

Abstract
Transforming growth factor β (TGF-β) is a pleiotropic cytokine expressed by a wide range of cell types and is known for hampering the effectiveness of cancer immune cell therapeutic approaches. We have designed a novel construct containing the extracellular domain of the TGF-β receptor II linked to a glycosylphosphatidylinositol (GPI) anchor (GPI-ecto-TβRII) lacking the transmembrane and cytoplasmic signaling domain of TGF-β receptor II (TβRII). T cells transduced with lentivirus expressing the GPI-ecto-TβRII construct show 5 to 15 times higher membrane expression compared with a previously established dominant-negative receptor carrying a truncated signaling domain. GPI-ecto-TβRII expression renders T cells unresponsive to TGF-β-induced signaling seen by a lack of SMAD phosphorylation upon exogeneous TGF-β treatment. Transduced T cells continue to express high levels of IFNγ and granulocyte-macrophage colony-stimulating factor (GM-CSF), among other cytokines, in the presence of TGF-β while cytokine expression in untransduced T cells is being markedly suppressed. Furthermore, T cells expressing GPI-ecto-TβRII constructs have been shown to efficiently capture and inactivate TGF-β from their environment. These results indicate the potential benefits of GPI-ecto-TβRII expressing cytotoxic T cells (CTLs) in future cell therapies.
Keywords: tumor growth factor beta, TGF-β, decoy receptor, cell therapy, T cell, cancer, tumor microenvironment
Graphical abstract
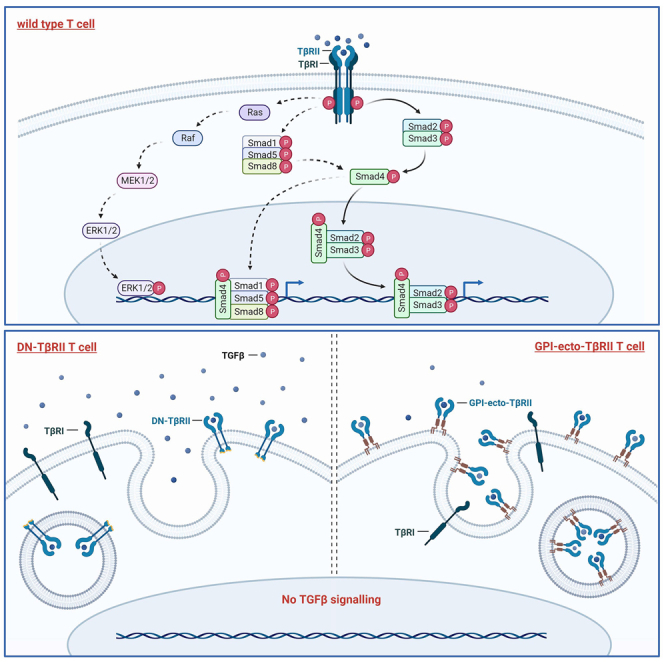
Connolly and colleagues demonstrate that GPI-anchored dominant-negative TGF-β decoy receptor enables transgenic T cells to be completely resistant to TGF-β and has the capacity to capture TGF-β from the environment, potentially benefiting a stimulatory bystander effect in the tumor microenvironment.
Introduction
Cellular immunotherapy is gaining increasing popularity as a potential cancer treatment. Cancer immune suppression, however, remains a major obstacle for immunotherapies’ efficacy.1,2 Although there has been remarkable progress in understanding tumor immune suppression, current therapeutics are still far from successfully counteracting the responsible mechanisms. Cancer immunoediting is the process whereby cancer cells overcome elimination and acquire traits that help escape immune surveillance. From this stage of equilibrium, the tumor gains malignancy by further exploiting several immunological strategies for its benefit, such as modifying responses of regulatory T cells (Tregs) or changing the cytokine environment at the tumor site. Tumor cell secretion of transforming growth factor β (TGF-β) is one predominant way to subvert the immune systems’ own machinery for tumor progression.3,4,5 TGF-β is involved in a wide range of cellular processes, such as cell growth, proliferation, differentiation, apoptosis, angiogenesis, and cell homeostasis. The role of TGF-β in the immune system is to limit effector responses in antigen-presenting cells (APCs) as well as memory and effector T cells.6
Soluble TGF-β dimers bind to the TGF-β receptor II (TβRII), which results in the recruitment of TGF-β receptor I (TβRI) and the formation of a tetramer complex involving two of each receptor. The subsequent phosphorylation of TβRII leads to activation of TβRI, which in turn initiates the signaling cascade by phosphorylation of SMAD protein complexes. Phosphorylated SMAD2/3 (p-SMAD2/3) and to a lesser degree p-SMAD1/5/8 translocate into the nucleus, where they bind to DNA and facilitate the expression of TGF-β-regulated genes.7,8,9 The effect of this signaling cascade is to induce growth arrest and apoptosis in cancer cells, which can ameliorate this process by carrying mutations in their genes for TGF-β receptors or SMAD proteins.10,11,12,13,14,15,16 To render cytotoxic T cells less responsive to TGF-β, a similar strategy was investigated by Wieser et al.17 and further used by Bollard and colleagues, as they expressed a truncated dominant-negative (DN) version of TβRII in T lymphocytes (CTLs) lacking the cytoplasmic kinase domain and most of the juxta membrane region.18,19,20 They showed that the expression of the DN TβRII renders CTLs resistant to the inhibitory effects of TGF-β in Hodgkin’s lymphoma. They observed a continued cytokine production and cytolytic activity in the presence of TGF-β.
Encouraged by the results obtained using the DN TβRII-decoy receptor, we have designed HA-tagged decoy receptors composed of the extracellular domain of TβRII (TβRII-ecto) attached to glycosylphosphatidylinositol (GPI) anchors. GPI-anchored proteins are a diverse group of proteins that are attached to the phospholipid bilayer via the post-translationally added GPI molecule. GPI anchors have no cytoplasmic domain, have no autonomous signaling function, and give their attached proteins distinct properties in lateral membrane sorting to lipid rafts.21 As we consider these features potentially beneficial for creating potent decoy receptor constructs, we designed GPI-anchored TβRII-ecto domains. The GPI anchors we chose to test derive from the naturally occuring GPI-anchored proteins CD48, CD55, CD90, and alkaline phosphatase (Alkphos). High surface expression of HA-CD48-GPI-TβRII, HA-CD55-GPI-TβRII, HA-CD90-GPI-TβRII, or HA-Alkphos-GPI-TβRII in Jurkat cells or ex vivo activated patient-derived T cells (ATCs) resulted in a profound inhibition of TGF-β-induced SMAD phosphorylation while not affecting proliferation or phenotype of transduced T cells in standard culture conditions. Although marginally fostering terminal differentiation in the presence of TGF-β, the expression of HA-GPI-ecto-TβRII constructs enables ATCs to maintain cytokine production. Furthermore, our data show that transduced Jurkat cells as well as ATCs exhibit a strongly enhanced ability to bind and take up TGF-β from their environment, which potentially turns them into potent TGF-β sinks in the tumor microenvironment.
Results
GPI-anchored TGFβR2 ecto domains (HA-GPI-ecto-TβRII) show high surface expression in Jurkat cells and activated primary CD3+ T cells
Jurkat cells and ATCs from healthy donors were transduced using lentivirus containing one of four different HA-tagged constructs of the GPI-anchored DN TGF-β receptor ecto domain (HA-GPI-ecto-TβRII), the truncated DN construct (HA-DN) (Figure 1A), or a GFP as a control. Each HA-GPI-ecto-TβRII construct is distinguished by its unique GPI anchor, derived from the naturally occurring GPI-anchored proteins CD48, CD55, CD90, and Alkphos. Cell surface expression could be detected in Jurkat cells and ATCs using flow cytometry 3 days post-transduction. Subsequent puromycin selection enabled us to acquire an almost homogeneous population with expression rates ranging from 62.4% to 96.1% (mean 80.75%) in Jurkat cells and from 81.9% to 97.9% (mean 93.59%) in ATCs (Figures 1B–1E). All HA-GPI-ecto-TβRII constructs show an increased surface expression compared with HA-DN, which, in ATCs, can go up to 15.22-fold in the case of HA-GPI-ecto-TβRII carrying the CD48-GPI anchor (Figures 1D and 1E). Confocal imaging of transduced Jurkat cells confirms an expression predominantly present on the cell surface, with little HA-GPI-ecto-TβRII detected in the cytoplasm (Figure 1F).
Figure 1.

GPI-ecto-TβRII is highly expressed on transduced Jurkat cells and ATCs
(A) Schematic representation of HA-DN and HA-GPI-ecto-TβRII construct design. The expression of any of the 4 tested GPI-ecto-TβRII constructs is several folds higher in Jurkat cells compared with HA-DN containing a natural transmembrane domain as seen in (B) flow cytometry or (C) displayed as mean fluorescence intensity (MFI) in bar graphs. (D) Flow cytometry dot plots or (E) MFI bar graphs of ATCs transduced with either HA-DN or one of four GPI-ecto-TβRII constructs. Bar graphs display the mean and SEM of at least 3 biological replicates. (F) Confocal microscopy of Jurkat cells transduced with either HA-DN or one of the four HA-GPI-ecto-TβRII constructs demonstrates transmembrane TβRII expression (red) in transduced cells.
Expression of HA-DN or HA-GPI-ecto-TβII decoy receptors inhibits TGF-β signaling in Jurkat cells and ATCs
The direct effects of the expression of TGF-β decoy receptors on TGF-βR signaling were analyzed using western blot (WB) or flow cytometry. For both methods, Jurkat cells or ATCs from three healthy donors were serum starved overnight and then treated with either 1 ng/mL TGF-β in a reverse time course experiment or with increasing concentrations of TGF-β (0, 1, 10, and 200 ng/mL) for 30 min. For WB, the cells (ATCs [Figure 2A] and Jurkat cells [Figure S1B]) were immediately lysed at 4°C after the last time point and prepared for SDS-PAGE and WB analysis. In both controls (wild-type [WT] and GFP), SMAD2/3 phosphorylation was detectable after 10 min, reaching a maximum at 30–60 min. No band for pSMAD2/3 was detected in cells expressing any of the tested TGF-β decoy receptors. ERK1/2 is a part of the non-canonical TGF-βR signaling pathway, and we observed a fast phosphorylation of this kinase upon TGF-β stimulation in our control cells, which was markedly reduced in TGF-β decoy receptor-positive cells. No differences were observed for Fyn, LCK, or Src phosphorylation (Figures 2A and S1B). Flow cytometric analysis of phosphorylation in Jurkat or ATCs, likewise, indicated protection from TGF-β receptor signaling in HA-DN or the HA-GPI-ecto-TβII-expressing cells compared with WT or GFP control (Figures 2B and S1C). As an internal control, the CD3+ population was divided into a TβRII-high gate (Figure 2B) and a TβRII-low gate (Figure S1C). The TβRII-low population contained most of the WT and GFP control cells as well as transduced cells failing to express the inserted decoy receptor. At 1 ng/mL TGF-β, TβRII-high control cells showed an increase in percentage pSMAD2/3-positive cells from 19.78% in WT and 21.5% in GFP. HA-DN or HA-GPI-ecto-TβII expression, however, protected cells from TGF-β-induced SMAD2/3 phosphorylation. Each of the tested decoy receptors differs from GFP controls at p < 0.001 (Figure 2B). Increasing the TGF-β concentration 10- or 200-fold also did not lead to any substantial increase in signaling. SMAD1/5/8 phosphorylation, which also is a target of the TβRII signaling pathway, almost completely mirrors this pattern seen for SMAD2/3. Similar to the WB data, pERK1/2 was more mildly reduced in TGF-β decoy receptor expressing cells.
Figure 2.

GPI-ecto-TβRII does interfere with TGF-β-induced SMAD2/3 signaling in Jurkat cells and ATCs
(A) TGF-β-dependent SMAD signaling is strongly disrupted by the expression of any of the chosen decoy receptors in ATCs as seen in WB. We see a time-dependent increase in p-SMAD2/3 or p-SMAD1/5 in WT or GFP-ctrl.-ATCs after the addition of 1 ng/mL TGF-β. These are hardly detectable in any of ATCs expressing decoy receptors. ERK signaling is not affected by HA-DN but by seems disrupted by HA-GPI-ecto-TβRII constructs. No changes in Fyn or LCK signaling were observed. (B) Flow cytometric analysis of p-SMAD2/3 and p-SMAD1/8 demonstrates a TGF-β dose-independent inhibition of SMAD signaling and a less pronounced inhibition in ERK-signaling in Jurkat cells expressing any of the decoy receptors. (C) TGF-β-dependent inhibition of calcium flux in stimulated Jurkat cells was significantly blocked by the expression of HA-GPI-ecto-TβRII constructs, particularly HA-CD55, HA-CD90, and HA-Alkphos. Bar graphs display the mean and SEM of at least 3 biological replicates.
As TGF-β has a known dampening effect on T cell receptor (TCR) stimulation-dependent calcium flux, we measured the cytoplasmic calcium flux in the presence of TGF-β and α-CD3 stimulation. Cells were stained using Fluo-8 for one hour, either in the presence of 100 ng/mL TGF-β or without. After the staining, the cells were activated by adding soluble α-CD3 antibody, and the resulting fluorescence was analyzed over time using a Hidex (Turku, Finland) plate reader. Although the two controls, WT and GFP cells, exhibited TGF-β-dependent inhibition of calcium release of approximately 21%, HA-DN and HA-CD48-GPI-ecto-TβRII expression partially protected Jurkat cells from this inhibition. An almost complete protection could be measured for cells transduced with the HA-CD55, HA-CD90, or the HA-Alkphos constructs. The differences to control cells are significant at p = 0.0116, p = 0.0052, and p = 0.0081, respectively, proving the GPI-anchored TGF-β receptor ecto domain to be superior compared with HA-DN (Figures 2C, S1D, and S1E).
Expression of HA-GPI-ecto-TβRII does not interfere with T cell differentiation in long-term culture
Transduced ATCs of three healthy donors were kept in culture in CTL media containing recombinant human interleukin-2 (rhIL-2) (100 U/mL) and Dynabeads Human T-Activator CD3/CD28 (Gibco, Thermo Fisher Scientific). Three, 6, 10, 14, and 20 days post-transduction, the subtype composition was analyzed using flow cytometry. The fraction of CD4+ T cells steadily decreased and that of CD8+ T cells increased for all control and transduced T cells, and the overall differentiation followed similar trends in both subpopulations for all tested cell types (Figures 3A, 3B, and S2). The changes in CD4-to-CD8 T cell ratio were due to the proliferation of CD4+ T cells decreasing steadily over the analyzed time period, while CD8+ T cell proliferation remained stable for all subpopulations (Figure 3C). This is true for control as well as the transduced T cells. Likewise, T cell activation, assessed by CD69 expression, remains stable for CD4+ and CD8+ T cells alike. A rapid 10-fold decline in the naive compartment and a slow reduction of central memory T (Tcm) cells after day 6 was accompanied by an 8-fold outgrowth of effector memory T (Tem) cells in the CD4+ ATCs and TEMRA cells in the CD8+ ATCs. The expression of TGF-β decoy receptors, however, mildly slows down the terminal differentiation of T cells, as seen for CD4+ Tcm and Tem cells as well as for CD8+-naive and TEMRA T cells. We did not observe any significant differences between the control and the TGF-β decoy receptor-expressing cells (Figure S2A). In terms of T cell exhaustion, we do not see any significant differences in the expression rates of PD1. A similar trend is observed for the two chosen differentiation markers CD57 and KLRG1 in any of the analyzed T cell subsets at any point of time (Figures S2B–S2D). T reg or natural killer T (NKT) cell proliferation is not affected by the expression of our constructs either (Figure S2E).
Figure 3.

Expression of TGF-β decoy receptor does not affect primary T cell differentiation per se
(A) Flow cytometry t-distributed stochastic neighbor embedding (tSNE) analysis of concatenated biological replicates shows a similar differentiation of control and decoy receptor expressing ATCs over time. (B and C) Line graphs of each of the T cell subtypes over time confirm a similar differentiation and proliferation pattern in GPI-ecto-TβRII-positive ATCs compared with WT and GFP control cells. Line graphs display the mean and SEM of 6 biological replicates.
Expression of HA-GPI-ecto-TβRII renders T cells resistant to TGF-β
The above-mentioned ATCs were cultured for 4 days in CTL media containing rhIL-2 (100 U/mL), Dynabeads Human T-Activator CD3/CD28, and a series of concentrations of rTGF-β (0, 1, 10, or 100 ng/mL). On day 4, cells were analyzed using flow cytometry. For simplicity and because of the similarity between all HA-GPI-ecto-TβRII-expressing cells, Figure 4B displays data obtained for the Alkphos-TβRII construct only. These are representative to all 4 GPI constructs tested. Except for a marginally elevated percentage of CD4+ T cells in HA-DN, we did not detect any remarkable changes in the CD4+/CD8+ T cell ratios between control and transduced T cells at any concentration of TGF-β. However, focusing on T cell differentiation, we observed an expected TGF-β-dependent maintenance of central memory in WT and GFP control cells (increase of 20%–25% in CD4+ and 17%–24% in CD8+) accompanied by a lower percentage of Tem and TEMRA cells (Figures 4A and 4B). T cells expressing any of the TGF-β decoy receptors (HA-DN or HA-GPI-ecto-TβRII) showed resistance to the addition of TGF-β, irrespective of what concentration we used. Compared with TGF-β-treated control cells, TGF-β-resistant cells contained a lower proportion of memory cells (resembling that of untreated control cells) and correspondingly higher proportions of Tem and to a lesser degree TEMRA cells. This effect was seen in CD4+ and CD8+ T cells alike and was independent of the tested TGF-β concentration (Figures 4A and 4B). TGF-β did not significantly affect the proliferation of any of the T cell subtypes at any tested concentration under the above-mentioned culture conditions (Ki67; Figure 4C). T cell activation was slightly increased in HA-DN CD8+ T cells, particularly in the naive, Tem, and TEMRA compartments. The HA-GPI-ecto-TβRII-expressing cells, however, resembled the WT and GFP control. Overall, CD69 expression was not majorly affected by the presence of TGF-β, and we saw no significant differences among all the remaining cells and subpopulations (Figure S3A). TGF-β increased PD1 expression in CD4+ T cells in WT and GFP control, particularly in the Tem and TEMRA compartments, indicating increased exhaustion. However, TGF-β decoy receptor-positive cells were protected from this effect (Figure S3B). No effect could be seen on the expression of CD57 or KLRG1 or the proportion of CD4+ T reg cells (Figures S3C–S3E). NKT cells displayed an enhanced proliferation when expressing any of the HA-GPI-ecto-TβRII constructs but not the HA-DN version indicating an intrinsic function of the GPI-associated expression of the decoy receptor (Figure S3E).
Figure 4.

TGF-β helps T cells maintain a central memory phenotype, whereas the expression of TGF-β decoy receptors inhibits this effect without affecting proliferation
(A) Flow cytometry tSNE analysis of concatenated biological replicates shows a higher ratio of Tcm and lower ratio of Tem and TEMRA cells in control cells in the presence of TGF-β. Decoy receptor expressing ATCs are unaffected even at highest tested TGF-β concentrations. (B) Line graphs of each of the T cell subtypes over time confirms the lack of TGF-β-dependent maintenance of Tcm cells in GPI-ecto-TβRII-positive ATCs compared with WT and GFP control cells. (C) Proliferation is similar in all tested ATCs. Line graphs display the mean and SEM of 6 biological replicates. (D) Heatmap presentation of cytokine levels measured in cell culture supernatants after ATC culture in the presence of different concentrations of TGF-β. Cytokine secretion of most inflammatory cytokines are significantly reduced in control ATCs, whereas the expression of HA-GPI-ecto-TβRII has a protective effect. Heatmaps display the data of 6 biological replicates (statistical significance in Figure S4B).
HA-GPI-ecto-TβII transduced T cells are protected from TGF-β-dependent cytokine secretion inhibition
The cell supernatants from the above-mentioned experiment were harvested on day 4 and analyzed for cytokine secretion using a Luminex multiplex assay. As expected, TGF-β significantly reduced the release of several inflammatory cytokines in WT and GFP control ATCs. Among those cytokines, IFNγ, granulocyte-macrophage colony-stimulating factor (GM-CSF), MIP1b, TNF-α, TNF-β, IL-3, IL-4, IL-5, IL-6, and IL-13 stood out. The expression of the HA-DN or any of the HA-GPI-ecto-TβRII decoy receptor significantly diminished the inhibitory effect of TGF-β in ATCs (Figures 4D and S4). Although we still observed a slight dose dependent reduction in cytokine release in cells expressing a decoy receptor, the overall concentration measured in the supernatants remained markedly higher compared with the control cell’s release. IFNγ and GM-CSF are two of the main effectors in supporting an inflammatory microenvironment during an anti-cancer immune response, and their maintenance by the decoy receptors in a TGF-β-rich microenvironment could be clinically relevant. The GFP control experienced a 5-fold inhibition in IFNγ release (from 17.4 to 3.4 ng/mL) at a concentration of just 1 ng/mL TGF-β, while IFNγ release of cells positive for TGF-β decoy receptors decreased by only 2-fold at maximum (from 28.8 to 15.3 ng/mL for HA-Alkphos, p = 0.0026). Similarly, GM-CSF production was reduced 3.8-fold in GFP control cells (from 7.3 to 1.9 ng/mL), whereas ATCs transduced with decoy receptors exhibited only a 1.3-fold reduction in average (from 6.4 to 4.3 ng/mL for HA-Alkphos, p = 0.00024). Interestingly, a highly significant change could also be observed for IL-13. TGF-β at 1 ng/mL did suppress IL-13 production up to 24 times (from 69.4 to 2.9 ng/mL) in GFP control cells. HA-Alkphos-transduced cells, for example, maintain 10.8 times higher IL-13 production at the same concentration of TGF-β (p = 0.0005) (Figures 4D and S4B). Additionally, two inhibitory cytokines, IL-10 and sCD40L, showed a similar degree of inhibition by TGF-β in control cells, and this inhibition is significantly reduced in cells positive for TGF-β decoy receptors (Figure S4).
HA-GPI-ecto-TβRII enables T cells to capture and inactivate TGF-β
To measure the cell’s capacity to capture and remove TGF-β from the microenvironment, we cultured Jurkat cells (Figure 5A) or ATCs (Figure 5B) for 6 h in the presence of different doses (1–10 ng/mL) of TGF-β in serum-free medium and measured the remaining TGF-β in the supernatant by Luminex analysis. At an initial concentration of 1 ng/mL, the measured TGF-β concentration in WT and GFP control Jurkat cells increased 3-fold to almost 3 ng/mL. At higher initial concentrations, the measured remaining TGF-β concentration remained almost constant (9 ng/mL). HA-DN-positive cells showed the capacity to take up TGF-β from the environment. At an initial concentration of 1 ng/mL, the HA-DN culture showed a 75% lower (p = 0.0015) and at 10 ng/mL a 50% lower (p = 0.0026) concentration compared with the GFP control (Figure 5A). Jurkat cells expressing any of the four HA-GPI-ecto-TβIIR constructs, however, demonstrated an even superior TGF-β uptake capacity. These cells were able to almost completely absorb 200 pg TGF-β (1 ng/mL in 200 μL) TGF-β added to the culture within the given time (p = 0.002 for CD48, p = 0.0005 for CD55, p = 0.0008 for CD90, and p = 0.0005 for Alkphos). Likewise, the higher dose of 2 ng (10 ng/mL in 200 μL) has been absorbed by approximately 90% (p = 0.0007 for CD48, p = 0.0005 for CD55, p = 0.0006 for CD90, and p = 0.0005 for Alkphos) (Figure 5A). ATCs expressing HA-GPI-ecto-TβIIR elicit a very similar capacity to take up soluble TGF-β. Hardly any TGF-β was detectable in the 200 pg doses, and approximately 90% of TGF-β was taken up in the 2 ng doses (p = 0.001 for CD48 and p = 0.0009 for CD55, CD90, and Alkphos) (Figure 5B). The TGF-β removed from the environment was likely degraded inside the cell.
Figure 5.

The expression of HA-GPI-ecto-TβRII enables T cells to capture and inactivate TGF-β from culture medium
(A and B) TGF-β-Luminex analysis of recombinant TGF-β-spiked cell culture supernatant after a 6 h incubation with Jurkat cells (A) or ATCs (B) expressing TGF-β decoy receptors. Bar graphs display the mean and SEM of 3 biological replicates.
In preliminary data, the supernatants of such 6 h incubations using either GFP control or HA-GPI-ecto-TβIIR (CD90) expressing Jurkat cells were given to fresh GFP control Jurkat cells for 30 min to test for subsequent TGF-β-dependent signaling. We observed reduced SMAD phosphorylation in Jurkat cells exposed to media preincubated with GPI-ecto-TβIIR-expressing cells compared with freshly spiked TGF-β-containing media. This further indicates TGF-β depletion by GPI-ecto-TβIIR-positive cells (data not shown).
Discussion
We designed and expressed the TβRII-ecto domain on a set of GPI anchors in Jurkat cells as well as ATCs and compared their TGF-β-modulating activities against the previously characterized truncated HA-DN-TβRII, with a GFP-expressing transduced control and an untransduced WT control. Both the GPI-anchored and truncated TβRII were designed to be decoy receptors competing against and preventing ligand activation of native TβRII. The surface expression of any HA-GPI-ecto-TβRII was significantly higher than compared with HA-DN-TβRII and, likewise, protected Jurkat and ATCs from canonical TGF-β dependent phosphorylation of SMAD proteins. Non-canonical TGF-βR signaling is mildly affected as seen in ERK phosphorylation.
The expression of GPI-anchored ectodomains of TβRII did not have any effect on the phosphorylation of TCR stimulation-related proteins such as Fyn and LCK. We had hypothesized that TGF-β binding to GPI-anchored ectodomains of TβRII would cause enhanced lipid raft clustering of surface proteins and hence increased stimulation of lipid-raft-associated TCRs or costimulatory proteins.22,23,24,25,26,27 This might still be the case for Toll-like receptor (TLR)-dependent amplification of TCR signaling, as TLRs are clustered to T cell lipid rafts.28 However, we did not further investigate this hypothesis in this study.
Calcium signaling in CTLs is directly dependent on TCR-antigen engagement and has been shown to be a crucial part of T cell activation.29,30 TGF-β curbs calcium flux in T cells by abrogating TCR signaling as part of its immunosuppressive effects.31,32 Adding recombinant TGF-β to Jurkat cells diminished the measurable calcium flux within cells by 20% in control cells, whereas cells expressing TGF-β decoy receptors were protected. Particularly HA-GPI-CD55, HA-GPI-CD90, and HA-GPI-Alkphos demonstrated superior protection compared with HA-DN-TβRII. We hypothesize the effect might be due to higher surface expression of the GPI-anchored decoys.
Consistent with this mechanism, we have shown that the HA-DN-TβRII is able to “capture” and deplete TGF-β from the environment. This is possibly facilitated by the overexpression of ecto domains on the cell surface, enabling the cells to bind, endocytose, and recycle soluble TGF-β. This “sink function” was greatly enhanced using cells expressing any of the HA-GPI-ecto-TβRII constructs, which we assume is due to an excessive clustering of GPI-anchored receptors in lipid rafts upon binding to TGF-β and subsequent pinocytosis, typically seen in GPI-anchored protein recycling events.33,34,35 Given TGF-β’s immunosuppressive activity, going far beyond impairing the cytotoxicity of adoptively added T cells at the tumor site, depleting TGF-β potentially has far-reaching positive implications for future immunotherapy. TGF-β dampens the immune response of professional APCs, natural killer (NK) cells, and B cells and stimulates Tregs and myeloid derived suppressor cells and thereby blocks myriad potential cancer-fighting tools.36 Therefore, a tumor microenvironment being drastically diminished in TGF-β could be infiltrated by bystander cells, leading to a more robust and widespread anti-cancer immune response benefiting antigen spreading and counteracting potential resistance.37
Under standard culture conditions, transduction of ATCs with GFP or any of the decoy receptors had no significant effect on the phenotype and differentiation of T cells over time. We observed a steady decline in CD4+ T cell percentage, which was compensated for by an equivalent enrichment in CD8+ T cells. Both subtypes, however, displayed a similar pattern in differentiation. Particularly naive cells and to a lesser degree Tcm cells declined in their percentages throughout the time of measurement, whereas CD45RA− and CD45RA+ (TEMRA) effector memory cells increasingly formed the majority of both subpopulations. Proliferation was not affected in any of the subpopulations analyzed. The media used contained 10% fetal bovine serum (FBS) and hence approximately 4 ng/mL bovine TGF-β. However, our data showed that this does not result in TGF-βR signaling in human T cells (data not shown). Once TGF-βR signaling does occur in T cells, differentiation is markedly decelerated,32 which we confirmed by adding recombinant human TGF-β1 to our T cell culture for 96 h. WT and GFP control cells elicited a significantly higher proportion of Tcm cells and lower proportions of Tem and TEMRA cells when cultured in the presence of TGF-β. Cells resistant to TGF-βR signaling, however, displayed a similar differentiation pattern as untreated control cells, irrespective of the kind of TGF-β decoy receptor expressed. Despite the well-established adverse effect of TGF-β on T cell proliferation,19,38,39,40 we did not observe any change under any condition tested. This might be due to the rather high concentration of IL-2 we chose to add to our standard culture media for ATCs (100 U/mL), which potentially counteracted the anti-proliferative effect of TGF-β. A similar IL-2-dependent abrogation of the anti-proliferative effect of TGF-β has been observed in CD28-ζ chimeric antigen receptor (CAR) T cells in which increased IL-2 secretion and autocrine IL-2 signaling caused TGF-β resistance.41
One additional immunoinhibitory effect of TGF-β in the tumor microenvironment is decreasing the production of inflammatory cytokines,42,43,44,45,46 and accordingly HA-DN-TβRII has previously been shown to prevent TGF-β-dependent cytokine downregulation in T cells.18,47 Here, we demonstrated that GPI-anchored TβRII ectodomains expressed in T cells, likewise, were able to maintain a significantly higher cytokine production in the presence of TGF-β compared with WT or GFP control. Important to note is that despite the lack of TβRII signaling, cytokine secretion never went far beyond the level we see in control cells without TGF-β inhibition. This points to the fact that the expression of a TGF-β decoy receptor does not seem to lead to an excessive stimulation of T cells, which is of utmost importance to maintain a regulated T cell response during cell therapy production and in the tumor microenvironment during cell therapy. In correlation to these observed characteristics, preliminary data of ours reveal a superior cancer-killing capacity of ATCs expressing GPI-TβRII constructs. In these experiments, CAR and GPI-TβRII co-expressing ATCs were able to kill antigen-expressing cancer cell spheroids markedly more efficiently (data not shown).
Overall, this novel GPI-anchored ecto domain of the TβRII proves to be a promising way to combat the adverse effect of TGF-β in cancer immunotherapy. Its superiority in cell surface expression, T cell activation, and TGF-β depletion from the microenvironment compared with its previously established cytoplasmic truncated counterpart makes GPI-ecto-TβRII a potent construct worthy of further investigation in the context of cancer immunotherapy and beyond.
Materials and methods
Cell lines
HEK293T and Jurkat cells were obtained from American Type Culture Collection (ATCC). Peripheral blood mononuclear cells (PBMCs) were purified from whole-blood samples acquired from healthy donors (Singapore General Hospital) (ethical approval: Tessa-IRB-2017-001, SingHealth).
HA-GPI-ecto-TβRII lentiviral plasmids
The human type II TGF-β receptor was truncated at nucleotide 549, thereby leaving only the extracellular domain. The corresponding DNA containing the 29 nt sequence of the influenza virus hemagglutinin peptide epitope HA1 plus an AGA linker and a GPI signaling sequence at the 3′ end was cloned into a pD2109-EF1 vector by ATUM (Newark, CA) (94560). The HA sequence was inserted after the signal sequence in the human TGF-βRII so that the HA epitope is retained near the amino terminus of the mature receptor. The presence of the HA tag does not affect ligand binding and allows the mutant construct to be distinguished from the WT TGF-βRII receptor with an anti-HA antibody. The GPI signaling sequences used were the ones from the naturally occurring GPI-anchored proteins CD48, CD55, CD90, or Alkphos.
Production of recombinant lentivirus
HEK293T cells were transiently transfected with vector DNA using FuGENE 6 transfection reagent (Promega) in DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% fetal calf serum (FCS; Gibco, Thermo Fisher Scientific). Eighteen hours after transfection, the transfection medium was replaced with fresh DMEM supplemented with 10% FCS and 2 mM caffeine and adjusted to pH 6.3 using hydrochloric acid and cultured for another 24 h. Fresh lentivirus supernatant was collected 24 and 48 h later, filtered through a 0.45 μm filter, and concentrated using Lenti-X Concentrator (Clontech Laboratories, Takara Bio, Kusatsu, Japan). The resulting viral pellet was resuspended in CTL medium (50% Click’s medium [EHAA]; Fujifilm Irvine Scientific; 50% RPMI; Gibco, Thermo Fisher Scientific) supplemented with 10% FCS and either stored at −80°C or used freshly for transduction of target cells.
Generation and transduction of ATCs
CD3-positive T cells were enriched from PBMCs using the Pan-T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The T cells were transferred to CTL medium and stimulated by adding Dynabeads Human T-Activator CD3/CD28 and rhIL-2 (100 IU/mL; Miltenyi Biotec). After 18–24 h in culture, T cells were transduced by adding concentrated viral resuspension supplemented containing the transduction enhancer LentiBOOST (pharmaceutical grade, final concentration 1:100; Sirion Biotec, Martinsried, Germany) and by spinoculation (32°C, 800 × g, 90 min). The cells were then transferred to the incubator and cultured overnight. The next day, the viral supernatant was replaced with fresh CTL supplemented with rhIL-2 (100 IU/mL) without removing the Dynabeads. Three days post-transduction, the surface expression of TβRII was measured using flow cytometry and puromycin (2 μg/mL; Gibco, Thermo Fisher Scientific) was added to the T cell culture if further experiments required a homogeneously transduced population.
For long-term culture of transduced ATCs, cells of at least three healthy donors were kept in culture in 24-well plates with 2 mL CTL media per well containing rhIL-2 (100 U/mL) and Dynabeads Human T-Activator CD3/CD28. Serial dilutions of TGF-β were added to the culture for the analysis of TGF-β resistance. CTL medium with all components was refreshed twice a week, and cells were split into new wells when necessary. After approximately 28 days, ATCs seemed completely exhausted and were discarded.
Flow cytometry
For immunophenotyping, cells were stained with fluorescein-conjugated monoclonal antibodies directed against CD3, CD25, CD4, CD8, CD45RA, CD69, KLRG1, and PD1 (Becton Dickinson) and FOXP3, CCR7, CD57, and CD56 (BioLegend) and Ki67 (Life Technologies) and TβRII (Miltenyi Biotec). Viability was assessed using Live/Dead fixable stain (Thermo Fisher Scientific). Cells were incubated with Live/Dead stain in PBS (1:1,000) for 10 min at 4°C, then washed with PBS and incubated with the respective antibodies diluted in staining buffer (PBS, 2 mM EDTA, 15 mM HEPES, 1:50 FCS) for 30 min at 4°C in the dark. After incubation, cells were washed with staining buffer and subsequently fixed and permeabilized for 30 min at 4°C using Fix/Perm solution (Permeabilization Buffer + Fixation/Perm diluent, 1:30; Invitrogen, Thermo Fisher Scientific). Cells were washed with Permeabilization Wash, and intracellular antibody mix was added to the cells overnight at 4°C in the dark. The next day, cells were washed using staining buffer, resuspended in PBS, and analyzed using a BD FACSymphony flow cytometer (Becton Dickinson).
For the analysis of the phosphorylation of proteins, fluorescein-conjugated monoclonal antibodies directed against p-SMAD2/3, pSMAD1/8, and pERK1/2 (Becton Dickinson) were used. Cells were stimulated as required by experimental setup and immediately spun down at 4°C before continuing all further steps on ice. Live/Dead cell stain was added (1:200) for 30 s and washed with cold PBS. This step was followed by a 30 min incubation in ice-cold Cytofix solution (Becton Dickinson), another wash, and a 30 min incubation in ice-cold Perm buffer III (Becton Dickinson). Subsequently, cells were resuspended in the antibody mix containing surface as well as intracellular antibodies in staining buffer and kept overnight (4°C in the dark). The next day, cells were washed using staining buffer, resuspended in PBS, and analyzed using a BD FACSymphony flow cytometer (Becton Dickinson).
Western blotting
Cell pellets were resuspended and lysed for 30 min on ice in RIPA lysis buffer supplemented with Protease/Phosphatase Inhibitor (1:100; Thermo Fisher Scientific). Protein concentration was determined using the BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific) when necessary. Lysate was denatured by adding 4× Laemmli buffer (Bio-Rad) supplemented with 10% β-mercapto-ethanol and subsequent heat treatment at 95°C for 10 min. Proteins were separated by SDS-PAGE using “any kDa – stain free” pre-cast gels (Bio-Rad) and blotted using the Trans-blot Turbo System (Bio-Rad). Blocking and antibody dilution were done in 5% milk, and the antibodies used were anti-pSMAD2/3, pSMAD1/5, pERK1/2, pLCK, pFyn, SMAD, and β-actin, as well as horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgG (Cell Signaling Technology).
Measurement of calcium flux in T cells
To measure differences in TCR engagement-dependent calcium flux, we used the Fluo-8 No Wash Calcium Assay Kit (Abcam, Cambridge, UK) following the manufacturer’s instructions. Jurkat cells were seeded on poly-L-lysin (Sigma-Aldrich, Merck, Darmstadt, Germany) coated black μCLEAR 96-well flat-bottom microplates (Greiner Bio-One, Kremsmünster, Austria) and spun down at 800 × g for 5 min. The cells were carefully preincubated with Fluo-8 from the kit with or without 100 ng/mL recombinant human TGF-β1 (R&D Systems) for 1 h at 37°C in serum-free RPMI medium. After this incubation time, we swiftly but carefully added one of the following compounds to the cells: anti-CD3 (5 μg/mL; eBioscience), ionomycin (1 μg/mL; Sigma-Aldrich, Merck), or EDTA (10 mM; Promega) and immediately started the measurement of fluorescence at 37°C using the Hidex Sense Microplate Reader.
Fluorescence staining for confocal microscopy
Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) and permeabilized with PBST (PBS with 0.1% Triton X-100; Bio-Rad). They were then blocked with 3% goat serum (Sigma-Aldrich, Merck) in PBST and incubated with the primary antibody mix for 2 h. Staining reagents and fluorescein-conjugated antibodies used were anti-TβRII (1:200; Miltenyi Biotec), anti-calnexin (1:500; Invitrogen, Thermo Fisher Scientific), and Hoechst BV421 (1:1,000; Thermo Fisher Scientific). Hoechst was added for 10 min as a nuclear counterstain, and cells were mounted on a poly-d-lysine-coated CellCarrier-384 black fluorescence-compatible plate. Images were acquired using a Zeiss LSM800 confocal microscope and analyzed using Zenblack software.
Measurement of cytokine production by Luminex multiplex assay
To assess the effect of the TGF-β decoy receptor on cytokine release in the presence of TGF-β, cell culture supernatants were measured using the immunology multiplex assay from Millipore (Milliplex Map human cytokine/chemokine magnetic bead panel; #HCYTMAG60PMX41BK). Supernatants of ATCs cultured in the presence of TGF-β for 4 days were harvested and analyzed using Flexmap 3D (Luminex) and Bio-plex Manager software (Bio-Rad). Data outside the range of standards, but within the asymptote of the equation, were extrapolated beyond the standard curve.
For the TGF-β capture assay, Jurkat cells or ATCs (1 × 105 cells/well in a 96-well plate) were incubated with serum-free growth medium containing increasing concentrations of recombinant TGF-β1 for 6 h. Supernatant was harvested and analyzed for remaining TGF-β using Bioplex Pro TGF-β Assays (Bio-Rad; #171W4001M). Dilutions were as recommended by the manufacturer and plates were read and analyzed as seen above.
Statistical analysis
Quantitative data were statistically analyzed assuming equal variance. The paired or unpaired Student’s t test was used to test for significance in each set of values. Mean values ± SD are given unless otherwise stated.
Acknowledgments
This study was fully funded by Tessa Therapeutics. The authors would like to express their gratitude to Ivan Horak for his continued support to publish this study.
Author contributions
J.E.C. conceived the idea and supervised the project. S.H.P. performed the majority of experiments, processed the experimental data, performed the analysis, drafted the manuscript, and designed the figures. K.A.B. performed experiments and the analysis. R.H. was involved in planning and supervising the work and aided in interpreting the results. D.L.V., M.R., and S.M.P.M. manufactured samples, performed experiments, and supported the work with their scientific assistance. All authors were involved in the submission of this manuscript.
Declaration of interests
All authors were employed by Tessa Therapeutics at the time the experiments were planned and conducted. The disclosed information is correct, no artificial intelligence (AI) or AI-assisted technologies were used to generate any of part of the manuscript, and no other situation of real, potential, or apparent conflict of interest is known to us.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2023.100730.
Supplemental information
Data and code availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material. Raw data of this study can be accessed upon reasonable request from the corresponding author.
References
- 1.Gajewski T.F., Meng Y., Harlin H. Immune suppression in the tumor microenvironment. J. Immunother. 2006;29:233–240. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro Franco P.I., Rodrigues A.P., de Menezes L.B., Pacheco Miguel M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020;216 doi: 10.1016/j.prp.2019.152729. [DOI] [PubMed] [Google Scholar]
- 3.Principe D.R., DeCant B., Mascariñas E., Wayne E.A., Diaz A.M., Akagi N., Hwang R., Pasche B., Dawson D.W., Fang D., et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016;76:2525–2539. doi: 10.1158/0008-5472.CAN-15-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouce R.H., Shaim H., Sekine T., Weber G., Ballard B., Ku S., Barese C., Murali V., Wu M.F., Liu H., et al. The TGF-beta/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia. 2016;30:800–811. doi: 10.1038/leu.2015.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z., Zhang L.J., Zhang H.R., Tian G.F., Tian J., Mao X.L., Jia Z.H., Meng Z.Y., Zhao L.Q., Yin Z.N., Wu Z.Z. Tumor-derived transforming growth factor-beta is critical for tumor progression and evasion from immune surveillance. Asian Pac. J. Cancer Prev. 2014;15:5181–5186. doi: 10.7314/apjcp.2014.15.13.5181. [DOI] [PubMed] [Google Scholar]
- 6.Yu Y., Feng X.H. TGF-beta signaling in cell fate control and cancer. Curr. Opin. Cel Biol. 2019;61:56–63. doi: 10.1016/j.ceb.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Massagué J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 8.Heldin C.H., Miyazono K., ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 9.Rooke H.M., Crosier K.E. The smad proteins and TGFbeta signalling: uncovering a pathway critical in cancer. Pathology. 2001;33:73–84. [PubMed] [Google Scholar]
- 10.Drabsch Y., ten Dijke P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012;31:553–568. doi: 10.1007/s10555-012-9375-7. [DOI] [PubMed] [Google Scholar]
- 11.Bellam N., Pasche B. Tgf-beta signaling alterations and colon cancer. Cancer Treat. Res. 2010;155:85–103. doi: 10.1007/978-1-4419-6033-7_5. [DOI] [PubMed] [Google Scholar]
- 12.Qiu W., Schönleben F., Li X., Su G.H. Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 2007;245:163–170. doi: 10.1016/j.canlet.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kretzschmar M. Transforming growth factor-beta and breast cancer: Transforming growth factor-beta/SMAD signaling defects and cancer. Breast Cancer Res. 2000;2:107–115. doi: 10.1186/bcr42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D., Kanuma T., Mizumuma H., Ibuki Y., Takenoshita S. Mutation analysis of the Smad6 and Smad7 gene in human ovarian cancers. Int. J. Oncol. 2000;17:1087–1091. doi: 10.3892/ijo.17.6.1087. [DOI] [PubMed] [Google Scholar]
- 15.Xu J., Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor beta signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA. 2000;97:4820–4825. doi: 10.1073/pnas.97.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S.J., Im Y.H., Markowitz S.D., Bang Y.J. Molecular mechanisms of inactivation of TGF-beta receptors during carcinogenesis. Cytokine Growth Factor. Rev. 2000;11:159–168. doi: 10.1016/s1359-6101(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 17.Wieser R., Attisano L., Wrana J.L., Massagué J. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol. Cel. Biol. 1993;13:7239–7247. doi: 10.1128/mcb.13.12.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bollard C.M., Rössig C., Calonge M.J., Huls M.H., Wagner H.J., Massague J., Brenner M.K., Heslop H.E., Rooney C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 19.Foster A.E., Dotti G., Lu A., Khalil M., Brenner M.K., Heslop H.E., Rooney C.M., Bollard C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 2008;31:500–505. doi: 10.1097/CJI.0b013e318177092b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bollard C.M., Tripic T., Cruz C.R., Dotti G., Gottschalk S., Torrano V., Dakhova O., Carrum G., Ramos C.A., Liu H., et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J. Clin. Oncol. 2018;36:1128–1139. doi: 10.1200/JCO.2017.74.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee S., Smith E.R., Hanada K., Stevens V.L., Mayor S. GPI anchoring leads to sphingolipid-dependent retention of endocytosed proteins in the recycling endosomal compartment. EMBO J. 2001;20:1583–1592. doi: 10.1093/emboj/20.7.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varshney P., Yadav V., Saini N. Lipid rafts in immune signalling: current progress and future perspective. Immunology. 2016;149:13–24. doi: 10.1111/imm.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horejsi V., Hrdinka M. Membrane microdomains in immunoreceptor signaling. FEBS Lett. 2014;588:2392–2397. doi: 10.1016/j.febslet.2014.05.047. [DOI] [PubMed] [Google Scholar]
- 24.Otáhal P., Angelisová P., Hrdinka M., Brdicka T., Novák P., Drbal K., Horejsí V. A new type of membrane raft-like microdomains and their possible involvement in TCR signaling. J. Immunol. 2010;184:3689–3696. doi: 10.4049/jimmunol.0902075. [DOI] [PubMed] [Google Scholar]
- 25.Davidson D., Bakinowski M., Thomas M.L., Horejsi V., Veillette A. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol. Cel. Biol. 2003;23:2017–2028. doi: 10.1128/mcb.23.6.2017-2028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinic J., Riehl A., Adler J., Parmryd I. The T cell receptor resides in ordered plasma membrane nanodomains that aggregate upon patching of the receptor. Sci. Rep. 2015;5 doi: 10.1038/srep10082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yashiro-Ohtani Y., Zhou X.Y., Toyo-Oka K., Tai X.G., Park C.S., Hamaoka T., Abe R., Miyake K., Fujiwara H. Non-CD28 costimulatory molecules present in T cell rafts induce T cell costimulation by enhancing the association of TCR with rafts. J. Immunol. 2000;164:1251–1259. doi: 10.4049/jimmunol.164.3.1251. [DOI] [PubMed] [Google Scholar]
- 28.Young M.H., Navarro G., Askovich P., Aderem A. T cell receptor signaling and Toll-like receptor signaling converge to amplify T cell responses. J. Immunol. 2016;196:128.5. [Google Scholar]
- 29.Lewis R.S. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 30.Trebak M., Kinet J.P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019;19:154–169. doi: 10.1038/s41577-018-0110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choudhry M.A., Sir O., Sayeed M.M. TGF-beta abrogates TCR-mediated signaling by upregulating tyrosine phosphatases in T cells. Shock. 2001;15:193–199. doi: 10.1097/00024382-200115030-00006. [DOI] [PubMed] [Google Scholar]
- 32.Chen C.H., Seguin-Devaux C., Burke N.A., Oriss T.B., Watkins S.C., Clipstone N., Ray A. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J. Exp. Med. 2003;197:1689–1699. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayor S., Riezman H. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cel Biol. 2004;5:110–120. doi: 10.1038/nrm1309. [DOI] [PubMed] [Google Scholar]
- 34.Sabharanjak S., Sharma P., Parton R.G., Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cel. 2002;2:411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 35.Sharma P., Sabharanjak S., Mayor S. Endocytosis of lipid rafts: an identity crisis. Semin. Cel Dev. Biol. 2002;13:205–214. doi: 10.1016/s1084-9521(02)00049-6. [DOI] [PubMed] [Google Scholar]
- 36.Haque S., Morris J.C. Transforming growth factor-beta: A therapeutic target for cancer. Hum. Vaccin. Immunother. 2017;13:1741–1750. doi: 10.1080/21645515.2017.1327107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gulley J.L., Madan R.A., Pachynski R., Mulders P., Sheikh N.A., Trager J., Drake C.G. Role of Antigen Spread and Distinctive Characteristics of Immunotherapy in Cancer Treatment. J. Natl. Cancer Inst. 2017;109 doi: 10.1093/jnci/djw261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fox F.E., Capocasale R.J., Ford H.C., Lamb R.J., Moore J.S., Nowell P.C. Transforming growth factor-beta inhibits human T-cell proliferation through multiple targets. Lymphokine Cytokine Res. 1992;11:299–305. [PubMed] [Google Scholar]
- 39.Delisle J.S., Giroux M., Boucher G., Landry J.R., Hardy M.P., Lemieux S., Jones R.G., Wilhelm B.T., Perreault C. The TGF-beta-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 2013;14:115–126. doi: 10.1038/gene.2012.63. [DOI] [PubMed] [Google Scholar]
- 40.Das L., Levine A.D. TGF-beta inhibits IL-2 production and promotes cell cycle arrest in TCR-activated effector/memory T cells in the presence of sustained TCR signal transduction. J. Immunol. 2008;180:1490–1498. doi: 10.4049/jimmunol.180.3.1490. [DOI] [PubMed] [Google Scholar]
- 41.Golumba-Nagy V., Kuehle J., Hombach A.A., Abken H. CD28-zeta CAR T Cells Resist TGF-beta Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018;26:2218–2230. doi: 10.1016/j.ymthe.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruffini P.A., Rivoltini L., Silvani A., Boiardi A., Parmiani G. Factors, including transforming growth factor beta, released in the glioblastoma residual cavity, impair activity of adherent lymphokine-activated killer cells. Cancer Immunol. Immunother. 1993;36:409–416. doi: 10.1007/bf01742258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mulé J.J., Schwarz S.L., Roberts A.B., Sporn M.B., Rosenberg S.A. Transforming growth factor-beta inhibits the in vitro generation of lymphokine-activated killer cells and cytotoxic T cells. Cancer Immunol. Immunother. 1988;26:95–100. doi: 10.1007/bf00205600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobie J.J., Wu R.S., Kurt R.A., Lou S., Adelman M.K., Whitesell L.J., Ramanathapuram L.V., Arteaga C.L., Akporiaye E.T. Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003;63:1860–1864. [PubMed] [Google Scholar]
- 45.Kao J.Y., Gong Y., Chen C.M., Zheng Q.D., Chen J.J. Tumor-derived TGF-beta reduces the efficacy of dendritic cell/tumor fusion vaccine. J. Immunol. 2003;170:3806–3811. doi: 10.4049/jimmunol.170.7.3806. [DOI] [PubMed] [Google Scholar]
- 46.Chen W., Wahl S.M. TGF-beta: the missing link in CD4+CD25+ regulatory T cell-mediated immunosuppression. Cytokine Growth Factor. Rev. 2003;14:85–89. doi: 10.1016/s1359-6101(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 47.Kloss C.C., Lee J., Zhang A., Chen F., Melenhorst J.J., Lacey S.F., Maus M.V., Fraietta J.A., Zhao Y., June C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018;26:1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material. Raw data of this study can be accessed upon reasonable request from the corresponding author.