

Abstract
Background:
Homozygous familial hypercholesterolaemia (HoFH) is a rare inherited disorder resulting in extremely elevated low-density lipoprotein cholesterol (LDL-C) levels and premature atherosclerotic cardiovascular disease (ASCVD). Current guidance about its management and prognosis stems from relatively small studies, mostly from western countries. The objective of this study was to assess the clinical and genetic characteristics as well as the impact of current practice on health outcomes of HoFH patients globally.
Methods:
The HoFH International Clinical Collaborators (HICC) registry collected data on patients with a clinical and/or genetic diagnosis of HoFH using a retrospective cohort study design.
Findings:
Overall, 751 patients (52% female) from 38 countries were included, with 75% reporting bi-allelic pathogenic variants. Median age of diagnosis was 12∙0 (IQR 5∙5–27∙0) years, with major manifestations of ASCVD or aortic stenosis already present in 9% at diagnosis of HoFH. Globally, pre-treatment LDL-C levels were 14∙7 (IQR 11∙6–18∙4) mmol/L, with 92% of patients subsequently receiving statins, 64% ezetimibe and 39% lipoprotein apheresis. On-treatment LDL-C levels were lower in high-income versus non-high-income countries (3∙93 [IQR 2∙6–5∙8] versus 9∙3 [IQR 6∙7–12∙7] mmol/L), with greater use of three or more lipid-lowering therapies (LLT) (66% versus 24%) and consequently more patients attaining guideline-recommended LDL-C goals (21% versus 3% respectively). A first major adverse cardiovascular event occurred a decade earlier in non-high-income countries, at a median age of 24∙5 (IQR 17∙0–34∙5) versus 37∙0 (IQR 29∙0–49∙0) years in high-income countries (adjusted hazard ratio: 1∙64 [95%CI 1∙13–2∙38]).
Interpretation:
Worldwide, patients with HoFH are diagnosed too late, undertreated and at high premature ASCVD risk. Greater use of multi-LLT regimens associates with lower LDL-C levels and better outcomes. Significant global disparities exist in treatment regimens, control of LDL-C levels and cardiovascular event-free survival, which demands a critical re-evaluation of global health policy to reduce inequalities and improve outcomes for all patients with HoFH.
Study registration:
Funding:
Detailed at end of paper (see Acknowledgements).
Keywords: Homozygous familial hypercholesterolaemia, low-density lipoprotein cholesterol, lipid-lowering therapy, atherosclerotic cardiovascular disease, aortic valve stenosis, lipoprotein apheresis
Introduction
Familial hypercholesterolaemia (FH) is an inherited disorder resulting from pathogenic variants in genes involved in the metabolism of low-density lipoproteins, leading to markedly elevated LDL-C levels and an increased risk of premature atherosclerotic cardiovascular disease (ASCVD) if not treated early and effectively.1 The most severe form of FH is homozygous FH (HoFH) which broadly comprises simple homozygous as well as compound and double heterozygous cases (see box: “Definition and Diagnosis”).
Definition and Diagnosis.
Patients with HoFH have extremely high plasma LDL-C levels that causes accelerated atherosclerotic cardiovascular disease (ASCVD). Manifestations of ASCVD most notably include fatal and non-fatal myocardial infarction as well as occlusive vascular disease requiring surgical or percutaneous revascularisation. Similarly, deposition of cholesterol in and around the aortic valve can cause severe (supra-)valvular aortic stenosis. Deposits of cholesterol in the skin and/or tendons, called xanthomas, are the hallmark of the disease. The development and severity of ASCVD and/or aortic stenosis determine prognosis in HoFH.
HoFH can be diagnosed clinically or genetically.
Clinical diagnosis:
-
Untreated LDL-C levels >13 mmol/L (500 mg/dL), or LDL-C ≥8 mmol/L (300 mg/dL) while on conventional LLT
AND
Presence of xanthomas before the age of ten years, or the presence of heterozygous FH in both parents1
Genetic diagnosis:
Identification of bi-allelic pathogenic variants at the LDLR, APOB, PCSK9 or LDLRAP1 gene locus
Patients with identical variants in both alleles of the same gene are simple homozygous. Patients with non-identical variants in both alleles of the same gene are compound heterozygous and patients with variants in two different FH-genes are termed double heterozygous. Autosomal recessive hypercholesterolaemia is a very rare form of HoFH caused by bi-allelic variants in LDLRAP1.9
Importantly, the phenotype of HoFH varies considerably and genetic testing has identified many patients with less severe phenotypes.2,10,11 Conversely, the absence of two pathogenic variants in the presence of a phenotype consistent with HoFH does not exclude the diagnosis.
The prevalence of HoFH was historically reported as 1 per million but has recently been estimated as 1 in ~300,000 persons worldwide,2–5 with a higher prevalence in populations with a founder effect.1 Plasma LDL-C levels may exceed 20 mmol/L depending on the variants carried; patients with an LDLR variant that leads to no residual functional protein (LDLR negative variant) in both alleles are generally the most severely affected. The magnitude and duration of exposure to extreme LDL-C levels largely determines prognosis.6 Combination of commonly used lipid-lowering therapies (LLT), such as statins and ezetimibe, are often insufficient to control such high LDL-C levels, with many patients requiring extracorporeal removal of LDL by means of lipoprotein apheresis. Therapies that decrease LDL-C levels irrespective of residual LDLR function have recently emerged,7,8 but their use is limited by cost and availability.
Our current view on the clinical characteristics and natural history of HoFH is largely based on studies of relatively small sample size comprising patients from high-income countries. Little is known about global differences in detection, management and cardiovascular outcomes in HoFH. To address these uncertainties, we created a global consortium of researchers and clinicians caring for HoFH patients. The objective of this study was to provide a contemporary, systematic assessment of the characteristics, diagnosis, treatment and outcomes of HoFH patients, both on a global scale and by country income status.
Methods
Participating centres and patient selection
The HoFH International Clinical Collaborators (HICC, NCT04815005) is a global consortium of clinicians and researchers involved in the care for HoFH patients. Patients were eligible for inclusion into the registry if they had received a clinical or genetic diagnosis of HoFH by the treating clinician.1 Where genetic testing was reported, patients were considered HoFH if they were found to be simple HoFH, compound heterozygous or double heterozygous, consistent with current guidelines.1
Data collection
The present study has a retrospective cohort design. To reflect contemporary data, only patients with HoFH who were alive and being followed up in, or after, 2010 were eligible for inclusion. Baseline was defined as the point at which HoFH was diagnosed, and follow-up was defined as years post diagnosis. The method of data entry, variables collected and definitions of lipid targets, cardiovascular outcomes and aortic valve stenosis are described in the Supplementary Methods. For comparison between affluent and less affluent regions of the world, countries were grouped according to the 2019 World Bank definition of income category (Table S1).12
Genetic data
Genetic information was curated to a uniform nomenclature and independently validated by four clinical and molecular genetics experts (JCD, LZ, LT and TF) who confirmed the pathogenicity and assessed the functionality of the variants as detailed in the Supplementary Materials.
Statistical analysis
Statistical analyses were performed using R software, version 4.0.3 (R Foundation for Statistical Computing, Vienna, Austria). The primary outcome in the survival analyses was major adverse cardiovascular events (MACE), defined as a composite of cardiovascular death, non-fatal myocardial infarction (MI), percutaneous coronary intervention (PCI) and coronary artery bypass grafting (CABG). Descriptive estimates are presented as median and interquartile range (IQR) or mean (95% CI). We used bootstrapping (10,000 randomized samples) to estimate the 95% CIs around mean estimates using the percentile method. Due to the descriptive nature of the study, we did not impute missing data and performed available case analyses without formal hypothesis testing. Comparisons of survival times free from events between groups of interest were assessed using the Kaplan-Meier method and log-rank tests. Details on the generation of proportional hazard models are provided in the Supplementary Materials.
Ethics
Individual contributors were responsible for meeting local standards set by their institutional review board or ethics committee and obtaining approval. The study was conducted according to International Standards of Good Clinical Practice.
Role of the funding source
The HICC is an investigator-initiated project supported by funding from the academic institutions of the collaborators. The European Atherosclerosis Society provided funding to support a registry coordinator. The funders had no role in study design, data collection, data analysis, data interpretation, writing the manuscript or decision to submit for publication. The writing committee takes final responsibility for the content of the manuscript and the decision to submit for publication.
Results
Patient characteristics
Individual-level data on 751 patients from 88 institutions across 38 countries representing all seven World Bank regions were available. Twenty countries were classified as high-income, 12 as upper-middle income and six as lower-middle income countries; countries and number of patients per country are listed in Table S1. Patient demographic, clinical and genetic characteristic at the time of inclusion are presented in Table 1, overall and stratified by country income status. Median age of diagnosis was 12∙0 (IQR 5∙5–27∙0) years, and 52% of patients were women. Race was reported in 527 patients; of these, 338 (64%) were White, 121 (23%) Asian, and 68 (13%) were Black or of mixed race. Patients from high-income countries, compared with those from non-high-income countries, were older at the time of diagnosis (16∙0 [IQR 6∙0–33∙0] versus 10∙0 [5∙0–20∙0] years) and had fewer physical stigmata such as xanthomas (64% versus 74%) at the time of diagnosis.
Table 1 –
Demographic, clinical and genetic characteristics and plasma lipid levels in HoFH patients, overall and stratified by country income status
| Overall | High-income countries | Non-high-income countries | |
|---|---|---|---|
| N=751 | N=398 | N=353 | |
| Age of FH diagnosis (years) | 12∙0 [5∙5–27∙0] 18∙0 (16∙8–19∙2) |
16∙0 [6∙0–33∙0] 20∙7 (18∙9–22∙5) |
10∙0 [5∙0–20∙0] 15∙1 (13∙6–16∙7) |
| Women | 389 (52∙1%) | 205 (51∙5%) | 184 (52∙9%) |
| Xanthomas at diagnosis | 516 (68∙7%) | 255 (64∙1%) | 261 (73∙9%) |
| Body mass index (kg/m2) | 24∙0 (23∙4–24∙6) | 24∙0 (23∙2–24∙8) | 24∙0 (23∙1–24∙9) |
| Diabetes mellitus | 23 (3∙6%) | 15 (5∙2%) | 8 (2∙3%) |
| Hypertension | 93 (14∙5%) | 41 (14∙0%) | 52 (14∙9%) |
| Chronic kidney disease | 6 (1∙2%) | 5 (2∙2%) | 1 (0∙4%) |
| Current smoker | 43 (7∙8%) | 25 (8∙7%) | 18 (6∙8%) |
| Previous smoker | 54 (9∙8%) | 31 (10∙8%) | 23 (8∙7%) |
| Lipids (mmol/L) | |||
| Untreated | |||
| Total cholesterol | 16∙2 [13∙1–20∙0] 16∙8 (16∙3–17∙2) |
15∙5 [12∙4–19∙3] 16∙4 (15∙8–17∙0) |
17∙2 [14∙6–20∙6] 17∙6 (16∙9–18∙2) |
| LDL-C | 14∙7 [11∙6–18∙4] 15∙2 (14∙8–15∙6) |
13∙5 [10∙4–17∙2] 14∙2 (13∙6–14∙9) |
15∙8 [12∙9–19∙2] 16∙2 (15∙6–16∙7) |
| HDL-C | 1∙00 [0∙78–1∙26] 1∙05 (1∙01–1∙09) |
1∙03 [0∙80–1∙27] 1∙05 (1∙00–1∙09) |
0∙93 [0∙70–1∙21] 1∙05 (0∙97–1∙13) |
| Triglycerides | 1∙20 [0∙88–1∙70] 1∙41 (1∙33–1∙50) |
1∙19 [0∙85–1∙65] 1∙38 (1∙27–1∙51) |
1∙23 [0∙90–1∙79] 1∙46 (1∙33–1∙60) |
| Most recent ** | |||
| Total cholesterol | 9∙0 [5∙8–13∙0] 9∙7 (9∙3–10∙1) |
6∙7 [4∙9–9∙1] 7∙4 (7∙0–7∙9) |
12∙3 [8∙9–15∙4] 12∙3 (11∙7–12∙9) |
| LDL-C | 7∙7 [4∙6–11∙5] 8∙3 (8∙0–8∙7) |
4∙9 [3∙0–7∙5] 5∙7 (5∙3–6∙1) |
10∙1 [7∙4–13∙2] 10∙5 (11∙0–10∙9) |
| LDL-C below guideline-recommended goals*** | 42 (7∙2%) | 38 (14∙6%) | 4 (1∙2%) |
| Lowest recorded level † | |||
| Total cholesterol | 7∙6 [4∙9–11∙1] 8∙7 (8∙2–9∙1) |
5∙6 [4∙1–7∙6] 6∙3 (5∙9–6∙7) |
10∙7 [7∙9–14∙7] 11∙3 (10∙7–11∙9) |
| LDL-C | 6∙6 [3∙6–10∙4] 7∙5 (7∙1–7∙9) |
3∙9 [2∙6–5∙8] 4∙7 (4∙3–5∙0) |
9∙3 [6∙7–12∙7] 9∙8 (9∙3–10∙3) |
| LDL-C below guideline-recommended goals*** | 64 (10∙9%) | 56 (21∙4%) | 8 (2∙5%) |
| Genetic information available †† | 565 (75∙2%) | 367 (92∙2%) | 198 (56∙1%) |
Data are shown as n (%) for categorical variables or as median [IQR]. In addition, numbers in italic describe quantitative variables as bootstrapped means (95%).Classification of high- and non-high-income countries is shown in Table S1. FH, familial hypercholesterolaemia; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol
This reflects the most recent measurement available after diagnosis and prior to data entry in the registry.
LDL-C below guideline-recommended goals is defined as an LDL-C level< 2∙5 mmol/L in primary prevention or < 1∙8 mmol/L in case of secondary prevention.
This reflects the lowest recorded LDL-C measurement between untreated (at diagnosis) and most recent measurement. When unavailable, the most recent measurement itself was considered the lowest.
For details see Table S2 in the online supplement.
Overall, untreated LDL-C levels were 14∙7 (IQR 11∙6–18∙4) mmol/L, and lower in patients from high-income countries than those from non-high-income countries (13·5 [IQR 10∙4–17∙2] versus 15·8 [IQR 12∙9–19∙2] mmol/L, respectively). The prevalence of modifiable risk factors for cardiovascular disease such as smoking (8%), obesity (15%), diabetes mellitus (4%) and hypertension (15%) was comparable between high and non-high-income countries. Among 505 patients with data on family pedigree available, 150 (30%) had a first-degree family member with HoFH who was also entered in the registry.
Genetics
A genetic confirmation of HoFH was available for 565 of 751 patients (75%), with a higher proportion in high-income compared to non-high-income countries (92% versus 56%). Of note, two non-high-income countries (South Africa and Brazil) accounted for over half (54%) of genetic diagnoses reported in this income group. Patients who had a genetic diagnosis had lower untreated LDL-C levels (14·2 [IQR 11·3–17·6] versus 16·1 [IQR 12·9–19·7] mmol/L) and presented less frequently with xanthomas at diagnosis (66% versus 76%). The allele combinations and classification by LDLR residual function are presented in Table S2 and the individual genetic variants are listed in Table S3. Among patients with genetic information available, the majority were either simple homozygous or compound heterozygous carriers of LDLR mutations (471 patients, 83%). These patients had higher untreated LDL-C levels (14·7 [IQR 11·8–18·1] mmol/L) compared with patients with autosomal recessive hypercholesterolaemia (28 patients [5%]; LDL-C 12·0 [IQR 11·3–14·3] mmol/L) and with those carrying any other bi-allelic combination including APOB and/or PCSK9 (66 patients [12%]; LDL-C 8·5 [IQR 6∙8–13∙2] mmol/L, Table S4). Of patients with bi-allelic variants in LDLR for whom the residual LDLR function was classified, 104 (23%) carried two LDLR-negative alleles and had higher untreated LDL-C levels compared with patients carrying any LDLR-defective allele (17·2 [IQR 14∙2–22∙2] versus 14·0 [IQR 11∙3–17∙1] mmol/L, Table S5).
Lipid-lowering therapy and LDL-C levels
Table 2 shows the type of LLT used at the time when the lowest on-treatment LDL-C levels were recorded. Nearly all patients (92%) were on statin therapy, usually high-intensity (311/379 [82%] defined as atorvastatin ≥40mg or rosuvastatin ≥20mg daily), where statin dosage was available. Ezetimibe was used by 72% of patients from high-income countries, while its use was 54% among patients from non-high-income countries. LLTs such as PCSK9 inhibitors, lomitapide and evinacumab were used infrequently and predominantly in patients from high-income countries. Among patients taking LLT, 78% were on combination therapy with two or more therapies and 42% used three or more types of LLT. Percentages of patients taking multi-LLT combinations were higher in high-income countries (Figure 1).
Table 2 –
Lipid-lowering therapy at the time of the lowest on-treatment LDL-C level recorded, overall and stratified by country income status
| Overall | High-income countries | Non-high-income countries | |
|---|---|---|---|
| N=534 | N=293 | N=241 | |
| Medication | |||
| Statins | 491 (91∙9%) | 262 (89∙4%) | 229 (95∙0%) |
| Ezetimibe | 342 (64∙0%) | 212 (72∙4%) | 130 (53∙9%) |
| PCSK9 inhibitors | 118 (22∙1%) | 76 (25∙9%) | 42 (17∙4%) |
| Lomitapide | 45 (8∙4%) | 40 (13∙7%) | 5 (2∙1%) |
| Evinacumab* | 13 (2∙4%) | 13 (4∙4%) | 0 |
| Mipomersen | 5 (0∙9%) | 0 | 5 (2∙1%) |
| Bile acid sequestrants | 33 (6∙2%) | 31 (10∙6%) | 2 (0∙8%) |
| Fibrates | 6 (1∙1%) | 2 (0∙7%) | 4 (1∙7%) |
| Other** | 17 (3∙2%) | 9 (3∙1%) | 8 (3∙3%) |
| Lipoprotein apheresis† | 243/621 (39∙1%) | 118/293 (39∙7%) | 125/328 (38∙1%) |
| Surgeries | |||
| Liver transplantation | 5 (0∙8%) | 4 (1∙3%) | 1 (0∙3%) |
| Age at liver transplantation (years) | 19∙4 (10∙5–30∙0) | 10, 16, 24, 36 | 11 |
| Ileal bypass surgery†† | 1 (0∙2%) | 1 (0∙3%) | 0 |
| Age at Ileal bypass surgery (years) | 21 | 21 | NA |
| Portacaval shunt surgery†† | 6 (1∙1%) | 0 | 6 (2∙9%) |
| Age at Portacaval shunt surgery (years) | 9∙7 (5∙7–14∙2) | NA | 5, 5, 7, 11, 12, 18 |
Data are shown as n (%) for categorical variables, as bootstrapped mean (95%CI) for quantitative variables. Classification of high- and non-high-income countries is shown in Table S1. NA, not applicable; PCSK9, proprotein convertase subtilisin/kexin type 9
Evinacumab is an investigational product that has been recently approved by FDA but is not yet approved by other regulatory agencies. It was given as compassionate use and/or open label extension as part of a clinical trial
Other therapies were red yeast rice, omega-3 fish oils and plant stanols
Apheresis includes all lipoprotein apheresis types including plasma exchange. For 87 patients from non-high-income countries it was only known that they were on lipoprotein apheresis but no additional information was available on other lipid-lowering therapies. Patients from non-high-income countries who are on apheresis were mainly from Turkey (n=87) and Lebanon (n=26).
Ileal bypass and portacaval shunt surgery are no longer considered treatments for HoFH, these entries reflect (abandoned) historic practice.
Figure 1 -. Untreated LDL-C levels and lowest on-treatment LDL-C levels achieved, as a function of number of LLTs (including apheresis).
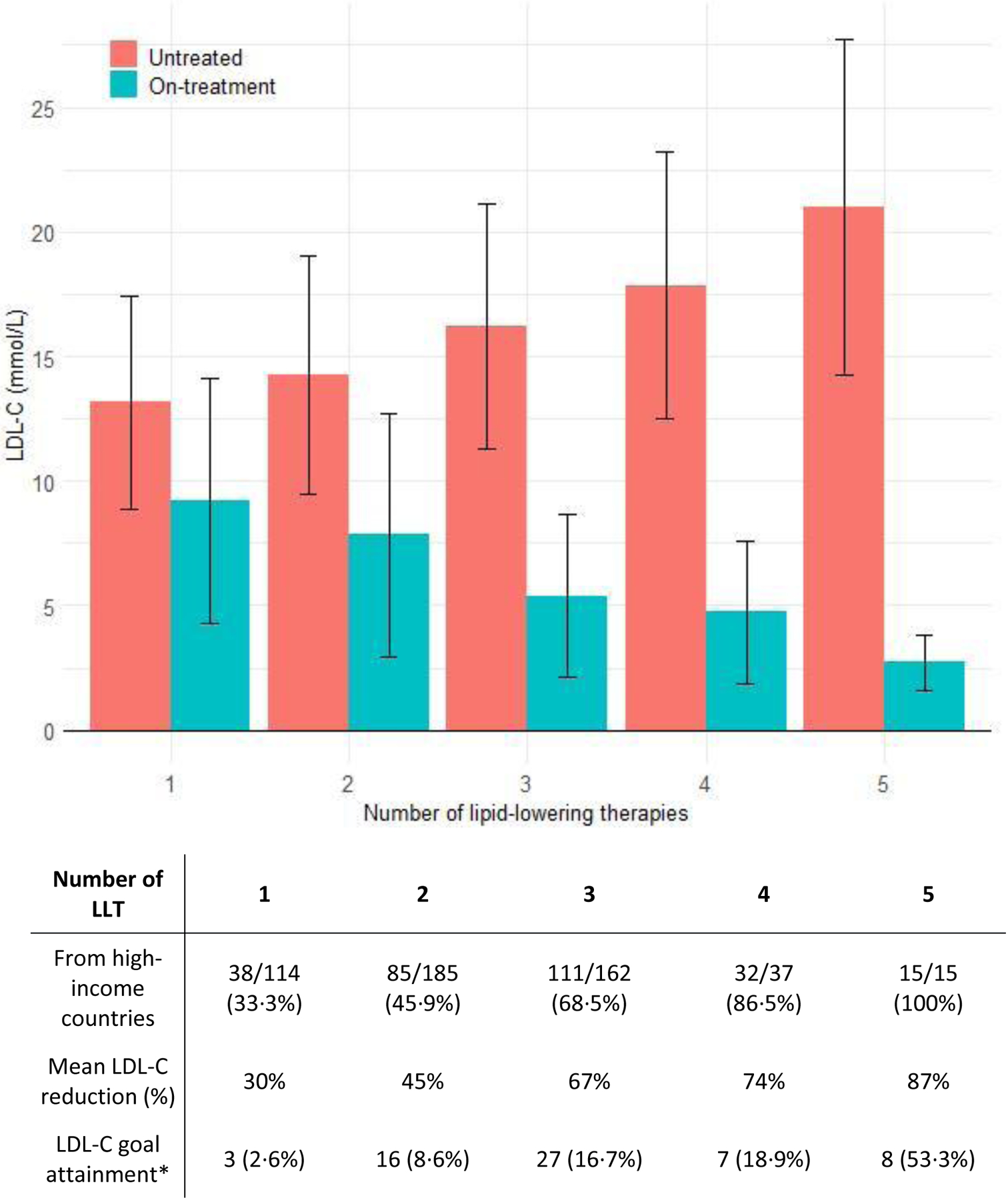
Data are shown as mean (±SD) or n (%), as appropriate. LLT included statins, ezetimibe, PCSK9 inhibitors, lipoprotein apheresis, lomitapide, evinacumab and mipomersen. Five patients who had undergone liver transplantation were excluded from this analysis. LDL-C, low-density lipoprotein cholesterol; LLT, lipid-lowering therapy
* LDL-C below guideline-recommended goals is defined as an LDL-C level < 2∙5 mmol/L in primary prevention or < 1∙8 mmol/L in case of secondary prevention.
Lipoprotein apheresis (including plasma exchange) was conducted in 243 patients (39%), initiated at a median age of 15∙0 [IQR 10∙0–28∙0] years, and performed weekly (25%) or biweekly (54%) in the majority of patients. Patients on apheresis had higher untreated LDL-C at diagnosis compared with patients who were not on apheresis (Table S6, 17·2 [IQR 13∙9–21∙4] versus 13·5 [IQR 11∙1–17∙1] mmol/L).
Figure 1 shows the untreated LDL-C levels and the lowest LDL-C levels achieved with the number of LLTs used, including apheresis. Fibrates, omega-3 fish oils, red yeast rice and plant stanols, which lower LDL-C levels modestly, were not included in this analysis. Five patients who had undergone liver transplantation were also excluded from this analysis. Despite multiple therapies, attainment of guideline-recommended LDL-C levels was low: overall, 12% of patients reached an LDL-C <2·6 mmol/L (primary prevention) or <1·8 mmol/L (secondary prevention). The LDL-C reduction was 30% in patients on monotherapy, 45% with 2 classes of LLT and over 65% in patients using ≥3 LLT (Figure 1). The percentage of patients who attained LDL-C goals increased with the number of LLTs, and were more frequently attained in patients from high-income countries compared to non-high-income countries (Table 1, 21% versus 3%). Only 5% of the overall population achieved the more recent lower LDL-C goals (<1·8 and <1·4 mmol/L, respectively).13
Cardiovascular Disease
Table 3 shows the proportion of patients reported to have cardiovascular disease overall and stratified by income. The median age at which MACE occurred was 31·0 [IQR 22·0–42·0] years, with 9% of patients already having suffered a non-fatal MI, having undergone PCI or CABG or with aortic valve stenosis at diagnosis of HoFH. There were 37 deaths of which 28 (76%) were from cardiovascular causes (median 28∙0 [IQR 17∙0–45∙5] years). The earliest recorded age at which angina pectoris, MI, CABG or PCI were reported were 4, 10, 5 and 10 years old, respectively. Among those with a recorded non-fatal coronary event, a recurrent coronary event occurred in 28% of patients (29/102), where reported. Peripheral artery and cerebrovascular disease occurred in 42 (6%) and 22 (3%) patients, respectively.
Table 3 –
Cardiovascular disease in the overall population and stratified by country income status
| Overall | High-income countries | Non-high-income countries | |
|---|---|---|---|
| N=751 | N=398 | N=353 | |
| Cardiovascular death* | 28 (3∙7%) | 10 (2∙5%) | 18 (5∙1%) |
| Unknown or non-cardiovascular death | 9 (1∙2%) | 6 (1∙5%) | 3 (0∙8%) |
| Age at cardiovascular death | 28∙0 [17∙0–45∙5] 31∙5 (25∙5–37∙6) Range 5–58 |
49∙5 [32∙0–50∙8] 37∙0 (26∙1–46∙6) |
24∙0 [17∙0–40∙3] 28∙4 (21∙2–36∙2) |
| Myocardial infarction | 90 (11∙9%) | 48 (11∙9%) | 42 (11∙9%) |
| Age at first MI | 37∙5 [30∙0–50∙0] 38∙8 (35∙6–42∙0) Range 10–68 |
39∙0 [32∙0–50∙0] 39∙9 (36∙2–43∙6) |
32∙5 [28∙5–42∙5] 35∙4 (29∙2–41∙9) |
| Angina pectoris | 95 (12∙5%) | 63 (15∙6%) | 32 (9∙0%) |
| Age at AP onset | 30∙0 [20∙0–39∙0] 30∙4 (27∙3–33∙7) Range 4–75 |
32∙0 [20∙8–42∙3] 33∙2 (29∙0–37∙5) |
24∙0 [20∙0–32∙0] 25∙3 (21∙6–29∙1) |
| CABG | 120 (15∙8%) | 60 (14∙9%) | 60 (16∙9%) |
| Age at first CABG | 30∙0 [22∙5–40∙0] 31∙5 (28∙9–34∙2) Range 5–69 |
32∙0 [28∙0–46∙0] 36∙7 (32∙9–40∙6) |
24∙0 [17∙3–32∙8] 26∙0 (23∙0–29∙0) |
| PCI | 91 (12∙1%) | 54 (13∙4%) | 37 (10∙2%) |
| Age at first PCI | 39∙5 [28∙0–48∙5] 38∙5 (35∙5–41∙5) Range 10–75 |
42∙5 [36∙3–52∙8] 42∙9 (39∙4–46∙6) |
30∙0 [21∙0–40∙0] 31∙2 (26∙8–35∙5) |
| Aortic valve replacement | 52 (6∙9%) | 36 (8∙9%) | 16 (4∙5%) |
| Age at first AVR | 31∙0 [24∙8–41∙0] 33∙0 (28∙6–37∙4) Range 5–69 |
31∙5 [27∙0–43∙8] 36∙1 (30∙3–42∙1) |
30∙0 [22∙0–35∙3] 27∙9 (21∙9–33∙5) |
| Peripheral artery disease | 42 (6∙2%) | 8 (2∙4%) | 34 (9∙8%) |
| Age at PAD diagnosis | 34∙5 [20∙5–47∙3] 35∙5 (27∙5–44∙0) Range 7–74 |
51∙0 [34∙5–64∙0] 49∙9 (35∙1–63∙7) |
21∙0 [17∙0–38∙0] 27∙8 (19∙8–36∙4) |
| Cerebrovascular disease** | 22 (2∙9%) | 18 (4∙5%) | 4 (1∙1%) |
| Age at first cerebrovascular disease event | 37∙0 [28∙0–48∙0] 40∙9 (33∙9–48∙7) Range 23–71 |
38∙0 [29∙0–53∙0] 42∙5 (34∙6–50∙8) |
28∙5 [27∙3–29∙8] 26, 31, NA, NA |
| Composite outcomes | |||
| MACE§ | 216 (28∙8%) | 110 (27∙2%) | 106 (29∙9%) |
| Age of first MACE | 31∙0 [22∙0–42∙0] 33∙0 (30∙9–35∙0) Range 5–75 |
37∙0 [29∙0–49∙0] 38∙1 (35∙4–40∙9) |
24∙5 [17∙0–34∙5] 26∙8 (24∙3–29∙3) |
| MACE+§§ | 267 (35∙6%) | 137 (34∙4%) | 130 (36∙7%) |
| Age of first MACE+ | 30∙0 [21∙0–41∙0] 32∙0 (30∙0–33∙9) Range 4–75 |
35∙5 [25∙0–48∙3] 36∙5 (33∙8–39∙2) |
24∙0 [17∙0–32∙0] 26∙2 (23∙9–28∙6) |
Data are shown as n (%) for the prevalence of cardiovascular events, and as median [IQR] and range (minimum, maximum) for ages at cardiovascular events. In addition, ages at cardiovascular events are shown in italic as bootstrapped mean (95%CI). MI, myocardial infarction; PCI, percutaneous coronary intervention; CABG, coronary artery bypass grafting; AP, angina pectoris; AVR, aortic valve replacement; NA, not available; MACE, major adverse cardiovascular event
Cardiovascular death was physician reported death from cardiovascular causes. “Sudden death” and periprocedural death due to cardiac surgery necessitated by consequences of hypercholesterolaemia was additionally considered cardiovascular death.
Cerebrovascular disease was defined as ischemic stroke, carotid artery stenting or carotid endarterectomy.
MACE is a composite of cardiovascular death, non-fatal MI, PCI and CABG.
MACE+ is a composite of cardiovascular death, non-fatal MI, PCI and CABG, AP, non-fatal ischemic stroke, carotid stenting, carotid endarterectomy and peripheral artery disease.
(Supra-)valvular aortic stenosis (any severity) was reported in 29% (216/751) of patients. Where echocardiographic data were available (n=265), 35 (13%) patients had mild, 25 (9%) moderate, and 7 (3%) severe aortic stenosis. Aortic valve replacement had been performed in 52 (7%) patients (median 31·0 [24·8–41·0] years; youngest 5 years).
Figure 2A shows MACE-free survival, with an earlier occurrence in patients managed in non-high-income compared to high-income countries (24·5 [IQR 17·0–34·5] versus 35·0 [IQR 25·0–49·0] years, respectively), with a crude ratio (HR) of 2·01 (95%CI 1·40–2·88). Stepwise attenuation of the HR for incident MACE is shown in Figure 3; adjustment for treatment with three or more types of LLT, age of diagnosis and sex reduced the HR to 1·64 (95%CI 1·13–2·38), suggesting that a fifth of the excess risk might be mitigated through early diagnosis and use of three or more LLTs.
Figure 2 – Survival-time free from major adverse cardiovascular events.
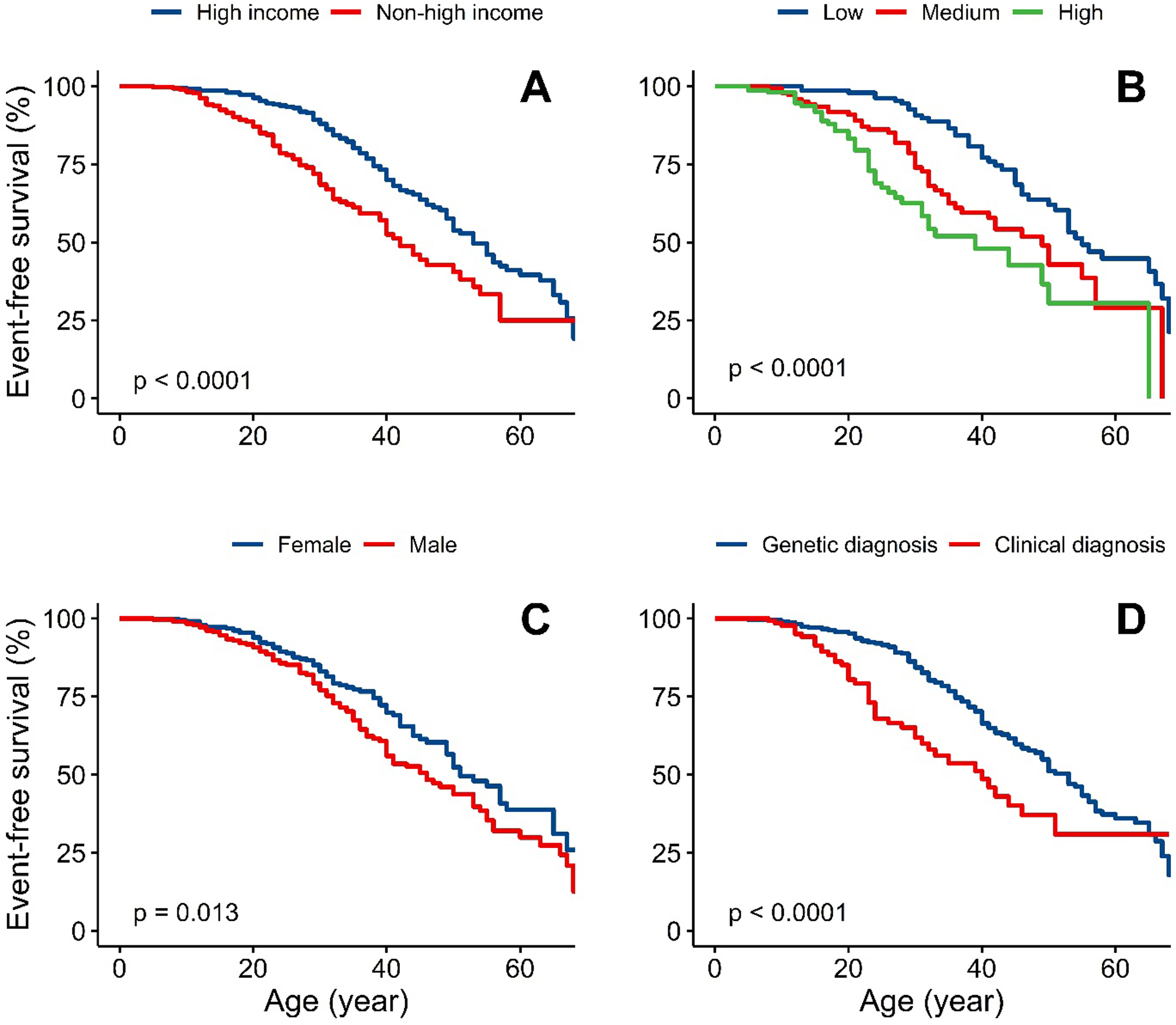
Panel: event-free survival stratified by A) High-income vs non-high-income countries B) Untreated LDL-C tertiles, lowest (4∙9–12∙5 mmol/L), middle (12∙6–17∙1 mmol/L) and highest (17∙1–36∙3 mmol/L) C) Sex D) Clinical diagnosis only versus genetic diagnosis
Statistical test for comparison between groups is Log-rank test.
Figure 3 – Forest plot showing unadjusted and adjusted hazard ratios for occurrence of major adverse cardiovascular events between specific groups of interest.
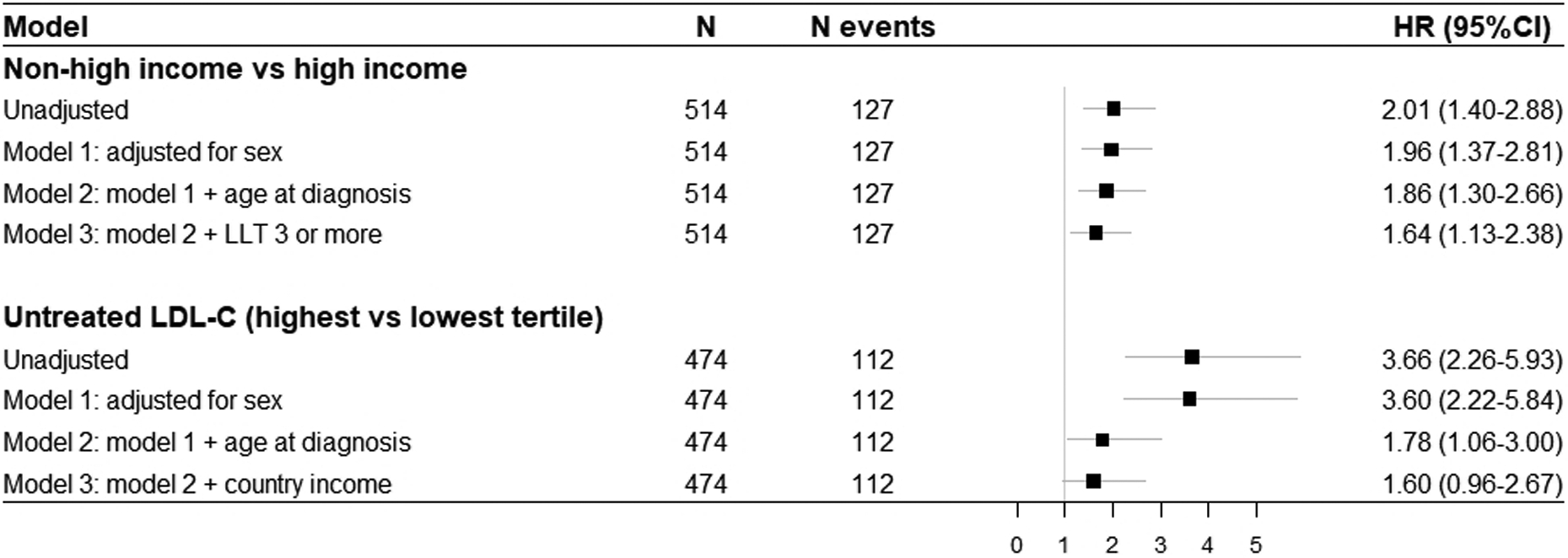
LDL-C, low-density lipoprotein cholesterol; LDLR, LDL receptor; LLT, lipid-lowering therapy; HR, hazard ratio; CI, confidence interval
Major adverse cardiovascular events were defined as cardiovascular death, myocardial infarction, coronary artery bypass grafting or percutaneous coronary intervention that occurred after the diagnosis of HoFH was made. Presented data are based on complete case analysis.
Figure 2B shows MACE-free survival stratified by tertiles of untreated LDL-C. A graded relationship was observed, with events occurring earlier among the highest tertile. Stepwise attenuation of the HR for incident MACE is shown in Figure 3; after adjustment for age of diagnosis and income status the HR for the highest versus lowest tertile with MACE fell from 3·60 (2·22–5·84) to 1·60 (0·96–2·67). Using country status as a proxy for use of multi-LLT regimens suggests that as much as half of the excess risk could be attenuated by early diagnosis and better treatment.
MACE-free survival was shorter in males (Figure 2C), despite similar demographic characteristics compared to females (Table S7). In sensitivity analyses, the coefficient for sex changed little after addition of smoking to the model: the coefficient for male sex changed from 0·63 to 0·67. Event-free survival was also shorter for patients with a clinical diagnosis of HoFH (no genetic data) versus those genetically confirmed (Figure 2D). In patients with bi-allelic LDLR variants, there was a trend towards shorter survival free from MACE in patients carrying two LDLR-negative alleles compared with those carrying LDLR-defective variants (p=0·21, Figure S1).
Discussion
The present study reports the largest international cohort of HoFH patients to date. Our findings show that, although a rare disease, HoFH occurs worldwide with severe manifestations of cardiovascular diseases very early in life, contributing significantly to premature deaths and disability among those affected. We found clinically meaningful treatment inequalities between countries, with patients in less affluent countries less likely to receive three or more LLTs, resulting in higher on-treatment LDL-C levels and over a decade shorter survival free from cardiovascular events.
Assuming a prevalence of HoFH of about 1 in 300,000 and a global population of 7 billion, we expect approximately 23,000 cases worldwide with the majority residing in less affluent parts of the world, often in regions with high consanguinity or with founder effects, where the condition remains largely underdiagnosed and untreated. Although manyfold larger than previous reports, the 751 patients included in this study thus only comprise ~3% of the estimated total population of HoFH patients worldwide, highlighting the pressing need to increase the identification of these patients using systematic screening and genetic testing for FH globally.14
Prior studies of smaller sample size have reported on the severe cardiovascular consequences of HoFH.2,5,10,11,15–18 Though in part confirmatory, the present report leverages data from 751 patients from 38 countries with a larger number of events, providing more robust information to better guide health-policy and improve patient care. We show that diagnosing HoFH in the second decade of life is too late, as by this age many patients have already experienced cardiovascular complications, supporting the need for more effective strategies to aid timely diagnosis, such as systematic cascade screening or universal screening at an early age. Despite the use of LLT, first MACE occurs early at a median age of 31 years, and in 4% even before the age of 18 years, in line with anecdotal evidence that cardiovascular events can occur in HoFH during childhood.19 Additionally, one third of patients had (supra-)valvular aortic stenosis, which frequently required surgical intervention. Hence, systematic and more frequent image-guided assessment of aortic (valve) pathology in addition to ASCVD should be implemented in care pathways for HoFH patients.1
Cumulative exposure to extreme elevations of LDL-C drives the premature onset of ASCVD6, therefore guidelines recommend starting intensive lipid lowering immediately from the time of HoFH diagnosis.1,20,21 The backbone of LLT to date has been high-intensity statin therapy with ezetimibe. However, in the present study, very few patients achieved current LDL-C recommendations with this approach. Use of three or more LLTs (nearly exclusive to patients managed in high-income countries) were associated with lower LDL-C levels and greater likelihood of goal achievement. Our finding that use of five LLTs lowered LDL-C by more than 85% demonstrates that reaching acceptable LDL-C levels, and consequently better outcomes, is possible if a combination of drugs is used. For many patients, especially those without residual LDLR function, therapeutic approaches independent of LDLR function can significantly improve LDL-C levels. These approaches include frequent lipoprotein apheresis16,22–24, although this option is invasive, not uniformly available25 and associated with reduced quality of life.26 Recently, medications such as lomitapide and evinacumab have emerged, which have been shown to reduce LDL-C independently of LDLR function7,8,27, and can be used in combination with PCSK9 inhibitors for patients with residual LDLR activity.28,29 Among those with the highest LDL-C levels, our study suggests that as much as half of excess risk could be attenuated through earlier diagnosis and greater use of multi-LLT combinations. Furthermore, as cardiovascular complications may already occur in childhood, it is imperative that existing and new LLTs are rapidly approved for use in the paediatric population.27
HDL-C levels in our cohort of HoFH patients were relatively low compared to those expected in a general population. The cause for this known observation is unclear; however the magnitude of the effect of lifelong exposure to extreme LDL-C levels dwarfs any meaningful impact of lower HDL-C levels on cardiovascular outcomes.30
Our study also offers insights into the important role of genetics in HoFH diagnosis. Nearly all (~90%) patients from high-income countries were genetically confirmed versus just over half (56%) from non-high-income countries. Of these, more than half resided in South Africa or Brazil, where some local institutions have access to genetic testing. Patients from non-high-income countries had, on average, a more severe phenotype at diagnosis (higher untreated LDL-C levels and greater prevalence of xanthomas), despite being diagnosed at a younger age. These differences may be an artefact of healthcare systems and approaches to case finding, including screening affected relatives and use of genetic testing. Thus, it is possible that only patients with the most severe phenotypes are diagnosed clinically in non-high-income countries, while those with a less severe phenotype are diagnosed clinically as “severe heterozygous FH” or remain undiagnosed. This possibility is supported by the fact that in our cohort double heterozygous patients, who have a less severe phenotype, were almost exclusively reported from high-income countries.
The global nature of our study not only allows for a comparison of the impact of current practice between high and non-high-income countries, but also provides an opportunity to explore potential determinants of health outcomes. The most striking finding in this regard is that event-free survival in HoFH is on average a decade shorter among patients managed in non-high-income countries. These patients had significantly higher risk of MACE, even after adjustment for age of diagnosis, sex and LLT. Patients managed in non-high-income countries had higher on-treatment LDL-C levels and were less likely to receive multi-LLT combinations. As on-treatment LDL-C levels are a major determinant of event-free survival for HoFH patients,18 it is likely that this could in part explain the excess risk. Thus the uneven global health burden from HoFH cannot be addressed until less affluent countries have access to effective and affordable LLT regimens starting in childhood, with inevitable implications for healthcare systems and the pharmaceutical industry.
This study has several limitations. Patients entered in the registry may not reflect clinical practice or phenotypes outside of participating centres. That said, as a rare condition, HoFH is mostly managed in specialist and/or academic centres, such as those participating in this registry. Inevitably, those diagnosed reflect local healthcare systems, impacting referrals to specialist clinics and thus availability for inclusion. To generate contemporary data, this registry only included patients alive in 2010 or later. Survival bias is thus inevitable because patients with less severe phenotypes survive longer and are consequently more likely to be included. Collection of retrospective data reduces granularity and completeness of some variables of interest and missing data may also reflect clinical practice at country or institution level. For example, data on Lp(a) levels were not included in this analysis since they were only available in one third of patients, mainly from high-income countries, and measured using different laboratory assays. Although we included participants from 38 countries, more clinicians from other countries and sites were invited to this initiative than those who ultimately participated. Some regions (e.g. much of Latin America and Africa) remain underrepresented and more information is needed to further reduce existing data gaps. Furthermore, a significant proportion of the total number of patients came from three countries: Italy, Turkey and South Africa. However, patients from these countries were comparable to others in their respective income group, and sensitivity analyses excluding these countries did not change results. Finally, the observational nature of the study including survival analyses does not allow assessment of causality and we cannot exclude the possibility of unmeasured variable and residual confounding on outcomes. Despite these limitations, the scale and global reach of this study offer important insights into the contemporary nature of HoFH and its management.
In conclusion, this study reports on the largest international cohort of HoFH patients to date and highlights global disparities that result in clinically significant differences in their care and health outcomes. Our data strongly support the fact that patients with HoFH require early diagnosis and initiation of treatment within the first decade of life as well as more intensive lipid lowering using three or more types of LLT as standard of care in order to prevent the serious consequences of extreme LDL-C exposure. As the greatest global burden resides in less affluent regions of the world, a critical reappraisal of healthcare policy and funding is required at a global level to improve health outcomes for all patients with HoFH.
Supplementary Material
Research in context.
Evidence before this study
Articles were identified by PubMed searches using terms related to “(homozygous) familial hypercholesterolaemia” and the reference list was expanded to include references cited in relevant articles. Articles published in English up to and including February 2021 were included.
While the prevalence of homozygous familial hypercholesterolaemia (HoFH) was traditionally estimated to be ~ 1 in 1,000,000, more recent studies have suggested a prevalence closer to 1 in 300,000 in populations not subject to gene founder or consanguinity effects. Given its rarity, guidance for screening and treatment has relied on expert opinion and studies of small sample size, derived mostly from patients of European ancestry or from high-income countries, prior to advances in treatment strategies. Such studies have suggested that the clinical consequences of HoFH likely relate to untreated low-density lipoprotein cholesterol (LDL-C) levels, type of genetic defect, and age at which treatments are started.
Added value of this study
The HoFH International Clinical Collaborators (HICC) registry (NCT04815005) is the first and only global HoFH registry. Initiated by physicians caring for HoFH patients in specialized centres across diverse healthcare settings, HICC offers a unique opportunity to not only provide a comprehensive assessment of the genetic profile and clinical characteristics of HoFH patients globally, but also to provide insights into the impact of policies and access to healthcare and use of effective medications on health outcomes. The present study shows that HoFH patients are often only diagnosed in the second decade of life with extreme LDL-C elevation and a prevalence of cardiovascular or aortic valve disease at diagnosis of almost one in ten. We found significant health inequalities in the management of patients with HoFH globally. Despite the development of newer, more effective therapies that have been demonstrated to result in significantly better control of LDL-C levels, guideline-recommended goal attainment is rare and largely restricted to patients from high-income countries. Patients from non-high-income countries have on average a more severe phenotype at diagnosis, are less likely to receive advanced treatments and have a decade shorter cardiovascular event-free survival compared to those from high-income countries.
Implications
The findings from HICC provide a framework to inform the development of clinical practice guidelines and public health policies concerning HoFH and help establish a uniform world-wide approach to the management of this high-risk condition. Greater awareness and changes in health policy, including restructuring approaches to screening and diagnosis, are urgently required to improve early detection and treatment of HoFH. This is particularly relevant to non-high-income countries where patients with HoFH require greater access to more effective combinations of lipid-lowering therapies, in order to improve health outcomes.
Acknowledgements
In-house funding at each Institution was used to cover effort of contributors for data collection and entry. The creation and the maintenance of the REDCap database and support of a study coordinator (CN) for bulk data entry was provided in house by MC at the University of Pennsylvania. Support of the registry coordinators (TRT and MLH) was provided in house by GKH at Amsterdam UMC, location AMC, supplemented by a grant from the European Atherosclerosis Society to MLH. KKR acknowledges support from the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre, United Kingdom. AJVV acknowledges support from the “Programa de Ayudas Beatriz Galindo” from the Ministry of Universities, Government of Spain, and University of Sevilla, Spain.
TF was partly supported by the Ministry of Health, Czech Republic, grant number NU20-02-00261.
Declaration of Interests
Authors listed alphabetically by last name. AFAAZ has nothing to disclose. MA Melih Aktan PENDING FORMS. MDA has nothing to disclose. RA has nothing to disclose. MAK has nothing to disclose. BBA has nothing to disclose. KAW reports lecture fees and personal fees from Abbot, Amgen, AstraZeneca, Merck, Pfizer and Sanofi. MA reports research grants from Novartis, Akcea, Amryt, Regeneron, Daiichi-Sankyo; consulting fees from Amryt, Akcea and Pfizer; lecture fees from Amryt, Akcea, Daiichi-Sankyo, Novartis, Amarin, Regeneron and Alfasigma; manuscript writing fees for Alfasigma; participation in advisory boards for Novartis, Amryt, Akcea, Amarin, Pfizer, Alfasigma and Daiichi-Sankyo. MHA has nothing to disclose. STA has nothing to disclose. AB has nothing to disclose. SB has nothing to disclose. SAB has nothing to disclose. KBT has nothing to disclose. FFB has nothing to disclose. VB has nothing to disclose. DJB reports research grants from Amgen, Amryt, AstraZeneca, Sanofi, and Regeneron; lecture fees and personal fees from Amgen, Sanofi-Aventis and Novartis; participation in advisory board for Amryt (Chair of the LOWER study steering committee) and being member of the executive committee of the Lipid and Atherosclerosis Society of South Africa. MB has nothing to disclose. JAB has nothing to disclose. EB reports personal fees from Sanofi-Aventis, Amgen and Amryt. LRB reports research grants from Amryt (paid to the institution). MB has nothing to disclose. PSB has nothing to disclose. PC has nothing to disclose. SC has nothing to disclose. DC has nothing to disclose. MC has nothing to disclose. ALC reports research grants from Sanofi, Amgen and Akcea; consulting and lecture fees for Kowa, Recordati, Novartis, Pfizer, Sanofi, Amgen, Merck, Akcea, Amarin, Aegerion, Genzyme, Bayer, Regeneron, Esperion and Daiichi-Sankyo. ABC has nothing to disclose. RC has nothing to disclose. MJC reports personal fees from Amgen and Sanofi; participation in advisory board for Sanofi. KC reports research grants from the Polish Ministry of Health; lecture fees from Sanofi, Amgen, Novartis, Polpharma and the Polish Cardiac Society; participation in advisory board for the Polish National FH Registry. AFGC reports consulting fees from Roelmi SpA; lecture fees from Menarini IFR and Fidia Farmaceutici. HC has nothing to disclose. MC reports institutional support for the conduction of clinical trials from Regeneron Pharmaceuticals, Akcea and REGENXBIO; consulting fees from Amryt Pharma; and support from NIH/NHLBI grant P01HL059407. AJC has nothing to disclose. SD reports honoraria for advisory board for Amryt and Sobi. EJD has nothing to disclose. JCD has nothing to disclose. SD has nothing to disclose. LD reports honoraria for public speaking for Amryt, Sanofi, Pfizer and Amger; personal and consulting fees from Amgen and Akcea. LD Lubomir Dlouhy PENDING FORMS. OSD reports personal and consulting fees from Amgen, Novartis, MSD, Sanofi, Daiichi-Sankyo. AD has nothing to disclose. D-LD has nothing to disclose. RD has nothing to disclose. JD has nothing to disclose. CFE has nothing to disclose. AE has nothing to disclose. SE has nothing to disclose. MSE has nothing to disclose. MVE has nothing to disclose. ACF has nothing to disclose. TF has nothing to disclose. GAF has nothing to disclose. TF reports personal fees from Novartis, Sanofi and Amgen; and that he was partly supported by the Ministry of Health, Czech Republic, grant number NU20-02-00261. AG report consulting and lecturing fees from Amgen, Sanofi, Regeneron, Mylan, Akcea, Novartis and MSD. IMG reports (non-financial) support from FH Portugal and Portugese Organization of Patients with Familial Hypercholesterolemia and being president of FH Portugal (unpaid). MG has nothing to disclose. JG reports research grants from the Canadian Institutes of Health Research. AG has nothing to disclose. SG-P Susanne Greber-Platzer PENDING FORMS.UG has nothing to disclose. MH-S reports research grants from Recordati and Kaneka; personal fees from Amgen, Astellas, Recardati, MSD and Sanofi; advisory board for New Amsterdam Pharma and Medicine Company; being chairperson Primary Hyperlipidemia, Research on Measures against Intractable Diseases by the Japanese Ministry of Health, Labor, and Welfare; being chairperson of the Working Group by Japan Atherosclerosis Society for Making Guidance of Familial Hypercholesterolemia; owning stock options of Liid Pharma. MLH has nothing to disclose. RAH reports speaker fees and consulting fees from Akcea-Ionis, Amgen, Arrowhead, HLS Therapeutics, Novartis, Pfizer and Sanofi. PH has nothing to disclose. MH has nothing to disclose. GKH reports research grants from the Netherlands Organization for Scientific Research (vidi 016.156.445), CardioVascular Research Initiative, European Union and the Klinkerpad fonds; institutional research support from Aegerion, Amgen, AstraZeneca, Eli Lilly, Genzyme, Ionis, Kowa, Pfizer, Regeneron, Roche, Sanofi, and The Medicines Company; speaker’s bureau and consulting fees from Amgen, Aegerion, Sanofi, and Regeneron until April 2019 (fees paid to the academic institution); and part-time employment at Novo Nordisk A/S, Denmark since April 2019. LCH reports participation on scientific advisory boards for the FH Foundation and the Rockefeller University IRB; owning mutual fund stock options. OH Osama Hussein PENDING FORMS. GI reports personal fees from Pfizer, Amgen, Sanofi, Amryt and Novartis; participation on advisory board for SOBI. AI has nothing to disclose. OI has nothing to disclose. LJ has nothing to disclose. BAK has nothing to disclose. WK reports research grants from Amgen; honoraria for lectures and speakers bureaus for Amgen, Meiji, AstraZeneca, Eli Lilly, Nichi-Iko and Abbott; support for attending online meetings from Amgen; Advisory Board for Meiji and Sanofi; receipt of medication for patients from Amgen, Meiji and Sanofi. N-TK has nothing to disclose. GK has nothing to disclose. MK has nothing to disclose. LGK Leyla G. Kaynar PENDING FORMS. IK has nothing to disclose. EK Erdal Kurtoglu PENDING FORMS. KSL has nothing to disclose. H-AL has nothing to disclose. T-TL has nothing to disclose. EL has nothing to disclose. ARML reports institutional research grants from Pfizer, Amgen, MSD, Sanofi-Aventis, Daichii-Sankyo, and Regeneron. RM has nothing to disclose. MM has nothing to disclose. RM lecturing fees from Abbott, Amgen, AstraZeneca, Boehringer-Ingelheim, Eli Lilly, Janssen, Novo Nordisk and Sanofi. OM has nothing to disclose. TN Tarik Naguib PENDING FORMS. EMM has nothing to disclose. HMN has nothing to disclose. EAN has nothing to disclose. GN has nothing to disclose. CN has nothing to disclose. M-NTN has nothing to disclose. HO Harika Okutan PENDING FORMS. OIO Osman I. Ozcebe PENDING FORMS. JP reports funding from the National Health and medical Research Council Australia. AP has nothing to disclose. CP reports personal honoraria for lectures from Sobi. ZP has nothing to disclose. LP has nothing to disclose. FJR reports consulting fees, lecturing fees and Advisory Board from Amgen, Sanofi-Aventis, Regeneron, Novartis and Lib Therapeutics outside the submitted work; member of the International Atherosclerosis Society. CR has nothing to disclose. KKR reports institutional research grants from Amgen, Sanofi, Daiichi-Sankyo, Regeneron and Pfizer; consulting fees and lecturing fees from Amgen, Sanofi, Novartis, Pfizer, AstraZeneca, Boehringer Ingelheim, Novo Nordisk, Kowa, Silence Therapeutics, New Amsterdam, Esperion, Daiichi-Sankyo, Bayer, Abbott, Resverlogix, Medicines Company, Lilly, Algorithm, MSD, Abbvie, Esperion and Viatris, outside the submitted work. AR has nothing to disclose. MDR has nothing to disclose. ZR has nothing to disclose. SHR has nothing to disclose. CR has nothing to disclose. JRvL Jeanine Roeters-van Lennep PENDING FORMS. IR has nothing to disclose. DR has nothing to disclose. FS has nothing to disclose. SA Saim Sag PENDING FORMS. OZS Osman Z. Salcioglu PENDING FORMS. TS has nothing to disclose. RDS reports research grants from Amgen, Kowa, Esperion, Novartis and Sanofi; consulting fees from Abbott, Amgen, AstraZeneca, Aché, Hypera, Novo Nordisk, Roche and Sanofi; personal fees from Abbott, Amgen, AstraZeneca, Aché, EMS, GETZ Pharma, Hypera, Novo Nordisk, Novartis, Merck, MSD, Pfizer, PTC, Roche and Sanofi; leadership in the International Atherosclerosis Society and IberoAmerican FH Network. FS has nothing to disclose. DS has nothing to disclose. NS has nothing to disclose. FKS has nothing to disclose. AS has nothing to disclose. MHS reports speaker fees from Sanofi, Janssen, Gilead and HLS therapeutics; Advisory Board for Novo Nordisk, Akcea, Gilead, HLS Therapeutics. FS has nothing to disclose. HS reports research grants from Amgen, MSD, Synageva, Amryt, Alexion and Akcea; consulting fees from Amgen, Alexion, Daiichi-Sankyo, Pfizer, Akcea; speaker fees from Amgen, Daiichi-Sankyo, Sanofi and Akcea. VS has nothing to disclose. CATS has nothing to disclose. ESGS reports consulting fees from Amgen, Sanofi, Regeneron, Esperion, Novartis, Ionis/Akcea (all paid to the institution). TMS reports honorarium and travel reimbursement for participating in an expert panel by Akcea. PS has nothing to disclose. AVS has nothing to disclose. PT has nothing to disclose. AT has nothing to disclose. LT reports support from the Ministry of Health of the Czech Republic (grant no. 16-20984A and NU20-02-00261). CT has nothing to disclose. TRT has nothing to disclose. T-HT has nothing to disclose. RU has nothing to disclose. AJVV reports participation in research grants to Imperial College London and/or European Atherosclerosis Society from Pfizer, Amgen, MSD, Sanofi-Aventis, Daiichi-Sankyo and Regeneron; personal fees for consulting from Bayer and Regeneron; and honoraria for lectures from Amgen, Mylan and Akcea; outside the submitted work. HV has nothing to disclose. ICV has nothing to disclose. MV has nothing to disclose. LW has nothing to disclose. GFW reports research grants from Arrowhead; consulting and speaker fees from Amgen, Novartis, Arrowhead, AstraZeneca; travel support from Amgen, Arrowhead and Kowa. AW reports research support for pharmaceutical trials of lipid lowering agents from Amgen, Regeneron and Novartis. PW has nothing to disclose. MY has nothing to disclose. MY Mehmet Yilmaz PENDING FORMS. HYY Hamiyet Yilmaz Yasar PENDING FORMS. SZ has nothing to disclose. MGZ has nothing to disclose. LZ has nothing to disclose. LZ has nothing to disclose.
HoFH Clinical Collaborators:
Australia: Jing Pang13, PhD, Prof Gerald F. Watts13, DSc Austria: Prof Susanne Greber-Platzer14, PhD, Martin Mäser15, MD, Prof Thomas M. Stulnig16, MD, Prof Christoph F. Ebenbichler17, MD Bahrain: Khalid Bin Thani18, MD Belgium: Prof David Cassiman19, PhD, Olivier S. Descamps20, PhD, Daisy Rymen21, PhD, Peter Witters21, PhD Brazil: Raul D. Santos22, PhD Canada: Liam R. Brunham23, PhD, Prof Gordon A. Francis23, MD, Jacques Genest24, MD, Prof Robert A. Hegele25, MD, Brooke A. Kennedy26, Isabelle Ruel24, PhD, Mark H. Sherman24, MD China: Long Jiang27, PhD, Luya Wang27, MD Croatia: Prof Željko Reiner28, PhD Czech Republic: Prof Vladimir Blaha29, PhD, Prof Richard Ceska30, PhD, Jana Dvorakova31, MD, Lubomir Dlouhy32, Pavel Horak30, PhD, Prof Vladimir Soska33, MD, Lukas Tichy34, PhD, Robin Urbanek35, MD, Prof Helena Vaverkova36, MD, Prof Michal Vrablik30, PhD, Stanislav Zemek37, MD, Lukas Zlatohlavek30, PhD Egypt: Prof Sameh Emil38, MD, Tarek Naguib39, Prof Ashraf Reda40, MD France: Sophie Béliard41, MD, Prof Eric Bruckert42, MD, Antonio Gallo42, PhD Greece: Prof Moses S. Elisaf43, PhD, Genovefa Kolovou44, PhD Israel: Hofit Cohen45, MD, Prof Ronen Durst46, MD, Prof Eldad J. Dann47, MD, Prof Avishay Elis48, MD, Osama Hussein49, MD, Prof Eran Leitersdorf50, MD, Daniel Schurr50, MD India: Nitika Setia51, PhD, Prof Ishwar C. Verma51, FRCP Iraq: Mohammed D. Alareedh52, PhD, Mutaz Al-Khnifsawi53, MD, Ali F. Abdalsahib Al-Zamili54, PhD, Sabah H. Rhadi55, PhD, Foaad K. Shaghee56 Italy: Prof Marcello Arca57, MD, Prof Maurizio Averna58, MD, Andrea Bartuli59, MD, Marco Bucci60, MD, Paola S. Buonuomo59, MD, Prof Paolo Calabrò61, PhD, Prof Sebastiano Calandra62, MD, Manuela Casula63, PhD, Prof Alberico L. Catapano63, PhD, Angelo B. Cefalù58, PhD, Arrigo F. Cicero64, PhD, Sergio D’Addato64, PhD, Laura D’Erasmo57, PhD, Alessia Di Costanzo57, PhD, Tommaso Fasano65, PhD, Marta Gazzotti63, MSc, Antonina Giammanco58, PhD, Gabriella Iannuzzo66, PhD, Anastasia Ibba67, MD, Emanuele A. Negri68, MD, Andrea Pasta8, MD, Chiara Pavanello69, PhD, Livia Pisciotta8, PhD, Claudio Rabacchi70, PhD, Carlo Ripoli71, MD, Tiziana Sampietro72, PhD, Francesco Sbrana72, MD, Fulvio Sileo73, MD, Patrizia Suppressa74, PhD, Prof Patrizia Tarugi70, PhD, Chiara Trenti68, MD, Maria G. Zenti75, PhD Japan: Mika Hori76,77, PhD Jordan: Mahmoud H. Ayesh78, MD Lebanon: Prof Sami T. Azar79, MD, Prof Fadi F. Bitar80, MD, Akl C. Fahed81, MD, Elie M. Moubarak82, MD, Prof Georges Nemer83, PhD Malaysia: Prof Hapizah Nawawi84, FRCPath Mexico: Ramón Madriz85, MD, Roopa Mehta86, PhD Netherlands: Arjen J. Cupido1, MD, Joep C. Defesche87, PhD, M. Doortje Reijman88, MD, Jeanine E. Roeters-van Lennep89, PhD, Prof Erik S. Stroes1, PhD, Albert Wiegman88, PhD, Linda Zuurbier87, PhD Oman: Khalid Al-Waili90, MD Pakistan: Fouzia Sadiq91, PhD Poland: Krzysztof Chlebus92, PhD Portugal: Prof Mafalda Bourbon93, PhD, Isabel M. Gaspar94, MD Serbia: Prof Katarina S. Lalic95, PhD Russia: Marat V. Ezhov96, DMSc, Prof Andrey V. Susekov97, PhD Slovenia: Urh Groselj98, PhD Taiwan: Prof Min-Ji Charng99, PhD Thailand: Prof Weerapan Khovidhunkit100, PhD Turkey: Melih Aktan101, MD, Prof Bulent B. Altunkeser102, MD, Sinan Demircioglu103, MD, Melis Kose104, PhD, Prof Cumali Gokce105, MD, Prof Osman Ilhan106, MD, Prof Meral Kayikcioglu107, MD, Leyla G. Kaynar108, MD, Prof Irfan Kuku109, MD, Erdal Kurtoglu110, MD, Harika Okutan111, MD, Osman I. Ozcebe112, MD, Zafer Pekkolay113, MD, Saim Sag114, PhD, Osman Z. Salcioglu115, MD, Prof Ahmet Temizhan116, MD, Mustafa Yenercag117, MD, Mehmet Yilmaz118, MD, Hamiyet Yilmaz Yasar119, MD Ukraine: Prof Olena Mitchenko120, MD United Kingdom: Alexander R. Lyons3, PhD, Christophe A. Stevens3, MSc United States: Julie A. Brothers121, MD, Lisa C. Hudgins122, MD, Christina Nguyen12, MBS Uzbekistan: Rano Alieva123, MD, Prof Aleksandr Shek123, MD Vietnam: Doan-Loi Do124,125, PhD, Ngoc-Thanh Kim124,125, MD, Hong-An Le126, MD, Thanh-Tung Le124, MD, Mai-Ngoc T. Nguyen124, PhD, Thanh-Huong Truong124,125, PhD
Affiliations
13. School of Medicine, University of Western Australia; Departments of Cardiology and Internal Medicine, Royal Perth Hospital, Perth, WA, Australia
14. Department of Pediatrics and Adolescent Medicine, Division of Pediatric Pulmonology, Allergology and Endocrinology, Medical University Vienna, Vienna, Austria
15. Department of Pediatrics, Academic Teaching Hospital, Landeskrankenhaus Feldkirch, Feldkirch, Austria
16. Third Department of Medicine and Karl Landsteiner Institute for Metabolic Diseases and Nephrology, Clinic Hietzing, Vienna Healthcare Group, Wokersbergernstrasse 1, 1130 Vienna, Austria
17. Department of Internal Medicine I, Medical University Innsbruck, Innsbruck, Austria
18. Salmaniya Medical Complex, Manama, Bahrain
19. Department of Hepatology and Center for Metabolic Diseases, University Hospital Leuven, Leuven, Belgium
20. Department of Internal Medicine, Centres hospitaliers Jolimont, Belgium
21. Department of Paediatrics and Center for Metabolic Diseases, University Hospital Leuven, Leuven, Belgium
22. Heart Institute (InCor) University of Sao Paulo and Hospital Israelita Albert Einstein, Sao Paulo, Brazil
23. Department of Medicine, University of British Columbia, Vancouver, British Columbia, Canada
24. Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada
25. Department of Medicine, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
26. Robarts Research Institute, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
27. Department of Atherosclerosis, Beijing Anzhen Hospital, Capital Medical University, The Key Laboratory of Remodeling–Related Cardiovascular Diseases, Ministry of Education, Beijing Institute of Heart, Lung and Blood Vessel Diseases, Beijing, China
28. Department of Internal medicine, University Hospital Centre Zagreb, School of Medicine University of Zagreb, Croatia
29. University Hospital Hradec Králové and Charles University, Faculty of Medicine in Hradec Králové, 3rd Department of Internal Medicine - Metabolism and Gerontology, Hradec Králové, Czech Republic
30. General University Hospital and 1st Faculty of Medicine, Charles University, 3rd Department of Internal Medicine – Endocrinology and Metabolism, Prague, Czech Republic
31. Department of Clinical Biochemistry, Haematology and Immunology, Na Homolce Hospital, Prague, Czech republic
32. Laboratory Medicine Center, Regional Hospital Liberec, Czech Republic
33. Department of Clinical Biochemistry, St. Anne’s University Hospital Brno, Czech Republic; 2nd Clinic of Internal Medicine, Faculty of Medicine, Masaryk University, Brno, Czech Republic
34. Center of Molecular Biology and Genetics, Department of Internal Medicine – Hematology and Oncology, University Hospital Brno and Faculty of Medicine, Masaryk University, Brno, Czech Republic
35. Lipid Clinic, Zlin, Czech Republic
36. Faculty of Medicine and Dentistry, Palacky University Olomouc and University Hospital Olomouc, Third Department of Internal Medicine – NRE, Olomouc, Czech Republic
37. Lipidová ambulance, Masarykovo namesti 155, Uherske Hradiste, Czech Republic
38. Military Academy, Cairo
39. Zagazig University, Zagazig
40. Menoufia University, Faculty of medicine, Cardiology department, Egypt
41. Department of Endocrinology and Nutrition, APHM, La Conception University hospital, Marseille, France
42. Department of Endocrinology and prevention of cardiovascular disease, Pitié-Salpêtrière University Hospital, Paris, France
43. Department of Internal Medicine, Medical School, University of Ioannina, Ioannina, Greece
44. Cardiometabolic Center, Lipid Clinic, LA apheresis Unit, Metropolitan Hospital, Athens, Greece
45. Bert W. Strassburger Lipid Center, the Chaim Sheba Medical Center, Tel Hashomer, Israel
46. Cardiology Dept. and Centre for Treatment and Prevention of Atherosclerosis, Hadassah Hebrew University Medical Centre, Jerusalem, Israel
47. Department of Hematology and Bone Marrow Transplantation, Rambam Health Care Campus, Haifa, Israel
48. Department of Internal Medicine, Beilinson Hospital, Rabin Medical Center, Petah Tikva, Israel
49. Internal Medicine department, Ziv Medical Center, Zfat, Israel
50. Internal Medicine department and Centre for Treatment and Prevention of Atherosclerosis, Hadassah Hebrew University Medical Centre, Jerusalem, Israel
51. Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi, India
52. Kufa University, college of medicine, Iraq
53. Al-Qadisiyah University, Faculty of Medicine, Department of Internal Medicine, Diwaniya City, Iraq
54. Al-Diwaniya teaching hospital, Iraq
55. Al-Hussain teaching hospital, Thi-Qar, Iraq
56. Jabir Ibn Hayyan Medical University, faculty of medicine, Iraq
57. Department of Translational and Precision Medicine, Sapienza University of Rome, Rome, Italy
58. Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties—University of Palermo, Italy
59. Rare Diseases and Medical Genetics, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
60. Internal Medicine Department, European Center of Excellence on Hypertension, Regional Reference Center for Dyslipidemias, SS Annunziata Hospital and University “G. d’Annunzio”, Chieti, Italy
61. Division of Cardiology, A.O.R.N. “Sant’Anna and San Sebastiano”, Caserta and Department of Translational Medical Sciences, University of Campania “Luigi Vanvitelli”, Naples, Italy
62. Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy
63. Department of Pharmacological and Biomolecular Sciences, University of Milan and IRCCS Multimedica, Milan, Italy
64. IRCCS Policlinico S. Orsola-Malpighi, Hypertension and cardiovascular risk factor research center, University of Bologna, Bologna, Italy
65. Clinical Chemistry and Endocrinology Laboratory, Department of Diagnostic Imaging and Laboratory Medicine, Azienda USL-IRCCS of Reggio Emilia, Reggio Emilia, Italy
66. Department of Clinical Medicine and Surgery, University “Federico II” of Naples, Naples, Italy
67. Pediatric Endocrine Unit and Newborn Screening Centre, Microcitemico Pediatric Hospital “A. Cao”, AO Brotzu, Cagliari, Italy
68. Department of Internal Medicine, Azienda USL-IRCCS of Reggio Emilia, Reggio Emilia, Italy
69. E. Grossi Paoletti Center, Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
70. Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
71. Pediatric Diabetology Unit, Pediatric and Microcytemia Department, AO Brotzu, Cagliari, Italy
72. Lipoapheresis Unit and Centre for Inherited Dyslipidaemias Fondazione CNR Toscana Gabriele Monasterio, Pisa, Italy
73. Division of Endocrinology, Papa Giovanni XXIII Hospital -ASST-PG23, Bergamo, Italy
74. Department of Internal Medicine and Rare Disease Centre “C. Frugoni” University Hospital of Bari, Bari, Italy
75. Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, UOC Endocrinologia, University Hospital of Verona, Verona, Italy
76. Department of Molecular Innovation in Lipidology, National Cerebral & Cardiovascular Center Research Institute, Japan
77. Department of Endocrinology, Research Institute of Environmental Medicine, Nagoya University, Japan
78. Department of Internal Medicine, King Abdullah University Hospital, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan
79. Department of Internal Medicine, American University of Beirut, Beirut, Lebanon
80. Division of Pediatric Cardiology, Department of Pediatrics, American University of Beirut Medical Center, Beirut, Lebanon
81. Division of Cardiology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA
82. National LDL Apheresis Center, Dahr El-Bashek Governmental University Hospital, Roumieh, Lebanon
83. Genomics and Translational Biomedicine Division, College of Health and Life Sciences, Hamad Bin Khalifa University, Doha, Qatar
84. Institute of Pathology, Laboratory and Forensic Medicine (I-PPerForM) and Faculty of Medicine, Universiti Teknologi MARA (UiTM), Sungai Buloh, Selangor, Malaysia
85. Servicio de Endocrinología, Unidad de Especialidades Médicas de la Secretaría de la Defensa Nacional, Ciudad de México, Mexico
86. Unidad de Investigación de Enfermedades Metabólicas, Instituto Nacional de Ciencias Médicas y Nutrición “Salvador Zubirán”, Mexico City, Mexico
87. Department of Clinical Genetics, Amsterdam UMC, Location AMC, University of Amsterdam, Amsterdam, the Netherlands
88. Department of Pediatrics, Amsterdam UMC, Location AMC, Amsterdam, the Netherlands
89. Department of Internal Medicine, Erasmus MC, Erasmus University Medical Center, Rotterdam, the Netherlands
90. Department of Clinical Biochemistry, Sultan Qaboos University Hospital, Muscat, Oman
91. Directorate of Research, Shifa Tameer-e-Millat University, Islamabad, Pakistan
92. 1st Department of Cardiology, Medical University of Gdansk, Poland and National Centre of Familial Hypercholesterolaemia in Gdańsk, Poland
93. Unidade de I&D, Grupo de Investigação Cardiovascular, Departamento de Promoção da Saúde e Prevenção de Doenças Não Transmissíveis, Instituto Nacional de Saúde Doutor Ricardo Jorge, Lisboa, Portugal; and BioISI – Biosystems & Integrative Sciences Institute, Faculdade de Ciências, Universidade de Lisboa, Lisboa, Portugal
94. Department of Cardiogenetics, Centro Hospitalar Lisboa Ocidental, Lisbon, Portugal and Lisbon Medical School, Genetics Laboratory, University of Lisbon, Lisbon, Portugal
95. Faculty of Medicine University of Belgrade, Clinic for Endocrinology, Diabetes and Metabolic Diseases, Belgrade, Serbia
96. National Medical Research Centre of Cardiology of Ministry of Health of the Russian Federation, Moscow, Russia
97. Academy for Postgraduate Medical Education, Faculty of Clinical Pharmacology and therapeutics, Ministry of Health, Moscow, Russian Federation, Russia
98. University of Ljubljana, Faculty of Medicine and UMC - University Children’s Hospital Ljubljana, Ljubljana, Slovenia
99. Division of Cardiology, Department of Medicine, Taipei Veterans General Hospital, Taipei, Taiwan
100. Endocrinology and Metabolism Unit, Department of Medicine, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Patumwan, Bangkok, Thailand
101. Istanbul University Istanbul Medical Faculty, Department of Hematology, Turkey
102. Selcuk University Medical Faculty, Department of Cardiology, Turkey
103. Necmettin Erbakan University Meram Medical Faculty, Department of Hematology, Turkey
104. Izmir Katip Çelebi University Medical Faculty Department of Pediatrics, Division of Inborn Errors of Metabolism, Turkey
105. Erdem Hospital, Department of Endocrinology and Metabolic Diseases, Turkey
106. Ankara University Medical Faculty Ibn-i Sina Hospital, Department of Hematology, Turkey
107. Ege University Medical Faculty, Department of Cardiology, Turkey
108. Erciyes University Medical Faculty, Department of Hematology, Turkey
109. Inonu University Medical Faculty, Department of Hematology, Turkey
110. Antalya Training and Research Hospital, Department of Hematology, Turkey
111. LOSANTE Children’s and Adult Hospital, Department of Hematology, Turkey
112. Hacettepe University Medical Faculty, Department of Hematology, Turkey
113. Dicle University Medical Faculty, Department of Endocrinology, Turkey
114. Uludag University Medical Faculty, Department of Cardiology, Turkey
115. Kanuni Sultan Suleyman Training and Research Hospital, Department of Pediatric Hematology, Turkey
116. Ankara State Hospital, Department of Cardiology, Turkey
117. Ordu University Medical Faculty, Department of Cardiology, Turkey
118. Sanko University, Department of Internal Diseases, Turkey
119. Tepecik Training and Research Hospital, Department of Endocrinology and Metabolism, Turkey
120. Dyslipidemia Department State Institution National Scientific Centre “The M.D. Strazhesko Institute of Cardiology National Academy of Medical Sciences of Ukraine”, Kyiv, Ukraine
121. Children’s Hospital of Philadelphia, Philadelphia, USA
122. The Rogosin Institute/Departments of Medicine and Pediatrics, Weill Cornell Medical College, New York, New York, USA
123. Republican Specialized Scientific and Practical Medical Center of Cardiology, Tashkent, Uzbekistan
124. Vietnam National Heart Institute, Bach Mai Hospital, Vietnam
125. Department of Cardiology, Hanoi Medical University, Vietnam
126. Vietnam National University, University of Medicine and Pharmacy, Vietnam
Data sharing statement
Data ownership for the data shared with the HICC registry remains the property of the individual contributors. Hence, the HICC Registry cannot share data with third parties without the respective contributors’ approval.
References
- 1.Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2014; 35: 2146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: Prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J 2015; 36: 560–5. [DOI] [PubMed] [Google Scholar]
- 3.Hu P, Dharmayat KI, Stevens CAT, et al. Prevalence of Familial Hypercholesterolemia among the General Population and Patients with Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020; 141: 1742–59. [DOI] [PubMed] [Google Scholar]
- 4.Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol 2020; 75: 2553–66. [DOI] [PubMed] [Google Scholar]
- 5.Di Taranto MD, Giacobbe C, Buonaiuto A, et al. A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia. J Clin Med 2020; 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017; 38: 2459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet (London, England) 2013; 381: 40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med 2020; 383: 711–20. [DOI] [PubMed] [Google Scholar]
- 9.Hegele RA, Borén J, Ginsberg HN, et al. Rare dyslipidaemias, from phenotype to genotype to management: a European Atherosclerosis Society task force consensus statement. Lancet Diabetes Endocrinol 2020; 8: 50–67. [DOI] [PubMed] [Google Scholar]
- 10.Bertolini S, Calandra S, Arca M, et al. Homozygous familial hypercholesterolemia in Italy: Clinical and molecular features. Atherosclerosis 2020; 312: 72–8. [DOI] [PubMed] [Google Scholar]
- 11.Alves AC, Alonso R, Diaz-Diaz JL, et al. Phenotypical, clinical, and molecular aspects of adults and children with homozygous familial hypercholesterolemia in iberoamerica. Arterioscler Thromb Vasc Biol 2020; : 2508–15. [DOI] [PubMed] [Google Scholar]
- 12.World Bank. Data World Bank national accounts. GNI per capita, Atlas method (current US$) - High income, Middle income, Low income https://data.worldbank.org/indicator/NY.GNP.PCAP.CD?locations=XD-XP-XM (accessed March 3, 2021). [Google Scholar]
- 13.Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur Heart J 2020; 41: 111–88. [DOI] [PubMed] [Google Scholar]
- 14.Sturm AC, Knowles JW, Gidding SS, et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol 2018; 72: 662–80. [DOI] [PubMed] [Google Scholar]
- 15.Alonso R, Díaz-Díaz JL, Arrieta F, et al. Clinical and molecular characteristics of homozygous familial hypercholesterolemia patients: Insights from SAFEHEART registry. J Clin Lipidol 2016; 10: 953–61. [DOI] [PubMed] [Google Scholar]
- 16.Stefanutti C, Pang J, Di Giacomo S, et al. A cross-national investigation of cardiovascular survival in homozygous familial hypercholesterolemia: The Sino-Roman Study. J Clin Lipidol 2019; 13: 608–17. [DOI] [PubMed] [Google Scholar]
- 17.D’Erasmo L, Minicocci I, Nicolucci A, et al. Autosomal Recessive Hypercholesterolemia: Long-Term Cardiovascular Outcomes. J Am Coll Cardiol 2018; 71: 279–88. [DOI] [PubMed] [Google Scholar]
- 18.Thompson GR, Blom DJ, Marais AD, Seed M, Pilcher GJ, Raal FJ. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur Heart J 2018; 39: 1162–8. [DOI] [PubMed] [Google Scholar]
- 19.Widhalm K, Benke IM, Fritz M, et al. Homozygous familial hypercholesterolemia: Summarized case reports. Atherosclerosis 2017; 257: 86–9. [DOI] [PubMed] [Google Scholar]
- 20.France M, Rees A, Datta D, et al. HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis 2016; 255: 128–39. [DOI] [PubMed] [Google Scholar]
- 21.Watts GF, Sullivan DR, Hare DL, et al. Integrated Guidance for Enhancing the Care of Familial Hypercholesterolaemia in Australia. Hear Lung Circ 2021; 30: 324–49. [DOI] [PubMed] [Google Scholar]
- 22.Luirink IK, Hutten BA, Greber-Platzer S, et al. Practice of lipoprotein apheresis and short-term efficacy in children with homozygous familial hypercholesterolemia: Data from an international registry. Atherosclerosis 2020; 299: 24–31. [DOI] [PubMed] [Google Scholar]
- 23.Beliard S, Gallo A, Duchêne E, et al. Lipoprotein-apheresis in familial hypercholesterolemia: Long-term patient compliance in a French cohort. Atherosclerosis 2018; 277: 66–71. [DOI] [PubMed] [Google Scholar]
- 24.Stefanutti C, Julius U, Watts GF, et al. Toward an international consensus—Integrating lipoprotein apheresis and new lipid-lowering drugs. J Clin Lipidol 2017; 11: 858–871.e3. [DOI] [PubMed] [Google Scholar]
- 25.EAS Familial Hypercholesterolaemia Studies Collaboration, Vallejo-Vaz AJ, De Marco M, et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries - The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 2018; 277: 234–55. [DOI] [PubMed] [Google Scholar]
- 26.Kayikcioglu M, Kuman-Tunçel O, Pirildar S, et al. Clinical management, psychosocial characteristics, and quality of life in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis in Turkey: Results of a nationwide survey (A-HIT1 registry). J Clin Lipidol 2019; 13: 455–67. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Omran T, Masana L, Kolovou G, et al. Real-World Outcomes with Lomitapide Use in Paediatric Patients with Homozygous Familial Hypercholesterolaemia. Adv Ther 2019; 36: 1786–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santos RD, Stein EA, Hovingh GK, et al. Long-Term Evolocumab in Patients With Familial Hypercholesterolemia. J Am Coll Cardiol 2020; 75: 565–74. [DOI] [PubMed] [Google Scholar]
- 29.Raal FJ, Hovingh GK, Blom D, et al. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol 2017; 5: 280–90. [DOI] [PubMed] [Google Scholar]
- 30.Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009; 302: 1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data ownership for the data shared with the HICC registry remains the property of the individual contributors. Hence, the HICC Registry cannot share data with third parties without the respective contributors’ approval.