

Abstract
Genome engineering technologies are powerful tools in cell-based immunotherapy to optimize or fine-tune cell functionalities. However, their use for multiple gene edits poses relevant biological and technical challenges. Short hairpin RNA (shRNA)-based cell engineering bypasses these criticalities and represents a valid alternative to CRISPR-based gene editing. Here, we describe a microRNA (miRNA)-based multiplex shRNA platform obtained by combining highly efficient miRNA scaffolds into a chimeric cluster, to deliver up to four shRNA-like sequences. Thanks to its limited size, our cassette could be deployed in a one-step process along with all the CAR components, streamlining the generation of engineered CAR T cells. The plug-and-play design of the shRNA platform allowed us to swap each shRNA-derived guide sequence without affecting the system performance. Appropriately choosing the target sequences, we were able to either achieve a functional KO, or fine-tune the expression levels of the target genes, all without the need for gene editing. Through our strategy we achieved easy, safe, efficient, and tunable modulation of multiple target genes simultaneously. This approach allows for the effective introduction of multiple functionally relevant tweaks in the transcriptome of the engineered cells, which may lead to increased performance in challenging environments, e.g., solid tumors.
Keywords: MT: Oligonucleotides: Therapies and Applications, miRNA, shRNA, multiplex, platform, gene expression modulation, knockdown, adoptive cell therapy, CAR, CAR T cell
Graphical abstract
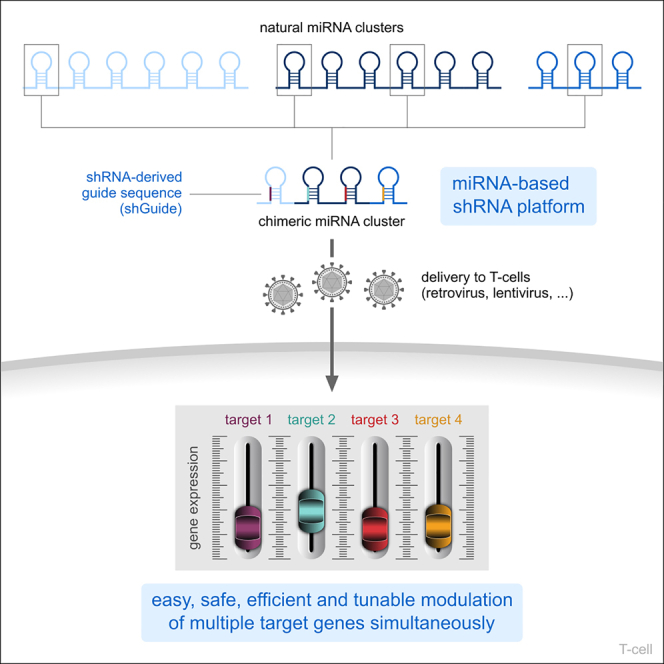
Rossi and colleagues developed a miRNA-based shRNA platform combining different natural miRNA scaffolds into a chimeric cluster and substituting the natural miRNAs with shRNA-derived guide sequences. The system allows for easy, safe, efficient, and tunable modulation of multiple target genes for adoptive cell therapy without the need for gene editing.
Introduction
In recent years, cellular immunotherapy has redrawn the treatment landscape for a growing number of human cancers. In particular, T cells engineered to express a chimeric antigen receptor (CAR) enabling antigen recognition, downstream signaling, and costimulatory activation warranted remarkable responses across different patient populations with hematological malignancies.1,2,3,4,5,6 However, these initial successes did not translate into solid tumor indications.7,8 Multiple mechanisms contribute to the poor clinical responses observed. The immunosuppressive properties of the tumor microenvironment (TME) impose metabolic pressure on the CAR T cells and promote the formation of dysfunctional effector cells and regulatory T cells (Treg).9,10 Next to those issues related to the TME, CAR T cells often exhibit poor long-term persistence and exhaustion, which limit the duration of potential responses.11 In addition, the often poor quality and quantity of T cells and the time, expense, and demanding logistics associated with manufacturing of autologous T cell products further limit the applicability of this approach to a broad patient population. Allogeneic CAR T cells may allow to circumvent some of the outstanding limitations that hamper the autologous approach; however, further challenges due to alloreactivity arise. Expression of the endogenous T cell receptor (TCR) and human leukocyte antigen (HLA) molecules on the surface of allogeneic T cells must be dealt with to prevent graft-versus-host disease and allorejection by the host immune system, respectively. To overcome all the above-mentioned issues, CAR T cells require multiple edits that go beyond the deployment of the CAR itself. The simultaneous downregulation of several immune checkpoint inhibitor receptors (e.g., PD-1, CTLA-4, LAG-3, TIM-3, and TIGIT) may be necessary to improve CAR T cell persistence and activity.12 In addition, the successful generation of allogeneic therapies relies on the tight control of the expression of surface molecules involved in alloreactivity and allorejection (TCR, major histocompatibility complex class I [MHC class I], MHC class II) or of their regulators (e.g., CIITA).13 The ability to modulate the levels of multiple target genes at once is therefore crucial to develop the next generation of adoptive cell therapies.
Gene editing technologies such as meganucleases,14 TALENs,15,16 megaTALs, and CRISPR-Cas917,18 granted unprecedented possibilities in T cell engineering. CRISPR-Cas9 technology, in particular, has been already explored for the editing of multiple genomic sites in T cells.19,20,21,22,23,24 Still, their use for multiple simultaneous gene edits poses relevant biological and technical challenges. The co-occurrence of all the desired edits within the same cell wanes for high-grade multiplexing,25 with heavy implications on the manufacturability of multi-edited products. The competition for a dwindling pool of endonucleases as the number of gRNAs scales up26 may contribute to this phenomenon, giving raise to unpredictable patterns of gRNA efficiency in a multiplex context. Furthermore, nuclease-based genome-editing strategies carry the risk of accidental off-target cleavages, mutational events, and chromosomal rearrangements, which is exacerbated by the simultaneous introduction of multiple edits.27 The consequence is the introduction of unintended, irreversible off-target genetic alterations in the product cells. If the risk of such off-target effects may be mitigated through a variety of strategies,28,29,30,31,32,33,34 dealing with mutational events and chromosomal rearrangements represents a bigger challenge. Indeed, such unwelcome eventualities are inherent to the biological mechanism behind gene editing itself, which relies on double-strand breaks (DSBs) and on the error-prone DSB repair machinery. CRISPR-Cas9 cleavage has indeed been linked to gross chromosomal aberrations in both embryos35,36,37 and stem cells.38,39 Remarkably, CRISPR-based editing of primary human T cells at the TRAC locus (Ch14q11.2) was shown to lead to chromosome 14 truncation at the cleavage site (functionally equivalent to chromosomal loss due to the acrocentric nature of chromosome 14) in 5.3% of the cells. Even more strikingly, when the TRAC and TCRB (Ch7q34) loci were co-edited, this percentage increased to 9%, concomitant with chromosome 7 truncation at the TCRB locus in 9.9% of the cells.40 This body of evidence is of particular interest for the adoptive cell therapy field, as TCR disruption is a prerequisite for the generation of allogeneic CAR T cell products.
In light of these limitations of gene-editing-based approaches, non-gene-editing technologies may represent a valid alternative for the simultaneous downregulation of multiple targets in adoptive cell therapy products. Small interfering RNAs (siRNAs), short hairpin RNAs (shRNAs), and microRNAs (miRNAs) are all technologies based on small non-coding RNAs that regulate gene expression post-transcriptionally. Hence, these approaches do not require a site-directed intervention on the target cell genome, greatly minimizing the risk of unintended genomic alterations. Among them, siRNAs are likely not suitable for the development of engineered immune cells, despite the technological advancements in the last decade that greatly increased their in vivo persistence41 and led to the approval of five siRNA-based drugs. Indeed, siRNAs are delivered in their mature, functionally active form. The available information in the context of T cells is scarce, but the impossibility to self-replenish the initial siRNA pool argues for a rapid dilution of the siRNAs in the fast-dividing activated T cells. Hence, by means of this approach only a transient biological effect is likely to be achieved, rather than the long-term gene knockdown (KD) required for effective T cell engineering. On the other hand, shRNAs and miRNAs are actively transcribed as precursors in the recipient cells, ensuring long lasting gene downregulation. They largely share the processing machinery responsible for their maturation: following transcription, their core hairpin structure is processed by RNase III enzymes (first Drosha, then DICER in the case of miRNAs, only DICER in the case of shRNAs), leading to the formation of a mature RNA duplex, which is in turn incorporated into the RNA-induced silencing complex (RISC) complex. Upon release of the passenger RNA strand, the RISC-guide strand riboprotein mediates target mRNA recognition and eventually gene KD.42,43,44 However, the bypassing of Drosha in shRNA processing may overload the cytoplasm with double-stranded RNA and hence lead to toxicity by obstructing the natural miRNA pathway.45,46 As synthetic miRNAs, in which the guide sequence has been swapped for an shRNA-based one directed against the gene of interest, closely exploit the natural pathway, they can overcome this issue,47 standing out as the most valid candidates among non-gene-editing technologies for immune cell engineering. Another valuable characteristic of miRNAs is their frequent co-occurrence within the same transcriptional unit. About 50% of conserved vertebrate miRNAs are organized in clusters,48,49 making them the ideal shRNA-derived guide (shGuide) sequence vector for multiplex applications. Indeed, miRNA systems for multiple gene targeting have already been used successfully to inhibit HIV-1 and HCV.50,51,52 Nonetheless, their applicability to the generation of clinically valuable engineered immune cells has been hampered by the relatively weak KD activity.53 In part, this relates to the poor understanding of the structural constraints, of crucial importance for processing and KD efficiency, within the more complex multiplex architecture.54,55
Here, we describe our work on an miRNA-based delivery system for shRNAs that, optimizing the miRNA scaffold selection and the chimeric cluster design, ensures high KD efficiency of multiple targets simultaneously. The system is built upon our clinically validated technology, which proved both safe and effective in two separate clinical trials (NCT04613557 and NCT04167696). As a result, we obtained an miRNA-based multiplex shRNA platform for easy, safe, efficient, and tunable gene modulation. We combine highly efficient miRNA scaffolds into an optimized chimeric cluster able to deliver up to four shRNA-like sequences in a simple cassette format that can be deployed together with other components, streamlining the generation of engineered CAR T cells. The plug-and-play design of the platform has been validated to allow the swap of the shGuide sequence in any of the scaffolds without affecting the system’s overall KD performance. The expression of each target gene can be fine-tuned, up to achieving a functional KO, all without the need for gene editing, ensuring a high safety profile. These characteristics hoist our miRNA-based multiplex shRNA platform as a valuable product-oriented solution for the simultaneous engineering of multiple parameters in immune cells for adoptive cell therapy.
Results
A chimeric cluster combining different miRNA scaffolds is required for KD of multiple gene targets by shRNA
Delivery of shRNAs using miRNA-based constructs is one of the most promising non-gene-editing approaches for cell engineering. However, to date its clinical application has been limited by the difficulty in obtaining adequate KD when targeting different genes simultaneously. We have previously successfully integrated a single miRNA scaffold, based on miR-196a-2, carrying an shRNA against a specific target gene into our CAR T cells and further validated the technology in two separate clinical trials (NCT04613557 and NCT04167696). To determine whether our construct could be used as a basis for a multiplex system, we combined four repeats of the miR-196a-2 scaffold into a 4-plex, with each scaffold carrying a validated shRNA against a different target gene (namely CD3ζ, encoding a TCR component whose loss impairs proper expression of the TCR on the T cell surface, TRAC; β2M, encoding a key component of MHC class I; and CD52). Contextually, 1-plex constructs for each target were also created. Primary T cells were activated and subsequently transduced with the different retroviral vectors. The KD efficiencies, relative to a no-shRNA control, were assessed both at protein level by flow cytometric analysis (Figure 1A) and at mRNA level by qPCR (Figure S1) for CD3ζ, TCRa/b, HLA ABC, and CD52, respectively. For all four genes examined, the KD efficiency decreased when the shRNAs were embedded into a 4-plex compared with the 1-plex. These results are in line with previous evidence showing that using tandem repeats of the same miRNA scaffold is not a viable approach to scale up the number of targets in a miRNA-based platform56 and call instead for a more refined design.
Figure 1.

Rationale and scaffold screening for the building of a multiplex miRNA-based shRNA platform
(A) Effect on KD efficiency of the use of the same scaffold in tandem repeats as evaluated by flow cytometry. The KD efficiency of four different shGuide sequences (expressed as mean fluorescence intensity [MFI]) was compared in a single miR196a-2 scaffold and in the context of a 4-plex made of repeats of the miR196a-2 scaffold transduced in CAR T cells. (B) Schematic representation of the four miRNA cluster most highly expressed in T cells with an average scaffold size below 250 bases. (C) KD efficiency in CAR T cells of the miRNA scaffolds belonging to the four clusters most highly expressed in T cells with an average scaffold size below 250 bases. KD efficiency was measured by flow cytometry as MFI relative to a control without shRNA, using validated shRNA-derived sequences. Bars represent mean ± standard deviation (SD) for three independent biological replicates. Each symbol superimposed to the bars represents a different peripheral blood mononuclear cell (PBMC) donor.
About 50% conserved vertebrate miRNAs are located within miRNA clusters.48,49 Relying on a naturally occurring sequence of miRNA scaffolds may therefore be a valid alternative for a multiplex miRNA-based platform. We aimed at building a multiplex shRNA expression system that can be easily embedded in expression vectors along with all the other components (such as the CAR, a purification tag, and other armoring elements) for CAR T cell generation, with a minimal increase in vector size. Hence, we analyzed in silico 48 human miRNA clusters57 and we selected the ones with an average scaffold size lower than 250 bp. The 11 clusters falling below this limit were considered for further evaluation (Table 1). As the multiplex miRNA-based platform was primarily aimed at the engineering of T cells, a second selection criterion was the high expression level of the clusters in this cell type. Based on expression data reported in the literature,58,59,60,61 we identified four clusters as potential candidates (Figure 1B): miR-106a-363, miR-17-92a-1 (carrying six scaffolds each), miR-106b-25, and miR-23-a-24-2 (carrying three scaffolds each). To evaluate whether the four miRNA clusters would be suitable for multiplexed expression of shRNAs, we replaced the original shGuide sequences in the miRNA scaffolds with shRNAs we preemptively validated (Figure 1C), targeted against β2M, CD3ζ, CD28, CD95, CIITA, and CD27. We subcloned the clusters into a retroviral vector encoding a second-generation anti human CD19-41BB CAR and a truncated CD34 (tCD34) tag (ahCD19-41BB-tCD34) and transduced the constructs in primary T cells. We monitored the KD efficiency of each target by flow cytometry. CIITA encodes for the MHC class II transactivator, a key coregulator that controls expression of HLA class II genes.62 We therefore used surface expression of HLA DR isotype (HLA DR), one of the main MHC class II proteins, as proxy to assess CIITA KD efficiency. For the other targets, we assessed HLA ABC, TCRa/b, CD28, CD95, and CD27 surface expression, respectively. Not surprisingly, not all scaffolds within the same cluster granted effective downregulation of the target genes, with several showing no activity (Figure 1C). This is in line with the well-consolidated notion that miRNAs belonging to the same cluster are subjected to complex, differential post-transcriptional regulation and can be expressed at very different levels.63,64,65,66 As none of the natural clusters tested could be used as a basis for the development of the shRNA platform, we decided to combine different scaffolds in a chimeric cluster. Within clusters, miRNA scaffolds do not behave as independent units,66 hence the results obtained from the screening of whole clusters may not be indicative of the activity of the single scaffolds in other contexts. We therefore reassessed all scaffolds from the four clusters individually (data not shown) and shortlisted six scaffolds (sc106a, sc20b, sc17, sc20a, sc106b, and sc93), based on their KD efficiency, to be used in the generation of a chimeric miRNA cluster. Importantly, the secondary structure of the scaffolds is key to ensure proper processing of the miRNAs.66 We therefore paid special attention to design the proper passenger sequence for each shGuide sequence-scaffold combination, making sure that all the structural features (e.g., mismatches, loops, and bulges) were identical to those in the wild-type scaffold.
Table 1.
Summary of the characteristics of the evaluated miRNA clusters
| Clusters | miRNAs | Genomic position | Location | Strand | Size (bp) | No. of miRNAs | Size/no. of miRNAs (bp) |
|---|---|---|---|---|---|---|---|
| cl1.4 | miR-181b-1-213 | chr1:197094625–197094905 | predicted gene | – | 280 | 2 | 140 |
| cl3.2 | miR-15b-16-2 | chr3:161605070–161605307 | SMC4L1 | + | 237 | 2 | 118.5 |
| cl7.1 | miR-25-106b | chr7:99529119–99529633 | MCM7 | – | 514 | 3 | 171 |
| cl11.1 | miR-192-194-2 | chr11:64415185–64415487 | predicted gene | – | 302 | 2 | 151 |
| cl13.1 | miR-16-1-15a | chr13:49521110–49521338 | DLEU2 | – | 228 | 2 | 114 |
| cl13.2 | miR-17-92a-1 | chr13:90800860–90801646 | mRNA | + | 786 | 6 | 131 |
| cl17.3 | miR-451-144 | chr17:24212513–24212762 | EST | – | 249 | 2 | 124.5 |
| cl19.1 | miR-23a-24-2 | chr19:13808101–13808473 | EST | – | 372 | 3 | 124 |
| cl19.2 | miR-181c-181d | chr19:13846513–13846825 | predicted gene | + | 312 | 2 | 156 |
| cl19.3 | miR-99b-125a | chr19:56887677–56888404 | intergenic | + | 727 | 3 | 242 |
| clX.5 | miR-106a-363 | chrX:133131074–133131974 | intergenic | – | 900 | 6 | 150 |
(−) Indicates that the miRNA cluster is located in the opposite orientation of the host gene.
Adapted from Muiños-Gimeno et al.57
KD efficiency is dependent on the length of the linker sequences flanking the miRNA scaffolds and of the shGuide sequence
To further understand which elements govern the KD efficiency of the miRNA scaffolds, we evaluated the role of scaffold and shGuide sequence length.
The linker sequences flanking the miRNA hairpins can create non-miRNA stem-loop secondary structures that can interact with and potentially alter the processing of the miRNAs.67 To evaluate the effect of linker sequences on the activity of the selected scaffolds, we made progressive deletions of the linkers flanking the stem-loop core of the miRNAs (Figure 2A). The full sequences are reported in Table S1. The full-length scaffolds and the shortened versions were subcloned into a retroviral vector expressing an anti-human BCMA-41BB CAR and a truncated CD34 (tCD34) tag (ahBCMA-41BB-tCD34). All scaffolds carried an shRNA-derived sequence targeting CD3ζ. The constructs were transduced into primary T cells to obtain CAR T cells, and their KD efficiencies were compared based on the residual surface expression of TCRa/b by flow cytometry (Figures 2B and S2A). For all scaffolds, the full-length constructs had equal or superior KD efficiency compared with their shorter counterparts. Therefore, we resolved to use all scaffolds in their full-length form in subsequent experiments.
Figure 2.

Identification of the optimal scaffold and shGuide sequence length
(A) Schematic representation of the deletion strategy used to identify the best length for each of the six candidate scaffolds selected for the multiplex platform development. (B) KD efficiencies of a shGuide sequence against the TCR component CD3ζ, obtained from the sequential deletions of each of the candidate scaffolds, measured in CAR T cells as surface expression of TCRa/b. The orange bars correspond to the full-length scaffolds, ultimately chosen for further experiments. (C) Schematic representation of the shGuide sequence lengths tested (in red) compared with the typical shGuide sequence length in miRNA scaffolds (in gray). (D) KD efficiencies of shGuide sequences spanning between 18 and 25 bases against CD3ζ and the HLA class I component β2M in the context of two different scaffolds (sc106a and sc20b), measured in CAR T cells as surface expression of TCRa/b and HLA ABC, respectively. The orange bars correspond to the shGuide sequence lengths ultimately chosen for further experiments. KD efficiency was measured by flow cytometry as MFI relative to a control without shRNA, using validated shRNA-derived sequences. Bars represent mean ± SD for three independent biological replicates. Each symbol superimposed to the bars represents a different PBMC donor. All p values refer to the comparison with the no-shRNA control. The absolute MFI values for the no-shRNA control samples are reported in Table S2.
The shGuide sequences we introduced in the miRNA scaffold are shRNA derived, and therefore have the typical shRNA length of 18–20 nucleotides. Naturally occurring mature miRNAs, instead, usually consist of 20–23 nucleotides. We wondered whether modifying the length of the shGuide sequences to make it resemble more that of a mature miRNA would help in achieving a better KD efficiency. We tested this hypothesis in two different scaffolds (namely sc106a and sc20b), in which we introduced shGuide sequences against two different genes (CD3ζ and β2M) spanning from 18 to 23 nucleotides (Figure 2C). The scaffolds were cloned into lentiviral vectors along with a tCD34 tag and transduced into primary T cells to obtain CAR T cells. The surface expression of TCRa/b and HLA ABC was assessed by flow cytometry to determine the KD efficiency of the CD3ζ- and β2M-targeted sequences, respectively. In general, we observed a progressive decline in KD efficiency when the shGuide sequence length increased above 20 nucleotides (Figures 2D and S2B). Hence, we decided to set the length of shGuide sequences to be used in the miRNA-based shRNA platform to 20 nucleotides.
A combination of two different scaffolds ensures efficient KD in a 2-plex miRNA platform
We have previously observed that the repetition of the miR-196a-2 scaffold leads to a decrease in KD efficiency (Figure 1A). To understand whether this behavior is common to other miRNA scaffolds, we combined two repeats of the sc106 or two repeats of the sc20b in a 2-plex, carrying shGuide sequences against CD3ζ, β2M, and CD95 in different configurations (Figures 3 and S3). We subcloned the constructs into a retroviral ahBCMA-41BB-tCD34 vector and we expressed them in primary T cells. We then monitored the surface expression of TCRa/b, HLA ABC, and CD95 by flow cytometry to determine the KD efficiency. Interestingly, when the 2-plex consisted of two repeats of sc106a, for all three genes tested the KD was nearly lost if the shGuide sequence was in the first scaffold. Conversely, we did not observe such an effect when the 2-plex consisted of two repeats of sc20b, which granted similar KD in all configurations (Figures 3 and S3). These results indicate that the loss of KD efficiency upon tandem repetition of the same scaffold is scaffold dependent. However, the fact that this effect can emerge already in a low-complexity system, such as a 2-plex, advocates against the use of scaffold repetition in higher-tier multiplex platforms.
Figure 3.

Effect of scaffold combination on KD efficiency in a 2-plex miRNA platform
Results of two different approaches followed to build a chimeric cluster: combining two different scaffolds or replicating the same scaffold multiple times. Combinations of sc106b and sc20b as well as repetitions of either sc106a or sc20b were tested in a 2-plex with validated shGuide sequences against CD3ζ, β2M, and CD95. Different configurations of the 2-plex were tested, alternating the position of the scaffolds and of the shGuide sequences. KD efficiencies were evaluated by flow cytometry by comparing the MFI of TCRa/b, HLA ABC, and CD95, respectively, in CAR T cells transduced with the 2-plex and with a no-shRNA control. MFIs are depicted as relative to the no-shRNA control. Bars represent mean ± SD for three independent biological replicates. Each symbol superimposed to the bars represents a different PBMC donor. All p values refer to the comparison with the no-shRNA control. The absolute MFI values for the no-shRNA control samples are reported in Table S2.
Conversely, in the same system, the combination of sc106a and sc20b yielded strong and consistent KD for all three target genes tested (CD3ζ, β2M, and CD95), irrespective of the order of the scaffolds and of the shGuide sequences (Figures 3 and S3). This evidence once more confirms the need for the combination of different scaffolds to build an effective and stable miRNA multiplex platform and poses a sc106a-sc20b 2-plex as a valid base for further expansion.
The combination of different scaffolds allows expansion to a highly efficient 4-plex miRNA-based shRNA platform
Building upon the encouraging results obtained with sc106a-sc20b 2-plex, we sought to expand our miRNA-based shRNA platform into a 4-plex, achieving true multiplex capability. In our original design for the 4-plex, we planned to include restriction sites between the scaffolds to allow for easy replacement of each scaffold individually. Eventually, we aim at incorporating a broad array of shGuide sequences in the platform in a plug-and-play fashion. This will likely require adapting the restriction sites from time to time to allow compatibility with the shGuide sequences and their corresponding passenger sequences. Changes in the restriction sites, however, may alter the secondary structure of the clusters. Secondary structure is a key determinant in the processing and maturation of miRNAs within a cluster,66 and its variation is likely to ultimately affect KD efficiency. To verify the impact of changes in the restriction sites on the cluster’s secondary structure, we designed four versions of the same cluster, carrying no restriction sites or the same three restriction sites in different order between the scaffolds. The in silico analysis of the cluster sequences using the RNAfold secondary structure prediction tool (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi) demonstrated that the order of the restriction sites could strongly affect the secondary structure of the cluster (Figure S4A). Likewise, maintaining the restriction sites in the same positions while swapping the shGuide and passenger sequences may also alter the secondary structure (Figure S4B). Hence, the presence of restriction sites between the scaffolds increases the complexity of the system, possibly affecting KD efficiency in an unpredictable way, and limits the plug-and-play characteristics of the platform, imposing additional constraints to the selection of the shGuide sequences. For these reasons, we ultimately resolved to omit the restriction sites between the scaffolds, opting for a more streamlined design with restriction sites only at the sides, which allow for removal and subcloning of the whole 4-plex cassette (Figure S4C).
To build the 4-plex platform, we expanded the sc106a-sc20b 2-plex by adding two additional scaffolds. Our preliminary data pointed at sc20a as a good candidate to work in combination with sc106a and sc20b (data not shown). Therefore, we included sc20a in the 4-plex either in position 3 or position 4. As fourth scaffold (in position 4 or position 3, respectively), we tested the three remaining candidates that emerged from our initial screening, namely sc93, sc106b, and sc17 (Figures 4 and S5). We inserted validated shGuide sequences against β2M in sc106a, CD3ζ in sc20b, CD28 in sc20a, and CD95 in sc93, sc106a, and sc17. We subcloned the constructs into both ahCD19-41BB-tCD34 and ahBCMA-41BB-tCD34 retroviral vectors and we expressed them in primary T cells. Flow cytometric analysis for HLA ABC, TCRa/b, CD28, and CD95, respectively, showed high KD efficiency for all the target genes irrespective of the 4-plex configuration, both in combination with the anti-CD19 CAR (Figures 4A and S5A) and with the anti-BCMA CAR (Figures 4B and S5B). Despite all three candidates for the fourth scaffold performing equally well, our choice fell on sc93 due to its smaller size, which allowed us to minimize the overall size of the cluster. We therefore decided to adopt the sc106a-sc20b-sc20a-sc93 configuration for further development.
Figure 4.

Selection of scaffolds for the expansion of the miRNA-based shRNA platform to a 4-plex
Starting with the sc106a-sc20b 2-plex as a basis, the platform was expanded into a 4-plex by the addition of two extra scaffolds. In a first cluster design, sc20a was added in third position, and sc93, sc106b, or sc17 in fourth position. In a second cluster design, sc93, sc106b, or sc17 were added in third position and sc20a in fourth position. Each position carried a different validated shGuide sequence (β2M, CD3ζ, CD28, and CD95). KD efficiency for each target was assessed by flow cytometry by monitoring the MFI of TCRa/b, HLA ABC, CD28, and CD95, respectively, in CAR T cells transduced with the 4-plex constructs or with a no-shRNA control. The different 4-plex combinations were tested in the context of both an anti-CD19 CAR (A) and an anti-BCMA CAR (B). MFIs are depicted as relative to the no-shRNA control. Bars represent mean ± SD for three independent biological replicates. Each symbol superimposed to the bars represents a different PBMC donor. All p values refer to the comparison with the no-shRNA control. The absolute MFI values for the no-shRNA control samples are reported in Table S2.
The 20b scaffold shows compatibility issues with selected shGuide sequences in the higher-complexity context of the 4-plex
To work as a plug-and-play platform, it is crucial for our system to be able to accommodate a variety of shGuide sequences while ensuring high KD efficiency. To ascertain that the chosen configuration of the 4-plex has this capability, we created a series of constructs in which we substituted the shGuide sequence in each of the scaffolds with in-house validated shGuide sequences against either CIITA (Figures 5A and S6A) or lymphocyte activation gene 3 (LAG-3) (Figures 5B and S6B). We subcloned the constructs into an ahCD19-41BB-tCD34 retroviral vector and we expressed them in primary T cells. LAG-3, an immune checkpoint receptor, is expressed at low levels in resting T cells, but is induced following T cell activation.68 Hence, we tested surface LAG-3 expression in our CAR T cells upon co-culture with NALM-6, a human pre-B acute lymphoblastic leukemia (ALL) cell line positive for CD19.
Figure 5.

Assessing the plug-and-play capability of the 4-plex miRNA-based shRNA platform
To test the plug-and-play capability of the 4-plex and its stability irrespective of the target, a fifth and sixth shGuide sequence were substituted in each position in the sc106a-sc20b-sc20a-sc93 cluster carrying validated shGuide sequences against β2M, CD3ζ, CD28, and CD95, in the context of an anti-CD19 CAR. Validated shRNA-derived sequences against CIITA (A), a key coregulator that controls expression of HLA class II genes, and LAG-3 (B) were used for the purpose. The plug-and-play capability of the system was assessed by flow cytometry by monitoring the MFI of all targets, including HLA DR, one of the main MHC class II proteins, as a proxy for CIITA and the immune checkpoint LAG-3, in CAR T cells transduced with the 4-plex constructs, or with a no-shRNA control. As LAG-3 is expressed at low levels in resting T cells, but is induced following T cell activation, surface LAG-3 expression was assessed upon co-culture for 24 h with NALM-6, a human pre-B ALL cell line positive for CD19. As a marked loss in KD efficiency was observed when the shGuide sequence against LAG-3 was in sc20b, to determine whether this is due to a positional effect of this scaffold, a construct with sc20b in position 4 (sc106a-sc20a-sc93-sc20b) was tested in comparison with the original cluster with sc20b in position 2 (sc106a-sc20b-sc20a-sc93) and a no-shRNA control. The comparison was made with both CIITA (C) and LAG-3 (D) shGuide sequences. KD efficiencies were assessed by flow cytometry by monitoring the MFI of HLA DR and LAG-3, the latter upon co-culture for 24 h with NALM-6 cells. MFIs are depicted as relative to the no-shRNA control. Bars represent mean ± SD for three independent biological replicates. Each symbol superimposed to the bars represents a different PBMC donor. All p values refer to the comparison with the no-shRNA control. The absolute MFI values for the no-shRNA control samples are reported in Table S2.
The substitution of the shGuide sequences in any of the scaffolds with the shGuide sequence against CIITA led to strong HLA DR downregulation and, as expected, did not influence the KD of the other three targets (Figures 5A and S6A). Interestingly, however, the combination of the shGuide sequence against LAG-3 with the sc20b completely abolished LAG-3 KD (Figures 5B and S6B). In line with the results obtained with the shGuide sequence against CIITA, the KD efficiency of the other targets was not affected by the substitution.
Analysis of the cluster sequence containing the LAG-3 shGuide in RNAfold did not demonstrate any obvious alteration in the cluster’s secondary structure compared with 4-plex constructs carrying shGuide sequences against CD3ζ or CIITA, with all the scaffolds having a predicted structure identical to their wild-type counterparts. We therefore reasoned that the unexpected behavior of sc20b in combination with the LAG-3 shGuide sequence could be due to a positional effect of sc20b within the cluster, causing detrimental cis interactions with the neighboring scaffolds. To test this hypothesis, we created a 4-plex construct in which sc20b carrying either LAG-3 or CIITA shGuide sequences was moved in position 4 (sc106a-sc20a-sc93-sc20b) and compared its KD efficiency for all targets to the 4-plex configuration with sc20b in position 2 (sc106a-sc20b-sc20a-sc93) (Figures 5C and S6C). As expected, the 4-plex constructs with the CIITA shGuide sequence in sc20b gave comparable, strong KD for HLA DR as well as for the other targets, irrespective of sc20b position. However, LAG-3 KD was completely abolished in both cluster configurations (Figures 5D and S6D). Hence, we concluded that the loss of KD efficiency of sc20b is not a direct consequence of its position in the 4-plex. Still, sc20b in combination with the LAG-3 shGuide sequence achieved good KD efficiency in a sc106a-sc20b 2-plex (data not shown). The higher structural complexity of the 4-plex may therefore put additional, hard to predict constraints on the compatibility of sc20b with some shGuide sequences.
The miRNA-based shRNA platform built upon sc106a-sc17-sc20a-sc93 efficiently knocks down four target genes, with broad compatibility with different shGuide sequences
We observed a dramatic loss in the KD efficiency of sc20b when combined with the LAG-3 shGuide sequence (Figures 5 and S6). As similar results with other shGuide sequences cannot be excluded, we decided to test sc106b and sc17, both of which already showed good KD efficiency (Figures 4 and S5), as replacements for sc20b. We created the 4-plex clusters with either sc106b or sc17 in position 2 (sc106a-sc106b-sc20a-sc93 and sc106a-sc17-sc20a-sc93, respectively) and compared them with the equivalent cluster containing sc20b (sc106a-sc20b-sc20a-sc93). For all three configurations, we tested both CIITA and LAG-3 shGuide sequences in position 2. We subcloned the constructs into an ahCD19-41BB-tCD34 retroviral vector and expressed them in primary T cells. In line with our previous results, both constructs containing sc106a and sc17 delivered similarly strong and consistent KD of all the target genes tested, including CIITA (Figures 6A and S7A) and LAG-3 (Figures 6B and S7B). We chose sc17 over sc106a as replacement for sc20b due to its smaller size, which allowed us to minimize the overall size of the cluster. As a result, we obtained an miRNA-based shRNA platform (sc106a-sc17-sc20a-sc93) able to efficiently knock down four target genes with broad compatibility with different shGuide sequences.
Figure 6.

Testing of an updated version of the 4-plex miRNA-based shRNA platform with true plug-and-play capability
As an alternative to the unstable sc106a-sc20a-sc93-sc20b configuration of the 4-plex, different designs were tested, in which sc20b was substituted by sc106b or sc17, in the context of an anti-CD19 CAR. The clusters carried validated shGuide sequences against β2M in position 1, CD28 in position 3, and CD95 in position 4. In position 2, where sc20b originally laid, validated shGuide sequences against CIITA (A) and LAG-3 (B) were introduced. KD efficiency was assessed for all targets by monitoring the surface expression of HLA ABC, HLA DR (respectively controlled by β2M and CIITA), LAG-3, CD28, and CD95. As LAG-3 is expressed at low levels in resting T cells, but is induced following T cell activation, surface LAG-3 expression was assessed upon co-culture for 24 h with NALM-6, a human pre-B ALL cell line positive for CD19. In the upper part of the panels, MFIs are depicted as relative to the no-shRNA control. Bars represent mean ± SD for three independent biological replicates. Each symbol superimposed to the bars represents a different PBMC donor. All p values refer to the comparison with the no-shRNA control. In the lower part of the panels, representative dot plots showing multiple simultaneous KD are depicted for the no-shRNA control and for the sc106a-sc17-sc93-sc20b 4-plex configuration. The absolute MFI values for the no-shRNA control samples are reported in Table S2.
The miRNA-based shRNA approach allows for the fine-tuning of target gene expression, adapting KD intensity to the target biology
One of the main advantages of shRNA over gene-editing technologies is the possibility to modulate the expression of the target genes. Indeed, gene-editing technologies, by directly ablating the target gene, can only yield an all-or-nothing effect, whereas shRNA allows for various KD intensities depending on the strength of the selected shGuide sequence.
Using our miRNA-based shRNA platform, for each target gene we screened several shGuide sequences and ranked them according to their KD efficiency. Representative screening results are depicted in Figure 7A for CD3ζ, CD95, and β2M, respectively. As a result, we obtained a broad range of KD efficiencies, up to levels that functionally match a KO. This allowed us to pick for each target the sequence that grants the most appropriate KD level in relation to the target biology. As an example, to avoid alloreactivity in allogeneic CAR T products, it is crucial to strongly impede the expression of the TCR. By selecting a shGuide sequence with a KD efficiency against CD3ζ of over 90% (in orange in Figure 7A, left panel), we were able to completely abolish TCR-mediated activation of the CAR T cells with OKT3, a monoclonal antibody against CD3 able to induce T cell activation69 (Figure 7B). Likewise, CD95 is a death receptor expressed in many cell types, including T cells. Its engagement by its ligand CD95L, often expressed by cancer cells, induces apoptosis in CAR T cells, hampering their tumor-controlling effect.70 By transducing our CAR T cells with a highly efficient shGuide sequence against CD95 (in orange in Figure 7A, middle panel), we were able to protect them from CD95-mediated apoptosis to an extent similar to that achieved with a CD95-blocking antibody (Figure 7C). These data demonstrate that our miRNA-based shRNA approach can reach functional KO-like effects. On the other hand, the complete ablation of some targets may be detrimental, depending on the target biology. An example is downregulation of HLA class I, one of the key strategies to prevent allorejection of allogeneic immune cell therapy. Complete loss of HLA class I surface expression leaves the engineered cells vulnerable to lysis by natural killer (NK) cells, calling for more sophisticated engineering approaches, such as overexpression of HLA E to compensate for HLA class I loss.71,72,73,74,75 To circumvent this issue, we selected a shGuide sequence against β2M, which can efficiently downregulate the target but still allows for ∼30% residual HLA class I expression (in orange in Figure 7A, right panel). This strategy allowed us to limit NK activation, measured as percentage of CD107a+ NK, in the presence of CAR T cells carrying the shGuide against β2M to levels similar to those measured in presence of CAR T cells with no shRNA, and thus normally expressing HLA class I. On the contrary, β2M KO in CAR T cells by CRISPR-Cas9 led to substantial activation of NK cells (Figure 7D, left panel). As a consequence, the viability of CAR T cells with β2M KD was comparable with that of CAR T cells with no shRNA, whereas the viability of CAR T cells with β2M KO was reduced to nearly 50% due to the NK cytotoxic effect (Figure 7D, right panel).
Figure 7.

Tunability and functional relevance of the KD achieved with the 4-plex miRNA-based shRNA platform
(A) Conversely from gene editing technologies, our shRNA-based approach allows for the fine-tuning of target gene expression. Indeed, shGuide sequences can be designed that lead to varying degrees of KD. The desired level of KD can therefore be achieved by selecting the appropriate shGuide sequence. Representative examples are shown here for CD3ζ, CD95, and β2M. KD efficiency was assessed by the surface expression of TCRa/b (controlled by CD3ζ), CD95, and HLA ABC (controlled by β2M), respectively, in the context of an anti-CD19 CAR. Our miRNA-based shRNA platform allows to modulate the target gene expression to achieve the desired biological outcome. (B) When CD3ζ was knocked down with a highly efficient shGuide sequence (orange bar in A, left panel), the resulting downregulation of the TCRa/b receptor rendered the CAR T cells insensitive to TCR-specific activation by OKT3, a monoclonal antibody against CD3 able to induce T cell activation, as assessed by the measure IFN-γ secretion, mimicking a functional KO. (C) Likewise, a highly effective KD of death receptor CD95 (orange bar in A, middle panel) protected CAR T cells from apoptosis upon incubation with its soluble ligand CD95L, with an effect comparable with that of a CD95 blocking antibody. (D) On the contrary, a shGuide sequence for β2M with less pronounced KD ability could efficiently downregulate HLA ABC (orange bar in A, right panel) while, at the same time, preserving expression levels that protected CAR T cells from NK cells. Indeed, the percentage of CD107a+ NK cells upon co-culture with CAR T cells knocked down for β2M was comparable with that of NK cells in presence of CAR T cells without shRNA. Conversely, β2M KO in CAR T cells led to a substantial increase in the percentage of CD107a+ NK cells upon co-culture (D, left panel). As a consequence, CAR T cells harboring the β2M KD maintained a viability comparable with control CAR T cells without shRNA, whereas the viability of CAR T cells with β2M KO was strongly reduced (D, right panel). Bars represent mean ± SD for three independent biological replicates. In (A), for screening purposes only one PBMC donor was tested, and the bars therefore refer to a single biological replicate.
Taken together, these data showcase the ability of our miRNA-based shRNA platform to fine-tune the target gene expression, from partial KD to a KO-like phenotype, modulating the intensity of the KD to match the requirements imposed by the biology of the target. Most importantly, the expression of each target in the multiplex can be modulated to different levels independently of the other targets, a result to date not achievable by means of gene editing technologies.
Discussion
By exploiting the natural miRNA pathway, synthetic miRNAs overcome the potential toxicity due to competition of the shRNAs with endogenous miRNAs and an eventual subsequent saturation of the RNAi machinery.46,76,77 Hence, gene KD using miRNA-based constructs stirred great expectations as a non-gene-edited cell engineering approach, leading to several clinical trials with CAR T cells incorporating miRNA-based KD approaches. In this respect, we proved the safety and effectiveness of this strategy in two clinical trials (NCT04613557 and NCT04167696),78,79 in which we assessed CAR T cells equipped with a miRNA-based cassette for the KD of a single gene. However, the simultaneous targeting of multiple genes will likely be required for the effective treatment of those cancer indications that are not yet successfully managed by adoptive cell therapy, particularly solid tumors. In this paper, we describe the successful establishment and optimization of a multiplex miRNA-based shRNA platform for the simultaneous KD of up to four different target genes, with high efficiency and consistency across multiple targets.
Multiplex miRNA-based systems have been actively investigated43,54,80,81 and were successfully used to inhibit HIV-1 and HCV.50,51,52 Still, to date multiplex miRNA approaches have shown limitations in their overall efficiency that still prevent their effective use in clinical applications for adaptive cell therapy. The reason partially resides on the far greater complexity of a miRNA-based RNAi system compared with shRNAs. Such complexity can be separated into several aspects, many not yet fully elucidated: (1) the structural constraints for efficient miRNA processing, (2) the interplay of different miRNA scaffolds within a cluster, and (3) the technical optimization of miRNA cassette expression in clinical products such as CAR T cells.
Most of the seminal work on miRNA-based silencing systems that paved the road for further applications is based on a few naturally occurring miRNA scaffolds, such as miR-30, or on their modified and optimized versions.54,82,83,84,85 Despite the high KD efficiency reached by some of these systems, the tandem repetition of the same scaffold proved ill-suited for multiple gene silencing, due to the risk of recombination and to the overall lower KD efficiency compared with their single-scaffold counterparts.86 Along this line, the data we present in this publication confirmed that, already in a low-complexity system, such as a 2-plex, scaffold repetition may have detrimental effects on KD efficiency (Figure 3). We were able to overcome this limitation by combining four different, carefully selected scaffolds into a chimeric miRNA cluster. This strategy allowed us to achieve high KD levels for all the target genes tested.
The scaffolds that showed the most promising KD efficiency in our screening share similar characteristics in their secondary structure. The miRNA secondary structure is of crucial importance for proper KD efficiency, as the machinery responsible for miRNA maturation recognizes and processes immature miRNAs mainly based on their structure rather than on their sequence. Extensive work in recent years has gone into defining these structural requirements,87,88,89,90,91,92 which can be summarized in three main aspects: optimal length of the lower stem favors Drosha processing, a destabilized 5′ end of the guide sequence allows for optimal RISC incorporation, and a mismatch in the middle of the upper stem helps in avoiding abortive processing by Drosha.54 All our scaffolds closely abide to these rules, with the only notable exception of sc20a, which lacks the internal mismatch in the upper stem, although with no impact on its KD efficiency for any of the shGuide sequences tested.
We observed that shGuide sequences of a length comprised between 18 and 20 bases yield the best KD (Figure 2D and S2B), and that the KD efficiency progressively declines with the increase of the shGuide sequence length beyond 20 bases. Processing at the 5′ end of miRNAs is likely to start precisely at the +1 position, due to accurate cleavage of the miRNA precursors by both Drosha and Dicer in human cells.43,93 On the other hand, and in support of our results, the 3′ end of mature miRNAs often stops between the +21 and +26 position, suggesting that efficient RNAi in mammalian cells can tolerate a few unpaired sequences to the target gene at the 3′ end of the mature miRNA.43 On the other hand, shGuide sequences shorter than 20 bases, despite granting efficient KD in our experimental setting (Figures 2D and S2B), are more susceptible to off-target effects, as the specificity of shRNAs and miRNAs is inversely correlated with both the overall sequence and seed length.94,95 Together, this evidence gives mechanistic support to our choice of selecting a shGuide sequence length of 20 bases for our miRNA-based shRNA platform.
During the optimization of our platform, we witnessed several scaffolds displaying poor KD efficiency in the context of their natural clusters (e.g., sc106a and sc17, Figure 1C), only to reveal themselves among the best candidates when evaluated independently or in a chimeric cluster (Figures 4 and 6); likewise, sc20b, which gave satisfactory results within its own natural cluster and in a 2-plex (Figures 1C and 3), suffered strongly reduced activity in the context of a 4-plex (Figure 5). These results can be explained by the fact that, within miRNA clusters, individual miRNAs do not behave as independent units, instead making the whole cluster a dynamic and interdependent system tied by intricate relationships.66 Indeed, although there is coordinated transcription of miRNAs within the same cluster, the mature miRNAs originating from these transcriptional units can be expressed at very different levels and can follow different expression patterns in response to physiological or pathological stimuli.66 This indicates the existence of regulatory mechanisms at the post-transcriptional level that lead to differential expression of individual cluster members. Some scaffolds may naturally have suboptimal characteristics, to the extent that they may rely on neighboring scaffolds for the recruitment of the processing machinery.96,97 Furthermore, the tridimensional folding of the transcribed cluster may give rise to local interaction or tertiary structures that limit its accessibility to the microprocessing machinery, as already demonstrated for the mir-17-92a cluster.67,98,99 This body of evidence supports our findings in highlighting the importance of proper scaffold evaluation in the design of clinically oriented approaches, as prior information may not be readily translatable to novel settings. To our knowledge, our work is the first report tackling the optimization of a chimeric miRNA cluster based on the definition of the best scaffold selection and position, as well as evaluating the stability of the system with a broad selection of shGuide sequences.
We embedded our miRNA-based shRNA platform into an all-in-one retroviral vector carrying from 5′ to 3′ the CAR, the purification tag, and the miRNA cassette in a polycistron under the control of a single promoter. With this configuration we were able to achieve satisfactory levels of both the proteins and the miRNAs, allowing efficient CAR T purification, potent effector function, and effective gene KD. Thanks to their relatively small size, miRNA cassettes can be embedded into advanced single-vector systems, allowing their simultaneous expression along with the CAR and the tag from the same mRNA. Careful evaluation of the positioning of the miRNA cassette within the transcriptional unit is crucial. Placing the miRNA cassette at 5′ of the mRNA diminishes the expression of the companion coding gene(s), as cleavage by Drosha removes the 5′mC necessary for the efficient translation of the mRNA.100 This effect can be partially mitigated by inserting an internal ribosome entry site (IRES) upstream of the coding region.101 However, protein translation from IRES sequences is often less efficient compared with cap-dependent translation.100 Alternatively, in line with our results, it has been demonstrated that positioning the miRNAs at the 3′ end of the mRNA produces sufficient levels of both the coding genes and the miRNAs simultaneously, despite the loss of the poly(A) tail caused by the miRNA processing.100
Current gene-editing strategies relying on ZFN, TALEN, and Cas9 all have in common a mechanism based on the induction of DSBs, which may lead to inadvertent off-target cleavages or chromosomal aberrations. To overcome these safety concerns, several innovative strategies are in active development that do not rely on double-strand breaks and thus offer a potentially safer route to genetic editing. Nuclease-inactive platforms including base and prime editors have recently been described.24 Another Cas-based approach relies on mutant, catalytically inactive forms of Cas9 or Cas12a (dCas9 and dCas12a) fused to effector domains that regulate transcription, either positively (CRISPR-mediated activation) or negatively (CRISPR-mediated inhibition).102,103 When dealing with multiplex gene editing, control mechanisms must be implemented to limit crosstalk between components. An effort in this direction is the use of Cas orthologs from different organisms, each recognizing a slightly different gRNA structure, thus enabling orthogonal assembly of the ribonucleoprotein complexes.104 A radically different concept is represented by technologies exploiting Cas13. This Cas allows for direct transcriptome engineering via RNA editing and KD without the requirement for permanent genetic manipulations.105,106,107,108 This approach is potentially safer than Cas9-based technologies as it can induce defined cellular phenotypes without introducing genomic alterations.
However, while a number of novel candidate technologies for advanced immune cell editing have been proposed, it will be critical to see how robust and safe they prove themselves in pre-clinical validation and eventually in clinical studies. Recent and upcoming developments will likely address most safety concerns revolving around gene editing, although their use to enable precise modulation of gene expression remains beyond reach. miRNA-based non-gene-edited technologies, on the other hand, have already demonstrated their value in the clinical settings.78,79 We proved the safety of our miRNA-based shRNA approach and its effectiveness in the generation of CAR T products in two distinct clinical programs, one for the creation of allogeneic CAR T cells and one for the downregulation of two NKG2D stress ligands simultaneously, targeting CD3ζ and MICA/B, respectively. The upgrade of the system to a multiplex plug-and-play platform that we detail here paves the road to exciting new outcomes. The simplicity and the small size of the miRNA cassette allows its deployment in a wide array of delivery systems, from lentiviral and retroviral vectors to transposons. The tunability of shRNA-mediated KD is suited, among others, for the targeting of genes whose complete KO would be detrimental for T cell function. The robustness and versatility of our approach makes it easily applicable to many aspects of CAR T engineering (e.g., targeting of immune checkpoints, enhancing immune evasion and persistence, managing exhaustion, optimizing cell metabolism, priming of the T cells to the TME) as well as to a variety of contexts beyond the CAR T field and beyond immuno-oncology, such as chronic inflammation and autoimmune diseases.
Still, to date, large amounts of information have been gathered on the properties and design requirement for shRNAs, but shRNA sequences often do not have optimal characteristics for use in miRNA-based scaffolds. As a consequence, a relatively large number of shRNA candidates needs to be screened for each target. The clinical implementation of our system would therefore benefit from a better understanding of the sequence requirements of shGuides and from miRNA-tailored design algorithms. Moreover, further research efforts are needed to expand the multiplexing capacity of the system to allow the simultaneous regulation of multiple targets in different pathways and consequently manipulate complex cellular behaviors.
Materials and methods
Cell lines and cell culture
HEK293T cells, Phoenix Eco and PG13 packaging cells, and the NALM-6 ALL cell line were sourced from ATCC. HEK293T, Phoenix Eco, and PG13 cells were maintained in Dulbecco’s modified Eagle’s medium: 4,500 mg/L glucose, 2 mM L-glutamine, 1 mM sodium pyruvate (Merck Life Sciences, Hoeilaart, Belgium) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA). NALM-6 cells were maintained in RPMI 1640 medium, 25 mM HEPES (Capricorn Scientific, Ebsdorfergrund, Germany) supplemented with 10% FBS. All cells were grown at 37°C in an atmosphere of 5% CO2/air. All cells were routinely screened for mycoplasma contamination using the PlasmoTest Mycoplasma Detection Kit (InvivoGen, Toulouse, France).
Construction of miRNA-containing lentiviral and retroviral vectors
All the sequences containing the miRNA-based scaffold were purchased as custom-synthesized gBlocks from Integrated DNA Technologies (Leuven, Belgium) and cloned into the recipient backbones using the In-Fusion Snap Assembly Master Mix (TaKaRa, Saint-Germain-en-Laye, France) following the manufacturer’s instructions. For lentiviral expression, a third-generation pCCL backbone was used. For retroviral expression, a pSFG backbone-based plasmid expressing a second-generation anti-human CD19-41BB or anti-human BCMA-41BB CAR and a truncated CD34 (tCD34) selection TAG was used. All backbones and the cloned sequences in the newly generated plasmids were verified by Sanger sequencing. The following shRNA-based sequences were used for target gene KD: CD3ζ (5′-AACAGACTCAACAACTCAGG-3′), TRAC (5′-GAAAGTTTAGGTTCGTATC-3′), β2M (5′-TTCATCCAATCCAAATGCGG-3′), CD52 (5′-TGAGAGTCCAGTTTGTATC-3′), CD28 (5′-AATGATCTTGATATTTGGTC-3′), CD95 (5′-ACAGACGTAAGAACCAGAGG-3′), CIITA (5′-TATTGTACAAGCTTAGCCTG-3′), CD27 (5′-ACAGCTTTCCCTGAGCCCAG-3′), LAG-3 (5′-TATTTGGACTGGGCTGCTGA-3′). The following readout proteins were used to assess the KD efficiency of the different shRNA-based sequences: CD3ζ or TCRa/b for CD3ζ, TCRa/b for TRAC, HLA ABC for β2M, CD52 for CD52, CD28 for CD28, CD95 for CD95, HLA DR for CIITA, CD27 for CD27, LAG-3 for LAG-3.
Lentiviral and retroviral vector production and titration
Lentiviral vectors were generated by transiently transfecting HEK293T cells with the appropriate lentiviral plasmid along with the packaging plasmids pRSV.Rev and pMD2.G (ViveBiotech, Donostia, Spain), using GeneJuice Transfection Reagent (Merck Life Sciences, Hoeilaart, Belgium) according to the manufacturer’s instructions. The lentiviral vectors were harvested 24 h post-transfection, titrated, and used immediately to transduce T cell. Retroviral vectors were generated by transiently transfecting Phoenix Eco packaging cells with the appropriate retroviral plasmids. The obtained vectors were collected at 48 and 72 h post-transfection, pooled, and used to transduce PG13 packaging cells expressing the gibbon ape leukemia virus env and the Mo-MuLV gag-pol proteins. The final retroviral vectors were collected at 48 and 72 h post PG13 transduction, pooled, titrated and stored at −80°C until needed for T cell transduction. During vector production, the transfection efficiency of Phoenix Eco cells and the transduction efficiency of PG13 cells was assessed by monitoring the tCD34 tag expression by flow cytometry. For each viral vector production, viral RNA was purified using the NucleoSpin viral RNA isolation kit (Macherey-Nagel, Dueren, Germany) and the viral titer was assessed using either the Lenti-X qRT-PCR Titration Kit or the Retro-X qRT-PCR Titration Kit (TaKaRa). All kits were used according to the manufacturer’s instructions. RNA detection was performed by real-time qPCR on a LightCycler LC480 instrument (Roche, Diegem, Belgium).
CAR T cell production
Peripheral blood mononuclear cells (PBMCs) were isolated either from apheretic material followed by red blood cell lysis or from whole blood of healthy donors (ImmuneHealth, CHU, Tivoli) by Ficoll density gradient (VWR, Leuven, Belgium) according to standard procedures. Specifically, whole blood was diluted three times with DPBS and added carefully onto the Ficoll layer in 50-mL tubes. Tubes were centrifuged at 500 × g and the intermediate layer carefully removed. The PBMCs were subsequently washed three times with DPBS, harvested, and activated in XVivo 15 medium (Lonza, Antwerp, Belgium) supplemented with 10% of Human Male AB Plasma-Derived Serum (Access Biologicals, Vista, CA), 1 GlutaMAX (Thermo Fisher Scientific), and 100 IU/mL IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany). After isolation, the cells were activated at a density of 2 × 106 cells/mL for 48 h in complete XVivo medium and R&D grade TransAct (Miltenyi Biotec) at 1/100 dilution, in an incubator at 37°C in an atmosphere of 5% CO2/air. Cells were subsequently harvested and transduced at 1 × 106 cells/well in 6-well plates coated with 8 μg/mL retronectin (TaKaRa) with the appropriate viral vector in the presence of 100 IU/mL IL-2 (Miltenyi Biotec) and 1 μM Akti-1/2 (Bio-Techne, Minneapolis, MN) and incubated for 2 days. The vector titers were adjusted to ensure the incorporation of ≤5 viral genomes per cell. On day 4 post activation, transduced cells were expanded in flask in complete XVivo 15 medium supplemented with 100 IU/mL IL-2 at a density between 0.33 and 0.66 × 106 cells/mL for a further 48 h. On day 6 post activation, cells were collected and washed with HBSS containing 1% human serum albumin (Octapharma, Brussels, Belgium) before staining for the tCD34 tag with the CD34 MicroBead Kit (Miltenyi Biotec) for positive selection. Cells were further expanded in complete XVivo 15 medium with 100 IU/mL IL-2 and harvested between days 10 and 12 post activation, depending on the experiment.
RNA extraction and target gene expression assessment by real-time qPCR
Total cellular RNA was extracted using the RNeasy Mini Kit (QIAGEN, Antwerp, Belgium) and genomic DNA was removed with the On-Column DNaseI Digestion Set (Merck Life Sciences) following the manufacturers’ instructions. Reverse-transcription and gene expression analysis were performed in a one-step real-time qPCR by means of the LightCycler 480 RNA Master Hydrolysis Probes (Roche) on a LightCycler LC480 instrument (Roche). TaqMan assays were purchased from (Thermo Fisher Scientific) for CD3ζ (Hs00609515_m1), TRAC (Hs01062241_m1), β2M (Hs99999907_m1), and CD52 (Hs00174349_m1).
Antibody staining and target gene expression assessment by flow cytometry
For flow cytometry analysis of extracellular markers, CAR T cells were washed once with PBS, and incubated with FVS575 live/dead dye for 15 min at room temperature in the dark. They were then washed with PBS and resuspended in Attune Focusing Fluid (Thermo Fisher Scientific). CAR T cells were then incubated in round-bottom 96-well plate wells with the appropriate mix of fluorescently labeled primary antibodies and incubated for 30 min at 4°C in the dark. After a wash with Attune Focusing Fluid, they were resuspended in Attune Focusing Fluid and analyzed on an Attune NxT Acoustic Focusing Flow Cytometer (Thermo Fisher Scientific) equipped with blue (488 nm), red (638 nm), violet (405 nm), and yellow (561 nm) excitation lasers. The following fluorescently labeled primary antibodies were used to assess target gene levels: anti-CD27, anti-CD28, anti-CD52 (BioLegend, Amsterdam, the Netherlands); anti-CD95, anti-LAG-3 (Thermo Fisher Scientific); anti-TCRa/b, anti-CD3ζ (Miltenyi Biotec); HLA ABC, HLA DR (BD Biosciences, Erembodegem, Belgium). The following fluorescently labeled primary antibodies were used for the gating of transduced CAR T cells: CD34 (BioLegend); CD4, CD8 (BD Biosciences). All antibodies were titrated prior to experimental use. Data analysis was performed with FlowJo v.10.
Functional assay for CD3ζ KD
Control CAR T cells and CAR T cells carrying the shGuide sequence against CD3ζ were treated with different concentrations of the anti-CD3 monoclonal antibody OKT3, as reported in Figure 7B. (Miltenyi Biotec). After the appropriate incubation time (24 h up to 120 h), the levels of IFN-γ released by the CAR T cells into the cell culture medium were measured as a readout for TCR-mediated T cell activation, using the Human IFN-gamma Quantikine ELISA Kit (R&D Systems, Abingdon, UK) according to the manufacturer’s instructions.
Functional assay for CD95 KD
Both control CAR T cells and CAR T cells carrying the shGuide sequence against CD95 were split in two groups, which were either left untreated or incubated with Ultra-LEAF Purified anti-human CD95 (FAS) blocking antibody (BioLegend) at a concentration of 1 μg/mL. Following a 30-min incubation, each group was further split in two and either left untreated or treated with Human FAS ligand, His Tag, active trimer (CD95L, Fisher Scientific, Geel, Belgium) at a concentration of 200 ng/mL. After 24 h, CD95L-induced apoptosis was assessed by flow cytometry, by determining the number of Annexin V-positive CAR T cells with the Dead Cell Apoptosis Kit with Annexin V (AF488 & PI) (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Functional assay for β2M KD
Primary human NK cells were primed overnight with cytokines (IL-2 and IL-15), before being exposed to target T cells at a 1:3 effector to target ratio in the presence of fluorochrome-conjugated CD107a (lysosome-associated membrane protein 1) antibody. As cell surface exposure of CD107a is extremely transient owing to the recycling of the granules, CD107a antibody was included throughout the duration of the stimulation period. One hour after co-culture seeding, the Golgi transmembrane transport inhibitor monensin (BioLegend) was added to the culture, to neutralize the acidification of the granules, thus preventing the degradation of re-internalized CD107a. After a total of 4 h of co-culture, cells were stained for viability, NK and T cell markers, and the percentage of CD107a+ among viable NK cells, corresponding to the fraction of NK that have degranulated during the co-culture, was analyzed by flow cytometry.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA). Data were subjected to one-way ANOVA followed by Holm-Sidak correction for multiple comparisons. All data shown are the result of three independent biological replicates, unless otherwise specified.
Data and code availability
This study includes no data deposited in external repositories.
Acknowledgments
The authors would like to thank Benjamin Le Calvé, Nancy Ramelot, Simon Bornschein, Jennifer Bolsée, and Benjamin Violle for helping with the experiments.
Author contributions
M.R. designed the study, analyzed the data, and wrote the manuscript. M.S. designed the study, analyzed the data, and performed the experiments. F.H., T.N., J.M., C.J.-H., and P.N. performed the experiments. C.L. helped with the discussion of the data and with the writing of the manuscript. E.B. conceived the study, helped with the discussion of the data, and with the writing of the manuscript.
Declaration of interests
All authors were employees of Celyad Oncology SA during the realization of this work. The research conducted within the manuscript may lead to the development of products which may be licensed by Celyad Oncology SA. The following patents and patent applications are associated to this work: WO2021224278, WO2022233982.
Supplemental information
References
- 1.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munshi N.C., Anderson L.D., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 3.Park J.H., Rivière I., Orlow S.J., Nagler A.R., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018;19:449–455. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A., et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 6.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W., et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner J., Wickman E., DeRenzo C., Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020;28:2320–2339. doi: 10.1016/j.ymthe.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel U., Abernathy J., Savani B.N., Oluwole O., Sengsayadeth S., Dholaria B. CAR T cell therapy in solid tumors: A review of current clinical trials. eJHaem. 2022;3:24–31. doi: 10.1002/jha2.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ali Hosseini Rad S.M., Halpin J.C., Mollaei M., Smith Bell S.W.J., Hirankarn N., McLellan A.D. Metabolic and mitochondrial functioning in chimeric antigen receptor (Car)—t cells. Cancers. 2021;13:1–23. doi: 10.3390/cancers13061229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou A.J., Chen L.C., Chen Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021;20:531–550. doi: 10.1038/s41573-021-00189-2. [DOI] [PubMed] [Google Scholar]
- 11.McLellan A.D., Ali Hosseini Rad S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019;97:664–674. doi: 10.1111/imcb.12254. [DOI] [PubMed] [Google Scholar]
- 12.Yap T.A., Parkes E.E., Peng W., Moyers J.T., Curran M.A., Tawbi H.A. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021;11:1368–1397. doi: 10.1158/2159-8290.CD-20-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y., Zhu Y., Kramer A., Fang Y., Wilson M., Li Y.R., Yang L. Genetic engineering strategies to enhance antitumor reactivity and reduce alloreactivity for allogeneic cell-based cancer therapy. Front. Med. 2023;10:1135468–1135516. doi: 10.3389/fmed.2023.1135468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacLeod D.T., Antony J., Martin A.J., Moser R.J., Hekele A., Wetzel K.J., Brown A.E., Triggiano M.A., Hux J.A., Pham C.D., et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017;25:949–961. doi: 10.1016/j.ymthe.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R., et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 16.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K., et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 17.Osborn M.J., Webber B.R., Knipping F., Lonetree C.l., Tennis N., DeFeo A.P., McElroy A.N., Starker C.G., Lee C., Merkel S., et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. 2016;24:570–581. doi: 10.1038/mt.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H., et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren J., Zhang X., Liu X., Fang C., Jiang S., June C.H., Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002–17011. doi: 10.18632/oncotarget.15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren J., Liu X., Fang C., Jiang S., June C.H., Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017;23:2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou F., Lu L., Liu J., Xia B., Zhang W., Hu Q., Liu W., Zhang Y., Lin Y., Jing S., et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019;10:4109. doi: 10.1038/s41467-019-11893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee Y.-H., Lee H.J., Kim H.C., Lee Y., Nam S.K., Hupperetz C., Ma J.S.Y., Wang X., Singer O., Kim W.S., et al. PD-1 and TIGIT downregulation distinctly affect the effector and early memory phenotypes of CD19-targeting CAR T cells. Mol. Ther. 2022;30:579–592. doi: 10.1016/j.ymthe.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciraolo E., Althoff S., Ruß J., Rosnev S., Butze M., Pühl M., Frentsch M., Bullinger L., Na I.K. Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome. Int. J. Mol. Sci. 2022;23:1–14. doi: 10.3390/ijms23063207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biederstädt A., Manzar G.S., Daher M. Multiplexed engineering and precision gene editing in cellular immunotherapy. Front. Immunol. 2022;13:1–20. doi: 10.3389/fimmu.2022.1063303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarty N.S., Graham A.E., Studená L., Ledesma-Amaro R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020;11:1–13. doi: 10.1038/s41467-020-15053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S., Voigt C.A. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res. 2018;46:11115–11125. doi: 10.1093/nar/gky884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazzarotto C.R., Malinin N.L., Li Y., Zhang R., Yang Y., Lee G., Cowley E., He Y., Lan X., Jividen K., et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tycko J., Wainberg M., Marinov G.K., Ursu O., Hess G.T., Ego B.K., Aradhana, Li A., Truong A., Trevino A.E., et al. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat. Commun. 2019;10:4063. doi: 10.1038/s41467-019-11955-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgens D.W., Wainberg M., Boyle E.A., Ursu O., Araya C.L., Tsui C.K., Haney M.S., Hess G.T., Han K., Jeng E.E., et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017;8:15178. doi: 10.1038/ncomms15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran F.A., Hsu P.D., Lin C.-Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guilinger J.P., Thompson D.B., Liu D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsai S.Q., Wyvekens N., Khayter C., Foden J.A., Thapar V., Reyon D., Goodwin M.J., Aryee M.J., Joung J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 36.Papathanasiou S., Markoulaki S., Blaine L.J., Leibowitz M.L., Zhang C.-Z., Jaenisch R., Pellman D. Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing. Nat. Commun. 2021;12:5855. doi: 10.1038/s41467-021-26097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alanis-Lobato G., Zohren J., McCarthy A., Fogarty N.M.E., Kubikova N., Hardman E., Greco M., Wells D., Turner J.M.A., Niakan K.K. Frequent loss of heterozygosity in CRISPR-Cas9–edited early human embryos. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2004832117. e2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weisheit I., Kroeger J.A., Malik R., Klimmt J., Crusius D., Dannert A., Dichgans M., Paquet D. Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep. 2020;31:107689. doi: 10.1016/j.celrep.2020.107689. [DOI] [PubMed] [Google Scholar]
- 39.Boutin J., Rosier J., Cappellen D., Prat F., Toutain J., Pennamen P., Bouron J., Rooryck C., Merlio J.P., Lamrissi-Garcia I., et al. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 2021;12:4922. doi: 10.1038/s41467-021-25190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahmad A.D., Reuveni E., Goldschmidt E., Tenne T., Liberman M., Horovitz-Fried M., Khosravi R., Kobo H., Reinstein E., Madi A., et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat. Biotechnol. 2022;40:1807–1813. doi: 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Badri P., Jiang X., Borodovsky A., Najafian N., Kim J., Clausen V.A., Goel V., Habtemariam B., Robbie G.J. Pharmacokinetic and Pharmacodynamic Properties of Cemdisiran, an RNAi Therapeutic Targeting Complement Component 5, in Healthy Subjects and Patients with Paroxysmal Nocturnal Hemoglobinuria. Clin. Pharmacokinet. 2021;60:365–378. doi: 10.1007/s40262-020-00940-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwarz D.S., Hutvágner G., Du T., Xu Z., Aronin N., Zamore P.D. Asymmetry in the Assembly of the RNAi Enzyme Complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 43.Wang T., Xie Y., Tan A., Li S., Xie Z. Construction and characterization of a synthetic MicroRNA cluster for multiplex RNA interference in mammalian cells. ACS Synth. Biol. 2015;5:1193–1200. doi: 10.1021/acssynbio.5b00180. [DOI] [PubMed] [Google Scholar]
- 44.Winter J., Jung S., Keller S., Gregory R.I., Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 45.Boudreau R.L., Martins I., Davidson B.L. Artificial MicroRNAs as siRNA Shuttles: Improved Safety as Compared to shRNAs In vitro and In vivo. Mol. Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grimm D. The dose can make the poison: lessons learned from adverse in vivo toxicities caused by RNAi overexpression. Silence. 2011;2:8. doi: 10.1186/1758-907X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maczuga P., Lubelski J., van Logtenstein R., Borel F., Blits B., Fakkert E., Costessi A., Butler D., van Deventer S., Petry H., et al. Embedding siRNA sequences targeting Apolipoprotein B100 in shRNA and miRNA scaffolds results in differential processing and in vivo efficacy. Mol. Ther. 2013;21:217–227. doi: 10.1038/mt.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altuvia Y., Landgraf P., Lithwick G., Elefant N., Pfeffer S., Aravin A., Brownstein M.J., Tuschl T., Margalit H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697–2706. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y., Luo J., Zhang H., Lu J. J. microRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol. 2016;33:2232–2247. doi: 10.1093/molbev/msw089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bourhill T., Arbuthnot P., Ely A. Successful disabling of the 5′ UTR of HCV using adeno-associated viral vectors to deliver modular multimeric primary microRNA mimics. J. Virol. Methods. 2016;235:26–33. doi: 10.1016/j.jviromet.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y.P., Haasnoot J., ter Brake O., Berkhout B., Konstantinova P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008;36:2811–2824. doi: 10.1093/nar/gkn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi J.G., Bharaj P., Abraham S., Ma H., Yi G., Ye C., Dang Y., Manjunath N., Wu H., Shankar P. Multiplexing seven miRNA-Based shRNAs to suppress HIV replication. Mol. Ther. 2015;23:310–320. doi: 10.1038/mt.2014.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boudreau R.L., Monteys A.M., Davidson B.L. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA. 2008;14:1834–1844. doi: 10.1261/rna.1062908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myburgh R., Cherpin O., Schlaepfer E., Rehrauer H., Speck R.F., Krause K.H., Salmon P. Optimization of critical hairpin features allows miRNA-based gene knockdown upon single-copy transduction. Mol. Ther. Nucleic Acids. 2014;3:e207. doi: 10.1038/mtna.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fowler D.K., Williams C., Gerritsen A.T., Washbourne P. Improved knockdown from artificial microRNAs in an enhanced miR-155 backbone: a designer’s guide to potent multi-target RNAi. Nucleic Acids Res. 2016;44:e48. doi: 10.1093/nar/gkv1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brake O.T., Hooft K. ’t, Liu Y.P., Centlivre M., Jasmijn von Eije K., Berkhout B. Lentiviral Vector Design for Multiple shRNA Expression and Durable HIV-1 Inhibition. Mol. Ther. 2008;16:557–564. doi: 10.1038/sj.mt.6300382. [DOI] [PubMed] [Google Scholar]
- 57.Muiños-Gimeno M., Montfort M., Bayés M., Estivill X., Espinosa-Parrilla Y. Design and evaluation of a panel of single-nucleotide polymorphisms in microRNA genomic regions for association studies in human disease. Eur. J. Hum. Genet. 2010;18:218–226. doi: 10.1038/ejhg.2009.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutiérrez-Vázquez C., Rodríguez-Galán A., Fernández-Alfara M., Mittelbrunn M., Sánchez-Cabo F., Martínez-Herrera D.J., Ramírez-Huesca M., Pascual-Montano A., Sánchez-Madrid F. miRNA profiling during antigen-dependent T cell activation: A role for miR-132-3p. Sci. Rep. 2017;7:3508. doi: 10.1038/s41598-017-03689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teteloshvili N., Smigielska-Czepiel K., Kroesen B.-J., Brouwer E., Kluiver J., Boots A.M.H., van den Berg A. T-cell Activation Induces Dynamic Changes in miRNA Expression Patterns in CD4 and CD8 T-cell Subsets. MicroRNA. 2015;4:117–122. doi: 10.2174/2211536604666150819194636. [DOI] [PubMed] [Google Scholar]
- 60.Brunner S., Herndler-Brandstetter D., Arnold C.R., Wiegers G.J., Villunger A., Hackl M., Grillari J., Moreno-Villanueva M., Bürkle A., Grubeck-Loebenstein B. Upregulation of miR-24 is associated with a decreased DNA damage response upon etoposide treatment in highly differentiated CD8 + T cells sensitizing them to apoptotic cell death. Aging Cell. 2012;11:579–587. doi: 10.1111/j.1474-9726.2012.00819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khuu C., Utheim T.P., Sehic A. The Three Paralogous MicroRNA Clusters in Development and Disease, miR-17-92, miR-106a-363, and miR-106b-25. Sci. Tech. Rep. 2016:1–10. doi: 10.1155/2016/1379643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.LeibundGut-Landmann S., Waldburger J.-M., Krawczyk M., Otten L.A., Suter T., Fontana A., Acha-Orbea H., Reith W. Mini-review: Specificity and expression of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol. 2004;34:1513–1525. doi: 10.1002/eji.200424964. [DOI] [PubMed] [Google Scholar]
- 63.Contrant M., Fender A., Chane-Woon-Ming B., Randrianjafy R., Vivet-Boudou V., Richer D., Pfeffer S. Importance of the RNA secondary structure for the relative accumulation of clustered viral microRNAs. Nucleic Acids Res. 2014;42:7981–7996. doi: 10.1093/nar/gku424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hooykaas M.J.G., Kruse E., Wiertz E.J.H.J., Lebbink R.J. Comprehensive profiling of functional Epstein-Barr virus miRNA expression in human cell lines. BMC Genom. 2016;17:644–713. doi: 10.1186/s12864-016-2978-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryazansky S.S., Gvozdev V.A., Berezikov E. Evidence for post-transcriptional regulation of clustered microRNAs in Drosophila. BMC Genom. 2011;12:371. doi: 10.1186/1471-2164-12-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vilimova M., Pfeffer S. Vol. 14. Wiley Interdiscip. Rev. RNA; 2023. (Post-Transcriptional Regulation of Polycistronic microRNAs). [DOI] [PubMed] [Google Scholar]
- 67.Chaulk S.G., Xu Z., Glover M.J.N., Fahlman R.P. MicroRNA miR-92a-1 biogenesis and mRNA targeting is modulated by a tertiary contact within the miR-17∼92 microRNA cluster. Nucleic Acids Res. 2014;42:5234–5244. doi: 10.1093/nar/gku133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anderson A.C., Joller N., Kuchroo V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Norman D.J. Mechanisms of Action and Overview of OKT3. Ther. Drug Monit. 1995;17:615–620. doi: 10.1097/00007691-199512000-00012. [DOI] [PubMed] [Google Scholar]
- 70.Arakaki R., Yamada A., Kudo Y., Hayashi Y., Ishimaru N. Mechanism of Activation-Induced Cell Death of T Cells and Regulation of FasL Expression. Crit. Rev. Immunol. 2014;34:301–314. doi: 10.1615/critrevimmunol.2014009988. [DOI] [PubMed] [Google Scholar]
- 71.Figueiredo C., Seltsam A., Blasczyk R. Class-gene-and group-specific HLA silencing by lentiviral shRNA delivery. J. Mol. Med. 2006;84:425–437. doi: 10.1007/s00109-005-0024-2. [DOI] [PubMed] [Google Scholar]
- 72.Gornalusse G.G., Hirata R.K., Funk S.E., Riolobos L., Lopes V.S., Manske G., Prunkard D., Colunga A.G., Hanafi L.A., Clegg D.O., et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017;35:765–772. doi: 10.1038/nbt.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoerster K., Uhrberg M., Wiek C., Horn P.A., Hanenberg H., Heinrichs S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for “Off-the-Shelf” Immunotherapy. Front. Immunol. 2021;11:1–14. doi: 10.3389/fimmu.2020.586168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Riolobos L., Hirata R.K., Turtle C.J., Wang P.-R., Gornalusse G.G., Zavajlevski M., Riddell S.R., Russell D.W. HLA Engineering of Human Pluripotent Stem Cells. Mol. Ther. 2013;21:1232–1241. doi: 10.1038/mt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torikai H., Reik A., Soldner F., Warren E.H., Yuen C., Zhou Y., Crossland D.L., Huls H., Littman N., Zhang Z., et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–1349. doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grimm D., Streetz K.L., Jopling C.L., Storm T.A., Pandey K., Davis C.R., Marion P., Salazar F., Kay M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 77.An D.S., Qin F.X.-F., Auyeung V.C., Mao S.H., Kung S.K.P., Baltimore D., Chen I.S.Y. Optimization and Functional Effects of Stable Short Hairpin RNA Expression in Primary Human Lymphocytes via Lentiviral Vectors. Mol. Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Al-Homsi A.-S., Anguille S., Deeren D., Nishihori T., Meuleman N., Abdul-Hay M., Morgan G.J., Brayer J., Braun N., Lonez C., et al. Immunicy-1: Targeting BCMA with Cyad-211 to Establish Proof of Concept of an shRNA-Based Allogeneic CAR T Cell Therapy Platform. Blood. 2021;138:2817. [Google Scholar]
- 79.Deeren D., Maertens J.A., Lin T.L., Beguin Y., Alcantar-Orozco E., Dheur M.-S., Breman E., Braun N., Lonez C., Gilham D., et al. Co-Expression of an shRNA Targeting MICA/Micb Improves the Clinical Activity of a NKG2D-Based CAR T in Patients with Relapsed/Refractory AML/MDS. Blood. 2021;138:408. [Google Scholar]
- 80.Seyhan A.A. A multiplexed miRNA and transgene expression platform for simultaneous repression and expression of protein coding sequences. Mol. Biosyst. 2016;12:295–312. doi: 10.1039/c5mb00506j. [DOI] [PubMed] [Google Scholar]
- 81.Rad S.M.A.H., Halpin J.C., Tawinwung S., Suppipat K., Hirankarn N., McLellan A.D. MicroRNA-mediated metabolic reprogramming of chimeric antigen receptor T cells. Immunol. Cell Biol. 2022;100:424–439. doi: 10.1111/imcb.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dickins R.A., Hemann M.T., Zilfou J.T., Simpson D.R., Ibarra I., Hannon G.J., Lowe S.W. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat. Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- 83.Silva J.M., Li M.Z., Chang K., Ge W., Golding M.C., Rickles R.J., Siolas D., Hu G., Paddison P.J., Schlabach M.R., et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- 84.Zeng Y., Cullen B.R. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32:4776–4785. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Knott S.R.V., Maceli A., Erard N., Chang K., Marran K., Zhou X., Gordon A., Demerdash O.E., Wagenblast E., Kim S., et al. A Computational Algorithm to Predict shRNA Potency. Mol. Cell. 2014;56:796–807. doi: 10.1016/j.molcel.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amendola M., Passerini L., Pucci F., Gentner B., Bacchetta R., Naldini L. Regulated and multiple miRNA and siRNA delivery into primary cells by a lentiviral platform. Mol. Ther. 2009;17:1039–1052. doi: 10.1038/mt.2009.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rice G.M., Shivashankar V., Ma E.J., Baryza J.L., Nutiu R. Functional Atlas of Primary miRNA Maturation by the Microprocessor. Mol. Cell. 2020;80:892–902.e4. doi: 10.1016/j.molcel.2020.10.028. [DOI] [PubMed] [Google Scholar]
- 88.Kwon S.C., Baek S.C., Choi Y.G., Yang J., Lee Y.S., Kim V.N., Woo J.S., Kim V.N. Molecular Basis for the Single-Nucleotide Precision of Primary microRNA Processing. Mol. Cell. 2019;73:505–518.e5. doi: 10.1016/j.molcel.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 89.Quick-Cleveland J., Jacob J.P., Weitz S.H., Shoffner G., Senturia R., Guo F. The DGCR8 RNA-Binding Heme Domain Recognizes Primary MicroRNAs by Clamping the Hairpin. Cell Rep. 2014;7:1994–2005. doi: 10.1016/j.celrep.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han J., Lee Y., Yeom K.H., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N. Molecular Basis for the Recognition of Primary microRNAs by the Drosha-DGCR8 Complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 91.Burke J.M., Kelenis D.P., Kincaid R.P., Sullivan C.S. A central role for the primary microRNA stem in guiding the position and efficiency of Drosha processing of a viral pri-miRNA. RNA. 2014;20:1068–1077. doi: 10.1261/rna.044537.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nguyen T.D., Trinh T.A., Bao S., Nguyen T.A. Secondary structure RNA elements control the cleavage activity of DICER. Nat. Commun. 2022;13:2138–2216. doi: 10.1038/s41467-022-29822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park J.-E., Heo I., Tian Y., Simanshu D.K., Chang H., Jee D., Patel D.J., Kim V.N. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475:201–205. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nedorezova D.D., Dubovichenko M.V., Belyaeva E.P., Grigorieva E.D., Peresadina A.V., Kolpashchikov D.M. Specificity of oligonucleotide gene therapy (OGT) agents. Theranostics. 2022;12:7132–7157. doi: 10.7150/thno.77830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nielsen C.B., Shomron N., Sandberg R., Hornstein E., Kitzman J., Burge C.B. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 2007;13:1894–1910. doi: 10.1261/rna.768207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fang W., Bartel D.P. The Menu of Features that Define Primary MicroRNAs and Enable De Novo Design of MicroRNA Genes. Mol. Cell. 2015;60:131–145. doi: 10.1016/j.molcel.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hutter K., Lohmüller M., Jukic A., Eichin F., Avci S., Labi V., Szabo T.G., Hoser S.M., Hüttenhofer A., Villunger A., Herzog S. SAFB2 Enables the Processing of Suboptimal Stem-Loop Structures in Clustered Primary miRNA Transcripts. Mol. Cell. 2020;78:876–889.e6. doi: 10.1016/j.molcel.2020.05.011. [DOI] [PubMed] [Google Scholar]
- 98.Chakraborty C., Sharma A.R., Sharma G., Doss C.G.P., Lee S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids. 2017;8:132–143. doi: 10.1016/j.omtn.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Du P., Wang L., Sliz P., Gregory R.I. A Biogenesis Step Upstream of Microprocessor Controls miR-17∼92 Expression. Cell. 2015;162:885–899. doi: 10.1016/j.cell.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu T., Chen P., Fu Q., Liu Y., Ishaq M., Li J., Ma L., Guo D. Comparative Studies of Various Artificial microRNA Expression Vectors for RNAi in Mammalian Cells. Mol. Biotechnol. 2010;46:34–40. doi: 10.1007/s12033-010-9264-7. [DOI] [PubMed] [Google Scholar]
- 101.Trujillo R.D., Yue S.-B., Tang Y., O’Gorman W.E., Chen C.-Z. The potential functions of primary microRNAs in target recognition and repression. EMBO J. 2010;29:3272–3285. doi: 10.1038/emboj.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nowak C.M., Lawson S., Zerez M., Bleris L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Res. 2016;44:9555–9564. doi: 10.1093/nar/gkw908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cox D.B.T., Gootenberg J.S., Abudayyeh O.O., Franklin B., Kellner M.J., Joung J., Zhang F. RNA editing with CRISPR-Cas13. Science (80- 2017;358:1019–1027. doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B.T., Shmakov S., Makarova K.S., Semenova E., Minakhin L., et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K., et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smargon A.A., Cox D.B.T., Pyzocha N.K., Zheng K., Slaymaker I.M., Gootenberg J.S., Abudayyeh O.A., Essletzbichler P., Shmakov S., Makarova K.S., et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol. Cell. 2017;65:618–630.e7. doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study includes no data deposited in external repositories.