

This cross-sectional study assesses whether synapses containing tau oligomers are excessively eliminated by microglia and astrocytes in brains of individuals with vs without dementia but similar neuropathologic changes at autopsy suggestive of early clinical stages of Alzheimer disease.
Key Points
Question
Are synapses containing tau oligomers excessively eliminated by glia in brains of individuals with dementia at early clinical stages of Alzheimer disease (AD)?
Findings
In this cross-sectional study assessing 40 postmortem brains, individuals with dementia but not those without dementia and identical intermediate Braak III to IV stages at autopsy had significant loss of synapses in the absence of neurofibrillary tangle deposition and increased engulfment of tau oligomer–containing synapses by microglia and astrocytes.
Meaning
These findings suggest a potential key role of microglia, astrocytes, and synaptic tau oligomers in the development of early cognitive impairment in AD.
Abstract
Importance
Factors associated with synapse loss beyond amyloid-β plaques and neurofibrillary tangles may more closely correlate with the emergence of cognitive deficits in Alzheimer disease (AD) and be relevant for early therapeutic intervention.
Objective
To investigate whether accumulation of tau oligomers in synapses is associated with excessive synapse elimination by microglia or astrocytes and with cognitive outcomes (dementia vs no dementia [hereinafter termed resilient]) of individuals with equal burdens of AD neuropathologic changes at autopsy.
Design, Setting, and Participants
This cross-sectional postmortem study included 40 human brains from the Massachusetts Alzheimer Disease Research Center Brain Bank with Braak III to IV stages of tau pathology but divergent antemortem cognition (dementia vs resilient) and cognitively normal controls with negligible AD neuropathologic changes. The visual cortex, a region without tau tangle deposition at Braak III to IV stages, was assessed after expansion microscopy to analyze spatial relationships of synapses with microglia and astrocytes. Participants were matched for age, sex, and apolipoprotein E status. Evidence of Lewy bodies, TDP-43 aggregates, or other lesions different from AD neuropathology were exclusion criteria. Tissue was collected from July 1998 to November 2020, and analyses were conducted from February 1, 2022, through May 31, 2023.
Main Outcomes and Measures
Amyloid-β plaques, tau neuropil thread burden, synapse density, tau oligomers in synapses, and internalization of tau oligomer–tagged synapses by microglia and astrocytes were quantitated. Analyses were performed using 1-way analysis of variance for parametric variables and the Kruskal-Wallis test for nonparametric variables; between-group differences were evaluated with Holm-Šídák tests.
Results
Of 40 included participants (mean [SD] age at death, 88 [8] years; 21 [52%] male), 19 had early-stage dementia with Braak stages III to IV, 13 had resilient brains with similar Braak stages III to IV, and 8 had no dementia (Braak stages 0-II). Brains with dementia but not resilient brains had substantial loss of presynaptic (43%), postsynaptic (33%), and colocalized mature synaptic elements (38%) compared with controls and significantly higher percentages of mature synapses internalized by IBA1-positive microglia (mean [SD], 13.3% [3.9%] in dementia vs 2.6% [1.9%] in resilient vs 0.9% [0.5%] in control; P < .001) and by GFAP-positive astrocytes (mean [SD], 17.2% [10.9%] in dementia vs 3.7% [4.0%] in resilient vs 2.7% [1.8%] in control; P = .001). In brains with dementia but not in resilient brains, tau oligomers more often colocalized with synapses, and the proportions of tau oligomer–containing synapses inside microglia (mean [SD] for presynapses, mean [SD], 7.4% [1.8%] in dementia vs 5.1% [1.9%] resilient vs 3.7% [0.8%] control; P = .006; and for postsynapses 11.6% [3.6%] dementia vs 6.8% [1.3%] resilient vs 7.4% [2.5%] control; P = .001) and astrocytes (mean [SD] for presynapses, 7.0% [2.1%] dementia vs 4.3% [2.2%] resilient vs 4.0% [0.7%] control; P = .001; and for postsynapses, 7.9% [2.2%] dementia vs 5.3% [1.8%] resilient vs 3.0% [1.5%] control; P < .001) were significantly increased compared with controls. Those changes in brains with dementia occurred in the absence of tau tangle deposition in visual cortex.
Conclusion and Relevance
The findings from this cross-sectional study suggest that microglia and astrocytes may excessively engulf synapses in brains of individuals with dementia and that the abnormal presence of tau oligomers in synapses may serve as signals for increased glial-mediated synapse elimination and early loss of brain function in AD.
Introduction
Loss of synapses is one of the earliest hallmarks of neurodegeneration in Alzheimer disease (AD) and the closest correlate of dementia severity,1,2,3 but the underlying mechanisms remain largely unclear. Clinicopathological correlation studies suggest that not everyone with amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain will inevitably develop synaptic and neuronal loss and symptoms of dementia during life4,5,6,7; we refer to this phenomenon as brain resilience to AD neuropathologic changes. Compared with brains from individuals with dementia, resilient brains with equivalent loads of Aβ-plaques and NFTs exhibit preservation of synaptic markers and fewer inflammatory microglial and astrocyte changes.8,9,10,11 Thus, the aberrant response of microglia or astrocytes may be the primary contributor to synaptic loss and clinical disease expression in AD. Neuroinflammation in AD has recently gained considerable interest, supported by the identification of several risk-factor genes expressed in microglia2 and novel insights into microglial-mediated synapse elimination.12,13,14,15,16 Additionally, studies suggest that tau oligomers can disrupt synaptic function17,18,19,20 and closely associate with cognitive deficits in AD,20,21,22,23,24,25,26 and members of our team and other studies have shown that tau oligomers abnormally accumulate in synapses of brains with dementia but not in resilient and control brains.8,27,28 Recent in vitro data also suggest that accumulation of tau oligomers in synapses triggers release of neurotransmitters that cause aberrant glial responses, with glial-mediated internalization of synapses3,29 and subsequent glial dysfunction that results in further accumulation and spreading of toxic tau.30,31,32
These emerging associations between glial phenotypic changes, tau oligomers in synapses, and loss of synaptic integrity in AD favor a disease model beyond Aβ-plaques and NFTs, in which early accumulation of tau oligomers in synapses may serve as a signal for microglia or astrocytes to engulf and eliminate synapses. The temporospatial relationships of the potential drivers of synaptic loss and dementia in AD remain unknown. For years, we lacked techniques with sufficient resolution to allow quantitative detection and study of individual synapses. Here, we took advantage of expansion microscopy (ExM), a developed and well-validated technique that achieves nanoscale imaging of 3-dimensional (3-D) tissue samples through physical magnification by polymer embedding and swelling,33,34,35,36 to evaluate the precise spatial relationships of microglia and astrocytes, tau oligomers, and synaptic elements in an informative cohort of age-matched symptomatic AD and resilient brains at equivalent intermediate stages of tau tangle pathology (Braak stage III-IV). The analysis of the visual cortex (a region not yet affected by classic NFT deposition at those stages) enabled us to address the following 4 key questions: (1) Does loss of synapses precede classic tau tangle appearance? (2) If so, are microglia or astrocytes associated with synaptic engulfment? (3) Are synapses that contain oligomeric tau preferentially internalized and eliminated by glia? (4) Most importantly, is the presence or absence of this tissue injury response associated with whether individuals with brain Aβ-plaques and NFTs manifest clinical symptoms of disease during their lifetime?
Methods
Human Brain Samples
This cross-sectional postmortem study included 40 human brains obtained from the Massachusetts Alzheimer Disease Research Center Brain Bank from July 1998 to November 2020. After a participant’s death, written consent was obtained from the legally authorized representative, in compliance with Massachusetts law. For deaths occurring outside of the hospital, a witnessed telephone conversation was held followed by completion of written consent, in keeping with hospital policy. Autopsies were performed according to standardized protocols,37 and tissue collection and use was approved by the local institutional review boards. Brains were scored by Thal phase for Aβ deposition (range, 0-5, with higher numbers indicating more advanced stages of amyloid deposits),38 Braak stage for NFTs (range, 0–VI, with higher numbers indicating more advanced stages of NFT deposition),39 and the Consortium to Establish a Registry for Alzheimer Disease (CERAD) scale for neuritic plaques (range, A-C, with C indicating frequent plaques),40 and divided into 3 groups. The first group comprised brains collected from 8 persons who were cognitively normal and whose postmortem examination demonstrated Braak stage 0 to II (controls). The second group comprised brains from 13 persons who were cognitively normal but whose postmortem examination demonstrated Braak stage III to IV (hereinafter termed resilient). The third group comprised brains from 19 persons who were cognitively impaired (either with mild cognitive impairment or mild dementia stages) and whose postmortem examination demonstrated Braak stage III to IV (hereinafter termed with dementia). A subset of 5 brains with dementia and 3 resilient brains fulfilled criteria for primary age-related tauopathy (5 brains with dementia and 1 resilient brain were definite primary age-related tauopathy and 2 resilient brains were possible primary age-related tauopathy).41 Cases with evidence of Lewy body pathology, phosphorylated TDP-43 aggregates, or other lesions different from classic AD pathology were excluded. Of 40 patients, 26 (4 controls, 6 resilient, and 16 with dementia) had undergone extensive antemortem cognitive assessments close to death as part of their longitudinal enrollment in the Uniform Data Set of the National Institute on Aging Alzheimer Disease Centers program.42 For the remaining 14 patients, cognitive status was assessed by review of clinical records. The 3 groups were matched for age, sex, and apolipoprotein E status. Resilient brains and brains with dementia were also matched for Thal, Braak, and CERAD scores. Quantitative assessments of Aβ plaques, neuropil threads, and γH2AX burden in the visual cortex (Brodmann areas 17 and 18) were conducted10 (eMethods and eFigure 1 in Supplement 1). Demographic characteristics and cognitive and neuropathologic data are summarized in the Table. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Table. Baseline Demographic, Clinical, and Neuropathologic Characteristics of 40 Human Brains Studied.
| Characteristic | Cognitive status group, median (range) | P valuea | ||
|---|---|---|---|---|
| Control (n = 8) | Resilient (n = 13) | Dementia (n = 19) | ||
| Age, mean (SD), y | 86.9 (7.4) | 86.2 (10.6) | 89.6 (6.3) | .92 |
| Sex, No. (%) | ||||
| Female | 4 (50) | 7 (54) | 8 (42) | .12 |
| Male | 4 (50) | 6 (46) | 11 (58) | |
| Years of education, mean (SD) | 17.5 (1.7) | 14.4 (1.7) | 17.4 (1.9) | .02 |
| Cognitive scoreb | .01 | |||
| MMSE, median (range)c | 29 (28-30) | 29.5 (29-30) | 26.5 (11-30) | <.001 |
| CDR-global, median (range)d | 0.3 (0-0.5) | 0 (0-0.5) | 1.0 (0-3.0) | <.001 |
| CDR-SoB, median (range)e | 0.5 (0-2.0) | 0 (0-0.5) | 6 (1.5-18.0) | .003 |
| WAIS-R subscore, median (range)f | 40.5 (36-48) | 46.5 (39-57) | 33 (18-46) | .006 |
| Boston Naming Test, median (range)g | 29 (28-30) | 27 (25-28) | 23 (8-30) | .009 |
| Trail Making Test, Part A, median (range)h | 35 (32-38) | 30 (26-44) | 46 (30-127) | .007 |
| Verbal fluency, animals, median (range)i | 18 (15-26) | 18 (15-22) | 11 (0-21) | <.001 |
| Verbal fluency, vegetables, median (range)j | 16 (9-22) | 15 (11-19) | 7 (2-12) | .01 |
| ApoE allele status, % | ||||
| ApoE2 allele | 8 | 7 | 13 | .34 |
| ApoE3 allele | 92 | 86 | 79 | |
| ApoE4 allele | 0 | 7 | 8 | |
| Brain weight, mean (SD), g | 1269 (224) | 1247 (162) | 1224 (123) | .14 |
| Neuropathology score | ||||
| Thal phase, median (range)k | 0 (0-4) | 2 (0-4) | 3 (0-5) | .92 |
| Braak stage, median (range)l | 1 (0-2) | 3 (3-4) | 4 (3-4) | .57 |
| CERAD score, median (range)m | 0 | 1 (0-2) | 1 (0-3) | .29 |
| Cerebral vascular composite score, mean (SD)n | 4.1 (1.7) | 2.8 (2) | 5.6 (2.5) | .008 |
| Postmortem interval, mean (SD), h | 23.5 (15.2) | 18 (14.2) | 19.8 (6.7) | .99 |
| Time of last clinical visit prior to death, mean (SD), y | 1.5 (2.2) | 0.6 (0.4) | 0.5 (0.4) | .99 |
| Duration of symptoms, mean (SD), y | NA | NA | 14.7 (6.5) | NA |
Abbreviations: CDR-global, Clinical Dementia Rating global; CDR-SoB: Clinical Dementia Rating-Sum of Boxes; CERAD, Consortium to Establish a Registry for Alzheimer Disease; MMSE: Mini-Mental State Examination; NA, not applicable; WAIS-R, Wechsler Adult Intelligence Scale-Revised.
P values are for differences between brains with dementia and resilient brains.
All antemortem cognitive measures were significantly worse in persons with dementia compared with persons with resilience, which were not significantly different from controls.
MMSE score ranges from 0 to 30, with 30 indicating better cognition.
CDR-global score ranges from 0 to 3, with 0 indicating better cognition.
CDR-SoB score ranges from 0 to 18, with 0 indicating better cognition.
WAIS-R score ranges from 0 to 93, with 93 indicating better cognition.
Boston Naming Test score ranges from 0 to 60, with 60 indicating better cognition.
Trail Making Test A score ranges from 0 to 100, with 0 indicating better cognition.
Verbal fluency (animals) score ranges from 0 to 77, with 77 indicating better cognition.
Verbal fluency (vegetables) score ranges from 0 to 77, with 77 indicating better cognition.
Thal phase includes no amyloid deposition (A0), amyloid in neocortex (A1), amyloid in allocortex and limbic region (A2), amyloid in diencephalon and basal ganglia (A3), amyloid in brainstem and midbrain (A4), and amyloid in cerebellum (A5).
Braak stage, includes Braak stages I and II when neurofibrillary tangle involvement is confined to the transentorhinal and entorhinal regions of the brain, stages III and IV when there is involvement of limbic regions including hippocampus, and stages V and VI when there is extensive neocortical involvement.
CERAD score includes no neuritic plaques (C0), sparse plaques (C1), moderate plaques (C2), and frequent plaques (C3).
Cerebral vascular composite score includes subscores for hypertensive cerebrovascular, atherosclerosis, cerebral atherosclerosis, occlusive atherosclerosis, and cerebral amyloid angiopathy score.
ExM
The use of ExM enables the physical expansion of tissue specimens isotropically in 3 dimensions to 4 to 5 times their original size,33 allowing optical resolution of fine glial processes and individual synapses.33,43 Previous studies have validated the isotropy of the expansion achieved by the ExM protocol at the nanoscale level in multiple tissue types, including brain tissue.44 We applied ExM following previously published protocols33,45,46 with minor modifications (eMethods in Supplement 1) and attained a mean (SD) tissue expansion factor of 4.6 (0.3), in line with prior publications33,45,46 (eFigure 2 in Supplement 1).
Confocal Imaging and 3-D Image Analyses
Expanded tissue sections were imaged using an Olympus FV3000 confocal microscope (resolution, 1024 × 1024 pixels). A z-stack of 0.46 μm was applied to optimize discrimination of true signal (synaptic puncta present in a minimum of 2 consecutive z-stack images) from artifacts.47 For synapse density measures, 3 to 4 fields of view per section were randomly selected in layer II of visual cortex in 2 nonadjacent sections (6-8 fields of view per case). Synapsin 1 and postsynaptic density protein 95 (PSD95) immunostaining were used to identify presynaptic and postsynaptic elements, respectively (Figure 1). For microglia, ionized calcium-binding adaptor protein molecule 1 (Iba1)–positive ameboid-shaped cells48 were selected. For astrocytes, glial fibrillary acidic protein (GFAP)–positive cells with clearly visible bodies and processes were selected. We imaged 2 to 3 Iba1-positive ameboid microglial cells and 3 to 4 GFAP-positive astrocytes per section. Quantifications of tau oligomeric complex 1 (TOC1)–positive tau oligomer–containing synapse densities and tau oligomer–containing synapses colocalized with Iba1-positive microglia or GFAP-positive astrocytes were conducted with Imaris software (BitPlane). Individual layers were created for each presynaptic and postsynaptic element, selecting a diameter of the largest sphere of 0.8 μm and a seed point diameter of 0.5 μm. To assess colocalizing presynaptic and postsynaptic “mature” puncta, presynaptic and postsynaptic layers were masked, and the setting voxels outside the surface distance were set to 0 μm. For colocalization analyses of synaptic puncta with microglia and astrocytes, cell bodies and processes were reconstructed in 3 dimensions, and distances from puncta to the cell surface were calculated; only negative distance values were considered puncta “inside” a glial cell. The numbers of puncta internalized by each microglia and astrocyte were normalized to individual cell volume and total number of puncta in each section.
Figure 1. Synapse Densities Across Groups and Correlation Analyses Between Synapse Densities and Cognitive Measures.
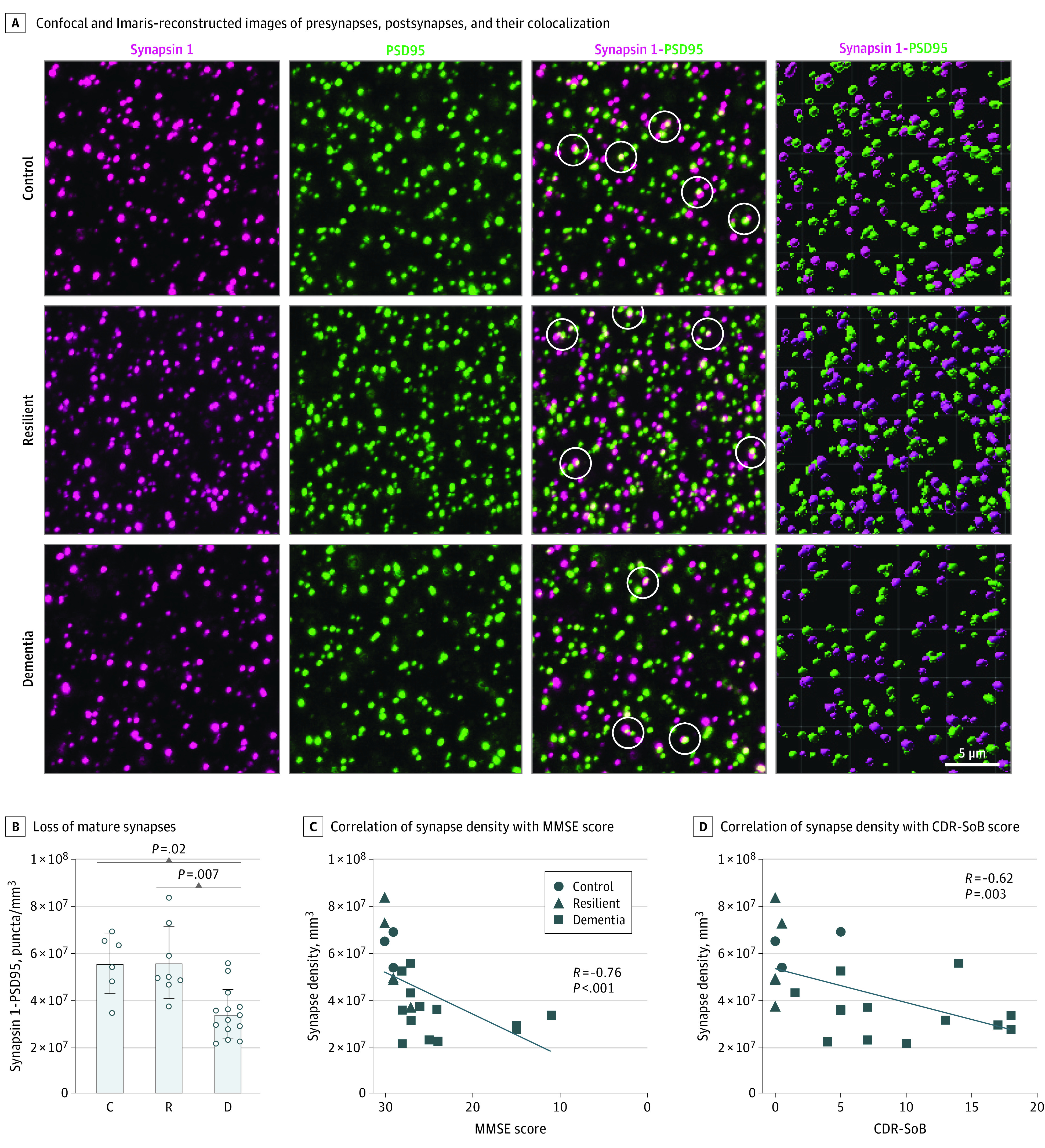
A, Representative images of synapsin 1–positive presynapses, postsynaptic density protein–positive postsynapses, and their colocalization after expansion microscopy (ExM) with confocal imaging in the first 3 columns and in Imaris 3-dimensional reconstructed images in the fourth column. White circles in the third column indicate colocalization. Loss of mature synapses (B) is significantly correlated with antemortem Mini-Mental State Examination (MMSE) score (C) and Clinical Dementia Rating-Sum of Boxes (CDR-SoB) score (D) in the visual cortex. Analyses shown were performed on 28 brains (6-8 fields of view per case). Synapse densities in quantification plots correspond to values obtained in expanded tissue sections and must be multiplied by approximately 100 (4.63) to account for the volume expansion factor of 4.6 achieved by the ExM protocol to extrapolate to pre-expanded tissue material. Control (indicated by C in panel B) comprised 6 brains with Braak stages 0 to II; resilient (R), 8 brains with Braak stages III to IV; dementia (indicated by D in panel B), 14 brains with Braak stages III to IV.
Western Blot Analysis of Synaptosome and Cytosol-Enriched Preparations
Synaptosome and cytosol-enriched fractions were prepared from frozen blocks containing the visual cortex following previously published protocols8,49,50 with minor modifications (eMethods in Supplement 1). Western blot analyses were conducted using reducing or denaturing and native conditions following previously published protocols51,52 with minor modifications (eMethods in Supplement 1). The list of antibodies used for immunohistochemistry and Western blot assays is in eTable 1 in Supplement 1.
Statistical Analysis
The D’Agostino-Pearson normality test was applied to test for Gaussian distribution. Multiple group analyses were performed using 1-way analysis of variance for parametric variables and the Kruskal-Wallis test for nonparametric variables. Post hoc analyses to assess for between-group differences were evaluated with the Holm-Šídák test. Correlation analyses were performed with Pearson tests when both variables were normally distributed, and with Spearman tests when at least 1 variable was not normally distributed. A 2-sided significance level was set at P < .05.
All statistical analyses and graphs were generated using GraphPad Prism, version 9.4.1 (GraphPad Software Inc). Data are presented as means and SDs for normally distributed variables, and medians and ranges for not normally distributed variables, as indicated. When applicable, CIs are provided. Analyses were conducted from February 1, 2022, through May 31, 2023.
Results
This cross-sectional study included 40 patients with AD whose mean (SD) age at death was 88 (8) years. Among them, 19 (48%) were female, and 21 (52%) were male (Table).
Timing of Synapse Loss
Detailed quantification procedures for assessing Aβ plaque and NFT loads across multiple regions in brains with dementia and resilient brains included here were previously published elsewhere.10 Burdens of Aβ plaque deposits, defined as the percentage of cortex occupied by Aβ plaques labeled by the 4G8 antibody (mean [SD], 1.5% [1.7%] vs 2.9% [3.7%]; P = .60) and neuropil threads, defined as neurites labeled by the AT8 antibody (mean [SD], 0.004% [0.008%] vs 0.003% [0.0025%]; P = .50) in the visual cortex did not significantly differ between brains with dementia and resilient brains (eFigure 1 in Supplement 1). As expected at Braak III to IV stages, no NFTs were present in the visual cortex of either resilient brains or brains with dementia. Brains with dementia showed a higher vascular composite score than resilient and control brains (Table). In agreement with previous results,10 the mean [SD] number of γH2AX-positive cells per square millimeter was significantly increased in brains with dementia (508 [461]) compared with resilient (168 [165]) and control brains (76 [87]; P = .001) (eFigure 1 in Supplement 1). Synapsin 1–positive presynaptic densities (by 43%), PSD95-positive postsynaptic densities (by 33%), and synapsin 1–positive and PSD95-positive densities (by 38%) were substantially decreased in brains with dementia compared with resilient and control brains (Figure 1 and eFigure 3 in Supplement 1). Similar differences were found when analyses were limited to the smaller subset of individuals with primary age-related tauopathy, indicating that early loss of synapses in the visual cortex of brains with dementia occurs regardless of presence or absence of Aβ plaques in the absence of NFT deposition. The studied brains showed a mean (SD) density of mature synapses of 5.6 × 107 (1.5 × 107) for resilient brains and 3.4 × 107 (1.05 × 107) for brains with dementia, whereas mean (SD) synapse densities in the subset of primary age-related tauopathy cases were comparable with synapse densities of 5.2 × 107 (6.4 × 106) for resilient primary age-related tauopathy, 3.6 × 107 (1.0 × 107) for primary age-related tauopathy cases with dementia, and 5.5 × 109/mm3 for controls. In agreement with prior studies, colocalized mature puncta in control brains represented approximately 65% of all puncta.53,54 Significant correlations were detected between mature puncta densities and antemortem Clinical Dementia Rating-Sum of Boxes (R = −0.62; P = .003), Mini-Mental State Examination score (R = −0.76; P < .001), Wechsler Adult Intelligence Scale-Revised scores (R = −0.85; P = .003), and number of γH2AX-positive cells (R = −0.5; P = .009) (Figure 1 and eFigure 3 in Supplement 1). Brains were well-matched for postmortem intervals (Table). No correlation was found between postmortem intervals and any of the variables studied, including synapse densities (R, 0.19 [95% CI, −0.23 to 0.55]; P = .36), 4G8 burden (R, 0.10 [95% CI, −0.32 to 0.5]; P = .63), AT8+ neuropil (R, 0.04 [95% CI, −0.37 to 0.44]; P = .84), p-tau181 (R, 0.32 [95% CI, −0.3 to 0.75]; P = .28), TOC1 (R, −0.10 [95% CI, −0.64 to 0.47]; P = .69), and GFAP (R, −0.16 [95% CI, −0.55 to 0.3]; P = .48).
These results reinforce the association between loss of synapses and decline in cognition at early AD clinical stages and point to the different tissue response (eg, loss vs preservation of synapses) between brains with dementia and resilient brains as the most likely anatomical basis for their widely divergent clinical phenotypes in the setting of equivalent burdens of Aβ plaques and NFTs.
Microglia and Astrocyte Engulfment of Synapses
Detailed analyses of proinflammatory and homeostatic markers of microglial cells and astrocytes in brains with dementia and resilient brains included here are published elsewhere.10 Quantification of synaptic puncta inside Iba1-positive ameboid microglia (mean [SD] for presynaptic puncta, 7.7% [2.8%] vs 1.7% [1.2%] vs 1.0% [0.7%]; P < .001; mean [SD] for postsynaptic puncta, 8.5% [0.7%] vs 1.9% [0.4%] vs 0.9% [0.2%]; P < .001]; mean [SD] for mature puncta, 13.3% [3.9%] vs 2.6% [1.9%] vs 0.9% [0.5%]; P < .001) and GFAP-positive astrocytes (mean [SD] for presynaptic puncta, 11.2% [6.4%] vs 2.8% [1.5%] vs 2.2% [1.9%]; P = .001; mean [SD] for postsynaptic puncta, 13.3% [7.6%] vs 1.8% [0.7%] vs 2.7% [2.8%]; P = .001; mean [SD] for mature puncta 17.2% [10.9%] vs 3.7% [4.0%] vs 2.7% [1.8%]; P = .001) indicated a significantly higher proportion of internalized synapsin 1–positive presynaptic, PSD95-positive postsynaptic, and synapsin 1–positive and PSD95-positive mature puncta in both microglia and astrocytes of brains with dementia compared with resilient and control brains (Figure 2 and eFigure 4 in Supplement 1).
Figure 2. Analyses of Engulfment of Synaptic Elements by Microglia and Astrocytes.
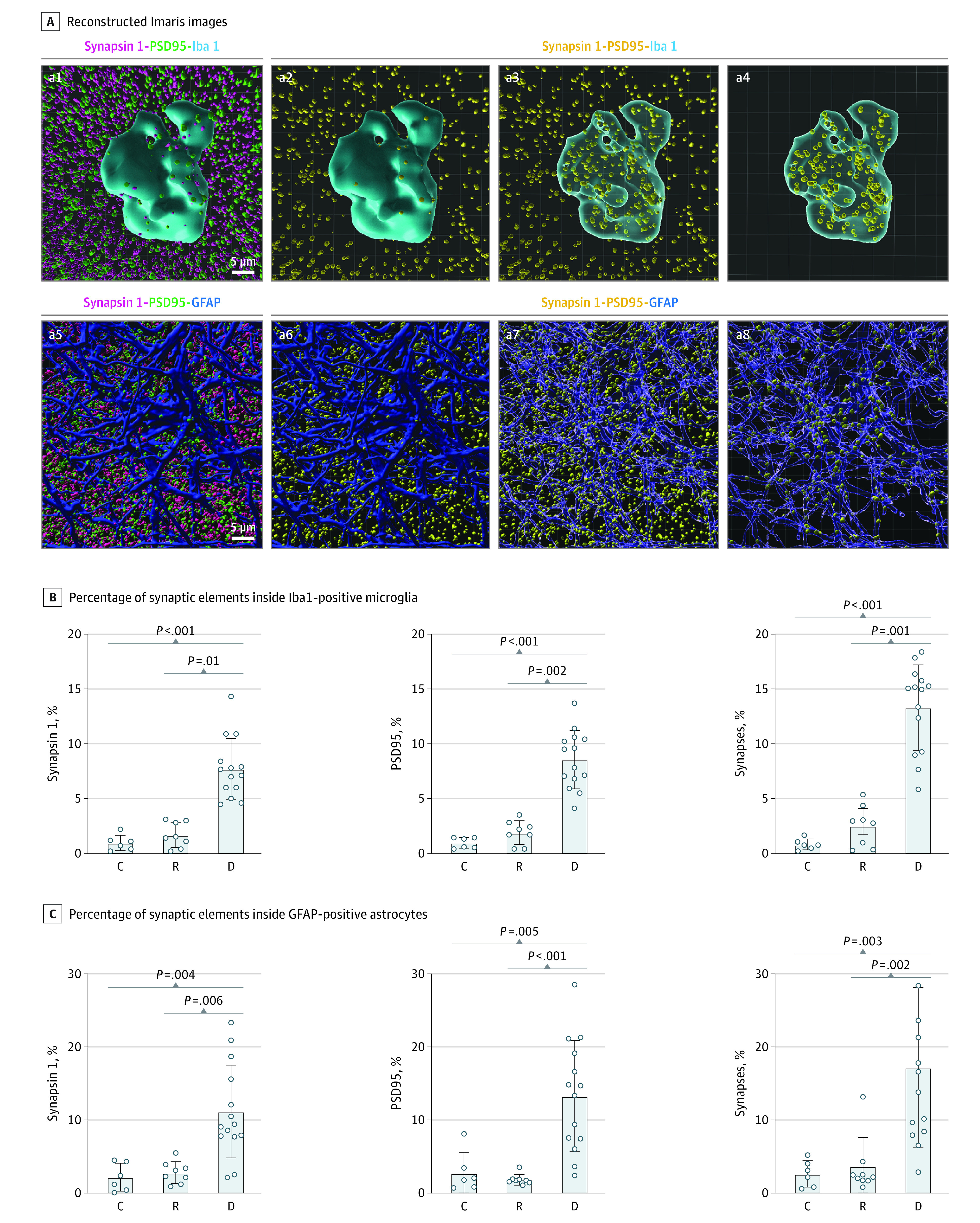
Representative Imaris 3-dimensional image reconstructions (A) showing internalized synaptic elements inside Iba1-positive ameboid microglial cells and glial fibrillary acidic protein (GFAP)–positive astrocytes, and quantification of engulfed synaptic elements inside Iba1-positive microglia (B) and GFAP-positive astrocytes (C). Reconstructed 3-dimensional Imaris images (A) showing glial cells in blue, synapsin 1–positive presynapses in magenta (a1, a5), postsynaptic density protein 95 (PSD95)–positive postsynapses in green (a1, a5), and colocalized synapsin 1–positive and PSD95-positive puncta in yellow (a2-a4, a6-a8), displaying colocalized synapses in yellow inside and outside of a microglial cell (a3) and an astrocyte (a7), and colocalized synapses in yellow only inside the microglia (a4) and astrocyte (a8). C indicates control; D, dementia; and R, resilient.
To rule out artifactual variations of the expansion factor within single astrocyte and microglial cells, we used the pan-astrocytic cytosolic marker aldehyde dehydrogenase 1 family member L1 (ALDH1L1) combined with GFAP, and the lysosomal marker lysosome-associated membrane protein 2 (LAMP2) combined with PSD95. We found that ALDH1L1 labeled the cytosol of GFAP-positive and GFAP-negative astrocytes (eFigure 5 in Supplement 1). Double immunostaining with LAMP2 and either GFAP or IBA1 antibodies convincingly demonstrated the colocalization of engulfed synaptic puncta and lysosomes in the cytoplasm of GFAP-positive astrocytes and Iba1-positive microglial cells (eFigure 6 in Supplement 1).
These results demonstrate that not only microglia but also astrocytes are capable of engulfing synapses in the human brain, and that glia-mediated excessive internalization of synapses occurs in the absence of overt NFT deposition and may be associated with early synaptic brain function loss. The reduced microglial- and astrocyte-mediated engulfment of synapses in resilient brains may be associated with the preserved cognition of these individuals.
Tau Hyperphosphorylation and Synaptic Accumulation of Tau Oligomers
We assessed early tau hyperphosphorylation sites (AT270/pTau [Thr181], pTau217 [Thr217], and AT180/pTau [Thr231]), and levels of total tau (Tau5), N-terminal tau (Tau12), and C-terminal tau (Tau46) by Western blot analyses of synaptosome-enriched fractions. Levels of pTau181 (ratio of AT270/Tau5) were significantly increased in synaptosomes derived from brains with dementia (median [range], 0.27 [0.14-0.40]) compared with resilient (median [range], 0.06 [0.007-0.10]) and control (median [range], 0.18 [0-0.50]; P = .02) brains (Figure 3 and eFigure 7 in Supplement 1). Tau oligomers were measured in whole-tissue homogenates and synaptosome-enriched fractions using the well-characterized antibody TOC1.55,56 Signal intensity levels of TOC1 in total homogenates did not significantly differ among the 3 groups, but brains with dementia contained a significantly higher TOC1 intensity (median [range], 96 147 [82 963-109 330] relative fluorescence units [RFUs]) in synapses compared with resilient (median [range], 50 834 [31 077-70 591] RFU) and control (median [range], 58 112 [36 361-79 862] RFU; P < .001) brains (Figure 3).
Figure 3. Measurement Oligomeric and Hyperphosphorylated Tau Species in Total Brain Homogenates and in Synaptosome Fractions.

Synaptosomes derived from brains with dementia show a significant increase in tau oligomeric complex 1 (TOC1)–positive tau oligomers and of hyperphosphorylated AT270 phosphorylated (p)Thr181–positive tau compared with resilient and control brains. Western blot (WB) analyses of TOC1 plus tau oligomers in total brain tissue homogenates (A) and synaptosome extractions (B) were conducted by quantifying the signal intensity of the full lane labeled oligomers for each case in the native TOC1 WB. The WB analyses of synaptosome-enriched fractions from the visual cortex (B-D) total tau, as measured with the middle domain total tau antibody (Tau5), did not differ across dementia, resilient, and control brains (D). Analyses were performed for 31 total homogenates (6 controls, 10 resilient, 15 dementia) and 21 synaptosome extractions (4 controls, 5 resilient, 12 dementia). Black arrowheads indicate the bandwidth quantified for each WB analysis and respective antibodies. C indicates control brain (Braak stage 0-II); D, dementia brain (Braak stage III-IV); and R, resilient brain (Braak stage III-IV); RFU, relative fluorescence unit.
These data suggest that tau hyperphosphorylation at Thr181 and accumulation of TOC1 plus tau oligomers in synapses may be early key pathological modifications of tau associated with the different fates of synapses and cognitive outcomes of individuals with dementia vs resilience at Braak III to IV stages.
Tau Oligomers in Presynaptic and Postsynaptic Compartments
Significant increases in TOC1-positive tau oligomers colocalized with bassoon-positive presynaptic puncta (mean [SD], 49.9% [5.5%] vs 28.2% [6.6%] vs 18.7% [2.3%]; P < .001) and with PSD95-positive postsynaptic puncta (mean [SD], 32.5% [6.7%] vs 17.3 [2.8%] vs 14.5% [3.1%]; P < .001) were found in brains with dementia compared with resilient and control brains (eFigure 8 in Supplement 1). This finding agrees with the aforementioned data showing significantly higher levels of TOC1-positive tau oligomers in synaptosome-enriched fractions derived from brains with dementia compared with resilient and control brains. The TOC1-positive tau oligomers in synapses (but not 4G8-positive plaques or AT8-positive neuropil burdens) negatively correlated with Mini-Mental State Examination scores (R = −0.58; 95% CI, −0.10 to −0.85; P = .03).
Synapses Containing Tau Oligomers Engulfed by Microglia and Astrocytes
We quantified the proportion of internalized TOC1-positive and bassoon-positive presynaptic puncta and TOC1-positive and PSD95-positive postsynaptic puncta by Iba1-positive ameboid microglia and GFAP-positive astrocytes in a subset of 10 representative cases (4 brains with dementia; 4 resilient brains matched for Aβ plaque, neuropil thread, and vascular burden; and 2 control brains; eTable 2 in Supplement 2). We found a significantly higher proportion of internalized tau oligomer–labeled presynaptic and postsynaptic puncta in both Iba1-positive ameboid microglia (dementia vs resilient vs control: 7.4% [1.8%] vs 5.1% [1.9%] vs 3.7% [0.8%]; P = .006 for internalized TOC1-positive and bassoon-positive puncta; and 11.6% [3.6%] vs 6.8% [1.3%] vs 7.4% [2.5%]; P = .001 for internalized TOC1-positive and PSD95-positive puncta) and GFAP-positive astrocytes (dementia vs resilient vs control: 7.0% [2.1%] vs 4.3% [2.6%] vs 4.0% [0.7%]; P = .001 for internalized TOC1-positive and bassoon-positive puncta; 7.9% [2.2%] vs 5.3% [1.8%] vs 3.0% [1.5%]; P = .001 for internalized TOC1-positive and PSD95-positive puncta) in brains with dementia compared with resilient brains (Figure 4). Internalized tau oligomer–containing synaptic puncta represented a large percentage of total puncta engulfed by microglia and astrocytes in brains with dementia (91% of presynaptic and 93% of postsynaptic puncta in Iba1-positive ameboid microglia, and 63% of presynaptic puncta and 60% of postsynaptic puncta in GFAP-positive astrocytes), indicating that tau oligomer–containing synaptic elements were preferentially engulfed in those brains.
Figure 4. Glial-Mediated Engulfment of Tau Oligomer–Tagged Synapses.
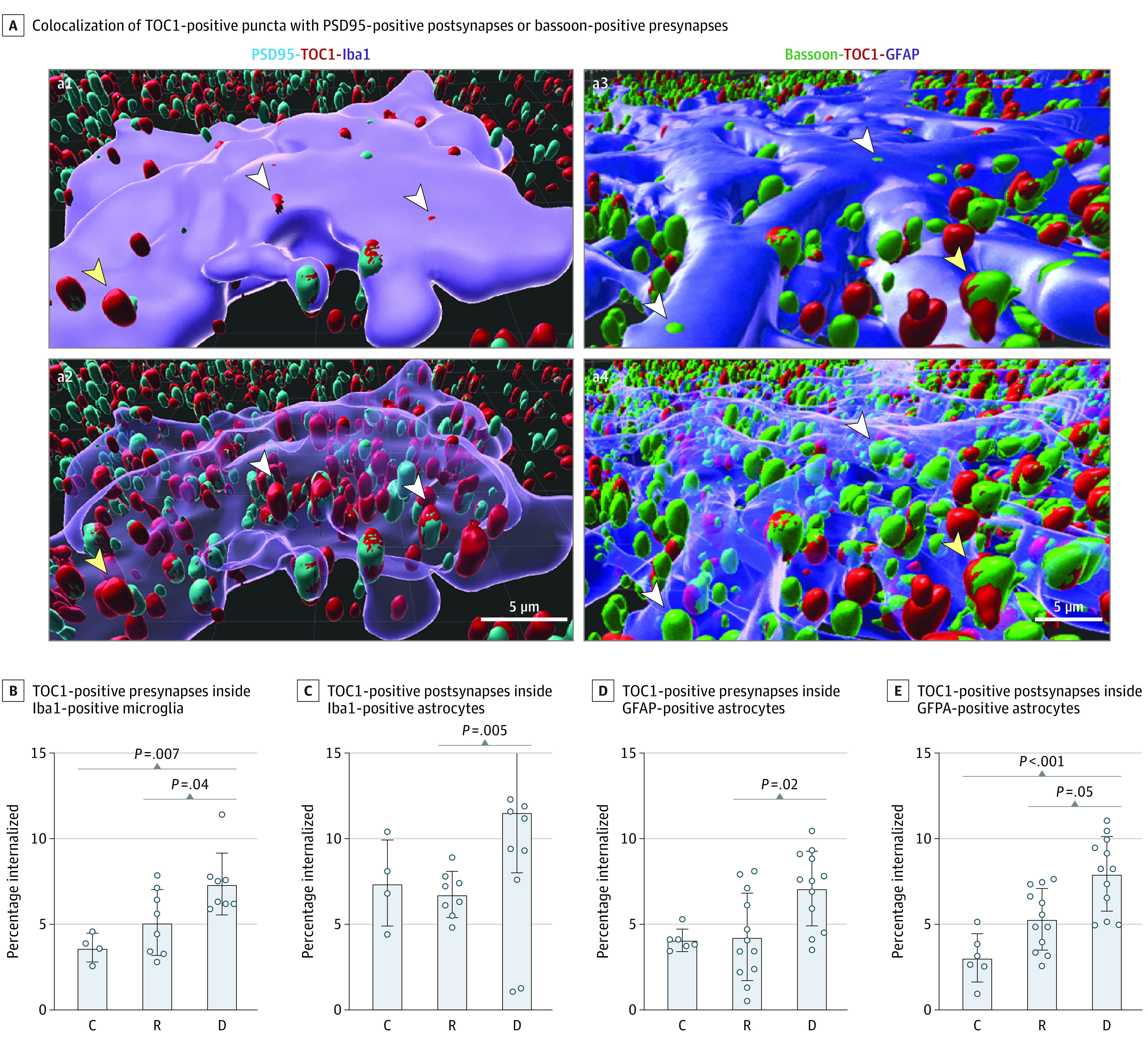
A, Representative 3-dimensional Imaris reconstructed images of postsynaptic density 95 (PSD95)–positive with tau oligomeric complex 1 (TOC1)–positive synapses inside an ionized calcium-binding adaptor protein molecule 1 (Iba1)–positive ameboid microglia before (a1) and after (a2) making the cell body transparent, and bassoon-positive with TOC1-positive synapses inside a glial fibrillary acidic protein (GFAP)–positive astrocyte before (a3) and after (a4) making the cell body transparent. White arrowheads indicate engulfed; yellow arrowheads, not engulfed. Assessments of percentages of internalized TOC1-positive presynapses (B) and TOC1-positive postsynapses (C) inside Iba1-positive ameboid microglia and GFAP-positive astrocytes (D and E). Analyses were performed for 20 ionized calcium-binding adaptor molecule 1–positive and 30 GFAP-positive cells from 10 brains (2 controls, 4 resilient, 4 dementia). C indicates control (Braak stage 0-II); D, dementia (Braak stage III-IV); and R, resilient (Braak stage III-IV).
Discussion
This cross-sectional study assessed an informative cohort of human brains carefully matched for intermediate (Braak III-IV) stages of tau pathology at autopsy but widely diverging antemortem cognitive statuses (dementia vs resilience). Most of the individuals with dementia had mild cognitive impairment or mild dementia (Table), giving us the opportunity to identify brain changes that, beyond Aβ plaques and NFTs, could be more closely associated with cognition at these early disease stages. We found that synapse densities in the visual cortex (a brain region not yet affected by NFTs in Braak III-IV stages) were already significantly reduced in brains with dementia, and were correlated with markers of early cellular damage (γH2AX) and antemortem cognitive scores. Moreover, we found that synapses were excessively internalized by both microglia and astrocytes in brains with dementia compared with resilient brains, and that early aberrant accumulation of TOC1-positive tau oligomers in synapses was associated with engulfment of synapses by microglia and astrocytes. To our knowledge, this is one of the first studies to report evidence of astrocyte engulfment of synapses in human brains, and to suggest that tau oligomers may be associated with glia-mediated synapse elimination in early AD.
Several in vitro and in vivo studies mimicking AD have shown that microglia and astrocytes can engulf synapses.13,57,58,59,60 Emerging human studies also suggest that microglia may play a role in the excessive elimination of presynaptic elements at high Braak AD stages.14,16 However, the potential involvement of astrocytes in synapse elimination in human AD has not yet been explored. Astrocytes are significantly more abundant than microglial cells, and most synapses are in close contact with astrocytes.61,62 Thus, the potential contribution of astrocytes to early synaptic loss in AD could be even greater than that of microglia. To assess individual synaptic elements and overcome the optical resolution limitations of conventional imaging techniques, we used ExM.33,45,46 Prior work44,45,46 with this emerging method has demonstrated that it provides isotropic expansion with maintenance of structural relationship, which allows for the use of light microscopy to reveal spatial relationships that were not otherwise directly observable and are critical when examining the interplay of glial cells with synapses. By using established methods for ExM, we obtained a mean (SD) expansion factor of 4.6 (0.3), which allowed us to attain an effective resolution of 25 to 30 nm, sufficient to study individual synaptic elements, tau oligomers, and their spatial relationships with microglia and astrocytes using confocal imaging.
We assessed synapse densities in layer II of the visual cortex, an integrating cortical layer for primary and higher-order visual information that becomes consistently affected in AD by Aβ plaques and tau tangle deposition when pathology progresses to higher Braak stages (V-VI)63,64,65,66 and evaluated synapse colocalization with Iba1-positive ameboid-shaped microglia and GFAP-positive astrocytes. Morphologic subtypes of microglia expressing similar phenotypic markers can display distinct neurobiological behaviors,67 and ameboid-shaped Iba1-positive cells are presumably the more neurotoxic microglial subset.68,69 Synapses were labeled using synapsin 1, a protein ubiquitously present on the surface of presynaptic vesicles,70 and PSD95, a protein found in the postsynaptic density of excitatory neurons.71 This synaptic marker combination labels 80% to 90% of all synapses of the human brain.72,73 We sampled a minimum of 6 fields of view in each brain to account for interindividual variability and the possibility of increased synapse loss in the vicinity of Aβ plaques.74 Our results showed that brains with dementia but not resilient brains had a significant loss of presynaptic and postsynaptic elements and colocalized mature synapses—presumably required for neuronal signal transmission75—in the visual cortex. Our colocalized synapse measures (approximately 5 × 109/mm3 in controls after accounting for the 4.6 volume expansion factor; Figure 1) were comparable with previously published work,72,76,77,78 including the use of electron microscopy79 and synaptic biomarker measurements in early AD dementia stages.80,81 The overall higher synapse densities (approximately 5 times) observed here when compared with initial studies using electron microscopy1,82 is likely attributable to the recently described “decrowding” phenomenon of ExM and may account for a more accurate estimate of the total synapse densities in the human brain.45 Of note, synapse densities in the subset of 8 brains with primary age-related tauopathy were equivalent to those found in Aβ plaque–containing brains, suggesting a common underlying mechanisms responsible for synaptic loss in AD and primary age-related tauopathy unrelated to Aβ.
We observed a significant increase in microglia- and astrocyte-engulfed synaptic elements, including mature synapses, in brains with dementia compared with resilient brains. We stringently considered only fully internalized synaptic elements as “engulfed synapses” to unequivocally exclude the likely physiologic contacts between glial cells and synaptic elements. The excessive internalization and elimination of mature and presumably still functioning synapses by microglia and astrocytes strongly suggests that aberrant glial responses are likely associated with synaptic loss and loss of brain function in dementia. Contrary to prior hypotheses,59 we did not observe an accumulation of “single” presynapses or postsynapses in brains with dementia but rather an overall loss of these likely “nonfunctional” synaptic elements, favoring the idea that the pathogenic role of glia in early disease stages may primarily involve active and excessive engulfment of synapses rather than defective elimination of dysfunctional synaptic elements.
Growing evidence suggests that tau oligomers may be earlier and better determinants of cognitive decline in AD than NFTs.83 Human brain studies have demonstrated that tau oligomers are increased in synapses of brains with dementia8,17,28,84,85,86 and that synaptic oligomers are particularly synaptotoxic and correlate with cognition in mouse models of tauopathy.20,87 Here, we investigated the possibility that accumulation of tau oligomers in synapses was associated with the enhanced elimination of synapses by microglia and astrocytes in human brains. In agreement with a recent study,17 we found that TOC1-positive tau oligomers were significantly increased in both presynapses and postsynapses in brains with dementia, and we observed the novel finding that tau oligomer–containing synapses were preferentially engulfed by microglia and astrocytes in those brains. This finding could theoretically induce a self-perpetuating cycle of glial-mediated elimination of tau oligomer–containing synapses, chronic glial cell dysfunction, enhanced glial accumulation of tau oligomers, and subsequent release or propagation of toxic oligomers to new synapses associated with the slowly progressive dementia syndrome characteristic of AD. A negligible accumulation of tau oligomers in the synapses of resilient brains was associated with suppressed glial inflammatory responses and anatomic preservation of synapses, and thus is likely associated with preserved cognition.
Limitations
Autopsy studies do not allow for drawing conclusions on specific mechanisms. We cannot rule out that tau oligomers directly damage synapses, and that glial engulfment of oligomer–containing synapses may be a mechanism to remove dysfunctional synaptic elements. Future studies are needed to better understand the specific pathways and molecular mechanisms that drive the interactions between synaptic tau oligomers and glial cells and their temporal relationship with brain function. Although brains were carefully matched for other coincident neuropathologic changes, brains with dementia showed a higher vascular composite score than resilient and control brains; thus, we cannot exclude with certainty a potential deleterious association with vascular factors on synapse function or glia-mediated synapse elimination. Our study included brains from individuals who died at a relatively advanced age (mean age at death, 88 years), which may limit generalizability of the results to younger ages.
Conclusions
Findings from this cross-sectional study suggest that synaptic loss in AD may be primarily associated with aberrant engulfment of synaptic elements by microglia and astrocytes, rather than with Aβ plaques or NFTs, and that abnormal accrual of tau oligomers in synapses may be a key signal targeting those synapses for elimination, leading to loss of brain function. These observations may be relevant for the development of in vivo biomarkers that may more accurately determine the future of asymptomatic individuals with Aβ plaques and NFTs in their brains and guide novel interventions that mimic resilient brains to prevent accumulation of tau oligomers in synapses and halt neurodegeneration (eg, loss of synapses) and clinical symptoms of dementia.
eMethods.
eReferences.
eTable 1. Antibodies Used in the Present Study
eTable 2. Baseline Demographic and Neuropathologic Characteristics of the Subset of N = 10 Human Brains Studied
eFigure 1. Neuropathologic Assessment of Amyloid-β and Tau Burdens in the Studied Brains
eFigure 2. Steps of Expansion Microscopy (ExM) Exemplified
eFigure 3. Synapse Densities Across Groups and Correlation Analyses Between Synapse Densities and Cognitive and Neuropathological Measures
eFigure 4. Confocal Images of Engulfment of Synaptic Elements by Microglia and Astrocytes
eFigure 5. Expanded Astrocyte Co-Stained With a Cytoskeletal and a Cytoplasmic Antibody
eFigure 6. Lysosomal Co-Staining of Engulfed Synaptic Elements in Representative Microglia and Astrocyte
eFigure 7. Quality of Extracted Synaptosomes and Measures of Truncated Tau Species in Synaptosome Fractions
eFigure 8. Association of Oligomeric Tau With Synaptic Elements
Data Sharing Statement
References
- 1.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457-464. doi: 10.1002/ana.410270502 [DOI] [PubMed] [Google Scholar]
- 2.Henstridge CM, Tzioras M, Paolicelli RC. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front Cell Neurosci. 2019;13:63. doi: 10.3389/fncel.2019.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogels T, Murgoci AN, Hromádka T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol Commun. 2019;7(1):109. doi: 10.1186/s40478-019-0754-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crystal H, Dickson D, Fuld P, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38(11):1682-1687. doi: 10.1212/WNL.38.11.1682 [DOI] [PubMed] [Google Scholar]
- 5.Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23(2):138-144. doi: 10.1002/ana.410230206 [DOI] [PubMed] [Google Scholar]
- 6.Gelber RP, Launer LJ, White LR. The Honolulu-Asia Aging Study: epidemiologic and neuropathologic research on cognitive impairment. Curr Alzheimer Res. 2012;9(6):664-672. doi: 10.2174/156720512801322618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snowdon DA; Nun Study . Healthy aging and dementia: findings from the Nun Study. Ann Intern Med. 2003;139(5, pt 2):450-454. doi: 10.7326/0003-4819-139-5_Part_2-200309021-00014 [DOI] [PubMed] [Google Scholar]
- 8.Perez-Nievas BG, Stein TD, Tai HC, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain. 2013;136(Pt 8):2510-2526. doi: 10.1093/brain/awt171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barroeta-Espar I, Weinstock LD, Perez-Nievas BG, et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol Dis. 2019;121:327-337. doi: 10.1016/j.nbd.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taddei RN, Sanchez-Mico MV, Bonnar O, et al. Changes in glial cell phenotypes precede overt neurofibrillary tangle formation, correlate with markers of cortical cell damage, and predict cognitive status of individuals at Braak III-IV stages. Acta Neuropathol Commun. 2022;10(1):72. doi: 10.1186/s40478-022-01370-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paasila PJ, Davies DS, Sutherland GT, Goldsbury C. Clustering of activated microglia occurs before the formation of dystrophic neurites in the evolution of Aβ plaques in Alzheimer’s disease. Free Neuropathol. 2020;1(0):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164-1178. doi: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- 13.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712-716. doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tzioras M, Daniels MJD, King D, et al. Altered synaptic ingestion by human microglia in Alzheimer’s disease. bioRxiv. Published online January 1, 2019:795930.
- 15.Bellenguez C, Küçükali F, Jansen I, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nature Genetics. 2022;54(4):412-436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paasila PJ, Fok SYY, Flores-Rodriguez N, et al. Ground state depletion microscopy as a tool for studying microglia-synapse interactions. J Neurosci Res. 2021;99(6):1515-1532. doi: 10.1002/jnr.24819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colom-Cadena M, Davies C, Sirisi S, et al. Synaptic oligomeric tau in Alzheimer’s disease—a potential culprit in the spread of tau pathology through the brain. Neuron. 2023;111(14):2170-2183.e6. doi: 10.1016/j.neuron.2023.04.020 [DOI] [PubMed] [Google Scholar]
- 18.Fein JA, Sokolow S, Miller CA, et al. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am J Pathol. 2008;172(6):1683-1692. doi: 10.2353/ajpath.2008.070829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerson J, Castillo-Carranza DL, Sengupta U, et al. Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in Htau mice. J Neurotrauma. 2016;33(22):2034-2043. doi: 10.1089/neu.2015.4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6(1):39. doi: 10.1186/1750-1326-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niewiadomska G, Niewiadomski W, Steczkowska M, Gasiorowska A. Tau oligomers neurotoxicity. Life (Basel). 2021;11(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shafiei SS, Guerrero-Muñoz MJ, Castillo-Carranza DL. Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci. 2017;9:83-83. doi: 10.3389/fnagi.2017.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerrero-Muñoz MJ, Gerson J, Castillo-Carranza DL. Tau oligomers: the toxic player at synapses in Alzheimer’s disease. Front Cell Neurosci. 2015;9:464. doi: 10.3389/fncel.2015.00464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pampuscenko K, Morkuniene R, Krasauskas L, Smirnovas V, Tomita T, Borutaite V. Distinct neurotoxic effects of extracellular tau species in primary neuronal-glial cultures. Mol Neurobiol. 2021;58(2):658-667. doi: 10.1007/s12035-020-02150-7 [DOI] [PubMed] [Google Scholar]
- 25.Wu M, Zhang M, Yin X, et al. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl Neurodegener. 2021;10(1):45. doi: 10.1186/s40035-021-00270-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wysocka A, Palasz E, Steczkowska M, Niewiadomska G. Dangerous liaisons: tau interaction with muscarinic receptors. Curr Alzheimer Res. 2020;17(3):224-237. doi: 10.2174/1567205017666200424134311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bilousova T, Miller CA, Poon WW, et al. Synaptic amyloid-β oligomers precede p-tau and differentiate high pathology control cases. Am J Pathol. 2016;186(1):185-198. doi: 10.1016/j.ajpath.2015.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh A, Allen D, Fracassi A, et al. Functional integrity of synapses in the central nervous system of cognitively intact individuals with high Alzheimer’s disease neuropathology is associated with absence of synaptic tau oligomers. J Alzheimers Dis. 2020;78(4):1661-1678. doi: 10.3233/JAD-200716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dejanovic B, Huntley MA, De Mazière A, et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. 2018;100(6):1322-1336.e7. doi: 10.1016/j.neuron.2018.10.014 [DOI] [PubMed] [Google Scholar]
- 30.Sanchez-Mejias E, Navarro V, Jimenez S, et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016;132(6):897-916. doi: 10.1007/s00401-016-1630-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romero-Molina C, Navarro V, Sanchez-Varo R, et al. Distinct microglial responses in two transgenic murine models of tau pathology. Front Cell Neurosci. 2018;12:421. https://www.frontiersin.org/articles/10.3389/fncel.2018.00421. doi: 10.3389/fncel.2018.00421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brelstaff JH, Mason M, Katsinelos T, et al. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv. 2021;7(43):eabg4980. doi: 10.1126/sciadv.abg4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen F, Tillberg PW, Boyden ES. Optical imaging: expansion microscopy. Science. 2015;347(6221):543-548. doi: 10.1126/science.1260088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tillberg PW, Chen F, Piatkevich KD, et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat Biotechnol. 2016;34(9):987-992. doi: 10.1038/nbt.3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallagher BR, Zhao Y. Expansion microscopy: a powerful nanoscale imaging tool for neuroscientists. Neurobiol Dis. 2021;154:105362. doi: 10.1016/j.nbd.2021.105362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karagiannis ED, Boyden ES. Expansion microscopy: development and neuroscience applications. Curr Opin Neurobiol. 2018;50:56-63. doi: 10.1016/j.conb.2017.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vonsattel JPG, Del Amaya MP, Keller CE. Twenty-first century brain banking: processing brains for research: the Columbia University methods. Acta Neuropathol. 2008;115(5):509-532. doi: 10.1007/s00401-007-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thal D, Rüb U, Schultz C, et al. Sequence of Aβ-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59(8):733-748. [DOI] [PubMed] [Google Scholar]
- 39.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 40.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): part II, standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479-486. doi: 10.1212/WNL.41.4.479 [DOI] [PubMed] [Google Scholar]
- 41.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755-766. doi: 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord. 2009;23(2):91-101. doi: 10.1097/WAD.0b013e318191c7dd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freifeld L, Odstrcil I, Förster D, et al. Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proc Natl Acad Sci U S A. 2017;114(50):E10799-E10808. doi: 10.1073/pnas.1706281114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gallagher B, Zhao Y. Nanoscale imaging of synaptic connections with expansion microscopy. Discoveries (Craiova). 2019;7(3):e101-e101. doi: 10.15190/d.2019.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarkar D, Kang J, Wassie AT, et al. Expansion revealing: decrowding proteins to unmask invisible brain nanostructures. bioRxiv. Published online January 1, 2020:2020.08.29.273540. doi: 10.1101/2020.08.29.273540 [DOI]
- 46.Asano SM, Gao R, Wassie AT, Tillberg PW, Chen F, Boyden ES. Expansion microscopy: protocols for imaging proteins and RNA in cells and tissues. Curr Protoc Cell Biol. 2018;80(1):e56-e56. doi: 10.1002/cpcb.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fogarty MJ, Hammond LA, Kanjhan R, Bellingham MC, Noakes PG. A method for the three-dimensional reconstruction of Neurobiotin™-filled neurons and the location of their synaptic inputs. Front Neural Circuits. 2013;7:153-153. doi: 10.3389/fncir.2013.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doorn KJ, Goudriaan A, Blits-Huizinga C, et al. Increased amoeboid microglial density in the olfactory bulb of Parkinson’s and Alzheimer’s patients. Brain Pathol. 2014;24(2):152-165. doi: 10.1111/bpa.12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181(4):1426-1435. doi: 10.1016/j.ajpath.2012.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeVos SL, Corjuc BT, Oakley DH, et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front Neurosci. 2018;12:267. doi: 10.3389/fnins.2018.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petry FR, Pelletier J, Bretteville A, et al. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PLoS One. 2014;9(5):e94251. doi: 10.1371/journal.pone.0094251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schägger H. Blue-native gels to isolate protein complexes from mitochondria. In: Pon LA, Shon EA, eds. Methods in Cell Biology. Vol 65. Academic Press; 2001:231-244, . [DOI] [PubMed] [Google Scholar]
- 53.Pfeiffer T, Poll S, Bancelin S, et al. Chronic 2P-STED imaging reveals high turnover of dendritic spines in the hippocampus in vivo. ELife. 2018;7:e34700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Südhof TC. The cell biology of synapse formation. J Cell Biol. 2021;220(7):e202103052. doi: 10.1083/jcb.202103052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis. 2013;37(3):593-602. doi: 10.3233/JAD-131235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patterson KR, Remmers C, Fu Y, et al. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286(26):23063-23076. doi: 10.1074/jbc.M111.237974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung WS, Clarke LE, Wang GX, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394-400. doi: 10.1038/nature12776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SY, Chung WS. The roles of astrocytic phagocytosis in maintaining homeostasis of brains. J Pharmacol Sci. 2021;145(3):223-227. doi: 10.1016/j.jphs.2020.12.007 [DOI] [PubMed] [Google Scholar]
- 59.Hulshof LA, van Nuijs D, Hol EM, Middeldorp J. The role of astrocytes in synapse loss in Alzheimer’s disease: a systematic review. Front Cell Neurosci. 2022;16:899251. https://www.frontiersin.org/articles/10.3389/fncel.2022.899251. doi: 10.3389/fncel.2022.899251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Damisah EC, Hill RA, Rai A, et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv. 2020;6(26):eaba3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. 2016;524(18):3865-3895. doi: 10.1002/cne.24040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Catalan NA, Doe CQ, Ackerman SD. The role of astrocyte-mediated plasticity in neural circuit development and function. Neural Dev. 2021;16(1):1. doi: 10.1186/s13064-020-00151-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Young H, Belbut B, Baeta M, Petreanu L. Laminar-specific cortico-cortical loops in mouse visual cortex. ELife. 2021;10:e59551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hage TA, Bosma-Moody A, Baker CA, et al. Synaptic connectivity to L2/3 of primary visual cortex measured by two-photon optogenetic stimulation. ELife. 2022;11:e71103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16(14):4491-4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stranahan AM, Mattson MP. Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast. 2010;2010:108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohm DT, Fought AJ, Martersteck A, et al. Accumulation of neurofibrillary tangles and activated microglia is associated with lower neuron densities in the aphasic variant of Alzheimer’s disease. Brain Pathol. 2021;31(1):189-204. doi: 10.1111/bpa.12902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Papageorgiou IE, Lewen A, Galow LV, et al. TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc Natl Acad Sci U S A. 2016;113(1):212-217. doi: 10.1073/pnas.1513853113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Au NPB, Ma CHE. Recent advances in the study of bipolar/rod-shaped microglia and their roles in neurodegeneration. Front Aging Neurosci. 2017;9:128. https://www.frontiersin.org/articles/10.3389/fnagi.2017.00128. doi: 10.3389/fnagi.2017.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mertens R, Melchert S, Gitler D, et al. Epitope specificity of anti-synapsin autoantibodies: differential targeting of synapsin I domains. PLoS One. 2018;13(12):e0208636. doi: 10.1371/journal.pone.0208636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoo KS, Lee K, Oh JY, et al. Postsynaptic density protein 95 (PSD-95) is transported by KIF5 to dendritic regions. Mol Brain. 2019;12(1):97. doi: 10.1186/s13041-019-0520-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sherwood CC, Miller SB, Karl M, et al. Invariant synapse density and neuronal connectivity scaling in primate neocortical evolution. Cereb Cortex. 2020;30(10):5604-5615. doi: 10.1093/cercor/bhaa149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kubota Y, Karube F, Nomura M, Kawaguchi Y. The diversity of cortical inhibitory synapses. Front Neural Circuits. 2016;10:27. https://www.frontiersin.org/article/10.3389/fncir.2016.00027. doi: 10.3389/fncir.2016.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82(4):756-771. doi: 10.1016/j.neuron.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Camporesi E, Nilsson J, Brinkmalm A, et al. Fluid biomarkers for synaptic dysfunction and loss. Biomark Insights. 2020;15:1177271920950319. doi: 10.1177/1177271920950319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Domínguez-Álvaro M, Montero-Crespo M, Blazquez-Llorca L, Insausti R, DeFelipe J, Alonso-Nanclares L. Three-dimensional analysis of synapses in the transentorhinal cortex of Alzheimer’s disease patients. Acta Neuropathol Commun. 2018;6(1):20-20. doi: 10.1186/s40478-018-0520-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cragg BG. The density of synapses and neurons in normal, mentally defective ageing human brains. Brain. 1975;98(1):81-90. doi: 10.1093/brain/98.1.81 [DOI] [PubMed] [Google Scholar]
- 78.Shapson-Coe A, Januszewski M, Berger DR, et al. A connectomic study of a petascale fragment of human cerebral cortex. bioRxiv. Published online January 1, 2021:2021.05.29.446289. doi: 10.1101/2021.05.29.446289 [DOI]
- 79.Domínguez-Álvaro M, Montero-Crespo M, Blazquez-Llorca L, et al. 3D analysis of the synaptic organization in the entorhinal cortex in Alzheimer’s disease. eNeuro. 2021;8(3):ENEURO.0504-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mecca AP, Chen MK, O’Dell RS, et al. In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimers Dement. 2020;16(7):974-982. doi: 10.1002/alz.12097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Colom-Cadena M, Spires-Jones T, Zetterberg H, et al. ; Synaptic Health Endpoints Working Group . The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1):21. doi: 10.1186/s13195-020-00588-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huttenlocher PR. Synaptic density in human frontal cortex—developmental changes and effects of aging. Brain Res. 1979;163(2):195-205. doi: 10.1016/0006-8993(79)90349-4 [DOI] [PubMed] [Google Scholar]
- 83.Kopeikina KJ, Hyman BT, Spires-Jones TL. Soluble forms of tau are toxic in Alzheimer’s disease. Transl Neurosci. 2012;3(3):223-233. doi: 10.2478/s13380-012-0032-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bjorklund NL, Reese LC, Sadagoparamanujam VM, Ghirardi V, Woltjer RL, Taglialatela G. Absence of amyloid β oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol Neurodegener. 2012;7(1):23. doi: 10.1186/1750-1326-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fritschi SK, Langer F, Kaeser SA, et al. Highly potent soluble amyloid-β seeds in human Alzheimer brain but not cerebrospinal fluid. Brain. 2014;137(Pt 11):2909-2915. doi: 10.1093/brain/awu255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin Y, Li F, Sonoustoun B, et al. APOE4 exacerbates α-synuclein seeding activity and contributes to neurotoxicity in Alzheimer’s disease with Lewy body pathology. Acta Neuropathol. 2022;143(6):641-662. doi: 10.1007/s00401-022-02421-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Calignon A, Spires-Jones TL, Pitstick R, Carlson GA, Hyman BT. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68(7):757-761. doi: 10.1097/NEN.0b013e3181a9fc66 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eReferences.
eTable 1. Antibodies Used in the Present Study
eTable 2. Baseline Demographic and Neuropathologic Characteristics of the Subset of N = 10 Human Brains Studied
eFigure 1. Neuropathologic Assessment of Amyloid-β and Tau Burdens in the Studied Brains
eFigure 2. Steps of Expansion Microscopy (ExM) Exemplified
eFigure 3. Synapse Densities Across Groups and Correlation Analyses Between Synapse Densities and Cognitive and Neuropathological Measures
eFigure 4. Confocal Images of Engulfment of Synaptic Elements by Microglia and Astrocytes
eFigure 5. Expanded Astrocyte Co-Stained With a Cytoskeletal and a Cytoplasmic Antibody
eFigure 6. Lysosomal Co-Staining of Engulfed Synaptic Elements in Representative Microglia and Astrocyte
eFigure 7. Quality of Extracted Synaptosomes and Measures of Truncated Tau Species in Synaptosome Fractions
eFigure 8. Association of Oligomeric Tau With Synaptic Elements
Data Sharing Statement