

Abstract
The marine environment is an excellent source of molecules that have a wide structural diversity and a variety of biological activities. Many marine natural products (MNPs) have been established as leads for anticancer drug discovery. Most of these compounds are alkaloids, including several chemical subclasses. In this review, we focus on the bis-indolyl alkaloid Nortopsentins and their derivatives with antiproliferative properties. Nortopsentins A–C were found to exhibit in vitro cytotoxicity against the P388 murine leukaemia cell line. Their structural manipulation provided a wide range of derivatives with significant anti-tumour activity against human cell lines derived from different cancer types (bladder, colon, gastric, CNS, liver, lung, breast, melanoma, ovarian, pancreatic, prostate, pleural mesothelioma, renal, sarcoma, and uterus). In vivo assays on animal models also proved that Nortopsentins and related bis-indolyl compounds have potent anti-inflammatory activity. These remarks set the foundation for future investigations into the development of new Nortopsentin derivatives as new anticancer and anti-inflammatory agents.
Keywords: natural products, marine alkaloids, nortopsentin derivatives, anticancer agents, anti-inflammatory agents
1. Introduction
Cancer is one of the scariest diseases in the human population, especially in developing countries, and the second leading cause of death worldwide [1]. Cancer cells are characterized by abnormal proliferation, unstoppable differentiation, invasion, and metastasis [2,3,4,5,6]. The inhibition of the proliferation pathways of cancer cells constitutes an effective strategy to treat this pathology. In the search for new anticancer drugs, medicinal chemists have set their sights on natural products, particularly the secondary metabolites of marine organisms, for several reasons. Firstly, living in such relatively closed surroundings characterized by high salt, high pressure, low temperature, hypoxia, and lack of light, marine organisms have developed, in their process of long-term evolution, a distinct metabolism system and immune system which are completely different from the terrestrial ones. Secondly, they have formed and accumulated large numbers of bioactive compounds with structural diversity and complexity, such as fatty acids, proteins, alkaloids, peroxides, coumarins, and terpenes [7,8,9]. Unsurprisingly, marine products tend to exhibit greater potency than terrestrial ones to be effective against predators because their release into the environment is quickly followed by a dilution by water [10,11,12,13,14,15,16].
The marine environment provides a wealth of marine invertebrates such as sponges, coelenterates, tunicates, bryozoans, red algae, acorn worms, and symbiotic bacteria. More than 300,000 species have been found in the ocean, and it is estimated that more than 1 million new species have not been found [16,17,18]. Among marine organisms, sponges have proved to be an abundant reserve of compounds with antibacterial, antiviral, anti-inflammatory, immunomodulatory, and antiproliferative activities [19,20]. The first marine-derived drug on the market was Cytosar®, whose active compound acting as an inhibitor of DNA polymerase, cytarabine (Ara-C), is a synthetic analogue of a C-nucleoside isolated from the Caribbean sponge Tethya crypta. Cytosar® was approved by the Food and Drug Administration (FDA) in 1969 for the treatment of leukaemia and lymphoma (Table 1) [17,18,21]. Another drug available on the market is Halaven®, which was accepted by the FDA in late 2010 as a chemotherapy agent for the treatment of metastatic breast cancer. Its active compound, Eribulin, is an antimitotic agent and an analogue of Halichondrin-B, a macrocyclic polyether initially extracted from Halichondria okadai sponge in 1986. It causes G2/M cell cycle arrest by acting on tubulin or microtubules [22,23,24].
Table 1.
Marketed drugs by FDA.
| Compound | Date of FDA Authorisation | Natural Source | Clinical Use |
|---|---|---|---|
| Cytarabine | 1969 | Sponge | Leukaemia |
| Fludarabine | 2004 | Sponge | Leukaemia |
| Trabectedin | 2015 | Tunicate | Ovarian cancer |
| Eribulin | 2010 | Sponge | Breast cancer |
| Brentuximab vedotin | 2011 | Mollusk/cyanobacterium | Lymphomas |
| Lurbinectedin | 2020 | Tunicate | Ovarian cancer |
| Polatuzumab vedotin | 2019 | Mollusk/cyanobacterium | Breast cancer |
| Enfortumab vedotin | 2019 | Mollusk/cyanobacterium | Urothelial cancer |
| Belantamab mafodotin | 2020 | Mollusk/cyanobacterium | Multiple myeloma |
Many marine natural products have been shown to have a good inhibitory effect on human cancer cell lines during activity screening [16,20,25], in particular alkaloids [16,26,27]. Besides antiproliferative activity, marine alkaloids possess a high degree of biological activities, such as anti-inflammatory, antiviral, antimalarial, anti-fungal, antibacterial, antiosteoporosis, and immunomodulatory activity [16,22,28,29,30,31,32,33,34,35]. Alkaloids can be classified in terms of their chemical structure into subclasses such as sterols, pyridoacridines, pyrazinones, pyrroles, isoquinolines, guadinines, aminoimidazoles, indoles, and bis-indoles [36]. In particular, bis-indolyl alkaloids and their synthetic derivatives, consisting of two indole moieties linked to each other via heterocyclic units or linear chains, are well known because of their broad spectrum of biological properties, including anti-microbial [37,38], anti-viral [39,40], and anti-tumour activities [41,42,43,44,45,46,47,48,49]. Herein, the anticancer activities of Nortopsentins and their derivatives are reviewed.
2. Results and Discussion
Among marine bis-indolyl alkaloids, Nortopsentins (Figure 1) represent promising lead compounds that have attracted remarkable attention due to their in vitro cytotoxicity. Many Nortopsentin analogues have been synthesised and showed interesting biological activities such as cytotoxic, anti-inflammatory, antiplasmodial, antibacterial, antifungal, and insecticidal [20,50,51,52,53].
Figure 1.

Nortopsentins.
Nortopsentins A–C, isolated from the Caribbean deep-sea sponge Spongosorites ruetzleri in 1988, have a characteristic 2,4-bis-indolyl-imidazole skeleton and showed significant antiproliferative and antifungal activity [52]. They exhibited in vitro cytotoxicity against the P388 murine leukaemia cell line (IC50 values of 7.6, 7.8, and 1.7 µM, respectively), antifungal activity against Candida albicans (MIC values of 3.1, 6.2, and 12.5 µM, respectively) [52] and anti-microbial activity against Bacillus subtilis [54,55]. In addition, tri- and tetramethylated derivatives of Nortopsentin B exhibited remarkable improvement in in vitro cytotoxicity against the P388 cells when compared to the activity of the parent compound (IC50 values of 0.9 and 0.34 μM, respectively) [52].
Catalytic hydrogenation of Nortopsentin A–C yielded the synthetic analogue D (sometimes referred to in the literature as Nortopsentin D).
In 1996, a new bis-indolyl alkaloid was isolated, Nortopsentin E [53,55,56]. Nortopsentin E was originally isolated from the axinellid sponge Dragmacidon in deep waters south of New Caledonia and later from the sponge Agelas dendromorpha. It is a structural variant of the Nortopsentin family bearing a complex central trisubstituted (4H)-imidazol-4-one with a 6-bromoindole at the C2 position and a 4-methyl-1H-imidazol-2-amine and 6-bromoindole at C5, whose total synthesis has recently been reported [53]. Surprisingly, while Nortopsentin E was inactive on KB tumour cells in vitro, its methylated derivative showed both high cytotoxicity on KB cell lines (IC50 0.014 µM) and antifungal activity against yeast [53,55,56].
2.1. Nortopsentin Derivatives as Antiproliferative Agents
2.1.1. Thiazoles
Several bis-indolyl-thiazole compounds 1 (Figure 2) were synthesised and tested against the National Cancer Institute (NCI, Bethesda, MD 20892, USA) full panel of 60 human cancer cell lines derived from nine cancer cell types and grouped into disease subpanels including leukaemia, non-small cell lung, colon, central nervous system, melanoma, ovarian, renal, prostate, and breast cancers. Many compounds showed GI50 values in the micromolar-submicromolar range. In particular, compounds 1a–j exhibited cytotoxic activities against a variety of human cancer cell lines. The compound 1a exhibited highly selective in vitro cytotoxicity against leukaemia (GI50 of 3.27 µM in K562, 5.31 µM in Molt-4) and ovarian cancer cell lines (GI50 8.14 µM in IGROV1) (Table 1). In many other human tumour cell lines, the GI50 of compound 1a exceeded 100 µM. It is worth noting that unlike the unsubstituted compound 1a, the bis-indolyl-thiazoles 1b–j showed broad effects on leukaemia, colon, CNS, and breast cancer panels, suggesting that substituents in the indole ring might result in a potency increase (Table 2) [57,58].
Figure 2.

Bis-indolyl-thiazole compounds 1.
Table 2.
In vitro inhibition of cancer cell lines growth in leukaemia, colon, CNS, ovarian, and breast cancer subpanels by compounds 1.
| Cell Line | GI50 (µM) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | |
| Leukaemia | ||||||||||
| CCRF-CEM | ND b | 14.6 | 10.9 | 10.1 | 2.11 | 2.40 | 27.7 | 2.58 | 2.66 | 2.99 |
| HL-60 (TB) | ND b | ND b | ND b | 0.95 | 2.43 | 2.76 | ND b | 3.76 | 4.13 | 3.86 |
| K-562 | 3.27 | 18.8 | 5.61 | 4.69 | 1.96 | 1.94 | 15.2 | 2.13 | 1.74 | 2.15 |
| MOLT-4 | 5.31 | 19.9 | 31.2 | 5.80 | 1.41 | 1.75 | 23.0 | 1.55 | 2.95 | 2.26 |
| RPMI82226 | ND b | 19.4 | 12.2 | 11.4 | 1.97 | 1.95 | 27.1 | 2.24 | 2.03 | 1.84 |
| Colon Cancer | ||||||||||
| HCT-15 | >100 | 15.2 | 17.8 | 8.5 | 1.81 | 2.51 | 7.54 | 2.70 | 0.81 | 2.84 |
| SW-620 | >100 | 16.5 | 25.6 | 12.5 | 1.52 | 2.14 | 7.00 | 3.57 | 5.50 | 2.89 |
| CNS Cancer | ||||||||||
| SF-295 | 33.6 | 14.6 | 9.23 | 4.81 | 1.98 | 2.71 | 73.5 | 4.56 | ND b | ND b |
| SF-268 | ND b | 18.3 | 32.1 | ND b | 1.52 | 2.44 | 26.0 | 2.69 | 13.8 | 1.80 |
| SNB-19 | >100 | 17.8 | 41.9 | 17.2 | 2.11 | 3.75 | >100 | 2.60 | 10.5 | 3.34 |
| U251 | >100 | 17.9 | 28.1 | 15.3 | 2.10 | 2.30 | 25.0 | 3.34 | 5.07 | 3.00 |
| Ovarian Cancer | ||||||||||
| IGROV1 | 8.14 | 13.0 | 30.5 | 14.4 | 1.85 | 1.70 | 81.5 | 2.96 | 4.61 | 2.43 |
| OVCAR-5 | >100 | 16.1 | 37.1 | 23.4 | 1.96 | 2.14 | 42.7 | 2.16 | 3.44 | 2.35 |
| Breast Cancer | ||||||||||
| MCF-7 | >100 | 16.7 | 27.2 | 6.5 | 2.13 | 2.70 | 54.1 | 0.88 | 4.36 | 3.82 |
| MDA-MB-435 | 33.1 | 14.9 | 25.6 | 4.3 | 2.53 | 2.27 | 7.70 | 4.54 | 14.6 | 3.97 |
| MDA-N | 83.0 | 19.2 | 31.6 | 2.9 | 1.88 | 2.09 | 8.27 | 2.86 | 6.84 | 3.77 |
| T-47D | >100 | 24.8 | 23.9 | 16.2 | 3.27 | 2.90 | 59.3 | 3.33 | 4.12 | 1.76 |
| BT-549 | >100 | 18.3 | 73.8 | 41.1 | 2.71 | 10.6 | 67.3 | 1.24 | 1.46 | 1.41 |
a Concentration (µM) that inhibits 50% net cell growth. b ND = Not Determined.
Another thiazole series 2a–m (Figure 3), bearing an indole and a 7-azaindole moiety, has been reported. These derivatives were tested by NCI against a panel of 60 human cancer cell lines.
Figure 3.

Thiazole series 2a–m.
Data revealed that these compounds 2a–m showed GI50 values in the micromolar–submicromolar range. The five most active compounds, 2c, 2d, 2e, 2g, and 2m, which did not show selectivity against any of the tumour subpanels, were further investigated in two additional cell lines, STO and MesoII, derived from human diffuse malignant peritoneal mesothelioma (DMPM), a tumour type not included in the NCI panel. Seventy-two hours of exposure to increasing concentrations of each compound resulted in dose-dependent cell proliferation inhibition in both cellular models. Compounds 2c, 2d, 2e, 2g, and 2m, exhibited comparable activity in STO cells with IC50 values ranging from 0.33 to 0.61 μM. By contrast, a variable growth inhibitory effect was induced by the different compounds in MesoII cells (IC50 values ranging from 4.11 to 25.12 μM). In addition, compounds 2c, 2e, and 2m, did not interfere with the growth of normal cells (Table 3). The anti-tumour activity of 2c, 2e, and 2m derivatives was then evaluated on STO cells xenotransplanted in athymic nude mice. The treatment with the different compounds resulted in marked tumour growth inhibition. Specifically, at the end of the experiment, a statistically significant tumour volume inhibition (TVI) compared with the control (73%, 75%, and 58%, for 2c, 2e, and 2m derivatives, respectively) was observed, and two complete responses (disappearance of tumour) were also identified in each treatment group (Table 4). Moreover, the compounds 2c, 2e, and 2m were well tolerated without any appreciable sign of toxicity. In vitro kinase assays revealed CDK1 inhibition exerted by the compounds with IC50 values of 0.89, 0.75, and 0.86 μM, respectively, for the derivatives 2c, 2e, and 2m (Table 5). These results were comparable to those reported for two well-known CDK1 inhibitors, roscovitine and purvanalol A. In addition, derivatives 2c, 2e, and 2m were able to inhibit GSK3β, but only at higher concentrations (IC50 values of 42.18, 40.18, and 35.68 μM, respectively) (Table 5). Further investigations revealed a marked time-dependent cell cycle arrest at the G2/M phase and an increase in the apoptotic rate by reducing the phosphorylated form of the antiapoptotic protein survivin. Moreover, the addition of compound 2m to paclitaxel-treated cells resulted in a synergistic cytotoxic effect due to an increased apoptotic response [20].
Table 3.
Cytotoxic activity of compounds 2c, 2d, 2e, 2g, and 2m in DMPM and normal cells.
| Compound | IC50 (µM) a | ||
|---|---|---|---|
| STO | MesoII | W138 | |
| 2c | 0.49 ± 0.07 | 25.12 ± 3.06 | >100 |
| 2d | 0.61 ± 0.14 | 16.77 ± 1.99 | 18.76 ± 3.21 |
| 2e | 0.43 ± 0.11 | 4.85 ± 0.64 | >100 |
| 2g | 0.54 ± 0.09 | 13.27 ± 0.74 | 15.44 ± 3.87 |
| 2m | 0.33 ± 0.07 | 4.11 ± 0.22 | >100 |
a Data are reported as IC50 values (concentration of drug required to inhibit growth by 50%) determined by MTS assay after 72 h of continuous exposure to each compound. The data represent mean values ± SD of at least three independent experiments.
Table 4.
Activity of derivatives 2c, 2e, and 2m on STO cells xenotransplanted in athymic nude mice.
| Compound | TVI (%) a | CR b | BWL (%) c | TOX d |
|---|---|---|---|---|
| 2c | 73 * | 2/8 | 4 | 0/8 |
| 2e | 75 ** | 2/8 | 1 | 0/8 |
| 2m | 58 * | 2/8 | 7 | 0/8 |
a Tumour volume inhibition (%) in treated vs. control mice, determined 17 days after the end of drug treatment (day 35). b Complete response, disappearance of tumour induced by treatment. c BWL, body weight loss induced by treatment (%). d Toxic death on treated animals. ** p < 0.01, * p < 0.05.
Table 5.
In Vitro kinase inhibitory properties of derivatives 2c, 2e, and 2m.
| Protein Kinase | IC50 (µM) a | ||
|---|---|---|---|
| 2c | 2e | 2m | |
| CDK1 | 0.89 ± 0.07 | 0.75 ± 0.03 | 0.86 ± 0.04 |
| GSK3β | 42.18 ± 3.28 | 40.18 ± 2.94 | 35.68 ± 1.69 |
a Concentration of drug required to inhibit by 50% (IC50) the activity of CDK1 and GSK3β. The data represent mean values ± SD of at least three independent experiments.
Thiazole Nortopsentin analogues of type 2n–q (Figure 4), in which the nitrogen atom of the indole and/or 7-azaindole moiety is substituted with a 2-methoxyethyl chain, and analogues 2r–2ao, in which indole nitrogen is substituted with alkylmorpholine or alkylpiperidine, were synthesized. Derivatives 2n–2q, 2t–w, 2z, 2ab, 2ae, and 2ak–am were tested by NCI on the full panel of approximately 60 human cancer cell lines and showed good antiproliferative activity with GI50 in the micromolar–nanomolar range. Compounds 2n, 2q, 2t–w, 2z, 2ab, 2ae, and 2ak–am were active against the total number of cell lines investigated, whereas compounds 2o and 2p were cytotoxic against a very high percentage of the tested cell lines (96% and 93%, respectively). Their action mechanism, investigated on human breast cancer MCF-7 cells, was pro-apoptotic, being associated with externalisation of plasma membrane phosphatidylserine and DNA fragmentation, accompanied by perturbation of the cell cycle progression. It was found that the derivatives 2n–p confined viable cells in the G2/M phase. Derivative 2n showed the most interesting in vitro anticancer activity, expressing lower GI50 values (0.03–12.6 µM) and, markedly, in vitro inhibited CDK1 activity with an IC50 value of 1.14 ± 0.09 µM, comparable to that reported for other indolyl-thiazolyl-7-azaindole derivatives or well-known CDK1 inhibitors, roscovitine and purvanalol A. Moreover, cytotoxicity assays on intestinal normal-like differentiated Caco-2 cells after treatment with compounds 2n–q in the 25–100 µM range revealed that these compounds were selectively cytotoxic to cancer cells (Table 6) [20].
Figure 4.

Thiazole series 2n–ao.
Table 6.
Cytotoxic activity and the selectivity index (SI) of the synthesized compounds 2n–q against MCF-7 cancer cell line.
| Compound | Intestinal Normal-like Caco-2 Cells LC50 (µM) a |
MCF-7 GI50 (µM) b | SI |
|---|---|---|---|
| 2n | >100 | 0.05 | ND c |
| 2o | 96.1 ± 3.1 | 2.09 | 46 |
| 2p | 71.4 ± 2.1 | 1.74 | 41 |
| 2q | 83.3 ± 2.5 | 1.79 | 46 |
a LC50 concentration which is lethal to 50% of the normal cells compared to untreated controls; b GI50 Concentration (µM) that inhibits 50% net cell growth; c ND, not determined.
Among the derivatives 2r–2ao, compound 2ak was the most active of the series, showing selectivity against leukaemia and colon cancer subpanels. In addition, 2ak was effective against the A498 cell line of the renal cancer subpanel (GI50 value of 20 nM) (Table 7). Cytotoxicity and selectivity experiments were performed for compounds 2r–2ao using the HepG2 cell line of human hepatoma, the MCF-7 cell line of human breast cancer, and the non-tumorigenic MCF 10A cell line by single-dose administration (10 µM). Most of the molecules 2r–2ao showed antiproliferative effects on both Hep G2 and MCF-7 cells. Some of these molecules (2r, 2ac, 2aa, 2ad, 2ae, 2am, 2an, and 2ao) showed cytotoxicity against the tumour cell lines without compromising non-tumorigenic MCF-10A cell viability. The N-alkylpiperidine substituted compounds (2u, 2v, 2w, 2ag, and 2ah) were very active against the MCF-7 cell line but were also cytotoxic over MCF-10A cell line. Among the alkylmorpholino derivatives, propyl- and ethyl-morpholino derivatives (2t and 2ae) were active over both Hep G2 and MCF-7 cells and non-toxic over non-tumorigenic cells, while compound 2af was active on cancer and non-cancer cell lines examined. On the other hand, butylmorpholino derivatives gave different results, being 2s, 2ai, 2aj, and 2ak cytotoxic, while 2x and 2y did not impair non-tumorigenic cells; in particular, the compound 2y showed selective toxicity on the Hep G2 cancer cell line (EC50 values of 3.25, 23.05, and 29.09 µM for Hep G2, MCF-7, and MCF-10A cell lines, respectively). Moreover, considering the overexpression of the enzyme glutaminase-1 (GLS-1) in hepatic cancer cell lines and its low expression in the MCF-7 cell line, enzymatic assays were also performed, revealing good inhibitory potency of the compound 2y over GLS-1 (IC50 value of 3.96 µM). This could explain the selective cytotoxicity shown by 2y on Hep G2 as opposed to MCF-7 and MCF-10A. Additional experiments on aggressive cancer cell lines with GLS-1 overexpression, like glioblastoma (U-87 MG), pancreatic cancer (MIA PaCa-2), osteosarcoma (Saos2), melanoma (A-375), and non-small lung cancer (A549) confirmed the cell growth inhibition potency for the compound 2y, with EC50 values in the micromolar range (Table 8). Data suggest that the decoration of the nitrogen atom of the indole and/or 7-azaindole moiety with 2-methoxyethyl, alkylmorpholine, or alkylpiperidine chain led to interesting biological results [20,59].
Table 7.
In vitro inhibition of cancer cell lines growth in leukaemia, colon, renal, and breast cancer subpanels by compounds 2t–w, 2z, 2ab, 2ae, and 2ak–am.
| Cell Line | GI50 (µM) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2t | 2u | 2v | 2w | 2z | 2ab | 2ae | 2ak | 2al | 2am | |
| Leukaemia | ||||||||||
| CCRF-CEM | 2.76 | 1.94 | 2.12 | 2.25 | 2.12 | 1.83 | 3.07 | 0.45 | 1.97 | 1.73 |
| HL-60(TB) | 2.11 | 1.88 | 1.77 | 1.86 | 1.93 | 1.72 | 2.52 | 1.67 | 1.80 | 1.70 |
| K-562 | 2.08 | 1.74 | 1.72 | 1.88 | 1.95 | 1.61 | 2.94 | 0.24 | 1.49 | 1.49 |
| MOLT-4 | 1.85 | 1.82 | 1.83 | 1.80 | 1.86 | 1.77 | 1.79 | 0.60 | 1.79 | 1.54 |
| RPMI82226 | 1.92 | 2.08 | 2.04 | 2.19 | 1.99 | 1.84 | 1.68 | 0.77 | 1.84 | 1.89 |
| SR | 2.30 | 1.89 | 2.05 | 1.84 | 2.01 | 1.68 | 2.29 | 0.27 | 1.70 | 1.58 |
| Colon Cancer | ||||||||||
| COLO-205 | 1.82 | 1.67 | 1.73 | 1.80 | 1.85 | 1.73 | 2.09 | 1.49 | 1.78 | 1.63 |
| HCC-2998 | 2.04 | 1.71 | 1.70 | 1.78 | 1.91 | 1.74 | 3.42 | 1.47 | 1.59 | 1.86 |
| HCT-116 | 1.85 | 1.70 | 1.75 | 1.68 | 1.78 | 1.60 | 3.00 | 0.28 | 1.83 | 1.37 |
| HCT-15 | 2.32 | 1.72 | 1.73 | 1.57 | 1.96 | 1.58 | 2.28 | 0.24 | 1.70 | 1.45 |
| HT29 | 2.22 | 1.75 | 1.68 | 1.81 | 2.18 | 1.56 | 2.85 | 0.31 | 1.70 | 1.59 |
| KM12 | 2.41 | 1.61 | 1.77 | 1.87 | 1.93 | 1.73 | 2.85 | 1.09 | 1.69 | 1.70 |
| SW-620 | 2.26 | 1.82 | 1.92 | 2.00 | 1.99 | 1.74 | 3.62 | 0.31 | 1.83 | 1.62 |
| Renal Cancer | ||||||||||
| 786-0 | 1.74 | 1.63 | 1.65 | 1.61 | 1.78 | 1.59 | 2.91 | 0.42 | 1.65 | 1.61 |
| A498 | 0.48 | 1.83 | 2.57 | 1.82 | 0.22 | 1.43 | 5.66 | 0.02 | 1.60 | 1.66 |
| ACHN | 2.50 | 1.66 | 1.70 | 1.62 | 1.79 | 1.66 | 2.82 | 1.50 | 1.73 | 165 |
| CAKI-1 | 1.88 | ND b | ND b | ND b | 1.80 | ND b | ND b | 1.39 | ND b | ND b |
| RXF 393 | 2.09 | 1.62 | 1.61 | 1.59 | 1.73 | 1.38 | 2.58 | 0.93 | 1.52 | 1.33 |
| SN12C | 3.16 | 1.68 | 1.74 | 1.75 | 1.91 | 1.60 | 3.15 | 6.05 | 1.53 | 1.61 |
| TK-10 | 3.48 | 2.22 | 1.85 | 1.59 | 5.25 | 1.51 | 3.28 | 4.25 | 1.58 | 1.53 |
| UO-31 | 1.37 | 1.18 | 1.28 | 1.38 | 1.43 | 1.38 | 2.20 | 1.11 | 1.46 | 1.43 |
| Breast Cancer | ||||||||||
| MCF-7 | 1.43 | 1.24 | 1.53 | 1.43 | 1.56 | 1.53 | 1.94 | 1.65 | 1.62 | 1.70 |
| MDA-MB-231/ATCC | 1.86 | 1.63 | 1.69 | 1.66 | 1.70 | 1.61 | 2.51 | 0.52 | 1.64 | 1.64 |
| HS 578T | 2.08 | 2.34 | 2.59 | 2.05 | 2.05 | 1.66 | 3.75 | 0.76 | 12.40 | 1.50 |
| BT-549 | 2.00 | 1.68 | 3.72 | 3.28 | 3.74 | 1.66 | 2.17 | 2.97 | 1.67 | 1.64 |
| T-47D | 2.07 | 1.22 | 1.77 | 1.75 | 2.12 | 1.45 | 1.52 | 12.0 | 15.70 | 1.65 |
| MDA-MB-468 | 2.52 | 1.88 | 1.61 | 1.86 | 1.95 | 1.68 | 2.20 | 0.97 | 1.51 | 1.50 |
a Concentration (µM) that inhibits 50% net cell growth. b ND = Not Determined.
Table 8.
Toxicity of the compound 2y on aggressive cancer cell lines with GLS-1 overexpression.
| Cell Line | EC50 ± SD (µM) a |
|---|---|
| U-87 MG | 3.63 ± 1.87 |
| MIA PaCa-2 | 6.98 ± 0.44 |
| Saos2 | 4.85 ± 1.19 |
| A-375 | 5.84 ± 0.43 |
| A549 | 8.47 ± 0.32 |
a EC50 values are expressed as mean ± SD.
A series of thiazole Nortopsentin analogues of type 3 (Figure 5), in which the imidazole moiety of Nortopsentins was replaced by a thiazole ring and one indole unit by a 5-azaindole ring, was synthesized. Derivatives 3a–p were active against the NCI full panel, showing good antiproliferative activity in the micro–submicromolar range. Thiazoles 3e, 3f, and 3p were particularly cytotoxic against the leukaemia subpanel (GI50 in the range 0.24–1.71 µM, 0.24–1.57 µM, and 0.35–2.13 µM, respectively) (Table 9 and Table 10). Compound 3b turned out to be the most active against the breast cancer subpanel (GI50 in the range 0.27–2.16 µM). Moreover, compounds 3b and 3f proved to be selective against the HCT-116 cell line of the colon cancer subpanel (GI50 of 0.93 µM and 0.18 µM, respectively) [60].
Figure 5.

Thiazole series 3.
Table 9.
In vitro inhibition of cancer cell lines growth by compounds 3a–h.
| Cell Line | GI50 (µM) a | |||||||
|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 3d | 3e | 3f | 3g | 3h | |
| Leukaemia | ||||||||
| CCRF-CEM | 2.02 | 1.78 | 1.83 | 2.15 | 1.30 | 0.36 | 1.74 | 1.96 |
| HL-60(TB) | 2.03 | 1.94 | 1.68 | 1.96 | 1.71 | 1.48 | 1.76 | 1.93 |
| K-562 | 1.76 | 2.09 | 0.72 | 1.90 | 0.24 | 0.24 | 0.46 | 1.70 |
| RPMI82226 | 2.00 | 1.76 | 1.83 | 1.84 | 1.58 | 1.57 | 1.77 | 1.98 |
| Colon Cancer | ||||||||
| HCC-2998 | 1.22 | 1.26 | 1.85 | 1.89 | 1.76 | 1.90 | 1.64 | 1.86 |
| HCT-116 | 1.61 | 0.93 | 1.69 | 1.90 | 1.21 | 0.18 | 1.63 | 1.66 |
| HCT-15 | 1.37 | 1.51 | 1.25 | 1.54 | 1.35 | 1.75 | 1.33 | 1.68 |
| HT29 | 1.82 | 1.98 | 1.20 | 1.43 | 1.59 | 1.60 | 1.44 | 1.47 |
| KM12 | 1.11 | 1.00 | 1.73 | 1.84 | 1.71 | 1.97 | 1.63 | 1.79 |
| SW-620 | 2.03 | 1.85 | 1.75 | 1.96 | 1.52 | 1.85 | 1.65 | 1.88 |
| Breast Cancer | ||||||||
| MCF-7 | 0.30 | 0.32 | 1.47 | 1.70 | 1.33 | 1.78 | 1.23 | 1.93 |
| MDA-MB-231/ATCC | 1.56 | 1.47 | 1.45 | 1.83 | 1.41 | 1.66 | 1.55 | 1.75 |
| HS 578T | 1.93 | 2.16 | 2.03 | 2.21 | 1.94 | 2.23 | 2.21 | 2.36 |
| BT-549 | 9.00 | 1.68 | 1.81 | 1.80 | 1.93 | 1.61 | 1.80 | 1.71 |
| MDA-MB-468 | 0.40 | 0.27 | 0.44 | 1.62 | 1.38 | 1.79 | 1.40 | 1.86 |
a Concentration (µM) that inhibits 50% net cell growth.
Table 10.
In vitro inhibition of cancer cell lines growth by compounds 3i–p.
| Cell Line | GI50 (µM) a | |||||||
|---|---|---|---|---|---|---|---|---|
| 3i | 3j | 3k | 3l | 3m | 3n | 3o | 3p | |
| Leukaemia | ||||||||
| CCRF-CEM | 1.89 | 1.75 | 1.44 | 2.40 | 2.45 | 2.33 | 2.24 | 0.42 |
| HL-60(TB) | 2.07 | 2.19 | 2.02 | 2.17 | 1.51 | 1.90 | 2.09 | 2.11 |
| K-562 | 0.37 | 1.48 | 2.05 | 1.85 | 1.72 | 1.78 | 0.98 | 0.35 |
| RPMI82226 | 1.77 | 1.86 | 1.25 | 2.14 | 2.08 | 2.02 | 2.48 | 2.13 |
| Colon Cancer | ||||||||
| HCC-2998 | 1.92 | 1.95 | 1.87 | 2.01 | 1.87 | 1.88 | 1.98 | 2.10 |
| HCT-116 | 1.61 | 1.22 | 1.71 | 1.73 | 1.77 | 1.77 | 1.66 | 1.56 |
| HCT-15 | 1.33 | 1.67 | 1.56 | 1.79 | 1.95 | 1.88 | 1.65 | 1.38 |
| HT29 | 1.35 | 1.69 | 2.07 | 2.01 | 2.23 | 1.53 | 1.71 | 1.57 |
| KM12 | 1.67 | 1.83 | 2.02 | 1.65 | 1.76 | 1.87 | 1.91 | 2.33 |
| SW-620 | 1.88 | 2.12 | 1.73 | 1.98 | 1.93 | 1.95 | 2.04 | 1.81 |
| Breast Cancer | ||||||||
| MCF-7 | 1.48 | 1.27 | 1.43 | 1.22 | 1.66 | 1.81 | 1.53 | 1.77 |
| MDA-MB-231/ATCC | 1.27 | 1.46 | 1.06 | 1.67 | 1.65 | 1.87 | 1.52 | 2.34 |
| HS 578T | 1.71 | 2.26 | 2.00 | 2.21 | 2.15 | 14.7 | 2.23 | 2.47 |
| BT-549 | 1.65 | 1.63 | 8.82 | 1.78 | 8.96 | 1.79 | 1.86 | 26.3 |
| MDA-MB-468 | 1.51 | 1.45 | 0.23 | ND b | 1.78 | 1.97 | 1.77 | 1.68 |
a Concentration (µM) that inhibits 50% net cell growth. b ND = Not Determined.
Thiazole derivatives 4a,b (Figure 6), in which indole and 7-azaindole units were switched compared to derivatives 2, were also reported. The derivatives 4a and 4b showed good antiproliferative activity against the NCI full panel (about 60 human tumour cell lines) with GI50 values ranging from low micromolar to nanomolar levels (0.03–13.0 and 0.04–14.2 μM, respectively) (Table 11). They also exhibited potent cytotoxicity on HepG2 hepatocarcinoma cells, a cell line not included in the NCI panel. Both compounds inhibited the HepG2 cells growth in a dose-dependent manner, with GI50 values of 1.69 and 0.21 µM for 4a and 4b, respectively. Under the same conditions, both compounds did not affect the viability of normal immortalised human liver cells Chang, proving to be selective towards tumour cells. The mechanism of action of these derivatives was pro-apoptotic, being associated with the externalisation of plasma membrane phosphatidylserine and mitochondrial dysfunction. Both compounds 4a and 4b caused a significant dose-dependent decrease in the percentage of cells in the G0/G1 and S phases, coupled with an increase in cells in the G2/M phase, and the appearance of a subG1-cell population [20].
Figure 6.

Thiazole derivatives 4a (NORA234), 4b.
Table 11.
Overview of the results of the in vitro anti-tumour screening for compounds 4a, 4b, 5a, 6a, and 7a–7g.
| Thiazole Derivative | N° Cell Lines Tested | N° Active Cell Lines | GI50 Range (µM) a |
|---|---|---|---|
| 4a | 60 | 60 | 0.03–13.0 |
| 4b | 59 | 59 | 0.04–14.2 |
| 5a | 54 | 48 | 0.81–27.7 |
| 6a | 59 | 59 | 0.93–4.70 |
| 7a | 58 | 29 | 4.11–6.89 |
| 7b | 59 | 59 | 4.45–5.98 |
| 7c | 59 | 59 | 4.63–5.28 |
| 7d | 60 | 60 | 4.87–7.50 |
| 7e | 60 | 42 | 4.03–6.11 |
| 7f | 59 | 9 | 4.01–7.56 |
| 7g | 59 | 59 | 4.64–5.59 |
a Concentration range that inhibits 50% net cell growth.
In addition, the compound 4a, called Nortopsentin 234 (NORA234) (Figure 6) was found to lead to an initial reduction in the proliferative and clonogenic potential of advanced colorectal cancer sphere cells (CR-CSphCs), followed by an adaptive response selecting the CR-CSphC-resistant compartment. Cells saved from the treatment with NORA234 expressed high levels of CD44v6, combined with constitutive activation of the Wnt pathway. In CR-CSphC-based organoids, NORA234 caused genotoxic stress with concomitant G2/M cell cycle arrest and activation of CHK1, driving the DNA damage repair of CR-CSphCs, regardless of the mutational background, microsatellite stability, and consensus molecular subtype. The synergic combination of NORA234 and a CHK1 inhibitor (rabusertib) targeted synthetic lethal inducing death in both CD44v6-negative and CD44v6-positive CRC stem cell fractions, apart from Wnt pathway activity. These data could provide a rationale for developing an effective strategy for the treatment of colorectal cancer (CRC) [61].
Two series of Nortopsentin thiazolyl analogues 5 and 6 (Figure 7) were also synthesized. Compared to derivatives 2, in derivatives 5, both indole units were replaced by 7-azaindole moieties, while in derivatives 6, one indole unit was replaced by a 6-azaindole unit. In these series, compounds 5a and 6a showed cytotoxic activity against a broad spectrum of human cancer cell lines included in the NCI panel, having GI50 values of 0.81–27.7 and 0.93–4.70 µM, respectively (Table 11). The indolyl-thiazolyl-pyrrolo[2,3-c]pyridine derivative 6a resulted in more active than thiazolyl-bis-pyrrolo[2,3-b]pyridines derivative 5a in terms of GI50. Interestingly, the compounds did not significantly compromise the vitality of intestinal normal-like differentiated Caco-2 cells, providing tumour cells as the main target of their cytotoxicity. Investigation of the mechanisms behind the antiproliferative activity in HCT-116 colon cancer cells showed that derivative 5a caused a dose-dependent increase in the apoptotic cell population, engaging the mitochondria-mediated pathway and causing cell cycle arrest at the G2/M phase. On the other hand, derivative 6a, at a concentration lower than its GI50, exhibited antiproliferative effects with a great accumulation of autophagic vacuoles without apparent signs of apoptosis. The arrest of the cell cycle at G1 phase proved the autophagic fate of the cells. Evidence indicates that autophagic cell death can be induced as an alternative to apoptosis with therapeutic finality in cancer cells that are resistant to apoptosis. It follows that thiazole compound 6a could be considered a lead compound for Nortopsentin derivatives with autophagic activity [20].
Figure 7.

Thiazole series 5,6.
Among thiazole analogues, derivatives 7 (Figure 8), in which one indole ring was replaced by a phenyl while the other one was replaced by a 7-azaindole, were also reported.
Figure 8.

Thiazole series 7.
Derivatives 7b,c,d, and g were active against all cancer cell lines tested by NCI, while derivatives 7a,e, and f were cytotoxic against a good percentage of the tested cell lines (50%, 70% and 15% respectively), having GI50 values in the micromolar to sub-micromolar/nanomolar range (Table 11). The most active compounds were the N-methyl derivatives, and four of them, 7b,7c,7d, and 7g, were further tested against pancreatic carcinoma (MiaPaCa-2) and malignant peritoneal mesothelioma (STO) showing IC50 values in the range 4.3–41.6 µM and 0.41–17.2 µM, respectively (Table 12). Kinase activity assays (CDK1/cyclin, CDK5/p25, or GSK3β) were also performed to explain the mechanism of action of this series of compounds. Only the compounds with the highest antiproliferative activity (7c and 7d) exhibited affinity for CDK1, with IC50 values of 0.41 and 0.85 µM, respectively. Such values were like those of roscovitine and purvanalol A, used as reference drugs (Table 13). Moreover, exposure of asynchronously growing STO cells to both compounds affected cell-cycle phase distribution, leading to a concentration-dependent accumulation of cells in the G2/M phase with a concomitant increase in the sub-G1 apoptotic cell population [62].
Table 12.
Cytotoxic activity of compounds 7b–d,7g in MiaPaCA-2 and STO cell lines.
| Compound | IC50 (µM) a | |
|---|---|---|
| MiaPaCA-2 | STO | |
| 7b | 41.6 ± 2.4 | 17.2 ± 2.9 |
| 7c | 5.7 ± 0.8 | 0.83 ± 0.04 |
| 7d | 4.3 ± 0.6 | 0.41 ± 0.06 |
| 7g | 39.6 ± 3.9 | 5.7 ± 0.8 |
a Concentration of drug required to inhibit growth by 50% as determined by SRB assay after 72 h continuous exposure to each compound; the data represent mean values ± SD of at least three independent experiments.
Table 13.
Kinase inhibition by compounds 7b–d,6g.
| Compound | IC50 (µM) a | ||
|---|---|---|---|
| CDK1 | CDK5 | GSK3β | |
| 7b | 33.27 ± 2.97 | >50.0 | >50.0 |
| 7c | 0.41 ± 0.08 | >50.0 | >50.0 |
| 7d | 0.85 ± 0.13 | >50.0 | >50.0 |
| 7g | 45.93 ± 4.19 | >50.0 | >50.0 |
| Purvanalol A | 0.73 ± 0.06 | >50.0 | >50.0 |
| Roscovitine | 0.59 ± 0.08 | >50.0 | >50.0 |
a Inhibitor concentration at which enzyme activity is decreased by 50%; data represent the mean ± SD of at least three independent experiments.
Thiazole derivatives 8 and 9 (Figure 9) combine an indole unit with a naphthalyl portion. Derivatives 8a, 9a, and 9b in particular displayed good antiproliferative activity against the MCF-7 cell line with GI50 values in the micromolar range (2.13, 3.26 and 5.14 µM, respectively, Table 14). Their mechanism of action was found to be pro-apoptotic, inducing early apoptosis in MCF-7 cells after 24 h of treatment without necrotic effects. They also caused a decrease in the percentage of cells in the G0/G1 and S phases, with a concomitant percentage increase in cells in the G2/M phase [63].
Figure 9.

Thiazole series 8,9.
Table 14.
GI50 values of the most active compounds 8a, 9a, and 9b on the MCF-7 cells.
| Thiazole Derivative | GI50 (μM) a |
|---|---|
| 8a | 2.13 ± 0.12 |
| 9a | 3.26 ± 0.19 |
| 9b | 5.14 ± 0.34 |
a Concentration (µM) that inhibits 50% net cell growth; data represent the mean ± SD of at least three independent experiments.
2.1.2. Thiadiazoles
A bis-indolyl-1,2,4-thiadiazole series 10 (Figure 10) was screened for in vitro cytotoxicity against six human cancer cell lines: prostate (PC3, DU145, and LnCaP), breast (MCF-7 and MDA-MB-231), and pancreas (PaCa2). In this series, indolyl-1,2,4-thiadiazole 10a was identified as the most potent compound with IC50 values of 14.6, 21.4, and 21.2 µM against the cancer cell lines LnCaP, PC3, and PaCa2, respectively (Table 15) [64].
Figure 10.

Bis-indolyl-1,2,4-thiadiazole derivatives 10.
Table 15.
Anticancer activity of the compound 10a against selected human cancer cell lines IC50 (μM)a.
| Compound | MDA-MB-231 | MCF-7 | LnCaP | DU145 | PC3 | PaCa2 |
|---|---|---|---|---|---|---|
| 10a | 67.9 | 32.1 | 14.6 | 369.8 | 21.4 | 21.2 |
a IC50 values were obtained using a dose–response curve by non-linear regression using a curve fitting program.
2.1.3. Thiophenes
Bis-indolyl-thiophene derivatives of type 11 (Figure 11), in which the imidazole moiety of Nortopsentin was replaced by a thiophene ring, were submitted to the NCI for evaluation of the full panel (about 60 human cancer cell lines). The most active compound was the derivative 11a, having GI50 values in the range of 0.34–19.0 µM. It was particularly powerful against the leukaemia subpanel, having GI50 in the range of 0.34–3.54 µM (Table 16) [63].
Figure 11.
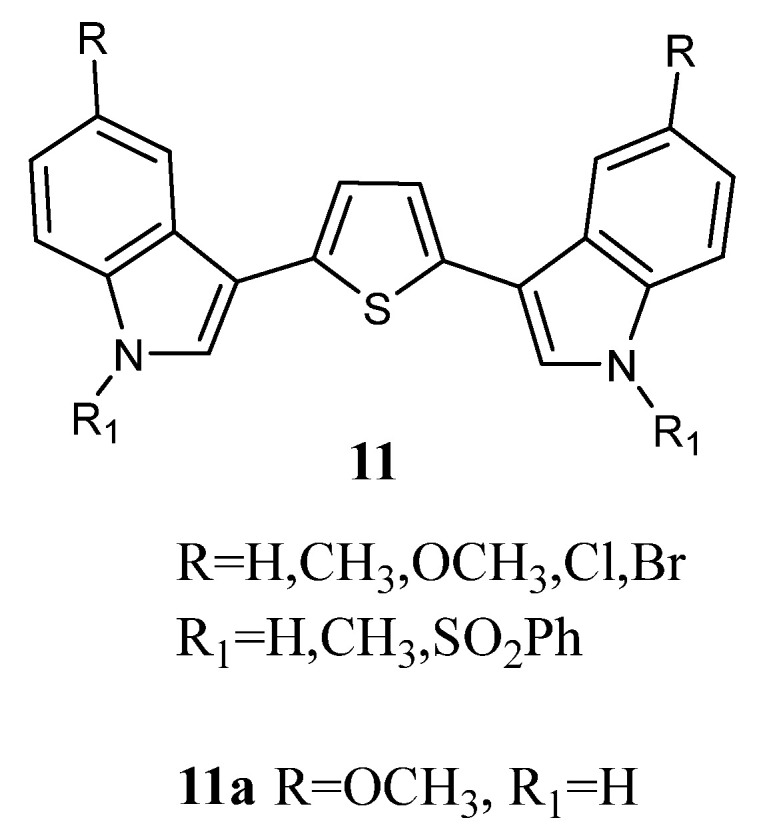
Bis-indolyl-thiophene derivatives 11.
Table 16.
In vitro activity of derivatives 12a–c towards 10 human tumour cell lines.
| Compound | IC50 (µg/mL) | Active/Total a | Tumour Selectivity b | ||
|---|---|---|---|---|---|
| 1 (µg/mL) | 10 (µg/mL) | 100 (µg/mL) | |||
| 12a | 27.1 | 0% | 0% | 70% | 0/10 |
| 12b | 21.1 | 0% | 10% | 80% | 1/10 |
| 12c | 17.1 | 0% | 20% | 70% | 2/10 |
a Responsive (T/C < 30%)/total cell lines. b Selective (individual IC70 < 1/3 mean IC70)/total cell lines.
2.1.4. Furans
Bis-indolyl-furan derivatives 12a–c (Figure 12) were screened for in vitro anti-tumour activity in a panel of 10 human tumour cell lines and showed mean IC50 values of 27.1, 21.1, and 17.1 µg/mL, respectively (Table 16). The most active candidate, 12c, was further screened in a panel of 29 cell lines, showing cytotoxicity against a high percentage of tested cell lines (76%) with mean IC50 values of 20.5 µg/mL (53.1 µM). Moreover, compound 12c was further tested by the NCI on a panel of approximately 60 tumour cell lines. Data revealed that compound 12c was cytotoxic against all cell lines investigated, displaying GI50 values at micromolar concentration and was particularly potent against the leukaemia subpanel, having GI50 values in the range of 1.63–6.46 µM (Table 17) [63].
Figure 12.

Bis-indolyl-furan derivatives 12.
Table 17.
In vitro inhibition of cancer cell lines growth in leukaemia subpanel by compounds 11a and 12c.
| Cell Line | GI50 (µM) a | |
|---|---|---|
| 11a | 12c | |
| Leukaemia | ||
| CCRF-CEM | 0.34 | 6.46 |
| HL-60(TB) | 2.27 | 1.98 |
| K-562 | 3.54 | 2.86 |
| MOLT-4 | 1.91 | >4.00 |
| RPMI82226 | 2.83 | 2.25 |
| SR | ND b | 1.63 |
a Concentration (µM) that inhibits 50% net cell growth. b ND = Not Determined.
2.1.5. Pyrroles
Bis-indolyl-pyrroles of type 13 (Figure 13) were investigated in vitro against human tumour cell lines and by ex-vivo clonogenic assay using human tumour xenografts. Screening in monolayer cultures of 42 human tumour cell lines derived from 15 different solid tumour types (bladder, colon, gastric, head-neck, liver, lung, mammary, melanoma, ovarian, pancreatic, prostate, pleural mesothelioma, renal, sarcoma, and uterus) revealed that the most active derivatives were 13a and 13b. Compounds 13a and 13b affected concentration-dependent inhibition of tumour cell growth with IC50 values in the range 0.22–6.34 μM and 0.11–2.65 μM, respectively, indicating pronounced cytotoxic potency (Table 18). Regarding compound 13a, selective activity was observed with submicromolar IC50 values against some cell lines, such as cell lines of bladder cancer (BXF 1218L, BXF 1352L), gastric cancer cell line (GXA MKN45), head-neck cell line (HNXF CAL27), two melanoma cell lines (MEXF 1341L; MEXF 276L), as well as LXFL 1121L (lung cancer), PAXF PANC-1 (pancreatic cancer), PRXF PC3M (prostate cancer), SXF SAOS-2 (sarcoma), and UXF 1138L (cancer of the uterine body) cell lines. Less sensitive cell lines were found among colon (HCT-116, HT-29), lung (LXFA 289L), ovarian (OVXF 899L), prostate (DU145), and renal cancer (RXF 393NL, RXF 486L). Compound 13b exhibited pronounced activity with submicromolar IC50 values in 32 cell lines.
Figure 13.

Bis-indolyl-pyrrole derivatives 13.
Table 18.
In vitro and ex vivo anti-tumour activity by derivatives 13a and b: In in vitro tumour cell lines (monolayer assay) and in ex vivo human xenografts (clonogenic assay).
| Cell Line | IC50 a (μM) | Tumour Histotype |
IC50 a (μM) | ||
|---|---|---|---|---|---|
| 13a | 13b | 13a | 13b | ||
| Bladder | |||||
| BXF 1218L | 0.72 | 0.32 | BXF 1218 | 2.45 | 2.35 |
| BXF 1352L | 0.68 | 0.41 | BXF 1228 | 2.89 | 2.86 |
| BXF T24 | 1.72 | 0.58 | |||
| Lung Cancer | |||||
| LXFA 289L | 6.34 | 2.39 | LXFA 1012 | 4.76 | 27.34 |
| LXFA 526L | 1.57 | 0.67 | LXFA 1584 | 2.07 | 2.73 |
| LXFA 629L | 2.28 | 1.36 | LXFA 297 | 54.90 | >100 |
| LXFL 1121L | 0.67 | 0.38 | LXFA 526 | 2.96 | 3.15 |
| LXFL 529L | 1.85 | 0.67 | LXFA 629 | 2.23 | 3.63 |
| LXFL H460 | 2.08 | 0.89 | LXFA 677 | 23.19 | 27.02 |
| LXFA 923 | 19.50 | 25.68 | |||
| LXFE 1422 | 5.90 | 1.72 | |||
| LXFL 1072 | 3.44 | 3.42 | |||
| LXFL 529 | 5.07 | 4.46 | |||
| LXFL 625 | 24.04 | 38.21 | |||
| Colon Cancer | |||||
| CXF 269L | 1.39 | 0.56 | CXF 1103 | 4.47 | 18.36 |
| CXF HCT116 | 3.24 | 1.64 | CXF 1729 | 3.49 | 7.77 |
| CXF HT29 | 5.20 | 2.65 | CXF 1783 | 37.57 | 40.70 |
| CXF RKO | 1.50 | 0.63 | CXF 280 | 26.18 | 35.70 |
| CXF 975 | 6.93 | 6.03 | |||
| Head and Neck | |||||
| HNXF CAL27 | 0.81 | 0.50 | HNXF 536 | 2.45 | 2.67 |
| HNXF 908 | 2.57 | 4.00 | |||
| Melanoma | |||||
| MEXF 1341L | 0.52 | 0.19 | MEXF 1539 | 19.90 | 15.31 |
| MEXF 276L | 0.22 | 0.11 | MEXF 276 | 1.44 | 1.75 |
| MEXF 462NL | 1.31 | 0.55 | MEXF 462 | 3.24 | 4.10 |
| MEXF 989 | 1.18 | 0.58 | |||
| Ovarian Cancer | |||||
| OVXF OVCAR3 | 1.46 | 0.58 | OVXF 1353 | 20.96 | 22.90 |
| OVXF 899L | 5.40 | 2.03 | OVXF 899 | 3.54 | 3.93 |
| Renal Cancer | |||||
| RXF 1183L | 1.13 | 0.58 | RXF 1220 | 6.34 | 3.16 |
| RXF 1781L | 1.77 | 0.66 | RXF 486 | 2.90 | 3.90 |
| RXF 393NL | 3.14 | 1.34 | RXF 631 | 2.98 | 2.81 |
| RXF 486L | 3.86 | 1.60 | |||
| Prostate Cancer | |||||
| PRXF 22RV1 | 1.46 | 0.63 | PRXF DU145 | 28.67 | 25.98 |
| PRXF DU145 | 4.45 | 1.96 | PRXF PC3M | 2.89 | 2.80 |
| PRXF LNCAP | 2.10 | 0.68 | |||
| PRXF PC3M | 0.85 | 0.32 | |||
| Mammary Cancer | |||||
| MAXF 401NL | 1.24 | 0.64 | MAXF 1322 | 16.80 | 9.49 |
| MAXF MCF-7 | 2.44 | 0.98 | MAXF 1384 | 34.48 | 33.28 |
| MAXF MDA-231 | 1.18 | 0.49 | MAXF 401 | 5.90 | 11.32 |
| Gastric Cancer | |||||
| GXA MKN45 | 0.84 | 0.53 | GXF 1172 | 6.27 | >100 |
| GXF 251L | 1.56 | 0.65 | GXF 251 | 7.10 | 25.50 |
| GXF 97 | 2.72 | 3.74 | |||
| Pancreatic Cancer | |||||
| PANC1 | 0.74 | 0.41 | PAXF 546 | 3.52 | 14.63 |
| 1657L | 2.68 | 1.02 | PAXF 736 | 2.99 | 2.10 |
| 546L | 2.70 | 1.16 | |||
| Pleural mesothelioma | |||||
| PXF 1118L | 2.50 | 0.88 | PXF 1752L | 3.71 | 4.05 |
| PXF 1752L | 0.89 | 0.43 | PXF 541 | 2.20 | 0.37 |
| PXF 698L | 1.86 | 0.86 | |||
| Sarcoma | |||||
| SXF SAOS2 | 0.72 | 0.33 | SXF 1186 | 5.71 | 6.17 |
| SXF TE671 | 1.60 | 0.53 | SXF 1301 | 3.54 | 23.95 |
| SXF 627 | 3.40 | 4.06 | |||
| Uterus Cancer | |||||
| UXF 1138L | 0.72 | 0.35 | |||
a Concentration (μM) that inhibits 50% net cell growth.
The anti-proliferative activity of 13a and 13b was also evaluated in cell suspensions prepared from 44 human tumour xenografts of 13 different tumour types (bladder, colon, gastric, head-neck, lung, mammary, melanoma, ovarian, pancreatic, prostate, pleural mesothelioma, renal, and sarcoma), which were cultured as solid tumours in serial passage on immune-deficient nude mice (Table 18). The results confirmed the concentration-dependent activity of 13a and 13b on cell lines with IC50 values in the range of 1.18–54.90 μM and 0.37–40.70 μM, respectively. Selectivity was encountered for 13a against 9 out of the 44 tumours tested, while these sensitive tumours were scattered among various tumour histotypes, like bladder, gastric, head and neck, lung cancer, melanoma, and pleuramesothelioma. Better tumour selectivity was displayed for compound 13b, with 14 out of 44 tumours. Sensitive cancer types were found among the bladder, head and neck, lung, pancreatic, prostate, renal cancer, melanoma, and pleuromesothelioma [16].
2.1.6. Oxazoles
Bis-indolyl-isoxazoles 14a–e (Figure 14), were screened for in vitro anti-tumour activity in a panel of 10 human tumour cell lines by using a monolayer cell survival and proliferation assay. All compounds showed cytotoxic activity, exhibiting mean IC50 values in the range of 9.6–44.5 µg/mL (Table 19). Moreover, the most active candidate 14a was also tested in a panel of 29 cell lines (derived from bladder, lung, colon, CNS, melanoma, ovarian, renal, prostate, mammary, gastric, pancreatic, pleural mesothelioma, and uterus tumours), displaying cytotoxicity against all tested cell lines with IC50 values in the range 4.2–40.6 µg/mL [63].
Figure 14.

Bis-indolyl-isoxazole derivatives 14.
Table 19.
In vitro activity of derivatives 14a–e towards 10 human tumour cell lines.
| Compound | IC50 (µg/mL) | Active/Total a | Tumour Selectivity b |
||
|---|---|---|---|---|---|
| 1 (µg/mL) | 10 (µg/mL) | 100 (µg/mL) | |||
| 14a | 9.6 | 0/10 (0%) | 2/10 (20%) | 10/10 (100%) | 1/10 |
| 14b | 44.5 | 0/10 (0%) | 0/10 (0%) | 7/10 (70%) | 0/10 |
| 14c | 17.3 | 0/10 (0%) | 1/10 (10%) | 7/10 (70%) | 2/10 |
| 14d | 24.5 | 0/10 (0%) | 0/10 (0%) | 7/10 (67%) | 1/10 |
| 14e | 43.2 | 0/10 (0%) | 0/10 (0%) | 3/10 (30%) | 1/10 |
a Responsive (T/C < 30%)/total cell lines. b Selective (individual IC70 < 1/3 mean IC70)/total cell lines.
2.1.7. Oxadiazole
1,3,4-Oxadiazoles
The 1,3,4-oxadiazole scaffold is common to many anticancer agents and ensures cytotoxic properties [65]. Certain 1,3,4-oxadiazole compounds have been identified as tubulin-binding agents and DNA intercalators. Synthesized bis-indolyl-1,3,4-oxadiazoles 15a–m (Figure 15) were studied for their cytotoxic activity against six human cancer cell lines: pancreas (AsPC1), prostate (DU145 and PC3), cervical (HeLa), breast (MDA-MB-231), and ovarian (OVCAR). Most of the compounds showed strong anticancer activities, with IC50 values in the micromolar to nanomolar range (Table 20). The structure-activity relationship study proved that a bromo substituent is decisive for imparting potent cytotoxicity. Bromo-substituted 1,3,4-oxadiazole 15b was the most active compound in the series, with IC50 values of 20 nM against prostate (DU145) and cervical (HeLa) cancer cell lines. In addition, N-alkylation promoted the selectivity of the compound towards a particular tumour type. Preliminary studies in MDA-MB-231 breast cancer cells indicated that the mechanism of action of 1,3,4-oxadiazoles 15a–m was pro-apoptotic [66].
Figure 15.

Bis-indolyl-1,3,4-oxadiazole derivatives 15.
Table 20.
Cytotoxicity of oxadiazoles 15a–m against a panel of human cancer cell lines: pancreas (AsPC1), prostate (DU145 and PC3), cervical (HeLa), breast (MDA-MB-231), and ovarian (OVCAR).
| Compounds | R | R1 | R2 | R3 | R4 | IC50 (µM) a | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AsPc1 | DU145 | PC3 | MDA-MB-231 | OVCAR | HeLa | ||||||
| 15a | H | H | H | H | H | 0.46 | 0.23 | 8.43 | 0.09 | 0.10 | 0.53 |
| 15b | Br | H | H | H | H | 0.20 | 0.02 | 0.25 | 0.24 | 0.15 | 0.02 |
| 15c | OCH3 | H | H | H | H | 0.06 | 0.72 | 0.11 | 0.90 | 0.38 | 66.79 |
| 15d | H | F | H | H | H | 0.31 | 9.79 | 0.29 | 4.34 | 0.31 | 6.86 |
| 15e | Br | H | H | F | H | 0.45 | 0.08 | 4.55 | 0.20 | 0.23 | 0.06 |
| 15f | OCH3 | H | H | F | H | 1.68 | 0.42 | 0.10 | 6.44 | 2.91 | 52.90 |
| 15g | H | F | H | F | H | 0.20 | 0.15 | 1.09 | 0.21 | 0.15 | 0.06 |
| 15h | H | H | CH3 | H | CH3 | 0.10 | 0.52 | 0.12 | 15.71 | 0.97 | 4.55 |
| 15i | H | H | 4-ClC6H4CH2 | H | H | 2.47 | 0.37 | 7.11 | 126.3 | 0.16 | 0.73 |
| 15j | H | H | 4-CH3OC6H4CH2 | H | H | 0.05 | 0.54 | 3.33 | 168.9 | 0.10 | 5.94 |
| 15k | Br | H | 4-ClC6H4CH2 | H | H | 0.43 | 0.11 | 0.24 | 0.86 | 0.27 | 8.10 |
| 15l | H | F | 4-ClC6H4CH2 | H | H | 1.60 | 0.14 | 0.05 | 2.58 | 0.24 | 0.96 |
| 15m | H | H | 4-CH3OC6H4CH2 | H | H | 0.07 | 0.10 | 0.14 | 17.22 | 0.11 | 1.20 |
a IC50 values were obtained using a dose–response curve by nonlinear regression using GraphPad Prism 5.0 for curve fitting. For all data, standard error of the mean (SEM) values were ±<10%.
Another 1,3,4-oxadiazole series of type 16 (Figure 16) was synthesised and evaluated for their anti-proliferative activity against lung (A549), breast (MDA-MB-231, MCF-7), and cervical (HeLa) cancer cell lines. The IC50 values of the compounds evaluated ranged between 1.8 and 42.3 µM (Table 20). The compounds 16e and 16h showed good cytotoxic activity with IC50 values of 1.8 µM and 2.6 µM on the breast cancer cell line MCF-7. Three compounds, 16e, 16f, and 16h, showed better cytotoxicity on the cervical cancer cell line (HeLa) with IC50 values of 9.23 µM, 9.4 µM, and 6.34 µM. The compound 16h showed good cytotoxicity with IC50 values of 3.3 µM on the lung cancer cell line A549. Moreover, compound 16e was recognised as a promising drug lead as it showed potent cytotoxicity with an IC50 value of 1.8 µM towards MCF-7 when compared to the standard drug doxorubicin (IC50 value of 0.98 µM) (Table 21). The compounds 16a, 16b, 16g, and 16i exhibited moderate cytotoxicity on cervical cancer cell line HeLa. The compound 16d proved moderate cytotoxicity on breast cancer cell line MCF-7. The compounds 16e and 16h displayed moderate cytotoxicity on breast cancer cell line MDA-MB-231 with an IC50 value of 12.17 µM and 10.23 µM (Table 21). In addition, no cytotoxicity was found on normal human embryonic kidney cells, HEK-293 [67]. The impact of these compounds on the colchicine-binding site of the tubulin polymer was also evaluated using molecular docking studies. They showed an effective role in the inhibition of mitotic spindle formation thereby altering tubulin polymerization. Moreover, a careful investigation of the binding pattern of ligands provided a few specific elements that are consistent with in vitro data. The presence of bromo atom on the indole ring of compound 16e and the H-bond (2.96 Å) of the methoxy group with Lys 254 might be an explanation of the most potent anti-proliferative activity of 16e compared to 16a, 16b, 16c, and 16d. Regarding compound 16h, the absence of an N-methyl group could be a justifiable reason for the higher potency of compound 16h compared to compound 16d. On the other hand, the NH of the free indole ring of compound 16f was observed to be involved in H-bond with Cy241 (3.15 Å) and Val238 (3.25 Å) which may be the cause of its exhibiting the least activity [67].
Figure 16.

Bis-indolyl-1,3,4-oxadiazole derivatives 16.
Table 21.
Cytotoxicity of oxadiazoles 16a–i against a panel of human cancer cell lines: lung (A549), breast (MDA-MB-231 and MCF-7), and cervical (HeLa).
| Compound | R | R1 | R2 | IC50 (µM) | |||
|---|---|---|---|---|---|---|---|
| A549 | MDA-MB-231 | MCF-7 | HeLa | ||||
| 16a | Br | CH3 | H | - a | - | 30.9 ± 0.48 | 14.8 ± 0.39 |
| 16b | Br | CH3 | Br | - | 38.9 ± 0.85 | 24 ± 0.5 | 19.5 ± 0.43 |
| 16c | H | CH3 | OCH3 | - | - | 32.2 ± 0.9 | 42.3 ± 0.96 |
| 16d | H | CH3 | NO2 | - | - | 19.2 ± 0.8 | 33.2 ± 0.45 |
| 16e | Br | CH3 | OCH3 | 24.2 ± 0.89 | 12.17 ± 1.1 | 1.8 ± 0.9 | 9.23 ± 0.58 |
| 16f | H | H | H | - | - | 24.5 ± 0.9 | 9.4 ± 0.37 |
| 16g | H | H | Br | - | - | - | 16.3 ± 0.33 |
| 16h | H | H | NO2 | 3.3 ± 0.85 | 10.23 ± 1.3 | 2.6 ± 0.89 | 6.34 ± 0.56 |
| 16i | Br | H | H | - | - | - | 19.8 ± 0.45 |
a- no activity.
1,2,4-Oxadiazoles
The 1,2,4-oxadiazole ring system is a five-membered heterocycle ring found in many molecules with significant biological activity, especially anti-tumour. This heterocycle is an amide and ester bioisostere that could improve the bioavailability and physiochemical properties of compounds bearing it. Thus, new indolyl-1,2,4-oxadiazol-7-azaindole Nortopsentin analogs 17 (Figure 17), in which the 1,2,4-oxadiazole ring replaced the imidazole central ring and a 7-azaindole portion substituted the indole, were prescreened against the HCT-116 cell line (colon rectal carcinoma). Two compounds bearing the 5-bromo-1-methyl-7-azaindole moiety (17a,b) showed the highest cytotoxic activity having IC50 < 10 µM (IC50 values of 1.93 and 3.55 µM, respectively; Table 22) [68].
Figure 17.

Indolyl-1,2,4-oxadiazol-7-azaindole derivatives 17.
Table 22.
Cytotoxicity of oxadiazoles 17a,b against a panel of human cancer cell lines: breast (MCF-7), colon (HCT-116 and CaCo2), and cervical (HeLa).
| Compound | R | R1 | IC50 (µM) a | |||
|---|---|---|---|---|---|---|
| MCF-7 | HCT-116 | CaCo2 | HeLa | |||
| 17a | Cl | Br | 0.65 ± 0.05 | 1.93 ± 0.06 | 1.06 ± 0.09 | 10.56 ± 0.98 |
| 17b | OCH3 | Br | 2.41 ± 0.23 | 3.55 ± 0.1 | 3.33 ± 0.25 | 13.96 ± 1.41 |
a IC50 value was calculated by plotting the percentage viability versus concentration on a logarithmic graph. Results are the mean values SD (standard deviation) of three separate experiments carried out in triplicate.
Interestingly, the substitution of the bromine atom on the 7-azaindole with fluorine or the absence of the halogen atom resulted in a decrease in the antiproliferative effect. The most active compounds 17a,b were selected for further investigations in additional human tumour cell lines, such as MCF-7 (breast cancer), HeLa (cervical cancer), and CaCo2 (colorectal carcinoma) cell lines, showing IC50 values in the micromolar and submicromolar range (Table 22). Compared to 17b, derivative 17a appeared more effective [62]. The mechanism of the anti-proliferative effect on MCF-7 was pro-apoptotic, being associated with the externalisation of plasma membrane phosphatidylserine, chromatin condensation, and membrane blebbing. The analysis showed that both compounds led to early apoptosis without causing necrosis. These compounds induced an increase in cells in the G0–G1 phase suggesting that they can act to promote DNA duplication. Moreover, the non-toxicity of these derivatives (17a,b) was confirmed by experiments on intestinal normal-like differentiated Caco2 cells [68].
2.1.8. Pyrazoles
Bis-indolyl-pyrazoles 18 (Figure 18), in which a pyrazole central ring substituted the imidazole ring of Nortopsentin, were screened by the NCI.
Figure 18.
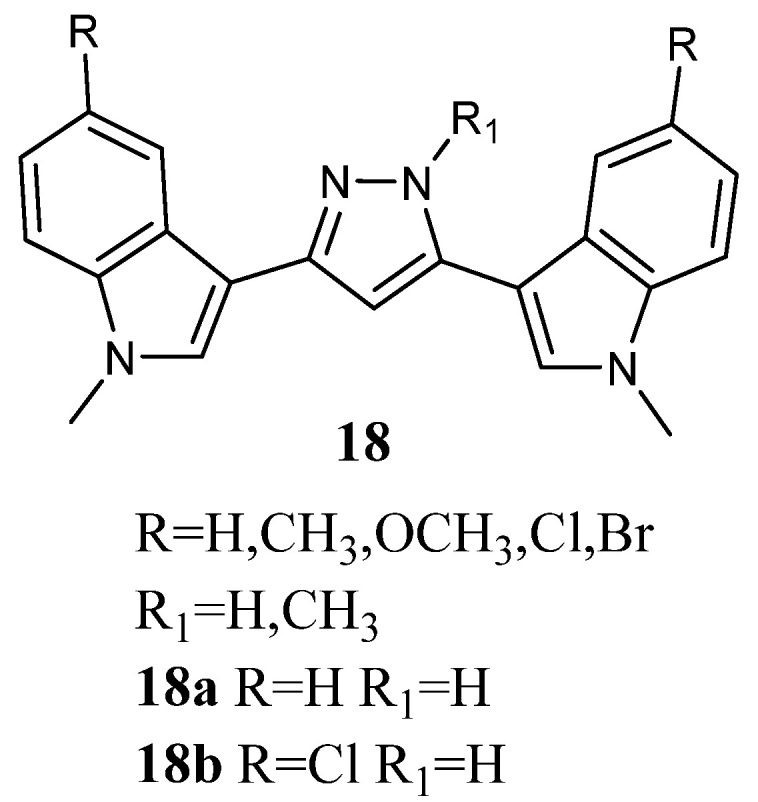
Bis-indolyl-pyrazole derivatives 18.
Among the investigated compounds, derivatives 18a and b were the most active, exhibiting antiproliferative activity against most of the human cell lines. The percentage of sensitive cell lines out of the total number of cell lines investigated was 90% and 100%, respectively, while MG_MID was 18.2 and 3.23 µM, respectively. Therefore, compound 18b, bearing a chlorine atom, was more active than the unsubstituted derivative 18a. Derivative 18b was cytotoxic against the totality of cell lines investigated at micromolar concentration, and it proved to be selective for the melanoma subpanel, having all the subpanel cell lines GI50 values in the range of 1.63–9.64 µM (Table 23). The most sensitive cell lines were UACC-62, LOX IMVI, and SK-MEL-5 (GI50 values of 1.63, 1.70, and 1.79 µM, respectively). It also showed selectivity for MOLT-4, SR, and K-562 (GI50 values of 1.55, 2.36, and 2.78 µM, respectively) of the leukaemia subpanel, HCC-2998 and COLO 205 (GI50 values of 1.71 and 2.22 µM, respectively) of colon cancer, CAKI-1 (GI50 value of 1.70 µM) of renal cancer, BT-549 (GI50 value of 2.03 µM) of breast cancer, and SF-539 (GI50 value of 1.81 µM) of CNS subpanel. Derivative 18a was particularly effective against the colon subpanel, having GI50 in the range of 4.58–19.0 µM. The most sensitive colon cell lines were KM12 and HCC-2998 (GI50 values of 4.58 and 4.74 µM, respectively). Compound 18a showed good selectivity for HOP-92 (GI50 2.06 µM) and NCIH460 (GI50 value of 4.48 µM) of the non-small-cell lung cancer subpanel and MCF-7 (GI50 value of 3.95 µM) of the breast cancer subpanel (Table 23) [60]. Experiments aimed at evaluating the ability of derivatives 18a and b to interact with DNA, revealed that they were unable to form a molecular complex with the macromolecule. In particular, linear flow dichroism spectra obtained by salmon testes DNA, where different concentrations of 18a and b were used, resulted in nearly overlapping to those recorded without compounds. Furthermore, the ability to interfere with the activity of the nuclear enzyme topoisomerase II, which catalyses the interconversion of different topological forms of DNA, was assayed. As to 18a, the results highlight that the inhibitory capability became detectable at about 50 µM, while in the case of 18b, it happened at 100 µM. Furthermore, both tested compounds presented a scored inhibition, which was considerably weaker than that of m-amsacrine used at 8 µM concentration. These results proved that DNA cannot be considered the main target of cell death, suggesting that other cellular molecular targets are engaged in the antiproliferative activity of 18a and b [63].
Table 23.
In vitro inhibition of cancer cell lines growth in leukaemia, NSCLC, colon, CNS, melanoma, renal, and breast cancer subpanels by compounds 18a and b.
| Cell Line | GI50 (µM) a | Cell Line | GI50 (µM) a | ||
|---|---|---|---|---|---|
| 18a | 18b | 18a | 18b | ||
| Leukaemia | CNS Cancer | ||||
| CCRF-CEM | 14.4 | 6.40 | SF-268 | 16.0 | 5.29 |
| HL-60(TB) | 15.4 | 4.45 | SF-295 | 17.1 | 2.52 |
| K-562 | 42.1 | 2.78 | SF-539 | 16.2 | 1.81 |
| MOLT-4 | 4.75 | 1.55 | SNB-19 | 27.0 | 4.09 |
| RPMI82226 | 37.8 | 6.79 | SNB-75 | 18.4 | 4.25 |
| SR | 3.46 | 2.36 | U251 | 12.9 | 3.37 |
| Non-small Cell Lung Cancer | Melanoma | ||||
| A549/ATCC | 16.3 | 3.24 | LOX IMVI | 5.38 | 1.70 |
| EKVX | 27.2 | 3.15 | MALME-3M | 93.1 | 9.64 |
| HOP-62 | 17.3 | 7.98 | M14 | >100 | 2.15 |
| HOP-92 | 2.06 | 1.86 | MDA-MB-435 | ND b | ND |
| NCI-H226 | 19.8 | 5.29 | SK-MEL-2 | 57.5 | 2.34 |
| NCI-H23 | 22.0 | 2.49 | SK-MEL-28 | >100 | 4.80 |
| NCI-H322M | >100 | 5.73 | SK-MEL-5 | 13.1 | 1.79 |
| NCI-H460 | 4.48 | 2.35 | UACC-257 | 80.6 | 4.27 |
| NCI-H522 | 22.1 | 1.75 | UACC-62 | 16.4 | 1.63 |
| Renal Cancer | |||||
| Colon Cancer | 786-0 | 14.1 | 3.41 | ||
| COLO 205 | 7.98 | 2.22 | A498 | 19.6 | 2.44 |
| HCC-2998 | 4.74 | 1.71 | ACHN | 7.67 | 3.23 |
| HCT-116 | 19.0 | 3.82 | CAKI-1 | >100 | 1.70 |
| HCT-15 | 8.68 | 3.01 | RXF 393 | 14.7 | 3.03 |
| HT29 | 5.35 | 3.57 | SN12C | 1.95 | ND |
| KM12 | 4.58 | 3.45 | TK-10 | 37.4 | 5.16 |
| SW-620 | >100 | 3.52 | UO-31 | 37.6 | 3.01 |
| Breast Cancer | |||||
| MCF-7 | 3.95 | 2.64 | |||
| NCI/ADR-RES | 27.6 | 2.25 | |||
| MDA-MB-231/ATCC | 8.06 | 2.95 | |||
| HS 578T | 18.5 | 3.27 | |||
| BT-549 | 15.9 | 2.03 | |||
| T-47D | 79.7 | 4.06 | |||
| MDA-MB-435 | ND | 2.99 | |||
a Concentration (µM) that inhibits 50% net cell growth. b ND = Not Determined.
2.1.9. Pyrazinones, Pyrazines, Pyrimidines, and Pyridines
Bis-indolyl-pyrazinone 19 and bis-indolyl-pyrazines 20a,b (Figure 19) showed inhibitory activity against a variety of human tumour cell lines with GI50 values that reached submicromolar levels. In particular, the pyrazinone derivative 19 was active against all tested cell lines (GI50 values range of 6.60–74.8 µM), except for the OVCAR-4 cell line; the pyrazine derivative 20a was active against all cell lines with GI50 values range of 2.47–15.5 µM, whereas the N-indolyl methylated compound 20b was the most active, showing GI50 values between 0.058 and 7.19 µM (Table 24) [55,63].
Figure 19.

Bis-indolyl-pyrazinone 19 and bis-indolyl-pyrazine derivatives 20a,b.
Table 24.
Overview of the results of the in vitro anti-tumour screening for compounds 19, 20a, and 20b.
| Derivative | N° Cell Lines Tested | N° Active Cell Lines | GI50 Range (µM) a |
|---|---|---|---|
| 19 | 17 | 17 | 6.60–74.8 |
| 20a | 17 | 16 | 2.47–15.5 |
| 20b | 16 | 16 | 0.058–7.19 |
a Concentration range that inhibits 50% net cell growth.
Novel indolyl-pyrazines 21 and indolyl-pyrimidines 22 (Figure 20) have been synthesized as potential anti-tumour agents. They were in vitro screened by NCI in a panel of 60 human tumour cell lines. Compounds 21a, 22a–d exhibited efficient cytotoxic activities with GI50 values in the low micromolar range against a variety of human cancer cell lines. Among these compounds, the pyrimidine 22a exhibited significant inhibitory activity against leukaemia SR, CNS Cancer SF-539, and breast cancer MDA-MB-435 cell lines with GI50 values of 0.22, 0.16, and 0.22 μM, respectively (Table 25). Derivatives 22b and c demonstrated good inhibitory effects against a variety of tumour cell lines with GI50 values less than 10 μM. Moreover, the pyrimidine 22b displayed selective cytotoxicity against IGROV1 tumour cell line with the GI50 value below 0.01 μM (Table 25). On the other hand, compound 22d was active against all tested cell lines with GI50 values in the range of 1.13–9.53 μM. Bis-indolyl-pyrazine 21a also exhibited good inhibitory effects against a variety of tumour cell lines with GI50 values less than 10 μM [69].
Figure 20.

Bis-indolyl-pyrazines 21 and indolyl-pyrimidines 22.
Table 25.
In vitro inhibition of cancer cell lines growth in leukaemia, CNS, ovarian and breast cancer subpanels by compounds 21a, 22a–d.
| Cell Line | GI50 (µM) a | ||||
|---|---|---|---|---|---|
| 21a | 22a | 22b | 22c | 22d | |
| Leukaemia | |||||
| CCRF-CEM | ND b | ND | 1.51 | 1.52 | 1.13 |
| RPMI82226 | 2.74 | 0.37 | 1.91 | 3.90 | 2.80 |
| SR | 3.24 | 0.22 | 4.24 | 11.3 | 9.53 |
| CNS Cancer | |||||
| SF-295 | 2.33 | 0.50 | 2.93 | 3.29 | 3.14 |
| SF-539 | 4.21 | 0.16 | 4.37 | 11.7 | 7.14 |
| SNB-19 | 1.15 | 0.56 | 4.81 | 7.88 | 5.26 |
| U251 | 3.37 | 0.36 | 3.86 | 6.46 | 4.19 |
| Ovarian Cancer | |||||
| IGROV1 | 3.24 | 0.25 | <0.01 | 1.14 | 1.86 |
| OVCAR-5 | 4.68 | 0.38 | >100 | 5.50 | 8.13 |
| Breast Cancer | |||||
| MCF-7 | 2.30 | 0.39 | 2.74 | 3.05 | 3.55 |
| MDA-MB-435 | 3.31 | 0.22 | 4.52 | 5.87 | 5.76 |
a Concentration (µM) that inhibits 50% net cell growth. b ND = Not Determined.
In addition, bis-indolyl-4-trifluoromethyl-pyridines 23 (Figure 21) were synthesized and tested against P388 (leukaemia) and A549 (lung cancer) cell lines. Only compound 23a was active, showing GI50 values of 4.3 and 1.7 µM, respectively [55].
Figure 21.

Bis-indolyl-4-trifluoromethyl-pyridines 23.
2.2. Nortopsentins and Bis-Indolyl Compounds as Anti-Inflammatory Agents
Many natural products, including polysaccharides, phenols, terpenoids, quinones, and alkaloids, have proven to be potential anti-inflammatory agents, among which emerge marine alkaloids derived from different marine sources, such as sponges, bryozoans, and fungus [70,71,72,73,74,75,76]. Nortopsentin A (Figure 1), Nortopsentin B (Figure 1), and Nortopsentin C (Figure 1), together with the structurally related bis-indolyl compounds Topsentin (Figure 22), Bromotopsentin (Figure 22), Topsentin monoacetate (Figure 22), Topsentin diacetate (Figure 22), Dragmacidin (Figure 22), Hamacanthin A (Figure 22), Hamacanthin B (Figure 22) have been found to have significant anti-inflammatory properties. These compounds have been screened by standard anti-inflammatory assays. In particular, when tested by mouse ear anti-inflammatory assay using phorbol myristate acetate (PMA), Topsentin showed greater potency (ED50 = 15 µg/ear) than the known anti-inflammatories hydrocortisone, Indomethacin, and Manoalide (ED50 of 20, 250, and 100 µg/ear, respectively). In addition, bis-indolyl compounds were tested in order to calculate the percentage of PMA-induced oedema inhibition. Data revealed that Topsentin, Bromotopsentin, Dragmacidin, Nortopsentin A, and Nortopsentin C displayed significant potency (Table 26), with inhibition percentages reaching 98.1 and 70.1% in the case of Nortopsentin A and C [77].
Figure 22.

Nortopsentin structurally related to bis-indolyl compounds.
Table 26.
Percentage of PMA-induced oedema inhibition in mouse ears by bis-indolyl compounds.
| Compound | Dose | % Oedema Inhibition |
|---|---|---|
| Topsentin | 50 µg/ear | 70.6 |
| Bromotopsentin | 50 µg/ear | 75.4 |
| Topsentin monoacetate | 50 µg/ear | 45.8 |
| Topsentin diacetate | 50 µg/ear | 42.6 |
| Dragmacidin | 50 µg/ear | 64.0 |
| Nortopsentin A | 50 µg/ear | 98.1 |
| Nortopsentin B | 50 µg/ear | 38.2 |
| Nortopsentin C | 50 µg/ear | 70.1 |
| Hamacanthin A | 50 µM | 50.0 |
| Hamacanthin B | 50 µM | 34.0 |
Topsentin also proved to be capable of inactivating bee venom phospholipase A2 with IC50 lower than hydrocortisone and Indomethacin (0.5 µM, >1 mM, and >1 mM, respectively). Bis-indolyl compounds were tested at a final concentration of 1 μΜ to determine the percentage of bee venom phospholipase A2 inactivation. The results of this test are shown in Table 27; the phospholipase A2 inactivation percentage reached 67% in the case of Topsentin.
Table 27.
The percentage of bee venom phospholipase A2 inactivation by bis-indolyl compounds.
| Compound | Final Concentration | % Inactivation |
|---|---|---|
| Topsentin | 1 µM | 67 |
| Bromotopsentin | 1 µM | 33 |
| Topsentin monoacetate | 1 µM | 42 |
| Topsentin diacetate | 1 µM | 32 |
| Dragmacidin | 1 µM | 27 |
| Nortopsentin A | 1 µM | 30 |
| Nortopsentin B | 1 µM | 27 |
| Nortopsentin C | 1 µM | 26 |
| Hamacanthin A | 1.6 µM | 37 |
| Hamacanthin B | 1.6 µM | 32 |
In consideration of the data presented, the aforementioned bis-indolyl compounds showed potent anti-inflammatory effects. Their mechanism of action appears to be the consequence of phospholipase A2 inactivation. Moreover, for Topsentin, the percentage of bee venom phospholipase A2 inactivation was found equal to 80% when tested at a final concentration of approximately 2 μΜ. In the mouse ear oedema inhibition assay, a dose of about 12 µg/ear of Topsentin achieved nearly 50% oedema inhibition, and doses of 100 µg/ear showed more than 90% oedema inhibition. Similarly, oedema inhibition percentages induced by Bromotopsentin ranged from about 20% for a dose of approximately 25 µg/ear to about 75% at a dose of 50 µg/ear [77,78].
Other inflammatory assays, such as mouse ear anti-inflammatory assay using resiniferatoxin (RTX), a neurogenic inflammation-producing compound, provided the ability of Nortopsentin C (Figure 1), Hamacanthin B (Figure 22), and Topsentin (Figure 22) to inhibit RTX-induced oedema. At a concentration of 50 µg/ear, oedema inhibition percentages of 98.4%, 96.9%, and 82%, respectively, for Nortopsentin C, Hamacanthin B, and Topsentin (Table 28), with ED50 values of 8 µg/ear, 1.5 µg/ear, and 20 µg/ear (Table 29) [48]. These data are consistent with the experiments previously discussed, supporting the claim that Nortopsentin and bis-indolyl analogues have potent anti-inflammatory activity through oedema inhibition.
Table 28.
RTX-induced oedema inhibition percentage by Nortopsentin C, Hamacanthin B, and Topsentin.
| Dose µg/ear |
% Oedema Inhibition | Dose µg/ear |
% Oedema Inhibition | |
|---|---|---|---|---|
| Nortopsentin C | Hamacanthin B | Topsentin | ||
| 50 | 98.4 | 96.9 | 50 | 82 |
| 25 | 90.2 | 81.6 | 25 | 41 |
| 12.5 | 86.8 | 86.8 | 10 | 31 |
| 6.25 | 45.1 | 46.0 | 5 | 12 |
| 3.12 | 5.9 | 68.0 | ||
Table 29.
ED50 values in oedema inhibition by Nortopsentin C, Hamacanthin B, and Topsentin.
| Nortopsentin C | Hamacanthin B | Topsentin | |
|---|---|---|---|
| ED50 | 8 µg/ear | 1.5 µg/ear | 20 µg/ear |
Moreover, Nortopsentin C (Figure 1) and the related compounds Dragmacidin (Figure 22), Hamacanthin A (Figure 22), Dragmacidin D (Figure 22), and Topsentin (Figure 22) were discovered to be neural nitric oxide synthase (bNOS) inhibitors. They inhibited rat bNOS activity with IC50 values of 27 µM, 20 µM, 7.5 µM, and 4 µM, respectively, while Tosentin showed minimal inhibitory effects. The bNOS inhibition percentage induced by these compounds at a concentration range of 1.0–50 µM was also measured. The most potent inhibitory activity was shown by Hamacanthin A, Dragmacidin D, Nortopsentin C, and Dragmacidin at the concentration of 50 µM with inhibition percentages of 99.58%, 99.56%, 98.77%, and 90.27%, respectively (Table 30). Furthermore, among these compounds, Nortopsentin C and Dragmacidin were the only ones that exhibited potent calcineurin inhibition with IC50 values of 11.4 µM and 10 µM [51]. This suggests that the likely target of Nortopsentin C and Dragmacidin is calmodulin, a co-factor common to bNOS and calcineurin. These experiments prove the usefulness of bis-indolyl compounds, including Nortopsentin C, in therapeutic applications for the treatment of neurodegenerative disorders and inflammatory reactions.
Table 30.
Rat bNOS inhibition percentage by Hamacanthin A, Dragmacidin D, Nortopsentin C, Dragmacidin, and Topsentin at a concentration range of 1.0–50 µM.
| Concentration µM |
bNOS Inhibition Percentage % | ||||
|---|---|---|---|---|---|
| Hamacanthin A | Dragmacidin D | Nortopsentin C | Dragmacidin | Topsentin | |
| 50 | 99.58 | 99.56 | 98.77 | 90.27 | 22.66 |
| 10 | 75.42 | 96.44 | 16.05 | 32.74 | -0.39 |
| 5 | 43.33 | 74.22 | 9.88 | 3.54 | 5.47 |
| 1 | 35.42 | 16.89 | 2016 | 38.94 | 5.08 |
Recent research also disclosed that some GSK3β inhibitors with the Nortopsentin-like scaffold (24a, 24b, 24c, 25a, and 25b, Figure 23, Table 31), exhibit inhibitory effects on microglial inflammation and oxidative neurotoxicity. After exposure to 100 ng/mL LPS for 24 h and preincubation with the compounds 24a, 24b, 24c, 25a, and 25b, a significant suppression of the LPS-induced NO production was observed in BV-2 cells (a well-known microglial inflammatory cellular model). Among these tested compounds, 24c and 25a at 20 μM exhibited the most potent anti-inflammatory properties, by showing an approximately 50% reduction in NO release, as compared to the LPS alone group. In addition, in order to evaluate the protective effects of compounds 24a, 24b, 24c, 25a, and 25b against glutamate-induced oxidative neuronal damage, HT-22 cells were treated with these compounds for 2 h before glutamate exposure. This experiment revealed that pretreatment with 1 and 10 μM of compounds 24a, 24b, and 24c induced potent protective effects with cell viability percentages greater than 80%. Compounds 25a and 25b at 10 μM also markedly relieved the oxidative neuronal damage in HT-22 cells, while no noticeable protection was noticed at a lower concentration (1 μM) [79].
Figure 23.

Aminopyrazole derivatives 24 and 25 with the Nortopsentin-like scaffold.
Table 31.
In vitro GSK3β inhibitory activities of aminopyrazole derivatives 24 and 25.
| Compound | R | R1 | IC50 (µM) a |
|---|---|---|---|
| 24a | 5-F | CH3 | 1.28 ± 0.18 |
| 24b | 5-Br | CH3 | 1.93 ± 0.22 |
| 24c | 5-OCH3 | CH3 | 1.76 ± 0.19 |
| 25a |
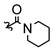
|
— | 1.46 ± 0.04 |
| 25b | H | — | 1.48 ± 0.30 |
a The data represent mean values ± SEM of at least three independent experiments.
The most promising compound, 24c, was selected for further in vivo study. Microglial activation and astrocyte proliferation in the brain of LPS-injected mice were measured by IBA-1 and GFAP immunofluorescence staining. Results indicated that compound 24c markedly reduced microglial activation and astrocyte proliferation, showing a potent anti-inflammatory effect. This new potent GSK3β inhibitor could be the starting point for the discovery of therapeutic agents to treat Alzheimer’s disease and other inflammation-associated neurological syndromes [79].
3. Conclusions
Synthesizing marine compound analogues appears to be a promising strategy to obtain new anticancer derivatives that act by intervening in specific cell processes. On the one hand, most studies on Nortopsentin derivatives have focused on the antiproliferative activity evaluation and action mechanisms commonly involved in cancer. Nortopsentin derivatives can induce cell cycle arrest and apoptosis in cancer cell lines and simultaneously impair cell viability. The driving element of their antiproliferative activity is mostly overexpressed target inhibition, such as the kinases CDK1 and GSK3β or enzymes like GLS-1. The most active compounds, showing GI50 values in the micromolar-submicromolar range in different human tumour cell lines, belong to the thiazole class, in which the structural manipulation of Nortopsentin was extended to one of the two indolyl portions that were replaced by a 7-azaindole ring. The five most active compounds, which were further investigated in two additional cell lines, STO and MesoII, derived from human diffuse malignant peritoneal mesothelioma (DMPM), exhibited IC50 values ranging from 0.33 to 0.61 μM in STO cells and from 4.11 to 25.12 μM in MesoII. Moreover, their anti-tumour activity was evaluated on STO cells xenotransplanted in athymic nude mice. The treatment with the different compounds resulted in marked tumour growth inhibition with two complete responses (disappearance of tumour) in each treatment group without any appreciable sign of toxicity. In vitro kinase assays revealed CDK1 inhibition exerted by the compounds with IC50 values lower than 1 μM. On the other hand, Nortopsentin and structurally related compounds also exhibit anti-inflammatory properties, such as anti-oedema and neuroprotection. In vivo anti-inflammatory assays on animal models disclose that Nortopsentins can powerfully inhibit oedema; in particular, Nortopsentin A, and Nortopsentin C, when tested by mouse ear anti-inflammatory assay using phorbol myristate acetate (PMA), displayed significant potency with inhibition percentages reaching 98.1 and 70.1%, respectively. The key factor behind this effect is phospholipase A2 inactivation. In addition, Nortopsentin C mightily inhibits rat neural nitric oxide synthase (bNOS) and calcineurin with IC50 values of 27 and 11.4 µM, respectively. Some Nortopsentin analogue GSK3β inhibitors, bearing an aminopyrazole central ring instead of the imidazole of the lead, also suppress LPS-induced NO production in mice, providing powerful anti-inflammatory effects. Overall, Nortopsentins emerge as new lead compounds for the development of novel anti-inflammatory agents, indicating the need to synthesise new Nortopsentin derivatives and investigate their antiproliferative activity as well as their anti-inflammatory potential.
Abbreviations
| CNS | Central Nervous System |
| Ara-C | Cytarabine |
| FDA | Food and Drug Administration |
| NCI | National Cancer Institute |
| DMPM | Diffuse Malignant Peritoneal Mesothelioma |
| TVI | Tumour Volume Inhibition |
| CDK | Cyclin-Dependent Kinase |
| GSK3β | Glycogen synthase kinase 3β |
| CR-CSphCs | Colorectal Cancer Sphere Cells |
| MG_MID | Mean Graph Midpoint |
| bNOS | Neural nitric oxide synthase |
| LPS | Lipopolysaccharide |
Author Contributions
Conceptualization, P.D. and E.G.; writing—original draft preparation, C.P., F.T. and G.P.; writing—review and editing, B.P., S.C. and D.C. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was partially supported by the following grants: PRIN2017, Prot. No. 2017E84AA4 (to P.D.) and AIRC-IG grant, No. 24444 (to E.G.).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics. CA Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Vermeulen K., Van Bockstaele D.R., Berneman Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuddihy A.R., O’Connell M.J. Cell-cycle responses to DNA damage in G2. Int. Rev. Cytol. 2003;222:99–140. doi: 10.1016/s0074-7696(02)22013-6. [DOI] [PubMed] [Google Scholar]
- 4.McGowan C.H. Running into problems: How cells cope with replicating damaged DNA. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2003;532:75–84. doi: 10.1016/j.mrfmmm.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Dash C., Lu J., Parikh V., Wathen S., Shah S., Chaudhari R.S., Adams-Campbell L. Disparities in colorectal cancer screening among breast and prostate cancer survivors. Cancer Med. 2021;10:1448–1456. doi: 10.1002/cam4.3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koutros S., Decker K.L., Baris D., Pardo L.A., Johnson A., Hosain G.M., Rothman N., Karagas M.R., Schwenn M.R., Silverman D.T. Bladder cancer risk associated with family history of cancer. Int. J. Cancer. 2021;148:2915–2923. doi: 10.1002/ijc.33486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sashidhara K.V., Kumar A., Kumar M., Srivastava A., Puri A. Synthesis and antihyperlipidemic activity of novel coumarin bisindole derivatives. Bioorg. Med. Chem. Lett. 2010;20:6504–6507. doi: 10.1016/j.bmcl.2010.09.055. [DOI] [PubMed] [Google Scholar]
- 8.Shahrisa A., Ghasemi Z., Saraei M. Synthesis of 2,6-bis(1H-indole-6-yl)-4H-pyran-4-onesviaLeimgruber-Batcho indole synthesis. J. Heterocycl. Chem. 2009;46:273–277. doi: 10.1002/jhet.80. [DOI] [Google Scholar]
- 9.Mauger A., Jarret M., Kouklovsky C., Poupon E., Evanno L., Vincent G. The chemistry of mavacurane alkaloids: A rich source of bis-indole alkaloids. Nat. Prod. Rep. 2021;38:1852–1886. doi: 10.1039/D0NP00088D. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi J., Murayama T., Ishibashi M., Kosuge S., Takamatsu M., Ohizumi Y., Kobayashi H., Ohta T., Nozoe S., Takuma S. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron. 1990;46:7699–7702. doi: 10.1016/S0040-4020(01)90065-1. [DOI] [Google Scholar]
- 11.König G.M., Wright A.D., Sticher O., Angerhofer C.K., Pezzuto J.M. Biological Activities of Selected Marine Natural Products. Planta Medica. 1994;60:532–537. doi: 10.1055/s-2006-959565. [DOI] [PubMed] [Google Scholar]
- 12.Firn R.D., Jones C.G. Natural products? a simple model to explain chemical diversity. Nat. Prod. Rep. 2003;20:382–391. doi: 10.1039/b208815k. [DOI] [PubMed] [Google Scholar]
- 13.Simmons T.L., Andrianasolo E., McPhail K., Flatt P., Gerwick W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005;4:333–342. doi: 10.1158/1535-7163.333.4.2. [DOI] [PubMed] [Google Scholar]
- 14.Zanchett G., Oliveira-Filho E.C. Cyanobacteria and cyanotoxins: From impacts on aquatic ecosystems and human health to anticarcinogenic effects. Toxins. 2013;5:1896–1917. doi: 10.3390/toxins5101896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reen F.J., Gutiérrez-Barranquero J.A., Dobson A.D.W., Adams C., O’Gara F. Emerging concepts promising new horizons for marine biodiscovery and synthetic biology. Mar. Drugs. 2015;13:2924–2954. doi: 10.3390/md13052924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou S., Huang G., Chen G. Synthesis and anti-tumor activity of marine alkaloids. Bioorg. Med. Chem. Lett. 2021;41:128009. doi: 10.1016/j.bmcl.2021.128009. [DOI] [PubMed] [Google Scholar]
- 17.Adrian T. Novel Marine-Derived Anti-Cancer Agents. Curr. Pharm. Des. 2007;13:3417–3426. doi: 10.2174/138161207782360500. [DOI] [PubMed] [Google Scholar]
- 18.Anjum K., Abbas S.Q., Shah S.A.A., Akhter N., Batool S., Hassan S.S.U. Marine sponges as a drug treasure. Biomol. Ther. 2016;24:347–362. doi: 10.4062/biomolther.2016.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pallela R., Ehrlich H., editors. Marine Sponges: Chemicobiological and Biomedical Applications. Springer; New Delhi, India: 2016. [DOI] [Google Scholar]
- 20.Kamel M.M., Abdel-Hameid M.K., El-Nassan H.B., El-Khouly E.A. Recent Advances in the Synthesis and Biological Applications of Nortopsentin Analogs. Chem. Heterocycl. Compd. 2020;56:499–502. doi: 10.1007/s10593-020-02687-4. [DOI] [Google Scholar]
- 21.Capalbo A., Lauritano C. Multiple Myeloma: Possible Cure from the Sea. Cancers. 2022;14:2965. doi: 10.3390/cancers14122965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuznetsov G., TenDyke K., Towle M.J., Cheng H., Liu J., Marsh J.P., Schiller S.E., Spyvee M.R., Yang H., Seletsky B.M., et al. Tubulin-based antimitotic mechanism of E7974, a novel analogue of the marine sponge natural product hemiasterlin. Mol. Cancer Ther. 2009;8:2852–2860. doi: 10.1158/1535-7163.MCT-09-0301. [DOI] [PubMed] [Google Scholar]
- 23.Srinivasan N., Dhanalakshmi S., Pandian P. Encouraging leads from marine sources for cancer therapy—A review approach. Pharmacogn. J. 2020;12:1475–1481. doi: 10.5530/pj.2020.12.202. [DOI] [Google Scholar]
- 24.Nakamoto S., Watanabe J., Ohtani S., Morita S., Ikeda M. Eribulin improved the overall survival from the initiation of first-line chemotherapy for HER2-negative advanced breast cancer: A multicenter retrospective study. BMC Cancer. 2022;22:31. doi: 10.1186/s12885-021-09137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi J. Search for New Bioactive Marine Natural Products and Application to Drug Development. Chem. Pharm. Bull. 2016;64:1079–1083. doi: 10.1248/cpb.c16-00281. [DOI] [PubMed] [Google Scholar]
- 26.Shin J., Seo Y., Cho K.W., Rho J.-R., Sim C.J. New bis(indole) alkaloids of the topsentin class from the sponge Spongosorites genitrix. J. Nat. Prod. 1999;62:647–649. doi: 10.1021/np980507b. [DOI] [PubMed] [Google Scholar]
- 27.Gul W., Hamann M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005;78:442–453. doi: 10.1016/j.lfs.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berlinck R.G.S., Britton R., Piers E., Lim L., Roberge M., da Rocha R.M., Andersen R.J. Granulatimide and isogranulatimide, aromatic alkaloids with g2 checkpoint inhibition activity isolated from the brazilian ascidian Didemnum granulatum: Structure elucidation and synthesis. J. Org. Chem. 1998;63:9850–9856. doi: 10.1021/jo981607p. [DOI] [Google Scholar]
- 29.Bitencourt M.A.O., Dantas G.R., Lira D.P., Barbosa-Filho J.M., de Miranda G.E.C., Santos B.V.d.O., Souto J.T. Aqueous and methanolic extracts of caulerpa mexicana suppress cell migration and ear edema induced by inflammatory agents. Mar. Drugs. 2011;9:1332–1345. doi: 10.3390/md9081332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou S., Huang G. Synthesis, and antimalarial and antibacterial activities of marine alkaloids. Chem. Biol. Drug Des. 2021;98:226–233. doi: 10.1111/cbdd.13892. [DOI] [PubMed] [Google Scholar]
- 31.Izumida M., Kotani O., Hayashi H., Smith C., Fukuda T., Suga K., Iwao M., Ishibashi F., Sato H., Kubo Y. Unique Mode of Antiviral Action of a Marine Alkaloid against Ebola Virus and SARS-CoV-2. Viruses. 2022;14:816. doi: 10.3390/v14040816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burattini S., Battistelli M., Verboni M., Falcieri E., Faenza I., Lucarini S., Salucci S. Morpho-functional analyses reveal that changes in the chemical structure of a marine bisindole alkaloid alter the cytotoxic effect of its derivatives. Microsc. Res. Tech. 2022;85:2381–2389. doi: 10.1002/jemt.24092. [DOI] [PubMed] [Google Scholar]
- 33.García-García P., Reyes R., Évora C., Delgado A., Fernández J.J., Daranas A.H. Osteoprotective effect of the marine alkaloid norzoanthamine on an osteoporosis model in ovariectomized rat. BioMedicine Pharmacother. 2022;147:112631. doi: 10.1016/j.biopha.2022.112631. [DOI] [PubMed] [Google Scholar]
- 34.Nuzzo G., Gallo C., Crocetta F., Romano L., Barra G., Senese G., Dell’isola M., Carbone D., Tanduo V., Albiani F., et al. Identification of the Marine Alkaloid Lepadin A as Potential Inducer of Immunogenic Cell Death. Biomolecules. 2022;12:246. doi: 10.3390/biom12020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhidkov M.E., Kaune M., Kantemirov A.V., Smirnova P.A., Spirin P.V., Sidorova M.A., Stadnik S.A., Shyrokova E.Y., Kaluzhny D.N., Tryapkin O.A., et al. Study of Structure–Activity Relationships of the Marine Alkaloid Fascaplysin and Its Derivatives as Potent Anticancer Agents. Mar. Drugs. 2022;20:185. doi: 10.3390/md20030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Torres V., Encinar J.A., Herranz-López M., Pérez-Sánchez A., Galiano V., Barrajón-Catalán E., Micol V. An Updated Review on Marine Anticancer Compounds: The Use of Virtual Screening for the Discovery of Small-Molecule Cancer Drugs. Molecules. 2017;22:1037. doi: 10.3390/molecules22071037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonvicini F., Locatelli A., Morigi R., Leoni A., Gentilomi G.A. Isatin Bis-Indole and Bis-Imidazothiazole Hybrids: Synthesis and Antimicrobial Activity. Molecules. 2022;27:5781. doi: 10.3390/molecules27185781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carbone A., Cascioferro S., Parrino B., Carbone D., Pecoraro C., Schillaci D., Cusimano M.G., Cirrincione G., Diana P. Thiazole Analogues of the Marine Alkaloid Nortopsentin as Inhibitors of Bacterial Biofilm Formation. Molecules. 2021;26:81. doi: 10.3390/molecules26010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji X., Guo J., Liu Y., Lu A., Wang Z., Li Y., Yang S., Wang Q. Marine-Natural-Product Development: First Discovery of Nortopsentin Alkaloids as Novel Antiviral, Anti-phytopathogenic-Fungus, and Insecticidal Agents. J. Agric. Food Chem. 2018;66:4062–4072. doi: 10.1021/acs.jafc.8b00507. [DOI] [PubMed] [Google Scholar]
- 40.Wang T., Li L., Zhou Y., Lu A., Li H., Chen J., Duan Z., Wang Q. Structural Simplification of Marine Natural Products: Discovery of Hamacanthin Derivatives Containing Indole and Piperazinone as Novel Antiviral and Anti-phytopathogenic-fungus Agents. J. Agric. Food Chem. 2021;69:10093–10103. doi: 10.1021/acs.jafc.1c04098. [DOI] [PubMed] [Google Scholar]
- 41.Carbone D., De Franco M., Pecoraro C., Bassani D., Pavan M., Cascioferro S., Parrino B., Cirrincione G., Dall’acqua S., Sut S., et al. Structural Manipulations of Marine Natural Products Inspire a New Library of 3-Amino-1,2,4-Triazine PDK Inhibitors Endowed with Antitumor Activity in Pancreatic Ductal Adenocarcinoma. Mar. Drugs. 2023;21:288. doi: 10.3390/md21050288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pecoraro C., De Franco M., Carbone D., Bassani D., Pavan M., Cascioferro S., Parrino B., Cirrincione G., Dall’acqua S., Moro S., et al. 1,2,4-Amino-triazine derivatives as pyruvate dehydrogenase kinase inhibitors: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2023;249:115134. doi: 10.1016/j.ejmech.2023.115134. [DOI] [PubMed] [Google Scholar]
- 43.Carbone D., De Franco M., Pecoraro C., Bassani D., Pavan M., Cascioferro S., Parrino B., Cirrincione G., Dall’acqua S., Moro S., et al. Discovery of the 3-Amino-1,2,4-triazine-Based Library as Selective PDK1 Inhibitors with Therapeutic Potential in Highly Aggressive Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023;24:3679. doi: 10.3390/ijms24043679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halawa A.H., El-Gilil S.M.A., Bedair A.H., Eliwa E.M., Frese M., Sewald N., Shaaban M., El-Agrody A.M. Synthesis of diverse amide linked bis-indoles and indole derivatives bearing coumarin-based moiety: Cytotoxicity and molecular docking investigations. Med. Chem. Res. 2018;27:796–806. doi: 10.1007/s00044-017-2103-7. [DOI] [Google Scholar]
- 45.Pecoraro C., Parrino B., Cascioferro S., Puerta A., Avan A., Peters G.J., Diana P., Giovannetti E., Carbone D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules. 2022;27:19. doi: 10.3390/molecules27010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song Y., Yang J., Yu J., Li J., Yuan J., Wong N.-K., Ju J. Chlorinated bis-indole alkaloids from deep-sea derived Streptomyces sp. SCSIO 11791 with antibacterial and cytotoxic activities. J. Antibiot. 2020;73:542–547. doi: 10.1038/s41429-020-0307-4. [DOI] [PubMed] [Google Scholar]
- 47.Khan S., Rehman W., Rahim F., Hussain R., Obaidullah A.J., Alotaibi H.F., Alanazi M.M., Khan M.U., Khan Y. Bis-indole based triazine derivatives: Synthesis, characterization, in vitro β-glucuronidase anti-cancer and anti-bacterial evaluation along with in silico molecular docking and ADME analysis. Arab. J. Chem. 2023;16:104970. doi: 10.1016/j.arabjc.2023.104970. [DOI] [Google Scholar]
- 48.Cascioferro S., Petri G.L., Parrino B., El Hassouni B., Carbone D., Arizza V., Perricone U., Padova A., Funel N., Peters G.J., et al. 3-(6-Phenylimidazo [2,1-b][1,3,4]thiadiazol-2-yl)-1H-Indole Derivatives as New Anticancer Agents in the Treatment of Pancreatic Ductal Adenocarcinoma. Molecules. 2020;25:329. doi: 10.3390/molecules25020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petri G.L., Cascioferro S., EL Hassouni B., Carbone D., Parrino B., Cirrincione G., Peters G.J., Diana P., Giovannetti E. Biological Evaluation of the Antiproliferative and Anti-migratory Activity of a Series of 3-(6-Phenylimidazo[2,1-b][1,3,4]thiadiazol-2-yl)-1H-indole Derivatives Against Pancreatic Cancer Cells. Anticancer Res. 2019;39:3615–3620. doi: 10.21873/anticanres.13509. [DOI] [PubMed] [Google Scholar]
- 50.Jacobs R.S., Pomponi S., Gunasekera S., Wright A. Anti-Neurogenic Inflammatory Compounds and Compositions and Methods of Use Thereof. 5955462A. U.S. Patent. 1999 September 21;
- 51.Longley R.E., Isbrucker R.A., Wright A.E. Use of imidazole and indole compounds as inhibitors of nitric oxide synthase. 6087363A. U.S. Patent. 2000 July 11;
- 52.Sakemi S., Sun H.H. Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991;56:4304–4307. doi: 10.1021/jo00013a044. [DOI] [Google Scholar]
- 53.Keel K.L., Tepe J.J. Total Synthesis of Nortopsentin D via a Late-Stage Pinacol-like Rearrangement. Org. Lett. 2021;23:5368–5372. doi: 10.1021/acs.orglett.1c01681. [DOI] [PubMed] [Google Scholar]
- 54.Sun H.H., Sakemi S., Gunasekera S., Kashman Y., Lui M., Burres N., McCarthy P. Bis-Indole Imidazole Compounds Which Are Useful Antitumor and Antimicrobial Agents. 4970226A. U.S. Patent. 1990 November 13;
- 55.Yang C.-G., Huang H., Jiang B. Progress in Studies of Novel Marine Bis(indole) Alkaloids. Curr. Org. Chem. 2004;8:1691–1720. doi: 10.2174/1385272043369656. [DOI] [Google Scholar]
- 56.Mancini I., Guella G., Pietra F., Waikedre J., Debitus C. From Inactive Nortopsentin D, a Novel Bis(indole) Alkaloid Isolated from the Axinellid SpongeDragmacidon sp. from Deep Waters South of New Caledonia, to a Strongly Cytotoxic Derivative. Helv. Chim. Acta. 1996;79:2075–2082. doi: 10.1002/hlca.19960790804. [DOI] [Google Scholar]
- 57.Jiang B., Gu X.-H. Syntheses and cytotoxicity evaluation of bis(indolyl)thiazole, bis(indolyl)pyrazinone and bis(indolyl)pyrazine: Analogues of cytotoxic marine bis(indole) alkaloid. Bioorg. Med. Chem. 2000;8:363–371. doi: 10.1016/S0968-0896(99)00290-4. [DOI] [PubMed] [Google Scholar]
- 58.Dembitsky V., Gloriozova T., Poroikov V. Novel Antitumor Agents: Marine Sponge Alkaloids, their Synthetic Analogs and Derivatives. Mini-Rev. Med. Chem. 2005;5:319–336. doi: 10.2174/1389557053175362. [DOI] [PubMed] [Google Scholar]
- 59.Carbone D., Vestuto V., Ferraro M.R., Ciaglia T., Pecoraro C., Sommella E., Cascioferro S., Salviati E., Novi S., Tecce M.F., et al. Metabolomics-assisted discovery of a new anticancer GLS-1 inhibitor chemotype from a nortopsentin-inspired library: From phenotype screening to target identification. Eur. J. Med. Chem. 2022;234:114233. doi: 10.1016/j.ejmech.2022.114233. [DOI] [PubMed] [Google Scholar]
- 60.Pecoraro C., Carbone D., Aiello D., Carbone A. Synthesis and cytotoxic activity of 3-[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-c]pyridine hydrobromides, analogues of the marine alkaloid nortopsentin. Arkivoc. 2022;2022:30–42. doi: 10.24820/ark.5550190.p011.640. [DOI] [Google Scholar]
- 61.Di Franco S., Parrino B., Gaggianesi M., Pantina V.D., Bianca P., Nicotra A., Mangiapane L.R., Iacono M.L., Ganduscio G., Veschi V., et al. CHK1 inhibitor sensitizes resistant colorectal cancer stem cells to nortopsentin. iScience. 2021;24:102664. doi: 10.1016/j.isci.2021.102664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mérour J.-Y., Buron F., Plé K., Bonnet P., Routier S. The Azaindole Framework in the Design of Kinase Inhibitors. Molecules. 2014;19:19935–19979. doi: 10.3390/molecules191219935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar D., Jain S.K. A Comprehensive Review of N-Heterocycles as Cytotoxic Agents. Curr. Med. Chem. 2016;23:4338–4394. doi: 10.2174/0929867323666160809093930. [DOI] [PubMed] [Google Scholar]
- 64.Kumar D., Kumar N.M., Chang K.-H., Gupta R., Shah K. Synthesis and in-vitro anticancer activity of 3,5-bis(indolyl)-1,2,4-thiadiazoles. Bioorg. Med. Chem. Lett. 2011;21:5897–5900. doi: 10.1016/j.bmcl.2011.07.089. [DOI] [PubMed] [Google Scholar]
- 65.Carbone D., Pecoraro C., Panzeca G., Xu G., Roeten M.S.F., Cascioferro S., Giovannetti E., Diana P., Parrino B. 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Nortopsentin Derivatives against Pancreatic Ductal Adenocarcinoma: Synthesis, Cytotoxic Activity, and Inhibition of CDK1. Mar. Drugs. 2023;21:412. doi: 10.3390/md21070412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar D., Arun V., Kumar N.M., Acosta G., Noel B., Shah K. A Facile Synthesis of Novel Bis-(indolyl)-1,3,4-oxadiazoles as Potent Cytotoxic Agents. ChemMedChem. 2012;7:1915–1920. doi: 10.1002/cmdc.201200363. [DOI] [PubMed] [Google Scholar]
- 67.Sreenivasulu R., Tej M.B., Jadav S.S., Sujitha P., Kumar C.G., Raju R.R. Synthesis, anticancer evaluation and molecular docking studies of 2,5-bis(indolyl)-1,3,4-oxadiazoles, Nortopsentin analogues. J. Mol. Struct. 2020;1208:127875. doi: 10.1016/j.molstruc.2020.127875. [DOI] [Google Scholar]
- 68.Cascioferro S., Attanzio A., Di Sarno V., Musella S., Tesoriere L., Cirrincione G., Diana P., Parrino B. New 1,2,4-Oxadiazole Nortopsentin Derivatives with Cytotoxic Activity. Mar. Drugs. 2019;17:35. doi: 10.3390/md17010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang B., Yang C.-G., Xiong W.-N., Wang J. Synthesis and cytotoxicity evaluation of novel indolylpyrimidines and indolylpyrazines as potential antitumor agents. Bioorg. Med. Chem. 2001;9:1149–1154. doi: 10.1016/S0968-0896(00)00337-0. [DOI] [PubMed] [Google Scholar]
- 70.Fernandes P.D., Zardo R.S., Figueiredo G.S., Silva B.V., Pinto A.C. Anti-inflammatory properties of convolutamydine A and two structural analogues. Life Sci. 2014;116:16–24. doi: 10.1016/j.lfs.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 71.Di X., Rouger C., Hardardottir I., Freysdottir J., Molinski T.F., Tasdemir D., Omarsdottir S. 6-Bromoindole Derivatives from the Icelandic Marine Sponge Geodia barretti: Isolation and Anti-Inflammatory Activity. Mar. Drugs. 2018;16:437. doi: 10.3390/md16110437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan H., Qiu H., Zhang K., Zhang P., Liang W., Yang M., Mou C., Lin M., He M., Xiao X., et al. Fascaplysin Derivatives Are Potent Multitarget Agents against Alzheimer’s Disease: In Vitro and in Vivo Evidence. ACS Chem. Neurosci. 2019;10:4741–4756. doi: 10.1021/acschemneuro.9b00503. [DOI] [PubMed] [Google Scholar]
- 73.Ahmad B., Shah M., Choi S. Oceans as a Source of Immunotherapy. Mar. Drugs. 2019;17:282. doi: 10.3390/md17050282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patra S., Praharaj P.P., Panigrahi D.P., Panda B., Bhol C.S., Mahapatra K.K., Mishra S.R., Behera B.P., Jena M., Sethi G., et al. Bioactive compounds from marine invertebrates as potent anticancer drugs: The possible pharmacophores modulating cell death pathways. Mol. Biol. Rep. 2020;47:7209–7228. doi: 10.1007/s11033-020-05709-8. [DOI] [PubMed] [Google Scholar]
- 75.Florean C., Dicato M., Diederich M. Immune-modulating and anti-inflammatory marine compounds against cancer. Semin. Cancer Biol. 2022;80:58–72. doi: 10.1016/j.semcancer.2020.02.008. [DOI] [PubMed] [Google Scholar]
- 76.Mitra S., Anand U., Sanyal R., Jha N.K., Behl T., Mundhra A., Ghosh A., Radha, Kumar M., Proćków J., et al. Neoechinulins: Molecular, cellular, and functional attributes as promising therapeutics against cancer and other human diseases. BioMedicine. 2022;145:112378. doi: 10.1016/j.biopha.2021.112378. [DOI] [PubMed] [Google Scholar]
- 77.McConnell O.J., Saucy G., Jacobs R., Gunasekera S.P. Use for Bis-Heterocyclic Compounds and Pharmaceutical Compositions Containing Same. 5290777A. U.S. Patent. 1995 November 7;
- 78.Keyzers R.A., Davies-Coleman M.T. Anti-inflammatory metabolites from marine sponges. Chem. Soc. Rev. 2005;34:355–365. doi: 10.1039/b408600g. [DOI] [PubMed] [Google Scholar]
- 79.Liu J.-G., Zhao D., Gong Q., Bao F., Chen W.-W., Zhang H., Xu M.-H. Development of Bisindole-Substituted Aminopyrazoles as Novel GSK-3β Inhibitors with Suppressive Effects against Microglial Inflammation and Oxidative Neurotoxicity. ACS Chem. Neurosci. 2020;11:3398–3408. doi: 10.1021/acschemneuro.0c00520. [DOI] [PubMed] [Google Scholar]