

Abstract
Pancreatic ductal adenocarcinoma (PDAC) has been classified into classical and basal-like transcriptional subtypes by bulk RNA measurements. However, recent work has uncovered greater complexity to transcriptional subtypes than was initially appreciated using bulk RNA expression profiling. To provide a deeper understanding of PDAC subtypes, we developed a multiplex immunofluorescence (mIF) pipeline that quantifies protein expression of six PDAC subtype markers (CLDN18.2, TFF1, GATA6, KRT17, KRT5, and S100A2) and permits spatially resolved, single-cell interrogation of pancreatic tumors from resection specimens and core needle biopsies. Both primary and metastatic tumors displayed striking intra-tumoral subtype heterogeneity that was associated with patient outcomes, existed at the scale of individual glands, and was significantly reduced in patient-derived organoid cultures. Tumor cells co-expressing classical and basal markers were present in >90% of tumors, existed on a basal-classical polarization continuum, and were enriched in tumors containing a greater admixture of basal and classical cell populations. Cell-cell neighbor analyses within tumor glands further suggested that co-expressor cells may represent an intermediate state between expression subtype poles. The extensive intra-tumoral heterogeneity identified through this clinically applicable mIF pipeline may inform prognosis and treatment selection for patients with PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the 3rd leading cause of cancer-related death in the United States(1). Patients commonly present with advanced disease and have limited treatment options that are generally restricted to two multi-agent chemotherapy regimens(2,3). At the genomic level, PDAC is characterized by mutations in KRAS, SMAD4, TP53 and CDKN2A, and only a modest percentage of tumors harbor therapeutically actionable targets(4–6). At the RNA expression level, numerous transcriptional subtypes have been identified. However, studies have characterized similar phenotypes among these subtypes, leading to a dichotomous framework in which tumors are classified as classical/progenitor (“classical”) or basal-like/quasimesenchymal/squamous (“basal”)(7–9). Notably, patients with basal tumors appear to have less chemotherapy responsive disease and shorter survival times(7,10). Based on these data, randomized clinical trials are currently underway for PDAC patients to evaluate expression subtyping in therapy selection (NCT04469556, NCT04683315).
Recently, we and others have uncovered greater complexity to transcriptional subtypes than was initially appreciated using bulk RNA expression profiling(10,11). Single-cell RNA sequencing (scRNA-seq) studies have now shown that a single tumor can harbor both classical and basal cells, resulting in a mixed or “hybrid” composition, and some tumors contain cells expressing both classical and basal markers within the same cell(12–14). However, the frequency, clinical implications, spatial architecture, and site differences of such mixed tumors and co-expressor cells are poorly understood(15). Furthermore, while scRNA-seq provides highly resolved and detailed information on patient tissue, it is unlikely to be feasible in routine clinical practice. In contrast, immunohistochemical staining is standard practice in clinical care(16), and several studies have examined single marker immunohistochemistry to identify PDAC expression subtypes(17,18). Nevertheless, single marker immunohistochemical approaches preclude detection of cell states defined by expression of multiple markers within a single cell, do not provide quantitative cell-level data and have limited capacity for spatial architecture assessment. Here, we present the translation of multi-gene transcriptomic signatures to a format that provides clinically feasible, multi-marker, spatially-resolved, single cell characterization of pancreatic tumors by creating a high-throughput pipeline that utilizes multiplex immunofluorescence, digital image analysis, and supervised machine learning.
Materials and methods
Tissue cohorts
We analyzed two patient cohorts with formalin-fixed paraffin-embedded (FFPE) tumor tissue. (Supplementary Fig. S1). The multi-institutional primary resection cohort comprised 305 non-neoadjuvant treated PDAC resection specimens with extensive clinicopathologic characterization (Supplementary Table.T1 & T2). For survival analyses, six cases were excluded due to metastatic disease at resection, and 10 cases were excluded due to 30-day or in-hospital mortality, leading to a cohort for analysis of 289 resected PDAC cases. Tissue from these tumors was analyzed by tissue microarrays (TMAs) designed to contain 2 representative 1 mm diameter cores from each tumor. Within this cohort, a matched set of 25 whole slide sections (WSS) was also evaluated to allow for TMA to whole slide tissue area comparisons. The metastatic cohort contained 37 core needle biopsies of metastatic PDAC from patients not included in the primary resection cohort, all of whom had FFPE tissue for protein analysis, 14 of whom had matched bulk RNA-seq data(6) and 10 of whom had matched scRNA-seq data(14) (Supplementary Table.T3). Patients treated at DF/BWCC signed written informed consent for participation in the study, whereas, at URMC and SCI, informed consent was waived as patients were identified retrospectively, according to institutional review board (IRB)-exempt protocols. The study design was approved by the IRB at each institution.
Organoid cohort
The organoid TMA cohort was developed from organoids derived from tissue specimens from 77 patients with metastatic (n=49) or localized PDAC (n=28) who underwent resection or biopsy between November 2015 and July 2019 at Dana-Farber/Brigham and Women’s Cancer Center (DF/BWCC)(6) (Supplementary Table.T4). Investigators obtained written, informed consent from patients at least 18 years old with pancreatic cancer for Dana-Farber/Harvard Cancer Center Institutional Review Board (IRB)-approved protocols 11–104, 17–000, 03–189, and/or 14–408 for tissue collection, molecular analysis, and organoid generation. Patient-derived organoids were cultured as previously described in Matrigel (Corning) with complete organoid media(14) for approximately 1–2 weeks, until multicellular organoids were apparent but still subconfluent. Organoids in Matrigel domes were washed once with PBS, then fixed with 10% neutral buffered formalin overnight. For each sample, 5 intact Matrigel domes with organoids were gently lifted from the culture plate with a cell scraper, transferred to an Eppendorf tube, washed with cold PBS, spun down, resuspended in Histogel (Thermo Fisher), and transferred to a cryomold (Sakura Finetek USA). Histogel blocks were further fixed in 10% neutral buffered formalin before embedding in paraffin. Two 1 mm diameter cores with representative areas of tumor cells were taken from each formalin-fixed paraffin-embedded (FFPE) organoid block to construct organoid TMA blocks. The study was conducted in accordance with the U.S. Common Rule. Organoid cultures were routinely tested for mycoplasma contamination.
Multiplex immunofluorescence
A multi-marker panel was developed to characterize tumor cell subtype in FFPE 4μm tissue sections using mIF. The panel comprised three basal protein markers (KRT17, KRT5, S100A2) and three classical protein markers (GATA6, TTF1, CLDN18.2; Supplementary Table.T5). DAPI was included for identification of nuclei and pan-cytokeratin for identification of epithelial cells. Secondary Opal polymer HRP anti-mouse and anti-rabbit antibodies and Opal fluorophores were used to detect primary antibodies. Primary antibodies were first optimized via immunohistochemistry on control tissue to confirm contextual specificity. Monoplex immunofluorescence and iterative multiplex fluorescent staining were then used to optimize staining order, antibody-fluorophore assignments, and fluorophore concentrations (Supplementary Table.T6, Fig.S2). Multiplex staining was performed using a Leica BOND RX Research Stainer (Leica Biosystems, Buffalo, IL) with sequential cycles of antigen retrieval, protein blocking, primary antibody incubation, secondary antibody incubation, and fluorescent labeling (Supplementary Table.T7).
For all cohorts, overview images of stained slides were acquired at 10X magnification using a Vectra Polaris imaging system (PhenoImager HT) (Akoya Biosciences, Waltham, MA) and regions of interest (ROIs) were selected for multispectral image acquisition at 20X. For the primary resection TMA-WSS comparison, 6–7 ROIs were selected per WSS case from tumor areas. All available tumor area was sampled for metastatic biopsies. After unmixing using a spectral library of single-color references, each image was inspected to ensure uniform staining quality and adequate tumor representation (only TMA cores containing >5% tumor were included). A supervised machine learning algorithm was trained to segment tissue into regions of tumor, stroma or empty/acellular (“other”) space with segmentation resolution, edge trimming and minimum segmentation size settings chosen to maximize the annotation of individual, complete tumor glands as separate tumor tissue regions within each ROI (inForm 2.4.1, Akoya Biosciences) (Supplementary Fig.S3A). After cell enumeration, a separate supervised machine learning algorithm was trained to segment individual cells into nuclear and cytoplasmic regions. Tumor cells were identified by the presence of pan-cytokeratin expression within tumor classified regions. Regions identified as stromal or ‘other’ were excluded from analysis. All images underwent manual inspection to ensure accurate tissue and cell segmentation and absence of tissue processing artifacts. Pan-cytokeratin-positive epithelial cells not representing invasive cancer were identified by a pathologist, confirmed by corresponding H&E and manually excluded from further analysis. Single-cell-level data were exported and further processed and analyzed using R (v3.6.2, R Foundation for Statistical Computing; Vienna, Austria) and RStudio (v1.2.5033, RStudio, PBC; Boston, MA) (Supplementary Fig.S3B).
Cell subtype classification
Raw fluorescence intensities for each marker were normalized using z-scoring to enable comparison across multi-institution samples and staining batches. We next determined minimum positivity thresholds for each marker in the relevant subcellular compartment by jointly assessing qualitative expression pattern and the intensity value for which more than 99% of stromal cells were identified as negative, since stromal cells do not express tumor subtyping markers (excluding GATA6), (Supplementary Table.8). For GATA6, we determined an intensity value for which more than 98% of stromal cells are considered negative, as a small proportion of fibroblasts may express this marker. Tumor cell expression subtypes were then defined based upon combinatorial marker positivity across the six markers, yielding 64 total combinations, including seven pure classical marker combinations, seven pure basal marker combinations, 49 co-expressed marker combinations, and 1 combination representing absent expression for all six markers. (Fig. 1A, Supplementary Table.T9). To perform tumor-level subtyping, subtype fractions were calculated for each tumor using the total number of basal and classical cells as the denominator. For our two-group classification, tumors with >50% of subtype informative cells expressing solely classical markers were deemed classical, while tumors with >50% of subtype informative cells expressing solely basal markers were deemed classical. For our three-group classification, tumors with >90% of subtype informative cells expressing classical markers only were deemed classical-predominant, tumors with >90% of subtype informative cells expressing basal markers only were deemed basal-predominant, and the remaining tumors were classified as mixed.
Figure 1. Single cell, protein-based expression subtype assessment in a large cohort of resected primary pancreatic tumors reveals marked intratumor heterogeneity.
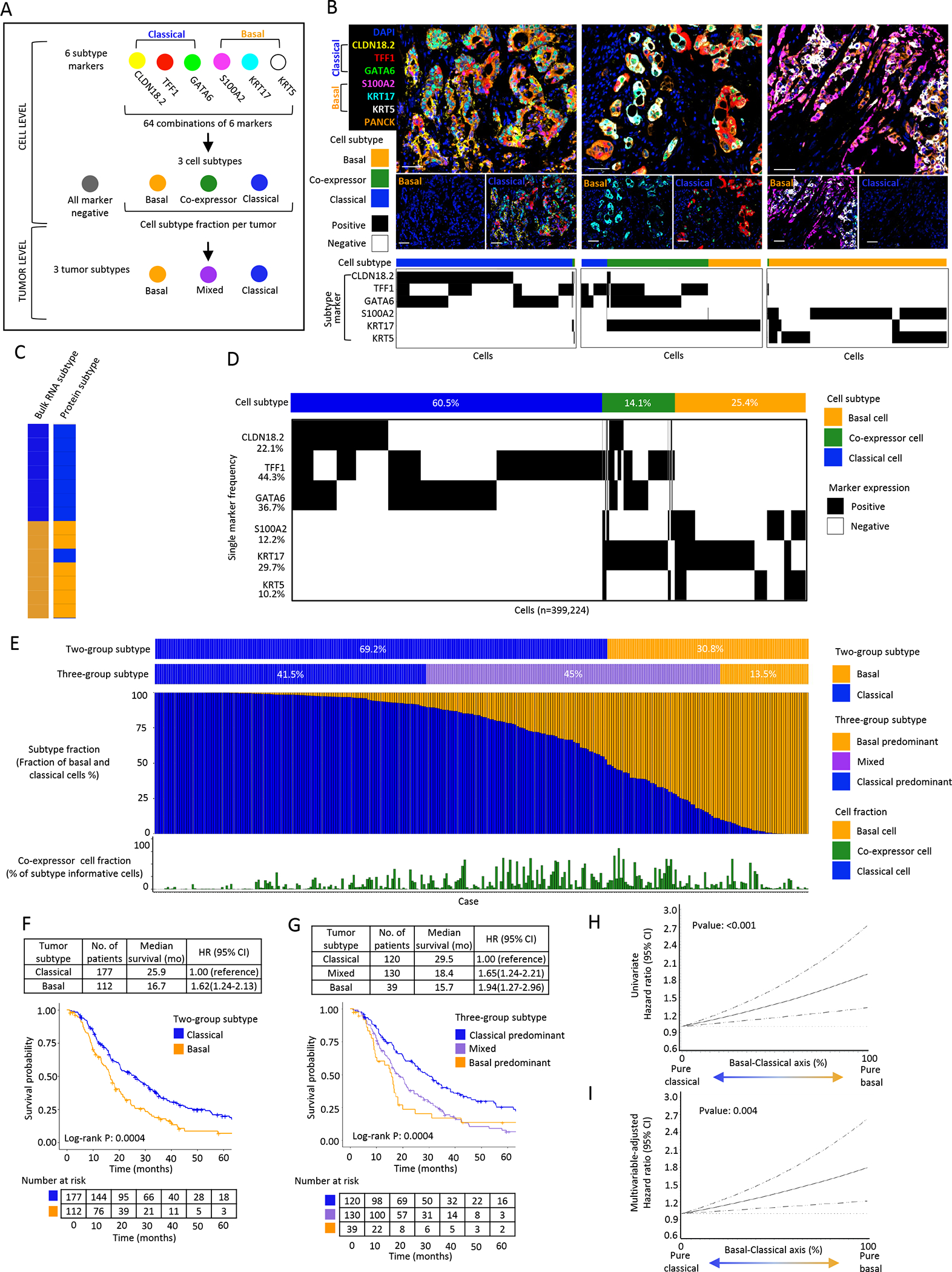
A: Schematic of multiplex immunofluorescence (mIF) panel analysis workflow to determine cell and tumor subtype. B: Representative mIF images of (left to right) classical, mixed and basal tumor expression subtypes. Plots depict single marker expression and subtype for individual epithelial cells in the corresponding tumor. Scale bar = 50μm. C: Bulk RNA-seq tumor subtype compared to protein tumor subtype in metastatic PDAC (n=14), with 13/14 assignments concordant. D: Cell subtype summary of 289 primary PDAC specimens. Cell subtype across 399,224 subtype informative tumor cells. Plot depicts cell level subtype marker combinations. Percentages represent overall cell positivity per marker. E: Tumor subtype summary across 289 primary PDAC specimens with each column representing a patient tumor. Tumors were classified using both two-group and three-group classification schemes. For each tumor, the fraction of basal and classical cells is shown, along with the fraction of co-expressor cells. F: Kaplan-Meier plot showing overall survival (OS) by tumor subtype using two-group classification. G: Kaplan-Meier plot showing OS by tumor subtype using three-group classification. H: Univariate restricted cubic spline regression curves for association of basal-classical axis score with OS (P:<0.001). I: Multivariable-adjusted restricted cubic spline regression curves for association of basal-classical axis score with OS (P:0.004). Spline regression curves adjusted for age, sex, pathologic N stage, tumor grade, lymphovascular invasion, and resection margin status.
Co-expressor cell polarization
The strength of marker expression in co-expressor cells was evaluated using a subtype polarization score. To generate this score, each subtype marker intensity across all subtype informative cells was split into percentiles. For each cell, basal percentile values across 3 basal markers and classical percentile values across 3 classical markers were summed respectively, producing a basal score (0–300) and classical score (0–300) per cell. The classical score was then subtracted from the basal score to produce an overall subtype score per cell. Cells with a positive subtype score were defined as classical polarized and those with a negative score were defined as basal polarized. Within each group, the 50th percentile was used as a cut-off to delineate strong and weak polarization classes. No co-expressor cells were found to have a subtype polarization score of 0 (Supplementary Fig. S4).
Gland-level spatial analysis
A machine learning algorithm was trained to detect CKPAN positive cells and define as tumor. Each separate and non-contiguous tumor region within an ROI was considered an individual tumor gland. To minimize bias from tangential or partial sectioning, glands containing fewer than five tumor cells were excluded from analysis. The fraction of basal, classical, co-expressor and unclassified cells was calculated within each gland. For direct neighbor analysis, classical, basal, and co-expressor cells were each separately designated as the referent cell, and the subtype of the two flanking neighbor cells was determined to generate observed neighbor cell subtype frequencies using the stringr (v1.4.0) package. These frequencies were then divided by the neighbor subtype frequencies that would be expected if cells were distributed randomly within a gland resulting in a difference score per gland unit. For domain-level analysis, regions of directly adjacent cells with identical subtypes were represented as a single subtype regardless of the number of involved cells. All glands with at least two transitions between different domains were then analyzed to generate the observed frequencies of domain-level organization.
RNA sequencing analysis
Bulk RNA-sequencing sample collection were performed as previously described(19). RNA-seq reads were aligned to GRCh38 and gene counts were generated using STAR Aligner (v2.6.1c)(20) as previously described(6). Normalized expression (TPM) values were generated using RSEM.(21) Heatmaps were generated using the transformed expression values (log2(TPM+1)) and subtype classifications were assigned via hierarchical clustering of cases based on expression of basal and classical gene sets. Heatmap plots were generated using R version 4.0.3 and pheatmap (v1.0.12) package. Single-cell RNA-seq sample collection and RNA subtyping of individual malignant cells from tumor samples were performed as previously described(11). Frequencies of cells classified as basal or classical were compared across metastatic samples analyzed by both scRNA-seq and mIF.
scRNA-seq pseudotime analysis
We performed a pseudotime analysis of data from Raghavan et al. which employed single-cell RNA sequencing of metastatic biopsies(14) and an independent dataset available from Peng et al(22), which included 57,530 cells derived from 24 PDAC samples from primary resections analyzed by scRNA-seq(22). This approach enabled both method and phenotype corroboration in two distinct datasets from primary and metastatic tumors. We accessed the processed Peng et al. counts data, which provided a ready comparison with our own single-cell RNAseq data. Separately for each data set, we normalized the data with Seurat(23), integrated the tumor cells by patient with Harmony(24) and generated a single-cell RNAseq dataset. We then generated unbiased UMAP representations of the normalized and integrated tumor cells, and subsequently overlaid subtype labels based on previously described gene sets for the classical, basal and co-expressor phenotypes(14,25).
We next separately analyzed both single-cell datasets using Monocle3(26), which is a standard method used to infer pseudo-temporal ordering (“pseudotime”) from scRNA-seq data. Given an input set of genes, approaches like Monocle3 learn an abstract unit of progress through the process that the input genes describe (e.g. differentiation trajectories). In this case we identified pseudo-temporal ordering for PDAC cancer cells from both datasets independently using an unbiased (variable genes) gene set. For the pseudotime analysis of the trajectories, we chose to begin this analysis at the root points in classical cell populations based on the hypothesis that tumor cells initially have a classical phenotype (the most commonly observed bulk tumor type in human PDAC) and may then progress to a basal phenotype with additional genetic or phenotypic alterations or perturbations such as chemotherapy. For the pseudotime analysis of the trajectories, we chose to begin this analysis at the root points in classical cell populations based on the hypothesis that tumor cells begin as classical (the most commonly observed bulk tumor type in human PDAC) and may progress to basal with additional genetic or phenotypic alterations or perturbations such as chemotherapy.
Statistical analysis
We used Cox proportional hazards regression to evaluate associations between two- or three-class tumor subtype classifications and DFS and OS. We also assessed these associations after dividing tumor subtype into quartiles to evaluate the full continuum of subtype percentage. The proportional hazards assumption was satisfied by evaluating the product of the exposure variable and time as a time-dependent variable in the Cox regression models (all P > 0.05). We performed a test for trend by including quartile-specific medians as a continuous variable and evaluated the Wald χ2 test for P values. In survival analyses, univariable analyses were performed to compute hazard ratios (HRs) and 95% confidence intervals (CI) for OS and DFS. We used the Kaplan–Meier method to provide median survival times and to graphically demonstrate survival curves. The log-rank test was used to evaluate statistical significance. We also adjusted for potential prognostic factors in multivariable models, which included age, sex, pathologic N stage (N0, N1, Nx), lymphovascular invasion (negative, positive, unknown), receipt of perioperative therapy, and resection margin status (R0, R1, R2, unknown). To evaluate relationships between tumor subtype, tumor grade and survival(27), multivariable models were further adjusted for tumor grade (well/moderately differentiated, poorly differentiated, unknown). Stratified analyses of survival for the two-group subtype classification were performed by adjuvant treatment received: (1) adjuvant gemcitabine alone or in combination with another agent (N=172 patients), (2) non-gemcitabine treatment programs or no adjuvant treatment (N=117 patients). Restricted cubic spline regression models with 3 knots were used to illustrate the relationships between basal-classical axis score and survival. The Wilcoxon rank-sum test and Fisher’s exact test were used to evaluate associations of tumor subtype fraction or two-class and three-class tumor subtype with tumor and patient characteristics, respectively. Two-sided P≤0.05 was considered statistically significant. All statistical analyses were performed with SAS 9.4 software (SAS Institute).
Data availability statement
De-identified single-cell RNA-seq data(14) used in this study are publicly available for download and visualization via the Single Cell Portal: https://singlecell.broadinstitute.org/single_cell/study/SCP1644. De-identified bulk RNA-seq data(19) used in this study have been deposited in dbGaP under accession number phs001652.v1.p1. All remaining data generated in this study are available upon request from the corresponding author.
Results
Single-cell multiplex immunofluorescence assay recapitulates PDAC tumor basal-classical transcriptomic subtyping
To conduct in situ, single cell, spatially resolved characterization of transcriptional subtypes in PDAC, we developed a multiplex immunofluorescence (mIF) panel containing 3 markers to identify basal cells (KRT5, KRT17, S100A2) and 3 markers to identify classical cells (GATA6, CLDN18.2, TFF1) (Supplementary Fig.S2; Supplementary Table.T5). Cell subtype was classified by combinatorial marker expression, yielding 64 possible marker combinations, including pure classical and pure basal cell subtypes, cells co-expressing both basal and classical markers termed “co-expressor cells”, and cells negative for all markers. (Fig. 1A–B; Supplementary Table.T9).
To assess the ability of our mIF assay to predict tumor subtyping classifications derived from bulk RNA sequencing (RNA-seq), we applied our assay to a cohort of 14 metastatic PDAC biopsies with existing RNA-seq data,(6) with 7 cases classified as classical and 7 cases as basal by bulk RNA-seq. By mIF (median 7,421 subtype-informative cells per case), 13 of 14 assignments were concordant with bulk RNA-seq (Fig. 1C). The single discordant case (Fig 1C; basal by bulk RNA-seq, classical by mIF) demonstrated strong RNA expression across transcripts from both basal and classical gene sets, and single-cell mIF measurements indicated that it contained large numbers of both basal and classical cells, so was difficult to reliably classify in a binary classification schema (Supplementary Fig.S5).
Single cell RNA-seq is increasingly being used to identify the diversity of cell types within complex tumors(22,28). Thus, we assessed tumor subtype assignment in a separate cohort of 10 cases by performing scRNA-seq from fresh tissue (3,062 total tumor cells(14)) and mIF from corresponding FFPE tissue (151,406 total tumor cells). Across these 10 tumors, we observed perfect concordance for tumor binary subtype classification. Thus, our protein-based mIF assay yielded tumor subtype classifications that were highly concordant with those derived from both bulk and single-cell RNA-seq.
Primary pancreatic tumors demonstrate substantial intra-tumoral expression subtype heterogeneity with relevance for patient outcomes
To characterize the single-cell subtype landscape of localized PDAC, we applied our mIF subtype assay to FFPE tissue microarrays (TMAs) containing 289 tumors from a multi-institutional primary resection cohort (Supplementary Table.T1)(19,29). Cumulatively, we identified 775,632 tumor cells, with 399,224 (51.4%) tumor cells classified as basal, classical or co-expressor cells (subtype informative cells). TFF1 was the most broadly expressed marker (positive in 44.3% of subtype-informative cells) while KRT5 was the least abundant marker (positive in 10.2% of subtype-informative cells) (Fig. 1D). Overall, 60.5% of subtype-informative cells were classified as classical, 25.4% were classified as basal and 14.1% of cells were classified as co-expressor cells, given that they showed expression of both classical and basal markers.
After defining basal-classical protein expression state at a single cell level, we next assessed subtype distribution at the tumor level (Fig. 1E). Using a two-group subtype classification schema to mimic prior bulk RNA-seq analyses, 69% of tumors were classified as classical and 31% of tumors as basal using a classification determined by the cell type comprising >50% of cells within the tumor. As previously reported(10,15), patients with basal tumors had significantly worse disease-free survival (DFS) and overall survival (OS) compared to those with classical tumors (Fig. 1F; Supplementary Table.T10). However, this two-group classification scheme failed to represent the large degree of intra-tumor subtype heterogeneity identified in our cohort (Fig. 1E), as very few tumors exhibited a purely classical or basal composition. Instead, most tumors were composed of both basal cells and classical cells, intermixed at varying proportions. We therefore created a three-group classification scheme based on a >90% tumor subtype purity cut-off, which resulted in classical-predominant (41.5%), mixed (45%), and basal-predominant (13.5%) tumors (Fig. 1E). Patients with mixed tumors demonstrated intermediate outcomes, with median OS time between those with basal and classical tumors (Fig. 1G; Supplementary Table.T10). We also classified cases by representing the percentage of classical or basal cells on a continuous scale, using a basal-classical axis score wherein a higher value indicated a greater basal cell fraction and a lower value indicated a greater classical cell fraction within a tumor. In quartile analyses (Supplementary Table.T11), higher percentages of basal cells and lower percentages of classical cells within a tumor were associated with worse patient survival, and this association was linear along the continuous scale of basal-classical axis score by cubic spline regression (Fig.1H&I, Supplementary Table.T11). In contrast, co-expressor cell fraction was not associated with patient outcomes (Supplementary Table.T12). Thus, these findings indicated that tumor expression subtypes exist on a continuum and that characterizing the frequency distribution of cell states within a tumor may provide greater insights into patient outcomes or therapeutic efficacy than bulk whole tumor classifications.
We subsequently assessed the association of the two-group subtype classification with OS and DFS by adjuvant treatment received (Supplementary Table.T2). Given the preponderance of patients receiving adjuvant gemcitabine alone or in combination, we divided the cohort into two groups: (1) receipt of adjuvant gemcitabine alone or in combination (N=172 patients), (2) receipt of other adjuvant treatment programs or no adjuvant treatment (N=117). Regardless of adjuvant treatment group, survival times were longer among patients with classical compared to basal tumors, although these differences were not statistically significant (all log-rank p>0.05; Supplementary Fig.S6). Sample sizes were less robust when stratifying by adjuvant therapy, and similar analyses should be considered in future studies.
Tumor expression subtype associations with patient clinicopathological characteristics and somatic genomic alterations
Given the large degree of heterogeneity in expression subtypes in patient tumors within our resection cohort, we explored the association of tumor subtype with clinicopathological and genomic alterations using our three-group classification schema (Supplementary Table.T13). As noted previously(27), we observed a stepwise increase in the percentage of tumors with poorly differentiated histology comparing classical-predominant, mixed, and basal-predominant tumors, although these differences were not statistically significant (P=0.10). No statistically significant associations were observed between tumor subtype and the 4 main genes somatically altered in PDAC (KRAS, CDKN2A, SMAD4 or TP53) (Supplementary Table.T14). However, as suggested in a previous study(12), enrichment of KRAS copy number gain was noted in basal-predominant tumors (P=0.003; Supplementary Table.T14).
Co-expressor cells are common, demonstrate a varied expression subtype marker composition, and exist on a spectrum of marker intensities associated with the prevalence of pure classical and pure basal cells
Unexpectedly, co-expressor cells were found in 266 (92%) of resected tumors (Fig. 1F). Thus, we sought to better understand the phenotypes and significance of these cells. First, we considered the relative frequency of the possible co-expressor cell marker combinations (Supplementary Fig.S7). Just three co-expression marker combinations accounted for 61% of all co-expressor cells and included the KRT17+TFF1+ (27%), KRT17+GATA6+ (22%) and KRT17+GATA6+TFF1+ (12%) marker combinations (Fig. 2A). Nine additional phenotypes accounted for a further 32% of co-expressor cells (range, 1–10%), and the remaining 35 phenotypes were rare and each individually represented <1% of co-expressor cells. These results indicate that co-expressor marker combinations do not follow a random distribution and particular combinations may be more stable or commonly adopted at the cellular level, with KRT17 expression particularly prevalent among co-expressor cells(30). We assessed the associations of the top two dominant co-expressor cell phenotypes (KRT17+TFF1+ and KRT17+GATA6+) with tumor subtype and patient survival. Results were consistent with the findings from the overall co-expressor population (data not shown), but additional studies will be needed to determine whether biologic differences are present between different co-expressor cell subtypes.
Figure 2. Single cell, protein-based expression subtype assessment identifies co-expressor cells that express classical and basal markers within the same cells.
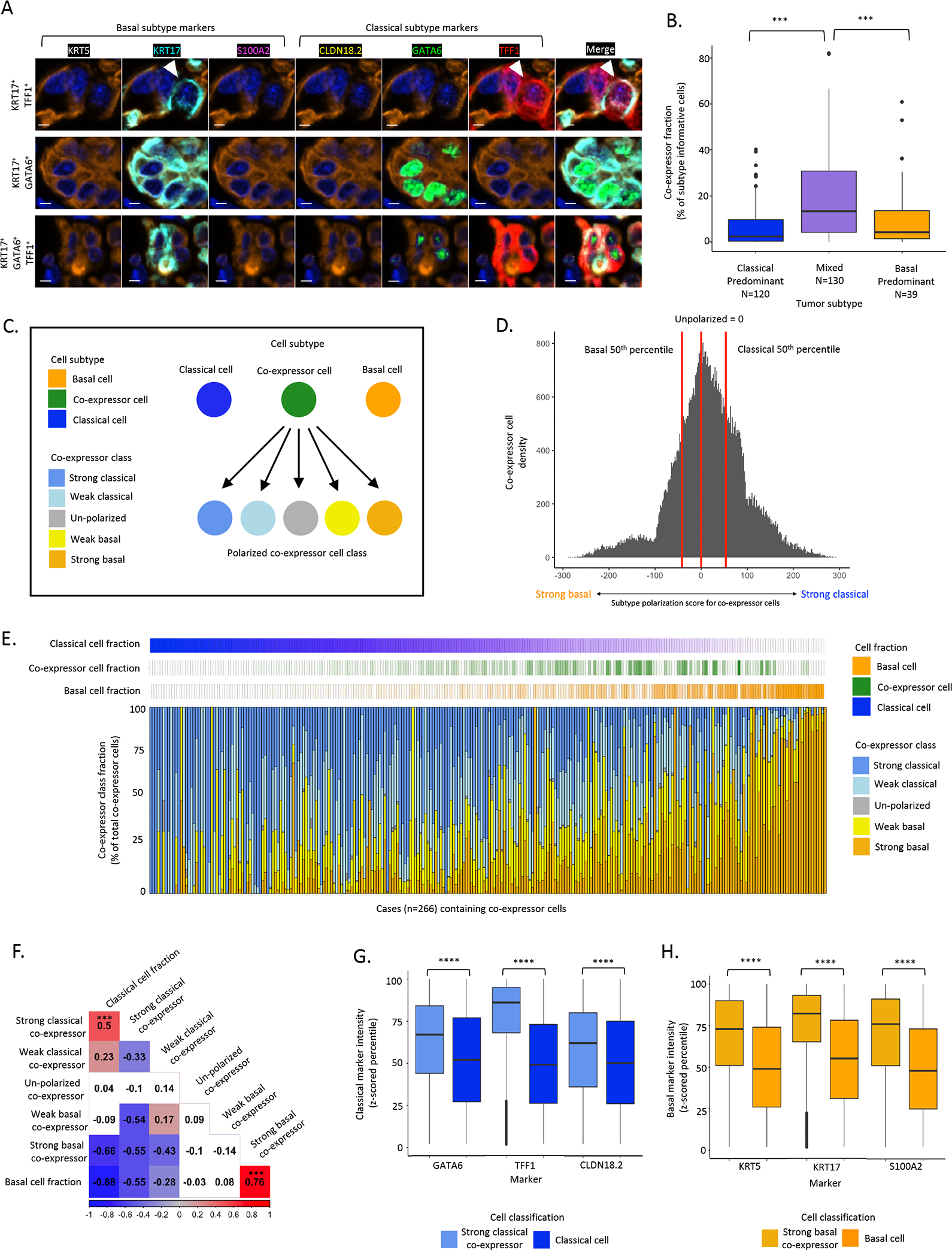
A: Representative mIF images of the top three most prevalent co-expressor cell phenotypes. Top: KRT17+TFF1+, Middle: KRT17+GATA6+, Bottom: KRT17+GATA6+TFF1+. Scale bar = 5μm. B: Higher co-expressor cell fraction in mixed compared to classical-predominant or basal-predominant tumors (Chi2 test). C: Schematic of co-expressor cell polarization. Basal marker and classical marker expression intensities assessed to calculate a subtype polarization score in co-expressor cells. D: Histogram of subtype polarization score for co-expressor cells with class cut-offs depicted. E: Across 266 resected tumors, co-expressor cell polarization was highly heterogeneous, with strong basal or strong classical marker polarization scores enriched in tumors with high fractions of pure basal or pure classical cell fractions, respectively. Each column represents one tumor. F: Correlations between co-expressor polarization classes and pure classical and basal cell fractions. Strong basal co-expressor cell abundance was positively correlated with pure basal fraction (Pearson correlation R=0.76, P<0.001, strong classical co-expressor cell abundance was positively correlated with pure classical cell fraction (Pearson correlation R=0.5, P<0.001). G: Classical marker expression intensity in strong classical co-expressor cells and pure classical cells (Mann-Whitney test). H: Basal marker expression intensity in strong basal co-expressor cells and pure basal cells (Mann-Whitney test). *** p value significant at <0.001, **** p value significant at <0.0001.
We next hypothesized that if co-expressor cells represented a cell type transitioning between classical and basal phenotypes, mixed tumors containing both classical and basal cells would be enriched for co-expressor cells. Indeed, a significantly higher fraction of co-expressor cells were present in mixed tumors compared to classical-predominant or basal-predominant tumors(14) (Fig. 2B). We next assessed the degree of classical or basal polarization within co-expressor cells to determine whether these cells lie on an expression subtype continuum with pure classical and basal cells (Fig. 2C). To accomplish this, we assessed IF marker intensity across co-expressor cell types and further classified co-expressor cells using continuous marker intensities, rather than solely positivity, placing each individual co-expressor cell on a 5-class polarization scale ranging from strong basal to strong classical (Fig. 2D, Supplementary Fig.S4). This finer-grained classification of co-expressor cells revealed a large degree of intra-tumor heterogeneity for co-expressor polarization classes (Fig. 2E). Furthermore, at the individual tumor level, strong basal co-expressor cell abundance was positively correlated with pure basal cell fraction (Pearson correlation R = 0.76, P<0.001) while strong classical co-expressor cell abundance was positively correlated with pure classical cell fraction (Pearson correlation R = 0.5, P<0.001) (Fig. 2F). These observations support a model in which expression subtypes within individual cells fall along a continuum ranging from pure classical to co-expressor to pure basal phenotypes.
Given the existence of co-expressor cell phenotypes across a continuum of polarization states, we anticipated that individual subtype marker intensities would follow distributions where intensity is strongest in the corresponding pure classical or basal phenotype and then decreases across co-expressor cell types. Unexpectedly, intensities for all three classical markers were higher in strong classical co-expressor cells than in pure classical cells (Fig. 2G), and the same pattern was observed for all three basal markers (Fig. 2H). Evaluation of pan-cytokeratin and DAPI intensities, which would not be expected to systematically vary between cells, revealed consistent expression across cell phenotypes, suggesting that the observed classical and basal marker intensity differences were not due to technical artifact. Taken together, these findings suggested that specification of cell phenotype at a protein level involves regulation of both the presence and level of marker expression, and that the highest levels of classical or basal marker expression do not occur in pure classical or basal cells.
Tumor subtype can be reliably determined from needle biopsies for clinical application
To explore whether smaller tissue fragments, such as those obtained clinically by needle biopsy, recapitulate tumor subtype fractions of the larger tissue mass, we first assessed the correlation of basal-classical axis score between two TMA cores from the same primary tumor (Fig. 3A). For this analysis, we performed matched core comparisons in cases which were represented by 2 cores across our primary resection cohort (n=440 cores from 220 tumors). We observed a strong correlation between the basal-classical axis score between cores (Pearson correlation R=0.67, P<0.0001), (Fig. 3B). To further provide confidence in the accuracy of subtype analysis in the context of low tumor cell numbers, we next compared the basal-classical axis score for 25 tumors with TMA cores (mean number of tumor cells, n=1754) and matched whole slide sections (WSS) (mean number of tumor cells, n=7543), which represented an average 8-fold increase in total tumor cells per sample (Fig. 3C). The basal-classical axis score for the two TMA cores was strongly correlated with the larger areas analyzed by WSS, with a Pearson correlation coefficient of 0.86 (P<0.001) (Fig. 3D).
Figure 3. Pancreatic cancer expression subtype concordance across tissue areas.
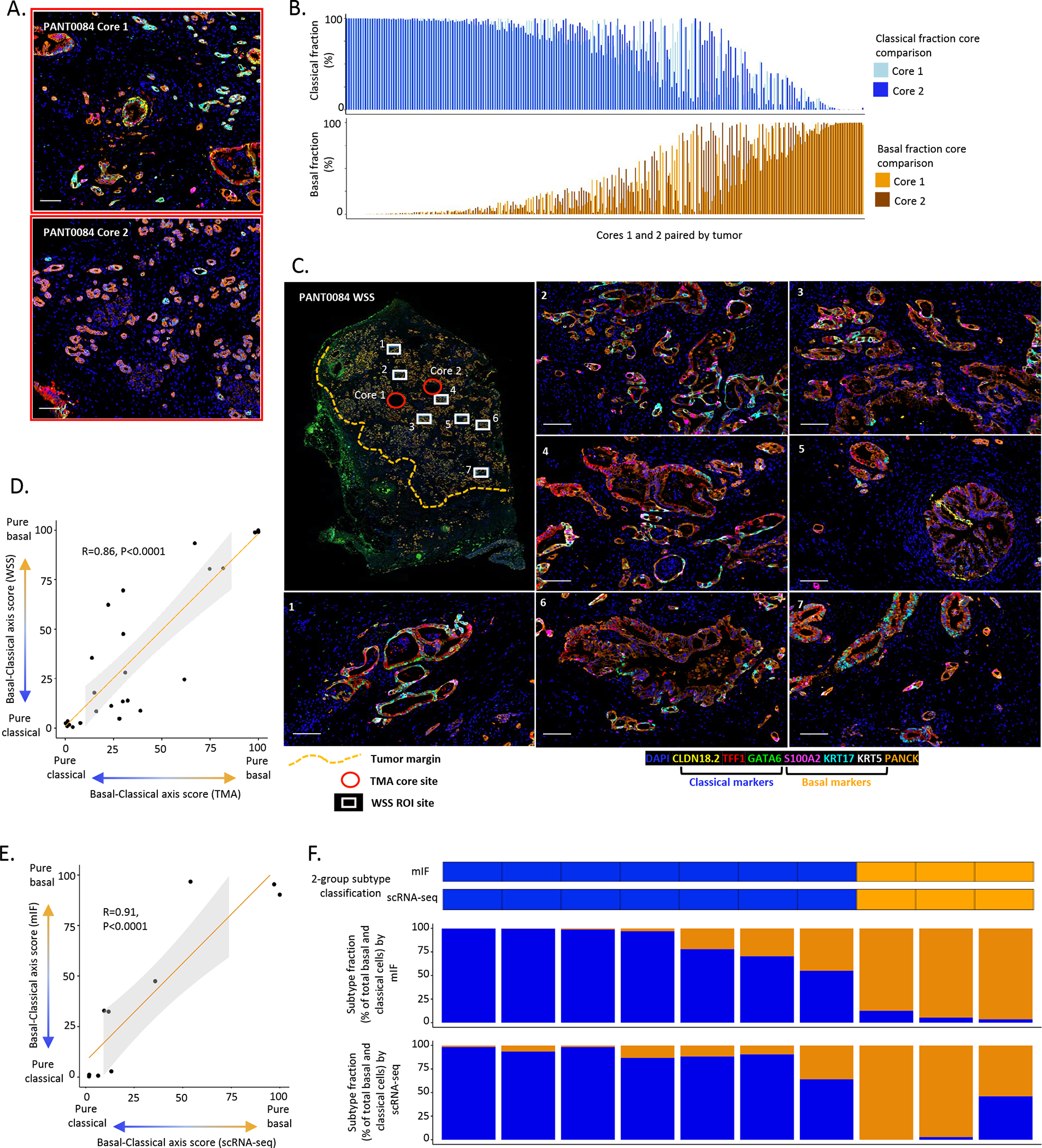
A: Representative mIF images from tissue microarray (TMA) core analysis. Case: PANT0084. Top: Core 1. Bottom: Core 2. (Also areas with red border in Fig.3D), scale bar = 100μm. B: Pairwise comparison for basal-classical axis score from 2 cores per tumor (N=220 tumors, with vertical bars representing the 2 cores for each patient). Strong positive correlation in basal-classical axis score between cores (Pearson correlation R=0.67, P<0.0001). C. Representative mIF images from ROI analysis in WSS. Case: PANT0084. Whole slide image. White boxes denote ROI sample sites for WSS analysis (7 sites), red circles denote sample sites for TMA analysis (2 sites) and yellow dash line denotes tumor area. Images 1–7, scale bar = 100μm. D: Strong positive correlation between basal-classical axis score as determined by mIF in TMA cores and whole slide sections (N=25, Pearson correlation R=0.86, P<0.0001). E. Strong positive correlation in basal-classical axis score between 2 core needle biopsy specimens from the same metastatic lesion analyzed using mIF and scRNA-seq (N=10, Pearson correlation R=0.91, P<0.0001). F: Tumor-level correlation analysis for two needle biopsy cores from the same metastatic lesion. Categorical tumor subtype call (basal: >50% basal fraction, classical: >50% classical fraction) and subtype fraction comparison.
To further assess the applicability of our subtyping assay to core needle biopsies, we analyzed a cohort of 10 patients with two biopsy cores taken from the same metastatic lesion during a single biopsy procedure. For each patient, one biopsy core was assessed using our mIF panel (mean number of tumor cells, n=16,398) and one biopsy was assessed by scRNA-seq (mean number of tumor cells, n=349). We observed a very high correlation of the basal-classical axis score derived from the two biopsy cores of the same metastatic lesion (Pearson correlation R=0.91, P<0.0001), (Fig. 3E&F). Taken together, these data suggest that tumor subtype fraction measured by mIF in a needle biopsy is highly correlated with that measured from a larger tumor volume and from another biopsy core of the same lesion.
Metastatic pancreatic cancer is enriched for co-expressor cells and basal tumors
Transcriptional profiling studies have reported a higher frequency of basal tumors in metastatic lesions than in primary lesions(7). To explore this further, we applied our mIF subtype assay to a cohort of metastatic PDAC biopsies (Supplementary Table.T3). In total we analyzed 405 ROIs across 37 metastatic tumor biopsies (511,795 total tumor cells) and compared the findings to our primary resection cohort (289 tumors, 775,632 total tumor cells). A comparable proportion of tumor cells were deemed subtype informative in metastatic biopsy specimens (289,854 subtype informative tumor cells, 56.6%).
We first evaluated the relative abundance of the 6 subtype markers between primary and metastatic sites. At the single marker level, all three basal markers were more abundant, while all three classical markers were less abundant (all P<0.0001) in tumor cells from metastatic compared to primary PDAC (Fig. 4A). Using the six markers in combination to identify classical, co-expressor and basal cells while profiling 689,078 tumor cells across the two cohorts, metastatic biopsies demonstrated a higher fraction of basal (39.4% vs 25.4%) and co-expressor (22.6% vs 14.1%) cells and a lower fraction of classical (38% vs 60.5%) cells compared to primary PDAC (Fig. 4B, Supplementary Table.T10). As seen in primary tumor specimens, co-expressor cells were present at a higher frequency within metastatic samples in mixed tumors (Fig. 4C). At the tumor level, metastatic lesions had a similar fraction of mixed tumors, but a higher fraction of basal tumors and a lower fraction of classical tumors when compared to primary cancers (Fig. 4D–E, Supplementary Table.T15). Thus, metastatic lesions had a greater fraction of co-expressor cells compared with primary tumors and were shifted towards a basal phenotype at the tumor level.
Figure 4. Comparison of single cell, protein-based expression subtype assessment between primary and metastatic pancreatic tumors and patient-derived organoids.
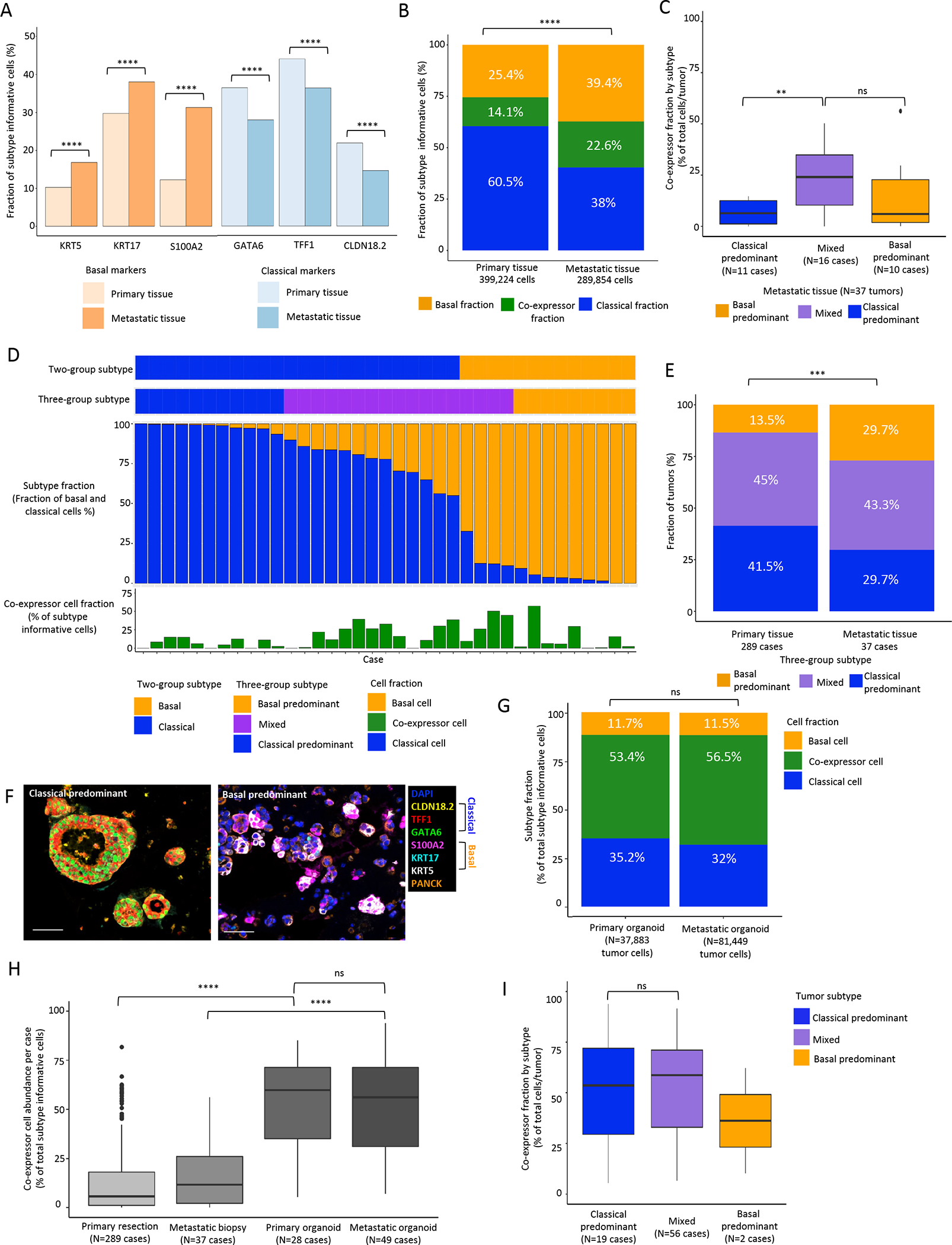
A: Positivity for classical and basal subtype markers within individual cells in primary and metastatic specimens. All classical markers are more commonly expressed in cells from primary tumors and all basal markers are more commonly expressed in cells from metastatic tumors (Mann-Whitney test). B: Comparison of aggregate cell subtype fractions in primary and metastatic tumors (Chi2 test). C: Co-expressor fraction among classical-predominant, mixed, and basal-predominant tumors. D: Tumor-level summary of tumor expression subtype and cell subtype fractions in metastatic PDAC (n=37). Top to bottom: tumor subtype using 2-group classification. Tumor subtype using 3-group classification. Cell subtype fraction per tumor. Co-expressor cell fraction per tumor. E: Tumor subtype distribution for primary and metastatic tumors (Chi2 test). *** P<0.001, **** P<0.0001. F: Representative mIF images of classical-predominant and basal-predominant organoids. Scale bar = 50μm. G: Cell subtype fraction between primary tissue, metastatic tissue, primary organoid and metastatic organoid specimens. Both organoid cohorts exhibited significantly higher co-expressor cell fractions than the corresponding primary resection and metastatic biopsy tissue cohorts (Mann-Whitney test). H: Co-expressor cell abundance in primary tissue, metastatic tissue, primary organoid and metastatic organoid specimens (Mann-Whitney test). I: Co-expressor fraction among classical-predominant, mixed, and basal-predominant tumors for patient-derived organoids. Co-expressor cell fraction is higher in mixed tumors from primary and metastatic sites (4C), but not in patient-derived organoids (4I) (Mann-Whitney test). Statistical testing was not performed between basal and mixed tumors for organoids due to insufficient cases for comparison. *** P<0.001, **** P <0.0001.
Cell subtype distributions are shifted in patient-derived organoids
The identification of different expression subtype fractions in primary versus metastatic tumors suggested that microenvironmental factors may influence tumor cell subtypes, as also suggested by recent work from us and others(31). To provide further evidence for this we performed subtype profiling on 77 patient-derived organoids with homogenized growth conditions, allowing for assessment of microenvironment effects on subtype specification (Supplementary Table.T4). After subtyping a median of 1760 cells per organoid with a median of 83.5% cells deemed subtype informative (Fig. 4F), we compared cell subtype fractions between those derived from primary versus metastatic tumors (Fig. 4G). We observed that the fraction of co-expressor cell containing glands was higher in metastatic (86.1%) compared to primary tumor organoids (70.5%), a trend also observed in tumor tissues from patients. Compared to primary resection and metastatic biopsy specimens, both organoid groups exhibiting a significantly higher abundance of co-expressor cells and a pronounced decrease in abundance of basal cells (Fig. 4H). We next assessed the distribution of co-expressor cell abundance by tumor subtype classification. While high co-expressor cell fraction was associated with mixed tumors in primary tissue and metastatic biopsies, this relationship was lost in patient derived organoids (Fig. 4I). Taken together, these findings suggest that the organoid culture microenvironment alters cancer cell subtype frequencies irrespective of tissue site of origin and that signals from the tumor microenvironment are likely to contribute to specifying cancer cell subtypes in vivo.
Gland-level analysis suggests cell state switching and implicates co-expressor cells as potential intermediates between basal and classical subtypes
To further investigate the concept of co-expressor cells representing a transition state between basal and classical cells, we performed spatial analysis of individual gland units, assessing gland subtype composition and constructing subtype neighbor maps (Fig. 5A). We began by delineating and classifying tumor glands according to cell phenotypes. We first assessed the distribution of co-expressor versus non-co-expressor cell containing glands, noting larger proportions of co-expressor containing glands in organoids compared to patient tissue and in metastatic lesions compared to primary tumors (Fig. 5B). Notably, by manual inspection of glands, many were heterogeneous in subtype composition, with most glands containing more than one cell subtype. We therefore performed a detailed analysis of gland composition and organization (Fig. 5C). Notably, glands composed of basal and classical cells but without accompanying co-expressor cells were extremely uncommon. Instead, glands were most commonly a mixture of cell subtypes with a large proportion of mixed basal-classical glands containing co-expressor cells.
Figure 5. Tumor gland expression subtype composition and organization further suggest co-expressor cells as an intermediate state between pure classical and pure basal cells.
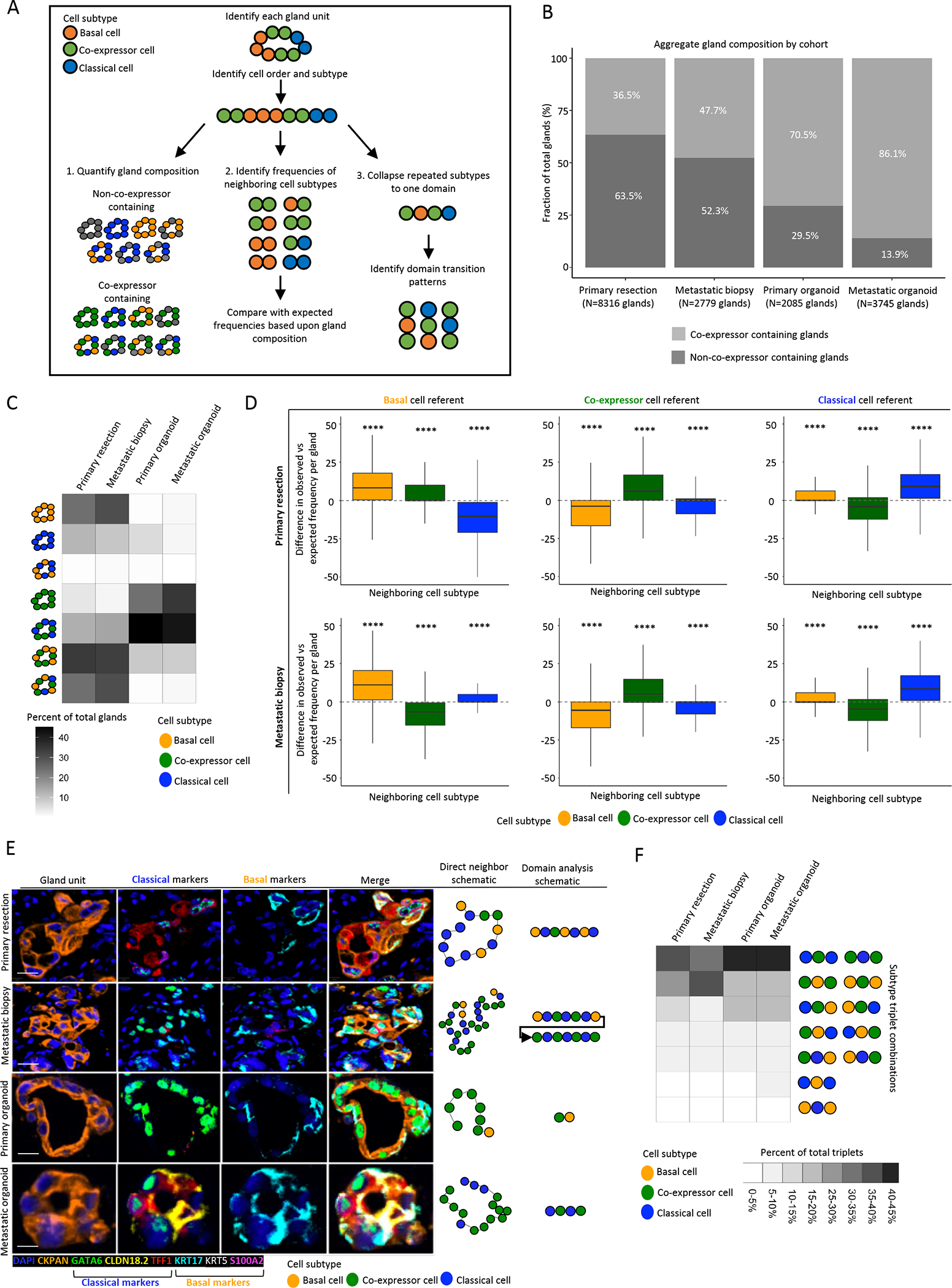
A: Schematic representing analysis workflow. Following gland-unit identification, cell order and subtype were determined. Gland composition identified, and direct neighbor and domain analysis conducted. B: Gland type distribution in primary tissue, metastatic tissue, primary organoid and metastatic organoid specimens. C: Distribution of gland composition for co-expressor containing and non-co-expressor containing glands by specimen type for gland units containing subtype informative cells, demonstrating gland units comprised of basal and classical cells rarely observed across cohorts and most gland units to be a mixture of expression subtypes. D: Direct neighbor analysis with comparison of expected and observed frequencies of subtypes by cell of reference and specimen type. Basal cells neighbor basal cells and classical cells neighbor classical cells more frequently than expected by chance. Difference between observed an expected frequencies per gland unit depicted (Mann-Whitney test between observed and expected frequencies) (Outliers removed from boxplots). E: Representative mIF images of gland-units by specimen type (top to bottom), scale bar = 10μm. Left to right: Gland-unit: DAPI and CKPAN, Classical markers: GATA6, CLDN18.2 and TFF1, Basal markers: KRT17, KRT5 and S100A2, Merge, Direct neighbor analysis schematic of cell subtypes in gland unit, Domain analysis schematic of concatenated domains of cell subtypes. E: Domain analysis of gland-units derived from primary resection, metastatic biopsy and patient-derived organoids. Domains of co-expressor cells are frequently flanked by domains of classical or basal cells, while domains of basal and classical cells are rarely directly adjacent. **** P<0.0001.
To measure the organization of cells within glands, we next analyzed the identity of the two neighboring cells that directly flanked each reference cell (basal, co-expressor or classical) (Fig. 5D). Basal cells were found next to basal cells and classical cells were found next to classical cells more than expected by chance, from which we inferred a likely clonal relatedness between adjacent cells. This trend was also observed in patient-derived organoids (Supplementary Fig.S8). To more directly test a model wherein co-expressor cells serve as a transition state between the basal and classical cell states, we analyzed the domain-level organization of classical, basal and co-expressor cells and focused on the pattern of subtype transitions between neighboring domains within a gland (Fig. 5E). In this analysis, one or more adjacent cells with identical subtypes were considered a single subtype domain. Across both the tissue and organoid cohorts, co-expressor domains were most commonly flanked on both sides by the same domain type, either classical or basal (Fig. 5F). However, co-expressor domains were also commonly flanked on one side by a classical domain and on the other side by a basal domain. Very rarely were direct transitions between basal and classical domains observed.
To further examine whether co-expressor cells represent a transition state between basal and classical cells, we performed pseudo-time analysis on two independent single cell RNA sequencing cohorts(11,22). We clustered malignant cells and identified pseudo-temporal ordering for PDAC cancer cells from both datasets. The Monocle3 algorithm placed a majority of the root points of the cell trajectory (terminal cell states predicted by the algorithm) in either the basal- or classical-enriched portion of the UMAP embedding (Figure S9A), suggesting that the co-expressing cells were not the primary terminal cell states. For the pseudotime analysis of the trajectories, we chose to begin this analysis at the root points in classical cell populations based on the hypothesis that tumor cells initially have a classical phenotype (the most commonly observed bulk tumor type in human PDAC) and may then progress to a basal phenotype with additional genetic or phenotypic alterations or perturbations such as chemotherapy. This pseudotime analysis placed the co-expressing cells between classical and basal cells on the pseudotime axis, further suggesting that the co-expressor phenotype represents an transitory cell state lying on a continuum between classical and basal cells (Figure S9B–C). In aggregate, these data suggested that classical, basal, and co-expressor cells were organized in a non-random manner within individual tumor glands and that co-expressor cells may represent a transitional phenotype between classical and basal subtype states.
Discussion
The current PDAC subtype classification schema is based on multi-gene transcriptional signatures that assign a categorical tumor subtype, the consensus of which has become a 2-class system of classical or basal-like(7–9,32). Herein, we described a quantitative multi-marker protein panel for the assessment of expression subtypes with single cell resolution and maintenance of spatial organization. Using this approach, we demonstrated that expression subtyping can be performed in the protein space, providing reliable expression subtype designations while using techniques readily translatable to real-time analysis of small volume tissue specimens for clinical care. With in situ resolution of thousands of individual cells per tumor, we identified profound intra-tumoral subtype heterogeneity in both primary and metastatic specimens, with very few tumors existing as purely basal or purely classical. Furthermore, most tumors contained a substantial fraction of “basal-classical” co-expressor cells, with individual co-expressor cells themselves sitting on a continuum of basal to classical phenotypes. Deeper spatial analyses suggested that although individual gland units often have a heterogeneous cell composition, basal and classical cells rarely existed adjacent to one another without intervening co-expressor cells, further implicating these cells as a plastic intermediate between the basal and classical cell poles. Importantly for clinical decision making, a continuous basal-classical axis score was linearly associated with survival among patients with resected PDAC, and in silico analyses suggested that the amount of tumor tissue typically present in core needle biopsies provides accurate expression subtype fractions compared to larger tissue areas.
Analysis of tumor cell protein expression is a standard technique practiced by clinical pathology laboratories, and one that can be completed from small FFPE samples obtained as part of routine clinical care. Extensive precedent exists for proteins serving as biomarkers in cancer patients for differential diagnosis, prognostication and treatment selection(33,34). Single protein IHC has been examined as a tool to identify pancreatic cancer expression subtypes, but such approaches have been semi-quantitative, subject to inter-observer variability, and lacking spatial resolution(18,35). In the current study, a protein-based assay utilizing spatially-resolved, multi-marker immunofluorescence demonstrated strong concordance with relative subtype fractions measured by bulk and dissociative single-cell RNA sequencing, respectively, and indicated the potential for quantitative protein marker measurements to provide clinical utility in expression subtyping.
Application of a multi-marker subtype protein assay to patients with localized and metastatic PDAC demonstrated similar percentages of patients with classical and basal tumors compared to previous bulk transcriptional studies(7–9,32). However, the ability to subtype each individual cell within a tumor exposed a striking degree of intra-tumor subtype heterogeneity, wherein both classical and basal cells were present in the vast majority of tumors. This mixed tumor subtype was suggested previously by observations from single-cell transcriptional analyses of dissociated tumors, where both basal and classical cell clusters were noted(12,14). In our large resection cohort, patients with mixed tumors had intermediate survival times, but a basal-classical axis score was associated with outcomes on a continuous scale, without a natural break-point to classify patients into two groups of worse versus better survival. The observation that subtype fractional abundance was continuously associated with survival outcomes was further supported by a recent study suggesting that patients with tumors having discordant expression subtype classification by multiple analytic methods at the bulk RNA level had intermediate survival outcomes compared with those uniformly classified as classical or basal-like/squamous(15). Our data would suggest that these discordant tumors are likely those that have a large admixture of both basal and classical cells within the same tumor. In ongoing and future studies where treatment selection is based upon expression subtype, it will be critical to assess continuous basal and classical expression subtype fractions, in addition to analyses based on two-group classifications. This more nuanced assessment of tumor subtype may allow for context-specific biomarker cut-points for treatment selection and for assessment of relative subtype switching at the time of therapy resistance.
In addition to identifying the frequent coexistence of classical and basal cells within the same tumor, we found that nearly 90% of primary tumors contained cells that co-expressed both classical and basal protein markers. These co-expressor cells are not readily detectable by bulk sequencing approaches, as bulk methods cannot differentiate pure basal and pure classical cells admixed in the same tumor from cells that express both basal and classical markers within the same cell. However, we recently detected co-expressor cells in a study of metastatic PDAC using a dissociative single cell RNA sequencing approach(14), supporting our protein mIF findings.
Several observations suggest that co-expressor cells may serve as a plastic intermediate between cells at the pure basal and classical poles. Co-expressor cells were highly enriched in mixed tumors, which contained the greatest degree of basal and classical cell admixture. Co-expressor cells were enriched in metastatic biopsies, which harbored more heterogeneous distributions of basal and classical cells than primary tumors. Quantification of a subtype polarization score showed that co-expressor cells with strong basal marker intensity were associated with a higher fraction of pure basal cells, while co-expressor cells with strong classical marker intensity were associated with a higher fraction of pure classical cells within a tumor. Basal and classical cells were infrequently found in the same gland without accompanying co-expressor cells. Analysis of cell domains within glands showed that domains of basal and classical cells were rarely directly adjacent but were more commonly found on opposite sides of co-expressor cell domains. Altogether, these findings suggest a potential lineage relationship across a basal-co-expressor-classical axis that underlies expression subtype organization within glands. Our findings also suggest that mIF-based gland-level analysis may serve as a complementary approach to scRNA-seq and spatial transcriptomic methods to examine cell-cell and tumor microenvironment interactions, and this approach may be informative for other solid tumor types. In addition, as therapies are explored to treat specific PDAC expression subtypes, a deeper understanding of co-expressor cell biology may prove helpful in preventing therapy resistance and facilitating the “push” of cells to a particular subtype pole with greater treatment responsiveness.
In three-dimensional organoid culture, we identified high co-expressor cell fractions regardless of whether organoids were derived from patient primary or metastatic lesions. In fact, the co-expressor cell fraction was nearly identical in primary- and metastatic-derived organoids, with more than half of all cells in organoid culture expressing both basal and classical markers. Interestingly, when assessing the distribution of glands containing co-expressor cells in primary tumors and metastases, we identified a comparable trend in patient tissue and organoids. These results suggest that culturing patient tumor samples in an environment different from their native in vivo setting can result in changes to cancer cell subtype fractions; however, spatial distribution will also be important to consider when assessing fidelity of model systems to patient tissue samples. Given these findings, experiments altering media conditions and incorporating non-epithelial cells in co-culture may allow further functional assessment of factors that shift cells along the basal-classical continuum(14). These findings also suggest using caution when interpreting drug sensitivity testing in patient-derived organoid cultures, as the effects of a given therapeutic agent may depend both upon a cell’s expression subtype(14) and the distribution of these subtypes within an organoid or tissue sample.
To assess the applicability of a protein-based subtyping algorithm to the small samples commonly obtained in clinical care, we compared tumor subtyping results in matched TMA cores and ROIs from whole slide sections and found a high degree of correlation. Thus, for studies of tumor subtypes related to patient outcomes or clinicopathologic covariates in large patient populations, appropriately constructed TMAs are likely sufficient to identify underlying relationships. However, in some cases with mixed tumors, a single ROI did not provide an accurate overview of the subtype fractions in the larger tumor specimen. Thus, it may be important in patients with highly basal-classical admixed tumors to interrogate larger tissue areas to ensure adequate sampling when considering therapy selection or clinical trial enrollment based on tumor subtype.
In conclusion, we demonstrated the utility of a multi-protein panel to reliably determine pancreatic cancer expression subtype fractions across multiple independent cohorts. We identified a large degree of intra-tumoral subtype heterogeneity that is significantly associated with patient outcomes. Furthermore, our single cell multiplex approach identified co-expressor cells that may represent a transitional cell type indicative of phenotypic plasticity during metastatic spread and development of therapy resistance.
Supplementary Material
Statement of significance.
A high-throughput pipeline utilizing multiplex immunofluorescence in pancreatic cancer reveals striking expression subtype intra-tumoral heterogeneity with implications for therapy selection and identifies co-expressor cells that may serve as intermediates during subtype switching.
Acknowledgments
The Hale Family Center for Pancreatic Cancer Research, Lustgarten Foundation Dedicated Laboratory program, NIH grant U01 CA210171, NIH grant P50 CA127003, Stand Up To Cancer, Pancreatic Cancer Action Network, Noble Effort Fund, Wexler Family Fund, Promises for Purple, and Bob Parsons Fund to B.M.W. The Hale Family Center for Pancreatic Cancer Research, Lustgarten Foundation Dedicated Laboratory program, NIH grants P50 CA127003, R01 CA248857, R01 CA205406, R01 CA169141, R35 CA197735, and U01 CA250549 to J.A.N. William Raveis Charitable Fund Physician-Scientist of the Damon Runyon Cancer Research Foundation (PST-15-18) to H.S. W.C.H. is a consultant for ThermoFisher, Solasta Ventures, MPM Capital, KSQ Therapeutics, Tyra Biosciences, Jubilant Therapeutics, RAPPTA Therapeutics, Function Oncology, Riva Therapeutics, Frontier Medicines and Calyx.
Footnotes
Conflict of interest:
B.M.W. declares research funding from Celgene, Eli Lilly & Company, Novartis, and Revolution Medicine, and consulting for Celgene, GRAIL, and Mirati. J.A.N. declares research funding from NanoString, Illumina, and Akoya Biosciences. K.P. declares One-time Pancreatic Advisory Board fee for Celgene (5/2019). One-time HCC Advisory Board fee for Eisai (11/2019). One-time Cholangiocarcinoma Advisory Board for Helsinn/QED (5/2021). R.F.D. declares Advisory Board for Exelixis INC. and Helsinn Healthcare. All other authors declare no potential conflicts of interest.
References
- 1.American Cancer Society. 2020 Cancer Estimates [Internet]. 2020. Available from: https://cancerstatisticscenter.cancer.org/module/yg6E0ZLc [Google Scholar]
- 2.Orth M, Metzger P, Gerum S, Mayerle J, Schneider G, Belka C, et al. Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiation Oncology. 2019;14:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adamska A, Domenichini A, Falasca M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. MDPI; 2017;18:1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aung KL, Fischer SE, Denroche RE, Jang G, Dodd A, Creighton S, et al. HHS Public Access Cancer : Early Results from the COMPASS Trial. 2019;24:1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aung KL, Fischer SE, Denroche RE, Jang G-H, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res. United States; 2018;24:1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. United States; 2018;8:1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. United States; 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. England; 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 9.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. United States; 2011;17:500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashid NU, Peng XL, Jin C, Moffitt RA, Volmar KE, Belt BA, et al. Purity Independent Subtyping of Tumors (PurIST), A Clinically Robust, Single-sample Classifier for Tumor Subtyping in Pancreatic Cancer. Clin Cancer Res. 2019/11/21. 2020;26:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghavan Srivatsan, Winter Peter S., Navia Andrew W., Williams Hannah L. et al. The tumor microenvironment drives transcriptional state, plasticity, and drug response in pancreatic cancer. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet. 2020; [DOI] [PubMed] [Google Scholar]
- 13.Nicolle R, Blum Y, Duconseil P, Vanbrugghe C, Brandone N, Poizat F, et al. Establishment of a pancreatic adenocarcinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer. EBioMedicine. 2020/07/03. Elsevier; 2020;57:102858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell. United States; 2021;184:6119–6137.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topham JT, Karasinska JM, Lee MKC, Csizmok V, Williamson LM, Jang GH, et al. Subtype-Discordant Pancreatic Ductal Adenocarcinoma Tumors Show Intermediate Clinical and Molecular Characteristics. Clin Cancer Res. United States; 2021;27:150–7. [DOI] [PubMed] [Google Scholar]
- 16.Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. Arch Pathol Lab Med. United States; 2018;142:1364–82. [DOI] [PubMed] [Google Scholar]
- 17.Roa-Peña L, Leiton CV, Babu S, Pan C-H, Vanner EA, Akalin A, et al. Keratin 17 identifies the most lethal molecular subtype of pancreatic cancer. Sci Rep. 2019;9:11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan K, Jang G-H, Grant RC, Wilson JM, Notta F, O’Kane GM, et al. The value of GATA6 immunohistochemistry and computer-assisted diagnosis to predict clinical outcome in advanced pancreatic cancer. Sci Rep. Laboratory Medicine Program, University Health Network, 200 Elizabeth Street, Toronto, ON, M5G 2C4, Canada. kai.duan@uhn.ca.; 2021. page 14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian ZR, Rubinson DA, Nowak JA, Morales-Oyarvide V, Dunne RF, Kozak MM, et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. United States; 2018;4:e173420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng J, Sun B-F, Chen C-Y, Zhou J-Y, Chen Y-S, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choudhary S, Satija R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 2022;23:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina, USA.; 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.N Kalimuthu S, Wilson GW, Grant RC, Seto M, O’Kane G, Vajpeyi R, et al. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut. England; 2020;69:317–28. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Väyrynen SA, Zhang J, Yuan C, Väyrynen JP, Dias Costa A, Williams H, et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin Cancer Res. United States; 2021;27:1069–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng Y, Zou M, Liu Y, Que K, Wang Y, Liu C, et al. Keratin 17 Suppresses Cell Proliferation and Epithelial-Mesenchymal Transition in Pancreatic Cancer. Frontiers in Medicine. 2020. page 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grünwald BT, Devisme A, Andrieux G, Vyas F, Aliar K, McCloskey CW, et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell. United States; 2021;184:5577–5592.e18. [DOI] [PubMed] [Google Scholar]
- 32.Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen W, Frankel WL. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod Pathol. United States; 2019;32:1–15. [DOI] [PubMed] [Google Scholar]
- 34.Rakha EA, Pinder SE, Bartlett JMS, Ibrahim M, Starczynski J, Carder PJ, et al. Updated UK Recommendations for HER2 assessment in breast cancer. J Clin Pathol. 2015;68:93 LP – 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muckenhuber A, Berger AK, Schlitter AM, Steiger K, Konukiewitz B, Trumpp A, et al. Pancreatic Ductal Adenocarcinoma Subtyping Using the Biomarkers Hepatocyte Nuclear Factor-1A and Cytokeratin-81 Correlates with Outcome and Treatment Response. Clin Cancer Res. United States; 2018;24:351–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
De-identified single-cell RNA-seq data(14) used in this study are publicly available for download and visualization via the Single Cell Portal: https://singlecell.broadinstitute.org/single_cell/study/SCP1644. De-identified bulk RNA-seq data(19) used in this study have been deposited in dbGaP under accession number phs001652.v1.p1. All remaining data generated in this study are available upon request from the corresponding author.