate the three-gene response signature. Adj, adjusted; DE, differential expression; FC, fold change; PFS, progression-free survival; PH, proportional hazard; Vd, bortezomib and dexamethasone; XVd, selinexor, bortezomib, and dexamethasone.
A Three-Gene Signature Predicts Selinexor Response
Next, we identified a gene expression signature for selinexor response. Each DE gene set identified through the nine unique comparisons was split into upregulated and downregulated subsets on the basis of logFC in the XVd arm, resulting in 17 candidate gene signatures for further analysis (nine upregulated and eight downregulated). We performed GSVA on the normalized, batch-corrected expression matrix to calculate a unique score for each of the 17 candidate gene signatures and selected the best performing model in the BOSTON cohort on the basis of a ranking procedure (Fig 2A, see Methods).
FIG 2.

Upregulation of the signature is associated with longer PFS in pre–Selinexor-treated MM. (A) Kaplan-Meier curve of low versus high expression of the three-gene Gene Set Variation Analysis signature in the XVd arm of the BOSTON cohort shows a significant association with longer PFS in patients with upregulation of the signature. (B) Using the Vd arm as a negative control, the three-gene signature is not associated with PFS. (C) Three-gene signature validates in the STORM cohort of patients with triple-class refractory MM who received selinexor as a single-agent treatment. (D) The signature also validates in 35 patients who received selinexor in a retrospective cohort at MSSM that is not part of a clinical trial and is a more heterogeneous patient population reflective of real-world treatment settings. (E) Signature does not validate in non-selinexor, standard-of-care–treated MMRF-CoMMpass samples. HR, hazard ratio; PFS, progression-free survival; PH, proportional hazard; MM, multiple myeloma; Vd, bortezomib and dexamethasone; XVd, selinexor, bortezomib, and dexamethasone.
The best-performing signature comprised WNT10A, DUSP1, and ETV7. It correlated with PFS (Spearman Rho = 0.46, P = .0007) and was upregulated in XVd patients with PFS ≥ 120 days. The signature was predictive when using a proportional hazard model (P Adj = 0.047, hazard ratio [HR] = 0.36 [95% CI, 0.14 to 0.84]; log-rank P = .017, Fig 2A). To further ensure that the predictive effect of the signature was not due to random variations, we also performed a permutation test and found that the signature was more predictive than a GSVA score composed of three randomly selected genes (10,000 permutations, P = .03). In patients who achieved a response of PR or better, higher signature expression was significantly associated with a longer duration of response (DOR), defined as the number of days from IMWG PR to progression (Appendix Fig A2A). Finally, an ordinal regression found that higher signature expression was significantly associated with deeper IMWG response category (P = .0247, R2 = 0.095, Spearman Rho = 0.317, Fig 3A), suggesting that the signature is associated with both duration and depth of response.
FIG 3.
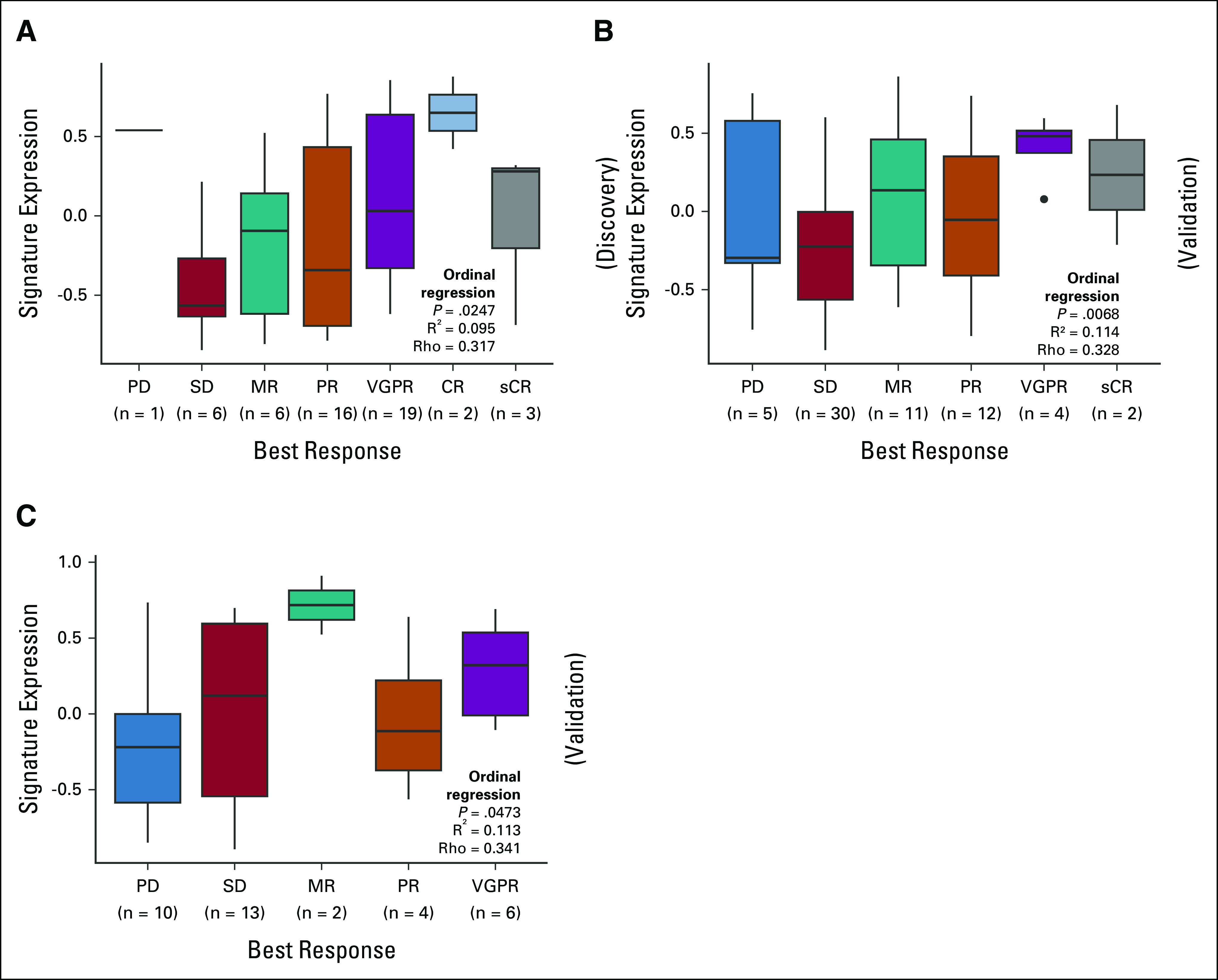
Higher signature expression is significantly associated with better depth of response. Higher three-gene signature expression is significantly associated with better International Myeloma Working Group response category via ordinal regression modeling for patients in the (A) BOSTON XVd, (B) STORM, and (C) MSSM cohorts. CR, complete response; MR, minor response; PD, progressive disease; PR, partial response; sCR, stringent complete response; SD, stable disease; VGPR, very good partial response; XVd, selinexor, bortezomib, and dexamethasone.
The same analysis was applied to the Vd arm as a negative control under the rationale that a signature specific to selinexor response would not accurately distinguish between long or short PFS in cohorts treated with non–selinexor-based therapy. Signature expression did not correlate with PFS or DOR in the Vd arm, as shown in Figure 2B and Appendix Figure A2B.
The Three-Gene Signature Is Validated in External Cohorts
STORM.
A log-rank test comparing signature expression higher or lower than the cutoff of zero in the STORM data set also validated the finding from BOSTON that higher signature expression is associated with PFS (log-rank P = .039; N = 64). We found that the linear association with PFS and the signature performed nominally well, despite not reaching statistical significance (P = .056, HR = 0.621 [95% CI, 0.483 to 0.389], Fig 2C). The poorer signature performance in a Cox regression may be explained by lower PFS as patients in the STORM cohort were triple-class refractory with more advanced disease to begin with. Ordinal regression analysis validated the association between signature expression and depth of response in the STORM cohort (P = .0068, R2 = 0.114, Spearman Rho = 0.328, Fig 3B).
MSSM.
Using Cox regression, we found that higher expression of the three-gene signature was significantly associated with survival (P = .0145, HR = 0.41 [95% CI, –0.467 to 0.551]). This result was also replicated via log-rank testing (P = .0023, Fig 2D). Furthermore, we found a statistically significant correlation between the signature expression and PFS via Spearman correlation (Rho = 0.4, P = .01). Higher expression of the signature was also significantly associated with a better IMWG response category via ordinal regression (P = .0473, R2 = 0.113, Spearman Rho = 0.341, Fig 3C).
MMRF-CoMMpass.
We additionally used the MMRF-CoMMpass data set (n = 767) as a negative control and found that the signature was not predictive of PFS in patients who were treated with non–selinexor-based, standard-of-care therapies. The signature did not show a significant association with PFS via Cox regression (P = .14, HR = 0.89 [95% CI, 0.73 to 1.04]) or log-rank testing (P = .32, Fig 2E). Taken together, these results suggest that the signature is specific to selinexor treatment response and does not reflect overall prognosis.
KING study in recurrent glioblastoma.
Finally, we sought to test whether the signature would be predictive in cohorts of patients with other cancer types treated with selinexor from other cancer types. To test this hypothesis, we obtained RNA-seq of tumor samples from 57 patients with recurrent glioblastoma who were treated with selinexor monotherapy as part of the phase II KING trial (ClinicalTrials.gov identifier: NCT01986348).11 Overall, we found that patients with higher expression of the signature experienced improved PFS, although statistical significance was not achieved (log-rank P = .078, Cox proportional hazard P = .0734, HR = 0.549, Appendix Fig A3A). However, patients who achieved a response of PR or better had significantly higher signature expression (Wilcoxon P = .0034, Appendix Fig A3B).
Gene Set Enrichment Analysis Reveals Response-Associated Activation in WNT, Apoptosis, and MAPK Signaling Pathways
We next identified pathways associated with response to Selinexor in the BOSTON XVd cohort using GSEA on the MSigDB Hallmark gene sets.18 Notably, we found downregulation of MYC targets, as well as upregulation of KRAS, apoptotic, and interferon signaling among significantly enriched pathways in XVd responders (Appendix Fig A4).
DISCUSSION
Selinexor is approved as a second-line therapy for MM, and its efficacy is well supported by clinical trials. However, there are currently no known biomarkers to guide the selection of patients whose tumors are more sensitive to selinexor-based therapy. Furthermore, although the mechanism of action is well characterized for selinexor, little is known about the correlates of response or resistance to selinexor-based therapy. Here, we describe the transcriptomic characteristics of patients who responded to selinexor therapy. Furthermore, we report the discovery of a robustly validated three-gene signature that is predictive of response to selinexor-based therapy in MM.
There is little literature to date on patient populations describing correlates to response in the context of selinexor therapy. Some studies have shown candidate biomarker activity in microRNAs as regulators of XPO1 and its targets, and certain mutations in XPO1 and XPO5 as prognostic markers for survival, but they have not been correlated to SINE drug sensitivity.7,24-27 Few studies have explored biomarkers in selinexor therapy, and fewer have validated their findings in external cohorts. One notable study found a signature on the basis of imputed activity comprising four master regulator proteins, IRF3, ARL2BP, ZBTB17, and ATRX.2 The signature we report lacks overlap with this signature because of differences in methodology. Specifically, the prior study relied on protein activity inferred from a network model originally trained in acute myeloid leukemia and does not directly correlate with RNA expression.2 Additional factors that may explain lower-than-expected overlap among the DE genes determined at different cutoffs include high interpatient heterogeneity, relatively lower sample sizes, and lack of negative controls. However, since our signature uses gene expression directly, it is more interpretable and easier to implement than previously cited signatures.2
The signature we report here is composed of three upregulated genes, ETV7, WNT10A, and DUSP1, that precisely and accurately predicts both depth and DOR to selinexor-based therapy in patients with MM. Furthermore, we present robust external validation and negative controls. This is the first robustly validated signature for selinexor response to date. Given the high rate of AEs in selinexor-based therapy, the discovery of a predictive gene signature holds tremendous potential for biomarker-guided selection of candidates for selinexor-based therapy that are more likely to respond. Since the signature was validated in multiple heterogeneous patient cohorts, including selinexor-dexamethasone monotherapy (STORM), and in various combination drug scenarios outside of clinical trials (MSSM), it is both flexible and applicable to a wide variety of real-world scenarios where selinexor may be administered in combination with other drugs. One limitation of the signature is that because it relies on a GSVA score, its accuracy is dependent on a cohort with multiple samples and prediction improves with greater sample sizes. However, since it is composed of only three genes, it also holds potential for fast and simple implementation in clinical settings, potentially via quantitative polymerase chain reaction–based assay development strategies or in combination with ex-vivo drug sensitivity assays.
The three genes comprising this signature, however, are not well characterized in the context of MM or the XPO1 mechanism of action. ETV7 is a transcription factor with oncogenic properties that has been described in the context of mammalian target of rapamycin signaling and as an interferon-stimulated gene.28-31 DUSP1 negatively regulates mitogen-activated protein kinase signaling via the dephosphorylation of extracellular-regulated kinase, which can subsequently phosphorylate ETV7.28,32 WNT10A participates in Wnt signaling, which is mediated by galectin-3, a known XPO1 target.3 All these processes converge on p38-mediated mammalian target of rapamycin signaling and are ultimately regulated by XPO1 targets.3,30,32,33 Further mechanistic studies are necessary to understand the biological role of these genes in the context of XPO1 inhibition.
Interferon signaling is responsible for the antiviral immune response and has antiproliferative properties. It has been shown to play an important role in apoptotic signaling in MM, mostly through IRF4, MYC, and BCL6.34-36 Here, we found a strong enrichment for upregulated interferon signaling and genes involved in interferon signaling in patients who responded to XVd therapy. We also found a corresponding enrichment of dysregulated MYC and apoptotic signaling. Interferon signaling has been found to modulate response to XPO1 inhibition by eltanexor to treat viral infection.37 Additionally, ETV7 has been identified as an interferon-stimulated gene.31 On the basis of these results and existing literature, we hypothesize that there may be a rationale for further evaluating the role of interferon signaling in MM selinexor response.
In conclusion, we report a novel gene expression signature for response to selinexor-based therapy in patients with MM. We have validated our findings in several external transcriptomic data sets of patients with MM treated with selinexor-based regimens. Ongoing in vitro and mechanistic studies will help determine whether they are causative of response or simply a correlative readout of some other selinexor response mechanism. This signature has important clinical significance as it could identify patients most likely to benefit from treatment with selinexor-based therapy, especially in earlier lines of therapy.
APPENDIX
TABLE A1.
Differential Expression–Based Signature Definitions

FIG A1.

Differentially expressed genes in all unique BOSTON comparisons. Volcano plots showing all differential expression genes identified across response comparisons in the BOSTON XVd cohort. (A) PFS120, (B) PFS200, (C) PFS300, (D) PFS90, (E) PR, (F) PR_PFS120, (G) VGPR, (H) VGPR_PFS120, and (I) VGPRR_PFS300. Gene symbols are labeled where Adj P < .05 is in red for overexpression and blue for underexpression in responders to XVd therapy. Adj, adjusted; FC, fold change; PFS, progression-free survival; PR, partial response; VGPR, very good partial response; XVd, selinexor, bortezomib, and dexamethasone.
FIG A2.

Signature expression tracks with longer DOR in BOSTON XVd. (A) Kaplan-Meier curve of higher versus lower signature expression as a function of DOR in patients who achieved a partial response or better in (A) the BOSTON XVd cohort shows a significant association, whereas there is no significant difference in (B) patients in the BOSTON Vd cohort of non-selinexor–based therapy. DOR, duration of response; Vd, bortezomib and dexamethasone; XVd, selinexor, bortezomib, and dexamethasone.
FIG A3.

Signature expression predicts selinexor response in recurrent glioblastoma. (A) Survival analysis of the KING recurrent glioblastoma cohort stratified by three-gene signature GSVA scores. (B) Three-gene signature GSVA scores in the KING cohort in responders (teal) versus nonresponders (orange). GSVA, Gene Set Variation Analysis; HR, hazard ratio; PFS, progression-free survival; PH, proportional hazard; PR, partial response.
FIG A4.

Functional enrichments in selinexor responders. Gene set enrichment analysis enrichments in selinexor, bortezomib, and dexamethasone responders across the various response cutoffs. Adj, adjusted; NFKB, nuclear factor kappa B; PFS, progression-free survival; PR, partial response; TNFA, tumor necrosis factor alpha; VGPR, very good partial response.
Deepu Madduri
Employment: Janssen Oncology
Stock and Other Ownership Interests: Roivant
Consulting or Advisory Role: Takeda, Janssen Oncology, Celgene, Legend Biotech, GlaxoSmithKline, Foundation Medicine
Joshua Richter
Consulting or Advisory Role: Amgen, Takeda, Celgene, Oncopeptides, Adaptive Biotechnologies, Karyopharm Therapeutics, Antengene, X4 Pharma, Sanofi, Secura Bio
Speakers' Bureau: Celgene, Janssen, Bristol Myers Squibb
Shambavi Richard
Honoraria: BMS, Janssen Oncology, Karyopharm Therapeutics
Consulting or Advisory Role: BMS, Karyopharm Therapeutics
Research Funding: BMS (Inst), Janssen Oncology (Inst)
Ajai Chari
Consulting or Advisory Role: Amgen, Janssen Oncology, Seattle Genetics, Karyopharm Therapeutics, Genzyme, OncoPeptides, Takeda, Antengene, GlaxoSmithKline, Secura Bio, Shattuck Labs, Genentech, AbbVie, Bristol Myers Squibb/Celgene
Research Funding: Celgene, Janssen, Amgen, Seattle Genetics, Takeda, Pharmacyclics
Hearn Jay Cho
Employment: The Multiple Myeloma Research Foundation
Research Funding: Bristol Myers Squibb, Takeda
Sundar Jagannath
Consulting or Advisory Role: Bristol Myers Squibb, Janssen, Karyopharm Therapeutics, Legend Biotech, Takeda, Sanofi
Travel, Accommodations, Expenses: Bristol Myers Squibb, Janssen Biotech
Christopher J. Walker
Employment: Karyopharm Therapeutics
Stock and Other Ownership Interests: Karyopharm Therapeutics
Patents, Royalties, Other Intellectual Property: Pending patents for biomarkers related to selinexor efficacy
Travel, Accommodations, Expenses: Karyopharm Therapeutics
Yossi Landesman
Employment: Karyopharm Therapeutics
Leadership: Karyopharm Therapeutics
Stock and Other Ownership Interests: Karyopharm Therapeutics
Consulting or Advisory Role: Advenchen Laboratories
Samir Parekh
Consulting or Advisory Role: Foundation Medicine
Research Funding: Amgen, Pfizer, Celgene, Karyopharm Therapeutics
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented as oral abstract at American Society of Hematology Annual Meeting, Atlanta, GA, December 11, 2021.
A.L. and S.P. contributed equally to this work.
PREPRINT VERSION
Preprint version available on MedRxiv, 2022 (doi: 10.1101/2022.02.25.22271401v3).
DATA SHARING STATEMENT
Raw sequencing data is deposited in SRA under the accession PRJNA827033. The code necessary to calculate Selinexor signatures is provided in a GitHub repository at https://github.com/Lagana-Lab/selinescore.
AUTHOR CONTRIBUTIONS
Conception and design: Yosef Landesman, Christopher J. Walker, Alessandro Lagana, Samir Parekh
Collection and assembly of data: Paula Restrepo, Yogita Ghodke-Puranik, Adolfo Aleman, Violetta Leschenko, David T. Melnekoff, Sarita Agte, Deepu Madduri, Joshua Richter, Shambavi Richard, Ajai Chari, Hearn Jay Cho, Sundar Jagannath, Christopher J. Walker, Yosef Landesman, Alessandro Laganá, Samir Parekh
Data analysis and interpretation: Paula Restrepo, Sherry Bhalla, Yogita Ghodke-Puranik, David T. Melnekoff, Joy Jiang, Alessandro Laganà
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Deepu Madduri
Employment: Janssen Oncology
Stock and Other Ownership Interests: Roivant
Consulting or Advisory Role: Takeda, Janssen Oncology, Celgene, Legend Biotech, GlaxoSmithKline, Foundation Medicine
Joshua Richter
Consulting or Advisory Role: Amgen, Takeda, Celgene, Oncopeptides, Adaptive Biotechnologies, Karyopharm Therapeutics, Antengene, X4 Pharma, Sanofi, Secura Bio
Speakers' Bureau: Celgene, Janssen, Bristol Myers Squibb
Shambavi Richard
Honoraria: BMS, Janssen Oncology, Karyopharm Therapeutics
Consulting or Advisory Role: BMS, Karyopharm Therapeutics
Research Funding: BMS (Inst), Janssen Oncology (Inst)
Ajai Chari
Consulting or Advisory Role: Amgen, Janssen Oncology, Seattle Genetics, Karyopharm Therapeutics, Genzyme, OncoPeptides, Takeda, Antengene, GlaxoSmithKline, Secura Bio, Shattuck Labs, Genentech, AbbVie, Bristol Myers Squibb/Celgene
Research Funding: Celgene, Janssen, Amgen, Seattle Genetics, Takeda, Pharmacyclics
Hearn Jay Cho
Employment: The Multiple Myeloma Research Foundation
Research Funding: Bristol Myers Squibb, Takeda
Sundar Jagannath
Consulting or Advisory Role: Bristol Myers Squibb, Janssen, Karyopharm Therapeutics, Legend Biotech, Takeda, Sanofi
Travel, Accommodations, Expenses: Bristol Myers Squibb, Janssen Biotech
Christopher J. Walker
Employment: Karyopharm Therapeutics
Stock and Other Ownership Interests: Karyopharm Therapeutics
Patents, Royalties, Other Intellectual Property: Pending patents for biomarkers related to selinexor efficacy
Travel, Accommodations, Expenses: Karyopharm Therapeutics
Yossi Landesman
Employment: Karyopharm Therapeutics
Leadership: Karyopharm Therapeutics
Stock and Other Ownership Interests: Karyopharm Therapeutics
Consulting or Advisory Role: Advenchen Laboratories
Samir Parekh
Consulting or Advisory Role: Foundation Medicine
Research Funding: Amgen, Pfizer, Celgene, Karyopharm Therapeutics
No other potential conflicts of interest were reported.
REFERENCES
- 1.Grosicki S, Simonova M, Spicka I, et al. : Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet 396:1563-1573, 2020 [DOI] [PubMed] [Google Scholar]
- 2.Chari A, Vogl DT, Gavriatopoulou M, et al. : Oral selinexor–dexamethasone for triple-class refractory multiple myeloma. N Engl J Med 381:727-738, 2019 [DOI] [PubMed] [Google Scholar]
- 3.Fung HYJ, Niesman A, Chook YM: An update to the CRM1 cargo/NES database NESdb. Mol Biol Cell 32:467-469, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chook YM, Fung HYJ: Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol 27:52-61, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azmi AS, Uddin MH, Mohammad RM: The nuclear export protein XPO1—From biology to targeted therapy. Nat Rev Clin Oncol 18:152-169, 2021 [DOI] [PubMed] [Google Scholar]
- 6.Tai Y-T, Landesman Y, Acharya C, et al. : CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 28:155-165, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neggers JE, Vanstreels E, Baloglu E, et al. : Heterozygous mutation of cysteine528 in XPO1 is sufficient for resistance to selective inhibitors of nuclear export. Oncotarget 7:68842-68850, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bahlis NJ, Kotb R, Sebag M, et al. : Selinexor in combination with bortezomib and dexamethasone (SdB) demonstrates significant activity in patients with refractory multiple myeloma (MM) including proteasome-inhibitor refractory patients: Results of the phase I stomp trial. Blood 128:977, 2016 [Google Scholar]
- 9.Lagana A, Bhalla S, Aleman A, et al. : A machine learning approach identifies a 30-gene model that predicts sensitivity to selinexor in multiple myeloma. Blood 134:3101, 2019 [Google Scholar]
- 10.Laganà A, Beno I, Melnekoff D, et al. : Precision medicine for relapsed multiple myeloma on the basis of an integrative multiomics approach. JCO Precis Oncol 10.1200/PO.18.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lassman AB, Wen PY, van den Bent MJ, et al. : A phase II study of the efficacy and safety of oral selinexor in recurrent glioblastoma. Clin Cancer Res 28:452-460, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobin A, Davis CA, Schlesinger F, et al. : STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29:15-21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao Y, Smyth GK, Shi W: featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923-930, 2014 [DOI] [PubMed] [Google Scholar]
- 14.Ritchie ME, Phipson B, Wu D, et al. : limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman GE, Schadt EE: variancePartition: Interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17:483, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leek JT, Johnson WE, Parker HS, et al. : The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28:882-883, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liberzon A, Subramanian A, Pinchback R, et al. : Molecular signatures database (MSigDB) 3.0. Bioinformatics 27:1739-1740, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korotkevich G, Sukhov V, Budin N, et al. : Fast gene set enrichment analysis. bioRxiv 10.1101/060012 [DOI] [Google Scholar]
- 20.Hänzelmann S, Castelo R, Guinney J: GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14:7, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franks JM, Cai G, Whitfield ML: Feature specific quantile normalization enables cross-platform classification of molecular subtypes using gene expression data. Bioinformatics 34:1868-1874, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrell FE: rms: Regression modeling strategies. 2020. https://CRAN.R-project.org/package=rms
- 23.Kumar S, Paiva B, Anderson KC, et al. : International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 17:e328-e346, 2016 [DOI] [PubMed] [Google Scholar]
- 24.Miloudi H, Bohers É, Guillonneau F, et al. : XPO1E571K mutation modifies exportin 1 localisation and interactome in B-cell lymphoma. Cancers (Basel) 12:2829, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumhardt JM, Walker JS, Lee Y, et al. : Recognition of nuclear export signals by CRM1 carrying the oncogenic E571K mutation. Mol Biol Cell 31:1879-1891, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leaderer D, Hoffman AE, Zheng T, et al. : Genetic and epigenetic association studies suggest a role of microRNA biogenesis gene exportin-5 (XPO5) in breast tumorigenesis. Int J Mol Epidemiol Genet 2:9-18, 2011 [PMC free article] [PubMed] [Google Scholar]
- 27.Martini S, Zuco V, Tortoreto M, et al. : miR-34a-Mediated survivin inhibition improves the antitumor activity of selinexor in triple-negative breast cancer. Pharmaceuticals (Basel) 14:523, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selvaraj N, Kedage V, Hollenhorst PC: Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells. Cell Commun Signal 13:12, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasighaemi P, Ward AC: ETV6 and ETV7: Siblings in hematopoiesis and its disruption in disease. Crit Rev Oncol Hematol 116:106-115, 2017 [DOI] [PubMed] [Google Scholar]
- 30.Harwood FC, Klein Geltink RI, O'Hara BP, et al. : ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci Adv 4:eaar3938, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pezzè L, Meškytė EM, Forcato M, et al. : ETV7 regulates breast cancer stem-like cell features by repressing IFN-response genes. Cell Death Dis 12:1-14, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sathe A, Guerth F, Cronauer MV, et al. : Mutant PIK3CA controls DUSP1-dependent ERK 1/2 activity to confer response to AKT target therapy. Br J Cancer 111:2103-2113, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Browne AJ, Göbel A, Thiele S, et al. : p38 MAPK regulates the Wnt inhibitor Dickkopf-1 in osteotropic prostate cancer cells. Cell Death Dis 7:e2119, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Gong B, Mahmoud-Ahmed AS, et al. : Apo2L/TRAIL and Bcl-2–related proteins regulate type I interferon–induced apoptosis in multiple myeloma. Blood 98:2183-2192, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agnarelli A, Chevassut T, Mancini EJ: IRF4 in multiple myeloma—Biology, disease and therapeutic target. Leuk Res 72:52-58, 2018 [DOI] [PubMed] [Google Scholar]
- 36.Dufva O, Pölönen P, Brück O, et al. : Immunogenomic landscape of hematological malignancies. Cancer Cell 38:380-399.e13, 2020 [DOI] [PubMed] [Google Scholar]
- 37.Liao Y, Ke X, Deng T, et al. : The second-generation XPO1 inhibitor eltanexor inhibits human cytomegalovirus (HCMV) replication and promotes type I interferon response. Front Microbiol 12:675112, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequencing data is deposited in SRA under the accession PRJNA827033. The code necessary to calculate Selinexor signatures is provided in a GitHub repository at https://github.com/Lagana-Lab/selinescore.