

Abstract
Fibroblast growth factors (FGFs) and their receptors (FGFRs) have been extensively investigated in solid malignancies, representing an attractive therapeutic target. In breast cancer, especially in estrogen receptor positive (ER+) subtype, FGFR signaling aberrations have been reported to contribute to proliferation, dedifferentiation, metastasis and drug resistance. However, clinical trials evaluating the use of FGFR inhibitors in breast cancer have had disappointing results. The different biological properties of distinct FGFR alterations and lack of established patient selection criteria, in addition to the early use of non-selective inhibitors, are possible reasons of this failure. Herein, we review the current knowledge regarding the role of FGFR signaling in endocrine resistance in breast cancer. We will also summarize the results from the clinical development of FGFR inhibitors in breast cancer, discussing future challenges to identify the correct cohorts of patients to enroll in trials testing FGFR inhibitors.
Keywords: FGFR1, breast cancer, endocrine resistance, tyrosine kinase inhibitors
Introduction
Breast cancer is the most frequently diagnosed malignant tumor in women, with more than 250,000 new cases expected each year (Siegel et al., 2020). About 80% of newly diagnosed breast cancers are estrogen receptor positive (ER+) (DeSantis et al., 2019) Endocrine therapies, such as selective ER modulators (SERMs), selective ER degraders (SERDs) and aromatase inhibitors (AIs) represent the mainstay of the treatment of ER+ breast cancer. The approvals of targeted therapies, such as cyclin-dependent kinases (CDK) 4/6 inhibitors (palbociclib, ribociclib and abemaciclib), anti-HER2 therapies (trastuzumab, pertuzumab, T-DM1, neratinib, lapatinib, tucatinib, trastuzumab deruxtecan), the phosphoinositide 3-kinase (PI3K) α inhibitor alpelisib and the mammalian target of rapamycin (mTOR) inhibitor everolimus, administered in combination with antiestrogens, have all improved the outcome of ER+ breast cancer patients (Álvarez-Fernández and Malumbres, 2020; André et al., 2019; Baselga et al., 2012; Modi et al., 2020; Murthy et al., 2020; Pernas and Tolaney, 2019). However, despite these advances, drug resistance still occurs in a significant fraction of patients (Hanker et al., 2020) leading to disease progression and ultimately death.
The oncogenic role of Receptor Tyrosine Kinases (RTKs) in ER+ breast cancer has been extensively investigated. Among them, the fibroblast growth factor receptors (FGFRs) family, consisting of four highly conserved transmembrane receptors (FGFR1–4) and another membrane-associated receptor lacking the intracellular domain (FGFR5, or FGFRL1), has been largely studied. FGFRs play various roles in normal physiology and development, such as embryogenesis, tissue development, immune surveillance and metabolism (Turner and Grose, 2010). FGFR signaling has been shown to be implicated in several oncogenic pathways, such as cancer cell proliferation, survival, migration, invasion and angiogenesis (Babina and Turner, 2017; Turner and Grose, 2010). More recently, various reports demonstrated a role of FGFR signaling in breast cancer biology and antiestrogen resistance (Sobhani et al., 2020). Thus, FGFR signaling represents a therapeutic target for the development of selective FGFR inhibitors.
This review will focus on genomic and non-genomic alterations that aberrantly activate FGFR signaling in breast cancer, particularly ER+ breast tumors. We will also summarize the current status of the clinical development of FGFR tyrosine kinase inhibitors (TKIs) in breast cancer, discussing their outcome, limitations and possible future directions.
FGFR signaling
Intracellular signal transduction induced by FGFRs follows the same canonical model observed for other RTKs. Membrane-bound FGFRs are activated by the binding of ligands (fibroblast growth factors, FGFs) to their extracellular domain (Schlessinger et al., 2000) (Figure 1). Mammalian FGF family is composed of 18 members that have critical functions in embryonic development and in adults, regulating cell proliferation, differentiation and migration (Beenken and Mohammadi, 2009). FGFRs have an extracellular domain, where ligands bind, a transmembrane domain and an intracellular domain involved in the activation of signaling cascades. The extracellular portion has three Immunoglobulin (Ig) like domains (D1-D3). FGFRs transcripts also undergo alternative splicing, affecting the composition of the D1-D3 domains, to generate different receptor isoforms. FGFs display varied binding specificity for different FGFRs isoforms (Beenken and Mohammadi, 2009). FGFs mainly act in a paracrine or autocrine fashion and the binding of FGF to FGFRs is mediated by heparin sulfate (HS) proteoglycans (HSPGs) which modulate the affinity of the ligand for their receptor (Makarenkova et al., 2009). In addition, FGF19, FGF21 and FGF23 act as endocrine ligands and require α- or β-Klotho cofactors to bind to FGFRs (Itoh et al., 2015). FGF-FGFR binding promotes receptor dimerization and phosphorylation of C-terminal tyrosines of the cytoplasmic domain (Eswarakumar et al., 2005). These phosphorylated tyrosines dock adaptor proteins which, in turn, activate downstream signaling pathways. The main adaptor protein of FGFRs is Fibroblast Growth Factor Receptor Substrate alpha (FRS2α) (Gotoh, 2008). Tyrosine phosphorylation of FRS2α promotes the recruitment of growth factor receptor-bound 2/son of sevenless (GRB2/SOS) complexes leading to activation of RAS/RAF/MEK/MAPK and PI3K/AKT. FGFRs can also induce FRS2α-independent activation of phospholipase Cγ (PLCγ). Phosphorylation of tyrosine 766 in FGFR creates a specific binding site for the SH2 domain of PLCγ, promoting its direct recruitment and activation and inducing protein kinase C (PKC) signaling (Peters et al., 1992). Finally, phosphorylation of C-terminal residues in FGFRs have been shown to directly activate members of Signal Transducer and Activator of Transcription (STAT) family, particularly Stat1 and Stat3, promoting their nuclear translocation and transcription of downstream target genes (Dudka et al., 2010; Hart et al., 2000). Various intracellular mechanisms that negatively regulate FGFR signaling activation have been described. The casitas B-lineage lymphoma (Cbl) protein is involved in ubiquitination of phosphorylated RTKs and consequent downregulation (Thien and Langdon, 2001). Also, Sprouty (SPRY) proteins can prevent Sos-mediated Ras activation (Kramer et al., 1999). MAPK phosphatases, such as MKP3 (Zhao and Zhang, 2001), and SEF family proteins (Tsang et al., 2002) have also been reported to downregulate FGFR-induced intracellular signaling.
Figure 1. FGFR signaling pathway in breast cancer.
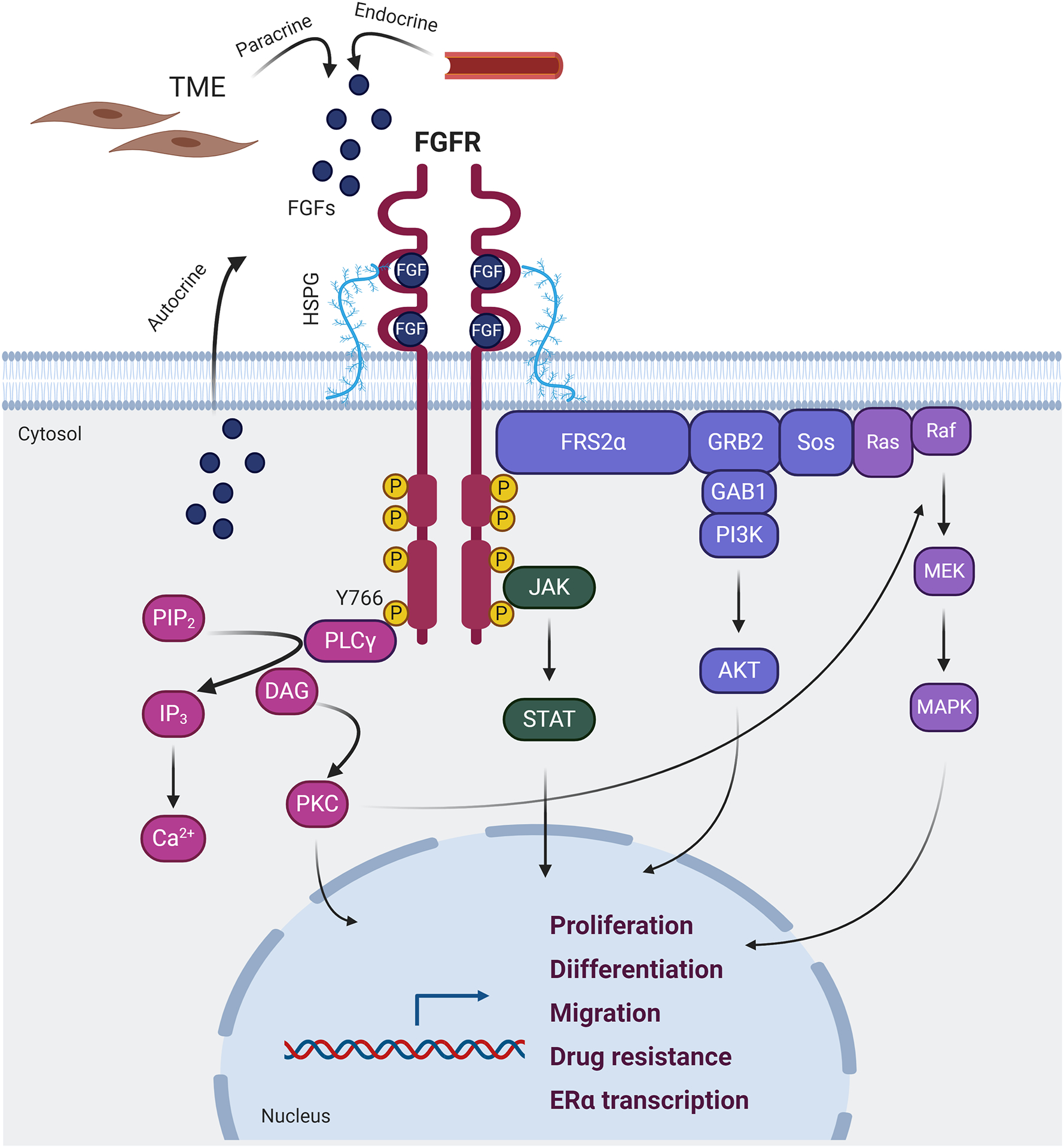
The binding of FGFs to the extracellular domain of FGFR induces dimerization of the receptor and phosphorylation of its intracellular C-terminal domain. The C-terminal phosphorylated tyrosine residues serve as anchors for adaptor proteins that, in turn, activate four distinct signaling cascades: RAS-RAF-MEK-MAPK, PI3K-AKT, JAK-STAT and PLCγ. The activation of these pathways ultimately promotes the expression of transcriptional profiles associated with cell proliferation, differentiation, migration, drug resistance and ERα signaling. TME: tumor microenvironment; HSPG: heparan sulfate proteoglycan; FGFs fibroblast growth factors; FGFR: fibroblast growth factor receptor; PLCγ: phospholipase Cγ; PIP2: phosphatidylinositol-4,5-biphosphate; DAG: diacylglicerol; IP3: inositol triphosphate; PKC: protein kinase C; FRS2α: FGFR substrate 2α; GRB2: growth factor receptor-bound 2; GAB1: GRB2-associated binding protein 1; SOS: son of sevenless; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; JAK: janus activated kinase; STAT: signal transducer and activator of transcription; ERα: estrogen receptor α.
In addition to canonical signaling induced by membrane-bound receptors, FGFRs have been shown to localize in the nucleus of cells in various tissues. Both clathrin-mediated endocytosis (CME) and clathrin-independent endocytosis (CIE) are involved in FGFR internalization from the plasma membrane after ligand binding (Figure 2) (Fannon and Nugent, 1996; Reilly et al., 2004; Sorokin et al., 1994). Internalization and nuclear translocation of FGFRs is mediated by importin-β (Reilly and Maher, 2001). ‘Retrotranslocation’ to nucleus of newly synthesized full-length FGFR protein from the ER/Golgi has also been described (Stachowiak and Stachowiak, 2016). In addition, a truncated FGFR1 variant, derived from the proteolytic activity of granzyme B on membrane-bound FGFR1, has been detected in breast cancer cell nuclei (Chioni and Grose, 2012). The role of nuclear FGFR1 in the context of neuronal development has been extensively characterized, demonstrating its direct involvement in promoting gene transcription associated with developmental pathways (Stachowiak et al., 2003). Finally, there is experimental evidence that FGFR1 and FGFR2 can localize in the nucleus of cancer cells, such as in medulloblastoma, breast, pancreatic and prostate cancer (Cerliani et al., 2011; Coleman et al., 2014; Formisano et al., 2017; Lee et al., 2019). These findings are concordant with reports showing that RTKs, such as EGFR, HER2, and insulin receptor (INSR), in addition to their signal transduction function as membrane-bound receptors, can localize in the nucleus and regulate gene transcription (Chervo et al., 2020; Hancock et al., 2019; Lo and Hung, 2006).
Figure 2. Mechanisms of FGFR1 nuclear translocation and nuclear FGFR1 activity in breast cancer.
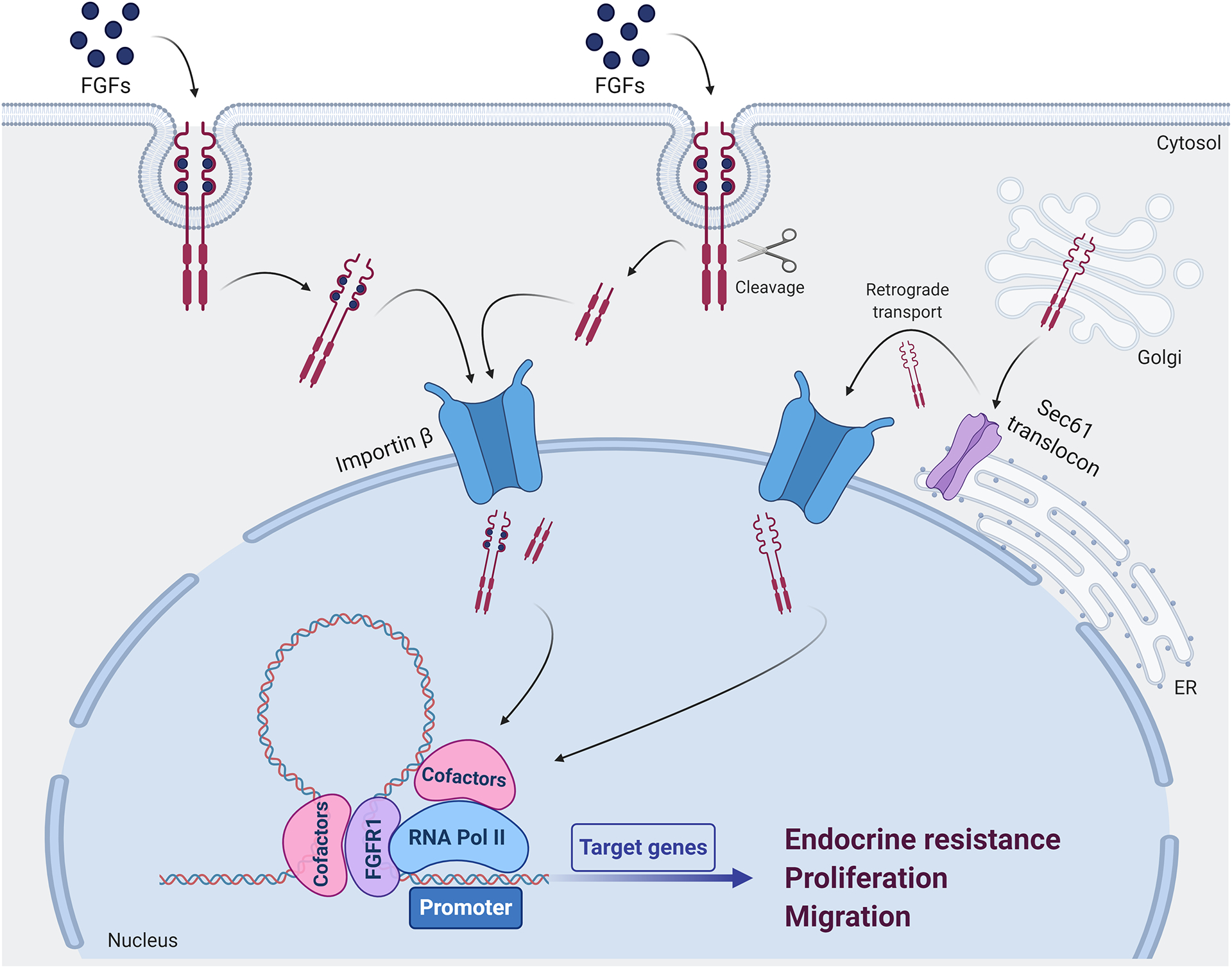
Following FGFs binding, full-length FGFR1 can undergo clathrin-mediated or clathrin independent endocytosis. In addition, upon FGFs binding, the intracellular domain of FGFR1 can be subjected to the proteolytic activity of granzyme B, which induces cleavage of the receptor recognizing the Asp432 residue. Both full-length and cleaved FGFR1 migrate into the nucleus via Importin β system. Furthermore, newly synthesized FGFR1 can be directed into the nucleus via a retrograde transport system involving the ER-associated Sec61 translocon and Importin β. In the nucleus, FGFR1 associates with RNA Polymerase II in a chromatin-bound complex of coregulators and contributes to regulate the transcription of genes associated with endocrine resistance, cell proliferation and migration. FGFs: fibroblast growth factors; ER: endoplasmic reticulum; RNA Pol II: RNA polymerase II.
FGFR alterations in breast cancer
Several genomic alterations occurring at FGFs and/or FGFRs genes have been reported in breast cancer, promoting activation of canonical FGFR signaling cascade and nuclear FGFR transcriptional activity. These alterations are summarized in Figure 3.
Figure 3. Mechanisms of FGFR signaling activation in breast cancer.
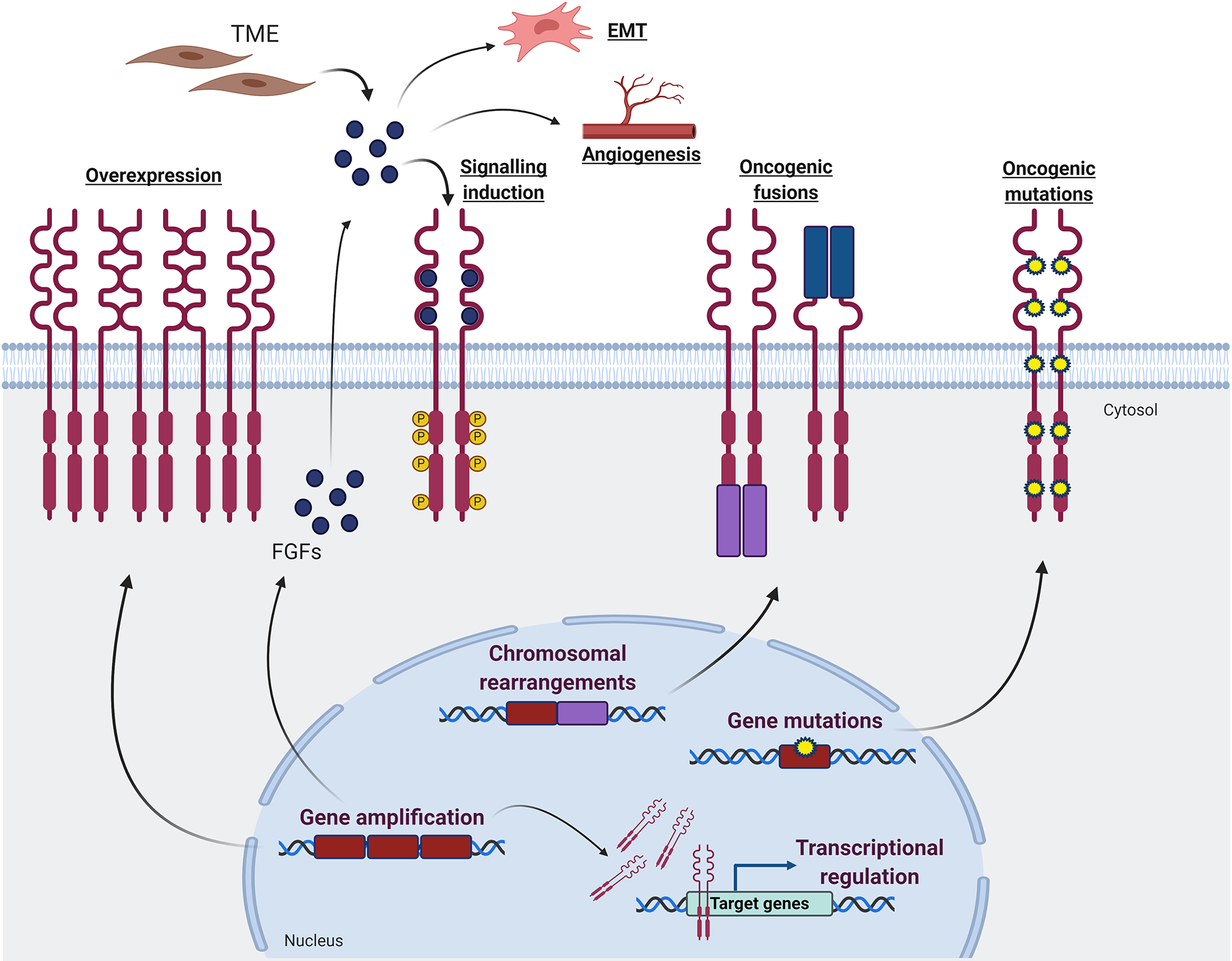
Various mechanisms contribute to enhanced oncogenic FGFR signaling in breast cancer. Gene amplification leads to overexpression of the membrane-bound receptors, ultimately enhancing the canonical signaling cascades induced by FGFRs activation. In addition, overexpression of nuclear FGFR1 contributes to transcriptional regulation. Gene amplification also leads to overexpression of FGF ligands, which in turn hyperactivate the FGFR signaling pathway. FGFs may also promote angiogenesis and epithelial-mesenchymal transition. Chromosomal rearrangements translate into oncogenic fusions, in which a protein partner can be fused at the FGFR C-terminal domain (violet fusion), promoting receptor dimerization and consequent activation of intracellular signaling cascades. In addition, FGFRs can be under the control of the promoter of a protein fused at the N-terminal domain (blue fusion), resulting in a ligand-independent activation. Point mutations can occur at both extra- and intracellular FGFRs domains, leading to ligand-independent receptor dimerization and/or constitutive activation of the C-terminal kinase domain. TME: tumor microenvironment; EMT: epithelial-mesenchymal transition; FGFs: fibroblast growth factors.
FGFR Amplification
The 8p11–12 locus, where the FGFR1 gene resides, is amplified in about 12% of breast cancers. The highest rate is registered among ER+ breast tumors, which display FGFR1 amplification in about 15% of cases (Cerami et al., 2012; Gao et al., 2013). FGFR1 amplification has been described as a strong predictor of poor prognosis in ER+ tumors (Elbauomy Elsheikh et al., 2007). Instead, FGFR2 amplification (genomic locus 10q26) is rare and found in ~ 2% of all breast cancers, with a slightly higher occurrence in ER negative/HER2 negative subtype (~3%) (Cerami et al., 2012). Reis-Filho et al performed siRNA FGFR1 studies to reveal that FGFR1 contributes to the survival of ER+/FGFR1-amplified breast cancer cells (Reis-Filho et al., 2006). However, later studies revealed that only tumors with high-level clonal FGFR1/2 amplification show addiction to the amplified receptor and exquisite sensitivity to FGFR TKIs (Pearson et al., 2016). Pearson et al demonstrated in FGFR2-amplified cancers that FGFR2 hyperactivates RAS/MAPK and PI3K signaling, likely through cooperation with other RTKs, such as ERBB3 and IGF1R (Kunii et al., 2008; Pearson et al., 2016). As a result, these tumors exhibit high dependency on FGFR2 and are also sensitive to FGFR inhibition. The less clear tumor dependence on FGFR1 amplification may be attributed to various reasons. First, low-level amplification may not translate to FGFR1 protein overexpression (Wynes et al., 2014). Second, the 8p11–12 amplicon that includes the FGFR1 locus is generally broad, implying the co-amplification of other genes contribute to oncogenesis. For instance, ZNF703, also in 8p11–12, has been shown to promote proliferation of human luminal breast cancer cells and its overexpression has been associated with a poor outcome in patients with ER+ breast cancers (Holland et al., 2011; Reynisdottir et al., 2013).
The rate of FGFR3 and FGFR4 amplification in breast cancer is negligible. The oncogenic role of these receptors is generally associated with their overexpression, activating mutations or amplification of their ligands. It has been recently demonstrated that FGFR4 overexpression is significantly higher in the HER2 enriched molecular subtype and may be associated with breast cancer dedifferentiation (Garcia-Recio et al., 2020). In this article, the authors reported an FGFR4-associated gene expression signature that is enriched in ER+ breast cancer metastases (Garcia-Recio et al., 2020).
FGFR activating mutations
In contrast to other tumors, such as urothelial cancer and intrahepatic cholangiocarcinoma (ICC), the rate of FGFRs mutations is very low in treatment naïve breast cancer. According to Project GENIE, only 413/11,746 (3.5%) breast cancers harbor FGFR1–4 mutations (AACR Project GENIE Consortium, 2017). FGFR1 N546K and K656E are the most frequent and best characterized. Both mutations are in the tyrosine kinase (TK) domain, constitutively activate the receptor, and result in oncogenic transformation (Hart et al., 2000; Lew et al., 2009). FGFR1 N546K has been detected in the liver biopsy of a patient with ER+ breast cancer progressing on antiestrogens (Mao et al., 2020) and in plasma tumor ctDNA in a patient with ER+ breast cancer progressing on endocrine therapy plus CDK4/6 inhibitors (Formisano et al., 2019), suggesting its causal role with drug resistance.
FGFR2 mutations occur in less than 1% of treatment naïve breast cancers (Cerami et al., 2012; Gao et al., 2013). However, acquired FGFR2 mutations have been reported with higher frequency in ER+ breast cancer that developed resistance to antiestrogens ± CDK4/6 inhibitors. Particularly, missense mutations of the TK domain, such as M538I, N549K (or N550K, corresponding to N549K in the canonical FGFR2 isoform), K569E and K660N (Formisano et al., 2019; Mao et al., 2020; O’Leary et al., 2018) have been detected. These mutations constitutively activate the FGFR kinase and confer transforming potential to it (Chen et al., 2007; Hart et al., 2000). The variant of unknown significance (VUS) FGFR2 V395D has been also identified in one post-progression plasma sample from a patient progressing on antiestrogens and CDK4/6 inhibitors (Formisano et al., 2019). In contrast to endometrial cancer, where FGFR2 mutations are detected in about 10% of cases (Helsten et al., 2016), FGFR2 mutations in the extracellular Ig II and Ig III loops, promoting increased ligand-receptor binding or constitutive receptor dimerization (Ibrahimi et al., 2001, 2004; Wilkie et al., 2002), have been rarely reported in breast cancer (AACR Project GENIE Consortium, 2017).
FGFR3 mutations are also very rare in breast cancer. Instead, FGFR4 mutations can be found in around 4% of primary breast cancers. They generally occur in the TK domain, such as K535 or E550. Mutations occurring in these residues were previously demonstrated to increase autophosphorylation, enhance Stat3 signaling and promote metastatic potential in preclinical models of rhabdomyosarcoma (Taylor et al., 2009). Razavi and colleagues found significant enrichment of FGFR4 mutations in metastatic biopsies of ER+/HER2−negative breast cancer compared to primary tumors (Razavi et al., 2018). These results are concordant with recent findings supporting the notion that FGFR4 signaling promotes metastasis in breast cancer (Garcia-Recio et al., 2020).
FGFR fusions
FGFR fusions, particularly involving FGFR2 and FGFR3 genes, are relatively frequent in glioblastoma, bladder cancer and ICC, but rare in breast cancer. Their detection is important as tumors bearing these alterations are exquisitely sensitive to FGFR TKIs. The most common FGFR3-fused gene is TACC3, encoding transforming acid coiled-coil containing protein 3. TACC3 replaces the final exon in the C-terminal domain of FGFR3, resulting in constitutive kinase activity of the fused protein. Several partners, fused to the C-terminus of FGFR2 and under control by the FGFR promoter, have been reported, such as BicC family RNA binding protein 1 (BICC1), pro coiled-coil domain-containing protein 6 (CCDC6), oral-facial-digital syndrome 1 protein (OFD1), Rho-interacting kinase (CIT), cell cycle and apoptosis regulator protein 2 (CCAR2). The protein products of these fusions exhibit enhanced dimerization and ligand-independent signaling (Parker et al., 2014; Wu et al., 2013).
More rarely, fusion partners locate at the N-terminal domain of FGFR. These include fusions of the prohibitin-containing protein ER lipid raft associated 2 (ERLIN2) with FGFR1 and of solute carrier family 45 member 3 (SLC45A3) with FGFR2 (Wu et al., 2013). A unique mechanism of activation has been described for the SLC45A3-FGFR2 fusion, where FGFR2 is regulated by the androgen-regulated promoter of SLC45A3, thereby promoting FGFR2 overexpression (Wu et al., 2013).
Oncogenic role of FGF ligands
Tumorigenic properties of FGF ligands have been previously investigated. FGF1, FGF2, FGF6, FGF8 and endocrine ligands FGF19 and FGF23 have been reported to be involved in tumor initiation and progression in prostate cancer [reviewed in (Corn et al., 2013)]. Recent evidence also supports a role of various FGFs in breast cancer. The chromosomal locus 11q13, containing FGF3, FGF4, FGF19 and CCND1, is amplified in about 15% of breast cancers (Cerami et al., 2012). CCND1 is considered the most important oncogene in the amplicon, particularly for ER+ breast tumors. In hepatocellular carcinoma (HCCs), 11q13 amplification positively correlates with FGF19, which has been shown to promote liver carcinogenesis through binding to its receptor, FGFR4, and to predict clinical response to the selective FGFR4 inhibitor fisogatinib (Blu-554) (Kim et al., 2019). Mao et al detected FGF3 amplification in 17 post-treatment ER+ breast cancer biopsies from women treated with antiestrogens. In 4/17 cases, FGF3 amplification was not detected in the corresponding pre-treatment biopsy. In 3/4 of these tumors FGF3 copy number gains occurred in the absence of CCND1 amplification (Mao et al., 2020).
FGF2 has been reported as one of the major soluble factors in tumor microenvironment (TME) of ER+ breast cancers, where it can confer resistance to antiestrogens and PI3K/mTOR inhibitors. Indeed, FGF2 stimulation activates MAPK signaling, resulting in downregulation of BCL-2-interacting mediator of cell death (Bim) and upregulation of Cyclin D1. These events prevent the apoptosis and the proliferative arrest induced by antiestrogens and PI3K/mTOR inhibitors (Shee et al., 2018). Further, extracellular-matrix bound FGF2 has been shown to induce ligand-independent ERα signaling through MAPK activation, also contributing to antiestrogen resistance (DiGiacomo et al., 2021). These findings shed light on the crucial role of TME-secreted FGFs in tumorigenesis, tumor progression and resistance to antiestrogens in breast cancer.
Nuclear FGFRs
Several reports support a role of nuclear FGFRs in breast cancer. Confocal microscopy and immunoprecipitation assays have demonstrated that FGFR2, STAT5 and Progesterone Receptor (PR) colocalize as a complex in the nucleus of breast cancer cells (Cerliani et al., 2011). Binding of FGFR2 to its nuclear partners is enhanced upon FGF2 and Medroxyprogesterone Acetate (MPA) stimulation. The FGFR2/PR/STA5 colocalization was also demonstrated in primary breast cancer biopsies from treatment-naïve patients. The FGFR2/STAT5/PR complex binds to Progesterone Response Elements (PRE) and Interfon-γ-activated sequences (GAS), regulating the expression of the genes containing these motifs in their regulatory regions (Cerliani et al., 2011).
FGFR1 can localize in the nucleus of breast cancer cells as a truncated peptide or as a full-length receptor (Figure 2). Loeb et al previously demonstrated that Asp432 in the juxtamembrane domain of FGFR1 is a direct substrate of granzyme B, which induces cleavage of the RTK (Loeb et al., 2006). Upon FGF10 stimulation, cleaved FGFR1 can accumulate in the nucleus of MCF-7 cells, thus promoting cell migration (Chioni and Grose, 2012). In addition, the full length receptor has been shown to physically associate with ERα in breast cancer cell nuclei, influencing ligand-independent ERα genomic distribution and transcription (Formisano et al., 2017). Using ChIP-Seq and Mass Spectrometry, we recently reported that FGFR1 associates with phosphorylated RNA-Polymerase II (Pol II) at gene promoters and this binding overlaps with the active transcription histone marks H3K27ac and H3K4me3 (Servetto et al., 2021). Integration of ChIP-Seq and RNA-Seq results further suggested that FGFR1 has a direct role in transcriptional regulation in breast cancer (Servetto et al., 2021).
Many questions about the role of nuclear FGFR1 in breast cancer remain unanswered. First, it is not clear whether the translocation of FGFR1 to the nucleus follows and/or requires receptor dimerization and activation at the plasma membrane. Second, it is unclear whether FGFR1-mediated gene transcription requires receptor’s TK activity. Next, the role of FGF ligands and FGFR1-interacting proteins on the modulation of nuclear FGFR1 activity have not been fully elucidated. Finally, it is unknown whether FGFR1 has a specific DNA-binding motif in its amino acid sequence, so that the receptor can directly bind DNA, or whether it needs other cofactors mediating the recruitment and tethering to target genomic loci.
Role of FGFRs in endocrine resistance
FGFRs overexpression, gene copy number alterations, mutations and fusions have been shown to promote antiestrogen resistance in breast cancer. FGFR1 amplification is associated with a worse prognosis in ER+ breast cancer patients (Elbauomy Elsheikh et al., 2007). Turner et al found significant correlation between FGFR1 gene copy number, mRNA expression and protein levels in ER+ breast cancer biopsies and cell lines, with a remarkable enrichment of FGFR1 amplification in the Luminal B subtype (Turner et al., 2010). These authors also demonstrated that siRNA-mediated FGFR1 knockdown in ER+/FGFR1-amplified breast cancer cells restored sensitivity to tamoxifen (Turner et al., 2010), further suggesting a role for FGFR1 in antiestrogen resistance.
Further studies have identified FGFR1 amplification as mechanism of intrinsic resistance to antiestrogens. Whole-exome sequencing of 155 ER+/HER2− early breast cancers from patients treated with the aromatase inhibitor letrozole for 10–21 days prior surgery, revealed that amplification of locus 8p11.12 was associated with high post-treatment Ki67 (Giltnane et al., 2017). Interestingly, FGFR1 IHC of paired pre- and post-treatment ER+/FGFR1-amplified breast cancer biopsies from patients treated with pre-operative letrozole revealed an increase in FGFR1 levels in post-treatment samples (Formisano et al., 2017), further suggesting that FGFR1 overexpression may represent an adaptive mechanism of escape to antiestrogen treatment. These findings were mimicked in vitro, where short-term and long-term estradiol deprivation (LTED) resulted in FGFR1 overexpression in ER+/FGFR1-amplified breast cancer cell lines. In addition to 8p11.12, amplification of 11q13, comprising FGF3, FGF4, FGF19 and CCND1 genes correlated with high post-letrozole Ki67 values (Formisano et al., 2017; Giltnane et al., 2017). Concordant with these findings, LTED also resulted in upregulation of FGF3, FGF4 and FGF19 in ER+ breast cancer cells (Formisano et al., 2017). Consistent with these data, in a cohort of 73 patients with ER+ metastatic breast cancer and treated with first line endocrine therapy, patients harboring FGFR1-amplified tumors (n=20) experienced worse time to progression (TTP) compared to those with non FGFR1-amplified tumors (n=53) (Drago et al., 2019). In the same study, patients with FGFR1-amplified tumors and treated with endocrine therapy plus CDK4/6 inhibitors experienced a worse TTP.
These clinical findings are supported by experimentally data. For example, Several reports demonstrated that ectopic overexpression of FGFR1 in ER+ breast cancer cells induces resistance to fulvestrant in vitro (Drago et al., 2019; Formisano et al., 2019; Mao et al., 2020). An Open Reading Frame (ORF) kinome screen performed in MCF-7 cells, evaluating 559 kinases, identified FGFR1 among the 15 candidate genes conferring resistance to fulvestrant and also among the 17 candidate genes promoting resistance to combination of fulvestrant and the CDK4/6 inhibitor ribociclib (Formisano et al., 2019). Concordant with these results, a genome-scale gain of function screen, evaluating 17,255 ORFs corresponding to 10,135 genes, revealed that genes involved in the FGFR signaling pathway significantly contribute to resistance to fulvestrant and the orally bioavailable SERD GDC-810 in MCF7 and T47D cells (Mao et al., 2020).
In addition to these findings, FGFR signaling has been associated with resistance to combination of antiestrogens with the PI3Kα inhibitor alpelisib or with CDK4/6 inhibitors. In a cohort of 26 patients with metastatic ER+ breast cancer treated with letrozole plus alpelisib, FGFR1 amplification was associated with absence of clinical benefit (Mayer et al., 2017). Constitutive overexpression of FGFR1 in the ER+/PIK3CAmut MCF7 cells significantly reduced sensitivity to alpelisb, corroborating the clinical findings (Mayer et al., 2017). Next, a subgroup analysis of 212 patients with ER+/HER2− metastatic breast cancer treated with letrozole plus ribociclib in the MONALEESA-2 trial revealed that 8p11.23 amplification (n=10) detected in ctDNA was associated with a worse progression free survival (PFS) compared to patients without 8p11.23 amplification (n=202; median PFS, 10.61 vs 24.84 months) (Formisano et al., 2019). FGFR1 mRNA levels were further investigated by NanoString 230-gene nCounter GX Human Cancer Reference panel in 197 tumor biopsies from patients enrolled in the MONALEESA-2 trial and treated with letrozole plus ribociclib. Notably, high FGFR1 mRNA levels (n=104) were associated with worse PFS, compared to to those patients with low FGFR1 mRNA (22.21 months vs not reached) (Formisano et al., 2019).
FGFR2 alterations have been also associated with antiestrogen resistance. The FGF7/FGFR2 axis has been shown to enhance PI3K/AKT-mediated phosphorylation of ERα on Ser118 and Ser167 and, as a result, confer resistance to tamoxifen in ER+ breast cancer cells (Turczyk et al., 2017). WES analysis of paired pre- and post-treatment metastatic tumor biopsies or liquid biopsies from 60 ER+ metastatic breast cancer patients treated with antiestrogens, revealed the emergence of FGFR2 amplification (3/60) and FGFR2 mutations (2/60), with one patient exhibiting both FGFR2 mutation and amplification (Mao et al., 2020). In this dataset, the rate of FGFR2 genomic alterations (6.7%) was higher than the rate in datasets of treatment-naïve ER+ primary breast cancer (less than 2%). Clonal evolution analysis revealed that the FGFR2 mutations (M538I and N550K) were acquired in the post-treatment biopsies. In mechanistic studies, overexpression of wild type or mutant FGFR2 in ER+ breast cancer cells hyperactivated MAPK signaling and induced cross resistance to fulvestrant and palbociclib (Formisano et al., 2019; Mao et al., 2020).
The only study reporting a role for FGFR3 in endocrine resistance was conducted by Tomlinson et al, who investigated FGFR3 expression in 429 ER+ primary breast cancer biopsies from patients treated with adjuvant tamoxifen (Tomlinson et al., 2012). FGFR3 protein levels were higher in patients who experienced disease relapse during the treatment with tamoxifen, suggesting that FGFR3 may also be involved in resistance to treatment.
Other studies have reported a high occurrence of FGFR4 alterations in metastatic breast cancer, suggesting a potential involvement in endocrine resistance. Levine et al (Levine et al., 2019) investigated the rate of FGFR4 mutations in three large datasets of metastatic ER+ breast cancers and sequencing data from Foundation Medicine (Lefebvre et al., 2016; Razavi et al., 2018; Robinson et al., 2017). In all these datasets, FGFR4 mutations were significantly enriched in metastatic compared to primary tumors. The same authors examined a cohort of 29 matched primary and metastatic biopsies, with the latter biospecimens collected at recurrence after endocrine therapy. RNA-Seq analysis of these tumor biopsies revealed a significant enrichment of FGFR4 mRNA levels in metastatic biopsies (Levine et al., 2019). In line with these data, FGFR4 mRNA and protein levels are highly enriched in LTED ER+ breast cancer cells compared to parental cells. In addition, Garcia-Recio et al (Garcia-Recio et al., 2020) showed that ectopic overexpression of FGFR4 in MCF7 and T47D cells promoted higher estrogen-independent growth compared to parental cells, also suggesting a potential causal role of FGFR4 in antiestrogen resistance.
Finally, our recent study proposed a role of nuclear FGFR1 in antiestrogen resistance. In a cohort of 155 primary ER+ breast cancer biopsies from patients treated for 10–21 days with pre-operative letrozole, nuclear FGFR1 levels, measured by IHC, positively correlated with on-letrozole Ki67 values (Servetto et al., 2021). In the same report, MCF-7 cells transduced with an FGFR1 expression vector containing a nuclear localization signal (NLS) exhibited higher estrogen-independent growth and reduced sensitivity to fulvestrant compared to control cells. MCF7 xenografts stably transduced with FGFR1-NLS also showed reduced sensitivity to fulvestrant compared to control tumors (Servetto et al., 2021).
Clinical development of FGFR inhibitors in ER+ breast cancer
FGFR targeted therapies mainly consist of small molecules tyrosine kinase inhibitors, tailored against the ATP-binding pocket in the FGFR TK domain. These inhibitors can be divided in two classes: multi-targeting TKIs, able to bind and inhibit various RTKs, and selective TKIs that are specific to the FGFR TK. These inhibitors have been tested in preclinical and clinical studies against various cancer types with or without FGFR alterations suggestive of oncogene dependence. A summary of clinical trials with FGFR TKIs is shown in Table 1.
Table 1:
FGFR TKIs tested or currently in clinical trials for advanced/metastatic breast cancer.
| Inhibitor | Phase | Description of treatment arms | Primary Endpoint | Main results | Clinical trial identifier |
|---|---|---|---|---|---|
| Non-selective inhibitors | |||||
| Dovitinib | II |
|
ORR |
|
NCT00958971 |
| II | ER+/HER2− patients.Stratification based on FGFR1, FGFR2 or FGF3 amplification and presence of visceral disease |
|
|
NCT01528345 | |
| I/II | Dovitinib + aromatase inhibitors in ER+/HER2− breast cancer (n=12) | CBR (at 24 weeks) | N/A (study terminated) | NCT01484041 | |
| Lucitanib | I/IIa | Advanced solid tumors:
|
|
In FGF aberrant breast cancer cohort:
|
NCT01283945 |
| II | ER+/HER2− MBC
|
ORR | In FGFR1amp cohort:
|
NCT02053636 | |
| Selective inhibitors | |||||
| AZD4547 | II | HER2−/FGFR1amp breast cancerd (n=8) | ORR | PR = 12.5% (n=1) | EudraCT No. 2011-003718-18 |
| II | Advanced solid tumors with FGFR aberrations (n=48):
|
ORR | In Breast cancer cohort: None of the patients reached PR or SD ≥ 6 months |
NCT02465060 | |
| AZD4547 | IIa | ER+/HER2− breast cancer, with FGFR1 polysomy or gene amplification
|
Safety | N/A (study terminated) | NCT01202591 |
| IIa | ER+/HER2− MBCf
|
|
N/A | NCT01791985 | |
| II | HER2− MBC with FGFR alterationsg
|
PFS | N/A (not recruiting; pending results) | NCT02299999 | |
| Infigratinib (NVP-BGJ398) | Ib | ER+/HER2−/FGFRaltered MBC
|
DLTs | N/A (active recruiting) | NCT04504331 |
| Ib | Advanced solid tumors with concomitant PIK3CA mutations and FGFR1–3 alterations
|
DLTs | N/A (recruitment completed) | NCT01928459 | |
| Zoligratinib (Debio-1347 | Ib | ER+/HER2−/FGFR amplified MBC
|
MTD | N/A (active, not recruiting) | NCT03344536 |
| Erdafitinib (JNJ-42756493) | Ib | ER+/HER2−/FGFR amplified MBCh
|
DLTs and MTD | N/A (active, not recruiting) | NCT03238196 |
| II | Advanced solid tumors or lymphomah Subprotocol K1-K2i: patients with FGFR amplification, mutations or fusions |
ORR | N/A (recruiting) | NCT02465060 | |
| Futibatinib (TAS-120) | II | Locally advanced/MBC with FGFR1–2 amplificationh
|
|
N/A (recruiting) | NCT04024436 |
| Rogaratinib (Bay 1163877) | I | Advanced ER+/HER2− FGFR1–3 positivel,m
|
|
N/A (recruiting) | NCT04483505 |
| Pemigatinib | I/II | Advanced solid tumors with FGF/FGFR alterations
|
|
N/A (recruiting) | NCT02393248 |
ER+: estrogen receptor positive; HER2: human epidermal growth factor receptor 2; FGFR: fibroblast growth factor receptor; FGF: fibroblast growth factor; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; MBC: metastatic breast cancer; ORR: overall response rate; PR: partial response; PFS: progression free survival; CBR: clinical benefit rate; SD: stable disease; CR: complete response; MTD: maximum tolerated dose; DLT: dose limiting toxicity; RP2D: recommended phase II dose; TEAEs: treatment emergent adverse events; N/A: not available;
Partial Response not confirmed at following radiological assessment;
FGF pathway amplified: NO, n=32 (68.1%); YES, n=15 (31.9%);
FGF pathway amplified: NO, n=33 (66.0%); YES, n=17 (34.0%);
Estrogen Receptor status not specified;
Patients progressed on previous endocrine therapy;
Patients not selected based on FGF/FGFR status;
Patients who received 6 to 8 cycles of chemotherapy, or having received at least 4 cycles of chemotherapy definitively stopped for toxicity reasons, and who are presenting a SD or PR at randomization, as per SAFIR02_BREAST study protocol;
At least one previous line of therapy in the metastatic setting;
Subprotocols of “The MATCH Screening Trial”;
Measurable disease;
Non-measurable disease;
FGFR1–3 positivity evaluated by RNA-Scopre and/or FISH. Cut-offs not available;
Patients progression on combination of aromatase inhibitor plus palbociclib;
Combination therapies with pemigatinib: gemcitabine + cisplatin; pembrolizumab; docetaxel; trastuzumab; INCMGA00012.
Multi-targeting TKIs
Dovitinib (TKI258, CHIR-258) is an orally bioavailable small molecule targeting Fms related receptor tyrosine kinase 3 (FLT3), KIT, Vascular Endothelial Growth Factor Receptors 1–3 (VEGFR1–3), FGFR1, FGFR3, Platelet Derived Growth Factor Receptors α-β (PDGFRα-β) and Colony Stimulating Factor Receptor 1 (CSF1R) (Lee et al., 2005). It was initially tested in renal cell carcinoma (RCC) without success (Angevin et al., 2013; Motzer et al., 2014). Andre et al (André et al., 2013) demonstrated the selective antiproliferative effect of dovitinib in FGFR1-amplified and FGFR2-amplified breast cancer cell lines compared to cells without FGFR amplification. However, in a cohort of 20 patients with ER+/FGFR1-amplified breast cancer, the clinical benefit of dovitinib, defined as complete response (CR) or partial response (PR) or stable disease (SD) (RECIST criteria) ≥24 weeks, was observed only in 3/20 (15%) (André et al., 2013). A more recent phase II clinical trial tested the combination of dovitinib plus fulvestrant versus placebo plus fulvestrant in postmenopausal ER+/HER2− breast cancer patients progressing on prior endocrine therapy (Musolino et al., 2017). Patients were randomized based on FGF pathway amplification status (defined as at least 6 copies of FGFR1, FGFR2 or FGF3, measured by qPCR in a tumor biopsy). In the group with FGF pathway amplification (n=31), dovitinib treatment (n=16) improved the median PFS compared to placebo (n=15; median PFS 10.5 vs 5.5 months, respectively; HR=0.64).
Lucitanib (E-3810) is a small molecule inhibitor active against VEGFR1–3, FGFR1–3, PDGFRα and CSF1R (Bello et al., 2011). In a phase I/IIa clinical trial testing the effect of lucitanib in advanced solid tumors, among breast cancer patients bearing FGFR pathway alterations (n=8, FGFR1-amplification; n=4, 11q amplification), 6 patients exhibited a partial response and 6 stable disease as best response by RECIST (Soria et al., 2014). In laboratory studies, single agent lucitanib had very modest activity against ER+/FGFR1-amplified breast cancer cells/tumors and PDXs (Formisano et al., 2017, 2019) but synergized with fulvestrant and CDK4/6 inhibitors (Formisano et al., 2019). Recent results of the FINESSE study revealed that lucitanib had modest antitumor activity in patients with ER+/FGFR1-amplified tumors, with an overall response rate (ORR) of 19% (Hui et al., 2020).
Ponatinib and Brivanib are two other multi-targeting TKIs that have shown promising antitumor activity in preclinical models of breast cancer, but they have not been tested clinically (Patel et al., 2010; Shao et al., 2019).
Selective FGFR TKIs
AZD4547 is an orally bioavailable selective inhibitor of FGFR1–3, with an enzymatic IC50 <5 nM (Gavine et al., 2012). Pearson et al tested the effect of AZD4547 in 8 patients with ER+/FGFR1-amplified breast cancer and 9 patients with FGFR2-amplified gastroesophageal cancers. Interestingly, only 1/8 patients with breast cancer and 3/9 patients with gastric cancer had a confirmed response to AZD4547 (Pearson et al., 2016). AZD4547 was also tested in 48 patients in the NCI-MATCH trial, which enrolled tumors harboring FGFR1 or FGFR2 amplification (n = 20), FGFR2 or FGFR3 single-nucleotide variants (SNVs) (n = 19), and FGFR1 or FGFR3 fusions (n = 9) (Chae et al., 2020). A partial response was observed in only 8% of patients, all presenting FGFR1–3 point mutations or fusions. Of the 48 enrolled patients, 16 had breast cancer, with FGFR1 amplification (n=11), FGFR2 amplification (n=2), and FGFR2 mutations (n=3). None of these patients reached PR or SD ≥6 months as best response. Two clinical trials (NCT01202591 and NCT01791985) have evaluated the effect of the combination of endocrine therapy and AZD4547 in metastatic ER+ breast cancer but results have not been reported at this time. Results from the SAFIR02 trial (NCT02299999), testing the effect of targeted treatment based on the identified genomic aberration in breast cancer, may clarify the role of AZD4547 in breast cancers harboring FGFR alterations.
Infigratinib (NVP-BGJ398) is an orally available FGFR1–3 TKI, mainly tested in urothelial cancer and ICC with FGFR2/3 aberrations (Javle et al., 2018; Nogova et al., 2017). The NCT04504331 trial is testing the effect of the combination of infigratinib with tamoxifen in patients with ER+ breast cancers harboring FGFR pathway alterations. Results from this trial are pending.
Zoligratinib (Debio-1347) is an FGFR1–3 TKI with promising antitumor activity in solid cancers with FGFR1–3 genetic alterations (Voss et al., 2019). This agent is currently tested in combination with fulvestrant in patients with metastatic ER+ breast cancer (NCT03344536).
Erdafitinib (JNJ-42756493, Balversa) is a pan-FGFR kinase inhibitor, recently approved for the treatment of patients with metastatic urothelial carcinoma harboring FGFR2/3 somatic alterations who had progressed on platinum-based chemotherapy (Loriot et al., 2019). Combination of fulvestrant, palbociclib and erdafitinib has shown relevant antitumor activity in ER+/FGFR1-amplified breast cancer cells and PDXs (Formisano et al., 2019). These results led to development of the phase Ib clinical trial (NCT03238196), testing the effect of the triple ER, CDK4/6 and FGFR blockade in ER+/FGFR-amplified breast cancer. Preliminary results of this trial suggests clinical activity of the triplet combination limited to tumors with high FGFR1 amplification (Mayer et al., 2021).
Futibatinib (TAS-120) is a third-generation irreversible pan-FGFR inhibitor that binds to a highly conserved cysteine in the ATP pocket of FGFR (C492 in the FGFR2-IIIb isoform) (Kalyukina et al., 2019). This drug displayed encouraging results in ICC harboring FGFR2 fusions, including patients who developed resistance to other FGFR TKIs such as NVP-BGJ398 and Debio-1347 (NCT02052778) (Goyal et al., 2019). Futibatinib has strong antiproliferative activity against MDA-MB-134 ER+/FGFR1-amplified and MFM-223 ER-/FGFR2-amplified breast cancer cells (Sootome et al., 2020). TAS-120 is currently tested in the phase II FOENIX-MBC2 clinical trial, which enrolls patients with locally advanced/metastatic breast cancer with FGFR1–2 amplification (NCT04024436). This trial includes the addition of fulvestrant in the cohort of ER+ breast cancer patients.
Rogaratinib (Bay 1163877) is a potent pan-FGFR inhibitor that has shown an antitumor effect in several preclinical cancer models with FGFR pathway alterations (Grünewald et al., 2019). In these studies, sensitivity to rogaratinib has correlated with FGFR mRNA expression levels. A phase I dose escalation trial (NCT04483505) is testing the combination of rogaratinib, palbociclib and fulvestrant in patients with ER+ breast cancer progressing on an aromatase inhibitor plus a CDK4/6 inhibitor, that were positive for FGFR1–3 as measured by RNA-scope and/or FISH.
Pemigatinib (INCB054828) is a potent selective FGFR1–3 inhibitor, approved for the treatment of patients with unresectable or metastatic ICC harboring FGFR2 fusions or rearrangements (Abou-Alfa et al., 2020). FIGHT-101 is a phase 1/2 trial (NCT02393248) testing the effect of pemigatinib in patients with solid tumors harboring genomic alterations in FGF ligands or FGFRs.
Challenges in targeting FGFR signaling
The clinical development of FGFR inhibitors in solid malignancies, including breast cancer, has been challenging for several reasons. First is the possibility that inadequate criteria have been used to select patients for study enrollment. FGFR amplification, by FISH, has been generally defined as ratio gene/centromere >2 or copy number >6. However, it has been shown that only tumors with high levels of FGFR1/2 amplification may rely on FGFR signaling and respond to FGFR TKIs (André et al., 2013; Pearson et al., 2016). Therefore, FGFR1 dependence – that would translate to increased sensitivity to FGFR antagonists – may be overestimated by the sole evaluation of FGFR1 gene copy number, whereas mRNA and protein levels may represent better predictive biomarkers of sensitivity to FGFR inhibitors (Malchers et al., 2014; Wynes et al., 2014). Previous studies reported that FGFR1 overexpression is not enough to induce oncogenic transformation of mammary epithelial cells but, instead, enhanced receptor dimerization is required to promote tumorigenesis (Welm et al., 2002; Xian et al., 2005). On the other hand, FGFR2 is a dominant oncogene, mainly due to its cooperation with other RTKs, such as HER3 and IGF1R (Pearson et al., 2016). Thus, the identification of FGFR2 amplification may represent a better predictive biomarker of sensitivity to FGFR TKIs rather than FGFR1 amplification.
Several studies have shown that FGFR TKIs have clinical efficacy in solid tumors harboring FGFR fusions and mutations (Chae et al., 2020; Voss et al., 2019). Although the rate of these alterations is negligible in primary breast cancers, their occurrence is higher in metastatic samples and in biopsies collected from patients progressing on antiestrogens (Formisano et al., 2019; Levine et al., 2019; Mao et al., 2020; Razavi et al., 2018), clearly supporting the need of tumor genomic characterization at disease progression.
The dissection of tumor heterogeneity and changes during tumor evolution also represent an important challenge to identify the fraction of patients with advanced disease that would respond to FGFR TKIs. It has been shown that tumors with clonal FGFR2 amplification are sensitive to FGFR inhibition (Pearson et al., 2016). Also, Mao et al (Mao et al., 2020) suggested that FGFR genomic aberrations are clonally acquired after treatment with antiestrogens, defining a subset of breast cancers highly dependent on FGFR signaling and hence sensitive to FGFR inhibition. More studies employing clonal evolutionary analysis, based on sequencing of pre- and post-treatment samples, are clearly needed to define the clonal evolution of FGFRs aberrations.
Inadequate dosing and drug-induced toxicities have also contributed to lack of clinical benefit from multi-kinase and FGFR selective TKIs. For example, hypertension, proteinuria, thrombotic microangiopathy, mainly due to VEGFR inhibition, resulted in dose reductions or early treatment discontinuation of lucitanib (Soria et al., 2014). More selective FGFR inhibitors are also burdened by FGFR inhibition-specific toxicities, such as hyperphosphatemia, stomatitis, diarrhea, dry eyes and skin (Loriot et al., 2019).
Conclusions
The role of FGFR signaling in breast cancer has been well characterized. In the last few years, it has become clear that the various FGFR alterations have different biological and oncogenic properties. These findings have important implications in the design of future clinical trials testing FGFR TKIs. Although FGFR copy number alterations represent the most common FGFR genomic alterations in breast cancer, by themselves they may not be good predictive biomarkers of sensitivity to FGFR TKIs. Conversely, the use of FGFR inhibitors, as monotherapy or in combination with other drugs, may represent a valid therapeutic option for patients with breast cancers harboring FGFR mutations and fusions. Future studies will help to define the subsets of patients with breast cancer highly dependent on FGFR signaling and, hence, worth to be treated with FGFR inhibitors.
Financial support:
UTSW Simmons Cancer Center P30 CA142543, CPRIT RR170061, NCI Breast SPORE P50 CA098131, Susan G. Komen Breast Cancer Foundation SAC100013 grant, a Breast Cancer Research Foundation grant, and NCI R01CA224899
Conflicts of Interest:
C.L.A receives or has received research grants from Pfizer, Lilly and Takeda; holds minor stock options in Provista; serves or has served in an advisory role to Novartis, Merck, Lilly, Daiichi Sankyo, Taiho Oncology, OrigiMed, Puma Biotechnology, Immunomedics, AstraZeneca, Arvinas and Sanofi; and reports scientific advisory board remuneration from the Susan G. Komen Foundation.
References
- AACR Project GENIE Consortium (2017). AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, Paulson AS, Borad MJ, Gallinson D, Murphy AG, et al. (2020). Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Fernández M, and Malumbres M (2020). Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 37, 514–529. [DOI] [PubMed] [Google Scholar]
- André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, et al. (2013). Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin. Cancer Res 19, 3693–3702. [DOI] [PubMed] [Google Scholar]
- André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al. (2019). Alpelisib for PIK3CA -Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med 380, 1929–1940. [DOI] [PubMed] [Google Scholar]
- Angevin E, Lopez-Martin JA, Lin C-C, Gschwend JE, Harzstark A, Castellano D, Soria J-C, Sen P, Chang J, Shi M, et al. (2013). Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin. Cancer Res 19, 1257–1268. [DOI] [PubMed] [Google Scholar]
- Babina IS, and Turner NC (2017). Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 17, 318–332. [DOI] [PubMed] [Google Scholar]
- Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. (2012). Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med 366, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken A, and Mohammadi M (2009). The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov 8, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello E, Colella G, Scarlato V, Oliva P, Berndt A, Valbusa G, Serra SC, D’Incalci M, Cavalletti E, Giavazzi R, et al. (2011). E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 71, 1396–1405. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerliani JP, Guillardoy T, Giulianelli S, Vaque JP, Gutkind JS, Vanzulli SI, Martins R, Zeitlin E, Lamb CA, and Lanari C (2011). Interaction between FGFR-2, STAT5, and progesterone receptors in breast cancer. Cancer Res. 71, 3720–3731. [DOI] [PubMed] [Google Scholar]
- Chae YK, Hong F, Vaklavas C, Cheng HH, Hammerman P, Mitchell EP, Zwiebel JA, Ivy SP, Gray RJ, Li S, et al. (2020). Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J. Clin. Oncol 38, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, and Mohammadi M (2007). A Molecular Brake in the Kinase Hinge Region Regulates the Activity of Receptor Tyrosine Kinases. Mol. Cell 27, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervo MF, Cordo Russo RI, Petrillo E, Izzo F, De Martino M, Bellora N, Cenciarini ME, Chiauzzi VA, Santa María de la Parra L, Pereyra MG, et al. (2020). Canonical ErbB-2 isoform and ErbB-2 variant c located in the nucleus drive triple negative breast cancer growth. Oncogene 39, 6245–6262. [DOI] [PubMed] [Google Scholar]
- Chioni A-M, and Grose R (2012). FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol 197, 801–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman SJ, Chioni A-M, Ghallab M, Anderson RK, Lemoine NR, Kocher HM, and Grose RP (2014). Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med 6, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corn PG, Wang F, McKeehan WL, and Navone N (2013). Targeting Fibroblast Growth Factor Pathways in Prostate Cancer. Clin. Cancer Res 19, 5856–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, Jemal A, and Siegel RL (2019). Breast cancer statistics, 2019. CA. Cancer J. Clin 69, 438–451. [DOI] [PubMed] [Google Scholar]
- DiGiacomo JW, Godet I, Trautmann-Rodriguez M, and Gilkes DM (2021). Extracellular Matrix-Bound FGF2 Mediates Estrogen Receptor Signaling and Therapeutic Response in Breast Cancer. Mol. Cancer Res 19, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago JZ, Formisano L, Juric D, Niemierko A, Servetto A, Wander SA, Spring LM, Vidula N, Younger J, Peppercorn J, et al. (2019). FGFR1 Amplification Mediates Endocrine Resistance but Retains TORC Sensitivity in Metastatic Hormone Receptor–Positive (HR + ) Breast Cancer. Clin. Cancer Res 25, 6443–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudka AA, Sweet SMM, and Heath JK (2010). Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. 70, 3391–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, Ellis IO, and Reis-Filho JS (2007). FGFR1 amplification in breast carcinomas: a chromogenic in situhybridisation analysis. Breast Cancer Res. 9, R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, and Schlessinger J (2005). Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149. [DOI] [PubMed] [Google Scholar]
- Fannon M, and Nugent MA (1996). Basic fibroblast growth factor binds its receptors, is internalized, and stimulates DNA synthesis in Balb/c3T3 cells in the absence of heparan sulfate. J. Biol. Chem 271, 17949–17956. [DOI] [PubMed] [Google Scholar]
- Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, Estrada MM, Hutchinson KE, Giltnane JM, Schwarz LJ, et al. (2017). Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER + Breast Cancer. Clin. Cancer Res 23, 6138–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, Sudhan DR, Guerrero-Zotano AL, Croessmann S, Guo Y, et al. (2019). Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun 10, 1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Recio S, Thennavan A, East MP, Parker JS, Cejalvo JM, Garay JP, Hollern DP, He X, Mott KR, Galván P, et al. (2020). FGFR4 regulates tumor subtype differentiation in luminal breast cancer and metastatic disease. J. Clin. Invest 130, 4871–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, et al. (2012). AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family. Cancer Res. 72, 2045–2056. [DOI] [PubMed] [Google Scholar]
- Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, Nixon MJ, Du L, Sanchez V, Ericsson PG, et al. (2017). Genomic profiling of ER+ breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci. Transl. Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh N (2008). Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci. 99, 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L, Shi L, Liu LY, Fece de la Cruz F, Lennerz JK, Raghavan S, Leschiner I, Elagina, Siravegna G, Ng RWS, et al. (2019). TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 9, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald S, Politz O, Bender S, Héroult M, Lustig K, Thuss U, Kneip C, Kopitz C, Zopf D, Collin M-P, et al. (2019). Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int. J. Cancer 145, 1346–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock ML, Meyer RC, Mistry M, Khetani RS, Wagschal A, Shin T, Ho Sui SJ, Näär AM, and Flanagan JG (2019). Insulin Receptor Associates with Promoters Genome-wide and Regulates Gene Expression. Cell 177, 722–736.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker AB, Sudhan DR, and Arteaga CL (2020). Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 37, 496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, and Donoghue DJ (2000). Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 19, 3309–3320. [DOI] [PubMed] [Google Scholar]
- Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, and Kurzrock R (2016). The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res 22, 259–267. [DOI] [PubMed] [Google Scholar]
- Holland DG, Burleigh A, Git A, Goldgraben MA, Perez‐Mancera PA, Chin S, Hurtado A, Bruna A, Ali HR, Greenwood W, et al. (2011). ZNF703 is a common Luminal B breast cancer oncogene that differentially regulates luminal and basal progenitors in human mammary epithelium. EMBO Mol. Med 3, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui R, Pearson A, Cortes J, Campbell C, Poirot C, Azim HA, Fumagalli D, Lambertini M, Daly F, Arahmani A, et al. (2020). Lucitanib for the Treatment of HR+/HER2− Metastatic Breast Cancer: Results from the Multicohort Phase II FINESSE Study. Clin. Cancer Res 26, 354–363. [DOI] [PubMed] [Google Scholar]
- Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, and Mohammadi M (2001). Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. U. S. A 98, 7182–7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi OA, Zhang F, Eliseenkova AV, Itoh N, Linhardt RJ, and Mohammadi M (2004). Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum. Mol. Genet 13, 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Ohta H, and Konishi M (2015). Endocrine FGFs: Evolution, Physiology, Pathophysiology, and Pharmacotherapy. Front. Endocrinol (Lausanne: ). 6, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, Ramanathan RK, Goyal L, Sadeghi S, Macarulla T, et al. (2018). Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol 36, 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, and Squire CJ (2019). TAS-120 Cancer Target Binding: Defining Reactivity and Revealing the First Fibroblast Growth Factor Receptor 1 (FGFR1) Irreversible Structure. ChemMedChem 14, 494–500. [DOI] [PubMed] [Google Scholar]
- Kim RD, Sarker D, Meyer T, Yau T, Macarulla T, Park J-W, Choo SP, Hollebecque A, Sung MW, Lim H-Y, et al. (2019). First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 9, 1696–1707. [DOI] [PubMed] [Google Scholar]
- Kramer S, Okabe M, Hacohen N, Krasnow MA, and Hiromi Y (1999). Sprouty: a common antagonist of FGF and EGF signaling pathways in Drosophila. Development 126, 2515–2525. [DOI] [PubMed] [Google Scholar]
- Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, Elbi C, and Lutterbach B (2008). FGFR2 -Amplified Gastric Cancer Cell Lines Require FGFR2 and Erbb3 Signaling for Growth and Survival. Cancer Res. 68, 2340–2348. [DOI] [PubMed] [Google Scholar]
- Lee JE, Shin S-H, Shin H-W, Chun Y-S, and Park J-W (2019). Nuclear FGFR2 negatively regulates hypoxia-induced cell invasion in prostate cancer by interacting with HIF-1 and HIF-2. Sci. Rep 9, 3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Lopes de Menezes D, Vora J, Harris A, Ye H, Nordahl L, Garrett E, Samara E, Aukerman SL, Gelb AB, et al. (2005). In vivo Target Modulation and Biological Activity of CHIR-258, a Multitargeted Growth Factor Receptor Kinase Inhibitor, in Colon Cancer Models. Clin. Cancer Res 11, 3633–3641. [DOI] [PubMed] [Google Scholar]
- Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria J-C, Massard C, Lévy C, Arnedos M, Lacroix-Triki M, et al. (2016). Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 13, e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine KM, Priedigkeit N, Basudan A, Tasdemir N, Sikora MJ, Sokol ES, Hartmaier RJ, Ding K, Ahmad NZ, Watters RJ, et al. (2019). FGFR4 overexpression and hotspot mutations in metastatic ER+ breast cancer are enriched in the lobular subtype. Npj Breast Cancer 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew ED, Furdui CM, Anderson KS, and Schlessinger J (2009). The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal 2, ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo H-W, and Hung M-C (2006). Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 94, 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb CRK, Harris JL, and Craik CS (2006). Granzyme B Proteolyzes Receptors Important to Proliferation and Survival, Tipping the Balance toward Apoptosis. J. Biol. Chem 281, 28326–28335. [DOI] [PubMed] [Google Scholar]
- Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, Fleming M, Rezazadeh A, Mellado B, Varlamov S, et al. (2019). Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med 381, 338–348. [DOI] [PubMed] [Google Scholar]
- Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA, and Mohammadi M (2009). Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci. Signal 2, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malchers F, Dietlein F, Schöttle J, Lu X, Nogova L, Albus K, Fernandez-Cuesta L, Heuckmann JM, Gautschi O, Diebold J, et al. (2014). Cell-Autonomous and Non–Cell-Autonomous Mechanisms of Transformation by Amplified FGFR1 in Lung Cancer. Cancer Discov. 4, 246–257. [DOI] [PubMed] [Google Scholar]
- Mao P, Cohen O, Kowalski KJ, Kusiel JG, Buendia-Buendia JE, Cuoco MS, Exman P, Wander SA, Waks AG, Nayar U, et al. (2020). Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER+ Metastatic Breast Cancer. Clin. Cancer Res 26, 5974–5989. [DOI] [PubMed] [Google Scholar]
- Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH, et al. (2017). A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clin. Cancer Res 23, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer IA, Haley BB, Abramson VG, Brufsky A, Rexer B, Stringer-Reasor E, Jhaveri KL, Sanders M, Ericsson-Gonzalez PI, Ye F, et al. (2021). Abstract PD1–03: A phase Ib trial of fulvestrant + CDK4/6 inhibitor (CDK4/6i) palbociclib + pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib in FGFR -amplified/ ER+/ HER2−negative metastatic breast cancer (MBC). In Poster Spotlight Session Abstracts, (American Association for Cancer Research; ), pp. PD1–03–PD1–03. [Google Scholar]
- Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, Andre F, Iwata H, Ito Y, Tsurutani J, et al. (2020). Trastuzumab Deruxtecan in Previously Treated HER2−Positive Breast Cancer. N. Engl. J. Med 382, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Porta C, Vogelzang NJ, Sternberg CN, Szczylik C, Zolnierek J, Kollmannsberger C, Rha SY, Bjarnason GA, Melichar B, et al. (2014). Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol. 15, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, Lin NU, Borges V, Abramson V, Anders C, et al. (2020). Tucatinib, Trastuzumab, and Capecitabine for HER2−Positive Metastatic Breast Cancer. N. Engl. J. Med 382, 597–609. [DOI] [PubMed] [Google Scholar]
- Musolino A, Campone M, Neven P, Denduluri N, Barrios CH, Cortes J, Blackwell K, Soliman H, Kahan Z, Bonnefoi H, et al. (2017). Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2− breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 19, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord J-P, Hidalgo M, Schellens JHM, Cassier PA, Camidge DR, Schuler M, et al. (2017). Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Stu. J. Clin. Oncol 35, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, André F, Loibl S, Loi S, Garcia-Murillas I, et al. (2018). The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 8, 1390–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BC, Engels M, Annala M, and Zhang W (2014). Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J. Pathol 232, 4–15. [DOI] [PubMed] [Google Scholar]
- Patel RR, Sengupta S, Kim HR, Klein-Szanto AJ, Pyle JR, Zhu F, Li T, Ross EA, Oseni S, Fargnoli J, et al. (2010). Experimental treatment of oestrogen receptor (ER) positive breast cancer with tamoxifen and brivanib alaninate, a VEGFR-2/FGFR-1 kinase inhibitor: A potential clinical application of angiogenesis inhibitors. Eur. J. Cancer 46, 1537–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, Kilgour E, Smith NR, Geh C, Rooney C, et al. (2016). High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 6, 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas S, and Tolaney SM (2019). HER2−positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol 11, 1758835919833519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, Mirda D, and Williams LT (1992). Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature 358, 678–681. [DOI] [PubMed] [Google Scholar]
- Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, Cai Y, Bielski CM, Donoghue MTA, Jonsson P, et al. (2018). The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 34, 427–438.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JF, and Maher PA (2001). Importin beta-mediated nuclear import of fibroblast growth factor receptor: role in cell proliferation. J. Cell Biol 152, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JF, Mizukoshi E, and Maher PA (2004). Ligand dependent and independent internalization and nuclear translocation of fibroblast growth factor (FGF) receptor 1. DNA Cell Biol. 23, 538–548. [DOI] [PubMed] [Google Scholar]
- Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, Grigoriadis A, Sarrio D, Savage K, Dexter T, et al. (2006). FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin. Cancer Res 12, 6652–6662. [DOI] [PubMed] [Google Scholar]
- Reynisdottir I, Arason A, Einarsdottir BO, Gunnarsson H, Staaf J, Vallon-Christersson J, Jonsson G, Ringnér M, Agnarsson BA, Olafsdottir K, et al. (2013). High expression of ZNF703 independent of amplification indicates worse prognosis in patients with luminal B breast cancer. Cancer Med. 2, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu Y-M, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. (2017). Integrative clinical genomics of metastatic cancer. Nature 548, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, and Mohammadi M (2000). Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6, 743–750. [DOI] [PubMed] [Google Scholar]
- Servetto A, Kollipara R, Formisano L, Lin C-C, Lee K, Sudhan DR, Gonzalez-Ericsson PI, Chatterjee S, Guerrero-Zotano A, Mendiratta S, et al. (2021). Nuclear FGFR1 regulates gene transcription and promotes antiestrogen resistance in ER+ breast cancer. Clin. Cancer Res clincanres.3905.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Li S, Li L, Lin K, Liu X, Wang H, Wang H, and Wang D (2019). Chemical genomics reveals inhibition of breast cancer lung metastasis by Ponatinib via c-Jun. Protein Cell 10, 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee K, Yang W, Hinds JW, Hampsch RA, Varn FS, Traphagen NA, Patel K, Cheng C, Jenkins NP, Kettenbach AN, et al. (2018). Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. J. Exp. Med 215, 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2020). Cancer statistics, 2020. CA. Cancer J. Clin 70, 7–30. [DOI] [PubMed] [Google Scholar]
- Sobhani N, Fan C, O. Flores-Villanueva P, Generali D, and Li Y (2020). The Fibroblast Growth Factor Receptors in Breast Cancer: from Oncogenesis to Better Treatments. Int. J. Mol. Sci 21, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sootome H, Fujita H, Ito K, Ochiiwa H, Fujioka Y, Ito K, Miura A, Sagara T, Ito S, Ohsawa H, et al. (2020). Futibatinib Is a Novel Irreversible FGFR 1–4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res. 80, 4986–4997. [DOI] [PubMed] [Google Scholar]
- Soria J-C, DeBraud F, Bahleda R, Adamo B, Andre F, Dientsmann R, Delmonte A, Cereda R, Isaacson J, Litten J, et al. (2014). Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 25, 2244–2251. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Mohammadi M, Huang J, and Schlessinger J (1994). Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J. Biol. Chem 269, 17056–17061. [PubMed] [Google Scholar]
- Stachowiak MK, and Stachowiak EK (2016). Evidence-Based Theory for Integrated Genome Regulation of Ontogeny--An Unprecedented Role of Nuclear FGFR1 Signaling. J. Cell. Physiol 231, 1199–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachowiak MK, Fang X, Myers JM, Dunham SM, Berezney R, Maher PA, and Stachowiak EK (2003). Integrative nuclear FGFR1 signaling (INFS) as a part of a universal “feed-forward-and-gate” signaling module that controls cell growth and differentiation. J. Cell. Biochem 90, 662–691. [DOI] [PubMed] [Google Scholar]
- Taylor JG, Cheuk AT, Tsang PS, Chung J-Y, Song YK, Desai K, Yu Y, Chen Q-R, Shah K, Youngblood V, et al. (2009). Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest 119, 3395–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thien CB, and Langdon WY (2001). Cbl: many adaptations to regulate protein tyrosine kinases. Nat. Rev. Mol. Cell Biol 2, 294–307. [DOI] [PubMed] [Google Scholar]
- Tomlinson DC, Knowles MA, and Speirs V (2012). Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 130, 2857–2866. [DOI] [PubMed] [Google Scholar]
- Tsang M, Friesel R, Kudoh T, and Dawid IB (2002). Identification of Sef, a novel modulator of FGF signalling. Nat. Cell Biol 4, 165–169. [DOI] [PubMed] [Google Scholar]
- Turczyk L, Kitowska K, Mieszkowska M, Mieczkowski K, Czaplinska D, Piasecka D, Kordek R, Skladanowski AC, Potemski P, Romanska HM, et al. (2017). FGFR2-Driven Signaling Counteracts Tamoxifen Effect on ERα-Positive Breast Cancer Cells. Neoplasia 19, 791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, and Grose R (2010). Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116–129. [DOI] [PubMed] [Google Scholar]
- Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, et al. (2010). FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 70, 2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MH, Hierro C, Heist RS, Cleary JM, Meric-Bernstam F, Tabernero J, Janku F, Gandhi L, Iafrate AJ, Borger DR, et al. (2019). A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res 25, 2699–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm BE, Freeman KW, Chen M, Contreras A, Spencer DM, and Rosen JM (2002). Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J. Cell Biol 157, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AOM, Patey SJ, Kan S-H, van den Ouweland AMW, and Hamel BCJ (2002). FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am. J. Med. Genet 112, 266–278. [DOI] [PubMed] [Google Scholar]
- Wu Y-M, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin S-F, et al. (2013). Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 3, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynes MW, Hinz TK, Gao D, Martini M, Marek LA, Ware KE, Edwards MG, Böhm D, Perner S, Helfrich BA, et al. (2014). FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin. Cancer Res 20, 3299–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian W, Schwertfeger KL, Vargo-Gogola T, and Rosen JM (2005). Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J. Cell Biol 171, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, and Zhang ZY (2001). The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem 276, 32382–32391. [DOI] [PubMed] [Google Scholar]