

Abstract
Tumors harboring homologous recombination deficiency (HRD) are considered optimal candidates for poly(ADP-ribose) polymerase 1 (PARP) inhibitor treatment. Such deficiency can be detected by analyzing breast cancer type (BRCA)1/2 gene mutations, as well as mutations in other genes of the homologous recombination pathway. The algorithmic measurement of the HRD effect by identifying genomic instability (GI) has been used as biomarker. As compared with the direct measurement of somatic gene alterations, this approach increases the number of patients who could benefit from PARP inhibitor treatment. In the present study, the performance of the Oncoscan CNV assay, accompanied by appropriate bioinformatic algorithms, was evaluated for its performance in GI calculation and was compared with that of a validated next-generation sequencing (NGS) test (myChoice HRD test). In addition, the clinical utility of the GI score (GIS) and BRCA1/2 tumor analysis were investigated in a cohort of 444 patients with ovarian cancer. For that reason, single nucleotide polymorphism (SNP) arrays and appropriate bioinformatics algorithms were used to calculate GIS in 29 patients with ovarian cancer with known GIS status using a validated NGS test. Furthermore, BRCA1/2 analysis results were compared between the aforementioned assay and the amplicon-based Oncomine™ BRCA Research Assay. BRCA1/2 analysis was performed in 444 patients with ovarian cancer, while GIS was calculated in 175 BRCA1/2-negative cases. The bioinformatics algorithm developed for GIS calculation in combination with NGS BRCA1/2 analysis (RediScore), and the OncoscanR pipeline exhibited a high overall agreement with the validated test (93.1%). In addition, the Oncomine NGS assay had a 100% agreement with the validated test. The BRCA1/2 mutation frequency was 26.5% in the examined patients with ovarian cancer. GIS was positive in 40% of the BRCA1/2-negative cases. The RediScore bioinformatics algorithm developed for GIS calculation in combination with NGS BRCA1/2 analysis is a viable and effective approach for HRD calculation in patients with ovarian cancer, offering a positive prediction for PARP inhibitor responsiveness in 55% of the patients.
Keywords: homologous recombination deficiency analysis, poly(ADP-ribose) polymerase 1 inhibitors, single nucleotide polymor-phism arrays, next generation sequencing, BRCA1, BRCA2
Introduction
Tumors with a deficiency in DNA repair by homologous recombination (HR) are considered optimal candidates for poly(ADP-ribose) polymerase 1 inhibitor (PARPi) treatment. Key components of the HR repair pathway are the breast cancer type (BRCA)1/2 proteins; their disruption inevitably leads to the disruption of the pathway, and therefore sensitivity to PARPi treatment (Fig. 1). Hence, PARPis have been approved for use in various tumor types based on somatic and/or germline BRCA1/2 mutation status as a biomarker of response prediction (1). This biomarker has revealed great robustness in terms of response prediction; however, it misses a high percentage of responsive cases. A variety of other proteins play significant roles in DNA repair by HR. Alterations in other HR repair genes, including partner and localizer of BRCA2, RAD51 paralog C (RAD51C) and RAD51D, have been associated with improved responses to PARPi (2,3). However, the modest mutation frequency of non-BRCA HR repair genes makes their contribution to PARPi sensitivity unclear, while for other genes there is evidence of absence of association (3).
Figure 1.

Mechanism of action of PARP inhibitors and comparison between HR proficient and HR deficient cells. HRD can be the result/cause of HRR pathway defects and/or genomic instability. PARP, poly (ADP) ribose polymerase; HR, homologous recombination; HRD, HR deficiency; HRR, HR repair.
In addition, in an exploratory subgroup analysis of the phase 3 PAOLA-1 trial, non-BRCA1 or BRCA2 HR repair genes failed to predict the benefit of olaparib plus bevacizumab in newly diagnosed advanced high-grade ovarian cancer (4).
Hence, the identification of genomic scars suggestive of HR deficiency (HRD) has provided an alternative method for detecting tumors with HRD pathway deficiencies, focusing on the identification of the effect of HRD on the genome rather than its cause.
Currently, the most common genomic scar analysis methodologies are based on the analysis of the percentage of genomic regions with loss of heterozygosity (LOH, %gLOH) or the combined use of LOH, telomeric allelic imbalance (TAI) and large-scale transitions (LSTs), giving rise to a genomic instability score (GIS). The validated next-generation sequencing (NGS)-based assay myChoice HRD test measures GIS derived from the unweighted sum of TAI, LST and LOH. Test positivity is consistent with a GIS score of ≥42 and/or the presence of a BRCA1/2 mutation. The clinical benefit of genomic scars analysis has been proven in several clinical trials using LOH alone or in combination with LST and TAI, indicating their utility as predictive biomarkers of response to platinum-based and PARPi treatment, as well as in the context of breast and ovarian cancer (5,6). The best predictive value is obtained by combining the evaluation of GI with tumor BRCA mutation analysis (7).
Several assays that measure the HRD status for treatment decision-making have also been developed. These include NGS assays measuring %gLOH and/or GIS, as well as genotyping arrays that evaluate single nucleotide polymorphisms (SNPs) and provide a viable cost-effective alternative for HRD computation. However, the lack of standardization among HRD tests emphasizes the significance of comparing existing testing methodologies (8–10).
In the present study, an SNP microarray assay (Oncoscan CNV assay), accompanied by appropriate bioinformatics algorithms, was evaluated for its performance in GIS calculation compared with the validated NGS test. In addition, the BRCA1/2 status in tumor samples from patients with ovarian cancer was assessed, and the clinical potential of the combined use of BRCA1/2 and GIS status was evaluated.
Materials and methods
Patients
A total of 29 formalin-fixed paraffin-embedded (FFPE) tumor tissue blocks obtained from patients undergoing tumor tissue analysis with the Myriad myChoice® CDx assay (Myriad Genetics) were used. HRD status, GIS and BRCA1/2 mutations were recorded in the Second Department of Surgery, Aretaieion Hospital, Medical School, National and Kapodistrian University of Athens (Athens, Greece) by an independent investigator and the results were revealed upon analysis completion. A total of 444 consecutive patients with ovarian cancer were tested for the presence of BRCA1/2 mutations between December 2021 and January 2023. The median age of disease diagnosis was 61 years (range, 29–89). Of those, GIS analysis was performed in 188 patients with negative BRCA1/2 results. The present study was approved (approval no, 502/07-01-2023) by the Ethics Committee of ‘Aretaieion’ University Hospital (Athens, Greece). Written informed consent was provided by all patients participating in the present study.
Tumor tissue processing and DNA isolation
Genomic DNA was extracted from the FFPE tumor biopsies using the MagMAX™ Total Nucleic Acid Isolation kit (cat no. AM1840; Thermo Fisher Scientific Inc.), according to the manufacturer's instructions. The nucleic acid isolation was conducted in FFPE tissues with >25% tumor cell content in a tumor area of >4 mm2, as indicated by an experienced pathologist. DNA concentration was measured with the Qubit™ 4 Fluorometer (cat no. Q33238; Thermo Fisher Scientific, Inc.).
BRCA1/2 NGS analysis
Analysis of the entire BRCA1 and BRCA2 gene coding region was performed using the Oncomine™ BRCA Research Assay (cat no. A32841; Thermo Fisher Scientific, Inc.). This is an amplicon-based targeted NGS assay that allows for the identification of all mutation types, namely single nucleotide variants, insertion-deletion mutation and exon or whole gene deletion or duplication events. Library preparation was performed using standard protocols, according to the manufacturer's instructions (Thermo Fisher Scientific, Inc.). Sequencing was carried out using the Next Generation Sequencing Platform Ion GeneStudio S5 Prime System (cat no. A38196; Thermo Fisher Scientific, Inc.). Run metrics were accessed in the Torrent Suite™ software, using the coverage analysis plug-in (v5.18.0.2; Thermo Fisher Scientific, Inc.). Data were filtered with a Q20 score threshold. Raw reads were mapped to the human reference genome assembly 19. Raw NGS data were uploaded to the Ion Reporter™ 5.18.4.0 Software (Thermo Fisher Scientific, Inc.) suite of bioinformatics tools. The analysis was performed using the manufacturer's workflow (Oncomine BRCA Research Somatic-530-w3.6-DNA). Furthermore, the commercial analysis software Sequence Pilot version 5.3.0 (JSI Medical Systems GmbH) was used for variant annotation.
SNP microarray assay and bioinformatics analysis
Hybridization was carried out via the OncoScan™ CNV Assay (cat no. 902695; Thermo Fisher Scientific, Inc.), as previously described (11–13). Briefly, arrays were washed and stained using a GeneChip Fluidics Station 450 (cat no. 00–0079; Thermo Fisher Scientific, Inc.) and scanned using a GeneChip Scanner 3000 7 G (cat no. 00–0218; Thermo Fisher Scientific, Inc.). The array fluorescence intensity data (CEL files) were converted to Oncoscan array data (OSCHP files) and analyzed using the Chromosome Analysis Suite 4.3 (ChAS v.4.3) software. Quality control calculations, including the median of the absolute values of all pairwise differences (MAPD), and SNP quality control of normal diploid markers were measured.
Allele-specific copy number analysis of tumors (ASCAT; v3.0.0) (14,15) was used to evaluate and calculate tumor purity, ploidy and allele-specific copy number profiles. Probe level data using logR ratio and B-allele frequency of autosomal markers were used as the input. Oncoscan-specific GC (Guanine-Cytocine) content and replication timing reference files were created and used for GC wave and replication timing correction. Segmentation data from ASCAT, along with the previously described algorithms (16) and definitions were used to calculate LOH (17), the number of telomeric-allelic imbalances (NTAI) (18) and LST (19). The NTAI score was calculated as the number of regions with allelic imbalance that extended to one of the sub-telomeres, did not cross the centromere and measured >11 Mb in length (20). The total value of GI was the sum of the three components and was given as a score (GIS). The GIS bioinformatics pipeline (RediScore v1.0) incorporated modifications adjusted to the Oncoscan array. The minimum number of probes/SNPs for a TAI region was adjusted to 126 so as to fit the OncoScan genome-wide resolution. Chromosome-specific ploidy was determined by major copy number fraction. To calculate the number of large-scale chromosomal breaks, the size of micro-variation S_micro was set to 15, fitting the SNP density in the array (21). The GIS bioinformatics algorithm was optimized (RediScore v2.0) using modifications of the LST algorithm. The LST score was adjusted by removing copy-neutral LOH segments and independently normalized by an estimation of whole-genome doubling events in the tumor sample, as previously described (22). The flowchart for the HRD status calculation is represented in Fig. 2. The oncoscanR method was executed as described in the GitHub repository (https://github.com/yannchristinat/OncoscanR); the normalized LST (nLS) score was then calculated for all samples analyzed.
Figure 2.

Flowchart of the homologous recombination deficiency status calculation algorithm using Oncoscan data for the measurement of the GIS score. GIS, genomic instability score; LOH, loss of heterozygosity; TAI, telomeric allelic imbalance; LSTs, large-scale transitions.
SNP down-sampling
Down-sampling of probe level data was performed at the prespecified proportions of 1, 5, 10, 20, 50 and 75% of the original 217,611 SNPs. SNP markers were down-sampled using a strategy of relaxing the density of the SNPs across the chromosomes by removing the between/intermediate SNPs, as appropriate (i.e. for 50% one every two SNPs was used, whereas for 10% one every 10 SNPs was used). Down-sampled probe level data were used for the calculation of the GIS score, simulating microarray assays of 10,000-165,000 markers and evaluating the effect of the numbers of SNP markers on HRD assessment, similar to Cristescu et al (9).
Statistical analysis
Statistical analysis was performed in R version 4.1.3 (The R Foundation). Pearson's correlation coefficient (r) and Lin's concordance correlation coefficient (CCC) were used to evaluate the GIS as a continuous variable. The concordance of the GIS score calculated from the microarray assay, and the clinically validated NGS assay was accessed at the dichotomous cut-off of 42 for RediScore v1.0 and v2.0 (GeneKor Medical S.A.), whereas the predefined cut-off of 15.5 was used for OncoscanR. Positive predictive value (PPV), negative predictive value (NPV), overall predictive agreement and Cohen's kappa index (κ) were calculated using the GIS score and HRD status from the validated NGS assay as a reference. The ROCit R library (The R Foundation) was used to assess the overall diagnostic ability of all methods as binary classifiers using a receiver operating characteristic (ROC) curve, as well as the measurements of sensitivity (Se), specificity (Sn), area under the curve (AUC) and the optimal cut-offs of the Youden index. 95% confidence intervals were used for all calculations. P<0.05 was considered to indicate a statistically significant difference.
Results
Validation of GIS analysis algorithm
A blind analysis of GIS using microarray data and NGS based BRCA1/2 mutation status was performed in 29 patients with HRD results available from the validated NGS assay. Out of the 29 patients, 20 (68.97%) had a positive result and were characterized as HR-deficient. Of those, a pathogenic or likely pathogenic BRCA1/2 mutation was identified in 6 cases. All BRCA1/2-positive cases were detected using the Oncomine assay used in the present study, which demonstrated 100% agreement with the validated test in BRCA1/2 analysis.
HRD status based on a GIS cut-off of 42 was evaluated for three different bioinformatics algorithms and compared with the validated NGS assay. Cohen's kappa (κ) was 0.696 [95% CI (0.424-0.968)] for RediScore v1.0, exhibiting a substantial agreement between assays. For RediScore v2.0, the κ was 0.848, [95% CI (0.647-1.000)], while for OncoscanR it was 0.828, [95% CI (0.602-1.000)] (Table I), showing a near perfect agreement for both algorithms. Positive and negative agreement between each algorithm and the validated NGS test is presented in Table I.
Table I.
Concordance characteristics on the validated NGS and the Oncoscan CNV assay on GIS score, as a categorical variable using alternative GIS analysis pipelines.
| Validated assay GIS score status | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| Assay | GIS (+/-) | GIS + | GIS - | PPV (%) | NPV (%) | OPA (%) | Kappa (κ) | r, P-value | CCC |
| RediScore v1.0 | GIS + | 17 | 1 | 94.0 | 72.7 | 86.2 | 0.696 | 0.831, | 0.830 |
| P<0.00001 | |||||||||
| GIS - | 3 | 8 | |||||||
| OncoscanR | GIS + | 20 | 2 | 90.9 | 100 | 93.1 | 0.828 | 0.899, | 0.298 |
| P<0.00001 | |||||||||
| GIS - | 0 | 7 | |||||||
| RediScore v2.0 | GIS + | 18 | 0 | 100 | 81.8 | 93.1 | 0.848 | 0.833, | 0.829 |
| P<0.00001 | |||||||||
| GIS - | 2 | 9 | |||||||
GIS, genomic instability score; GIS +, positive, GIS -, negative; PPV, positive predictive value; NPV, negative predictive value; OPA, overall predictive agreement; κ, Cohen's kappa index; r, Pearson's correlation coefficient; CCC, Lin's concordance correlation coefficient.
In addition, ROC curve analysis was used to calculate a Youden index cut-off and performance with an adjusted cut-off for each algorithm (Fig. 3). High sensitivity and specificity were observed for all methods tested. Optimal cut-off values based on Youden's index were highly similar to the predefined cut-off values, which confirms the high agreement level and robustness of the different GIS calculation pipelines.
Figure 3.

Empirical and binormal ROC curves, AUC and performance characteristics using the commercial food and drug administration approved next-generation sequencing assay as a reference for homologous recombination deficiency positive/negative groups. The Youden's cut off is displayed (green point) relative to the predefined cut-off (blue point) on the empirical ROC curve. (A) The RediScore v1 algorithm, (B) the OncoscanR algorithm and (C) the RediScore v2 algorithm. ROC, receiver operating characteristic; AUC, area under the curve; TPR, true positive rate; FPR, false positive rate; Se, sensitivity; Sp, specificity.
Furthermore, r and CCC were used to evaluate the GIS as a continuous variable (Fig. 4). The GIS score calculated by RediScore v1.0 and v2.0 revealed an excellent linear correlation (r=0.831 and r=0.833, respectively) and high agreement with the GIS score value from the FDA-approved NGS assay (CCC=0.830 and CCC=0.829, respectively). The nLST value of OncoscanR revealed an excellent linear correlation (r=0.899) with the GIS score value from the FDA-approved NGS assay but a poor agreement (CCC=0.290) with the GIS value, which was expected since the nLST is only one of the three components (scars) of the GIS score.
Figure 4.

GIS correlation determined using the food and drug administration approved commercial NGS assay and the Oncoscan assay with three different analysis pipelines. (A) The RediScore v1 algorithm, (B) the OncoscanR algorithm and (C) the RediScore v2 algorithm. GIS, genomic instability score; NGS, next-generation sequencing.
SNP down-sampling performance on GIS calculation
Recent evidence has suggested that the high density of SNPs may affect the LOH measurement by fitting noisy neighboring loci; therefore, the effect of down-sampling on the accuracy of calculating genomic metrics was investigated. Down-sampled SNP markers on probe-level data were exported from ChAS and used to calculate the sum of LOH, LST and TAI, as demonstrated in Fig. 2. The highest correlation with the clinically validated HRD test was observed when 10% of the SNPs were used, indicating this factor as the optimal for SNP down-sampling (Fig. 5). The difference in the GIS score values between the several down-sampled versions of each sample revealed a moderate positive correlation (r=0.534, P=0.0028) with the MAPD value of the sample.
Figure 5.
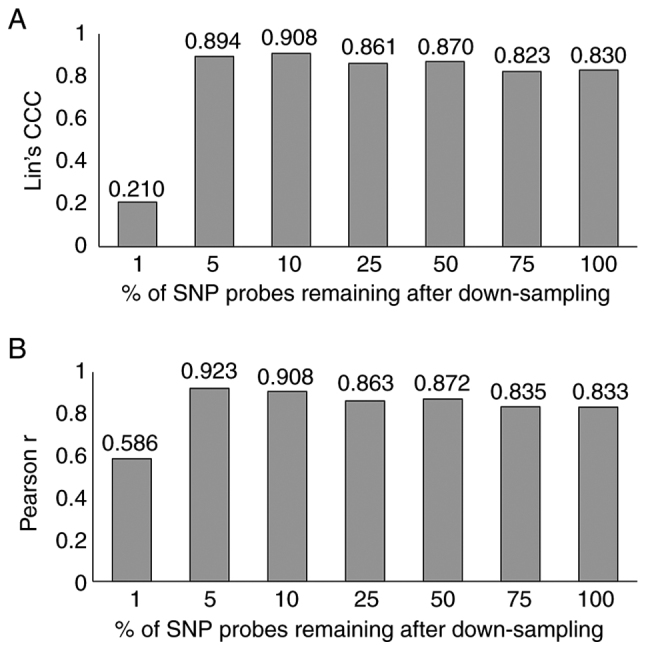
Homologous recombination deficiency score correlation determined using the food and drug administration approved commercial next-generation sequencing assay and the Oncoscan assay following SNP down-sampling. (A) Lin's CCC and (B) Pearson r correlation. CCC, concordance correlation coefficient; SNP, single nucleotide polymorphism.
HRD calculation in clinical samples
BRCA1/2 analysis was performed in FFPE tissue samples from 444 patients with ovarian cancer (Fig. S1 and Table SI). Adequate DNA quantity (>1.5 ng/µl) was obtained in 421 cases. Successful sequencing results from 408 (96.91%) samples were received and included in the study. A mutation in the BRCA1/2 genes was detected in 26.47% (108/408) of the tumor tissues assessed. A total of 79 patients carried a tumor mutation in the BRCA1 gene, 28 patients carried a BRCA2 mutation, and 1 patient carried both mutations simultaneously. The variant of uncertain significance (VUS) rate in our cohort was extremely low, accounting for 1.47% of the tumors assessed. The most common mutations detected were the BRCA1 c.3607C>T (16 cases) and the c.5266dupC (11 cases), which was in accordance with the Romanian origin of our cohort that shows a high prevalence of such mutations. The vast majority of mutations were truncating, leading to protein disruption, while a large genomic rearrangement involving the entire BRCA2 gene was detected in one patient.
Among patients with negative/VUS BRCA1/2, 188 requested GIS analysis testing. Of those, evaluable arrays results (MAPD <0.3) were obtained in 175 cases. The Rediscore v.2.0 was used for GIS calculation, showing a positivity rate of 40% (70/175). Given that the negative/VUS cases represent 73.53% of the initially tested cohort, if 100 patients with ovarian cancer were sequentially tested for BRCA1/2 genes and GIS, 26.47% of them could receive PARP inhibitor treatment based on BRCA testing, while an additional 29.41% could be treated based on GIS analysis. Therefore, the percentage of patients with an approved predictive biomarker for PARPi treatment would increase to 55.88%.
Discussion
PARPis have revolutionized treatment strategies for various types of cancer, including ovarian cancer. However, predictive biomarkers indicative of increased response probability are essential, in order to avoid unnecessary toxicity that is frequently observed in these patients (23). PARPis target DNA repair-related PARP proteins. The inhibition of such molecules is lethal to tumor cells whose HR pathway is dysfunctional. This may be caused by alterations in the numerous genes implicated in this pathway, with BRCA1 and BRCA2 playing a crucial role and being the most frequently altered HR genes (24). BRCA1/2 gene mutation analysis has been extensively used to predict PARPi response, demonstrating the efficacy of BRCA1/2 genes as biomarkers. However, the mutation rate among patients with ovarian cancer is restricted to ~25%, and PARPi responses have also been observed in patients without BRCA1/2 gene alterations, indicating that additional biomarkers are required to identify more patients who could benefit from treatment. Furthermore, the predictive value of alterations in other HR genes has not been demonstrated for ovarian cancer. Therefore, an indirect approach for detecting HRD was deemed reasonable, as it does not require identifying the origin of the deficiency while capturing its effect on the tumor genome. This method has demonstrated clinical utility and predictive value in ovarian cancer, and can detect up to 50% of cases (5). An NGS-based commercial test, has been extensively evaluated in clinical trials; it determines the combined status of LOH, TAI and LST, for the calculation of GIS, along with BRCA1/2 gene mutation analysis (4,25,26). Positivity is considered a GIS score of ≥42 and/or a BRCA1/2 somatic or germline mutation (27). This approach has exhibited predictive value in ovarian cancer, leading to the approval of olaparib + bevacizumab in newly diagnosed cases with HRD, namely BRCA1/2 mutations and/or high GIS (6).
Several alternative NGS assays have been developed for accessing GIS calculation, exhibiting a substantial-to-near perfect agreement with the results obtained using the validated NGS test, suggesting the feasibility and reliability of HRD testing in diagnostic laboratories (28,29).
Genotyping SNP-based microarrays are a reference methodology for genome-wide analysis of copy number changes, that can identify regions of gain or loss of chromosomal regions and LOH (9,27,30). Therefore, they could represent a low-cost alternative to NGS commercial assays. However, the bioinformatics approach used for GIS calculation is essential for obtaining high sensitivity and specificity in predicting HRD status.
In the present study, SNP array data obtained by the Oncoscan Assay, combined with NGS-based BRCA1/2 analysis, was used to calculate HRD status in patients with ovarian cancer.
Three different bioinformatics algorithms for GIS calculation from Oncoscan data (Rediscore v1.0, RediScore v2.0 and OncoscanR) were evaluated. The Rediscore v1.0 algorithm, which calculates the sum of LOH, LST and TAI, exhibited substantial agreement with the validated NGS test for GIS score calculation. The RediScore v2.0 algorithm, which includes in the algorithm the nLST analysis (instead of LST) in addition to LOH and TAI, exhibited a near perfect agreement with the validated NGS test; the same was true for nLST calculated by OncoscanR. Rediscore v2.0 and OncoscanR had an identically high overall agreement with the validated NGS test (93.1%). All positive samples were detected by OncoscanR (PPV, 100%), while all negative samples were identified by Rediscore v2.0 (NPV 100%). The high agreement rate of the SNP assay used indicated its feasibility in GIS calculation, in accordance with previous studies (30).
In addition, as previously reported, SNP down-sampling could contribute to an increase in overall agreement with the validated test, with the best GIS value agreement obtained when the 10% of the SNP arrays are used for GIS calculation (9).
A limitation of the present study was the absence of DNA extracted from the same sample for cross-test comparison of the derived GIS results. In addition, increasing the number of samples used could strengthen the statistical validity of the assay comparison results.
The RediScore v2.0 algorithm, which is less prone to false negative results, and thus unsuitable PARPi therapy administration, was selected for further evaluation in patients with ovarian cancer. In a cohort of 408 patients with ovarian cancer, 26.5% were positive for BRCA1/2, which was in accordance with previous studies evaluating BRCA1/2 status in this tumor type. Furthermore, among the 175 cases negative for BRCA1/2 mutations, an increased GIS score was identified in 70 cases. The addition of GIS to BRCA1/2 analysis could increase the number of patients who could benefit from PARPi treatment by 29.4% in the entire cohort, as 55.9% of the patients would be BRCA1/2-or GIS-positive and thus eligible for PARPi treatment based on the combined array and NGS data (Fig. 6). Therefore, joint analysis appears to be the most appropriate approach for HRD detection and is used in our laboratory by the name of RediScore.
Figure 6.
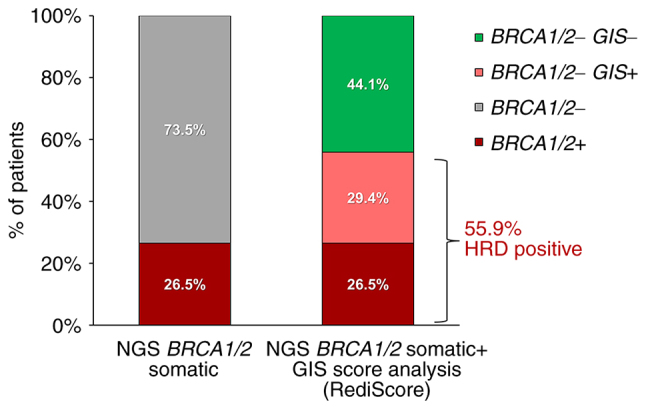
Clinical utility of combined GIS and BRCA1/2 analysis in ovarian cancer. The diagram shows the increase in the HRD detection obtained by the combination of BRCA1/2 and GIS analysis. GIS, genomic instability score; HRD, homologous recombination deficiency; NGS, next-generation sequencing; BRCA1/2+, BRCA1 or BRCA2 positive patients; BRCA1/2-, BRCA1 or BRCA2 negative patients; BRCA1/2-GIS+, GIS positive cases and BRCA1/2 negative cases; BRCA1/2-GIS-, GIS and BRCA1/2 negative cases.
In conclusion, SNP arrays are an efficient method for GIS calculation, as they exhibit high agreement with the validated NGS assay. The RediScore bioinformatics algorithm developed for GIS calculation in combination with NGS-based BRCA1/2 analysis is a viable and effective approach for HRD calculation. In addition, the clinical utility of HRD analysis was revealed in a cohort of patients with clinical ovarian cancer, offering a positive prediction for PARPi responsiveness in >50% of the patients.
Supplementary Material
Acknowledgements
Not applicable.
Funding Statement
Funding: No funding was received.
Availability of data and materials
BRCA1/2 NGS raw data are accessible through the Sequence Read Archive (SRA) under the PRJNA946802 BioProject (https://www.ncbi.nlm.nih.gov/sra/PRJNA946802) and Microarray (OncoScan) raw and processed data are accessible through the Gene Expression Omnibus (GEO) under the PRJNA947093 BioProject (Series GSE227800; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227800). The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
AT, EP and GN were responsible for conception and design. GT was responsible for analysis and interpretation of the data, AT, CP and EP drafted the manuscript. KP, TF, MG, AM, KPM, BG, VA, DLS, OT, FP, CM and CP collected the samples for analysis, provided clinical data and revised the manuscript critically for intellectual content. AT, EP, CP, KP, TF and GT confirm the authenticity of all the raw data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The present study was approved (approval no. 502/07-01-2023) by the Ethical Committee of ‘Aretaieion’ University Hospital (Athens, Greece).
Patient consent for publication
Written informed consent was provided by all patients participating in the present study.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Topatana W, Juengpanich S, Li S, Cao J, Hu J, Lee J, Suliyanto K, Ma D, Zhang B, Chen M, Cai X. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J Hematol Oncol. 2020;13:118. doi: 10.1186/s13045-020-00956-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swisher EM, Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, Coleman RL, Aghajanian C, Konecny GE, O'Malley DM, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2) Nat Commun. 2021;12:2487. doi: 10.1038/s41467-021-22582-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–1154. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pujade-Lauraine E, Brown J, Barnicle A, Wessen J, Lao-Sirieix P, Criscione SW, du Bois A, Lorusso D, Romero I, Petru E, et al. Homologous recombination repair gene mutations to predict olaparib plus bevacizumab efficacy in the first-line ovarian cancer PAOLA-1/ENGOT-ov25 trial. JCO Precis Oncol. 2023;7:e2200258. doi: 10.1200/PO.22.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, Chang DK, Garsed DW, Jonkers J, Ledermann JA, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31:1606–1622. doi: 10.1016/j.annonc.2020.08.2102. [DOI] [PubMed] [Google Scholar]
- 6.Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, Fujiwara K, Vergote I, Colombo N, Mäenpää J, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381:2416–2428. doi: 10.1056/NEJMoa1911361. [DOI] [PubMed] [Google Scholar]
- 7.Stewart MD, Merino Vega D, Arend RC, Baden JF, Barbash O, Beaubier N, Collins G, French T, Ghahramani N, Hinson P, et al. Homologous recombination deficiency: Concepts, definitions, and assays. Oncologist. 2022;27:167–174. doi: 10.1093/oncolo/oyab053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Sullivan Coyne G, Karlovich C, Wilsker D, Voth AR, Parchment RE, Chen AP, Doroshow JH. PARP inhibitor applicability: Detailed assays for homologous recombination repair pathway components. Onco Targets Ther. 2022;15:165–180. doi: 10.2147/OTT.S278092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cristescu R, Liu XQ, Arreaza G, Chen C, Albright A, Qiu P, Marton MJ. Concordance between single-nucleotide polymorphism-based genomic instability assays and a next-generation sequencing-based homologous recombination deficiency test. BMC Cancer. 2022;22:1310. doi: 10.1186/s12885-022-10197-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ngoi NYL, Tan DSP. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: do we need it? ESMO Open. 2021;6:100144. doi: 10.1016/j.esmoop.2021.100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, Tinkler-Hundal E, Southward K, Medlow P, McGreeghan-Crosby K, et al. Cross-laboratory validation of the OncoScan® FFPE assay, a multiplex tool for whole genome tumour profiling. BMC Med Genomics. 2015;8:5. doi: 10.1186/s12920-015-0079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowe LR, Thaker HM, Opitz JM, Schiffman JD, Haddadin ZM, Erickson LK, South ST. Molecular inversion probe array for the genetic evaluation of stillbirth using formalin-fixed, paraffin-embedded tissue. J Mol Diagn. 2013;15:466–472. doi: 10.1016/j.jmoldx.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Moorhead M, Karlin-Neumann G, Falkowski M, Chen C, Siddiqui F, Davis RW, Willis TD, Faham M. Allele quantification using molecular inversion probes (MIP) Nucleic Acids Res. 2005;33:e183. doi: 10.1093/nar/gni177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross EM, Haase K, Van Loo P, Markowetz F. Allele-specific multi-sample copy number segmentation in ASCAT. Bioinformatics. 2021;37:1909–1911. doi: 10.1093/bioinformatics/btaa538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imanishi S, Naoi Y, Shimazu K, Shimoda M, Kagara N, Tanei T, Miyake T, Kim SJ, Noguchi S. Clinicopathological analysis of homologous recombination-deficient breast cancers with special reference to response to neoadjuvant paclitaxel followed by FEC. Breast Cancer Res Trea. 2019;174:627–637. doi: 10.1007/s10549-018-05120-9. [DOI] [PubMed] [Google Scholar]
- 17.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2:366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 20.Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, Kalva S, Potter J, Tran TV, Chen J, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16:475. doi: 10.1186/s13058-014-0475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popova T, Manié E, Stern MH. Genomic signature of homologous recombination deficiency in breast and ovarian cancers. Bio-Protocol. 2013;3 doi: 10.21769/BioProtoc.814. [DOI] [Google Scholar]
- 22.Christinat Y, Ho L, Clément S, Genestie C, Sehouli J, Gonzalez Martin A, Denison U, Fujiwara K, Vergote I, Tognon G, et al. Normalized LST is an efficient biomarker for homologous recombination deficiency and Olaparib response in ovarian carcinoma. medRxiv. 2022 doi: 10.1200/PO.22.00555. 2022.08.22.22278669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiggans AJ, Cass GKS, Bryant A, Lawrie TA, Morrison J. Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer. Cochrane Database Syst Rev. 2015;2015:CD007929. doi: 10.1002/14651858.CD007929.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: Current practice and future directions. Br J Cancer. 2016;115:1157–1173. doi: 10.1038/bjc.2016.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabatier R, Rousseau F, Joly F, Cropet C, Montégut C, Frindte J, Cinieri S, Guerra Alía EM, Polterauer S, Yoshida H, et al. Efficacy and safety of maintenance olaparib and bevacizumab in ovarian cancer patients aged ≥65 years from the PAOLA-1/ENGOT-ov25 trial. Eur J Cancer. 2023;181:42–52. doi: 10.1016/j.ejca.2022.11.029. [DOI] [PubMed] [Google Scholar]
- 26.Labidi-Galy SI, Rodrigues M, Sandoval JL, Kurtz JE, Heitz F, Mosconi AM, Romero I, Denison U, Nagao S, Vergote I, et al. Association of location of BRCA1 and BRCA2 mutations with benefit from olaparib and bevacizumab maintenance in high-grade ovarian cancer: Phase III PAOLA-1/ENGOT-ov25 trial subgroup exploratory analysis. Ann Oncol. 2023;34:152–162. doi: 10.1016/j.annonc.2022.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Re. 2016;22:3764–3773. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doig KD, Fellowes AP, Fox SB. Homologous recombination repair deficiency: An overview for pathologists. Mod Pathol. 2023;36:100049. doi: 10.1016/j.modpat.2022.100049. [DOI] [PubMed] [Google Scholar]
- 29.Fumagalli C, Betella I, Ranghiero A, Guerini-Rocco E, Bonaldo G, Rappa A, Vacirca D, Colombo N, Barberis M. In-house testing for homologous recombination repair deficiency (HRD) testing in ovarian carcinoma: A feasibility study comparing AmoyDx HRD focus panel with myriad myChoiceCDx assay. Pathologica. 2022;114:288–294. doi: 10.32074/1591-951X-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stronach EA, Paul J, Timms KM, Hughes E, Brown K, Neff C, Perry M, Gutin A, El-Bahrawy M, Steel JH, et al. Biomarker assessment of HR deficiency, tumor BRCA1/2 mutations, and CCNE1 copy number in ovarian cancer: Associations with clinical outcome following platinum monotherapy. Mol Cancer Res. 2018;16:1103–1111. doi: 10.1158/1541-7786.MCR-18-0034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BRCA1/2 NGS raw data are accessible through the Sequence Read Archive (SRA) under the PRJNA946802 BioProject (https://www.ncbi.nlm.nih.gov/sra/PRJNA946802) and Microarray (OncoScan) raw and processed data are accessible through the Gene Expression Omnibus (GEO) under the PRJNA947093 BioProject (Series GSE227800; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227800). The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.