

Summary
Altered myeloid inflammation and lymphopenia are hallmarks of severe infections. We identified the upregulated EN-RAGE gene program in airway and blood myeloid cells from patients with acute lung injury from SARS-CoV-2 or other causes across 7 cohorts. This program was associated with greater clinical severity and predicted future mechanical ventilation and death. EN-RAGEhi myeloid cells express features consistent with suppressor cell functionality, including low HLA-DR and high PD-L1. Sustained EN-RAGE program expression in airway and blood myeloid cells correlated with clinical severity and increasing expression of T cell dysfunction markers. IL-6 upregulated many EN-RAGE program genes in monocytes in vitro. IL-6 signaling blockade by tocilizumab in a placebo-controlled clinical trial led to rapid normalization of EN-RAGE and T cell gene expression. This identifies IL-6 as a key driver of myeloid dysregulation associated with worse clinical outcomes in COVID-19 patients and provides insights into shared pathophysiological mechanisms in non-COVID-19 ARDS.
Subject areas: Clinical genetics, Molecular medicine, Immune response
Graphical abstract
Highlights
-
•
The myeloid EN-RAGE program upregulated by IL-6 predicts ARDS mortality
-
•
EN-RAGEhi cells traffic to the airways in COVID-19 and non-COVID-19 ARDS
-
•
EN-RAGEhi cells express low HLA-DR and high PD-L1 that may impair T cell responses
-
•
IL-6R blockade in COVID-19 patients rapidly normalizes EN-RAGE expression and T cells
Clinical genetics; Molecular medicine; Immune response
Introduction
Altered myeloid cell expression states, including the accumulation of cells with hallmarks of myeloid-derived suppressor cells (MDSCs), are consistent features in the blood of COVID-19 patients, and serve as a hallmark of severity.1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18 Monocytes and granulocytes associated with increased COVID-19 severity exhibit low expression of human leukocyte antigen DR isotype (HLA-DR) and high expression of hallmark MDSC genes such as S100A12 (extracellular newly identified receptor for advanced glycation end products (EN-RAGE)),1,4,19 and can impair T cell activation via contact-dependent (i.e., programmed death ligand 1 (PD-L1)) and soluble mechanisms, including interleukin-10 (IL-10), transforming growth factor β (TGF-β), arginase 1, indoleamine 2,3-dioxygenase (IDO)-dependent tryptophan metabolism, and reactive oxygen and nitrogen species (reviewed by Schrijver et al., Gabrilovich and Nagaraj, Vegila et al., and Hedge et al.20,21,22,23). Presence of MDSCs in severe COVID-19 patients, and patients with other severe infections, correlates with reduced T cell numbers, and can impair T cell proliferation and interferon gamma (IFN-γ) production ex vivo.24,25,26,27,28,29 Reduced T cell proliferation and tissue sequestration can in turn contribute to the lymphopenia observed in COVID-19 patients with severe disease, which increases with the severity of respiratory failure and is prognostic for higher mortality.30 Thus, understanding the role of myeloid suppressor cells and the pathways leading to their dysregulation is of critical importance in COVID-19 and other infections.
The pathways driving myeloid inflammation and the mechanistic connection of maladaptive cellular programs to severe disease and response to drug interventions are not yet understood. IL-6 is a key regulator of inflammation and has been proposed as a potential driver of dysfunctional myeloid immune response in cancer, COVID-19, and other diseases.10,12,21,27,31,32,33,34 IL-6 treatment in vitro leads to differentiation of hematopoietic stem cells into CD14+ monocytes expressing a similar expression program to that observed in COVID-19 patient monocytes, including high expression of EN-RAGE and low expression of HLA-DR.10 Circulating IL-6 levels positively correlate with this severity-associated myeloid state in COVID-19 and other severe infections.10 It has been suggested that treatment with IL-6-blocking antibodies normalizes alterations in the myeloid compartment that are associated with disease severity,12 a hypothesis requiring placebo-controlled trials to test. Samples from hospitalized COVID-19 patients treated with tocilizumab (a monoclonal antibody against IL6R/Actemra) in a placebo-controlled study (COVACTA, n = 438 hospitalized COVID-19 patients with hypoxemia randomized 2:1 tocilizumab:placebo) provided a unique opportunity to evaluate the role of IL-6 in shaping myeloid inflammation in patients.35 A meta-analysis by the World Health Organization concluded that IL-6 antagonists reduce 28-day mortality in patients hospitalized for COVID-19.36
Here, we used single-cell RNA sequencing (scRNA-seq) of peripheral blood mononuclear cells (PBMCs) and bronchoalveolar lavage (BAL) fluid cells collected from COVID-19 patients, to identify a myeloid cell program shared across tissue compartments, defined by high expression of the EN-RAGE gene, among other inflammatory markers. Myeloid EN-RAGE gene and serum protein levels correlate with increased clinical severity in COVID-19 patients.1,4,19 EN-RAGE is a secreted alarmin that promotes inflammation through binding to the receptor for advanced glycation end products and Toll-like receptor 4, and is also chemotactic for leukocytes (reviewed in the study by Foell et al.37). Expression of this program was associated with more severe disease in our discovery cohorts. We replicated this finding using data from the COMET observational study, which includes sampling from endotracheal aspirates (ETA), whole blood (WB), and PBMCs2,38,39 of COVID-19 patients. Combined single-cell profiling of RNA and cell surface proteins (CITE-seq) characterized the immunosuppressive expression program in EN-RAGE signature expressing cells and connected it to cell surface phenotypes of impaired myeloid antigen presentation and T cell dysfunction. The EN-RAGE expression program was associated with several measures of COVID-19 clinical severity and outcomes and was also observed in patients with acute respiratory distress syndrome (ARDS) from other causes, identifying a targetable pathway relevant to ARDS. Finally, blocking IL-6 signaling using tocilizumab in an interventional setting reduced expression of the EN-RAGE signature and normalized T cell numbers in COVID-19 patients from the COVACTA trial, providing mechanistic insight into the therapeutic response to COVID-19 patients to IL-6 blockade.
Results
A pan-myeloid EN-RAGE signature in blood and airway samples is associated with severe COVID-19
We used publicly available scRNA-seq data from COVID-19 patients to define a gene program that classifies a shared myeloid state across airway and blood samples. We seeded the program by the expression of the gene encoding EN-RAGE (S100A12), which has been implicated in several myeloid populations in the peripheral blood associated with COVID-19 severity.2,10,40,41 EN-RAGE expression is upregulated by IL-6,42 is elevated in the airways of ARDS patients as well as other lung diseases,43 and serum levels correlate with COVID-19 severity.1,4 We confirmed upregulation of the S100A12 gene in myeloid cells from both BAL and PBMC samples (Figure 1A). We identified a set of 84 genes that correlate (Pearson’s r > 0.5) with EN-RAGE expression across myeloid cells from BAL fluid and PBMCs from COVID-19 patients (see STAR methods for a description of the derivation of the signature)7,11 (Figure 1B; Table S1). This gene program showed strong coordinated expression across airway and blood samples and predominant expression in many myeloid cell types (monocytes, neutrophils, and macrophages), although dendritic cells (DCs) and non-classical monocytes showed lower overall expression (Figure S1). The method used to identify the EN-RAGE signature relies on coordinate expression both within and across myeloid cell populations. Because the workflows used for analyzing scRNA-seq data will divide cells both by cell type and cell state, we wanted to examine the expression of EN-RAGE signature genes within individual subpopulations of cells. Looking within classical monocytes from PBMC and whole blood, neutrophils from BAL and whole blood, and inflammatory macrophages from BAL, we find robust correlation (mean Pearson correlation coefficient 0.51–0.61) of signature genes with the S100A12 gene (Figure 1C). Increased expression of the EN-RAGE program in neutrophil, monocyte, and macrophage subsets was associated with greater clinical severity in both blood and airway samples (Figures 1D, S1A, and S1B). It is well known and also shown here that there is blood neutrophilia and an influx of inflammatory neutrophils and monocytes into acutely injured airways that is associated with increased clinical severity (Figure 1E). Therefore, both increased number of EN-RAGE-expressing cells and increased level of program expression is associated with greater clinical severity.
Figure 1.

Identification of the severity-associated EN-RAGE myeloid signature in COVID-19 airway (BAL) and peripheral (PBMC) samples
(A) Expression of S100A12 in either PBMC or BAL. Each point represents a patient. Blue = healthy (BAL n = 3; PBMC, n = 3), yellow = moderate/severe (BAL, n = 3; PBMC, n = 8; hospitalized −/+ supplemental O2), red = critical (BAL, n = 6; PBMC, n = 10; requiring mechanical ventilation), with severity defined within each dataset by the authors. Significance was tested using a t test across the indicated groups (n.s. = p > 0.05, ∗ = p < 0.05; ∗∗ = p < 0.01; ∗∗∗ = p < 0.001). Boxes represent the upper and lower quartile of the sample distribution, with the center line showing the median. Whiskers show the most extreme point up to 1.5x the inter-quartile range of the sample distribution.
(B) Pairwise Pearson correlation between all genes and S100A12 in either PBMC11 or BAL.7 Each point represents a gene measured in both BAL and PBMC samples. Cutoffs for identifying genes in the signature are shown as dotted red lines. Genes that are part of the signature are shown in red, while all other genes are shown in black. Selected signature genes are identified by gene symbol.
(C) Pairwise Pearson correlation between genes in the EN-RAGE signature in selected myeloid populations. Each violin represents the distribution of pairwise correlation coefficients across samples within a given myeloid population.
(D) Pseudo-bulk expression profiles of EN-RAGE signature in PBMC and BAL. Each point represents a patient. Groups are colored as in panel A above. Significance was tested using a t test across the indicated groups (n.s. = p > 0.05, ∗ = p < 0.05; ∗∗ = p < 0.01; ∗∗∗ = p < 0.001).
(E) Frequencies of myeloid populations in either PBMC or BAL. Each point represents a patient sample, colored as in panel (A).
The EN-RAGE program scores correlated with the scores of a previously defined MS1 (monocyte state 1)9 gene set, which was associated with increased severity in COVID-19 and sepsis patients9,10 (Spearman’s ρ = 0.64 and 0.95 in pseudo-bulk expression profiles of blood and airway monocytes and neutrophils (5 cohorts) and ρ = 0.65 in whole lung myeloid cells (one cohort) (all p < 0.0001)). Seven genes (CLU, CYP1B1, LILRA5, NAMPT, S100A12, S100A8, and VCAN) are shared between the 84 genes in the EN-RAGE program and the 23 genes in the MS1 signature identified in COVID-19 patients,10 which is a significant overlap (p value = 7e−14, hypergeometric test). Expression of the EN-RAGE program was more highly intercorrelated than the MS1 signature across sample types (Figure S2A). While MS1 had strong pairwise correlations in PBMC myeloid cells, many genes were no longer correlated when measured in WB or ETA (Figures S2B−S2M). Because of the reduced performance of MS1 outside of PBMCs, we used the EN-RAGE program (denoted EN-RAGEhi in subsequent analyses) to more specifically evaluate myeloid cell state in bulk RNA-seq from whole blood and airway samples, which have greater cellular complexity than PBMC samples.
EN-RAGE signature is associated with acute lung injury from diverse causes
We next hypothesized that the EN-RAGE myeloid program may also be a feature of non-COVID-19 lung injury. To test this hypothesis, we scored the EN-RAGE program in samples from the COMET observational cohort, where 75 patients with either COVID-19 or acute lung injury from other causes were followed longitudinally (Table 1, patient characteristics).2,38,39 WB, PBMC, and ETA were sampled and profiled by scRNA-seq. This cohort offers rich clinical and molecular phenotyping to allow single-cell dissection of the connection between the airways and the blood at the mRNA and protein level, and how this relates to clinical outcomes. EN-RAGE signature expression was highest in monocytes, macrophages, and neutrophils, consistent with our previous analyses (Figures 2 and S2). EN-RAGEhi myeloid cells were present in the blood and airways in both COVID-19 and non-COVID-19 acute lung injury patients, highlighting the generality of this program (Figure 2C).
Table 1.
Patient characteristics
| COMET |
COMET |
COMET |
COVACTA | |
|---|---|---|---|---|
| All | SARS-CoV-2+ | SARS-CoV-2− | ||
| n (%) | 75 | 57 (76%) | 18 (24%) | 404 |
| Age median (IQR) | 54 (42, 69) | 48 (42, 66) | 66 (51, 76) | 63 (53, 70) |
| Sex n (%) | ||||
| Male | 52 (69%) | 43 (75%) | 9 (50%) | 284 (70%) |
| Female | 23 (31%) | 14 (25%) | 9 (50%) | 120 (30%) |
| Race n (%) | ||||
| White | 20 (27%) | 11 (19%) | 9 (50%) | 233 (58%) |
| Black/African American | 4 (5%) | 3 (5%) | 1 (6%) | 58 (14%) |
| Asian | 13 (17%) | 9 (16%) | 4 (22%) | 37 (9%) |
| Other/Mixed/Unknown | 38 (51%) | 34 (60%) | 4 (22%) | 76 (19%) |
| Baseline NIH Ordinal Scale n (%) | ||||
| 3 No supplemental O2 | 14 (19%) | 12 (21%) | 2 (11%) | 15 (4%) |
| 4 Supplemental O2 | 25 (33%) | 18 (32%) | 7 (39%) | 109 (27%) |
| 5 Non-invasive/high flow O2 | 14 (19%) | 9 (16%) | 5 (28%) | 114 (28%) |
| 6 Mechanical ventilation (MV) | 3 (4%) | 3 (5%) | 0 (0%) | 58 (14%) |
| 7 MV + additional organ support | 19 (25%) | 15 (26%) | 4 (22%) | 107 (27%) |
| SOFA maximal median (IQR) | 4 (1, 10) | 4 (1, 11) | 4 (1, 9) | NA |
| ICU admission n (%) | 26 (35%) | 30 (53%) | 11 (61%) | 234 (58%) |
| Mortality n (%) | 10 (13%) | 6 (11%) | 4 (22%) | 83 (20%) |
| ARDS AECC definition n (%) | 38 (51%) | 26 (46%) | 12 (67%) | NA |
| ARDS Berlin definition n (%) | 23 (31%) | 18 (32%) | 5 (28%) | NA |
| Hospitalization, days median (IQR) | 8 (8, 18) | 11 (3, 20) | 5 (4, 7) | 22 (9, 28) |
| ICU duration, days median (IQR) | 2 (0, 9) | 14 (4, 26) | 5 (2, 4) | 14 (0, 27) |
| Ventilator-free days median (IQR) | 28 (18, 28) | 28 (16, 28) | 28 (26, 28) | 19 (0, 28) |
NA, not available. COVACTA data are for subjects with blood RNA-seq data included in this manuscript. COVACTA ICU admission frequency is at time of baseline sampling.
Figure 2.

Replication of EN-RAGE severity association across sample types in both COVID-19 and non-COVID-19 acute lung injury (COMET cohort)
(A) UMAP plots with cell type annotations. Each point represents a single cell, colored by cell type. Each panel shows a different sample type, as indicated. PBMC: peripheral blood mononuclear cells, WB, whole blood; ETA, endotracheal aspirates.
(B) UMAP plots showing EN-RAGE signature score. Each point represented a single cell colored by the expression signature value.
(C) Pseudo-bulk expression profiles within myeloid cells. Each point represents the pseudo-bulk gene expression signature score for a cell type in a patient sample. PBMC severity: Moderate = no supplemental O2, severe = supplemental O2 and critical = mechanical ventilation. Whole blood severity: Mild/Moderate = 0 days on ventilator and no more than 1 day in ICU, Severe patients had ≥1 day on ventilator. ETA: Critical = VFDS = 0 (ventilation for ≥28 days or death), severe ETA = VFDS>0. Significance was tested using a t test across the indicated groups (n.s. = p > 0.05, ∗∗ = p < 0.01; ∗∗∗ = p < 0.001). Sample numbers per cohort: PBMC healthy, n = 11; PBMC COVID− moderate, n = 4; PBMC COVID− severe, n = 6; PBMC COVID− critical, n = 3; PBMC COVID+ moderate, n = 12; PBMC COVID+ severe, n = 10; PBMC COVID+ critical, n = 14. WB healthy, n = 14; WB COVID− mild, n = 4; WB COVID− severe, n = 1; WB COVID+ mild, n = 8; WB COVID+ severe, n = 5. ETA COVID− moderate/severe, n = 2; ETA COVID− critical, n = 1; ETA COVID+ moderate/severe, n = 5; ETA COVID+ critical, n = 8. Boxes represent the upper and lower quartile of the sample distribution, with the center line showing the median. Whiskers show the most extreme point up to 1.5x the inter-quartile range of the sample distribution.
(D) Frequencies of myeloid populations in either PBMC or BAL. Each point represents a patient sample, colored as in panel (C).
EN-RAGE signature expression correlates with clinical severity and is prognostic for worse clinical outcomes
In COVID-19 patients, EN-RAGEhi myeloid cells were associated with increased clinical severity at presentation, as defined by the extent of respiratory support required at study enrollment, in monocytes (PBMC, Figures 2C and S1C) and neutrophils (whole blood, Figure 2C). The severity association of monocyte EN-RAGE score observed in PBMC was not observed in the smaller number of samples available from whole blood. An association with severity is not found in ETA samples, perhaps since this sample type is only obtained from critically ill patients on mechanical ventilation and so a milder severity group is lacking.
We next asked whether higher EN-RAGE signature expression predicts worse patient outcomes. In PBMC samples from COVID-19 and non-COVID-19 patients at COMET study enrollment, the EN-RAGE program score in PBMC monocytes was associated not only with greater baseline clinical severity (NIH COVID-19 severity ordinal score) but also worse clinical outcomes (ICU admission p < 0.01 and maximal NIH ordinal scale: Spearman ρ = 0.30 p = 0.02) (Figures 3A and S3A−S3C). However, these associations were no longer significant once the EN-RAGE score was adjusted for baseline ordinal severity score (all p > 0.05, Figure S3G), possibly because of the limited numbers of patients in this cohort and the increased risk of severe outcomes in patients presenting with greater severity. EN-RAGE program expression was not significantly associated with age (Spearman ρ = −0.02, p = 0.88) nor with days from symptom onset to study enrollment (Spearman ρ = 0.09, p = 0.57). EN-RAGE score was higher in patients who presented with or later developed ARDS compared with those who did not by either AECC or Berlin diagnostic criteria (p < 0.01 for AECC definition, p < 0.05 for Berlin definition) (Figures S3D and S3E). EN-RAGE score was higher in ARDS (AECC definition) resulting from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection or other etiology (p < 0.05) (Figure 3B), but the difference was not significant in the smaller patient subgroups defined using the more stringent Berlin ARDS definition (Figure S3F).
Figure 3.

EN-RAGE signature expression correlates with disease severity and immunosuppressive gene expression in myeloid cells (COMET cohort)
(A) Spearman correlations between pseudo-bulk EN-RAGE signature score in PBMC monocytes and NIH ordinal severity score, maximal NIH severity score, SOFA organ failure score, and plasma IL-6 and IL-10 protein levels at study enrollment.
(B) PBMC monocyte EN-RAGE gene score is higher in patients who develop ARDS (AECC definition) in COVID-19 patients; n = 46. Medians are indicated. ∗ t test p < 0.05.
(C) Longitudinal changes in EN-RAGE signature in bulk ETA RNA-seq. Each point represents a patient sample from COVID-19 (n = 16 patients, n = 276 samples) and non-COVID (n = 3 patients, n = 6 samples) patients requiring mechanical ventilation. Samples from the same patient are linked by dotted lines. Points are colored by severity of disease. For illustrative purposes, linear regression trend lines for signature scores over time, grouped by severity level are shown as solid lines. Slopes were significantly different using a linear mixed model, p < 0.05. MV, mechanical ventilation.
(D) Table of Spearman correlation coefficients between pseudo-bulk EN-RAGE signature score and genes encoding myeloid effector functions within monocyte or neutrophil populations across endotracheal aspirates (ETA), whole blood, and PBMCs from the COMET cohort. Positive correlations are shaded red and negative correlations shaded blue, with increasing darkness of shading indicating two-tailed p values of p < 0.05, p < 0.01, and p < 0.001.
(E) PBMC myeloid EN-RAGE gene score correlates with plasma IL-6 and IL-10 protein; n = 46.
(F) Correlation of pseudo-bulk expression signature for EN-RAGE genes compared to pseudo-bulk expression values of IL-10 and PD-L1 in monocytes in COMET tracheal aspirate samples. Each point represents the expression value in a cell type in a single sample; n = 40. Log2 gene expression, Spearman correlation coefficients, and two-tailed p values are shown.
Leveraging longitudinal sampling from ventilated patients in the COMET cohort, we found a significant association between the temporal trajectory of airway EN-RAGE expression in each patient and patient outcomes (Figure 3C). To test this, we stratified patients into two groups by the number of ventilator-free days and compared the slope of the regression lines of the expression of the EN-RAGE signature in bulk RNA-seq from ETA samples in the two groups. Airway EN-RAGE expression decreased over time in survivors with fewest days of ventilation and increased in patients who died or had ≥28 days of ventilation (p < 0.05 linear mixed model, ETA, Figure 3C). Worse clinical outcomes were accompanied by sustained elevated airway levels of EN-RAGEhi cells as well as higher baseline levels in the blood.
EN-RAGE program expression in myeloid cells is associated with increased markers of immunosuppression in blood and airways
The EN-RAGE program score was also associated with expression of genes characteristic of MDSCs, suggesting one path through which EN-RAGEhi cells may contribute to clinical severity. Specifically, EN-RAGE program expression was correlated with metrics of suppressed myeloid and lymphoid states across another five COVID-19 cohorts,2,5,7,11,12,38,39 spanning ETA, BAL, lung, PBMC, and blood samples (complete results in Table S2). For example, in COMET PBMC monocytes, EN-RAGE signature scores in pseudo-bulk expression profiles correlated with high expression of CD14 (Spearman ρ = 0.65, p < 0.001), CCR2 (ρ = 0.25, p < 0.05) and PTGER2 (ρ = 0.38, p < 0.001) in pseudo-bulk profiles of monocytes and DCs (Figure 3D). CCR2 and PTGER2 are two receptors important for myeloid cell recruitment to the infected lung via CCL2 and prostaglandins, respectively. EN-RAGE program expression correlated with low expression of MHC class II genes, suggesting reduced capacity for antigen presentation (in PBMC monocytes, HLA-DRA: ρ = −0.45 and HLA-DRB1: ρ = −0.48, p < 0.001, Figure 3D and Table S2). In blood monocytes, there was a positive correlation with STAT3, a key transcription factor regulating MDSC gene expression (ρ = 0.79, p < 0.001, Figure 3D). MDSCs can suppress T cells using context-specific mechanisms across sites of infection or malignancy, and the mechanisms used can also differ depending on whether they originate from the monocytic or granulocytic lineage.13,21 In blood monocytes, EN-RAGE program expression was positively correlated with expression of genes encoding effectors that can suppress T cells through reactive oxygen species (CYBB/PHOX) and prostaglandins (PTGER2), and TGFβ1, but inconsistent associations with arginase (ARG1) and tryptophan depletion (IDO1) across cohorts (Figure 3D and Table S2). Granulocytic EN-RAGEhi cells had similar associations as their monocytic counterparts, with notable differences including stronger correlations with PDL1 and TGFβ1 and little to no association with reactive oxygen species (CYBB/PHOX) and PTGER2 compared with EN-RAGEhi monocytes (Figure 3D and Table S2). Consistent with MDSCs characterized in other infections and cancers,21,23,44 EN-RAGEhi cells expressed higher levels of multiple potential mediators of immunosuppression (PDL1, CYBB/PHOX, and TGFβ1) with some genes preferentially expressed by monocytic lineages (i.e., CYBB/PHOX ρ > 0.4, Table S2) or granulocytic lineages (i.e., TGFβ1 ρ > 0.6, Table S2) (Table S2). EN-RAGE program expression in PBMC myeloid cells was modestly correlated with plasma protein levels of IL-6, a potential driver of MDSCs, and IL-10, a potential mediator of T cell suppression (Spearman ρ = 0.41, p = 0.005 and ρ = 0.29, p = 0.05, respectively; Figure 3E).
These associations were also largely observed in airways samples, suggesting an overall consistent phenotype in both the blood and the infected lung (Figure 3D). As in the blood, EN-RAGE program expression in monocytes and neutrophils was positively correlated with STAT3 in ETA samples but lacked the correlations with CCR2 and HLADR, showed stronger correlations with CCR5, and inconsistent relationships with CYBB and CD14 (Figure 3D and Table S2). EN-RAGEhi monocyte and neutrophil cells in lung and airway samples (ETA, BAL, and lung postmortem autopsy tissue) had increased association with PDL1, IL10, TGFβ, IDO, and IL1β, which can suppress T cell function, compared to the blood (Figures 3D and 3F; Table S2). Peripheral blood myeloid cells expressed lower levels of IL10 and IL1β and had weaker correlation with the EN-RAGE signature when transcripts were detected, illustrating the importance of sampling infected tissues and establishing blood correlates of tissue immune responses (Figure 3D and Table S2). Across four patient cohorts, airway EN-RAGEhi monocytes and neutrophils consistently expressed multiple markers of inflammation with autoregulatory functions that can be immunosuppressive, including IL10, PDL1, TGFβ1, IL1β, and IDO (Table S2).
Characterization of EN-RAGEhi myeloid and T cell phenotypes
To connect the EN-RAGE expression program to cellular phenotypes, we used CITE-seq data from PBMCs in COMET to relate cell surface protein expression and EN-RAGE program expression. The EN-RAGE signature was most highly expressed in CD14+CD16loHLA-DRlo classical monocytes (cM) (Figures 4A and 4B), confirming the reduced antigen presentation capacity suggested by scRNA-seq data (Table S2). EN-RAGE signature scores on cMs positively correlated across patients with cM surface expression of 19 of the 188 CITE-seq measured proteins (FDR<0.05, Figure 4C), including three markers characteristic of MDSCs: PD-L1 (CD274, Spearman ρ = 0.43, FDR = 0.008), podoplanin (PDPN, ρ = 0.52, FDR = 0.001), and CD38 (ρ = 0.49, FDR = 0.002), which is IL-6 inducible in tumors.45,46 Conversely, EN-RAGE signature scores on cM were negatively correlated with 19 markers (FDR<0.05, Figure 4C), including HLA-DR (ρ = −0.73, FDR<0.0001), the T cell costimulatory protein CD40L (ρ = −0.36, FDR = 0.03), the LFA subunit CD11A involved in trafficking and activation (ITGAL, ρ = −0.52, FDR = 0.002), and two markers of granulocytic MDSCs (SIGLEC7: ρ = −0.52, FDR 0.001; CD244: ρ = −0.50, FDR = 0.002) (Figure 4C).
Figure 4.

Characterization of myeloid and T cell immunosuppression phenotypes (COMET PBMC cohort)
(A) EN-RAGE gene set score is most highly expressed in classical monocytes (cM) in PBMC CITE-seq data. ncM = non-classical monocytes, progen = progenitor cells. Cells were defined by marker genes as previously described.39
(B) CD14, CD16, and HLA-DR surface protein expression across cell lineages in PBMC CITE-seq data.
(C−E) Spearman correlations between EN-RAGE gene set expression in classical monocytes and protein expression on (C) classical monocytes, (D) CD4+ T cells, and (E) CD8+ cells; n = 128 samples, including 11 healthy controls. FDR<0.05 for all correlations, panel of 188 proteins measured.
(F–L) Pseudo-bulked surface protein expression in 128 PBMC samples from 60 patients over 14 days in patients grouped by clinical outcomes. Classical monocyte expression of (F) EN-RAGE gene signature, (G). HLA-DR protein, (H) PD-L1 protein. CD8+ T cell expression of (I) PD-1 and (J) LAG3. CD4+ T cell expression of (K) PD-1 and (L) LAG3. Blue line denotes the linear regression trend for gene expression over time. Red line denotes expression level in healthy controls. Vent. duration = days of mechanical ventilation in survivors. n = 128 samples from 60 patients (429, 505 cells). Pearson correlation coefficients (r) and p values are indicated.
EN-RAGE program scores in cMs also negatively correlated with the level of the activation marker CD40 on CD4+ T cells (Figure 4D), consistent with the negative correlation of the expression of the cognate ligand CD40L on cMs with their EN-RAGE program (Figure 4C). Conversely, EN-RAGE program scores in cMs were positively correlated with two proteins expressed by exhausted CD4+ and CD8+ T cells: podoplanin/PDPN (Spearman ρ = 0.37, FDR = 0.04) and TNFSF14/LIGHT (ρ = 0.39, p = 0.03) (Figures 4D and 4E). These data support the EN-RAGE program activity in cMs correlating with distinct CD8+ and CD4+ T lymphocytes activation states across patients.
Moreover, the dynamic changes in EN-RAGE program expression and markers of myeloid activation and T cell dysfunction were associated with clinical outcomes. Patients with PBMC CITE-seq data were categorized into 4 outcome groups based on survival and the duration of mechanical ventilation. In monocytes from patients with more severe outcomes compared with patients not requiring ventilation, HLA-DR trended lower than healthy (indicated by red line) and failed to recover to healthy levels over 14 days (Figure 4F), and trends of sustained higher PD-L1 and EN-RAGE score in some greater severity groups remained higher in ventilated patients (Figures 4G and 4H). T cell marker gene expression does not differ from levels compared with healthy controls in patients with better clinical outcomes, while a trend toward increasing above healthy levels in non-survivors at day 14 was observed. On CD8+ and CD4+ T cells, markers of T cell dysfunction or exhaustion (PD-1, LAG3, TIGIT, CTLA4, and BTLA4) showed greater increases over 14 days in patients with worsening outcomes (>7 days mechanical ventilation and/or death) compared with non-ventilated patients (Figures 4I−4L, and S4, Pearson correlation p < 0.05, not significant for PD-1 on CD4+ T cells). Therefore, the EN-RAGE myeloid expression program correlates with markers of a suppressive cell surface phenotype on monocytes, and with reduced activation of CD4+ and CD8+ T cells, as indicated by increasing expression of T cell dysfunction markers over time (Figures 4I−4L and S4). The relationship between cellular phenotypes and clinical recovery could not be assessed as samples were not collected at or following hospital discharge.
IL-6 induces the EN-RAGE program in monocytes in vitro
Several lines of evidence led us to hypothesize that IL-6 can be a regulator of the EN-RAGE program. First, IL-6 treatment of HSPCs promotes upregulation of the MS1 gene signature in monocytes,10 which correlates with the EN-RAGE program (Spearman ρ ≥ 0.64 in monocytes, p < 0.0001 across 5 cohorts as described previously). Moreover, blood monocyte EN-RAGE program expression correlates with plasma IL-6 protein levels and with levels of monocyte STAT3 mRNA, a transcription factor activated by IL-6 signaling (Figures 3D and 3E; Table S2). To test this hypothesis, we treated human primary monocytes with IL-6 in vitro followed by RNA-seq.
IL-6 treatment altered the expression of 36 of 84 EN-RAGE program genes (Benjamini-Hochberg FDR<0.05, Figure 5A; Table S3), as well as of IL10, IL1β, CYBB, and CCR2 (Figure 5B; Table S3), all features of EN-RAGEhi monocytes in COVID-19 patients (Figure 3D). This IL-6-regulated subset was strongly correlated with the full EN-RAGE signature (Pearson’s correlation coefficient 0.98). Using gene set enrichment analysis, we found that IL-6 could partially upregulate the expression of EN-RAGE and MS1 signature genes (Benjamini-Hochberg adjusted p < 0.001 and p < 0.01, respectively; Figure 5C). There were no overlapping genes with a previously reported IL-6 or IL-1β transcriptional response signature.47 Our data suggest that IL-6 is sufficient to upregulate the expression of many EN-RAGE program genes in monocytes in vitro, including markers of a potentially T cell suppressive phenotype in patients.
Figure 5.

EN-RAGE signature genes are induced by in vitro treatment of monocytes with IL-6. Monocytes stimulated with IL-6 for 24 h compared with media, selected genes visualized as a heatmap using unsupervised clustering
(A) EN-RAGE signature genes.
(B) Genes associated with potential T cell suppressive functionality, from Table S3.
(C) FGSEA analysis of EN-RAGE and MS1 signatures in IL-6-treated monocytes. Bars represent the normalized enrichment scores of how much each gene set is regulated by IL-6 treatment (BH adjusted p value for MS1 < 0.01 and EN-RAGE <0.001).
The myeloid EN-RAGE program correlates with expression programs of suppressive myeloid cells and impaired T cells and with increased clinical severity in an interventional COVID-19 clinical trial
We next asked if blocking IL-6 signaling in patients correlated with a change in EN-RAGE program expression, leveraging data from COVACTA, a double-blind randomized clinical trial of tocilizumab (anti-IL6R/Actemra) in hospitalized COVID-19 patients with hypoxemia.35 Consistent with our findings in other cohorts, bulk RNA-seq expression profiles from whole blood collected at baseline from 438 patients showed higher normalized enrichment scores for the EN-RAGE program in patients requiring positive pressure ventilation at baseline compared with those who did not (Figures 6A and 6B, FGSEA Benjamini-Hochberg FDR <0.05). The EN-RAGE gene set was also enriched in patients needing future mechanical ventilation or progressing to death even when controlling for the association with baseline severity (Figures 6C and 6D, FGSEA Benjamini-Hochberg FDR<0.05). Treatment of COVID-19 patients with tocilizumab led to faster reduction in EN-RAGE signature expression (Figure 6E). Increased EN-RAGE program expression at baseline predicted 28-day mortality when continuous eigengenes were used in a Cox proportional hazards model adjusting for age, treatment arm, and baseline severity ordinal scale (EN-RAGE signature: HR = 2.5 (1.5–4.2 CI, p = 0.00056)). To identify patients with the worst outcomes, we generated a linear predictor of mortality risk combining EN-RAGE expression, age, treatment, and baseline ordinal severity score, and calculated a cut point of the linear predictor with maximal combined sensitivity and specificity for predicting 28-day mortality (Youden J index). This identified a high-risk subset comprising one-third of patients with significantly higher mortality rates in the high-risk group (Figure 6F, log rank p < 0.0001). The IL-6-regulated 36 gene subset (identified in Figure 5) showed similar prognostic value as the full 84 gene signature when continuous eigengenes were used to predict 28-day mortality in a Cox proportional hazards model adjusting for age, treatment arm, and baseline ordinal severity scale (HR = 2.4 (1.5–3.9 CI, p = 0.00028)). The prognostic relationship of the EN-RAGE signature with mortality was maintained even when adjusting for blood monocytes, neutrophils, or lymphocyte frequency (Figure S5A; FDR<0.05, FGSEA). Moreover, lower expression of gene sets classifying CD8+ and CD4+ T lymphocytes (CIBERSORT48) was associated with worse clinical severity and outcomes (Figures 6B–6D, (Benjamini-Hochberg FDR<0.05). Furthermore, consistent with COMET whole blood scRNA-seq, EN-RAGE program scores were positively correlated with expression of PDL1, IL10, and IL1β in whole blood at day 1 in COVACTA patients (Figure 7A), as well as with serum protein levels of EN-RAGE, IL-6, IL-10, IL-1β, and ARG1 (Figure S7A, t-test p < 0.05). Examination of T cell genes that were associated with EN-RAGE program expression in scRNA-seq data (Figure 7A) revealed that high myeloid EN-RAGE program expression in COVACTA bulk RNA-seq was associated with low expression of T cell effectors (granzyme, perforin, and lymphotoxin (GZMB, GZMM, PRF1, and LTA), cytotoxicity (FASL), and IFNG), activation markers (KLRK1/NKG2D), and dysfunction markers (CTLA4, LAG3, CD160). Many of these genes were expressed at a lower level in patients with more severe disease (IFNG, FASLG, CTLA4, LAG3, TIGIT, TBX21/Tbet, XCL1; Figure 7B, Benjamini-Hochberg FDR<0.05 and fold change < log2 -0.5). Higher EN-RAGE program expression was also correlated with lower lymphocytes (ρ = −0.5) and monocytes (ρ = −0.2) and increased neutrophils (ρ = 0.5) (Figure S7A, all p < 0.001). Thus, the COVACTA cohort shows similar features to those observed in COMET, including the expression of the EN-RAGEhi myeloid program and its correlation with greater clinical severity and worse clinical outcomes.
Figure 6.

Severity-associated EN-RAGE gene set is associated with poor outcome and decreased by IL-6 blockade in COVID-19 patients (COVACTA cohort)
(A) Heatmap of EN-RAGE gene set associations with NIH ordinal scale severity at baseline (Severity NIH), need for new mechanical ventilation in patients not ventilated at baseline (Mech Vent), 28-day mortality (Death), treatment with tocilizumab or placebo at day 3 or day 7 relative to baseline, and tocilizumab (TCZ) vs. placebo at day 7 relative to baseline.
(B−E) Gene set enrichment analyses (GSEA) for gene sets associated with the myeloid cell programs EN-RAGE, MS1,10 and MDSC61 and T cells (CIBERSORT48). Normalized enrichment scores are shown, with red shading for FDR<0.05. TCZ = tocilizumab. For (C) and (D), analyses were adjusted for baseline severity by incorporating baseline ordinal score as a covariate in our model. F. Kaplan-Meier curve showing differences between high and low risk patients for 28-day mortality. The cut point used to determine high- vs. low-risk patients was determined using Youden’s J index on the ROC curve of a linear predictor defined using a combined risk score incorporating EN-RAGE expression, age, treatment arm, and baseline ordinal severity score. Significance was determined using the log rank test.
(G and H) Tocilizumab treatment normalizes EN-RAGE and CD8+ T cell gene expression to healthy levels more rapidly than placebo in survivors. Only patients with measurements for all three time points are included. Lines represent the mean expression value for the gene set signature score across tocilizumab- or placebo-treated subjects. Error bars represent the 95% confidence interval around the mean. Patients are split into those that were discharged before 28, those that remained hospitalized, or subjects that died by day 28. Average signature scores are shown across the first 7 days of treatment. CTRL = healthy controls, SOC = standard of care drug therapy; significance testing was performed using t-test comparing each day to baseline, split by study arm ∗ = p < 0.05, ∗∗ = p < 0.01, ∗∗∗ = p < 0.001, ∗∗∗∗ = p < 0.0001.
Figure 7.

IL-6 blockade reduces potential T cell suppressive factors and normalizes T cells in COVID-19 patients (COVACTA cohort)
(A) The EN-RAGE program is positively correlated with IL-6-induced suppressive myeloid genes (IL-10, IL-1b, PD-L1) and inversely correlated with T cell genes (FASL, LKRK1, TIGIT, CD160, IFNG, IL7R, CTLA4, XCL1, HAVCR2, TBX21, PRF1, GZMB, LAG3, GZMM, PDCD1, FOXP3). Spearman correlation coefficients are shown for bulk whole blood gene expression.
(B and C) Volcano plots of reduced expression of T cell genes shown in (A) (shown in red) in patients with greater baseline severity (requiring positive pressure ventilation) versus lower baseline severity (not requiring positive pressure ventilation) (B) and increased expression of T cell genes following 7 days treatment with tocilizumab (C). Dotted lines indicate absolute >0.5 log2 fold change and FDR<0.05.
(D) Working model.
IL-6 blockade decreases the myeloid EN-RAGE state and increases T cells in COVID-19 patients
Finally, to understand the role of IL-6 in patients, we examined the effect of tocilizumab treatment on the EN-RAGE myeloid state in COVID-19 patients and effect on T cells using longitudinal samples from the COVACTA study.14,35 Blockade of IL-6 signaling reduced expression of many genes in the EN-RAGE program and increased T cell signature expression after 3 or 7 days of tocilizumab treatment compared with placebo (Figures 6A, 6E, and S5B−S5D; Benjamini-Hochberg FDR<0.05). Consistent with the EN-RAGE program expression, MDSC and MS1 signatures were elevated in patients with worse baseline clinical severity and needing future mechanical ventilation or who died, and decreased following tocilizumab treatment (Figures 6B–6E, Benjamini-Hochberg FDR<0.05). EN-RAGE program gene expression was sustained over the first 7 days in patients with worse clinical outcomes at day 28 (hospitalized or non-survivor vs. discharged, Figure 6G). The EN-RAGE program was reduced following tocilizumab treatment more than in placebo treatment over the first 7 days, particularly in patients with better clinical outcomes (Figure 6G, S5B, and S6A). At the same time, CD8 T cell-associated gene expression rapidly increased after tocilizumab treatment, again particularly in patients that showed clinical improvement (Figures 6H, S6B, and 7C). Similar results were observed for the MS1 severity-associated myeloid and CIBERSORT CD4+ T cell gene sets (Figures S6C, and S6D). This corresponded with a more rapid increase in blood lymphocytes and decrease in neutrophil cell counts in tocilizumab-treated patients who were discharged by 28 days compared with placebo (Figures S6E−S6J). When we compared the change between day 7 and baseline in EN-RAGE program expression with the change in measured blood cellularity, patients with a greater decrease in EN-RAGE had a greater increase in blood lymphocytes and monocytes and decreases in neutrophils (Figures S7B−S7D). This was more pronounced in patients treated with tocilizumab compared with placebo.
The reduction in EN-RAGE program following IL-6 blockade corresponded with normalization of potential mediators and correlates of T cell suppression. Patients with a greater decrease in EN-RAGE program expression from baseline to 7 had greater decreases in serum EN-RAGE, IL-10, IL-1β, and ARG1 protein levels, as indicated by a positive slope of the correlation line (Figure S7E−S7H). IL-6R blockade with tocilizumab resulted in a greater decrease in EN-RAGE program expression, as indicated by a greater offset in the regression in the tocilizumab-treated arm compared with placebo, and a greater decrease in this set of serum proteins, as indicated by more green vs. red dots in the bottom left quadrant (Figures S7E−S7H). This correlated with increased expression of the T cell genes IFNG, FASLG, CTLA4, and XCL1 in tocilizumab-treated patients (Figure 7C). In sum, blockade of IL-6 signaling in COVID-19 patients rapidly normalizes the severity-associated myeloid and T cell states, identified using both patient scRNA-seq data and the IL-6 in vitro model, to levels in healthy individuals, which correlates with clinical improvement by 28 days.
Discussion
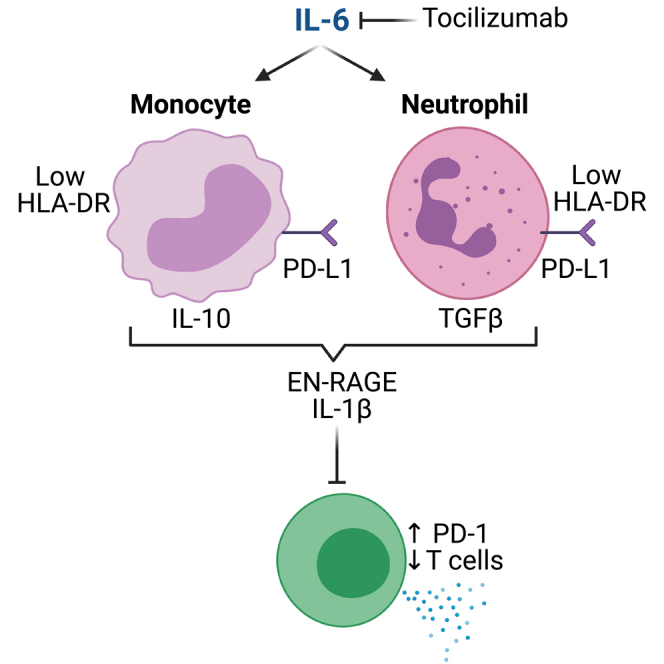
While altered myeloid states are hallmarks of COVID-19 disease severity,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18 the pathways driving maladaptive myeloid inflammation have not been clearly defined. This study supports a working model whereby IL-6 differentiates myeloid cells from both monocytic and granulocytic lineages to a suppressive phenotype characterized by low antigen presentation on HLA-DR and increased expression of multiple factors that can suppress T cells (IL-10, PD-L1, and TGF-β1) (Figure 7D). We define a pan-myeloid EN-RAGE program of coordinately expressed genes in the airways and blood of COVID-19 patients that is prognostic for severe outcomes and is robust across 7 cohorts.2,5,6,7,11,12,14,38,39 EN-RAGEhi cells express multiple phenotypic hallmarks of MDSCs by cell surface protein analysis: decreased capacity for antigen presentation and co-stimulation through HLA-DR and CD40, and increased potential to suppress T cells through PD-L1. This was associated with sustained elevated expression of markers on T cells dysfunction such as PD-1 in patients with prolonged mechanical ventilation or who died. COVID-19 patients with higher EN-RAGE signature expression had a greater risk of future mechanical ventilation and mortality, and EN-RAGEhi myeloid cell impairment of optimal T cell-mediated immunity is one potential mechanism. EN-RAGEhi cells also express genes that can increase or skew inflammation or promote lung barrier permeability (i.e., S100A12, IL-1β, and TGF-β1), consistent with MDSCs expressing both pro- and anti-inflammatory genes.22
These data demonstrate the importance of IL-6 in altering immune cell phenotypes in COVID-19 patients and provide a potential mechanism for the therapeutic benefit of tocilizumab in patients hospitalized with COVID-19.36 Tocilizumab is approved for treating hospitalized adult COVID-19 patients requiring supplemental oxygen and receiving corticosteroids by the US Food and Drug Administration and the European Medicines Agency. IL-6 antagonists significantly reduced mortality compared with usual care in a large meta-analysis using data from 27 trials.36 While COVACTA did not meet the primary endpoint of improving clinical status on day 28, tocilizumab demonstrated clinically meaningful benefits, such as shortening hospital stay by 8 days, compared with the placebo arm.35 Therapeutic blockade of IL-6 signaling with tocilizumab attenuates expression of the EN-RAGE signature in blood cells and normalizes T cell numbers, correlating with clinical improvement.
COVID-19 patients with lymphopenia are at higher risk for worse clinical outcomes in the COVACTA cohort.30 In addition to being reduced in numbers, suppressed T cell phenotypes have been described in severe COVID-19 patients. Polyfunctional Th1 and Th17 CD4+ and CD8+ cell subsets are underrepresented in SARS-CoV-2 infection, with less proliferation and impaired IFN-γ and IL-2 secretion following restimulation in vitro.49,50 T cells with increased expression of activation (OX40, CD69) and exhaustion (PD-1, TIGIT, and TIM1) markers have been observed in some40,51 but not all cohorts52 and functional exhaustion of CD8+ T cells has been reported.53,54,55 In single-cell analysis of the COMET cohort, a phenotype of sustained elevated expression of PD-1, TIGIT, and LAG3 was observed on T cells in patients requiring longer mechanical ventilation and non-survivors. As tocilizumab increased the number of circulating T cells, the observed increase in markers of T cell function such as IFNγ and CD69 may result from increased T cell abundance rather than increased functionality per cell. Differential regulation of T cell genes suggests a potential effect of tocilizumab on T cell quality in addition to quantity. IL-6R blockade increases markers of early polyfunctional T cells (IFNG, XCL1, CD69) and recently activated/less exhausted cells (IL7R) while markers associated with broader spectrum of T cell functional states (i.e., cytotoxic T cell markers T-bet (TBX21), perforin (PRF1), and granzymes (GZMM, GZMB)) are less affected. Natural killer (NK) cells are important antiviral effector cells that can be suppressed by MDSCs20 and can express markers of dysfunction in COVID-19.52 A potential connection between EN-RAGEhi myeloid cells and NK cells or the suppressive regulatory T cell phenotype observed in severe COVID-19 patients56 remains to be characterized.
Suppressive myeloid cells are one potential mechanism underlying impaired T cell immunity in severe COVID-19. MDSCs express mediators of T cell suppression in a context-dependent manner, depending on the type of infection or tumor.21,23 MDSCs express multiple genes through which they may potentially suppress T cells. Moreover, differential correlations between the EN-RAGE signature and genes encoding immunosuppressive mediators were observed between cell lineage (granulocytic vs. monocytic) and compartment (blood vs. airways). MDSCs are often identified by low HLA-DR expression as a marker of impaired antigen presentation capacity.24,27,28,29,49 Interestingly, while some features of EN-RAGEhi cells were consistent between blood and airways (i.e., high PD-L1), the expression of other genes was tissue dependent, and the low expression of HLA-DR in the blood was lost in airway samples. This is consistent with the observation that HLA-DRlo MDSCs were not found in ETA airway samples by flow cytometry.27 These data suggest that EN-RAGEhi myeloid cells home to the infected lung and adopt a tissue-specific phenotype, with increasing expression of IL-10 and IDO and decreasing expression of CCR2 and CYBB, highlighting the importance of characterizing immune responses at the site of infection. An alternative hypothesis is that IL-6 activity drives both hyperinflammatory responses causing disease severity, as well as a homeostatic MDSC differentiation program as a counter-regulatory consequence of a hyperinflammatory state, as supported by their increased frequency in some autoimmune conditions.57,58 The EN-RAGEhi myeloid state may underlie disease severity through additional mechanisms. For example, EN-RAGEhi cells express higher IL-1β and TGF-β1 which can increase endothelial and epithelial cell permeability, respectively, and potential mechanisms whereby EN-RAGEhi cells could affect barrier integrity and edema in patients with acute lung injury remain to be explored.10,59,60
This study connects immune cell phenotypes in the blood with the myeloid EN-RAGE state in the airways using paired quantification of mRNA and surface proteins at single-cell resolution in PBMCs and ETA single-cell transcriptomes from the COMET study and longitudinal blood mRNA expression data from the large Phase 3 COVACTA study. The inclusion of patients with acute lung injury from other causes in the COMET cohort reveals that EN-RAGEhi myeloid cells are relevant to insults in addition to SARS-CoV-2 infection, and identifies a targetable pathway relevant to ARDS. IL-6 predicts severe patient outcomes30 and has been hypothesized to be a driver of myeloid differentiation in severe infections.10,12 We show that tocilizumab treatment rapidly normalized myeloid and T cell states to healthy control levels, which was correlated with improved clinical outcomes. This study establishes the importance of IL-6 in driving differentiation of the severity-associated EN-RAGEhi myeloid state in patients. COVID-19 and cancer share risk factors, such as age and metabolic syndrome, and underlying immunobiology, such as MDSCs and T cell dysfunction, making it appealing to speculate that EN-RAGEhi myeloid cells may contribute to pathology in multiple diseases.
Limitations of the study
IL-6 alone does not significantly induce the entire EN-RAGE program, which is consistent with the partial IL-6R-dependent induction of the severity-associated MS1 program by sepsis patient serum using an in vitro system.10 The effect of IL-6R blockade in reducing the EN-RAGE signature expression in patients was observed in survivors and not non-survivors (Figure 6F), suggesting other potential factors besides IL-6 that regulate expression of the full severity-associated transcriptional program. We defined the EN-RAGE gene signature that performs well across blood, PBMC, and airway samples in order to study a pan-myeloid program that is prognostic for severe outcomes. Analyses focused on monocytes or neutrophils separately in peripheral and lung samples could reveal unique programs associated with clinical severity. As fresh cells were not collected in COMET or COVACTA to enable functional studies, future studies using additional cohorts will be required to understand the breadth of effect of EN-RAGEhi cells on inflammation in patients. There is limited ability to infer T cell functionality from bulk RNA-seq data, and the expression of T cell genes and surface proteins correlating with myeloid EN-RAGE program expression could indicate T cell suppression or hyperactivation. Future studies using multi-dimensional flow cytometry data will enable more precise definition of how IL-6 alters the T cell phenotype in COVID-19, both via myeloid activation and acting directly on T cells. While the EN-RAGE program is expressed in myeloid cells regardless of ARDS etiology, randomized placebo-controlled studies of tocilizumab are needed to assess the effect of IL-6R blockade on the EN-RAGE program in patients with ARDS caused by insults other than SARS-CoV-2.
Consortia
UCSF COMET Consortium co-authors: Yumiko Abe-Jones, Michael Adkisson, K. Mark Ansel, Saurabh Asthana, Alexander Beagle, Sharvari Bhide, Cathy Cai, Saharai Caldera, Maria Calvo, Sidney A. Carrillo, Suzanna Chak, Stephanie Christenson, Zachary Collins, Spyros Darmanis, Angela Detweiler, Catherine DeVoe, Walter Eckalbar, Jeremy Giberson, Ana Gonzalez, Gracie Gordon, Paula Hayakawa Serpa, Alejandra Jauregui, Chayse Jones, Serena Ke, Divya Kushnoor, Tasha Lea, Deanna Lee, Aleksandra Leligdowicz, Yale Liu, Salman Mahboob, Lenka Maliskova, Michael Matthay, Elizabeth McCarthy, Priscila Muñoz-Sandoval, Norma Neff, Viet Nguyen, Nishita Nigam, Randy Parada, Maira Phelps, Logan Pierce, Priya Prasad, Sadeed Rashid, Gabriella Reeder, Nicklaus Rodriguez, Bushra Samad, Andrew Schroeder, Cole Shaw, Alan Shen, Austin Sigman, Pratik Sinha, Matthew Spitzer, Sara Sunshine, Kevin Tang, Luz Torres Altamirano, Alexandra Tsitsiklis, Erden Tumurbaatar, Vaibhav Upadhyay, Alexander Whatley, Andrew Willmore, Michael Wilson, Juliane Winkler, Kristine Wong, Kimberly Yee, Michelle Yu, Mingyue Zhou, and Wandi S. Zhu.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Carrie Rosenberger (e-mail: rosenberger.carrie@gene.com).
Resource availability
This study did not generate new unique reagents or material.
Experimental model and study participant details
COMET cohort
The COVID-19 Multi-Phenotyping for Effective Therapies (COMET) cohort collected PBMCs, whole blood, ETA and plasma longitudinally from hospitalized patients presenting with COVID-19 symptoms. This study included 75 patients with samples collected in 2020, of whom 57 were positive (76%) for SARS-CoV-2, along with 11 healthy controls.2,38,39 Table 1 summarizes the patient characteristics. All-cause mortality occurred within 30 days for 9 of 10 subjects. Ventilator-free days (VFDS) were assessed at D28, with fatal cases assigned 0. NIH COVID-19 ordinal severity score and Sequential Organ Failure Assessment (SOFA) scores were assessed at study enrollment (Day 0), when baseline samples were collected, with a 9 days median time from symptom onset (4–13 IQR). Severity groups were defined as follows. PBMC: Moderate = no supplemental O2, severe = supplemental O2 and critical = mechanical ventilation. Whole blood: Mild/Moderate = 0 days on ventilator and no more than 1 day in ICU, Severe patients had ≥1 day on ventilator. ETA: Critical = VFDS = 0 (ventilation for ≥28 days or death), severe ETA = VFDS>0.
COVACTA tocilizumab clinical trial
A total of 438 hospitalized COVID-19 patients were randomized 2:1 for anti-interleukin-6 receptor antibody, tocilizumab, or placebo and included in the modified intention to treat population (tocilizumab: 294, placebo: 144). Hospitalized patients were ≥18 years of age with COVID-19 pneumonia confirmed by a positive SARS-CoV-2 PCR test and evidenced by X-ray or computed tomography (CT) scan. Eligible patients had a blood oxygen saturation of ≤93% or partial pressure of oxygen/fraction of inspired oxygen of <300 mm/Hg. Details of the COVACTA study design have been published (Clinical trials.gov NCT04320615).35 Population demographics for the 404 patients with available blood RNA-seq data are described in Table 1.
COVID-19 clinical severity
Within the COVACTA clinical trial, the NIH COVID-19 ordinal severity scale was used to assess disease severity: 3 = hospitalized not requiring supplemental O2, 4 = supplemental O2, 5 = non-invasive/high flow O2, 6 = mechanical ventilation, 7 = mechanical ventilation + additional organ support (e.g., vasopressors, renal replacement therapy, ECMO), 8 = death. In the COMET cohort of PBMC samples, moderate = no supplemental O2 (NIH ordinal scale 3–4), severe = supplemental O2 (5–6) and critical = mechanical ventilation (7). The maximal NIH ordinal severity scale recorded during hospitalization was calculated for each patient in COMET. Ventilator-free days were calculated over 28 days, with fatal patients scored as 0. ARDS was diagnosed using the American-European Consensus Conference (AECC) definition79 or Berlin definition.80 The sequential organ failure assessment (SOFA) score was calculated at study enrollment.
Study approval
The COMET study was approved by the Institutional Review Board: UCSF Human Research Protection Program (HRPP) IRB# 20–30497 and informed consent was obtained for patients. COVACTA was conducted in accordance with Good Clinical Practice guidelines of the International Council for Harmonisation E6 and the Declaration of Helsinki or local laws and regulations, whichever afforded greater protection. Informed consent was obtained from the patient or their legally authorized representative prior to participation. The studies were approved by institutional review boards or ethics committees at each site.
Method details
Reanalysis of published scRNA-seq data
Raw count data from Schulte-Schrepping et al.11 were downloaded from http://fastgenomics.org. For analysis of the PBMC data collected using 10x Genomics droplet-based capture, and whole blood collected using Rhapsody microwell-based capture, the raw count data was reprocessed using scripts written in the R programming language81 and packages from the Bioconductor project (https://www.bioconductor.org). Briefly, counts were normalized using the computeLibraryFactors method from the scater R package82 and dimensionality reduction was performed using PCA followed by UMAP projection. For calculating PCA, the top 5000 most variable genes were selected, accounting for overall expression level using the modelGeneVar method from the scran R package.83 The UMAP projection was calculated using the first 10 principal components. Graph-based clustering was performed using the Louvain algorithm on the same 10 principal components. Marker genes for each cluster were determined using pairwise t-tests, calculated using the pairwiseTTest method from the scran R package, and were used to manually assign broad cell type labels to each cluster. Clusters from the same broad cell type classification were merged. The myeloid populations (monocytes, DCs and neutrophils) were separately re-clustered using the same procedures: the 5000 most variable genes were selected, used for PCA calculation, graph-based clustering and UMAP projection. Marker genes were calculated for each cluster, and clusters were manually assigned using the marker gene lists.
Raw sequencing reads from Liao et al.7 were downloaded from SRA (SRA: PRJNA608742). Sequencing data were then processed using cellranger v4.0.0 (10x Genomics, Pleasanton, CA, USA) using GRCh38 as the reference genome, and gene models from GENCODE (v27) for assigning reads to genes. The emptyDrops method was used from the DropletUtils R package71 to identify barcodes that corresponded to cellular droplets. Any barcode with fewer than 200 UMIs or 100 genes detected was removed. This allowed us to retain neutrophil-containing droplets, as these are mostly removed by default processing using cellranger. Droplets with high abundance of mitochondrial RNA (>10%) were removed. This yielded 90592 cells for further analysis. The same procedures described above were used to normalize, cluster and annotate cells into broad populations (macrophage, neutrophil, T cell, epithelial cell, B cell). The myeloid populations (macrophages and neutrophils) were selected and reclustered into more fine-grained populations using the same strategy as above.
Data from Silvin et al.12 were retrieved from the European Genome Archive (EGA) under accession number ArrayExpress: E-MTAB-9221 as FASTQ files. Sequencing data were processed as detailed above for SRA: PRJNA608742, using the same parameters for identifying cellular droplets and filtering droplets with high mitochondrial RNA abundance. Normalization, clustering and manual annotation was performed as described above. Count data from Delorey et al.5 were downloaded from the Broad Single Cell Portal using accession number GEO: GSE162911. We used the cell annotations as defined within that dataset, selecting cells labeled as Myeloid using the Cluster field defined by the authors. Data from Grant et al.6 were retrieved from the Gene Expression Omnibus (GEO) under accession number GEO: GSE155249. Normalization and clustering were performed as above. Annotations from the original publication were used to group cells into broad lineages: epithelial cells, B cells, T cells, dendritic cells, mast cells, macrophages and mixed myeloid cells.
Derivation of the EN-RAGE gene signature
We generated pseudobulk expression profiles for myeloid sub-populations in both PBMC (monocyte, DC, and neutrophil subsets) and tissue (macrophage and neutrophil subsets), with one profile generated per cell population, per patient, using the method described below. Genes with low expression levels in at least one tissue were excluded, using the filterByExpr function from the edgeR package, removing genes with fewer than 2 counts in greater than 50% of samples within a subcluster, and less than 15 reads across all samples. Pseudobulk profiles were then normalized and log transformed, using the “cpm” function from the edgeR R package, yielding log2-normalized count per million (logCPM) values. On a per-gene basis, logCPM values are close to normally distributed across patients for genes with sufficient read depth, so we used Pearson’s correlation coefficient to calculate correlations with the S100A12 gene expression values. We selected an arbitrary cutoff of r > 0.5 in both datasets to identify our gene set of interest. Selecting a higher threshold yielded far fewer genes. Using Spearman’s correlation coefficient, the majority of these genes have correlation coefficients >0.5 (77 genes in the PBMC samples, 81 in the BAL samples), suggesting our selection strategy is robust to parametric or non-parametric methods for calculating correlations. Two-sided unpaired t-tests were used to compare gene signature scores between cell types and across severity groups in pseudobulk scRNA-seq datasets.
Pseudobulk expression profile calculation
The aggregateAcrossCells method from the scater R package82 was used to calculate pseudobulk expression profiles for each cell population in a sample. This method uses the sum of raw counts to determine an estimate of the aggregate expression of that cell type in a sample. The TMM method from the edgeR R package84 was used to normalize the pseudobulk count data within each dataset. Signature score calculation in pseudo-bulk data was performed using the GSDecon package (https://github.com/JasonHackney/GSDecon).
Identification of EN-RAGE gene expression induced by IL-6 in vitro
PBMCs were isolated from 50 mL of heparinized blood of four healthy donors by Ficoll-Paque. Primary human monocytes were purified from PBMC by Miltenyi Pan Monocyte Isolation Kit and cultured in RPMI with 10% heat-inactivated FBS, 10 mM HEPES and L-glutamine. The primary human monocytes were stimulated with 10 ng/mL IL-6 + 18.7 ng/mL IL6R for 24 h, and RNA-seq performed on bulk cells. Unsupervised clustering using average linkage cluster difference and Euclidean point distance metrics were used to generate heat maps. RNA was isolated using the Qiagen RNeasy 96 kit (Qiagen catalog: 74182). Total RNA was quantified with Qubit RNA HS Assay Kit (Thermo Fisher Scientific catalog: Q32852) and quality was assessed using RNA ScreenTape on 4200 TapeStation (Agilent Technologies catalog: 5067–5576). cDNA library was generated from 2 ng of total RNA using Smart-Seq V4 Ultra Low Input RNA Kit (Takara catalog: 634894). 150 picograms of cDNA was used to make sequencing libraries by Nextera XT DNA Sample Preparation Kit (Illumina catalog: FC-131-1024). Libraries were quantified with Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific catalog: Q32851) and the average library size was determined using D1000 ScreenTape on 4200 TapeStation (Agilent Technologies catalog: 5067–5582). Libraries were pooled and sequenced on the Illumina NovaSeq 6000 to generate 30 million single-end 50 base pair reads for each sample. Sequencing reads were filtered and aligned using HTSeqGenie v4.4.2.85 GSNAP v2013-11-01 was used for alignment, through the HTSeqGenie wrapper, against the GENCODE 27 Basic gene model on the human genome assembly GRCh38. Only reads with unique genomic alignments were analyzed. Normalized CPM (Counts Per Million) were used as a normalized measure of gene expression, calculated using method provided in edgeR.84
Comet data analysis
COMET data is available on GEO (GEO: GSE163668 (whole blood), GEO: GSE163426 (tracheal aspirate), GEO: GSE168453 (PBMC)). Generation of PBMC scRNA-seq and Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) libraries was previously published,39 and the complete protocol is available on protocols.io (https://www.protocols.io/view/10x-citeseq-protocol-covid-19-patient-samples-tetr-bqnqmvdw). CITE-seq data was generated for 188 unique cell surface antigens, and collected from 128 samples collected at day 0, day 7 and/or day 14 from 60 patients. Single cell RNA-seq data generated from PBMC collected at day 0 from 49 patients, whole blood collected at day 0 for 18 patients, and 41 ETA samples collected longitudinally from 16 patients were included in this study. Bulk gene expression from endotracheal aspirates was generated from 182 samples collected longitudinally from 19 patients. Plasma cytokine and paired ETA scRNA-seq data was available for only 7 samples, precluding correlation analyses. Student’s t test was used to calculate p values for comparisons of EN-RAGE gene set scores and clinical severity. Spearman correlations and two-tailed p values were calculated to examine relationships between biomarkers and clinical severity and EN-RAGE gene set vs. myeloid gene expression.
Raw RNA-seq reads (GEO: GSE163426 38) were processed to count matrices using the kallisto software (v0.46.1, including bias correction). Gene models used were from ENSEMBL (v99), and included all protein coding genes, ribosomal RNA genes, and sequences for the ERCC spike-in standards. Samples that failed to generate at least 1 million reads were excluded. Eigengene scores were calculated for the EN-RAGE gene set using the GSDecon package (https://github.com/JasonHackney/GSDecon). To evaluate changes in EN-RAGE signature expression and outcomes, we performed mixed effects linear modeling using the lmer function from the lme4 R package.86 Patients were split into groups based on 28-day mortality, or spending longer than 28 days on ventilation compared to those who survived or spent fewer than 28 days on ventilation. The longitudinal change in EN-RAGE signature was compared across these groups, with patient being treated as a random effect in the model. The difference in slope of change in signature over time was modeled as the interaction between sampling day and severity groups defined above. For significance testing, we examined the 95% confidence intervals for the interaction term in our model.
COMET PBMC CITE-seq analysis
For PBMC CITE-seq analysis, the raw scRNA-seq counts were normalized using the ‘LogNormalized’ method implemented in the ‘NormalizeData’ function from the Seurat R package69; with this method, the feature counts are divided by the total counts for each cell, multiplied by a scale factor of 10,000, and then natural-log transformed. The raw ADT counts were normalized using the ‘CLR’ method implemented in Seurat’s ‘NormalizeData’ function; with this method, a centered log ratio transformation is applied. The EN-RAGE and MS1 gene set scores were calculated for each cell using the ‘score_genes’ function implemented in the scanpy Python package.77 All of the pseudobulk values for each cell population in a sample (i.e., sample + cell type)—that is, the mRNA expression, ADT expression, and gene set scores—were calculated as the average across all cells in that population. Correlations between gene set scores and ADT expression were on pseudobulk data and refer to a population of cells across patients.
COVACTA data analysis
Serum IL-6 was quantified using a validated in vitro diagnostic method (Roche Cobas; Roche Diagnostics, Indianapolis, IN) at central laboratories (PPD). Complete blood counts were measured using standard clinical chemistry and haematology methods available at local hospital laboratories. IFNγ and IL10 were measured by qualified immunoassays (Simpleplex, ProteinSimple, San Jose, CA, USA) at central laboratories (Covance). The Olink Explore platform was used to measure 1472 serum proteins (Olink, Uppsala, Sweden). RNA was isolated from blood PaxGene (Qiagen, Hilden Germany) samples by Q2 Solutions. 1.25 μg of RNA was used for generating sequencing libraries with the TruSeq Stranded mRNA Library Prep kit (Illumina, San Diego, CA, USA). The libraries were sequenced by Illumina NovaSeq by 50 bp single-end reads at a read depth of 50 million reads per sample.
RNA-seq reads were mapped to the human genome (v38) using gsnap followed by filtering of reads mapping to ribosomal genes.87 Read counts mapping to annotated genes (Gencode v27) were then generated using summarizeOverlaps.78 For the time point comparisons (baseline to D7) patients were subset to those subjects with measurements at both time points before differential expression analysis with DESeq2.62 To determine associations to baseline severity subjects were grouped into those with severity score <4 (Moderate) and ≥ 4 (Severe). For association to mechanical ventilation, gene expression differences were calculated by DESeq2 for subjects not mechanically ventilated at baseline who progress to needing ventilation by day 28 vs. who don’t. Association to mortality was assessed by comparing subjects who die by day 28 vs. who don’t. For analysis of both MV and death, DESeq2 models were adjusted for age, baseline severity scores and treatment arm. Unfiltered DESeq2 outputs were ranked by log2 fold change and then the FGSEA Bioconductor package was used to calculate enrichment scores.70 EN-RAGE gene signature scores were calculated using the GSDecon package (https://github.com/JasonHackney/GSDecon). We used the ggplot2 package to generate visualizations. T cell genes with a relationship to EN-RAGE state in COMET scRNA-seq data were examined in COVACTA, and limited to the subset of T cell suppressive genes that were predominantly expressed by T cells in COMET whole blood transcriptome data to permit analysis of bulk gene expression data.
Quantification and statistical analysis
Cox Proportional Hazard models were used to assess the prognostic value of EN-RAGE eigengene scores for mortality censored at day 28. Continuous eigengene scores were used as main predictor of interest, and age, baseline ordinal scale, and categorical treatment arm were included as covariates, along with 28-day censored mortality from baseline events were used for both the prognostic model assessing the effect of EN-RAGE. Additionally, a linear predictor to establish an optimal Youden’s J cutoff for EN-RAGE score was found via a ROC on 28-day mortality. Kaplan-Meier curves and log rank tests were calculated using the optimized cut-off to split patients into high and low risk groups.
Acknowledgments
This study was supported with funding from Roche, Inc. and federal funds from the Department of Health and Human Services; Administration for Preparedness and Response; Biomedical Advanced Research and Development Authority, under OT number: HHSO100201800036C. The findings and conclusions herein are those of the authors and do not necessarily represent the views of the Department of Health and Human Services or its components. C.J.Y. is further supported by the NIH grants R01AR071522, R01AI136972, U01HG012192, and the Chan Zuckerberg Initiative, and is an investigator at the Chan Zuckerberg Biohub and is a member of the Parker Institute for Cancer Immunotherapy (PICI). G.C.H. was supported by the National Science Foundation Undergraduate Research Fellowship Program 1650113. C.S.C. is further supported by NIH R35HL140026. C.H. is further supported by a K23 from NHLBI K23 HL133495.
Author contributions
C.M.R., J.A.H., and H.S. conceived of and designed the overall study. J.A.H., H.S., H.V.H., C.O., L.O., X.G., A.C., and C.M.R. performed and interpreted computational analyses. X.G. performed in vitro experiments. X.G., N.W., A.Q., D.C., A.C., D.F.C., A.J.C., T.C., G.K.F., A.A.R., A.R., J.T., K.H., N.F.K., M.F.K., D.J.E., K.K., A.S., Z.L., C.S.C., P.G.W., R.G., E.M., A.B., B.S.Z., C.L., C.M.H., M.G.P.v.d.W., G.C.H., T.G., R.B., D.S.L., J.R.G., Y.S., R.P., A.O., A.W., C.J.Y., UCSF COMET Consortium, T.R., J.M.M., F.C., A.T., M.B., L.T., I.O.R., A.R., S.B.K., R.N.B., and C.M.R. facilitated the COMET or COVACTA studies and provided data and/or critical feedback on methods and results. C.M.R., J.A.H., and H.S. wrote the first draft of the manuscript. All authors reviewed and approved the final manuscript.
Declaration of interests
J.A.H., H.S., J.V.H., C.O., L.O., X.G., N.W., A.Q., D.C., A.C, D.F.C., T.R., J.M.M., F.C., A.T., M.B., L.T., A.R., S.B.K., R.N.B., and C.M.R. were employees of Genentech, Inc. at the time of this study and own equity in Roche. The COMET study was supported in part by Genentech funding. C.J.Y. is a scientific advisory board member for and holds equity in Related Sciences and ImmunAI, a consultant for and holds equity in Maze Therapeutics, and a consultant for TRex Bio. C.J.Y. has received research support from Chan Zuckerberg Initiative, Chan Zuckerberg Biohub, and Genentech. C.S.C. has received research funding from Roche-Genentech for an unrelated project as well as from NIH, DOD, and Quantum Leap Healthcare Collaborative. C.S.C. is a consultant for Vasomune, Quark, and GEn1E Lifesciences. C.H. is a consultant for Spring Discovery but does not have any financial interest in the company nor is the work related to what is covered in this manuscript. A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and, until 31 July 2020, was a scientific advisory board member of Thermo Fisher Scientific, Syros Pharmaceuticals, Asimov, and Neogene Therapeutics. A.R. is a named inventor on multiple patents related to single-cell and spatial genomics filed by or issued to the Broad Institute.
Published: September 1, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107813.
Contributor Information
Carrie M. Rosenberger, Email: rosenberger.carrie@gene.com.
UCSF COMET Consortium:
Yumiko Abe-Jones, Michael Adkisson, K. Mark Ansel, Saurabh Asthana, Alexander Beagle, Sharvari Bhide, Cathy Cai, Saharai Caldera, Maria Calvo, Sidney A. Carrillo, Suzanna Chak, Stephanie Christenson, Zachary Collins, Spyros Darmanis, Angela Detweiler, Catherine DeVoe, Walter Eckalbar, Jeremy Giberson, Ana Gonzalez, Gracie Gordon, Paula Hayakawa Serpa, Alejandra Jauregui, Chayse Jones, Serena Ke, Divya Kushnoor, Tasha Lea, Deanna Lee, Aleksandra Leligdowicz, Yale Liu, Salman Mahboob, Lenka Maliskova, Michael Matthay, Elizabeth McCarthy, Priscila Muñoz-Sandoval, Norma Neff, Viet Nguyen, Nishita Nigam, Randy Parada, Maira Phelps, Logan Pierce, Priya Prasad, Sadeed Rashid, Gabriella Reeder, Nicklaus Rodriguez, Bushra Samad, Andrew Schroeder, Cole Shaw, Alan Shen, Austin Sigman, Pratik Sinha, Matthew Spitzer, Sara Sunshine, Kevin Tang, Luz Torres Altamirano, Alexandra Tsitsiklis, Erden Tumurbaatar, Vaibhav Upadhyay, Alexander Whatley, Andrew Willmore, Michael Wilson, Juliane Winkler, Kristine Wong, Kimberly Yee, Michelle Yu, Mingyue Zhou, and Wandi S. Zhu
Supplemental information
Summary statistics (log2 fold change, p-value and FDR) are shown for genes identified in the EN-RAGE signature, myeloid function genes, and T cell modulatory genes.
Data and code availability
-
•
RNA-seq and Olink data have been deposited at The European Genome-Phenome Archive (EGA). They are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Details of the COVACTA study design are available as NCT04320615 on Clinical trials.gov.
References
- 1.Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Combes A.J., Courau T., Kuhn N.F., Hu K.H., Ray A., Chen W.S., Chew N.W., Cleary S.J., Kushnoor D., Reeder G.C., et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591:124–130. doi: 10.1038/S41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng H., Rao A.M., Dermadi D., Toh J., Murphy Jones L., Donato M., Liu Y., Su Y., Dai C.L., Kornilov S.A., et al. Multi-cohort analysis of host immune response identifies conserved protective and detrimental modules associated with severity across viruses. Immunity. 2021;54:753–768.e5. doi: 10.1016/j.immuni.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDonald L., Alivernini S., Tolusso B., Elmesmari A., Somma D., Perniola S., Paglionico A., Petricca L., Bosello S.L., Carfì A., et al. COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight. 2021;6 doi: 10.1172/jci.insight.147413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delorey T.M., Ziegler C.G.K., Heimberg G., Normand R., Yang Y., Segerstolpe Å., Abbondanza D., Fleming S.J., Subramanian A., Montoro D.T., et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grant R.A., Morales-Nebreda L., Markov N.S., Swaminathan S., Querrey M., Guzman E.R., Abbott D.A., Donnelly H.K., Donayre A., Goldberg I.A., et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 8.Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184:1895–1913.e19. doi: 10.1016/j.cell.2021.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyes M., Filbin M.R., Bhattacharyya R.P., Sonny A., Mehta A., Billman K., Kays K.R., Pinilla-Vera M., Benson M.E., Cosimi L.A., et al. Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro. Sci. Transl. Med. 2021;13:9599. doi: 10.1126/scitranslmed.abe9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silvin A., Chapuis N., Dunsmore G., Goubet A.-G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/J.CELL.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rowlands M., Segal F., Hartl D. Myeloid-Derived Suppressor Cells as a Potential Biomarker and Therapeutic Target in COVID-19. Front. Immunol. 2021;12:2435. doi: 10.3389/FIMMU.2021.697405/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shivram H., Hackney J.A., Rosenberger C.M., Teterina A., Qamra A., Onabajo O., Mcbride J., Cai F., Bao M., Tsai L., et al. Tocilizumab treatment leads to early resolution of myeloid dysfunction and lymphopenia in patients hospitalized with COVID-19. iScience. 2023;26 doi: 10.1016/j.isci.2023.107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaSalle T.J., Gonye A.L.K., Freeman S.S., Kaplonek P., Gushterova I., Kays K.R., Manakongtreecheep K., Tantivit J., Rojas-Lopez M., Russo B.C., et al. Longitudinal characterization of circulating neutrophils uncovers distinct phenotypes associated with severity in hospitalized COVID-19 patients. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/S41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bost P., de Sanctis F., Canè S., Ugel S., Donadello K., Castellucci M., Eyal D., Fiore A., Anselmi C., Barouni R.M., et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat. Commun. 2021;12:1428. doi: 10.1038/s41467-021-21702-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kvedaraite E., Hertwig L., Sinha I., Ponzetta A., Hed Myrberg I., Lourda M., Dzidic M., Akber M., Klingström J., Folkesson E., et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2018587118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei H. A single transcript for the prognosis of disease severity in COVID-19 patients. Sci. Rep. 2021;11:12174. doi: 10.1038/S41598-021-91754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrijver I.T., Théroude C., Roger T. Myeloid-Derived Suppressor Cells in Sepsis. Front. Immunol. 2019;10:327. doi: 10.3389/fimmu.2019.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veglia F., Sanseviero E., Gabrilovich D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021;21:485–498. doi: 10.1038/S41577-020-00490-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegde S., Leader A.M., Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. 2021;54:875–884. doi: 10.1016/j.immuni.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agrati C., Sacchi A., Bordoni V., Cimini E., Notari S., Grassi G., Casetti R., Tartaglia E., Lalle E., D’abramo A., et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19) Cell Death Differ. 2020;27:3196–3207. doi: 10.1038/s41418-020-0572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darcy C.J., Minigo G., Piera K.A., Davis J.S., McNeil Y.R., Chen Y., Volkheimer A.D., Weinberg J.B., Anstey N.M., Woodberry T. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit. Care. 2014;18:R163. doi: 10.1186/cc14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dean M.J., Ochoa J.B., Sanchez-Pino M.D., Zabaleta J., Garai J., del Valle L., Wyczechowska D., Baiamonte L.B., Philbrook P., Majumder R., et al. Severe COVID-19 Is Characterized by an Impaired Type I Interferon Response and Elevated Levels of Arginase Producing Granulocytic Myeloid Derived Suppressor Cells. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.695972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falck-Jones S., Vangeti S., Yu M., Falck-Jones R., Cagigi A., Badolati I., Österberg B., Lautenbach M.J., Åhlberg E., Lin A., et al. Functional monocytic myeloid-derived suppressor cells increase in blood but not airways and predict COVID-19 severity. J. Clin. Invest. 2021;131 doi: 10.1172/JCI144734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reizine F., Lesouhaitier M., Gregoire M., Pinceaux K., Gacouin A., Maamar A., Painvin B., Camus C., le Tulzo Y., Tattevin P., et al. SARS-CoV-2-Induced ARDS Associates with MDSC Expansion, Lymphocyte Dysfunction, and Arginine Shortage. J. Clin. Immunol. 2021;41:515–525. doi: 10.1007/S10875-020-00920-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sacchi A., Grassi G., Bordoni V., Lorenzini P., Cimini E., Casetti R., Tartaglia E., Marchioni L., Petrosillo N., Palmieri F., et al. Early expansion of myeloid-derived suppressor cells inhibits SARS-CoV-2 specific T-cell response and may predict fatal COVID-19 outcome. Cell Death Dis. 2020;11:921. doi: 10.1038/s41419-020-03125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tom J., Bao M., Tsai L., Qamra A., Summers D., Carrasco-Triguero M., McBride J., Rosenberger C.M., Lin C.J.F., Stubbings W., et al. Prognostic and Predictive Biomarkers in Patients with Coronavirus Disease 2019 Treated with Tocilizumab in a Randomized Controlled Trial. Crit. Care Med. 2022;50:398–409. doi: 10.1097/CCM.0000000000005229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bunt S.K., Yang L., Sinha P., Clements V.K., Leips J., Ostrand-Rosenberg S. Reduced Inflammation in the Tumor Microenvironment Delays the Accumulation of Myeloid-Derived Suppressor Cells and Limits Tumor Progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh K., Lee O.-Y., Shon S.Y., Nam O., Ryu P.M., Seo M.W., Lee D.-S. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013;15:R79. doi: 10.1186/bcr3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber R., Groth C., Lasser S., Arkhypov I., Petrova V., Altevogt P., Utikal J., Umansky V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell. Immunol. 2021;359 doi: 10.1016/J.CELLIMM.2020.104254. [DOI] [PubMed] [Google Scholar]
- 35.Rosas I.O., Bräu N., Waters M., Go R.C., Hunter B.D., Bhagani S., Skiest D., Aziz M.S., Cooper N., Douglas I.S., et al. Tocilizumab in Hospitalized Patients with Severe Covid-19 Pneumonia. N. Engl. J. Med. 2021;384:1503–1516. doi: 10.1056/NEJMOA2028700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.WHO Rapid Evidence Appraisal for COVID-19 Therapies REACT Working Group. Shankar-Hari M., Vale C.L., Godolphin P.J., Fisher D., Higgins J.P.T., Savović J., Savovic J., Tierney J., Baron G., et al. Association Between Administration of IL-6 Antagonists and Mortality Among Patients Hospitalized for COVID-19: A Meta-analysis. JAMA. 2021;326:499–518. doi: 10.1001/JAMA.2021.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foell D., Wittkowski H., Vogl T., Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 2007;81:28–37. doi: 10.1189/JLB.0306170. [DOI] [PubMed] [Google Scholar]
- 38.Sarma A., Christenson S.A., Byrne A., Mick E., Pisco A.O., DeVoe C., Deiss T., Ghale R., Zha B.S., Tsitsiklis A., et al. Tracheal aspirate RNA sequencing identifies distinct immunological features of COVID-19 ARDS. Nat. Commun. 2021;12:5152. doi: 10.1038/s41467-021-25040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Wijst M.G.P., Vazquez S.E., Hartoularos G.C., Bastard P., Grant T., Bueno R., Lee D.S., Greenland J.R., Sun Y., Perez R., et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 2021;13:eabh2624. doi: 10.1126/scitranslmed.abh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., et al. Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/J.CELL.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aschenbrenner A.C., Mouktaroudi M., Krämer B., Oestreich M., Antonakos N., Nuesch-Germano M., Gkizeli K., Bonaguro L., Reusch N., Baßler K., et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. 2021;13:1–25. doi: 10.1186/S13073-020-00823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasegawa T., Kosaki A., Kimura T., Matsubara H., Mori Y., Okigaki M., Masaki H., Toyoda N., Inoue-Shibata M., Kimura Y., et al. The regulation of EN-RAGE (S100A12) gene expression in human THP-1 macrophages. Atherosclerosis. 2003;171:211–218. doi: 10.1016/J.ATHEROSCLEROSIS.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 43.Lorenz E., Muhlebach M.S., Tessier P.A., Alexis N.E., Duncan Hite R., Seeds M.C., Peden D.B., Meredith W. Different expression ratio of S100A8/A9 and S100A12 in acute and chronic lung diseases. Respir. Med. 2008;102:567–573. doi: 10.1016/J.RMED.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medina E., Hartl D. Myeloid-Derived Suppressor Cells in Infection: A General Overview. J. Innate Immun. 2018;10:407–413. doi: 10.1159/000489830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisemann T., Costa B., Peterziel H., Angel P. Podoplanin Positive Myeloid Cells Promote Glioma Development by Immune Suppression. Front. Oncol. 2019;9:187. doi: 10.3389/fonc.2019.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karakasheva T.A., Waldron T.J., Eruslanov E., Kim S.B., Lee J.S., O’Brien S., Hicks P.D., Basu D., Singhal S., Malavasi F., Rustgi A.K. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. 2015;75:4074–4085. doi: 10.1158/0008-5472.CAN-14-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bell L.C.K., Meydan C., Kim J., Foox J., Butler D., Mason C.E., Shapira S.D., Noursadeghi M., Pollara G. Transcriptional response modules characterize IL-1β and IL-6 activity in COVID-19. iScience. 2021;24:101896. doi: 10.1016/j.isci.2020.101896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou R., To K.K.-W., Wong Y.-C., Liu L., Zhou B., Li X., Huang H., Mo Y., Luk T.-Y., Lau T.T.-K., et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity. 2020;53:864–877.e5. doi: 10.1016/J.IMMUNI.2020.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meckiff B.J., Ramírez-Suástegui C., Fajardo V., Chee S.J., Kusnadi A., Simon H., Eschweiler S., Grifoni A., Pelosi E., Weiskopf D., et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4+ T Cells in COVID-19. Cell. 2020;183:1340–1353.e16. doi: 10.1016/J.CELL.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Files J.K., Boppana S., Perez M.D., Sarkar S., Lowman K.E., Qin K., Sterrett S., Carlin E., Bansal A., Sabbaj S., et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J. Clin. Invest. 2021;131 doi: 10.1172/JCI140491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/S41591-020-0944-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19) Front. Immunol. 2020;11:827. doi: 10.3389/FIMMU.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science (1979. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng M., Gao Y., Wang G., Song G., Liu S., Sun D., Xu Y., Tian Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020;17:533–535. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galván-Peña S., Leon J., Chowdhary K., Michelson D.A., Vijaykumar B., Yang L., Magnuson A.M., Chen F., Manickas-Hill Z., Piechocka-Trocha A., et al. Profound Treg perturbations correlate with COVID-19 severity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/PNAS.2111315118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo C., Hu F., Yi H., Feng Z., Li C., Shi L., Li Y., Liu H., Yu X., Wang H., et al. Myeloid-derived Suppressor Cells Have a Proinflammatory Role in the Pathogenesis of Autoimmune Arthritis. Ann. Rheum. Dis. 2016;75:278–285. doi: 10.1136/ANNRHEUMDIS-2014-205508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z., Zhu F., Wang J., Tao Q., Xu X., Wang H., Xiong S., Wang Y., Zhai Z. Increased CD14+HLA-DR−/low Myeloid-Derived Suppressor Cells Correlate With Disease Severity in Systemic Lupus Erythematosus Patients in an iNOS-Dependent Manner. Front. Immunol. 2019;10:1202. doi: 10.3389/FIMMU.2019.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fahey E., Doyle S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019;10:1426. doi: 10.3389/fimmu.2019.01426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pittet J.-F., Griffiths M.J., Geiser T., Kaminski N., Dalton S.L., Huang X., Brown L.A., Gotwals P.J., Koteliansky V.E., Matthay M.A., Sheppard D. TGF-β is a critical mediator of acute lung injury. J. Clin. Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angelova M., Charoentong P., Hackl H., Fischer M.L., Snajder R., Krogsdam A.M., Waldner M.J., Bindea G., Mlecnik B., Galon J., Trajanoski Z. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64. doi: 10.1186/S13059-015-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/S13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/NAR/GKV007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blighe K., Rana S., Lewis M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.18.0. 2023. https://github.com/kevinblighe/EnhancedVolcano
- 65.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/BIOINFORMATICS/BTW313. [DOI] [PubMed] [Google Scholar]
- 66.Wickham H. Springer International Publishing; 2016. ggplot2: Elegant Graphics for Data Analysis. [DOI] [Google Scholar]
- 67.Tang Y., Horikoshi M., Li W. Ggfortify: Unified interface to visualize statistical results of popular r packages. R J. 2016;8:474–489. doi: 10.32614/RJ-2016-060. [DOI] [Google Scholar]
- 68.Therneau T.M., Grambsch P.M. 2000. Modeling Survival Data: Extending the Cox Model. [DOI] [Google Scholar]
- 69.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/J.CELL.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast Gene Set Enrichment Analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 71.Lun A.T.L., Riesenfeld S., Andrews T., Dao T.P., Gomes T., Marioni J.C. EmptyDrops: Distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019;20:63–69. doi: 10.1186/S13059-019-1662-Y/FIGURES/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gordon M., Lumley T. 2022. Forestplot: Advanced Forest Plot Using “Grid” Graphics.https://gforge.se/packages/ [Google Scholar]
- 73.Kassambara A., Kosinski M., Biecek P., Fabian S. 2021. Survminer: Drawing Survival Curves Using “Ggplot2.https://rpkgs.datanovia.com/survminer/index.html [Google Scholar]
- 74.Sing T., Sander O., Beerenwinkel N., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 75.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/NCOMMS14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 77.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013;9 doi: 10.1371/JOURNAL.PCBI.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bernard G.R., Artigas A., Brigham K.L., Carlet J., Falke K., Hudson L., Lamy M., Legall J.R., Morris A., Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 80.ARDS Definition Task Force. Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., Caldwell E., Fan E., Camporota L., Slutsky A.S. Acute respiratory distress syndrome: The Berlin definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 81.R Core Team . R Foundation for Statistical Computing; 2020. R: A Language and Environment for Statistical Computing.https://www.r-project.org/ [Google Scholar]
- 82.McCarthy D., Campbell K., Lun A., Wills Q. scater: pre-processing, quality control, normalisation and visualisation of single-cell RNA-seq data in R. Bioinformatics. 2016;33:1179–1186. doi: 10.1101/069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lun A.T.L., McCarthy D.J., Marioni J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122. doi: 10.12688/f1000research.9501.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pau G R.J. 2013. HTSeqGenie: A NGS Analysis Pipeline. R package version 4.22.0.https://bioconductor.org/packages/HTSeqGenie/ [Google Scholar]
- 86.Bates D.W., Mächler M., Bolker B.M., Walker S.C. Fitting Linear Mixed-Effects Models Using lme4. BMJ Qual. Saf. 2015;24:1–3. doi: 10.18637/JSS.V067.I01. [DOI] [Google Scholar]
- 87.Wu T.D., Reeder J., Lawrence M., Becker G., Brauer M.J. GMAP and GSNAP for genomic sequence alignment: Enhancements to speed, accuracy, and functionality. Methods Mol. Biol. 2016;1418:283–334. doi: 10.1007/978-1-4939-3578-9_15/FIGURES/8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary statistics (log2 fold change, p-value and FDR) are shown for genes identified in the EN-RAGE signature, myeloid function genes, and T cell modulatory genes.
Data Availability Statement
-
•
RNA-seq and Olink data have been deposited at The European Genome-Phenome Archive (EGA). They are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Details of the COVACTA study design are available as NCT04320615 on Clinical trials.gov.