

Abstract
Aggregation of TAR DNA-binding protein 43 kDa (TDP-43) is thought to drive the pathophysiology of amyotrophic lateral sclerosis and some frontotemporal dementias. TDP-43 is normally a nuclear protein that in neurons translocates to the cytoplasm and can form insoluble aggregates upon activation of the integrated stress response (ISR). Viruses evolved to control the ISR. In the case of Herpesvirus 8, the protein ORF57 acts to bind protein kinase R, inhibit phosphorylation of eIF2α and reduce activation of the ISR. We hypothesized that ORF57 might also possess the ability to inhibit aggregation of TDP-43. ORF57 was expressed in the neuronal SH-SY5Y line and its effects on TDP-43 aggregation characterized. We report that ORF57 inhibits TDP-43 aggregation by 55% and elicits a 2.45-fold increase in the rate of dispersion of existing TDP-43 granules. These changes were associated with a 50% decrease in cell death. Proteomic studies were carried out to identify the protein interaction network of ORF57. We observed that ORF57 directly binds to TDP-43 as well as interacts with many components of the ISR, including elements of the proteostasis machinery known to reduce TDP-43 aggregation. We propose that viral proteins designed to inhibit a chronic ISR can be engineered to remove aggregated proteins and dampen a chronic ISR.
Introduction
Tar DNA binding protein 43 (TDP-43) is a RNA binding protein (RBP) that accumulates in multiple neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia-TDP-43 (FTD-TDP) and Alzheimer’s disease (ad) (1,2). TDP-43 normally resides in the nucleus but translocates to the cytoplasm during stress (3). Cytoplasmic TDP-43 interacts with other disease-linked RBPs, undergoes post-translational modifications and forms insoluble aggregates (4,5). Mutations in TDP-43 are sufficient to cause ALS, and mutations in other proteins linked to ALS and FTD-43 (e.g. C9orf72 or PGRN) are also known to lead to TDP-43 pathology. The causes of TDP-43 linked neurodegeneration appear to be complex. One major mechanism of TDP-43 induced degeneration result from the chronic stress arising from the prolonged accumulation of cytoplasmic TDP-43 and cytoplasmic TDP-43 aggregates. These aggregates can also form under non-integrated stress response (ISR) mediated pathways (5,6). Another potential mechanism of degeneration arises from the loss of nuclear TDP-43, with a concomitant loss of nuclear splicing (7–13). Many therapeutic approaches focus on inhibiting the accumulation of cytoplasmic TDP-43 aggregates and inhibit nuclear to cytoplasmic translocation of TDP-43 (14–16), including by modulating phosphorylation of eIF2α (17,18).
Chronic stress elicits an ISR, which inhibits RNA translation and catabolism. The ISR is beneficial in addressing acute stresses, but in human disease a chronic, persistent stress response can itself contribute to the disease process (19). In neurodegenerative disease chronic activation of the ISR leads to prolonged accumulation of RNA–protein complexes, termed stress granules (SGs). These SGs act as a conduit in a pathway that produces cytoplasmic TDP-43 aggregates. The ISR pathway can be activated by four kinases that respond to exogenous and endogenous stressors: general control nonderepressible 2 (GCN2) kinase (20), heme-regulated eIF2α kinase (HRI) (21), PKR-like endoplasmic reticulum kinase (PERK) (22) and protein kinase R (PKR) (23). Both PERK and PKR have been shown to play important roles in the pathophysiology of neurodegenerative diseases, with inhibitors of these enzymes able to reduce the pathophysiology in many models of neurodegenerative diseases, including models of ALS and frontotemporal dementia (FTD), which express TDP-43 (24–26). Despite this promise, inhibitors of PERK and PKR have failed in the clinic because of peripheral toxicity (27,28).
An alternative approach to modulating the ISR for therapeutic purposes might be to take advantage of proteins naturally evolved to modulate proteostasis or inhibit the ISR. Prior studies have examined the plant Arabidopsis protein HSP104, which acts as a powerful disaggregase of disease linked proteins (29–32). However, HSP104 has not been adapted by nature to regulate the mammalian proteostasis machinery. In contrast, viral proteins evolved to target many elements of the mammalian ISR in order to maintain protein synthesis and allow production of viral proteins. For instance, Herpesvirus 8 produces the protein ORF57, which inhibits PKR activity and the ISR by binding and inhibiting both PKR and PKR activating protein, and is adapted to function in neurons (33–36). By inhibiting the ISR, ORF57 also allows a neuron to maintain normal translation, splicing and RNA stability, despite the impact of the virus on a cell (37–39). We hypothesized that, although the ability of ORF57 to inhibit the ISR is problematic for our bodies in the case of a viral infection, ORF57 expression might be beneficial in the case of a chronic neurodegenerative disease.
In this study, we used a genetically engineered SH-SY5Y neuroblastoma cell line to model the actions of TDP-43 in neurodegenerative diseases. Two SH-SY5Y cell line were engineered: one constitutively expresses wild type TDP-43, while the other expresses a tetracycline inducible TDP-43 construct lacking its nuclear localization signal. Of note, the inducible TDP-43 lines, termed TDP-43ΔNLS, readily forms cytoplasmic aggregates in response to induction plus stress, modeling what occurs in neurons of patients with ALS, FTD-TDP or ad. We investigated the effects of ORF57 on aggregate formation and dispersal in the TDP-43ΔNLS SH-SY5Y line. We demonstrate that ORF57 interacts with proteins robustly involved in TDP-43 aggregation, promotes the removal of TDP-43 aggregates and reduces TDP-43-mediated neurodegeneration. These results suggest that ORF57 might be able to be utilized as a therapeutics for neurodegenerative diseases exhibiting TDP-43 pathology.
Results
ORF57 is protective against oxidative stress in human neuroblastoma cells exhibiting TDP-43 pathology
We model the disease related cytoplasmic TDP-43 aggregates by overexpressing a TDP-43 construct lacking a functional nuclear localization signal, termed TDP-43ΔNLS (40). In cell culture, this construct yields abundant cytoplasmic TDP-43 which aggregates in response to activation of the ISR with sodium arsenite (SA) treatment (40). ORF57 inhibits the ISR through known interactions with PKR (33). SH-SY5Y cells expressing only endogenous (wildtype) TDP-43 do not exhibit robust cytoplasmic TDP-43 inclusions with the same sodium arsenite stress (Supplementary Material, Fig. S1).
We proceeded to examine how ORF57 affects the ISR in SH-SY5Y cells with inducible expression of TDP-43ΔNLS. The SH-SY5Y cells were transduced with ORF57::mCherry or mCherry expressing lentiviruses and the transgene expression level was quantified by mRNA levels (Fig. 1A and B) and by fluorescence (Fig. 2A). After infection, TDP-43ΔNLS was induced by treatment with doxycycline (1 μm, 24 h), and then the SH-SY5Y cells expressing TDP-43ΔNLS ± ORF57 were subjected to SA stress (300 μm, 90 min). SA stress activates PKR to phosphorylate eIF2α (p-eIF2α) and halts global translation. We confirmed that these effects of SA stress were relieved by ORF57 expression. The TDP-43ΔNLS SH-SY5Y cells exhibited increased levels of p-eIF2α in response to SA stress (Fig. 1C and D), this was also observed in wildtype SH-SY5Y cells (Supplementary Material, Fig. S2A and B). Treatment with SA also induced abundant cytoplasmic TDP-43 granules (Fig. 2A, F, and G). We found that ORF57 expression relieved multiple ISR associated phenotypes. ORF57 expression reduced p-eIF2α by 45% (P < 0.0001) in SH-SY5Y TDP-43ΔNLS cells following exposure to SA (Fig. 1C and D). We found that ORF57 increased protein synthesis when measured with the SUnSET assay (P < 0.05) (Fig. 1E and F), however this increase was small when compared with non-stressed TDP-43 overexpressing cells (Supplementary Material, Fig. S3). We also observed that ORF57 increased expression of TDP-43ΔNLS (Supplementary Material, Fig. S4 A, B), this is not surprising given ORF57’s demonstrated effects on protein synthesis. The increase in total protein synthesis was also observed by the SUnSET assay in wildtype SH-SY5Y cells (Supplementary Material, Fig. S2C–F). ORF57 also protected against apoptosis in TDP-43ΔNLS cells (Fig. 1G–J). Cleaved caspase-3 levels were significantly reduced in ORF57 compared with mCherry expressing cells in both stress and basal conditions (P < 0.05), demonstrating that ORF57 is protective against TDP-43ΔNLS induced apoptosis. Similar effects were observed in wildtype SH-SY5Y cells expressing ORF57 (Fig. 1I and J).
Figure 1.

ORF57 protects against oxidative stress in TDP-43𑀏NLS overexpressing SH-SY5Y cells. Experiments conducted in mCherry and ORF57 mCherry express ing SH-SY5Y TDP-43ΔNLS cells with and without SA stress. (A) Constructs for mCherry and ORF57::mCherry lentivirus. (B) Overexpression of ORF57 in SH-SY5Y cells by rt-qPCR. (C) Western blot for phosphorylated eIF2α and total eIF2α. (D) Quantification of C. (E) SUnSET of mCherry and ORF57 expressing TDP-43ΔNLS cells with SA stress. (F) Quantification of E. (G) Western blot of cleaved caspase-3 in TDP-43ΔNLS cells. (H) Quantification of G. (I) Western blot of cleaved caspase-3 in non-overexpressing SH-SY5Y cells. (J) Quantification of I.
Figure 2.

Stress granule assembly and TDP-43 aggregation dysregulated by ORF57.
(A) Immunocytochemistry in SH-SY5Y TDP-43ΔNLS. DAPI (405 nm). mCherry or ORF57::mCherry (594 nm). TDP-43ΔNLS::GFP (488 nm). G3BP1 (647 nm). Merge, overlay of all images. White arrows indicate overlap between ORF57 and TDP-43 aggregates. (B) Representation of small TDP-43ΔNLS aggregates 10–200 Aggrecount units. (C) Representation of large TDP-43ΔNLS aggregates 200–400 Aggrecount units. (D) Quantification of G3BP1 aggregates in each cell condition from A. (E) Mander’s coefficient of fraction of mCherry overlapping with TDP-43 signal. (F) Quantification of small TDP-43ΔNLS aggregates. (G) Quantification of large TDP-43ΔNLS aggregates. Two-way ANOVA with Bonferroni post hoc comparison, *P < 0.05, **P < 0.01, ***P < 0.0001.
SG formation was also reduced with ORF57 expression. Imaging was performed on induced SH-SY5Y TDP-43ΔNLS cells expressing ORF57::mCherry or mCherry alone. G3BP1 positive SGs were reduced > 20% (P < 0.05) in cells expressing ORF57 compared with those expressing mCherry under SA stress (Fig. 2A and D). As described previously, SA stress also caused cytoplasmic TDP-43 to aggregate (Fig. 2A–C, F, G). We observed that ORF57 appeared to be colocalizing with TDP-43 (Fig. 2A, white arrows) and therefore we analyzed colocalization of mCherry with TDP-43 signals. The fraction of the mCherry signal in both mCherry and ORF57::mCherry that overlapped with the unstressed TDP-43 signal was <0.9 (Fig. 2E). However, once stress was added the fraction of the mCherry signal that overlapped with TDP-43 was dependent on ORF57 expression. Stress caused the TDP-43 to form cytoplasmic inclusions without altering the mCherry signal, therefore the fraction of mCherry overlapping with TDP-43 signal was reduced to 0.137 ± 0.158. Meanwhile, the ORF57::mCherry signal was changed in response to stress and the fraction of ORF57::mCherry overlapping with the TDP-43 signal was 0.396 ± 0.106 (P < 0.05). This indicates that ORF57 colocalizes with aggregated TDP-43.
Co-expressing ORF57 overall increased the number of TDP-43 aggregates and changed the structure of the TDP-43 aggregate. ORF57 caused TDP-43 to form large amorphous aggregates that were almost completely absent in mCherry control cells (Fig. 2A–C, F, G). Parameters were made to distinguish these aggregates for quantification using the Aggrecount macro (41), marking the small aggregates from 10 to 200 Aggrecount units and the large aggregates 200–400 units. According to these parameters, small and large aggregates were quantified. Under stress, small aggregates of TDP-43 were increased by ~30% (P < 0.05) in ORF57 expressing cells compared with mCherry expressing cells. Large aggregates of TDP-43 were also increased in ORF57 cells under stress compared with mCherry expressing cells (P < 0.05).
ORF57 reduces TDP-43 aggregation
We hypothesized that the large TDP-43 aggregates represent a more diffuse state, and therefore contain less insoluble and more soluble TDP-43. In order to assess this, we fractionated TDP-43 into RIPA-soluble and RIPA-insoluble fractions and immunoblotted these fractions using a semi non-denaturing gel. Use of semi-non-denaturing electrophoresis (allowing visualization of secondary and tertiary structures associated with aggregation) combined with use of urea to solubilize the insoluble fraction and potential post-translational changes in TDP-43 leads to observation of shifts in the molecular weights of the insoluble TDP-43 fractions (Fig. 3A). Expression of ORF57 improved TDP-43 solubility during stress, increasing the amount of soluble and decreasing the amount of insoluble TDP-43 (Fig. 3A–C). Under basal conditions, ~87% of TDP-43 was in the soluble fraction in both ORF57 and mCherry treated cells (Supplementary Material, Fig. S5). With SA stress, the amount of soluble TDP-43 in mCherry cells was reduced to 59% and the amount of insoluble TDP-43 was increased by 41%. ORF57 expression prevented the TDP-43 from becoming insoluble. In ORF57 cells, 75% of TDP-43 was in the soluble fraction and only 26% was insoluble, P < 0.05) (Fig. 3A–C). This increase in soluble TDP-43 could also explain the higher TDP-43ΔNLS::GFP fluorescence observed in Figure 2A. These results indicate that ORF57 inhibits transition of TDP-43 to the insoluble fraction, which occurs in response to SA stress.
Figure 3.

TDP-43 aggregates are more soluble with ORF57. Experiments conducted in mCherry and ORF57 mCherry expressing SH-SY5Y TDP-43ΔNLS cells with SA stress. (A) Western blot of TDP-43ΔNLS::GFP RIPA-soluble and in-soluble fractions. (B) Quantification of %RIPA-Soluble TDP-43ΔNLS::GFP calculated as soluble TDP-43 divided by the sum of soluble and insoluble TDP-43. (C) Quantification of %RIPA-insoluble TDP-43ΔNLS::GFP. (D) Time course experiment measuring TDP-43ΔNLS::GFP aggregate recovery at 15, 30 min, 1 , 2 , 3.5 and 6.5 h after SA removal. (E) Imaging of TDP-43ΔNLS using FRAP. (F) Quantification of D with exponential one-phase decay best fit curves. (G) Analysis of TDP-43ΔNLS mobility using FRAP. mCherry controls cells had a mobile fraction of 0.03 and a t1/2 of 111 s; ORF57::mCherry expressing cells had a higher mobile fraction of 0.2 and a t1/2 of 68 s. Unpaired t-tests, *P < 0.05. Scale bars 20 μm.
Next, we hypothesized that the ability of ORF57 to increase soluble and reduce insoluble TDP-43 might lead to more rapid resolution of TDP-43 granules upon removal of stress. To investigate this, we conducted a time course experiment to quantify the kinetics of TDP-43 granule resolution after SA stress. SH-SY5Y TDP-43ΔNLS cells expressing ORF57 or mCherry were subjected to SA stress (300 μm, 90 min), and then SA was removed. The total number of TDP-43 aggregates/cell were quantified at 15 , 30 min, 1 , 2 , 3.5 and 6.5 h after removal of SA stress. The SH-SY5Y TDP-43ΔNLS cells expressing ORF57 exhibited a faster resolution of TDP-43 granules than mCherry expressing cells (Fig. 3D and E). The t1/2 for recovery was 0.84 h for the ORF57 cells versus 2.06 h for the mCherry cells, which is a 2.45-fold (P < 0.017) difference using a one-phase decay nonlinear regression model (Fig. 3D and F). This data indicates that ORF57 protects against insoluble TDP-43 aggregation in this model.
The diffusion of TDP-43ΔNLS was also evaluated using fluorescent recovery after photobleaching (FRAP). Using live-cell imaging we photobleached the aggregates under sodium arsenite stress and assessed the recovery of the fluorescent signal. Higher recovery indicates that the TDP-43 is soluble and mobile, while less fluorescent recovery indicates that the TDP-43 is insoluble and immobile. We observed that after photobleaching the cells expressing ORF57::mCherry exhibit increased fluorescent recovery compared with cells only expressing mCherry (Fig. 3E and G). The aggregates were photobleached for five iterations with 100% laser power resulting in a 67% and a 74% average bleaching depth in fluorescence for ORF57 and mCherry treated cells, respectively. mCherry controls cells had a mobile fraction of 0.03 and a t1/2 of 111 s; ORF57::mCherry expressing cells had a higher mobile fraction of 0.2 and a t1/2 of 68 s (Fig. 3 E and G). These results indicate that ORF57 promotes the solubility of TDP-43 in the presence of sodium arsenite stress.
ORF57 binds to stress pathway proteins involved in TDP-43 pathogenesis
In order to gain insight into the mechanisms through which ORF57 might affect TDP-43 granule dynamics, we investigated the ORF57 interactome in SH-SY5Y cells expressing ORF57 under basal and stressed conditions. Prior literature shows that ORF57 binds to PKR (Gene ID: EIF2AK2), but does not explore whether ORF57 might also bind other proteins linked to protein aggregation.
Using the mCherry-tagged TDP-43ΔNLS SH-SY5Y cell line, we were able to explore the ORF57 protein interaction with proteins involved in the ISR as well as those involved in TDP-43 aggregation. The cells were transduced with mCherry or ORF57::mCherry and immunoprecipitated with anti-RFP nanobody. Following washing and elution, the resulting eluant was analyzed by mass spectrometry (IP-MS) under basal overexpression of cytoplasmic TDP-43 conditions as well as SA-induced cellular stress (Fig. 4A). Under basal conditions, IP-MS revealed binding of ORF57 with 196 proteins (FDR-adjusted P-value < 0.05 and log2 fold-change > 0.58 versus the mCherry control) (Fig. 4B). Importantly, these interactions included PKR, encoded by the EIF2AK2 gene, which is a known ORF57 interactor and initiator of the ISR (Fig. 4B). This interaction was validated with immunoprecipitation of red fluorescent protein (RFP) and probing for PKR, which demonstrated selective binding of ORF57 to PKR in both stress conditions (Supplementary Material, Fig. S6). Enriched Gene Ontology (GO) biological process (BP) functional annotation terms revealed basal conditions binding of ORF57 to proteins involved in stress response (SRP) -dependent co-translational protein targeting to membrane, mRNA metabolism, RNA translation and RNA processing (Fig. 4B). These functional terms are consistent with known functions of ORF57 as a regulator of viral protein synthesis.
Figure 4.

ORF57 interactome. (A) Outline of proteomic approach using SH-SY5Y cells expressing GFP::TDP-43ΔNLS. (B) High-confidence ORF57-bound proteins observed under basal conditions and the GO-BP functional annotations terms associated with these enriched proteins. Functional annotation terms were determined using ShinyGO Version 0.76.3 (81) (C) High-confidence ORF57-bound proteins observed in presence of SA and the GO-BP functional annotations terms associated with these enriched proteins. Enriched proteins in basal and stress conditions were considered significant when they have a Log2 fold-change > 0.58 and an FDR adjusted P-value < 0.05.
Under SA-induced cellular stress, ORF57 revealed binding with 144 proteins (FDR-adjusted P-value < 0.05 and log2 fold-change > 0.58 versus mCherry-SA control) (Fig. 4C; Supplementary Material, Table S1). There was high overlap of ORF57 bound-proteins between basal and SA conditions (133 proteins, Venn diagram) (Fig. 5A). There were 28 proteins that interacted with ORF57 >4-fold either under TDP-43 overexpression or with the addition of stress (Table 2). Expectedly many of these proteins are involved in the antiviral response: TRIM25, ZC3HAV1, PABPC4, HELZ2, SYNCRIP, DHX30, YBX3, EIF2AK2, RBMX and DHX37. While other proteins are involved in SG assembly and disease pathology including TARDBP, HNRNPA2B1, FUS, TAF15, HNRNPDL, PABPC1, DHX30, ELAVL1, MTDH, HNRNPA3, EIF2AK2, HNRNPR and MATR3. These high interactors help to indicate the primary function of ORF57 under conditions of TDP-43 pathology.
Figure 5.
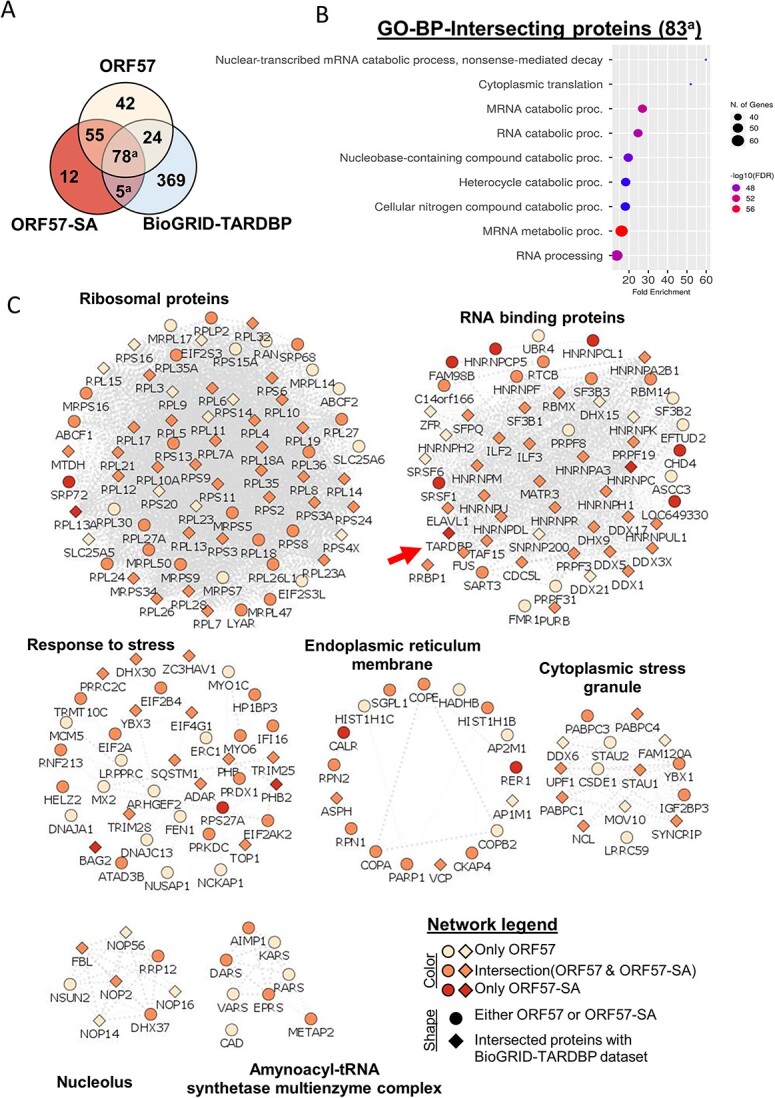
ORF57 interacts with the TDP-43 protein network. (A) Venn diagram using ORF57, ORF57-SA and the TDP-43 BioGRID-TARDBP) datasets. (B) Functional annotations terms for overlapping proteins between ORF57-SA and BioGRID-TARDBP datasets. (C) Multi-module representation of ORF57-bound protein networks (Using Cytoscape, STRING-db and ClusterMaker2. Node title describes gene names, node shape describes whether the protein was found in either ORF57 datasets (circle) or BioGRID-TARDBP (diamond), and color indicates whether the gene was only found in either ORF57, ORF57-SA or overlapped between them.
Table 2.
Proteins that interact highly with ORF57
| Protein |
Log-2 fold change mCherry vs ORF57 |
Log-2 fold change mCherry-SA vs ORF57-SA |
|---|---|---|
| HNRNPA2B1 | 3.521 | 3.248 |
| TRIM25 | 3.506 | 1.640 |
| FUS | 3.462 | 2.391 |
| ZC3HAV1 | 3.235 | 2.782 |
| RTRAF | 2.894 | 2.329 |
| PABPC4 | 2.888 | 1.609 |
| TAF15 | 2.859 | 3.249 |
| HELZ2 | 2.611 | 2.291 |
| SYNCRIP | 2.517 | 1.693 |
| HNRNPDL | 2.497 | 1.643 |
| PABPC1; PABPC3 |
2.450 | 1.630 |
| RPL19 | 2.401 | 1.397 |
| DHX30 | 2.345 | 2.465 |
| ELAVL1 | 2.336 | 1.337 |
| MTDH | 2.297 | 1.816 |
| YBX3 | 2.286 | 0.802 |
| HNRNPA3 | 2.234 | 1.803 |
| EIF2AK2 | 2.189 | 2.113 |
| RRP12 | 2.175 | 1.633 |
| ASPH | 2.117 | 1.630 |
| RBMX | 2.101 | 0.962 |
| RPL14 | 2.081 | 0.983 |
| HNRNPR | 2.074 | 1.064 |
| DHX37 | 2.062 | 1.791 |
| RTCB | 2.046 | 1.384 |
| RPL5 | 2.023 | 1.471 |
| MATR3 | 1.887 | 2.045 |
Note. Proteins that interact with ORF57 > a 4-fold increase in binding either in the absence or presence of SA stress. All proteins have FDR-adjusted P-value < 0.05.
The addition of SA stress also changed many proteins binding to ORF57. There were four proteins that significantly changed their ORF57 interaction in the presence of SA stress that were unchanged in mCherry. Three proteins (PRDX1, TRIM28 and EIF2S3) significantly increased their ORF57 binding while one protein (NUSAP1) significantly decreased its binding with ORF57 with stress (Table 3).
Table 3.
Unique proteins whose ORF57 interactions are significantly altered in the presence of stress
| Protein |
Log-2 fold change ORF57 vs ORF57-SA |
FDR-adjusted P-values |
|---|---|---|
| PRDX1 | 1.494 | 1.25E-06 |
| TRIM28 | 0.771 | 2.73E-03 |
| EIF2S3; EIF2S3L; F8W810 |
0.635 | 1.27E-03 |
| NUSAP1 | -1.169 | 1.18E-03 |
Note. Proteins that significantly change their interaction with ORF57 in the presence of SA stress. These proteins are not significantly changed in mCherry versus mCherry-SA comparisons.
The ORF57 interactome network showed 17 proteins that bind to ORF57 only under stress conditions (Table 4). The 17 proteins have diverse functions within the cell which highlights the ability of ORF57 to target many different biological processes. The ORF57 interactome network was visualized using Cytoscape and String-db; this network was then sub-clustered to create multiple modules (Fig. 5C). Importantly, the network of proteins specific to the ORF57-SA interactome includes TDP-43 (gene name TARDBP) (Fig. 5C, red arrow), validated interaction by immunoprecipitation and probing for TDP-43 (Supplementary Material, Fig. S6). We confirmed by IP that TDP-43 binds ORF57::mCherry robustly in the presence of stress. The ORF57-SA specific interactome network also includes other proteins known to be implicated in the ISR such as, CAL and RER1 and RBPs such as HNRNPC, CL1, HRNPCP5, SRSF1, RPL13A and RPS27A (Fig. 5C). The ORF57 interactomes show unique GO BP functional annotation terms from ORF57-SA that were not observed under basal conditions including: protein targeting to endoplasmic reticulum (ER) and mRNA catabolic processes. Proteins such as CALR and RER1 are ER proteins that explain the ER-associated functional GO terms displayed in Figure 4C. The module ‘Response to stress’ contains many proteins that are associated with proteostasis (e.g. SQSTM1, DNAJ13, TRIM28, RPS27A and others) and the ISR (e.g. EIF2AK2, EIF2A, etc.). This module could contribute strongly to the ability of ORF57 to inhibit the ISR and reduce TDP-43 aggregation. There have been at least 42 genes that have mutations which cause ALS disease (42). In our proteomics study, we found ORF57 to interact with seven of these ALS implicated proteins in our TDP-43 pathology cells (Table 5). Sodium arsenite stress significantly increased ORF57’s interaction with TARDBP but not with the other six ALS proteins. Stress also significantly increased TARDBP’s interaction with mCherry. Overall, ORF57 interacted with 196 and 144 proteins for basal and stress conditions, respectively (Supplementary Material, Table S1).
Table 4.
Proteins that are unique interactors of ORF57 in the presence of stress
| Protein | Log-2 fold change compared to mCherry-SA | FDR-adjusted P-values |
|---|---|---|
| HNRNPC; HNRNPCL1; HNRNPCL2; HNRNPCL3; HNRNPCL4 | 0.616 | 0.04018 |
| CALR | 0.688 | 0.02346 |
| BAG2 | 0.702 | 0.00102 |
| RER1 | 0.807 | 0.01270 |
| RPS27A | 0.959 | 0.00530 |
| SRSF1 | 0.961 | 0.00267 |
| PHB2 | 0.974 | 0.00139 |
| DNAAF5 | 0.708 | 0.00507 |
| TARDBP | 0.721 | 0.00227 |
| SRP72 | 1.244 | 0.00043 |
| CHD4 | 1.328 | 0.00035 |
| FAM98B | 1.809 | 0.00022 |
| RPL13A | 1.818 | 0.00001 |
Note. Proteins that interact significantly more in the presence of SA stress with ORF57 than mCherry.
Table 5.
Amyotrophic lateral sclerosis disease linked proteins that interact significantly with ORF57
| Protein |
Log-2 fold change mCherry-SA vs ORF57-SA |
Log-2 fold change ORF57 vs ORF57-SA |
|---|---|---|
| HNRNPA2B1 | 3.248* | -0.090 |
| VCP | 0.877* | 0.429* |
| SQSTM1 | 0.863* | 0.0526 |
| TARDBP | 0.721* | 4.64* |
| FUS | 2.391* | -0.333 |
| TAF15 | 3.249* | 0.446 |
| MATR3 | 2.04* | -0.371 |
Note. Interactions between ALS-causing genes and ORF57. *FDR-adjusted P-value < 0.05.
Discussion
Aggregation of TDP-43 plays important roles in the pathophysiology of multiple diseases, including ALS, FTD-TDP and ad. The experiments described previously used neuronal SH-SY5Y cells to demonstrate that ORF57 binds to TDP-43, proteins linked to TDP-43 proteostasis, proteins involved in the ISR, translation proteins and PKR. We further demonstrated that expressing ORF57 elicited a striking 2.45-fold increase in the rate of TDP-43 granule dispersion and a 40% reduction in levels of cleaved caspase 3, which suggest that ORF57 is neuroprotective. We also observed increased protein synthesis in the presence of induced TDP-43 and changes in the morphology of TDP-43 cytoplasmic granules. Taken together these data suggest that TDP-43 might be able to act as an agent that can disperse TDP-43 granules and inhibit TDP-43-mediated neurodegeneration.
The studies of the ORF57 protein interactome network provide strong insights into the mechanisms through which ORF57 might impact on TDP-43 aggregation. ORF57 was previously shown to inhibit PKR and reduce SG accumulation (33), but these studies were not done in the context of TDP-43 aggregation. Multiple studies suggest that TDP-43 first consolidates in SGs, and then subsequently proceeds to aggregate (5,43,44). Our proteomic studies of ORF57 show that this protein binds to TDP-43, as well as other known binders of TDP-43, and regulate TDP-43 proteostasis (45). Some of these proteins, such as SQSTM1 and VCP, are directly involved with clearance of TDP-43 through autophagy and these proteins exhibit mutations associated with TDP-43-opathies (46–54). Other proteins are chaperones with well documented roles in proteostasis, such as DNAJA1, DNAJC13 and BAG2, and the ubiquitin transferase TRIM25 (55–58). Our proteomics confirm binding of ORF57 to known interactors SRSF1 (59) and PABPC1 (60,61), which are RBPs also known to associate with TDP-43 and regulate translation (62,63). Binding to stress proteins is readily apparent in the ORF57 interactome sub-module titled ‘Response to stress,’ which includes many classic stress linked proteins including EIF2A, EIF2AK2, EIF4G1 and EIF2B4 (Fig. 5C). ORF57 also showed a strong ability to inhibit phosphorylation of EIF2α, which is consistent with prior studies showing that it binds PKR and inhibits PKR activity (33–36). The presence in the ORF57 network of other RBPs that are known risk factors for neurodegenerative diseases, including FUS, HNRNPA2B1, MATR3 and TAF15 raises the possibility that the beneficial actions of ORF57 might extend beyond TDP-43 (64–68) and that ORF57 may be able to reduce Fus-mediated pathology which is observed in FTLD. These many interactions point to broad mechanisms through which ORF57 could impact to reduce aggregation of TDP-43 and promote granule resolution.
The clearest actions of ORF57 lie in the resolution of TDP-43 granules and the reduction in cleaved caspase 3, suggesting that ORF57-mediated reduction of TDP-43 aggregation is associated with a reduction in cell death. The protein interaction network for ORF57 shows that it interacts with many proteins known to regulate SG formation. However, this interactome impacts on SGs and translational control in a manner that appears to be complex. For instance, although ORF57 increased removal of TDP-43 aggregates, it actually increased the number of granules, with about a 3.5-fold increase in the number of large, diffuse looking granules (Fig. 2G and J). The diffuse impression of the SGs was consistent with solubility studies of TDP-43 showing that ORF57 increased the ratio of soluble to insoluble TDP-43 by about 50% (Fig. 3A and B). One possibility is that ORF57 stabilized soluble TDP-43 in SGs, reducing their exit to form insoluble TDP-43 aggregates.
Multiple studies have investigated putative links between infection with Herpes simplex virus type I (HSV-1) and neurodegenerative diseases, including AD and ALS. Relevant pathological studies suggest that HSV-1 can increase production of β-amyloid, tau phosphorylation and local inflammation (69–72); however, HSV-1 infection has not been observed to induce intracellular TDP-43 or tau aggregates (42,69,71,73,74). Selective transduction of ORF57 differs from studies of intact viruses because we used one particular gene, which was specifically selected for its potential to reduce TDP-43 aggregation. The work presented in this study demonstrates that proteins produced by Herpes viruses, such as ORF57, have the ability to limit the accumulation of protein aggregates through mechanisms shown previously.
A limitation of this study is the use of immortalized cell lines over-expressing TDP-43. The approach of over-expressing ΔNLS TDP-43 is required when using neuroblastoma lines because TDP-43 does not form cytoplasmic aggregates under conditions of endogenous expression. Future studies will need to examine the actions of ORF57 in neuronally differentiated iPSC lines from patients who had sporadic or familial ALS, which do appear to exhibit TDP-43 cytoplasmic translocation and granule formation spontaneously or in response to stress.
Viruses evolved to control the translational machinery in order to induce the host cells to produce their viriomes. ORF57 provides one example of a protein that functions explicitly in regulating the host translational machinery, but there are many other proteins in other viruses that serve the same function. Our studies suggest that these pathways share strong overlap with pathways involved in protein aggregation processes that occur in neurodegenerative diseases. The work presented in this article shows how viral proteins might also be engineered to yield new therapeutic approaches for neurodegenerative disease.
Materials and Methods
Cell culture
Two human neuroblastoma, SH-SY5Y, cell lines were used for this study: regular SH-SY5Y and SH-SY5Y with a doxycycline-inducible TDP43 with its nuclear localization signal mutated (TDP-43ΔNLS). The TDP-43ΔNLS was a gift provided by Glenn Larsen, Ph.D., Aquinnah Pharmaceutical Inc.; requests for use of the cell line will be made at the determination of Aquinnah Pharmaceuticals, Inc. Both cell lines were maintained in complete media containing Dulbecco’s Modified Eagle Medium with high glucose (Corning 10-013-CV), 10% tet-tested fetal bovine serum, 1% penicillin streptomycin (Gibco Pen Strep, # 15070-063), 1% non-essential amino acids (Gibco MEM NEAA, # 11140-050), and 1% Glutamine. TDP-43ΔNLS overexpression was induced with 1 μg/ml doxycycline added to complete media for 24 h.
ORF57 cloning
The ORF57 DNA plasmid was bought from Addgene (pCDNA4.TO-ORF57-2xCSTREP Plasmid #136217). ORF57 was amplified for Gibson Assembly (NEBuilder® HiFi DNA Assembly Master Mix #E2621) using ORF57_F (5′-3′ggatctggagctctcgagaattctcacgcgtCATATGGCCACCatggtacaagcaatgatag) and ORF57_R (5′-3′ CTTGATGATGGCCATGTTATCCTCCTCGCCCTTGCTCACACTAGTGGAACCACCACCACCagaaagtggataaaagaataaaccc). This protein has been optimized for expression in mammalian cells by addition of a SFFV promoter, Kozak sequence and ATG start codon. The amplified fragments were assembled into a lentiviral packaging plasmid, pHR-SFFV-mCherry, with a C-terminus mCherry tag. mCherry tag was chosen for its monomeric nature and resistance to aggregation, as much of the ORF57 studies performed were tracking pathological aggregation.
Lentivirus
Plasmids pHR-SFFV-ORF57::mCherry (ORF57::mCherry) and pHR-SFFV-mCherry (mCherry) were produced using the MACHEREY-NAGEL NucleoBond Xtra Maxi kit for transfection-grade plasmid DNA (#740414.50) per the protocol.
HEK-293 T cells were transfected with FuGene HD reagent (Promega # E2311) and PSP plasmid, VSV-G plasmid, and OptiMEM media, at a 1:1:3 ratio, respectively. Lentivirus was harvested from the media at 72 h concentrated by LentiX concentrator (Takara # 631232) per the protocol. The concentrated lentivirus was resuspended in one-tenth of the original media volume using 1× phosphate buffered saline (PBS) with 25 mm HEPES (Sigma-Aldrich # H3375), pH 7.4.
Lentiviral aliquots of ORF57::mCherry and mCherry were diluted 1:10 and cells were infected using a double infection protocol. Infected cells were allowed to grow in the incubator for 48 h. After 48 h, cells were passaged then re-infected 24 h later, cells were incubated for 48 h more before experiments.
qPCR
RNA was extracted using Qiagen RNAeasy Mini Prep Plus Kit and eluted in 30 μl RNAse-free water. About 1 μg of RNA was amplified into cDNA using Applied Biosystems High Transcription cDNA Synthesis Kit (Thermo Fisher # 4368814). cDNA was amplified using SSO Advanced SyBr Green on the QuantFlex 12 K. See Table 1 for primers. Ct levels of ORF57 were normalized with GAPDH and fold change was determined. Ct values above 40 were recorded as 40 for calculations.
Table 1.
rt-qPCR primers for ORF57
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| ORF57 | GCACTGATCAAACAGGTGGC | CTTGACCTCGCCAAGAAGGT |
| GAPDH | CTTTGGTATCGTGGAAGGACTCATG | CCAGTAGAGGCAGGGATCATGTTC |
Sodium arsenite treatment and time course
Sodium arsenite (SA) has been shown to be effective in vitro to induce the ISR and SG formation by phosphorylation of eIF2α (75,76). Cells were stressed with 300 μm SA in complete media for 90 min.
Time course experiment for SA stress, TDP-43ΔNLS cells expressing mCherry or ORF57::mCherry were seeded into eight-chamber slides at ~80% confluency. Cells were induced with doxycycline for 24 h, then 300 μM SA stress was added for 90 min. The cells were washed in DBPS and allowed to recover in complete media for 15 , 30 min, 1, 2, 3.5 and 6.5 h. Cells were fixed at each time point and ICC performed.
BCA assay
Cells were lysed in RIPA buffer containing 50 mm Tris–HCl, 150 mm NaCl, 1% NP40, 0.5 mm EDTA, 0.1% sodium deoxycholate, 0.1% SDS. To RIPA buffer, fresh PhosSTOP (Millipore Sigma # 490684500), proteinase inhibitor, and 1 mm Pefabloc SC (Millipore Sigma # 30827-99-7) was added directly before addition to cells. Cells were lysed by homogenization on ice and then centrifuged at 10 000×g to pellet cell debris. Supernatant was collected and used for Pierce BCA Protein Assay Kit protocol (ThermoFisher #23225) per protocol. Absorbance was then measured using Softmax Plate Reader. Data were analyzed to quantify protein levels of each sample.
Immunoblot
Cell lysates were thawed on ice. About 10 μg of each sample was added to a solution of 1× NuPAGE LDS Sample Buffer (Novex #1249698) and 1× Bolt Sample Reducing Agent (Novex #B009). Samples were boiled for 5 min at 95°C. Boiled samples were then loaded into Invitrogen Blot 4–12% Bis-Tris Protein Gel (Invitrogen #NW04125BOX) in 1× MOPS solution. Gel was transferred to a nitrocellulose membrane using the iBlot template #1. Once transferred, the membrane was blocked in 5% non-fat milk in 0.05% Tween in Tris-Buffered Saline (TBST), or 5% BSA in TBST (for phosphorylated proteins) for 1 h while gently shaking. Membrane was then incubated in primary antibody (anti-phospho-eIf2α (rabbit, 1:500 dilution, Cell Signaling Technologies #119A11), anti-eIF2α (rabbit, 1:500, Cell Signaling Technologies #9722), anti-TDP43 (rabbit, 1:4000, ProteinTech #12892-1-AP), anti-puromycin (mouse, 1:1000, EMD Millipore #MABE343), anti-actin (mouse, 1:35 000, Millipore Sigma #MAB1501), anti-phospho-PKR (pThr451, rabbit, 1:1000, Invitrogen #44-668G), anti-PKR (rabbit, 1:4000, ProteinTech #18244-1-AP) diluted in TBST overnight at 4°C while gently shaking. Membrane was washed three times in TBST for 10 min each. Membrane was then incubated in secondary antibody, donkey anti-rabbit or donkey anti-mouse HRP, diluted 1:10 000 in TBST for 1 h. Secondary antibody solution was removed and membrane was washed three more times in TBST for 10 min each. Membranes were incubated in Super Signal West Pico PLUS stable peroxide and luminol/enhancer (Thermo Scientific #34580) prior to imaging. Membranes were imaged using BioRad software. Signal accumulation mode was used. Images chosen for analysis were analyzed on ImageJ software to detect density of bands of interest in each lane.
SUnSET assay
To assess changes in global translation a SUnSET assay was performed. In this assay, puromycin replaces amino acids in mRNA being actively transcribed and translation is aborted (77). Cells were co-incubated with 10 μg/ml puromycin and 300 μm SA diluted in complete media for 90 min. As a negative control, cells were pre-treated for 30 min with 30 μg/ml cycloheximide (CHX) diluted in complete media. After CHX treatment, the pre-treatment was removed and replaced with 10 μg/ml puromycin diluted in complete media for 90 min. Following all treatments, cells were lysed in RIPA buffer and processed according to the western blot protocol aforementioned with primary antibody against puromycin (mouse, 1:1000, Sigma Aldrich #MABE343).
Soluble/insoluble TDP43 fractionation
Following BCA, 150 μg of cell lysate were brought up to the same volume of 125 μl RIPA buffer. Lysates were sonicated in water bath for three 30 s intervals for a total of 90 s of sonication. Lysates were centrifuged at 100 000×g for 30 min. Supernatant was separated as soluble fraction. Pellet was resuspended in 100 μl RIPA buffer. Resuspended pellet was sonicated and centrifuged as previously mentioned. Wash step was pipetted off and the insoluble fraction (pellet) was resuspended in 40 μl of 8 m urea (2 m Thiourea, 4% CHAPS, 30 mm Tris HCl, pH 8.5). Equal volumes of the soluble and insoluble fractions were then prepared and run as semi non-denaturing western blot. Cell lysates were combined with NuPAGE LDS Sample Buffer (Novex #1249698) and run directly on Invitrogen Blot 4–12% Bis-Tris Protein Gel (Invitrogen #NW04125BOX) in 1× MOPS solution, Density of the whole lane was determined with ImageJ/Fiji. Primary antibody used was anti-GFP (rabbit, 1:2000, ProteinTech #50430-2-AP). Secondary antibody used was donkey anti-rabbit diluted 1:10 000 in TBST.
Immunocytochemistry
Cells were fixed in 0.5 ml 4% PFA/PBS. PFA solution was removed and slides were washed in PBS. Cells were permeabilized in 0.5 ml PBS/0.1% Triton-X-100 (PBS-T). Cells were then blocked with 10% donkey serum (DS) in PBS for 1 h. Cells were then incubated in primary antibody (anti-G3BP1 (rabbit, 1:300 dilution, Millipore Sigma #07–1801), anti-eIF3ɳ (mouse, 1:250 dilution, Santa Cruz #137214), anti-TDP43 C-terminus (rabbit, 1:1000, ProteinTech #12892-1-AP) and anti-G3BP1 (mouse, 1:250, ProteinTech #6648-1-Ig)) diluted in 5% DS in PBS overnight at 4°C. After overnight incubation, the cells were washed with PBS, then incubated in secondary antibody (donkey anti-rabbit Alexa Fluor 647 (1:500), donkey anti-mouse Alexa Fluor 488 (1:500)) diluted in PBS for 1 h at room temperature. After secondary antibody incubation, cells were washed in PBS. Nuclei were stained with DAPI diluted 1:10 000 in PBS for 5 min. DAPI was removed and cells were washed two final times in PBS for 10 min each. Prolong Gold Antifade mounting media (Invitrogen # P36930) was added before coverslips. Cells were imaged on Keyence microscope. Images were analyzed to find aggregates per cell of stained proteins using Aggrecount v1.1 in ImageJ/Fiji. Mander’s coefficient was calculated by converting images to 16-bit and then using JACoP in ImageJ/Fiji.
TDP-43 time course
TDP-43ΔNLS cells expressing either mCherry or ORF57::mCherry were seeded in triplicate in an eight-chamber slide. TDP-43 was induced with doxycycline for 24 h and then the cells were stressed with 300 μm sodium arsenite for 90 min. The cells were then rinsed with DPBS and then allowed to recover in complete media for 0 , 15, 30 min, 1 , 2 , 3.5 or 6.5 h. The cells were fixed and imaged per the immunocytochemistry protocol described previously.
Fluorescence recovery after photobleaching
TDP-43ΔNLS cells expressing either mCherry or ORF57::mCherry were seeded in a 96-well plate, black walled with #1.5 glass bottom. TDP-43ΔNLS was induced with doxycycline for 24 h and then the cells were stressed with 300 μm sodium arsenite for 90 min in Leibovitz’s L-15 Medium (ThermoFisher, Cat# 11415064) CO2-independent media at 37°C. The cells were live imaged on Zeiss LSM700 confocal microscope (Carl Zeiss, Germany) with sodium arsenite present using the Plan-Apochromat 20×/0.8 M27 objective lens at room temperature. Images were acquired with a frame size of 256 × 256 and a scanning speed 968.14 ms with 4.06 Airy units equaling a 7 μm section. Six ROIs were chosen from three frames and bleached for five iterations at a scan speed of 2 (177.32 μs) at 100% 488 laser power resulting in >% fluorescence signal loss. Three pre-bleach images were taken, then 300 images were taken immediately after photobleaching. The fluorescent intensity for each ROI was quantified in Zen 2.3 SP1 and the values normalized to the baseline fluorescent intensity, average mean intensities for aggregates were between 25 and 75. To analyze this data, raw values were input into EasyFRAP-web an online FRAP analyzer (78,79). The data were double normalized to baseline and background fluorescence. The normalized data were then fit with a curve using a single curve fit and the mobile fraction, t1/2 and r2 values calculated.
Statistical analysis
Statistical analysis was performed in GraphPad/Prism v9. Two-way ANOVAs and Bonferroni post hoc tests were used to compare means between conditions when there was a significant interaction. Unpaired t-tests were used for analysis of SUnSET and TDP-43 fractionation immunoblots. P-value of 0.05 was used for significance and mean ± standard deviation was reported for ANOVAs. The time course experiment was analyzed using exponential one-phase decay with a least squares regression. The best-fit curves were compared between data sets to find if there was one curve that adequately fit both data sets. Results were visualized in graphs made from GraphPad/Prism.
Immuno-purification and sample preparation for LC–MS/MS
Cells expressing either mCherry or ORF57::mCherry were plated in four replicates in six-well plates. Once these cells reached confluency, a subset of these cells were treated with 300 μm of sodium arsenite for 90 min to induce cellular stress. Subsequently, these cells were lysed in IP buffer: 10 mm Tris/Cl pH 7.5, 150 mm NaCl, 0.5 mm EDTA, 0.5% Nonidet™ P40 Substitute, 1% CHAPS and the protein levels were quantified using BCA assay.
Equal amounts of proteins were immunoprecipitated overnight at 4°C using the RFP-trap magnetic agarose beads, as recently described by our laboratory (Chromotek, RFP-TRAP Agarose, Cat#rta) (64). The following day, these beads were washed two times with lysis buffer (IP buffer) and they were then washed twice with cold-phosphate buffer saline (pH 7.4). Subsequently, these beads were stored at −80°C for further analysis.
Beads were suspended in 200 μl of ammonium bicarbonate (50 mm) to perform on bead-digestion. The suspended beads were incubated for 30 min at room temperature with both 40 mM chloroacetamide and 10 mM TCEP to alkylate and to reduce the proteins, respectively. Then, the reaction was quenched using 20 mM DTT. Subsequently, these beads were digested overnight at 37°C with 1 μg of MS-grade trypsin (Thermo Fisher Scientific, Cat#90058). The following day, the digested peptides were collected into new tubes and the trypsin-digestion reaction was quenched with formic acid (final concentration of 1%). These peptides were desalted using Pierce C18 columns (Thermo Fisher Scientific, Cat#89870) following manufacturer’s protocol. Finally, desalted peptides were stored at −80°C for further proteomics analysis.
The pull-downed proteins from the mCherry IP were analyzed by immunoblot. Briefly, the beads were washed with IP-lysis buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet™ P40 Substitute, 1% CHAPS) on day 2, the proteins were eluted with LDS-loading buffer (1×) containing 0.65% β-mercapto ethanol. The eluted samples were boiled for 5 min at 95°C. Half of the total RFP eluate (30 μl) or 10 μg of lysate were run on a 4–12% Tris-Bis gel. Blots were probed for α-PKR (rabbit, 1:5000 dilution, ThermoFisher #18244-1-AP), α-TDP-43 (rabbit, 1:2000 dilution, Proteintech Cat#10782-2-AP) and α-mCherry (goat, 1:30 000 dilution, MBS# MBS448057) per the immunoblot protocol described previously.
HPLC-ESI MS/MS and data analysis
The LC–MS/MS analysis was performed as previously described (64). Briefly, desalted peptides were re-dissolved in 1% formic acid and fractionated using C18 PepMap pre-column (3 μm, 100 Å, 75 mm × 2 cm) hyphenated to a RSLC C18 analytical column (2 mm, 100 Å, 75 μm × 50 cm). High performance nanoflow liquid chromatography-Orbitrap tandem mass spectrometry (LC–MS/MS) were performed using the Easy nLC 1200 system coupled to Q-Exactive HF-X MS (Thermo Scientific).
Raw data were searched against the Human proteome (the 2018_04 Uniprot release of UP000005640_9606) using MaxQuant software (version 1.6.7.0), with match-between runs activated. All other settings were left at default. The Mass Spectrometry Downstream Analysis Pipeline (MS-DAP, version 1.0.2), which is available at https://github.com/ftwkoopmans/msdap; was used for quality control and differential testing (80). Peptides present in ≥75% of sample replicates per contrast and proteins identified by >2 peptides, were used for differential testing. If the peptides did not match by ≥75% of sample replicates between groups, then the analysis was left blank. The Variance Stabilizing Normalization and mode between protein algorithms were used for normalization. Statistical testing was performed with the DEqMS package after rollup to proteins. All obtained raw data, and the complete quality control and differential testing report have been deposited to the ProteomeXchange Consortium via the PRIDE (81,82) (partner repository with the dataset identifier PXD039246. Enriched proteins from the comparisons mCherry versus ORF57 and mCherry-SA versus ORF57-SA, using a FDR-adjusted P-value < 0.05 and Log2 fold-change > 0.58 versus the mCherry controls, were cross-referenced with the human TARDBP interactome (BioGRID, accessed on 11.21.2022). Additionally, these enriched proteins were used to build protein networks using Cytoscape Version 3.9.1 and this network was sub-clustered with the ClusterMaker2 Version 2.3.2 plugin. Functional annotation terms were determined using ShinyGO Version 0.76.3 (83).
Supplementary Material
Acknowledgements
Authors thank Aquinnah Pharmaceuticals Inc., for use of the NLS-TDP-43 SH SY5Y cell line.
Conflict of Interest statement. B.W. is Co-Founder of Aquinnah Pharmaceuticals Inc.
A.S.K. is a scientific advisor for and holds equity in Senti Biosciences and Chroma Medicine, and is a co-founder of Fynch Biosciences and K2 Biotechnologies.
Contributor Information
Chelsea J Webber, Departments of Pharmacology, Physiology and Biophysics, Boston University, Boston, MA 02215, USA.
Caroline N Murphy, Departments of Pharmacology, Physiology and Biophysics, Boston University, Boston, MA 02215, USA.
Alejandro N Rondón-Ortiz, Departments of Pharmacology, Physiology and Biophysics, Boston University, Boston, MA 02215, USA; Center for Network Systems Biology, Boston University, Boston, MA 02215, USA; Department of Biology, Boston University, Boston, MA 02215, USA.
Sophie J F van der Spek, Departments of Pharmacology, Physiology and Biophysics, Boston University, Boston, MA 02215, USA.
Elena X Kelly, Departments of Pharmacology, Physiology and Biophysics, Boston University, Boston, MA 02215, USA.
Noah M Lampl, Center for Network Systems Biology, Boston University, Boston, MA 02215, USA.
Giulio Chiesa, Biological Design Center, Boston University, Boston, MA 02215, USA; Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA.
Ahmad S Khalil, Biological Design Center, Boston University, Boston, MA 02215, USA; Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA.
Andrew Emili, Center for Network Systems Biology, Boston University, Boston, MA 02215, USA; Department of Biochemistry, Boston University, Boston, MA 02115, USA; Department of Biochemistry, Oregon Health Sciences University, Portland, OR 97239, USA.
Benjamin Wolozin, Departments of Pharmacology, Physiology and Biophysics, Boston University, Boston, MA 02215, USA; Center for Systems Neuroscience, Boston University, Boston, MA 02115, USA; Center for Neurophotonics, Boston University, Boston, MA 02115, USA; Department of Neurology, Boston University, Boston, MA 02115, USA.
Funding
BrightFocus Foundation (AN2020002) and NIH (AG080810, AG050471, AG056318, AG06493 and AG072577) to B.W. Rainwater Charitable Foundation to A.R-O. US National Institutes of Health (NIH) (grant no. R01EB029483), Department of Defense Vannevar Bush Faculty Fellowship (no. N00014-20-1-2825) and Schmidt Science Polymath Award (no. G-22-63292) to A.S.K.
References
- 1. Neumann, M., Sampathu, D.M., Kwong, L.K., Truax, A.C., Micsenyi, M.C., Chou, T.T., Bruce, J., Schuck, T., Grossman, M., Clark, C.M. et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 2. Igaz, L.M., Kwong, L.K., Xu, Y., Truax, A.C., Uryu, K., Neumann, M., Clark, C.M., Elman, L.B., Miller, B.L., Grossman, M. et al. (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol., 173, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chou, C.C., Zhang, Y., Umoh, M.E., Vaughan, S.W., Lorenzini, I., Liu, F., Sayegh, M., Donlin-Asp, P.G., Chen, Y.H., Duong, D.M. et al. (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci., 21, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li, Y.R., King, O.D., Shorter, J. and Gitler, A.D. (2013) Stress granules as crucibles of ALS pathogenesis. J. Cell Biol., 201, 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., Shorter, J., Da Cruz, S. and Cleveland, D.W. (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron, 102, 339–357.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mann, J.R., Gleixner, A.M., Mauna, J.C., Gomes, E., DeChellis-Marks, M.R., Needham, P.G., Copley, K.E., Hurtle, B., Portz, B., Pyles, N.J. et al. (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron, 102, 321–338 e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ling, J.P., Pletnikova, O., Troncoso, J.C. and Wong, P.C. (2015) TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science, 349, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma, X.R., Prudencio, M., Koike, Y., Vatsavayai, S.C., Kim, G., Harbinski, F., Briner, A., Rodriguez, C.M., Guo, C., Akiyama, T. et al. (2022) TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature, 603, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown, A.L., Wilkins, O.G., Keuss, M.J., Hill, S.E., Zanovello, M., Lee, W.C., Bampton, A., Lee, F.C.Y., Masino, L., Qi, Y.A. et al. (2022) TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature, 603, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeong, Y.H., Ling, J.P., Lin, S.Z., Donde, A.N., Braunstein, K.E., Majounie, E., Traynor, B.J., LaClair, K.D., Lloyd, T.E. and Wong, P.C. (2017) Tdp-43 cryptic exons are highly variable between cell types. Mol. Neurodegener., 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prudencio, M., Humphrey, J., Pickles, S., Brown, A.L., Hill, S.E., Kachergus, J.M., Shi, J., Heckman, M.G., Spiegel, M.R., Cook, C. et al. (2020) Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Invest., 130, 6080–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Melamed, Z., Lopez-Erauskin, J., Baughn, M.W., Zhang, O., Drenner, K., Sun, Y., Freyermuth, F., McMahon, M.A., Beccari, M.S., Artates, J.W. et al. (2019) Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci., 22, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Theunissen, F., Anderton, R.S., Mastaglia, F.L., Flynn, L.L., Winter, S.J., James, I., Bedlack, R., Hodgetts, S., Fletcher, S., Wilton, S.D. et al. (2021) Novel STMN2 variant linked to amyotrophic lateral sclerosis risk and clinical phenotype. Front. Aging Neurosci., 13, 658226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu, H., Lu, S., Gasior, K., Singh, D., Vazquez-Sanchez, S., Tapia, O., Toprani, D., Beccari, M.S., Yates, J.R., 3rd, Da Cruz, S. et al. (2021) HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science, 371, eabb4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chou, C.C., Alexeeva, O.M., Yamada, S., Pribadi, A., Zhang, Y., Mo, B., Williams, K.R., Zarnescu, D.C. and Rossoll, W. (2015) PABPN1 suppresses TDP-43 toxicity in ALS disease models. Hum. Mol. Genet., 24, 5154–5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo, L., Kim, H.J., Wang, H., Monaghan, J., Freyermuth, F., Sung, J.C., O'Donovan, K., Fare, C.M., Diaz, Z., Singh, N. et al. (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell, 173, 677–692 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim, H.J., Raphael, A.R., LaDow, E.S., McGurk, L., Weber, R.A., Trojanowski, J.Q., Lee, V.M., Finkbeiner, S., Gitler, A.D. and Bonini, N.M. (2014) Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet., 46, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghadge, G.D., Sonobe, Y., Camarena, A., Drigotas, C., Rigo, F., Ling, K.K. and Roos, R.P. (2020) Knockdown of GADD34 in neonatal mutant SOD1 mice ameliorates ALS. Neurobiol. Dis., 136, 104702. [DOI] [PubMed] [Google Scholar]
- 19. Wolozin, B. and Ivanov, P. (2019) Stress granules and neurodegeneration. Nat. Rev. Neurosci., 20, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wek, S.A., Zhu, S. and Wek, R.C. (1995) The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol., 15, 4497–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McEwen, E., Kedersha, N., Song, B., Scheuner, D., Gilks, N., Han, A., Chen, J.J., Anderson, P. and Kaufman, R.J. (2005) Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem., 280, 16925–16933. [DOI] [PubMed] [Google Scholar]
- 22. Harding, H.P., Zhang, Y., Bertolotti, A., Zeng, H. and Ron, D. (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell, 5, 897–904. [DOI] [PubMed] [Google Scholar]
- 23. Srivastava, S.P., Kumar, K.U. and Kaufman, R.J. (1998) Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem., 273, 2416–2423. [DOI] [PubMed] [Google Scholar]
- 24. Zu, T., Guo, S., Bardhi, O., Ryskamp, D.A., Li, J., Khoramian Tusi, S., Engelbrecht, A., Klippel, K., Chakrabarty, P., Nguyen, L. et al. (2020) Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. U. S. A., 117, 18591–18599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hughes, D. and Mallucci, G.R. (2019) The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J., 286, 342–355. [DOI] [PubMed] [Google Scholar]
- 26. LaRocca, T.J., Mariani, A., Watkins, L.R. and Link, C.D. (2019) TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol. Dis., 132, 104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen, H.M., Wang, L. and D'Mello, S.R. (2008) A chemical compound commonly used to inhibit PKR, 8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one, protects neurons by inhibiting cyclin-dependent kinase. Eur. J. Neurosci., 28, 2003–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu, Q., Zhao, B., Gui, J., Katlinski, K.V., Brice, A., Gao, Y., Li, C., Kushner, J.A., Koumenis, C., Diehl, J.A. et al. (2015) Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc. Natl. Acad. Sci. U. S. A., 112, 15420–15425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackrel, M.E., DeSantis, M.E., Martinez, B.A., Castellano, L.M., Stewart, R.M., Caldwell, K.A., Caldwell, G.A. and Shorter, J. (2014) Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell, 156, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jackrel, M.E., Yee, K., Tariq, A., Chen, A.I. and Shorter, J. (2015) Disparate mutations confer therapeutic gain of Hsp104 function. ACS Chem. Biol., 10, 2672–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. March, Z.M., Sweeney, K., Kim, H., Yan, X., Castellano, L.M., Jackrel, M.E., Lin, J., Chuang, E., Gomes, E., Willicott, C.W. et al. (2020) Therapeutic genetic variation revealed in diverse Hsp104 homologs. elife, 9, e57457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shorter, J. (2017) Designer protein disaggregases to counter neurodegenerative disease. Curr. Opin. Genet. Dev., 44, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma, N.R., Majerciak, V., Kruhlak, M.J. and Zheng, Z.M. (2017) KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PLoS Pathog., 13, e1006677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma, N.R., Majerciak, V., Kruhlak, M.J., Yu, L., Kang, J.G., Yang, A., Gu, S., Fritzler, M.J. and Zheng, Z.M. (2019) KSHV RNA-binding protein ORF57 inhibits P-body formation to promote viral multiplication by interaction with Ago2 and GW182. Nucleic Acids Res., 47, 9368–9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kirshner, J.R., Lukac, D.M., Chang, J. and Ganem, D. (2000) Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol., 74, 3586–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang, Y., Cesarman, E., Pessin, M.S., Lee, F., Culpepper, J., Knowles, D.M. and Moore, P.S. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science, 266, 1865–1869. [DOI] [PubMed] [Google Scholar]
- 37. Majerciak, V., Yamanegi, K., Nie, S.H. and Zheng, Z.M. (2006) Structural and functional analyses of Kaposi sarcoma-associated herpesvirus ORF57 nuclear localization signals in living cells. J. Biol. Chem., 281, 28365–28378. [DOI] [PubMed] [Google Scholar]
- 38. Yuan, F., Gao, Z.Q., Majerciak, V., Bai, L., Hu, M.L., Lin, X.X., Zheng, Z.M., Dong, Y.H. and Lan, K. (2018) The crystal structure of KSHV ORF57 reveals dimeric active sites important for protein stability and function. PLoS Pathog., 14, e1007232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han, Z. and Swaminathan, S. (2006) Kaposi's sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J. Virol., 80, 5251–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walker, A.K., Spiller, K.J., Ge, G., Zheng, A., Xu, Y., Zhou, M., Tripathy, K., Kwong, L.K., Trojanowski, J.Q. and Lee, V.M. (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol., 130, 643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klickstein, J.A., Mukkavalli, S. and Raman, M. (2020) AggreCount: an unbiased image analysis tool for identifying and quantifying cellular aggregates in a spatially defined manner. J. Biol. Chem., 295, 17672–17683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghasemi, M. and Brown, R.H., Jr. (2018) Genetics of amyotrophic lateral sclerosis. Cold Spring Harbor perspectives in medicine, 8, a024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wolozin, B. (2019) The evolution of phase-separated TDP-43 in stress. Neuron, 102, 265–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y.J., Vanderweyde, T., Citro, A., Mehta, T., Zaarur, N., McKee, A., Bowser, R., Sherman, M. et al. (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One, 5, e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pourhaghighi, R., Ash, P.E.A., Phanse, S., Goebels, F., Hu, L.Z.M., Chen, S., Zhang, Y., Wierbowski, S.D., Boudeau, S., Moutaoufik, M.T. et al. (2020) BraInMap elucidates the macromolecular connectivity landscape of mammalian brain. Cell Syst, 11, 208. [DOI] [PubMed] [Google Scholar]
- 46. Fecto, F., Yan, J., Vemula, S.P., Liu, E., Yang, Y., Chen, W., Zheng, J.G., Shi, Y., Siddique, N., Arrat, H. et al. (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol., 68, 1440–1446. [DOI] [PubMed] [Google Scholar]
- 47. Lee, Y., Jonson, P.H., Sarparanta, J., Palmio, J., Sarkar, M., Vihola, A., Evila, A., Suominen, T., Penttila, S., Savarese, M. et al. (2018) TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J. Clin. Invest., 128, 1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niu, Z., Pontifex, C.S., Berini, S., Hamilton, L.E., Naddaf, E., Wieben, E., Aleff, R.A., Martens, K., Gruber, A., Engel, A.G. et al. (2018) Myopathy with SQSTM1 and TIA1 variants: clinical and pathological features. Front. Neurol., 9, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pankiv, S., Clausen, T.H., Lamark, T., Brech, A., Bruun, J.A., Outzen, H., Overvatn, A., Bjorkoy, G. and Johansen, T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem., 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 50. van der Zee, J., Van Langenhove, T., Kovacs, G.G., Dillen, L., Deschamps, W., Engelborghs, S., Matej, R., Vandenbulcke, M., Sieben, A., Dermaut, B. et al. (2014) Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol., 128, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gitcho, M.A., Strider, J., Carter, D., Taylor-Reinwald, L., Forman, M.S., Goate, A.M. and Cairns, N.J. (2009) VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem., 284, 12384–12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim, N.C., Tresse, E., Kolaitis, R.M., Molliex, A., Thomas, R.E., Alami, N.H., Wang, B., Joshi, A., Smith, R.B., Ritson, G.P. et al. (2013) VCP is essential for mitochondrial quality control by PINK1/parkin and this function is impaired by VCP mutations. Neuron, 78, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Seguin, S.J., Morelli, F.F., Vinet, J., Amore, D., De Biasi, S., Poletti, A., Rubinsztein, D.C. and Carra, S. (2014) Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ., 21, 1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yasuda, K., Watanabe, T.M., Kang, M.G., Seo, J.K., Rhee, H.W. and Tate, S.I. (2022) Valosin-containing protein (VCP) regulates the stability of fused in sarcoma (FUS) granules in cells by changing ATP concentrations. FEBS Lett., 596, 1412–1423. [DOI] [PubMed] [Google Scholar]
- 55. Tedesco, B., Vendredy, L., Timmerman, V. and Poletti, A. (2023) The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy, 19, 1619–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zarouchlioti, C., Parfitt, D.A., Li, W., Gittings, L.M. and Cheetham, M.E. (2018) DNAJ proteins in neurodegeneration: essential and protective factors. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 373, 20160534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lima, R.S., Carrettiero, D.C. and Ferrari, M.F.R. (2022) BAG2 prevents tau hyperphosphorylation and increases p62/SQSTM1 in cell models of neurodegeneration. Mol. Biol. Rep., 49, 7623–7635. [DOI] [PubMed] [Google Scholar]
- 58. Gomez-Tortosa, E., Baradaran-Heravi, Y., Dillen, L., Choudhury, N.R., Aguero Rabes, P., Perez-Perez, J., Kocoglu, C., Sainz, M.J., Ruiz Gonzalez, A., Tellez, R. et al. (2022) TRIM25 mutation (p.C168*), coding for an E3 ubiquitin ligase, is a cause of early-onset autosomal dominant dementia with amyloid load and parkinsonism. Alzheimers Dement., 19, 2805–2815. [DOI] [PubMed] [Google Scholar]
- 59. Majerciak, V., Yamanegi, K., Allemand, E., Kruhlak, M., Krainer, A.R. and Zheng, Z.M. (2008) Kaposi's sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J. Virol., 82, 2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alvarado-Hernandez, B., Ma, Y., Sharma, N.R., Majerciak, V., Lobanov, A., Cam, M., Zhu, J. and Zheng, Z.M. (2022) Protein-RNA Interactome analysis reveals wide Association of Kaposi's sarcoma-associated herpesvirus ORF57 with host noncoding RNAs and Polysomes. J. Virol., 96, e0178221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Massimelli, M.J., Majerciak, V., Kruhlak, M. and Zheng, Z.M. (2013) Interplay between polyadenylate-binding protein 1 and Kaposi's sarcoma-associated herpesvirus ORF57 in accumulation of polyadenylated nuclear RNA, a viral long noncoding RNA. J. Virol., 87, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Azpurua, J., El-Karim, E.G., Tranquille, M. and Dubnau, J. (2021) A behavioral screen for mediators of age-dependent TDP-43 neurodegeneration identifies SF2/SRSF1 among a group of potent suppressors in both neurons and glia. PLoS Genet., 17, e1009882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Peng, G., Gu, A., Niu, H., Chen, L., Chen, Y., Zhou, M., Zhang, Y., Liu, J., Cai, L., Liang, D. et al. (2022) Amyotrophic lateral sclerosis (ALS) linked mutation in Ubiquilin 2 affects stress granule assembly via TIA-1. CNS Neurosci Ther, 28, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiang, L., Lin, W., Zhang, C., Ash, P.E.A., Verma, M., Kwan, J., van Vliet, E., Yang, Z., Cruz, A.L., Boudeau, S. et al. (2021) Interaction of tau with HNRNPA2B1 and N(6)-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell, 81, 4209–4227 e4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johnson, J.O., Pioro, E.P., Boehringer, A., Chia, R., Feit, H., Renton, A.E., Pliner, H.A., Abramzon, Y., Marangi, G., Winborn, B.J. et al. (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci., 17, 664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kwiatkowski, T.J., Jr., Bosco, D.A., Leclerc, A.L., Tamrazian, E., Vanderburg, C.R., Russ, C., Davis, A., Gilchrist, J., Kasarskis, E.J., Munsat, T. et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- 67. Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K.J., Nishimura, A.L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D., Wright, P. et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Couthouis, J., Hart, M.P., Shorter, J., DeJesus-Hernandez, M., Erion, R., Oristano, R., Liu, A.X., Ramos, D., Jethava, N., Hosangadi, D. et al. (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. U. S. A., 108, 20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eimer, W.A., Vijaya Kumar, D.K., Navalpur Shanmugam, N.K., Rodriguez, A.S., Mitchell, T., Washicosky, K.J., Gyorgy, B., Breakefield, X.O., Tanzi, R.E. and Moir, R.D. (2018) Alzheimer's disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron, 99, 56–63 e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wozniak, M.A., Frost, A.L. and Itzhaki, R.F. (2009) Alzheimer's disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimers Dis., 16, 341–350. [DOI] [PubMed] [Google Scholar]
- 71. Cairns, D.M., Rouleau, N., Parker, R.N., Walsh, K.G., Gehrke, L. and Kaplan, D.L. (2020) A 3D human brain-like tissue model of herpes-induced Alzheimer's disease. Sci. Adv., 6, eaay8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wozniak, M.A., Itzhaki, R.F., Shipley, S.J. and Dobson, C.B. (2007) Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci. Lett., 429, 95–100. [DOI] [PubMed] [Google Scholar]
- 73. Tran, D.N., Bakx, A., van Dis, V., Aronica, E., Verdijk, R.M. and Ouwendijk, W.J.D. (2022) No evidence of aberrant amyloid beta and phosphorylated tau expression in herpes simplex virus-infected neurons of the trigeminal ganglia and brain. Brain Pathol., 32, e13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cabrera, J.R., Rodriguez-Izquierdo, I., Jimenez, J.L. and Munoz-Fernandez, M.A. (2020) Analysis of ALS-related proteins during herpes simplex virus-2 latent infection. J. Neuroinflammation, 17, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Arimoto, K., Fukuda, H., Imajoh-Ohmi, S., Saito, H., and Takekawa, M. (2008) Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nature Cell Biology, 10, 1324–1332. [DOI] [PubMed] [Google Scholar]
- 76. Sama, R.R.K., Ward, C.L., Kaushansky, L.J., Lemay, N., Ishigaki, S., Urano, F., and Bosco, D.A. (2013) FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. Journal of Cellular Physiology, 228, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schmidt, E.K., Clavarino, G., Ceppi, M. and Pierre, P. (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods, 6, 275–277. [DOI] [PubMed] [Google Scholar]
- 78. Rapsomaniki, M.A., Kotsantis, P., Symeonidou, I.E., Giakoumakis, N.N., Taraviras, S. and Lygerou, Z. (2012) easyFRAP: an interactive, easy-to-use tool for qualitative and quantitative analysis of FRAP data. Bioinformatics, 28, 1800–1801. [DOI] [PubMed] [Google Scholar]
- 79. Koulouras, G., Panagopoulos, A., Rapsomaniki, M.A., Giakoumakis, N.N., Taraviras, S. and Lygerou, Z. (2018) EasyFRAP-web: a web-based tool for the analysis of fluorescence recovery after photobleaching data. Nucleic Acids Res., 46, W467–W472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Koopmans, F., Li, K.W., Klaassen, R.V. and Smit, A.B. (2022) MS-DAP platform for downstream data analysis of label-free proteomics uncovers optimal workflows in benchmark data sets and increased sensitivity in analysis of Alzheimer's biomarker data. J. Proteome Res., 22, 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Deutsch, E.W., Csordas, A., Sun, Z., Jarnuczak, A., Perez-Riverol, Y., Ternent, T., Campbell, D.S., Bernal-Llinares, M., Okuda, S., Kawano, S. et al. (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res., 45, D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Perez-Riverol, Y., Bai, J., Bandla, C., Garcia-Seisdedos, D., Hewapathirana, S., Kamatchinathan, S., Kundu, D.J., Prakash, A., Frericks-Zipper, A., Eisenacher, M. et al. (2022) The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res., 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ge, S.X., Jung, D. and Yao, R. (2020) ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics, 36, 2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.