

Abstract
Objective
Mitochondrial pyruvate is a critical intermediary metabolite in gluconeogenesis, lipogenesis, and NADH production. As a result, the mitochondrial pyruvate carrier (MPC) complex has emerged as a promising therapeutic target in metabolic diseases. Clinical trials are currently underway. However, recent in vitro data indicate that MPC inhibition diverts glutamine/glutamate away from glutathione synthesis and toward glutaminolysis to compensate for loss of pyruvate oxidation, possibly sensitizing cells to oxidative insult. Here, we explored this in vivo using the clinically relevant acetaminophen (APAP) overdose model of acute liver injury, which is driven by oxidative stress.
Methods
We used pharmacological and genetic approaches to inhibit MPC2 and alanine aminotransferase 2 (ALT2), individually and concomitantly, in mice and cell culture models and determined the effects on APAP hepatotoxicity.
Results
We found that MPC inhibition sensitizes the liver to APAP-induced injury in vivo only with concomitant loss of alanine aminotransferase 2 (ALT2). Pharmacological and genetic manipulation of neither MPC2 nor ALT2 alone affected APAP toxicity, but liver-specific double knockout (DKO) significantly worsened APAP-induced liver damage. Further investigation indicated that DKO impaired glutathione synthesis and increased urea cycle flux, consistent with increased glutaminolysis, and these results were reproducible in vitro. Finally, induction of ALT2 and post-treatment with dichloroacetate both reduced APAP-induced liver injury, suggesting new therapeutic avenues.
Conclusions
Increased susceptibility to APAP toxicity requires loss of both the MPC and ALT2 in vivo, indicating that MPC inhibition alone is insufficient to disrupt redox balance. Furthermore, the results from ALT2 induction and dichloroacetate in the APAP model suggest new metabolic approaches to the treatment of liver damage.
Keywords: Acetaminophen, Diabetes, Glutathione, Liver, Metabolism, Oxidative stress
Graphical abstract
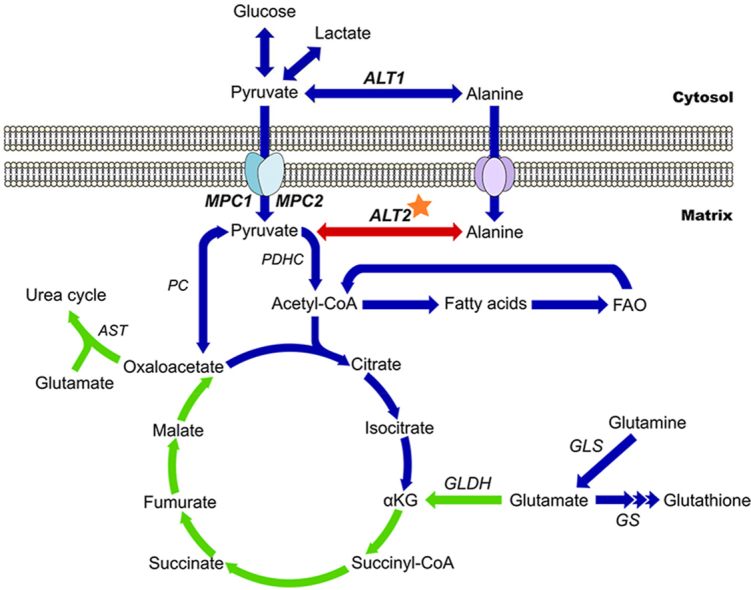
Schematic representing tricarboxylic acid cycle flux early during APAP toxicity in the different conditions tested. Under normal conditions, the tricarboxylic acid (TCA) cycle and mitochondrial metabolism proceeds through all arrows. Blue arrows indicate pathways or steps that are diminished with inhibition of both the MPC and ALT2 (DKO). Green arrows indicate pathways that are amplified in DKO. Red arrows indicate pathways or steps that are amplified specifically in MPC2 inhibition. Star shows dexamethasone target. αKG, α-ketoglutarate. ALT, alanine aminotransferase. AST, aspartate aminotransferase. GLDH, glutamate dehydrogenase. GLS, glutaminase. GS, glutathione synthase. MPC, mitochondrial pyruvate carrier. PC, pyruvate carboxylase. PDHC, pyruvate dehydrogenase complex.
Highlights
-
•
The MPC is a metabolic disease target but inhibition disrupts redox balance in cells.
-
•
ALT2 compensates for MPC loss in the liver in vivo, preserving redox homeostasis.
-
•
Combined MPC/ALT2 deletion impairs glutathione synthesis and worsens APAP toxicity.
-
•
ALT2 induction protects against APAP-induced injury.
1. Introduction
Acetaminophen (APAP; a.k.a. paracetamol) is one of the most widely used antipyretic and analgesic drugs in the world. In a cross-sectional survey of the US population, for example, >20% of both adults and children reported use of APAP at least once in the preceding seven days [1,2] and similar data have been published from other countries [[3], [4], [5]]. However, APAP overdose can cause severe liver injury leading to acute liver failure (ALF) and death [6].
Oxidative stress is believed to be the central driver of APAP hepatotoxicity [[7], [8], [9]]. Cytochrome P450 enzymes (P450s), particularly CYP2E1, convert APAP to the reactive intermediate N-acetyl-p-benzoquinone imine (NAPQI) [10,11] which depletes the antioxidant glutathione and then binds to mitochondrial proteins [7,[12], [13], [14]]. The mitochondrial protein alkylation coincides with and likely causes reduced respiration [15] and an increase in mitochondrial reactive oxygen species (ROS) production [7,16]. The redox imbalance that results from combined glutathione depletion and increased ROS rapidly promotes phosphorylation and activation of the c-Jun N-terminal kinases (JNK) 1/2 [17,18], which translocate to mitochondria and further reduce mitochondrial respiration via SHP1-mediated inhibition of Src [19,20]. Other kinases are also activated by the oxidative stress and may exacerbate the injury [[21], [22], [23]]. Eventually, the mitochondrial membrane permeability transition occurs [24], mitochondria swell, and their membranes rupture [25]. The latter releases intermembrane and matrix proteins into the cytosol, including endonucleases which then cleave nuclear DNA [26]. Ultimately, the damaged hepatocytes die by necrosis [27,28]. Thus, anything that upsets mitochondrial metabolism or cellular redox regulation would be expected to alter APAP toxicity.
The carbon backbone of pyruvate can enter the mitochondrial TCA cycle via either pyruvate carboxylation or oxidation and is an important substrate for gluconeogenesis, lipid synthesis, and NADH generation for the electron transport chain (ETC) [29]. Most pyruvate is produced in the cytosol as a product of glycolysis or conversion from lactate and is taken up by mitochondria via the mitochondrial pyruvate carrier (MPC) complex. Interestingly, in cultured cells and in hepatic tumors, genetic deletion of the MPC or pharmacological MPC inhibition not only blocks pyruvate oxidation [30,31], but also decreases glutathione synthesis and disrupts redox balance [32,33]. Thus, due to the role of mitochondrial dysfunction in APAP-induced liver injury and the critical importance of glutathione as a nucleophile and antioxidant in resistance to APAP, we hypothesized that preventing mitochondrial pyruvate uptake by disrupting the MPC would worsen mitochondrial damage and oxidative stress after APAP overdose and therefore exacerbate APAP toxicity.
2. Materials and methods
2.1. Animals
WT male C57BL/6J mice between the ages of 7–8 weeks were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and used for experiments at 8–10 weeks of age. The generation of mice harboring a floxed allele of the gene encoding ALT2 (Gpt2) derived from Gpt2 germline KO mice generated by the Knockout Mouse Project Repository (KOMP) (project ID CSD24977) has recently been described [34]. Similarly, the generation of Mpc2 floxed mice using ES cells obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM) (International Knockout Mouse Consortium [IKMC] project 89918, catalog no. MAE-1900) has been described [35]. Transgenic mice expressing Cre under control of the albumin promoter (B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J; Jackson Laboratory stock number: 003574) were bred with these animals to create liver-specific KO mice. Combined liver-specific Gpt2/Mpc2 double KO (DKO) mice were then generated by intercrossing the two liver-specific single gene knockouts. Littermate mice not expressing Cre (fl/fl mice) were used as controls. Male mice were used in all experiments because female mice are resistant to APAP hepatotoxicity [36]. All MPC2 KO and DKO mice and their WT littermate controls were used at 10–16 weeks of age. All Gpt2 (ALT2) KO mice were used between 14 and 28 weeks of age.
Animals were kept in a temperature-controlled 12 h light/dark-cycle room with ad libitum access to food and water. For BCLA time course and dose–response experiments, fed WT mice were treated with BCLA dissolved in 1× phosphate-buffered saline (PBS) at the indicated doses or PBS alone and liver tissue was collected at the indicated time points. For APAP experiments, mice were fasted overnight, then treated i.p. with 300 mg APAP per kg body mass (mg/kg) dissolved in PBS. Some WT mice were pre-treated i.p. with the specified doses of BCLA or PBS or with 100 mg/kg dexamethasone (Dex) for the indicated times before APAP. Others received 30 mg/kg MSDC, 318 mg/kg NAC, or appropriate vehicle 2 h after APAP. NAC and NAC vehicle (0.9% saline) were administered i.v. All other treatments were given i.p. Blood and liver tissue were collected at 6 h post-APAP. Some liver pieces were fixed in 10% phosphate-buffered formalin for histology, while others were flash-frozen in liquid nitrogen and stored at −80 °C until biochemical analyses. All study protocols were approved by the Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences and Washington University in St. Louis. The Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines were consulted during manuscript preparation and we have attempted to conform to those recommendations.
2.2. Biochemistry
Alanine aminotransferase (ALT) was measured in serum using a kit from MedTest Dx, Inc. (Canton, MI, USA), according to the manufacturer's instructions. Lactate dehydrogenase (LDH) was measured in serum as previously described [37]. For hepatic ALT activity, frozen liver tissue was homogenized in 25 mM HEPES buffer with 5 mM EDTA (pH 7.4), 0.1% CHAPS, and protease inhibitors using a bead homogenizer (Thermo Fisher Scientific, Waltham, MA). After centrifugation at 10,000×g and 4 °C for 5 min, ALT was measured in the supernatant using the MedTest kit mentioned above. Protein concentration in the lysates was measured using the bicinchoninic acid (BCA) assay and was used to normalize hepatic ALT activity. Ketones were measured using the EnzyChrom Ketone Body Assay (BioAssay Systems, Hayward, CA USA) according to the manufacturer's instructions.
2.3. Cell culture
Primary mouse hepatocytes were isolated from WT mice using a collagenase perfusion technique, as previously described [38], and used in experiments no more than 24 h after isolation. HepG2 cells overexpressing CYP2E1 were purchased from Hera Biolabs (Lexington, KY USA) and grown in Williams' Medium E with 5% fetal bovine serum, glutamate, penicillin/streptomycin, and insulin. Regular HepG2 cells were purchased from the American Type Culture Collection (Manassas, VA USA) and grown in the same medium. Cells were treated as described in the results section.
2.4. Histology
Formalin-fixed tissue was embedded in paraffin wax and 5 μm sections were mounted on glass slides for hematoxylin and eosin (H&E) staining. H&E staining was performed using a standard protocol. Necrosis was quantified by two independent, blinded, fellowship-trained hepatobiliary pathologists. Slides were imaged using a Labomed Lx400 microscope with a 16 MP Atlas digital camera and Pixel Pro software (Labo America, Fremont, CA USA).
2.5. Western blotting
Western blotting was performed as we previously described [39]. The primary antibodies against phospho-JNK (Cat. No. 4668), JNK (Cat. No. 9252), and MPC2 (Cat. No. 46141) were purchased from Cell Signaling Technology (Danvers, MA, USA) and used at 1:1,000 dilution in TBS-T with 5% milk. The Cyp2e1 antibody was purchased from Proteintech (Rosemont, IL, USA) (Cat. No. 19937-1-AP). The ALT2 antibody was from Sigma (St. Louis, MO, USA) (Cat. No. HPA051514). Secondary antibodies were purchased from Li-Cor Biotechnology (Lincoln, NE, USA) and Cell Signaling Technology. Total protein was stained with Coomassie blue. All blots were imaged using a Li-Cor Odyssey instrument (phospho-JNK and JNK) or an Invitrogen iBright FL1500 (all other blots and protein images).
2.6. Plasma metabolomics for amino acids and pyruvate
Amino acids were extracted from 20 μL of plasma with 200 μL of methanol containing Try-d8 (1600 ng), Phe-d8 (160 ng), Tyr-d4 (160 ng), Leu-d3 (320 ng), Ile-13C6,15N (160 ng), Met-d3 (160 ng), Val-d8 (400 ng), Glu-d3 (2400 ng), Thr-13C4 (320 ng), Ser-d3 (400 ng), Asp-d3 (3200 ng), Ala-d4 (1600 ng), Asn d3,15N2 (3200 ng), Gly-d2 (16000 ng), Gln-13C5 (3200 ng), Pro-d7 (800 ng), Cit-d4 (80 ng), His 13C6 (400 ng), Arg-13C6 (1600 ng), Orn-13C5 (400 ng), Lys-d8 (400 ng), and pyruvic acid-13C5 (80 ng) as the internal standards for Try, Phe, Tyr, Leu, Ile, Met, Val, Glu, Thr, Ser, Asp, Ala, Asn, Gly, Gln, Pro, Cit, His, Arg, Orn, Lys, and pyruvic acid (Pyr) respectively. The sample aliquots for pyruvic acid were derivatized with o-phenylenediamine to improve mass spectrometric sensitivity. Quality control (QC) samples were prepared by pooling aliquots of study samples and injected after every four study samples to monitor instrument performance throughout these analyses.
The analysis was performed on a Shimadzu 20AD HPLC system and a SIL-20AC autosampler coupled to 4000Qtrap mass spectrometer (AB Sciex) operated in positive multiple reaction monitoring (MRM) mode. Data processing was conducted with Analyst 1.6.3 (Applied Biosystems). The relative quantification data for all analytes were presented as the peak ratio of each analyte to the internal standard. The Metabolomics Facility of Washington University School of Medicine performed the analysis.
2.7. qPCR
RNA was extracted from liver tissue using the RNeasy Mini Kit (Qiagen, Vinlo, Netherlands). mRNA levels for Gclc (Fwd, 5′ to 3′): ATC TGC AAA GGC GGC AAC; Rev, 5′ to 3′: ACT CCT CTG CAG CTG GCT C), Gsr (Fwd, 5′ to 3′: ATG CCT ATG TGA GCC GCC TG; Rev, 5′ to 3′: TGC GAA TGT TGC ATA GCC GT), Gpt2 (Fwd, 5′ to 3′: AAC CAT TCA CTG AGG TAA TCC GA; Rev, 5′ to 3′: GGG CTG TTT AGT AGG TTT GGG TA) and Mpc2 (Fwd, 5′ to 3′: ACC TAC CAC CGA CTC ATG GAT; Rev, 5′ to 3′: TGT AAA GCG GCC TCA ATT TCT) were measured by real-time quantitative PCR using the Applied Biosystems PowerSYBR Green PCR Master Mix (Thermo Fisher, Waltham, MA USA) according to the manufacturer's instructions. The data were normalized to Gapdh and expressed as arbitrary units (AU) relative to the appropriate control group.
2.8. Statistical analyses
Sample sizes for the pharmacologic treatment experiments were determined a priori and are indicated in the caption for each figure. In each case, the difference in serum ALT between the vehicle control and drug-treated groups was the primary comparison of interest and the experiments were designed to achieve a minimum of 80% power at α = 0.05 using our historical ALT data to predict variation. Sample sizes for the KO and DKO animals were determined by litter size, frequency, and availability for this project. Secondary analyses were carried out on the same samples. Statistical significance was assessed using either Student's t-test or one-way analysis of variance (ANOVA) with post-hoc Dunn's test or Tukey's test. For non-normally distributed data, the Mann–Whitney U-test or the Kruskal–Wallis test were used. All statistical analyses were performed using SigmaPlot 12.5 (Systat, San Jose, CA, USA) or Prism 10.0 (GraphPad, Boston, MA, USA). P-values are indicated in the figures or figure captions.
3. Results
3.1. MPC inhibition and deletion have no effect on APAP hepatotoxicity
Prior work has suggested that enhanced hepatic pyruvate metabolism after APAP overdose may have hepatoprotective effects. For example, genetic deletion of pyruvate dehydrogenase kinase 4 (PDK4), which inhibits mitochondrial pyruvate oxidation, protected mice from APAP-induced injury [40]. To further test the effects of enhancing pyruvate oxidation, we post-treated mice with either vehicle or the PDK inhibitor dichloroacetate (DCA) 2 h after APAP in order to increase pyruvate flux through the pyruvate dehydrogenase complex (PDHC). Consistent with the prior work using PDK4 KO mice, DCA reduced APAP hepatotoxicity as indicated by serum ALT (Figure 1A). Given this finding, one might predict that loss of mitochondrial pyruvate metabolism would exacerbate APAP-induced liver injury. The mitochondrial pyruvate carrier (MPC) catalyzes the transport of pyruvate into the mitochondrial matrix and is composed of two proteins (MPC1 and MPC2) that form a heterodimeric complex in the inner mitochondrial membrane [29]. Thus, we treated liver-specific MPC2 KO mice with APAP to determine whether loss of mitochondrial pyruvate metabolism might affect the response to APAP mediated hepatotoxicity. Consistent with prior reports [35,41], MPC2 deletion resulted in instability and degradation of MPC1 (Figure 1B), effectively leading to complete loss of the MPC complex. However, MPC deficiency had no effect on APAP hepatotoxicity at 6 h after treatment, within the timeframe when mitochondrial metabolism is most critical [7,8]. There were no significant differences in serum ALT or hepatic necrosis, total glutathione, oxidized glutathione, APAP-protein binding, or JNK activation between MPC2 KO mice and WT littermate controls (Figure 1C–K).
Figure 1.

Neither MPC inhibition nor liver-specific knockout worsen liver injury or glutathione depletion after APAP overdose. (A) WT male mice 8–10 weeks old were treated with 300 mg/kg APAP followed by 250 mg/kg dichloroacetate (DCA) or PBS vehicle (Veh) 2 h later. Serum ALT was measured at 6 h post-APAP. (B–K) Male livers-specific MPC KO mice and WT littermates 10–16 weeks old were treated with 300 mg/kg APAP or PBS vehicle. Serum and liver tissue were collected 6 h later. (B) MPC1, MPC2, β-actin, and ALT2 were measured in liver homegenates by immunoblot. (C) Serum ALT was measured. (D) 5 μm-thick liver sections were glass-mounted and stained with hematoxylin and eosin (H&E) to measure necrosis. Representative images are shown at 100× magnification. (E) Necrosis was quantified in the liver sections. (F) Total glutathione (GSH + GSSG) was measured in liver tissue. (G) Oxidized glutathione was measured in liver tissue and expressed as a proportion of the total glutathione. (H) APAP-protein adducts were measured in liver tissue. (I) Densitometric analysis was performed on the ALT2 immunoblot in (B). (J) Phosphorylated and total JNK (p-JNK and JNK, respectively) were measured in liver homogenates by immunoblot. (K) Densitometric analysis was performed on the JNK blots in (J). Two-tailed t-test, p = 0.026. (J) Immnuoblots for phosphorylated (p-JNK) and total JNK in liver. (K) Densitometry of K. Comparison of two means was performed by two-tailed t-test. Comparison of three or more groups was performed by ANOVA with post-hoc Tukey's test for multiple comparisons. ∗p < 0.05, ∗∗∗∗p < 0.0001. In all panels, lines and error bars indicate mean ± SE. Circles show individual data points and sample sizes. Box plots show medians and IQR. Open circles: Veh. Filled circles: APAP.
Although uptake via the MPC is the primary source of mitochondrial pyruvate, we have previously shown that cytosolic and mitochondrial alanine-pyruvate cycling catalyzed by alanine transaminase enzymes can partially circumvent the loss of MPC activity [35]. Indeed, shRNA-mediated knockdown of alanine transaminase 2 (ALT2), which catalyzes the transamination of alanine and α-ketoglutarate to form pyruvate and glutamate in the mitochondrial matrix, further attenuates mitochondrial pyruvate flux in MPC2-deficient hepatocytes [35]. In the present study, we found that ALT2 protein abundance was 1.9 ± 0.2-fold greater in livers from MPC2 KO mice compared to WT mice after APAP treatment (Figure 1B and I), suggesting that mitochondrial alanine-pyruvate cycling might be compensating for loss of the MPC during APAP hepatotoxicity as well. Though we cannot rule out that injury may have also contributed to ALT2 induction at this point, the data induction of ALT2 in APAP toxicity may have been protective either way.
3.2. ALT2 inhibition and deletion have no effect on APAP hepatotoxicity
The idea that increased ALT2 might have prevented increased injury in the MPC2 KO mice led us to determine if increasing mitochondrial alanine flux is protective against APAP-induced injury more generally. We pre-treated WT mice for 24 h with a large dose of dexamethasone (Dex) followed by APAP. Large doses of Dex are known to strongly induce ALT2 expression in the liver [42,43]. Importantly, western blotting confirmed an increase in hepatic ALT2 with Dex treatment and the gold-standard for injury, histology, revealed that Dex administration was indeed protective (Figure 2A–C).
Figure 2.

Neither ALT2 inhibition nor ALT2 knockout worsen liver injury after APAP overdose. (A–C) WT male mice 8–10 weeks old were treated with 100 mg/kg dexamethasone (Dex) followed by 300 mg/kg APAP 24 h later. Liver tissue was collected 6 h post-APAP. (A) ALT2 induction in liver homogenates was measured by immunoblot. (B) 5 μm-thick liver sections were glass-mounted and stained with hematoxylin and eosin (H&E) to measure necrosis. Representative images are shown at 100× magnification. (C) Necrosis was quantified in the liver sections. (D–H) WT male mice 8–10 weeks old were treated with 100 mg/kg BCLA followed by 300 mg/kg APAP 24 h later. Serum and liver tissue were collected 6 h post-APAP. ALT activity was measured in (D) serum and (E) liver tissue homogenates. (F) 5 μm-thick liver sections were glass-mounted and stained with H&E to measure necrosis. Representative images are shown at 100× magnification. (G) Necrosis was quantified in the liver sections. (I–M) Male liver-specific ALT2 KO mice and WT littermate controls 14–28 weeks old were treated with 300 mg/kg APAP or PBS vehicle. Serum and liver tissue were collected at 6 h post-APAP. (I) ALT2 was measured in liver homogenates by immunoblot. (J) Serum ALT was measured in APAP and PBS treated WT and KO mice. (K) Serum LDH was measured in APAP and PBS treated WT and KO mice. (L) 5 μm-thick liver sections were glass-mounted and stained with H&E to measure necrosis. Representative images are shown at 100× magnification. (M) Necrosis was quantified in the liver sections. Comparison of two means was performed by two-tailed t-test. Comparison of three or more groups was performed by ANOVA with post-hoc Tukey's test for multiple comparisons. ∗p < 0.05, ∗∗∗p < 0.001. In all panels, lines and error bars indicate mean ± SE. Circles show individual data points and sample sizes. Box plots show medians and IQR.
We next used a pharmacological approach to suppress mitochondrial alanine metabolism. β-chloro-l-alanine (BCLA) is an alanine analog that inhibits the enzyme's activity [44]. Although it has been extensively used in studies in vitro, we are not aware of any data in the literature characterizing the dose–response and kinetics of BCLA inhibition of ALT activity in the liver in vivo. Thus, we first performed timing and dose–response studies to optimize dosing. The results revealed that BCLA can inhibit ALT activity in the liver in mice, that the effect is greater at 24 h post-BCLA treatment than an earlier time point, and that ≥50% inhibition can be achieved with a dose of 100 mg/kg (Suppl. Fig. 1). Based on these data, we adopted a 24 h single-dose (Figure 2D–H) and 48 h two-dose pre-treatment regimen with 100 mg/kg BCLA to study the role of ALT in APAP toxicity. Consistent with its action as an ALT inhibitor, BCLA reduced both serum and hepatic ALT activity after APAP overdose (Figure 2D and E). However, like MPC2 deletion, BCLA had no effect on injury as indicated by the alternative serum liver injury biomarker LDH and by hepatic necrosis (Figure 2F–H). Results from the two-dose 48 h BCLA pre-treatment regimen were nearly identical (data not shown).
BCLA did not completely block hepatic ALT activity (Figure 2E). In addition, it is possible that the drug cannot reach ALT2 within the mitochondrial matrix. Thus, to ensure complete inhibition of ALT2, we next used liver-specific ALT2 KO mice (Figure 2I). However, like MPC2 KO, there was no significant effect of ALT2 deletion on APAP-induced liver injury, as indicated by serum ALT (Figure 2J) and LDH (Figure 2K) and by hepatic necrosis (Figure 2L,M). Although it may seem surprising that ALT2 KO itself did not reduce serum ALT activity, this observation is consistent with reports that ALT1 is the dominant isoform released into serum [[45], [46], [47]], accounting for ≥90% of ALT activity in circulation after injury [47]. Thus, we continued to use serum ALT as our primary measure of liver injury. We also confirmed all ALT results with histological analysis. Altogether, these data indicate that neither MPC2 deletion nor ALT2 deficiency alone alters APAP hepatotoxicity but do not rule out the possibility of complementary effects.
3.3. Combined MPC2/ALT2 deficiency worsens APAP-induced liver injury
To determine if deletion of both MPC2 and ALT2 worsened APAP hepatotoxicity, we generated liver-specific ALT2/MPC2 double KO (DKO) mice (Suppl. Fig. 2). In contrast to mice with only single deletion of MPC2 or ALT2, DKO mice had greater serum ALT activity and hepatic necrosis after APAP overdose (Figure 3A–C). Importantly, they also had lower total hepatic glutathione levels (Figure 3D). Glutathione reaches its nadir with nearly complete loss by approximately 0.5 h post-APAP and then recovers over the following 6 h [12]. Thus, these 6 h data suggest impaired glutathione re-synthesis, possibly due to the diversion of glutamate to α-ketoglutarate (αKG) and away from glutathione synthesis as we had initially hypothesized would happen with MPC inhibition. Furthermore, these effects occurred despite less APAP-protein binding in the DKO mice (Figure 3E). Because protein alkylation is the initiating event in APAP hepatotoxicity, even greater injury may have been observed had the level of protein binding been the same as in WT. Indeed, when we normalized serum ALT to hepatic APAP-protein adducts, the difference in means was even greater with a 4.9 ± 0.2-fold increase in ALT in the DKO animals (mean ± SE) (Figure 3F).
Figure 3.

MPC2/ALT2 double-knockout signficantly worsens liver injury and prevents glutathione recovery after APAP overdose. Male liver-specific MPC2/ALT2 DKO mice 10–16 weeks old were treated with 300 mg/kg APAP. Serum and liver tissue were collected 6 h later. (A) Serum ALT activity was measured in the WT and DKO mice. (B) Necrosis was quantified from H&E-stained liver sections. (C) Representative liver sections from WT and DKO mice at 100× magnification. (D) Total glutathione (GSH + GSSG) was measured in liver tissues. (E) APAP-protein adducts (APAP-Prot) were measured in liver tissue homogenates. (F) ALT was normalized to APAP-Prot. (G) CYP2E1 levels were measured in liver homogenates by immunoblot. Total protein on the immunoblot membrane was stained using Coomassie blue. (H) Densiometric analysis was performed on the CYP2E1 immunoblot in (G). (I) NADPH was measured in liver tissue homogenates. (J) NADP+ was also measured in liver tissue and NADPH was normalized to NADP+ to determine their ratio. (K) Oxidized glutathione (GSSG) was measured and expressed as the proportion of total glutathione. Comparison of two means was performed by two-tailed t-test. Comparison of three or more groups was performed by ANOVA with post-hoc Tukey's test for multiple comparisons. ∗p < 0.05, ∗∗p < 0.01. In all panels, lines and error bars indicate mean ± SE. Circles show individual data points and sample sizes. Box plots show medians and IQR.
The reason for the reduced adducts in DKO animals may have been depletion of NADPH, which is necessary for the P450 catalysis that produces NAPQI because the P450 catalytic cycle uses electrons from NAPDH transferred via the enzyme P450 reductase. The protein levels of CYP2E1 – the primary enzyme that converts APAP to NAPQI [48] – were the same between genotypes (Fig. G,H), demonstrating that the lower NAPQI production and protein binding was not due to altered CYP2E1 expression. Furthermore, lower NADPH levels have been reported in MPC1-deficient cells in vitro [33] and we observed NADPH depletion in our DKO animals after APAP overdose as well (Figure 3I,J).
3.4. The mechanism of exacerbated injury in DKO mice may be related to diversion of glutamate away from glutathione
Compensatory glutaminolysis at the expense of glutathione synthesis has been previously reported in MPC1-deficient hepatocytes in vitro [32] and in other conditions in which mitochondrial pyruvate metabolism was impaired [49]. Thus, we compared basal total hepatic glutathione between DKO and WT mice and between HepG2 cells treated with the combination of another MPC inhibitor, UK-5099, and BCLA or vehicle to mimic combined MPC and ALT2 KO. Interestingly, glutathione was 22 ± 5% (mean ± SE) lower in liver tissue from DKO animals (Figure 4A) and almost undetectable in the UK/BCLA co-treated cells (Figure 4B). Targeted plasma metabolomics also revealed elevated urea cycle intermediates (citrulline, arginine, and ornithine) in the untreated DKO mice (Table 1). While steady state metabolite concentration is not equivalent to metabolic flux, the results are consistent with increased disposal of ammonia from glutamate and other amino acids that are being utilized to compensate for loss of pyruvate and alanine entry into the TCA cycle. Unfortunately, urea, potentially a more direct endpoint for urea cycle activity, could not be measured in these samples because APAP overdose also causes kidney damage, resulting in blood urea concentrations 2-3-fold greater than control values [50]. It would be difficult to disentangle the contributions of the urea cycle and kidney damage to elevated urea levels.
Figure 4.

Combined inhibition of the MPC and ALT2 reduces glutathione synthesis in vivo and in vitro. (A) Basal total glutathione was measured in liver tissue homogenates from untreated male 10–16 weeks old liver-specific MPC2/ALT2 DKO mice. (B) HepG2 cells were treated with 25 μM UK-5099 and 250 μM BCLA or DMSO vehicle (Veh) control for 24 h and total glutathione was measured. (C) HepG2 cells were treated with either vehicle or 500 μM BSO for 24 h and total glutathione was measured. Other HepG2 cells were treated with 500 μM BSO for 24 h followed by vehicle or the combination of 25 μM UK-5099 and 250 μM BCLA for another 24 h and total glutathione was measured in the cells. (D–E) HepG2 cells overexpressing CYP2E1 were co-treated with vehicle, 25 μM UK-5099 and 250 μM BCLA, APAP and vehicle, or APAP and 25 μM UK-5099 and 250 μM BCLA for 48 h. (D) Serum LDH release was measured in the cells. (E) Dead cells were stained with propidium iodide and the cells were imaged by either phase contrast or fluorescence imaging (red fluorescence indicates propidium iodide positivity). Scale bars: 200 μm for 100× magnification, 50 μm for 400×. Comparison of two means was performed by two-tailed t-test. Comparison of three or more means was performed by ANOVA with post-hoc Tukey's test for multiple comparisons. ∗∗∗∗p < 0.0001. In all panels, lines and error bars indicate mean ± SE. Circles show individual data points and sample sizes. Box plots show medians and IQR. For the cell culture studies (B–E), each data point shows the mean of multiple technical replicates from one experiment. Experiments were performed across multiple days.
Table 1.
Targeted plasma metabolomics data.
| Amino acid | WT | DKO | p-value |
|---|---|---|---|
| Arginine | 0.20 ± 0.02 | 0.36 ± 0.04 | 0.003 |
| Citrulline | 1.4 ± 0.1 | 4.4 ± 0.5 | <0.001 |
| Ornithine | 0.21 ± 0.02 | 0.31 ± 0.03 | 0.033 |
No other metabolites were significantly different between genotypes.
Alternatively or in addition to increased ammonia disposal, altered expression of glutathione synthesizing or metabolizing enzymes could explain glutathione depletion in the DKO animals. To address these possibilities, we measured mRNA for glutamate cysteine ligase (Gcl), the rate-limiting enzyme in glutathione synthesis, and for glutathione reductase (Gsr) in liver tissue from WT, MPC2 KO, ALT2 KO, and DKO mice. Importantly, there were no differences in the expression of either between any of the genotypes (Suppl. Fig. 3).
Importantly, post-treatment of cells with UK/BCLA after glutathione depletion with buthionine sulfoximine (BSO) completely prevented glutathione re-synthesis over the next 24 h (Figure 4C). Treatment with 500 μM BSO for 24 h almost fully depleted glutathione. Cells post-treated with Veh control for an additional 24 h after removal of BSO recovered approximately 30% of control glutathione content, but cells post-treated with the combination of 25 μM UK-5099 and 250 μM BCLA instead of vehicle failed to re-synthesize any (Figure 4C). Furthermore, APAP toxicity was dramatically increased in HepG2 cells overexpressing CYP2E1 when APAP was co-administered with UK and BCLA (Figure 4D,E), similar to the DKO mice. Importantly, CYP2E1-overexpressing HepG2 cells were previously validated for the study of APAP hepatotoxicity, with formation of APAP-protein adducts, mitochondrial damage, and cell death occurring similar to primary mouse and human hepatocytes [51], and our preliminary dose–response and time course data were consistent with those earlier results (Suppl. Fig. 5). Collectively, the data presented are consistent with increased conversion of glutamate and other amino acids to α-keto acids to feed the TCA cycle, leading to reduced glutamate availability for glutathione synthesis.
3.5. NAC still protects in MPC2 KO and DKO mice
NAC is currently the standard-of-care treatment for APAP overdose in humans. It is generally believed to protect against APAP hepatotoxicity by providing cysteine for re-synthesis of glutathione which can then scavenge NAPQI and ROS. However, the doses of NAC given to APAP overdose patients (140 mg/kg loading, followed by additional lower doses) greatly exceed those required to replenish glutathione. In addition, it has been reported that similar doses can increase flux through the PDHC in mice [52]. It has therefore been proposed that large doses of NAC like those administered clinically protect against APAP in part by increasing pyruvate oxidation [52,53] – an effect that would require the MPC. To test this hypothesis more directly than in prior work, we examined NAC protection in DKO mice and WT littermate controls. Interestingly, NAC protected equally well in the two genotypes (Figure 5A), despite using the same large dose (318 mg/kg) employed in the prior work [53]. To our surprise, NAC also supported glutathione re-synthesis in the DKO mice similar to the WT animals (Figure 5B). These data suggest that while excess NAC may or may not increase PDHC flux, this is not required for NAC-mediated protection after APAP hepatotoxicity.
Figure 5.

N-acetyl-l-cysteine still protects in MPC2/ALT2 double-knockout mice. Male liver-specific MPC2/ALT2 DKO mice and WT littermates 10–16 weeks old were treated with 300 mg/kg followed by 318 mg/kg NAC 2 h later. Serum and liver tissue were collected at 6 h post-APAP. (A) Serum ALT was measured. (B) Total glutathione was measured in liver tissue homogenates. Means were compared using a one-way ANOVA with post-hoc Tukey's test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Bars represent mean ± SE of n = 5 mice per group for WT Veh and DKO Veh. N = 6 mice per group for WT NAC and DKO NAC.
4. Discussion
There is much interest in the development of MPC-targeted drugs to treat metabolic conditions in humans, including diabetes and liver disease [54,55], with some early-stage clinical trials completed [56] and a phase 3 trial currently underway (ClinicalTrials.gov ID: NCT03970031). Because pyruvate can be converted to glucose through a series of steps that begins with conversion to oxaloacetate within mitochondria, inhibiting mitochondrial pyruvate uptake could reduce hyperglycemia and its downstream adverse effects. Indeed, obesity- and streptozotocin-induced hyperglycemia is attenuated in mice with liver-specific MPC KO and after MPC inhibitor treatment [35,57]. MPC inhibitors also hold promise in treating nonalcoholic steatohepatitis (NASH), as MPC deletion or inhibition protects mice from liver injury and fibrosis in preclinical NASH models [[57], [58], [59]]. However, given the central importance of pyruvate in mitochondrial intermediary metabolism, there are valid concerns regarding the safety of MPC inhibition.
For example, alterations in glutathione synthesis and redox status that have been observed in cell culture studies [32,33] could predispose patients to oxidative insults [55]. The cause of these changes in glutathione during MPC inhibition appears to be an increase in glutaminolysis (conversion of glutamine to α-KG through glutamate). Entry of amino acids, particularly alanine and glutamine/glutamate, into the TCA cycle can compensate for MPC deficiency [35,[60], [61], [62]]. However, increased TCA flux of glutamine and glutamate could also lead to decreased glutamate availability for glutathione synthesis, which could be detrimental in some contexts. Indeed, two recent studies have demonstrated that MPC inhibition leads to impaired glutathione synthesis and recycling of oxidized glutathione in cultured cells [32,33]. Interestingly, however, these effects were not observed in liver tissue from MPC1 KO mice in vivo [32]. Based on our data, the explanation for this discrepancy between cells and mice is likely compensation for loss of MPC activity by ALT2 in the animals. ALT2 was roughly 2-fold elevated in our MPC2 KO mice and there was no evidence of worsened glutathione depletion in those animals after APAP overdose. However, when both MPC2 and ALT2 were absent, hepatic glutathione decreased at baseline compared to WT mice and re-synthesis after APAP-induced depletion was severely impaired. Furthermore, the phenotype we observed in cells treated with both UK-5099 and the ALT inhibitor BCLA was much more severe than in prior studies using the MPC inhibitor alone. Altogether, our results suggest that loss of both the MPC and ALT2 is required to alter glutathione synthesis in hepatocytes in vivo. Thus, MPC inhibitors are likely insufficient to sensitize patients to oxidative insults, except possibly in rare cases of individuals with one of several recessive Gpt2 polymorphisms that drastically reduce enzyme activity [63,64].
Although one might think fatty acid oxidation (FAO) can serve as a source of acetyl-CoA for the TCA cycle and potentially also compensate for MPC loss, FAO is inhibited in APAP toxicity [[65], [66], [67], [68]] resulting in lower ketones (Suppl. Fig. 6) among other effects and there was no difference in serum ketones between DKO and WT mice in the present study (Suppl. Fig. 6). Furthermore, FAO cannot serve as an anaplerotic substrate for the TCA cycle to support anabolic functions like lipogenesis or gluconeogenesis. It is therefore unlikely that FAO can fully compensate for impaired mitochondrial pyruvate and alanine metabolism during APAP-induced injury.
Another novel finding from our study is that inducing expression of ALT2 with a large dose of dexamethasone can reduce APAP-induced liver injury. The mechanism of protection with ALT2 induction could be enhanced glutathione re-synthesis due to the glutamate/glutamine-sparing effect of pyruvate, as similar large doses of other glucocorticoids have been shown to increase hepatic glutathione [69]. Although treatment of liver damage was not the focus of this study and 24 h pre-treatment with Dex is not clinically feasible in the context of acute APAP overdose, this result adds to a growing body of literature indicating that improving pyruvate entry and flux in the TCA cycle is protective against liver injury. For example, it was recently demonstrated that mice deficient in PDK4, which phosphorylates and inhibits the PDHC, had much lower liver injury and oxidative stress after APAP overdose than WT controls [40]. This finding is consistent with the present data showing that a chemical inhibitor of PDK activity, DCA, also reduced liver injury after APAP. Importantly, greater glutathione re-synthesis was observed in the PDK4-deficient mice [40], which is consistent with a glutathione-sparing effect of increased pyruvate oxidation. Another group reported that exogenous pyruvate and ethyl pyruvate are also protective against APAP and NAPQI toxicity in mice and HepG2 cells, respectively [70]. Thus, targeting pyruvate metabolism may be a promising novel approach to treat APAP overdose in patients in combination with NAC.
The primary mechanism by which NAC protects against APAP hepatotoxicity is providing cysteine for the rapid re-synthesis of glutathione [14,71,72]. However, it is often claimed that NAC is beneficial in APAP overdose patients even when administered late after overdose, when APAP bioactivation and ROS production have already occurred [73,74]. There is also modest evidence that NAC is beneficial for patients with non-APAP acute liver damage, presumably unaccompanied by glutathione depletion [75]. An alternative hypothesis to explain how NAC protects in these scenarios is via increased pyruvate oxidation. For example, Zwingmann and Bilodeau demonstrated that NAC increases glucose flux through the PDHC and TCA cycle and protects against oxidative stress even in glutathione-replete mice [52]. The mechanism by which NAC had this effect was unclear from their study but could have been conversion of the excess cysteine to pyruvate. It was later demonstrated that when equimolar doses of NAC and the tripeptide glutathione were administered to mice separately after APAP overdose, glutathione was more effective than NAC at reducing liver injury and it increased TCA cycle flux to a greater degree [53]. Furthermore, increasing the NAC dose 3-fold to provide the same number of amino acids as glutathione equalized their effects [53]. Based on this, it was concluded instead that amino acids derived from NAC are used to synthesize TCA cycle substrates which then enhance TCA cycle flux [53]. This latter hypothesis is consistent with our observation that the same large dose of NAC used in those studies protected equally well in WT and DKO mice that cannot use cytosolic cysteine-derived pyruvate. In addition, the fact that NAC also restored hepatic glutathione in DKO mice is consistent with the possibility that some excess cysteine was instead converted to glutamate, which could then be used in both glutathione re-synthesis and the TCA cycle. However, a definitive mechanism by which NAC protects late after APAP overdose or in glutathione-replete subjects cannot be determined from our data and additional experiments are needed.
Finally, it is notable that ALT activity has been the primary serum biomarker of liver injury since 1955 [76,77] but its metabolic and physiological roles in liver injury and disease are rarely considered, even though it is an important metabolic enzyme within the tissue [77]. Interestingly, it was recently reported that hepatic ALT1 and ALT2 contribute to hyperglycemia in diabetic mice by converting alanine to pyruvate which is then used in gluconeogenesis [34,78], providing possibly the first evidence that ALT2 has pathophysiological significance in liver-related metabolic diseases. The current study adds to our emerging awareness that ALT is more than a passive biomarker of injury by demonstrating its complementary role with the MPC in maintenance of mitochondrial intermediary metabolism and preservation of redox balance.
5. Conclusions
Overall, our data suggest that hepatocytes have metabolic redundancies to preserve TCA cycle flux and defend against oxidative stress even in the context of impaired mitochondrial pyruvate metabolism (see Graphical Abstract), which is an important metabolic substrate of the TCA cycle. However, if all sources of mitochondrial pyruvate are impeded, hepatocytes switch to metabolism of amino acids to maintain flux at the cost of reduced synthesis of the antioxidant glutathione. As the amino acid glutamate is shunted into the TCA cycle, less is available for glutathione synthesis, rendering hepatocytes more susceptible to oxidative insult. Conversely, stimulation of mitochondrial pyruvate flux was protective. These findings have implications for the safety and future development of MPC inhibitors to treat metabolic diseases and for future treatment of APAP hepatotoxicity.
Financial Support
This study was funded in part by a 2018 Pinnacle Research Award from the AASLD Foundation (MRM); the Arkansas Biosciences Institute (MRM), which is the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000; and the National Institutes of Health grants T32 GM106999 (JHV), R01 DK104735 (BNF), R01 DK117657 (BNF), UL1 TR003107 (LPJ), and SB1 DK079387 (LPJ). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any other funding agency.
Author contributions
Conceptualization: McGill, Finck. Data curation: McGill. Formal analysis: McGill. Funding acquisition: McGill, Finck, James. Investigation: Yiew, Vazquez, Kennon-McGill, Price, Allard, Yee, McCommis, McGill. Methodology: McGill, Finck, Yiew, Martino. Project administration: McGill, Finck, Yiew. Resources: McGill, Finck. Software: McGill. Supervision: McGill, Finck. Validation: McGill, Yiew. Visualization: McGill, Finck. Writing – Original draft: McGill, Finck, Yiew. Writing – review & editing: All authors.
Declaration of Competing Interest
LPJ is Chief Medical Officer of Acetaminophen Toxicity Diagnostics (ATD), LLC, which is developing a diagnostic test for APAP overdose. MR McGill is a consultant for ATD and has received research funding from GlaxoSmithKline which sells APAP. BNF is a shareholder and member of the scientific advisory board of Cirius Therapeutics, which is developing an MPC inhibitor to treat nonalcoholic steatohepatitis. The remaining authors declare that they have no conflicts related to the content of this article. These companies had no role whatsoever in the funding, conceptualization, design, execution, or reporting of the study.
Acknowledgements
We thank the Division of Laboratory Animal Medicine (DLAM) at UAMS for expert animal care and Jennifer D. James, BS, HT/ASCP, HTL, QIHC in the Experimental Pathology Core Laboratory for assistance with immunohistochemistry.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2023.101808.
Abbreviations
- ALF
acute liver failure
- ALT
alanine aminotransferase
- APAP
acetaminophen
- Dex
dexamethasone
- DKO
double ALT2/MPC knockout
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- JNK
c-Jun N-terminal kinase
- KO
knockout
- LDH
lactate dehydrogenase
- MPC
mitochondrial pyruvate carrier
- NAPQI
N-acetyl-p-benzoquinone imine
- P450
cytochrome P450
- PDHC
pyruvate dehydrogenase complex
- PDK4
pyruvate dehydrogenase kinase 4
- TCA
tricarboxylic acid
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Kaufman D.W., Kelly J.P., Rosenberg L., Anderson T.E., Mitchell A.A. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. J Am Med Assoc. 2002;287(3):337–344. doi: 10.1001/jama.287.3.337. [DOI] [PubMed] [Google Scholar]
- 2.Vernacchio L., Kelly J.P., Kaufman D.W., Mitchell A.A. Medication use among children <12 years of age in the United States: results from the Slone Survey. Pediatrics. 2009;124(2):446–454. doi: 10.1542/peds.2008-2869. [DOI] [PubMed] [Google Scholar]
- 3.Walsh A., Edwards H., Fraser J. Over-the-counter medication use for childhood fever: a cross-sectional study of Australian parents. J Paediatr Child Health. 2007;43(9):601–606. doi: 10.1111/j.1440-1754.2007.01161.x. [DOI] [PubMed] [Google Scholar]
- 4.Barakat-Haddad C., Siddiqua A. Prévalence et facteurs prédictifs de l’automédication chez les adolescents aux Émirats arabes unis. East Mediterr Health J. 2017;23(11):744–753. doi: 10.26719/2017.23.11.744. [DOI] [PubMed] [Google Scholar]
- 5.Mehuys E., Crombez G., Paemeleire K., Adriaens E., Van Hees T., Demarche S., et al. Self-Medication with over-the-counter analgesics: a survey of patient characteristics and concerns about pain medication. J Pain. 2019;20(2):215–223. doi: 10.1016/j.jpain.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Stravitz R.T., Lee W.M. Acute liver failure. Lancet. 2019:869–881. doi: 10.1016/S0140-6736(19)31894-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGill M.R., Hinson J.A. The development and hepatotoxicity of acetaminophen: reviewing over a century of progress. Drug Metabol Rev. 2020 doi: 10.1080/03602532.2020.1832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramachandran A., Jaeschke H. A mitochondrial journey through acetaminophen hepatotoxicity. Food Chem Toxicol. 2020;140 doi: 10.1016/j.fct.2020.111282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iorga A., Dara L. Cell death in drug-induced liver injury. Adv Pharmacol. 2019;85:31–74. doi: 10.1016/BS.APHA.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell J.R., Jollow D.J., Potter W.Z., Davis D.C., Gillette J.R., Brodie B.B. Acetaminophen induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Therapeut. 1973;187(1):185–194. [PubMed] [Google Scholar]
- 11.Dahlin D.C., Miwa G.T., Lu A.Y.H., Nelson S.D. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Isotopenpraxis. 1984;20(1):1327–1331. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGill M.R., Lebofsky M., Norris H.R.K., Slawson M.H., Bajt M.L., Xie Y., et al. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol. 2013;269(3):240–249. doi: 10.1016/j.taap.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jollow D.J., Mitchell J.R., Potter W.Z., Davis D.C., Gillette J.R., Brodie B.B. Acetaminophen induced hepatic necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp Therapeut. 1973;187(1):195–202. [PubMed] [Google Scholar]
- 14.Mitchell J.R., Jollow D.J., Potter W.Z., Gillette J.R., Brodie B.B. Acetaminophen induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Therapeut. 1973;187(1):211–217. [PubMed] [Google Scholar]
- 15.Meyers L.L., Beierschmitt W.P., Khairallah E.A., Cohen S.D. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol. 1988;93(3):378–387. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- 16.Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Therapeut. 1990;255(3):935–941. [PubMed] [Google Scholar]
- 17.Gunawan B.K., Liu Z.X., Han D., Hanawa N., Gaarde W.A., Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131(1):165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 18.Henderson N.C., Pollock K.J., Frew J., Mackinnon A.C., Flavell R.A., Davis R.J., et al. Critical role of c-jun (NH2) terminal kinase in paracetamol-induced acute liver failure. Gut. 2007;56(7):982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Win S., Than T.A., Min R.W.M., Aghajan M., Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology. 2016;63(6):1987–2003. doi: 10.1002/hep.28486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Win S., Min R.W.M., Chen C.Q., Zhang J., Chen Y., Li M., et al. Expression of mitochondrial membrane-linked SAB determines severity of sex-dependent acute liver injury. J Clin Invest. 2019;129(12):5278–5293. doi: 10.1172/JCI128289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramachandran A., Mcgill M.R., Xie Y., Ni H.M., Ding W.X., Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58(6):2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma M., Gadang V., Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol. 2012;82(5):1001–1007. doi: 10.1124/mol.112.079863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iorga A., Donovan K., Shojaie L., Johnson H., Kwok J., Suda J., et al. Interaction of RIPK1 and A20 modulates MAPK signaling in murine acetaminophen toxicity. J Biol Chem. 2021;296 doi: 10.1016/j.jbc.2021.100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kon K., Kim J.S., Jaeschke H., Lemasters J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40(5):1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 25.Placke M.E., Ginsberg G.L., Wyand D.S., Cohen S.D. Ultrastructural changes during acute acetaminophen-induced hepatotoxicity in the mouse: a time and dose study. Toxicol Pathol. 1987;15(4):431–438. doi: 10.1177/019262338701500407. [DOI] [PubMed] [Google Scholar]
- 26.Bajt M.L., Cover C., Lemasters J.J., Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94(1):217–225. doi: 10.1093/toxsci/kfl077. [DOI] [PubMed] [Google Scholar]
- 27.Gujral J.S., Knight T.R., Farhood A., Bajt M.L., Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci: Off J Soc Toxicol. 2002;67(2):322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- 28.McGill M.R., Sharpe M.R., Williams C.D., Taha M., Curry S.C., Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122(4):1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCommis K.S., Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J. 2015:443–454. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bricker D.K., Taylor E.B., Schell J.C., Orsak T., Boutron A., Chen Y.C., et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;336(6090):96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herzig S., Raemy E., Montessuit S., Veuthey J.L., Zamboni N., Westermann B., et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;336(6090):93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 32.Tompkins S.C., Sheldon R.D., Rauckhorst A.J., Noterman M.F., Solst S.R., Buchanan J.L., et al. Disrupting mitochondrial pyruvate uptake directs glutamine into the TCA cycle away from glutathione synthesis and impairs hepatocellular tumorigenesis. Cell Rep. 2019;28(10):2608–2619.e6. doi: 10.1016/j.celrep.2019.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gansemer E.R., McCommis K.S., Martino M., King-McAlpin A.Q., Potthoff M.J., Finck B.N., et al. NADPH and glutathione redox link TCA cycle activity to endoplasmic reticulum homeostasis. iScience. 2020;23(5) doi: 10.1016/j.isci.2020.101116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martino M.R., Gutiérrez-Aguilar M., Yiew N.K.H., Lutkewitte A.J., Singer J.M., McCommis K.S., et al. Silencing alanine transaminase 2 in diabetic liver attenuates hyperglycemia by reducing gluconeogenesis from amino acids. Cell Rep. 2022;39(4) doi: 10.1016/j.celrep.2022.110733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCommis K.S., Chen Z., Fu X., McDonald W.G., Colca J.R., Kletzien R.F., et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metabol. 2015;22(4):682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du K., Williams C.D., McGill M.R., Jaeschke H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol Appl Pharmacol. 2014;281(1):58–66. doi: 10.1016/j.taap.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vazquez J.H., Kennon-Mcgill S., Byrum S.D., Mackintosh S.G., Jaeschke H., Williams D.K., et al. Proteomics indicates lactate dehydrogenase is prognostic in acetaminophen-induced acute liver failure patients and reveals altered signaling pathways. Toxicol Sci. 2022;187(1):25–34. doi: 10.1093/toxsci/kfac015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z., Fitzgerald R.L., Averna M.R., Schonfeld G. A targeted apolipoprotein B-38.9-producing mutation causes fatty livers in mice due to the reduced ability of apolipoprotein B-38.9 to transport triglycerides. J Biol Chem. 2000;275(42):32807–32815. doi: 10.1074/jbc.M004913200. [DOI] [PubMed] [Google Scholar]
- 39.Clemens M.M., Kennon-McGill S., Apte U., James L.P., Finck B.N., McGill M.R. The inhibitor of glycerol 3-phosphate acyltransferase FSG67 blunts liver regeneration after acetaminophen overdose by altering GSK3β and Wnt/β-catenin signaling. Food Chem Toxicol. 2019;125:279–288. doi: 10.1016/j.fct.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan L., Ramachandran A., Akakpo J.Y., Woolbright B.L., Zhang Y., Jaeschke H. Mice deficient in pyruvate dehydrogenase kinase 4 are protected against acetaminophen-induced hepatotoxicity. Toxicol Appl Pharmacol. 2020;387 doi: 10.1016/j.taap.2019.114849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colca J.R., McDonald W.G., Cavey G.S., Cole S.L., Holewa D.D., Brightwell-Conrad A.S., et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)-Relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013;8(5) doi: 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reagan W.J., Yang R.Z., Park S., Goldstein R., Brees D., Gong D.W. Metabolic adaptive ALT isoenzyme response in livers of C57/BL6 mice treated with dexamethasone. Toxicol Pathol. 2012;40(8):1117–1127. doi: 10.1177/0192623312447550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vazquez J.H., Clemens M.M., Allard F.D., Yee E.U., Kennon-Mcgill S., MacKintosh S.G., et al. Identification of serum biomarkers to distinguish hazardous and benign aminotransferase elevations. Toxicol Sci. 2020;173(2):244–254. doi: 10.1093/toxsci/kfz222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregermen R.I., Christensen H.N. Enzymatic and non-enzymatic dehydrochlorination of β-chloro-l-alanine. J Biol Chem. 1956;220(2):765–774. doi: 10.1016/S0021-9258(18)65302-5. [DOI] [PubMed] [Google Scholar]
- 45.Miyazaki M., Rosenblum J.S., Kasahara Y., Nakagawa I., Patricelli M.P. Determination of enzymatic source of alanine aminotransferase activity in serum from dogs with liver injury. J Pharmacol Toxicol Methods. 2009;60(3):307–315. doi: 10.1016/j.vascn.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 46.Yang R.Z., Park S., Reagan W.J., Goldstein R., Zhong S., Lawton M., et al. Alanine aminotransferase isoenzymes: molecular cloning and quantitative analysis of tissue expression in rats and serum elevation in liver toxicity. Hepatology. 2009;49(2):598–607. doi: 10.1002/hep.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rafter I., Gråberg T., Kotronen A., Strömmer L., Mattson C.M., Kim R.W., et al. Isoform-specific alanine aminotransferase measurement can distinguish hepatic from extrahepatic injury in humans. Int J Mol Med. 2012;30(5):1241–1249. doi: 10.3892/ijmm.2012.1106. [DOI] [PubMed] [Google Scholar]
- 48.McGill M.R., Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharmaceut Res. 2013;30(9) doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cappel D.A., Deja S., Duarte J.A.G., Kucejova B., Iñigo M., Fletcher J.A., et al. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metabol. 2019;29(6):1291–1305.e8. doi: 10.1016/j.cmet.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stern S.T., Bruno M.K., Hennig G.E., Horton R.A., Roberts J.C., Cohen S.D. Contribution of acetaminophen-cysteine to acetaminophen nephrotoxicity in CD-1 mice: I. Enhancement of acetaminophen nephrotoxicity by acetaminophen-cysteine. Toxicol Appl Pharmacol. 2005;202(2):151–159. doi: 10.1016/j.taap.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 51.Bai J., Cederbaum A.I. Adenovirus mediated overexpression of CYP2E1 increases sensitivity of HepG2 cells to acetaminophen induced cytotoxicity. Mol Cell Biochem. 2004;262(1–2):165–176. doi: 10.1023/B:MCBI.0000038232.61760.9e. [DOI] [PubMed] [Google Scholar]
- 52.Zwingmann C., Bilodeau M. Metabolic insights into the hepatoprotective role of N-acetylcysteine in mouse liver. Hepatology. 2006;43(3):454–463. doi: 10.1002/hep.21075. [DOI] [PubMed] [Google Scholar]
- 53.Saito C., Zwingmann C., Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51(1):246–254. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colca J. NASH (nonalcoholic steatohepatitis), diabetes, and macrovascular disease: multiple chronic conditions and a potential treatment at the metabolic root. Expet Opin Invest Drugs. 2020:191–196. doi: 10.1080/13543784.2020.1715940. [DOI] [PubMed] [Google Scholar]
- 55.McCommis K.S., Finck B.N. The hepatic mitochondrial pyruvate carrier as a regulator of systemic metabolism and a therapeutic target for treating metabolic disease. Biomolecules. 2023 doi: 10.3390/biom13020261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harrison S.A., Alkhouri N., Davison B.A., Sanyal A., Edwards C., Colca J.R., et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase IIb study. J Hepatol. 2020;72(4):613–626. doi: 10.1016/j.jhep.2019.10.023. [DOI] [PubMed] [Google Scholar]
- 57.McCommis K.S., Hodges W.T., Brunt E.M., Nalbantoglu I., McDonald W.G., Holley C., et al. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology. 2017;65(5):1543–1556. doi: 10.1002/hep.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rauckhorst A.J., Gray L.R., Sheldon R.D., Fu X., Pewa A.D., Feddersen C.R., et al. The mitochondrial pyruvate carrier mediates high fat diet-induced increases in hepatic TCA cycle capacity. Mol Metabol. 2017;6(11):1468–1479. doi: 10.1016/j.molmet.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hodges W.T., Jarasvaraparn C., Ferguson D., Griffett K., Gill L.E., Chen Y., et al. Mitochondrial pyruvate carrier inhibitors improve metabolic parameters in diet-induced obese mice. J Biol Chem. 2022;298(2) doi: 10.1016/j.jbc.2021.101554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang C., Ko B., Hensley C.T., Jiang L., Wasti A.T., Kim J., et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56(3):414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gray L.R., Sultana M.R., Rauckhorst A.J., Oonthonpan L., Tompkins S.C., Sharma A., et al. Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metabol. 2015;22(4):669–681. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feruson D., Eichler S.J., Yiew N.K.H., Colca J.R., Cho K., Patti G.J., et al. Mitochondrial pyruvate carrier inhibition initiates metabolic crosstalk to stimulate branched chain amino acid catabolism. Mol Metabol. 2023;70 doi: 10.1016/j.molmet.2023.101694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Celis K., Shuldiner S., Haverfield E.V., Cappell J., Yang R., Gong D.W., et al. Loss of function mutation in glutamic pyruvate transaminase 2 (GPT2) causes developmental encephalopathy. J Inherit Metab Dis. 2015;38(5):941–948. doi: 10.1007/s10545-015-9824-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ouyang Q., Nakayama T., Baytas O., Davidson S.M., Yang C., Schmidt M., et al. Mutations in mitochondrial enzyme GPT2 cause metabolic dysfunction and neurological disease with developmental and progressive features. Proc Natl Acad Sci USA. 2016;113(38):E5598–E5607. doi: 10.1073/pnas.1609221113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen C., Krausz K.W., Shah Y.M., Idle J.R., Gonzalez F.J. Serum metabolomics reveals irreversible inhibition of fatty acid β-oxidation through the suppression of PPARα activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem Res Toxicol. 2009;22(4):699–707. doi: 10.1021/tx800464q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhattacharyya S., Pence L., Beger R., Chaudhuri S., McCullough S., Yan K., et al. Acylcarnitine profiles in acetaminophen toxicity in the mouse: comparison to toxicity, metabolism and hepatocyte regeneration. Metabolites. 2013;3(3):606–622. doi: 10.3390/metabo3030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGill M.R., Li F., Sharpe M.R., Williams C.D., Curry S.C., Ma X., et al. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch Toxicol. 2014;88(2):391–401. doi: 10.1007/s00204-013-1118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clemens M.M., Kennon-McGill S., Vazquez J.H., Stephens O.W., Peterson E.A., Johann D.J., et al. Exogenous phosphatidic acid reduces acetaminophen-induced liver injury in mice by activating hepatic interleukin-6 signaling through inter-organ crosstalk. Acta Pharm Sin B. 2021 doi: 10.1016/j.apsb.2021.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Speck R.F., Schranz C., Lauterburg B.H. Prednisolone stimulates hepatic glutathione synthesis in mice. Protection by prednisolone against acetaminophen hepatotoxicity in vivo. J Hepatol. 1993;18(1):62–67. doi: 10.1016/S0168-8278(05)80010-8. [DOI] [PubMed] [Google Scholar]
- 70.Nagatome M., Kondo Y., Kadowaki D., Saishyo Y., Irikura M., Irie T., et al. Ethyl pyruvate attenuates acetaminophen-induced liver injury in mice and prevents cellular injury induced by N-acetyl-p-benzoquinone imine, a toxic metabolite of acetaminophen, in hepatic cell lines. Heliyon. 2018;4(2) doi: 10.1016/j.heliyon.2018.e00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miners J.O., Drew R., Birkett D.J. Mechanism of action of paracetamol protective agents in mice in vivo. Biochem Pharmacol. 1984;33(19):2995–3000. doi: 10.1016/0006-2952(84)90599-9. [DOI] [PubMed] [Google Scholar]
- 72.Corcoran G.B., Wong B.K. Role of glutathione in prevention of acetaminophen-induced hepatotoxicity by N-acetyl-L-cysteine in vivo: studies with N-acetyl-D-cysteine in mice. J Pharmacol Exp Therapeut. 1986;238(1):54–61. [PubMed] [Google Scholar]
- 73.Harrison P.M., Keays R., Bray G.P., Alexander G.J.M., Williams R. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet. 1990;335(8705):1572–1573. doi: 10.1016/0140-6736(90)91388-Q. [DOI] [PubMed] [Google Scholar]
- 74.Keays R., Harrison P.M., Wendon J.A., Forbes A., Gove C., Alexander G.J.M., et al. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: a prospective controlled trial. Br Med J. 1991;303(6809):1026–1029. doi: 10.1136/bmj.303.6809.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chughlay M.F., Kramer N., Spearman C.W., Werfalli M., Cohen K. N-acetylcysteine for non-paracetamol drug-induced liver injury: a systematic review. Br J Clin Pharmacol. 2016:1021–1029. doi: 10.1111/bcp.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karmen A., Wroblewski F., LaDue J.S., Wróblewski F., LaDue J.S. Transaminase activity in human blood. J Clin Invest. 1955;34(1):126–133. doi: 10.1172/JCI103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McGill M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI Journal. 2016;15:817–828. doi: 10.17179/excli2016-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okun J.G., Rusu P.M., Chan A.Y., Wu Y., Yap Y.W., Sharkie T., et al. Liver alanine catabolism promotes skeletal muscle atrophy and hyperglycaemia in type 2 diabetes. Nat Metab. 2021;3(3):394–409. doi: 10.1038/s42255-021-00369-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.