

Abstract
Background
Accurate identification of brain tumor molecular subgroups is increasingly important. We aimed to establish the most accurate and reproducible ependymoma subgroup biomarker detection techniques, across 147 cases from International Society of Pediatric Oncology (SIOP) Ependymoma II trial participants, enrolled in the pan-European “Biomarkers of Ependymoma in Children and Adolescents (BIOMECA)” study.
Methods
Across 6 European BIOMECA laboratories, we evaluated epigenetic profiling (DNA methylation array); immunohistochemistry (IHC) for nuclear p65-RELA, H3K27me3, and Tenascin-C; copy number analysis via fluorescent in situ hybridization (FISH) and MLPA (1q, CDKN2A), and MIP and DNA methylation array (genome-wide copy number evaluation); analysis of ZFTA- and YAP1-fusions by RT-PCR and sequencing, Nanostring and break-apart FISH.
Results
DNA Methylation profiling classified 65.3% (n = 96/147) of cases as EPN-PFA and 15% (n = 22/147) as ST-ZFTA fusion-positive. Immunohistochemical loss of H3K27me3 was a reproducible and accurate surrogate marker for EPN-PFA (sensitivity 99%–100% across 3 centers). IHC for p65-RELA, FISH, and RNA-based analyses effectively identified ZFTA- and YAP—fused supratentorial ependymomas. Detection of 1q gain using FISH exhibited only 57% inter-center concordance and low sensitivity and specificity while MIP, MLPA, and DNA methylation-based approaches demonstrated greater accuracy.
Conclusions
We confirm, in a prospective trial cohort, that H3K27me3 immunohistochemistry is a robust EPN-PFA biomarker. Tenascin-C should be abandoned as a PFA marker. DNA methylation and MIP arrays are effective tools for copy number analysis of 1q gain, 6q, and CDKN2A loss while FISH is inadequate. Fusion detection was successful, but rare novel fusions need more extensive technologies. Finally, we propose test sets to guide future diagnostic approaches.
Keywords: brain tumors, biomarkers, Ependymoma, neuro-oncology, paediatric
Key Points.
We evaluated and cross-validated ependymoma biomarkers in a large prospective clinical trial cohort.
Accurate biomarker evaluation is critical to the success of clinical trials and patient care.
We propose core and core plus biomarker test sets for future molecular stratification.
Importance of the Study.
High-risk pediatric ependymoma has a poor prognosis and is devastating at relapse. Molecularly defined ependymoma types need to be accurately and reliably linked to biomarkers to predict clinical outcomes and design clinical trials. Here, we evaluated and cross-validated ependymoma biomarkers in a large prospective clinical trial cohort highlighting the importance of systematic evaluation of different methods. We provide evidence to guide test selection to support the molecular stratification of pediatric ependymoma and deliver insights into the rationalization of biomarkers for use in resource-limited settings.
The management of ependymoma in children and young adults is complex and the clinico-bio-pathological correlates of outcome remain poorly understood. Overall, prognosis remains poor in most patients and at relapse is dismal.1 Over half of the patients ultimately die from the disease and survivors face significant long-term sequelae. Half of the cases occur under the age of five years, a time in which the infant brain is undergoing rapid development and therefore at heightened risk of harm from medical interventions.2–7
Prior to the last decade, ependymomas were defined by anatomical location. However, the advent of the DNA methylation-based classification of ependymal tumors has improved our understanding by delineating multiple distinct tumor types and subtypes.2,3 The latest World Health Organisation Classification of Tumors of the CNS (WHO CNS5)3,8 now defines ependymoma by both anatomical and molecular characteristics. It is critical to facilitate the identification, prognostication, and stratification of ependymoma by linking these molecular tumor types to robustly validated biomarkers.
The extent of tumor resection represents the most reproducible clinical prognostic factor to date, with gross total resection correlated with improved survival in multiple studies.1,9–14 Despite this, many patients with gross total resection experience relapse, calling for validation of previously proposed biomarkers7,15–19 in a prospective multicentre clinical trial setting. Additionally, work is needed to understand the best way to measure the accuracy and reproducibility of these biomarkers.
An aim of the SIOP Ependymoma II clinical trial is to identify and validate prognostic biomarkers within the collaborative “Biomarkers of Ependymoma in Children and Adolescents (BIOMECA)” study.20 In this first prospective BIOMECA study, we compare molecular pathology methods across the first 147 consecutive cases from the SIOP Ependymoma II trial across 6 European laboratories. We aimed to determine the most accurate and reproducible methods for the analysis of predefined high-priority biomarkers in a clinical trial context, while also considering their application in resource-limited settings.
The methods evaluated include epigenetic profiling via EPIC 850K methylation arrays; immunohistochemistry (IHC) for nuclear p65-RELA, H3K27me3, and Tenascin-C (TNC); copy number analysis via fluorescent in situ hybridization (FISH) and MLPA (multiplex ligation-dependent probe amplification; 1q, CDKN2A), and MIP (molecular inversion probe; whole genome) and DNA methylation array (whole genome); the analysis of ZFTA- and YAP1-fusions by RT-PCR, sequencing, Nanostring, and break-apart FISH.
Methods
Patients and Clinical Specimens
The first 147 consecutively enrolled cases in the SIOP Ependymoma II clinical trial (trials.gov identifier: NCT02265770) from the United Kingdom, France, Spain, Czech Republic, and Ireland were included. All patients had a newly diagnosed ependymoma, confirmed by central neuropathological review according to the revised WHO 2016 classification.20 All analyses were performed on whole sections of formalin-fixed paraffin-embedded (FFPE) primary samples. Nottingham 2 Research Ethics Committee of the National Health Service Health Research Authority gave ethical approval for this work (Reference: 15/EM/0103). Written consent was obtained before study enrollment.
Evaluation of Methods and Techniques
To evaluate reproducibility, techniques were conducted across 6 European BIOMECA national reference laboratories (Supplementary Table 1). Each marker was analyzed for inter-center concordance using Cohen’s or Fleiss’s kappa. K values >0.41 indicate moderate agreement, values >0.61 indicate substantial agreement.21 DNA methylation profiles were used as criterion standard (CS) for specificity, sensitivity, and accuracy (Supplementary Methods). p65 immunohistochemistry for the diagnosis of supratentorial ependymoma with RELA fusions was assessed against a standard criterion comprised of DNA methylation profiling and identification of fusions via PCR and/or targeted sequencing approaches. Where no methylation profiling result was available cases were excluded from sensitivity and specificity measurements.
Fluorescent In Situ Hybridization (1q25, ZFTA-, and YAP1-Fusions)
FISH for chromosome 1q gain was performed using commercial 1q25/1p36 probes on FFPE sections (4μm) to manufacturers’ instructions in the United Kingdom and France (Supplementary Methods). FISH for ZFTA- and YAP1-fusions was performed on interphase nuclei as previously described.16
DNA Extraction and Copy Number Analysis
Extracted DNA was assessed for copy number variation via multiplex ligation-dependent probe amplification assays (MLPA; chromosome 1q and CDKN2A; UK), molecular inversion probe assays (MIP; whole genome; Bonn), and EPIC 850K methylation array (whole genome; DKFZ) (Supplementary Methods).
Immunohistochemistry (H3K27me3, TNC, and Nuclear p65-RelA)
Whole FFPE sections (4 μm) immunostained in 3 BIOMECA centers (Supplementary Table 1; Supplementary Methods). H3K27me3, TNC, and nuclear p65-RelA staining were double-scored as positive or negative.
EPIC 850K DNA Methylation Array
DNA methylation array was performed in the UK BIOMECA laboratory in conjunction with University College London Genomics, London (Supplementary Methods) and at the German Cancer Research Centre (DKFZ), Heidelberg as previously described.2,18 Array data were analyzed using the Heidelberg Brain Tumor Methylation Classifier (www.molecularneuropathology.org, version 12 (V12). A classifier score of 0.9 was applied as a cutoff for confident methylation class prediction.
RT-PCR, Sequencing, and Nanostring (ZFTA- and YAP-Fusion)
In the Como BIOMECA laboratory, RT-PCR was performed to detect common variants of ZFTA-RELA fusions (type 1, exon 2–2; and type 2, exons 3–2), YAP1-MAMLD1 (exons 5–3 or 6–2), ZFTA-MAML2 (exons 5–2), and ZFTA -YAP1 (exons 5–1) (Supplementary Methods). ZFTA-RELA fusion transcript was investigated by TaqMan real-time PCR. Data were analyzed with Sequencing Analysis Software (Applied Biosystems).
In the Bonn BIOMECA laboratory, presence of ZFTA- and YAP1-MAMLD1 fusions was examined by RT-PCR as previously described.22,23 Further molecular analysis of gene fusions was implemented with the Nanostring fusion panel. Four ZFTA-like classified cases were examined further with the Next-generation mRNA gene fusion panel using the TruSight Fusion Panel (Illumina, San Diego, CA, USA) as previously described.24 Sequencing data were analyzed by the Arriba tool (https://github.com/suhrig/arriba).25
Results
Case Cohort
One hundred and forty-seven tumors accrued from 2 national centers and 3 partner centers were included (Table 1). There was an even gender balance (males, n = 78, 53%, females n = 69, 47%). Median age at diagnosis was 40 months (Range: 5–225). A total of 76% were infratentorial (posterior fossa; PF, n = 111), 22% supratentorial (ST, n = 32), and 3% spinal (SP, n = 4). (Supplementary Table 4).
Table 1.
Case cohort and patient characteristics
|
PF = posterior fossa, ST = supratentorial, SP = spinal | |||||
|---|---|---|---|---|---|
| Overall, n = 147 | PF, n = 111 | SP, n = 4 | ST, n = 32 | ||
| Country | UK | 65 (44%) | 54 (49%) | 4 (100%) | 7 (22%) |
| France | 62 (42%) | 42 (38%) | 0 (0%) | 20 (62%) | |
| Spain | 16 (11%) | 12 (11%) | 0 (0%) | 4 (12%) | |
| Czech Republic | 3 (2%) | 2 (1.8%) | 0 (0%) | 1 (3.1%) | |
| Ireland | 1 (0.7%) | 1 (0.9%) | 0 (0%) | 0 (0%) | |
| Gender | M | 78 (53%) | 64 (58%) | 0 (0%) | 14 (44%) |
| F | 69 (47%) | 47 (42%) | 4 (100%) | 18 (56%) | |
| Age (months) | ≥36 | 80 (54%) | 51 (46%) | 4 (100%) | 25 (78%) |
| ≤36 | 67 (46%) | 60 (54%) | 0 (0%) | 7 (22%) | |
Methylation Profiling
Application of v12.5 of the Heidelberg Brain Tumor Methylation Classifier resulted in a calibrated score ≥0.9 in 91.1% (134/147) of all cases (PFA: 96/134, PFB: 10/134, ST-ZFTA: 22/134, ST-YAP1: 1/134, SP-MPE: 3/134, ST-PLAGL1: 2/134; Figure 1). Eleven cases (7.5%) did not reach the cutoff of 0.9. However, manual inspection of the t-SNE showed that 7/11 of these samples clustered within (1/11) or close to (6/11) the cluster of their best prediction (4/11: PFA, 2/11: ST-ZFTA, 1/11: ST-YAP1) (Supplementary Figure 1). In 2 cases (1.4%) no score was generated.
Figure 1.
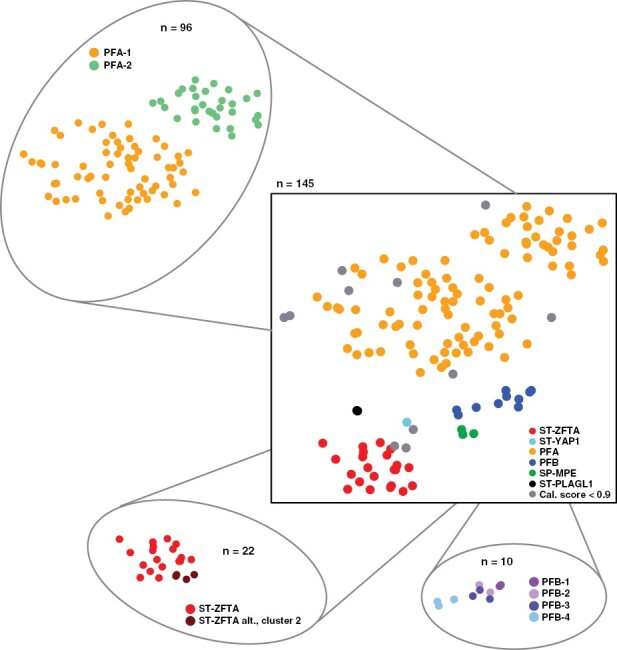
DNA-methylation profiling results. t-SNE plot visualizing DNA-methylation based clustering of the BIOMECA cohort. Central t-SNE: Molecular groups, satellite t-SNEs: molecular subgroup for PFA, ST-ZFTA and PFB. Samples are colorized according to the best available prediction.
PFA can be further stratified into 2 main subgroups (PFA-1/2) and 9 subtypes (1a – f and 2a – c).22 All 96 PFA cases also had a score ≥0.9 for 1 of the 2 PFA subgroups, with 67.8% (65/96) PFA-1, and 32.2% (31/96) PFA-2. These frequencies reflect those in the original study describing PFA subclassification (Figure 1).22 PFB subtyping resulted in confident prediction scores for 10/10 cases (PFB1: 2/10, PFB2: 2/10, PFB3: 3/10, PFB4: 3/10) (Figure 1).23 Recently, we have described further ST-ZFTA heterogeneity, with additional subgroups characterized by various histological appearances and alternative ZFTA-fusions.5 Out of the 22 patients predicted as ST-ZFTA, 18 had calibrated scores ≥0.9 for classic ST-ZFTA, which normally harbor ZFTA-RELA fusions, while 4 were stratified into the alternative ZFTA alt., cluster 2 (Figure 1).
Evaluation of Methods to Assess Copy Number Variation
DNA methylation assays.—
Methylation array-derived Copy Number Variation plots were analyzed with a focus on previously described copy number alterations within the respective molecularly defined types (Figure 2).2,5,22,23,26 Gain of chr. 1q was present in 4/22 ST-ZFTA, 14/96 PFA and 1/10 PFB, respectively (Figure 2; Supplementary Table 4). CDKN2A loss and chromothripsis on chr. 11 were restricted to ST-ZFTA, while chr. 22 loss was present in ST-ZFTA (6/22), PFB (7/10) and PFA (4/96), as previously described (Figure 2; Supplementary Table 4).2,5,22,23 As previously described,22 chr. 1q-gains were enriched in PFA1c, representing a particularly aggressive form of PFA (Supplementary Figure 1B; and Table 6).
Figure 2.

Evaluation of methods to assess copy number alterations.
Molecular inversion prob assays.—
High-resolution, quantitative MIP arrays revealed chr. 1q gain in 13/96 PFA, 1/10 PFB and 4/22 ST-ZFTA (Figure 2). MIP analysis identified 4/10 PFB and 1/3 SP-MPE as exhibiting whole chr.1 gain. Loss of CDKN2A and 13q, and chromothripsis at chr. 11, was not observed in PFA or PFB using MIP. Chromothripsis at chr.11 was observed in one ST-ZFTA, while loss of 13q was observed in one ST-ZFTA and one SP-MPE (Figure 2). CDKN2A loss was detected exclusively in 11 ST-ZFTA, with homozygous loss observed in 2/11 (#9-10, Figure 2; Supplementary Figure 2). Loss of 6q was observed in 4/96 PFA and 2/10 PFB. Co-occurrence of 1q/6q was documented in 2 cases (PFA, #3 and #12, Figure 2; Supplementary Figure 3). Loss at chr.22 was detected in 3/96 PFA, 2/10 PFB, and 5 ST-ZFTA.
MIP assays provided information regarding cytogenetic alterations/pattern and ploidy. A polyploid cytogenetic pattern was observed in 9/10 PFB and 3/3 SP-MPE (Figure 2) and demonstrated mostly numerical alterations (8/10 PFB). PFAs revealed 66 balanced, 19 structural, and 11 numerical cytogenetic alterations (n = 96; Figure 2). In contrast, ST-ZFTA showed a more equal mix of 7 balanced, 9 structural, and 6 numerical cytogenetic alterations (n = 22; Figure 2).
For 1q gain, MIP assays demonstrated sensitivity and specificity of 94.7% and 100%, respectively. Test accuracy was 99.2% compared with methylation-based assessments (Table 2).
Table 2.
Summary of Accuracy for Key BIOMECA Tests Under Evaluation Stratified by Center and Test Type. BIOMECA Tests With Equivocal or No Result Removed From Calculation. For Gain of Chromosome 1q Only Partial Chromosomal Gains Included in Analysis—Cases With Whole Chromosome Excluded as Known to Only be Detectable With MIP. Test Accuracy Calculated by True Positives and True Negatives Divided by All Test Results. Confidence Intervals Calculated Via the Exact Binomial Approach.
| Parameter | Criterion Standard (CS) | BIOMECA Test | Center | Positive CS and BIOMECA test (N) | Positive CS/ Negative BIOMECA test (N) | Sensitivity (95% CI) | Negative CS and BIOMECA test (N) | Negative CS and Positive BIOMECA test (N) | Specificity (95% CI) | No result (BIOMECA test) (N) | BIOMECA Accuracy (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PFA Diagnosis in confirmed PF ependymoma | MA | H3K27me3 IHC | Austria | 94 | 1 | 99.0 (94.3–100) | 10 | 0 | 100 (69.2–100) |
1 | 99.1 |
| Germany | 94 | 0 | 100 (96.2–100) | 8 | 0 | 100 (63.2–100) |
4 | 100 | |||
| UK | 94 | 1 | 99.0 (94.3–100) | 9 | 1 | 90.0 (55.5–99.8) | 1 | 98.1 | |||
| TNC IHC | Austria | 94 | 2 | 97.9 (92.7–99.8) | 6 | 4 | 60 (26.2–87.9) | 0 | 94.3 | ||
| France | 88 | 8 | 91.7 (84.2–96.3) | 7 | 3 | 70 (34.8–93.3) | 0 | 89.6 | |||
| Chromosome 1q Gain across all molecular diagnoses | MA | 1q FISH | UK | 9 | 6 | 60.0 (32.3–83.7 | 93 | 4 | 95.9 (89.8–98.9) | 17 | 91.1 |
| France | 15 | 3 | 83.3 (58.6–96.4) | 67 | 12 | 84.8 (75.0–91.9) | 32 | 84.5 | |||
| MLPA | UK | 18 | 1 | 94.7 (74–99.9) | 109 | 0 | 100 (96.7–100) | 1 | 99.2 | ||
| MIP | Germany | 18 | 1 | 94.7 (74–99.9) |
110 | 0 | 100 (96.7–100) | 0 | 99.2 | ||
| Diagnosis of ST EPN with gene fusions with RELA | MA plus targeted profiling for RELA | p65 IHC | Germany | 18 | 0 | 100 (81.5–100) | 6 | 0 | 100 (54.1–100) | 1 | 100 |
| France | 16 | 2 | 88.9 (65.6–98.6) | 6 | 1 | 85.7 (42.1–99.6) | 0 | 88 |
CS, Criterion Standard; MA, DNA Methylation Array; PF, Posterior Fossa; ST, Supratentorial; IHC, Immunohistochemistry; FISH, Fluorescent In Situ Hybridization; CI, Confidence Interval.
Multiplex-ligation dependent probe amplification (MLPA) Assay.—
Chr. 1q gain and CDKN2A loss were assessed using MLPA. After adjusting the analysis for whole chr.1 gain identified via MIP all 3 DNA-based techniques (MIP, methylation array, and MLPA) demonstrated 98.4% concordance for assessing chr. 1q gain (n = 134; Fleiss’ k = 0.958, p = 0) and 90.3% concordance for CDKN2A (n = 134; Fleiss’ k = 0.655, p = 0). MLPA did not yield a CDKN2A result in 8 cases. MIP and DNA methylation profiles for CDKN2A demonstrated 98.5% concordance (n = 134; Cohen’s k = 0.91, P < .0001).
MLPA had sensitivity and specificity of 94.7% and 100%, respectively for 1q gain. Test accuracy was 99.2% compared with methylation-based assessments (Table 2), identical to those for MIP.
Fluorescent In Situ Hybridisation.—
Chr. 1q FISH demonstrated low inter-center concordance (n = 134; 57.5%, Cohen’s k = 0.152, p = .0191). For cases classified using DNA methylation array, gain at chr. 1q was reported as 11.2% (15/134) and 21.6% (29/134) in the UK and France respectively. However, concordant observation was only reported in 8.2% (11/134; 3 PFA, 2 PFB, 6 ST-ZFTA; Figure 2). As similarly reported by Andreiuolo et al.,27 a significant number of cases, 26% (35/134, France) and 12.7% (17/134, United Kingdom) showed technical failure. The center in France determined 16.4% (22/134) UK cases to have failed compared to just 5.2% (7/134) UK cases determined to have failed in the UK center. This may reflect differences in tissue processing protocols in respective centers.
In addition to the discordant results and technical failures experienced via FISH, measures of accuracy were also poor. In the United Kingdom, FISH for 1q gain was associated with a sensitivity and specificity of 60.0% and 95.9% respectively, while the same measures in France were 83.3% and 84.8%. Accuracy was 91.1% in the UK and 84.5% in France (Table 2).
Evaluation of H3K27me3 and Tenascin-C Immunohistochemistry
In all PF cases with a classifier calibration score ≥ 0.9 H3K27me3 and TNC expression were assessed via IHC to investigate utility as a surrogate marker for PFA/PFB.
H3K27me3 expression demonstrated 92.5% inter-center concordance with agreement in 94.8% (91/96) PFA and 70% (7/10) PFB cases (n = 106; Fleiss’ k = 0.732, p < .000; Figure 3). Specifically, PFA demonstrated a loss of H3K27me3 expression, while PFB cases retained expression. Across 3 centers, loss of H3K27me3 had a sensitivity of 99%, 100%, and 99% and specificity of 100%, 100%, and 90% for diagnosing PFA in PF ependymoma. Test accuracy ranged from 98.1% to 100% (Table 2). Of 11 cases of PF ependymoma with classifier score <0.9, H3K27me3 staining identified 63.6% (7/11) as PFA. Seven of these eleven cases clustered close to the clusters of their respective best prediction, demonstrating that visual inspection of t-SNE or other dimensional reduction visualizations are useful in cases with ambiguous classification scores.
Figure 3.
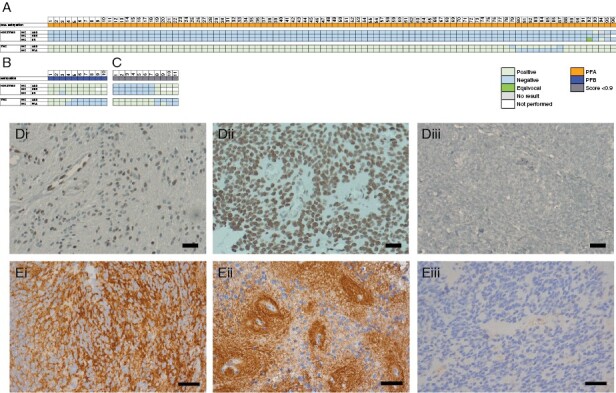
Evaluation of IHC for H3K27me3 and TNC as potential surrogate PFA markers. Posterior fossa tumors classed as PFA (A) and PFB (B) via DNA-methylation array (score ≥0.9) and the IHC result as assessed per center. (C) H3K27me3 and TNC results in cases not classified by DNA-methylation array. (D) Representative H3K27me3 staining in a PFA (i) and PFB (ii) case. (E) Representative TNC pericellular (i) and perivascular (ii) expression. (D-E) Magnification X40, scale bars 50μm. Representative negative controls (Diii, H3K27me3; Eiii, TNC).
TNC expression demonstrated a 91.7% inter-center concordance between the 2 centers performing the analysis (n = 106; Cohen’s k = 0.561, p < .000; Figure 3). Concordant-positive TNC staining was observed in 90.6% (87/96) of PFA and only 30% (3/10) of PFB. 60% (6/10) of PFB demonstrated concordant negative staining for TNC, while one case was discordant between centers. Positive staining for TNC as a predictor of PFA diagnosis in PF ependymoma had sensitivities of 97.9% and 91.7% and specificities of 60% and 70%. Test accuracy was 94.3% and 89.6%.
Assessing these markers together, concordant loss of H3K27me3 and simultaneous expression of TNC were observed in 86.5% (82/96) of PFA (Figure 3A).
Assessment of Methods Used for the Detection of ZFTA- and YAP1-Fused Ependymomas
The detection of molecularly defined ZFTA- and YAP-fused ependymomas was assessed using IHC, FISH, RT-PCR, sequencing, and Nanostring technology.
IHC for nuclear p65-RelA protein was assessed on 23 ZFTA- and YAP1-fused tumors, repeated across 2 centers (Figure 4A). ST-ZFTA demonstrated an 86.4% (19/22) inter-center concordance (n = 22; Cohen’s k = 0.582, p = .0058; Figure 4A). In total, 68.2% (16/22) of these cases demonstrated concordant positive staining for nuclear p65-RelA protein and in 3/22 cases, concordant negative staining. While only one ST-YAP1 case (case #23; Figure 4A) was identified in this cohort, this was negative for nuclear p65-RelA in both centers.
Figure 4.
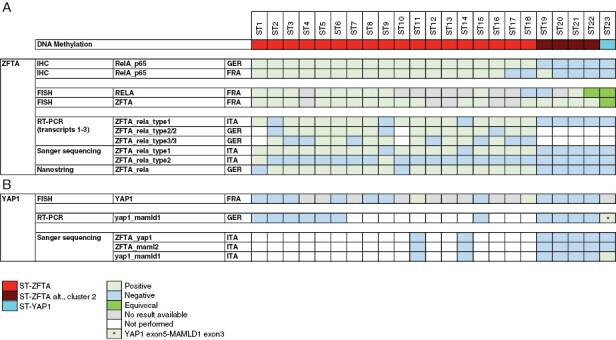
Comparison of methods used to assess ZFTA- (A) and YAP1- (B) fusions in supratentorial tumors.
FISH revealed 45.5% (10/22) ST-ZFTA cases with rearrangements at both the RELA and ZFTA loci (Figure 4A). One case gave an equivocal result, and 2 showed a rearrangement at the ZFTA, but not the RELA, locus. FISH failed in 9 cases assessed for rearrangement at the RELA locus, and 5 cases at the ZFTA locus. 41% (9/22) ST-ZFTA cases demonstrated concordance of positive nuclear p65-RelA IHC staining with simultaneous rearrangements at both the RELA and ZFTA loci via FISH.
RT-PCR with subsequent Sanger sequencing and Nanostring assay were used to confirm the presence of ZFTA-RELA fusions. Fusion transcripts were detected in 78.3% (18/22) molecularly defined cases in 2 centers. ZFTA-RELA transcripts 1–3 were detected in all classic ST-ZFTA cases (#1–18, Figure 4A) via DNA methylation array, with no such fusion transcripts detected in the 4 cases classified as ZFTA alt., cluster 2 (cases #19–22, Figure 4A). IHC for nuclear p65-RelA in the ZFTA alt. cases were concordantly negative in 3/4 cases in 2 centers. Interestingly, in all 4 ZFTA alt. cases, a rearrangement at the ZFTA locus was observed via FISH analysis, while rearrangement at the RELA locus was observed in one case, plus one equivocal result. RNA sequencing of these 4 ZFTA alt. cases identified fusions of ZFTA-NCOA2 in case #19, ZFTA-NCOA1 in case #20, and ZFTA-MAML2 in cases #21 and #22 (Supplementary Figure 3).
Case #23 (Figure 4), classified as a YAP1 tumor, demonstrated an equivocal result via FISH analysis for rearrangement at both the ZFTA and RELA locus, but was not positive for nuclear p65-RelA IHC and was negative for all ZFTA-RELA transcripts via RT-PCR, Sanger sequencing and Nanostring assay. FISH analysis for YAP1 failed in this case. However, RT-PCR detected a YAP1-MAMLD1 fusion (YAP1-exons 5/MAMLD1-exon3), which was confirmed by Sanger sequencing (Figure 4B).
When a combination of DNA methylation profile and fusion transcript analysis by targeted sequencing or PCR were combined as the criterion standard, p65 IHC had sensitivities of 100% and 88.9% and specificities of 85.7% and 100% in identifying supratentorial fusions which contained RELA as the partner with ZFTA (Table 2).
Discussion
Our study aimed to establish the most accurate and reproducible techniques for measuring key ependymoma biomarkers across 147 consecutive samples from SIOP Ependymoma II trial participants enrolled in the pan-European “Biomarkers of Ependymoma in Children and Adolescents (BIOMECA)” study. BIOMECA is the first pan-European study that has evaluated and cross-validated ependymoma biomarkers in a large prospective clinical trial cohort.
We were able to show that H3K27me3 IHC is both accurate and reproducible for the diagnosis of PFA ependymoma in a clinical trial setting. Additionally, DNA methylation profiling, MIP, and MLPA are all effective techniques for assessing key copy number changes in this disease. Combinations of IHC, PCR, and targeted sequencing are suitable for the delineation of fusion gene status in supratentorial ependymomas. Our study suggests that TNC is not a useful marker for PF ependymoma, and that FISH should be abandoned as a technique for the assessment of copy number status in ependymoma.
The integration of tumor-specific histopathology and molecular profiling is gaining pace, the updated 2021 WHO CNS5 classification of CNS tumors now lists ten molecularly defined types of ependymal tumors.3 Understanding the role of prognostic biomarkers for each of these entities is essential for the evolution of precision medicine and tailored therapy for ependymoma, and to understand how markers can be rationalized for use in both clinical trials and limited resource settings.
All cases included in this study were diagnosed according to local neuropathology review before confirmation by central review and DNA methylation profiling.2,3,28,29 DNA methylation profiling resulted in a confident diagnosis of a molecularly defined type for 91.1% of all patients. For the 7.5% of cases unable to be confidently profiled, manual inspection of the t-SNE plots placed 7/11 cases close to their best prediction and only 2/147 cases could not be profiled, highlighting DNA methylation profiling as the criterion standard for this assessment. The importance of DNA methylation profiling was also highlighted by the 2 supratentorial tumors diagnosed as ependymoma, which clustered with neuroepithelial tumors with PLAGL1 fusions,30 demonstrating that a classifier is a tool under continuous development.
A global reduction of H3K27me3 expression in EPN-PFA is used as a surrogate marker for this molecular group31–33 and is now recommended as an essential diagnostic criterion in the updated 2021 WHO CNS5 classification.3 Our data aligns strongly with this recommendation. 95.7% of PFA cases demonstrated concordant loss of H3K27me3 expression across 3 centers. The reproducibly high sensitivity and specificity across the 3 centers also robustly support the use of this marker for the diagnosis of PFA tumors. H3K27me3 expression represents a useful biomarker for settings with limited resources or where methylation profiling is not possible. In contrast, previous suggestions that TNC expression is a surrogate marker for PFA ependymomas34–36 are not confirmed by our data. TNC expression was found in a substantial fraction of PFB tumors and was associated with low specificity.
RELA encodes the p65-RelA protein which shows nuclear accumulation upon pathological activation of the NFkB signaling pathway.18,26 Other studies have investigated the potential of IHC to predict ZFTA-RELA fusion status in comparison to using RT-PCR and Sanger sequencing or Nanostring.37,38 Detection of the fusion is an essential diagnostic criterion as per 2021 WHO CNS5 classification of supratentorial ZFTA-fused ependymomas, and p65-RelA IHC is now listed as a desirable marker as part of the diagnostic pathway.3 Our data demonstrate significant inter-center concordance using IHC and corresponding directly to those cases where fusion transcripts were detected using RT-PCR and sequencing or Nanostring, and additionally classified as ST-ZFTA using DNA methylation array. We demonstrated sensitivities of 88.9 and 100% for the identification of fusions with RELA as a partner to ZFTA using p65 IHC, although confidence intervals are wide in view of the low number of these cases. Where atypical fusions with partners other than RELA are present, p65-RelA IHC cannot help in predicting molecular class. However, as in most cases ZFTA is fused to RELA, the detection of nuclear accumulation of p65-RelA by IHC represents an easy, cost-effective, and reliable surrogate marker for most ST-ZFTA cases.39
FISH has long been established in most diagnostic pathology laboratories around the world for the assessment of genomic rearrangements.40 Here we found that the break-apart FISH technique failed to detect RELA and ZFTA fusions in 9/22 (41%) cases classed as ST-ZFTA. This may be a consequence of both RELA and ZFTA being located only 1.9Mbp apart on chromosome band 11q13,18 making the interpretation and subsequent analysis difficult. This failure rate is higher than that observed by Pages et al.(38), where approximately 10% of supratentorial cases did not yield a result using break-apart FISH. Similarly, although only one YAP1-fused ependymoma case was identified in this study, FISH failed to detect the fusion. We do not recommend FISH as a primary approach for classifying supratentorial ependymomas.
Chromosome 1q gain and CDKN2A loss have been proposed as independent markers of worse prognosis in pediatric PFA and ST-ZFTA ependymoma respectively.7,12,15,34,41–43 Traditionally, FISH has been the primary technique to assess chromosome 1q gain however, the optimal method for detection of 1q gain has been debated, with reports that 20% of cases cannot be assessed by FISH on FFPE tissue.34 This study does not support FISH as a reliable method for assessment of 1q gain owing to the high failure rate observed by 2 centers (13%–26%) and low (57%) inter-center concordance. Additionally, the sensitivity associated with FISH in detecting 1q gain when compared to methylation-based techniques was just 60% and 83.3%. While FISH protocols may be optimized and standardized within a center, variation of tissue processing between centers may significantly impact systematic biomarker assessments. This cannot be avoided in a study where tissue samples are collected in multiple international centers, explaining some of the discordance observed in this study.
In assessing chromosome 1q gain and CDKN2A loss, we compared copy number analysis via 3 molecular methodologies: DNA methylation array, MIP, and MLPA. Concordance for the identification of gain at chromosome 1q (98.4%) and loss of CDKN2A (90.3%) using these 3 methods was high. Furthermore, whole genome-wide copy number analysis was enabled with DNA methylation array and MIP. Sensitivity, specificity, and accuracy for both MIP and MLPA were comparably high when compared with DNA methylation profiling and based on this measure alone all 3 techniques are appropriate for the identification of 1q gain in PFA ependymoma. However, the high-resolution, genome-wide MIP technology revealed quantitative copy number information in all tumors enrolled in this study. This technology works with DNA input down to 20ng in contrast to DNA methylation profiling which needs significantly more. Additionally, in contrast to MIP, DNA methylation-based CN calls cannot be adjusted for diploidy. This adjustment, however, is mandatory for exact copy number calls, particularly in the complex polyploid genomes occurring in PFB and MPE. DNA methylation array analysis was unable to identify some chromosomal losses in complex genomes such as PFB but, critically, was able to reliably detect gains of chromosome 1q in PFA tumors. Similarly, the distinction between hemizygous and homozygous CDKN2A deletion is more secure after diploid correction, however, CDKN2A deletions were correctly identified in all samples using DNA methylation. The ability of DNA methylation profiling to reliably detect 1q gain and CDKN2A deletions, alongside its wider availability, makes it the preferred tool for molecular stratification in urgently needed clinical trials in poor outcome ependymoma subgroups. MLPA is a similar DNA-based method as MIP; however, it scores only around 20 probes compared to high-resolution MIP with more than 300 000 probes distributed over the genome. In this study, no technical failures were reported for MIP compared to a small number with MLPA (6.1%) and DNA methylation array (2%).
From our data, we propose the concept of applying techniques in a CORE and CORE Plus model, which aligns with the 2021 WHO CNS5 classification’s essential and desirable criteria for ependymoma diagnosis. CORE tests represent those that can currently be used to stratify and inform clinical trials and diagnosis and include immunohistochemistry and DNA methylation profiling. CORE Plus tests have additional advantages for challenging cases and for use in the research setting and comprise of MIP and RNA-NGS sequencing.
All ependymoma subgroups can be profiled using CORE techniques. Using IHC initially as recommended enables a cost-effective, well-established, and available technique. IHC can be reliably used as a surrogate means to detect ZFTA-RELA-fused (nuclear p65-RelA) and PFA (loss of H3K27me3) ependymomas. There were very few cases where IHC did not align with DNA methylation array. In these situations, we would recommend accepting the DNA methylation result given this is the criterion standard (Supplementary Figure 5). DNA methylation profiling represents a powerful tool for classification of ependymoma where histopathological features may converge on more than one possible molecularly defined tumor type, examples include tumors with BCOR internal tandem duplication, astroblastomas with MN1 alteration or tumors with PLAGL1 fusions.5,30,44 Methylation array also provides important prognostic information about copy number changes and therefore should be combined with IHC as a CORE test. However, it is recognized that access to DNA methylation arrays varies, especially in low and middle-income countries, so continued development of techniques not based on complex molecular methodologies to confidently classify brain tumors is vital.
While CN information, particularly the important ependymoma biomarkers 1q gain and CDKN2A, can be reliably obtained with DNA methylation array, in cases where there are complex cytogenetic patterns or paucity of tissue, high-resolution, quantitative MIP arrays can be utilized. However, access is currently less widely available to centers that may participate in clinical trials of the future. Therefore, while MIP is not part of our core set recommendations, in the instances outlined above it can be used as a non-mandatory CORE Plus assessment. Similarly, if rare fusion events must be detected in supratentorial ependymomas, RNA-NGS sequencing should also be included as core plus assessment.
Biological systems are complex and multidimensional. Measuring multiple biomarkers and taking a variability-reductionist approach to interpreting outcomes will provide better information for future treatment stratification. Considering the relative rarity of ependymoma, it is of paramount importance that future prospective trials utilize a standardized and reliable set of diagnostic and prognostic markers. The BIOMECA study makes recommendations for standardizing ependymoma biomarkers across clinical trials in the years to come and provides insight into how core and core plus approaches may assist the selection of appropriate tests for use in resource-limited settings.
Supplementary Material
Acknowledgments
We thank clinical research teams from contributing CCLG centers and Tissue Bank for sample provision and clinical information, and Birmingham CRUK clinical trials unit for study support. We thank Monika Mauermann for technical assistance. We thank the Microarray Unit of the Genomics & Proteomics Core Facility, German Cancer Research Center (DKFZ) and University College London (UCL) Genomics, London, for providing excellent services regarding methylation arrays.
Prior presentation: Presented as oral abstract at International Society for Paediatric Neuro-Oncology meeting, Hamburg, Germany, 12-15 June 2022. https://doi.org/10.1093/neuonc/noac079.160
Contributor Information
Rebecca J Chapman, Children’s Brain Tumour Research Centre, University of Nottingham, Nottingham, UK.
David R Ghasemi, Hopp Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany; Division of Pediatric Neuro-oncology, German Cancer Research Center (DKFZ) and German Consortium for Translational Cancer Research (DKTK), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Felipe Andreiuolo, Department of Neuropathology, DGNN Brain Tumor Reference Center, University of Bonn, Bonn, Germany; Instituto Estadual do Cerebro Paulo Niemeyer, Rio de Janerio, Brazil; IDOR Institute, Rio de Janeiro, Brazil.
Valentina Zschernack, Department of Neuropathology, DGNN Brain Tumor Reference Center, University of Bonn, Bonn, Germany.
Arnault Tauziede Espariat, Departement de Neuropathologie, Hopital Sainte-Anne, Paris, France; INSERM Unit 981 and Department of Pediatric and Adolescent Oncology, Gustave Roussy, Villejuif, France.
Francesca R Buttarelli, Department of Radiological, Oncological and Anatomo-Pathological Sciences, Sapienza University of Rome, Rome, Italy.
Felice Giangaspero, Department of Radiological, Oncological and Anatomo-Pathological Sciences, Sapienza University of Rome, Rome, Italy; IRCCS Neuromed, Pozzilli, Italy.
Jacques Grill, INSERM Unit 981 and Department of Pediatric and Adolescent Oncology, Gustave Roussy, Villejuif, France.
Christine Haberler, Division of Neuropathology and Neurochemistry, Department of Neurology, Medical University of Vienna, Vienna, Austria.
Simon M L Paine, Department of Neuropathology, Nottingham University Hospital, Nottingham, UK.
Ian Scott, Department of Neuropathology, Nottingham University Hospital, Nottingham, UK.
Thomas S Jacques, Developmental Biology and Cancer Programme, UCL GOS Institute of Child Health, London, UK; Department of Histopathology, Great Ormond Street Hospital for Children, London, UK.
Martin Sill, Hopp Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany.
Stefan Pfister, Hopp Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany; Division of Pediatric Neuro-oncology, German Cancer Research Center (DKFZ) and German Consortium for Translational Cancer Research (DKTK), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
John-Paul Kilday, Children’s Brain Tumour Research Network (CBTRN), Royal Manchester Children’s Hospital, Manchester, UK; The Centre for Paediatric, Teenage and Young Adult Cancer, Institute of Cancer Sciences, University of Manchester, Manchester, UK.
Pierre Leblond, Institute of Hematology and Pediatric Oncology (IHOPe), Leon Berard Comprehensive Cancer Center, Lyon, France.
Maura Massimino, Paediatric Unit, Fondazione Istituto Di Ricovero e Cura a Carattere Scientifico, Istituto Nazionale dei Tumori, Milano, Italy.
Hendrik Witt, Hopp Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany; Division of Pediatric Neuro-oncology, German Cancer Research Center (DKFZ) and German Consortium for Translational Cancer Research (DKTK), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Piergiorgio Modena, Genetics Unit, Pathology Department, Ospedale S. Anna, Como, Italy.
Pascale Varlet, Departement de Neuropathologie, Hopital Sainte-Anne, Paris, France; INSERM Unit 981 and Department of Pediatric and Adolescent Oncology, Gustave Roussy, Villejuif, France.
Torsten Pietsch, Department of Neuropathology, DGNN Brain Tumor Reference Center, University of Bonn, Bonn, Germany.
Richard G Grundy, Children’s Brain Tumour Research Centre, University of Nottingham, Nottingham, UK.
Kristian W Pajtler, Children’s Brain Tumour Research Centre, University of Nottingham, Nottingham, UK; Hopp Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany; Division of Pediatric Neuro-oncology, German Cancer Research Center (DKFZ) and German Consortium for Translational Cancer Research (DKTK), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Timothy A Ritzmann, Children’s Brain Tumour Research Centre, University of Nottingham, Nottingham, UK.
Funding
SIOP Ependymoma II clinical trial: Cancer Research UK (CRUK) (UK). BIOMECA: Children with Cancer UK (CwCUK). RJC: CRUK and CwCUK. TAR: National Institute for Health Research Funded Academic Clinical Lecturer and Fighting Ependymoma. KWP and DRG: “Ein Kiwi gegen Krebs.” DRG: German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes).
Conflict of interest statement
The authors declare no conflict of interest.
Author Contribution
Experimental design: RJC, FA, FG, JG, CH, JPK, PB, MM, HW, PM, TP, PV, RGG, KWP, and TAR. Implementation: RJC, FA, FB, ATE, CH, PM, TP, PV, HW, SMLP, IS, TJ, and TAR. Data analysis: RJC, DRG, TAR, VZ, FA, SMLP, ATE, CH, PM, PV, and MS. Data interpretation: RJC, DRG, TAR, RGG, TP, KWP. Manuscript preparation: RJC, TAR, DRG, RGG, TP, and KWP
Dedication
We dedicate this paper in memoriam to Professor Felice Giangasparo. His tragically early demise has taken from us a perspicacious, talented Clinician Scientist and Humanitarian. He will be much missed by colleagues, friends and family.
References
- 1. Ritzmann TA, Rogers HA, Paine SML, et al. A retrospective analysis of recurrent pediatric ependymoma reveals extremely poor survival and ineffectiveness of current treatments across central nervous system locations and molecular subgroups. Pediatr Blood Cancer. 2020;67(9):67–79. [DOI] [PubMed] [Google Scholar]
- 2. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. WHO Classification of Tumours Editorial Board. Central Nervous System Tumours, WHO Classification of Tumours, 5th Edition, Volume 6. 5th Edition. (Cree IA, Lokuhetty D, Peferoen LAN, White VA, eds.). Lyon :International; Agency for Research on Cancer; 2021. [Google Scholar]
- 4. Arabzade A, Zhao Y, Varadharajan S, et al. ZFTA–RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov. 2021;11(9):2200–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng T, Ghasemi DR, Okonechnikov K, et al. Cross-species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion–positive supratentorial ependymomas. Cancer Discov. 2021;11(9):2230–2247. [DOI] [PubMed] [Google Scholar]
- 6. Kupp R, Ruff L, Terranova S, et al. ZFTA translocations constitute ependymoma chromatin remodeling and transcription factors. Cancer Discov. 2021;11(9):2216–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Godfraind C, Kaczmarska JM, Kocak M, et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. 2012;124(2):247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ellison DW, Aldape KD, Capper D, et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 2020;30(5):863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grill J, le Deley MC, Gambarelli D, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: A multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19(5):1288–1296. [DOI] [PubMed] [Google Scholar]
- 10. Grundy RG, Wilne SA, Weston CL, et al. Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. Eur J Cancer. 2010;46:120–123.
- 11. Merchant TE, Li C, Xiong X, et al. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009;10(3):258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Massimino M, Miceli R, Giangaspero F, et al. Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro Oncol. 2016;18(10):1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sato M, Gunther JR, Mahajan A, et al. Progression-free survival of children with localized ependymoma treated with intensity-modulated radiation therapy or proton-beam radiation therapy. Cancer. 2017;123(13):2570–2578. [DOI] [PubMed] [Google Scholar]
- 14. Zapotocky M, Beera K, Adamski J, et al. Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer. 2019;125(11):1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Korshunov A, Witt H, Hielscher T, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28(19):3182–3190. [DOI] [PubMed] [Google Scholar]
- 16. Pages M, Lacroix L, Tauziede-Espariat A, et al. Papillary glioneuronal tumors: Histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun. 2015;3:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Cottman M, Schiffman JD.. Molecular inversion probes: A novel microarray technology and its application in cancer research. Cancer Genet. 2012;205(7–8):341–355. [DOI] [PubMed] [Google Scholar]
- 18. Pietsch T, Wohlers I, Goschzik T, et al. Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol. 2014;127(4):609–611. [DOI] [PubMed] [Google Scholar]
- 19. Witt H, Gramatzki D, Hentschel B, et al. DNA methylation-based classification of ependymomas in adulthood: Implications for diagnosis and treatment. Neuro Oncol. 2018;20(12):1616–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leblond P, Massimino M, English M, et al. Toward improved diagnosis accuracy and treatment of children, adolescents, and young adults with ependymoma: The International SIOP Ependymoma II Protocol. Front Neurol. 2022;13:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watson PF, Petrie A.. Method agreement analysis: A review of correct methodology. Theriogenology. 2010;73(9):1167–1179. [DOI] [PubMed] [Google Scholar]
- 22. Pajtler KW, Wen J, Sill M, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018;136(2):211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cavalli FMG, Hübner JM, Sharma T, et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. 2018;136(2):227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zschernack V, Jünger ST, Mynarek M, et al. Supratentorial ependymoma in childhood: More than just RELA or YAP. Acta Neuropathol. 2021;141(3):455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uhrig S, Ellermann J, Walther T, et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021;31(3):448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parker M, Mohankumar KM, Punchihewa C, et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature. 2014;506(7489):451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andreiuolo F, Ferreira C, Puget S, Grill J.. Current and evolving knowledge of prognostic factors for pediatric ependymomas. Future Oncol. 2013;9(2):183–191. [DOI] [PubMed] [Google Scholar]
- 28. Neumann JE, Spohn M, Obrecht D, et al. Molecular characterization of histopathological ependymoma variants. Acta Neuropathol. 2020;139(2):305–318. [DOI] [PubMed] [Google Scholar]
- 29. Pickles JC, Stone TJ, Jacques TS.. Methylation-based algorithms for diagnosis: Experience from neuro-oncology. J Pathol. 2020;250(5):510–517. [DOI] [PubMed] [Google Scholar]
- 30. Sievers P, Henneken SC, Blume C, et al. Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol. 2021;142(5):827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bayliss J, Mukherjee P, Lu C, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med. 2016;8(366):8–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Panwalkar P, Clark J, Ramaswamy V, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017;134(5):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang RR, Kuo JS.. Reduced H3K27me3 is a new epigenetic biomarker for pediatric posterior fossa ependymomas. Neurosurgery. 2017;81(1):N7–N8. [DOI] [PubMed] [Google Scholar]
- 34. Andreiuolo F, le Teuff G, Bayar MA, et al. Integrating Tenascin-C protein expression and 1q25 copy number status in pediatric intracranial ependymoma prognostication: a new model for risk stratification. PLoS One. 2017;12(6):e0178351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Araki A, Chocholous M, Gojo J, et al. Chromosome 1q gain and tenascin-C expression are candidate markers to define different risk groups in pediatric posterior fossa ependymoma. Acta Neuropathol Commun. 2016;4(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gessi M, Diomedi Camassei F, Elefante G, et al. Role of immunohistochemistry in the identification of supratentorial C11ORF95-RELA fused ependymoma in routine neuropathology. Am J Surg Pathol. 2019;43(1):56–63. [DOI] [PubMed] [Google Scholar]
- 38. Pagès M, Pajtler KW, Puget S, et al. Diagnostics of pediatric supratentorial RELA ependymomas: Integration of information from histopathology, genetics, DNA methylation and imaging. Brain Pathol. 2019;29(3):29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaunmuktane Z, Capper D, Jones DTW, et al. Methylation array profiling of adult brain tumours: Diagnostic outcomes in a large, single centre. Acta Neuropathol Commun. 2019;7(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andreiuolo F, Mazeraud A, Chrétien F, Pietsch T.. A Global view on the availability of methods and information in the neuropathological diagnostics of CNS tumors: Results of an international survey among neuropathological units. Brain Pathol. 2016;26(4):551–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dyer S, Prebble E, Davison V, et al. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Am J Pathol. 2002;161(6):2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jünger ST, Mynarek M, Wohlers I, et al. Improved risk-stratification for posterior fossa ependymoma of childhood considering clinical, histological and genetic features – a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol Commun. 2019;7(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ritzmann TA, Chapman RJ, Kilday JP, et al. SIOP ependymoma I: final results, long-term follow-up, and molecular analysis of the trial cohort—A BIOMECA Consortium Study. Neuro Oncol. 2022;24(6):936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fukuoka K, Kanemura Y, Shofuda T, et al. Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun. 2018;6(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.