

SUMMARY
SORL1 is implicated in the pathogenesis of Alzheimer’s disease (AD) through genetic studies. To interrogate the roles of SORL1 in human brain cells, SORL1-null induced pluripotent stem cells (iPSCs) were differentiated to neuron, astrocyte, microglial, and endothelial cell fates. Loss of SORL1 leads to alterations in both overlapping and distinct pathways across cell types, with the greatest effects in neurons and astrocytes. SORL1 loss induces a neuron-specific reduction in apolipoprotein E (APOE) and clusterin (CLU) and altered lipid profiles. Analyses of iPSCs derived from a large cohort reveal a neuron-specific association between SORL1, APOE, and CLU levels, a finding validated in postmortem brain. Enhancement of retromer-mediated trafficking rescues tau phenotypes observed in SORL1-null neurons but does not rescue APOE levels. Pathway analyses implicate transforming growth factor β (TGF-β)/SMAD signaling in SORL1 function, and modulating SMAD signaling in neurons alters APOE RNA levels in a SORL1-dependent manner. Taken together, these data provide a mechanistic link between strong genetic risk factors for AD.
In brief
Lee et al. use SORL1-null iPSCs to identify genes and pathways altered by loss of SORL1 across brain cell types. These studies reveal that SORL1 has a neuron-specific role in regulating APOE and CLU levels and implicate TGF-β-mediated SMAD signaling in this process.
Graphical Abstract
INTRODUCTION
SORL1 belongs to both the vacuolar protein sorting 10 containing receptor family and the low-density lipoprotein (LDL) receptor family. Single-nucleotide polymorphisms (SNPs) at the SORL1 locus are associated with AD in genome-wide association studies (GWASs).1,2 Importantly, several SORL1 coding variants have been identified in a subset of patients with early-onset Alzheimer’s disease (EOAD),3–6 and variants leading to premature termination codons are found exclusively in Alzheimer’s disease (AD) cases.5 Further, reduced expression of SORL1 has been observed in the cerebrospinal fluid (CSF) and postmortem brain of individuals with late-onset AD.7–9 Together, these data suggest that SORL1 plays a causal role in AD.10
SORL1 has a major role in intracellular trafficking of various cargos, including amyloid precursor protein (APP), which is mediated in part through its role in retromer complex-mediated trafficking (reviewed in Small and Gandy11). In the absence of SORL1, APP fails to be transported out of the endosome and this leads to an increase in generation of Aβ.12–15 In induced pluripotent stem cell (iPSC)-derived neurons, loss of SORL1 leads to impaired endosomal trafficking and autophagy and, ultimately, increased Aβ levels.16,17 Consistent with these findings, loss of SORL1 in AD mouse models leads to an increase in Aβ levels in the brain.18
In addition to SORL1, GWASs have identified over 70 loci associated with AD risk,2 including variants in two apolipoprotein genes, APOE and CLU. The strongest genetic risk factor for late-onset Alzheimer’s disease (LOAD) is the ε4 genotype of apolipoprotein E (APOE), a protein that has a primary function in cholesterol metabolism in its role as a lipoprotein.19–21 In addition, APOE has been implicated in a variety of AD-relevant processes, including oligomerization of Aβ, enhancement of neurodegeneration in response to pathologic tau, impairment in endocytic trafficking, and dysfunction of microglia (reviewed in Martens et al.22). Recent studies have implicated a neuron-specific role for APOE in immune response pathways.23,24
SORL1 is also referred to as LR11, or LDL receptor with 11 class A ligand-binding repeats, and the presence of these repeats suggests it also may function as a lipoprotein receptor. Previous studies have shown that SORL1 can regulate the uptake of various lipoproteins in Chinese hamster ovary (CHO) cells,25 and exogenously overexpressed SORL1 in HEK293 cells can bind to APOE in an isoform-dependent manner.26 However, to understand if and how SORL1 and APOE may interact to affect AD risk, it is important to use a more physiologically relevant system wherein SORL1 is expressed at endogenous levels in cell types found in the brain. Moreover, the consequences of SORL1 and APOE interaction, if any, are unknown.
Here, we performed transcriptomic profiling comparing wild-type (WT) to SORL1-null iPSC lines that were differentiated to a variety of brain cell types (neurons, astrocytes, microglia, endothelial/epithelial cells). This screen revealed that neurons and astrocytes are most sensitive to loss of SORL1 and that overlapping and distinct pathways are altered across cell types. We found that loss of SORL1 leads to a significant reduction of APOE and CLU protein levels specifically in neurons but not in other tested cell types. In addition, loss of SORL1 in neurons led to an increase in Aβ levels and phosphorylation of tau, which is rescued by enhancing retromer or autophagy function in SORL1-null neurons. However, reduced APOE levels were not rescued with these treatments. Pathway analyses of multiple datasets implicated transforming growth factor β (TGF-β)/SMAD signaling in SORL1 biology, and stimulation and inhibition of this pathway modulated APOE RNA and protein levels. We further interrogated the relationship between APOE, CLU, and SORL1 in a set of 50 iPSC lines derived from the Religious Order Studies and Memory and Aging Project (ROSMAP) aging cohorts. Intriguingly, following differentiation, natural variation in SORL1 levels was strongly associated with both APOE and CLU protein levels in neurons but not in astrocytes. Analyses of single-nucleus RNA sequencing (snRNA-seq) from brain tissue revealed a significant association of SORL1 with both APOE and CLU expression in neurons, supporting the relevance of this relationship in the aged human brain. Last, SORL1 mutant neurons displayed altered lipidomic profiles and lipid droplet (LD) accumulation, phenotypes also observed in previously published studies of APOE knockout (KO) and APOE ε4 iPSC derivatives.27,28 Taken together, we demonstrate that AD-relevant SORL1 loss of function results in neuron-specific reduction in APOE and CLU and dysregulated lipid homeostasis.
RESULTS
Astrocytes and neurons are most sensitive to loss of SORL1
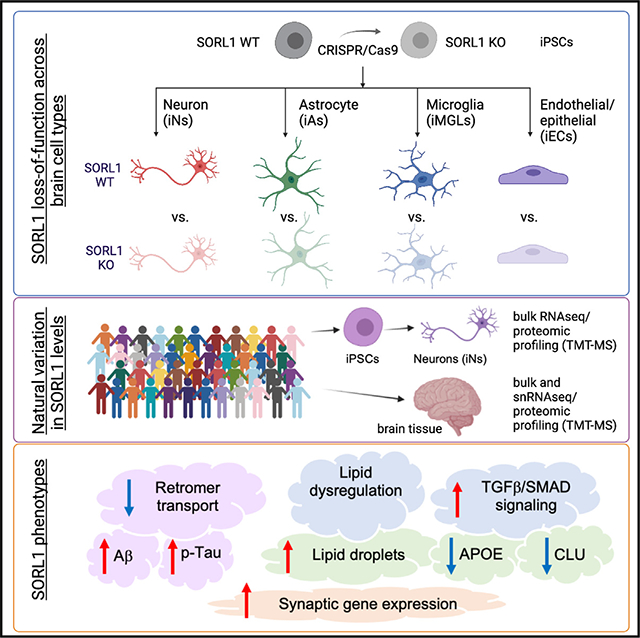
SORL1 is expressed in many cell types of the brain, and an unanswered question in the field is which cell type(s) are most sensitive to a reduction in levels of SORL1. Here, we compared gene expression in SORL1 KO cells to WT cells across cell types. We utilized a previously described pair of WT and SORL1 homozygous KO iPSC lines derived from a healthy male.16 Paired WT and KO iPSCs were differentiated into neurons (iNs), astrocytes (iAs), microglia (iMGLs), and endothelial/epithelial cells (iECs) following published protocols (Figures 1A and S1A–S1E). In brief, pure (>95%) cultures of iNs or iAs were generated using transduction of lentivirus expressing either NGN2 (a transcription factor that induces neuronal fate) or both SOX9 and NFIB (transcription factors shown to induce astrocyte fate), using a Tet-inducible system.29,30 These protocols generate brain cell types that have been extensively characterized.31–34 iMGLs were differentiated using a protocol that utilizes a hematopoietic stem cell intermediary to generate microglia-like cells, >95% of which express the microglial marker IBA1, as previously described.35,36 iECs were generated by differentiation into endothelial/epithelial-like cells as described.37–39 Once the differentiations were complete (at iN day 21 [D21], iA D21, iMGL D40, and iEC D14), samples were harvested for transcriptomic analysis and immunostaining. As previously described, cell cultures expressed expected cell-type-specific markers, as assayed by immunostaining and RNA sequencing (RNA-seq) (Figures 1B and 1C; Table S1).
Figure 1. APOE and CLU RNA and protein levels are reduced in SORL1 KO neurons.
(A) Schematic of the experimental design. SORL1 KO iPSCs were generated using CRISPR-Cas9 as previously published,16 and paired KO and WT iPSCs were differentiated into neurons (iNs), astrocytes (iAs), microglia (iMGLs), and endothelial/epithelial cell (iEC) fates.
(B) Representative immunocytochemistry images of iN, iA, iMGL, and iEC cultures. Scale bars, 50 μm.
(C) Heatmap of RNA expression (Z score) of cell-type-specific markers.
(D and E) Cultures were analyzed via RNA-seq and differential expression analyses performed within each cell type. DEGs were identified using Sleuth40 by performing a Wald test (false discovery rate [FDR] q < 0.05 and b < −0.5 or b > 0.5). The numbers of DEGs between SORL1 WT and KO in each cell type are shown as a table (D) and as a Venn diagram (E). Complete datasets can be found in Tables S1.
(F) Dot plot showing significantly overrepresented Gene Ontology (GO) pathways in iNs, iAs, iMGLs, and iECs comparing SORL1 KO and WT within each cell type. Dots are sized based on the number of DEGs in the indicated pathway and colored based on adjusted p value.
(G) Venn diagram of LOAD GWAS genes that are differentially expressed in SORL1 KO neurons, astrocytes, and microglia. Genes with adjusted p <0.05 are included in the diagram.
(H) Representative western blot of iNs, iAs, iMGLs, and iECs showing protein expression levels of SORL1, APOE, and GAPDH.
(I–L) Quantification of APOE/GAPDH in iNs, iAs, iMGLs, and iECs.
(M–P) Secreted APOE and CLU values were measured using MSD ELISA, and the values were normalized to WT within each comparison. For each panel, three independent differentiations were performed. In each round of differentiation, three or four wells were included for each group. Comparisons only performed within cell type; paired Student’s t test (two tailed), p values listed.
To determine which cell type is most sensitive to loss of SORL1, we performed differential expression analyses to compare WT to SORL1 KO cells within each fate. Neurons and astrocytes showed the highest number of differentially expressed genes (DEGs) compared to iPSCs, microglia, and endothelial cells (Figures 1D and 1E; Table S1). Enrichment analyses identified pathways that are differentially enriched in each cell type (Table S1). Some pathway alterations were shared across cell types, while others were specific to a single cell type (Figures 1E, 1F, and S1F). In parallel to RNA-seq, unbiased proteomic profiling also was performed in each cell type to support the RNA-seq analyses but were not powered or designed for discovery (Table S1). Pathways consistently altered in both RNA and protein datasets include “extracellular matrix organization” between neurons, astrocytes, and microglia; “pathway-restricted SMAD phosphorylation” between neurons and astrocytes; and gene sets relating to cytokine production between astrocytes and microglia (Figure S1F; Table S1). Cell-type-specific enrichment was observed in terms such as “lysosome localization” in neurons and “regulation of lipid localization” in astrocytes (Figures 1F and S1F). These analyses provide a preliminary view into the shared and cell-type-specific genes and pathways altered with SORL1 loss of function across human brain cell types.
APOE and CLU levels are reduced in SORL1 KO neurons
We next investigated whether any genes implicated in LOAD GWAS were differentially expressed. Intriguingly, several LOAD candidate genes showed disrupted expression in SORL1-null iNs, iAs, or iMGLs (Figures 1G and S2A–S2O). Due to the introduction of a premature stop codon, nonsense-mediated decay results in reduced SORL1 levels across all cell types (Figure S2C). Microglia expressed the highest RNA levels of SORL1 in WT cells and consequently the highest levels of RNA in the KO (likely due to the rates of nonsense-mediated decay). Protein SORL1 levels were undetectable by western blot (WB) in all KO cell types (Figure 1H). BIN1, CD2AP, and PICALM, genes with known trafficking roles, and two apolipoproteins, APOE and CLU (APOJ), showed cell-type-specific changes in their expression levels (Figures 1G, S2B, S2D–S2G, and S2N). With the loss of SORL1, APOE and CLU RNA levels were reduced in neurons but elevated in astrocytes (Figures S2F and S2G). APOE and CLU were not affected by SORL1 loss in iPSCs, microglia, and endothelial/epithelial cells, suggesting cell-type-dependent relationships. In SORL1 KO neurons, RNA levels of additional apolipoproteins also were reduced, including APOC1 and APOL4, while, in SORL1 KO astrocytes, APOE and CLU were the only apolipoproteins elevated as a result of loss of SORL1 (Table S1).
Western blot of iNs, iAs, iMGLs, and iECs showed that iMGLs showed the highest expression of SORL1, consistent with the RNA-seq results (Figure 1H). Consistent with RNA-seq, APOE protein levels were significantly decreased in SORL1 KO neurons (Figure 1I). In comparison, there were no detectable differences in APOE protein levels in SORL1 KO astrocytes, microglia, or endothelial cells (Figures 1J–1L). Secreted APOE levels in the media also showed a dramatic reduction (Figure 1M). The impact of loss of SORL1 was not isolated to APOE, as secreted CLU levels also were reduced in SORL1 KO iNs (Figure 1N). Secreted APOE and secreted CLU were not significantly changed in SORL1 KO astrocytes (Figures 1O and 1P). This cell-type dependency of regulation could explain why changes in APOE and CLU have not been previously reported and underscores the importance of studying the cell-type-specific roles of AD genes of interest.
SORL1 KO neurons show elevated Aβ and increased phosphorylation of tau
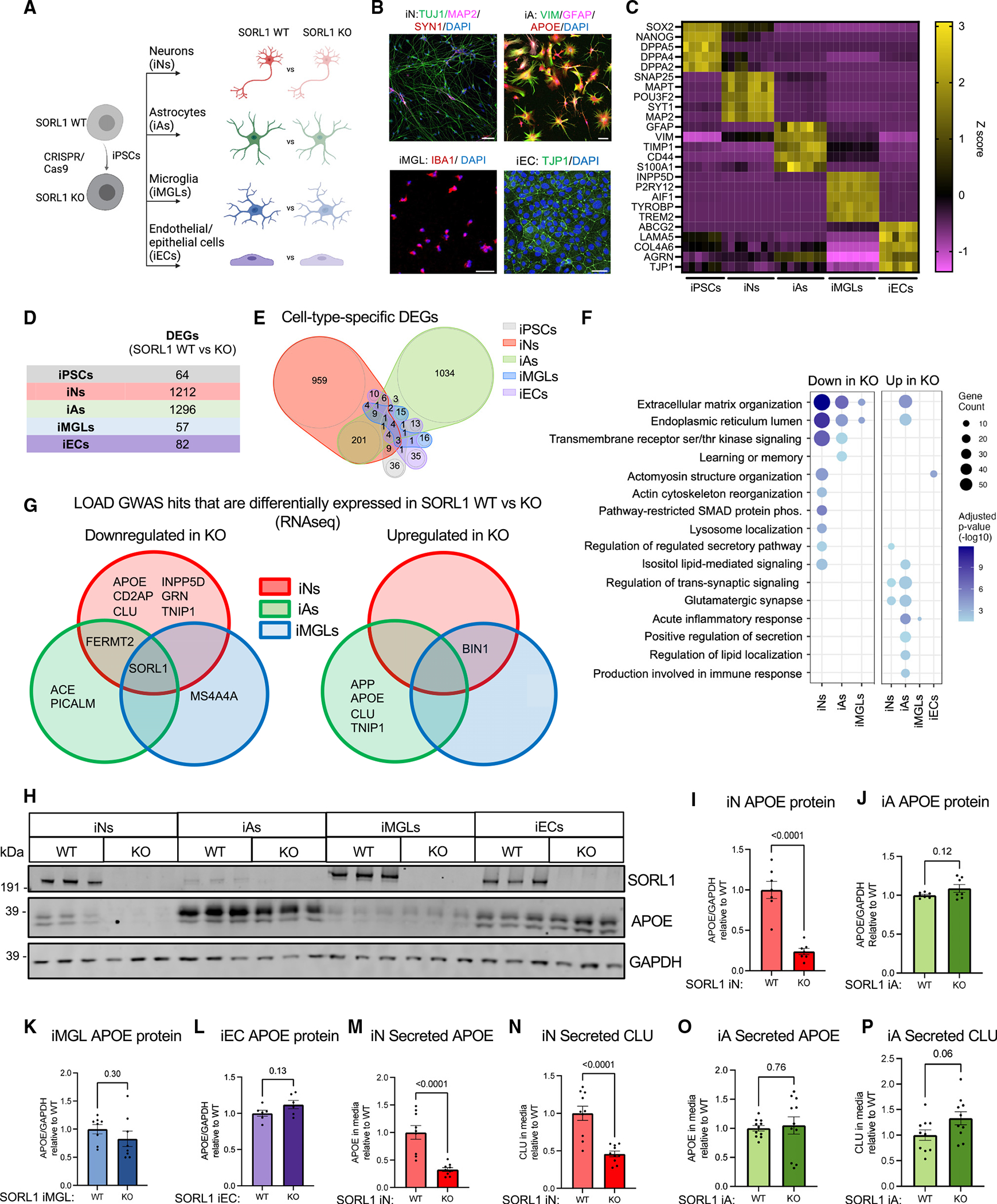
To test the reproducibility of findings in SORL1 KO iNs, we next used CRISPR-Cas9 editing to generate a second isogenic pair of SORL1-null iPSCs (Figure 2A). The second cell line was generated from a previously described female iPSC line.31 A guide RNA (gRNA) targeting a different exon was employed to generate cell lines with an indel introduced at the SORL1 locus that results in nonsense-mediated decay with no off-target mutations observed (Figures S3A–S3E). The two pairs of WT and SORL1 KO iPSC lines were differentiated into neurons (iNs), and all four lines showed robust differentiation to neuronal fate with no major differences in morphology as depicted by immunostaining with neuronal markers (Figure 2B). iN lysates from both cell line pairs show that protein expression of SORL1 is eliminated in SORL1 KO lines (Figure 2C). Consistent with our observations in line 1 iNs, intracellular and extracellular APOE and CLU protein levels were dramatically reduced in SORL1 KO iNs in line 2 (Figures 2C–2F and S4A–S4C). These data in a second genetic background strengthen the findings that SORL1 regulates levels of neuronal APOE and CLU.
Figure 2. SORL1 KO neurons express elevated Aβ and increased phosphorylation of tau.
(A) Overview of the experimental design to generate SORL1 KO neurons. SORL1 KO iPSCs were generated using CRISPR-Cas9 and were differentiated into neurons using induced expression of NGN2.
(B) Representative immunocytochemistry images of D21 iNs showing the expression of neuronal markers. Scale bars, 20 μm.
(C) Representative western blot of iN D21 protein lysates from line 1 and line 2.
(D–F) Quantifications within cell line pair 2 of APOE/GAPDH in lysates (D), APOE in the media (E), and CLU in the media (F) normalized to total protein are shown.
(G) Aβ42 levels in the media normalized to protein concentration for cell line pairs 1 and 2.
(H) Quantification of p-tau (p202/205)/total tau ratio in iN D21 protein lysates from western blot data. SORL1 KO iNs from pairs 1 and 2 both show increased phosphorylation of tau. Data in (D)–(H) show mean ± SEM from three independent differentiations. Within each round of differentiation, three wells were included for each group. Values are normalized to WT within paired lines. Unpaired Student’s t test (two tailed), p values are shown.
(I) Dot plot of molecular pathway over-representation in up- or downregulated DEGs in both lines 1 and 2. All pathway enrichments have an adjusted p value <0.05).
(J–L) Heatmap of a subset of genes that are differentially expressed in SORL1 WT vs. KO iNs and their relative expression levels (Z score) from selected GO terms in (I). Full datasets can be found in Table S2.
Multiple studies have shown that APP is a cargo of SORL1 in its neuronal retromer role and loss of SORL1 leads to retention of APP in endosomes, resulting in an increase in Aβ generation.13,16,18 Furthermore, it was previously shown that, in SORL1 KO neurons, retention of APP in early endosomes leads to enlarged endosomes and increased Aβ levels.16 As predicted, SORL1 KO iNs show an increase in Aβ42 levels (Figure 2G). All Aβ peptides detected by MSD ELISA were elevated in SORL1 KO iNs, without alteration of the Aβ42/40 ratio or protein levels of full-length APP, suggesting that SORL1 affects the generation of Aβ but does not alter the processivity of gamma-secretase (Figure S4D–S4M). In addition to Aβ, the second neuropathological hallmark of AD is the accumulation of intracellular tau tangles. Prior to overt tangle formation, an elevation of phosphorylation of tau occurs. SORL1 KO iNs showed an increase in phosphorylated tau levels (as measured by AT8) relative to total tau (Figures 2C and 2H).
We next examined neurons derived from an isogenic set of APOE KO, ε4/ε4, and ε3/ε3 iPSCs. SORL1 protein levels and p-ta u/tau levels were not altered across these iNs (Figures S5A–S5C), and knockdown of APOE using short hairpin RNA (shRNA) did not affect SORL1 levels (Figures S5D–S5G). Importantly, p-tau levels were not altered in APOE KO neurons, suggesting that the reduction in APOE observed in SORL1 KO iN is not sufficient to induce the p-tau phenotype.
Loss of SORL1 in human neurons induces a downregulation of genes associated with endocytosis, SMAD signaling, and extracellular matrix organization and an upregulation of genes encoding synaptic proteins
To identify pathways affected by loss of SORL1 and therefore potentially contributing to the increase in phosphorylated tau and reduction of APOE in SORL1 KO iNs, DEGs were identified that are concordantly altered between SORL1 WT and KO in both genetic backgrounds (full dataset for line 2 in Table S2, enriched Gene Ontology [GO] pathways in SORL1 KO vs. WT iNs in both line 1 and line 2 in Table S3). GO analysis revealed biological processes associated with SORL1 levels including terms also identified in the initial screen of DEGs across cell types, such as “extracellular matrix organization,” “regulation of pathway-restricted SMAD protein phosphorylation,” and “glutamatergic synapse,” as well as “regulation of endocytosis,” “endocytic vesicle,” and “protein secretion” (Figures 2I–2K). Importantly, each of the terms in Figure 2I also were replicated in an independently published RNA-seq dataset of SORL1 KO vs. WT neurons, which was generated using a different, non-virally transduced method for differentiation to neuronal fate41 (Table S3). Interestingly, several terms associated with lipids, such as “inositol lipid-mediated signaling” and “negative regulation of lipid biosynthetic process,” were downregulated in KO neurons, which supports the potential role of SORL1 in lipid metabolism in neurons (Figure 2L).
Enhancing retromer function via R33 treatment rescues elevated p-tau but not APOE
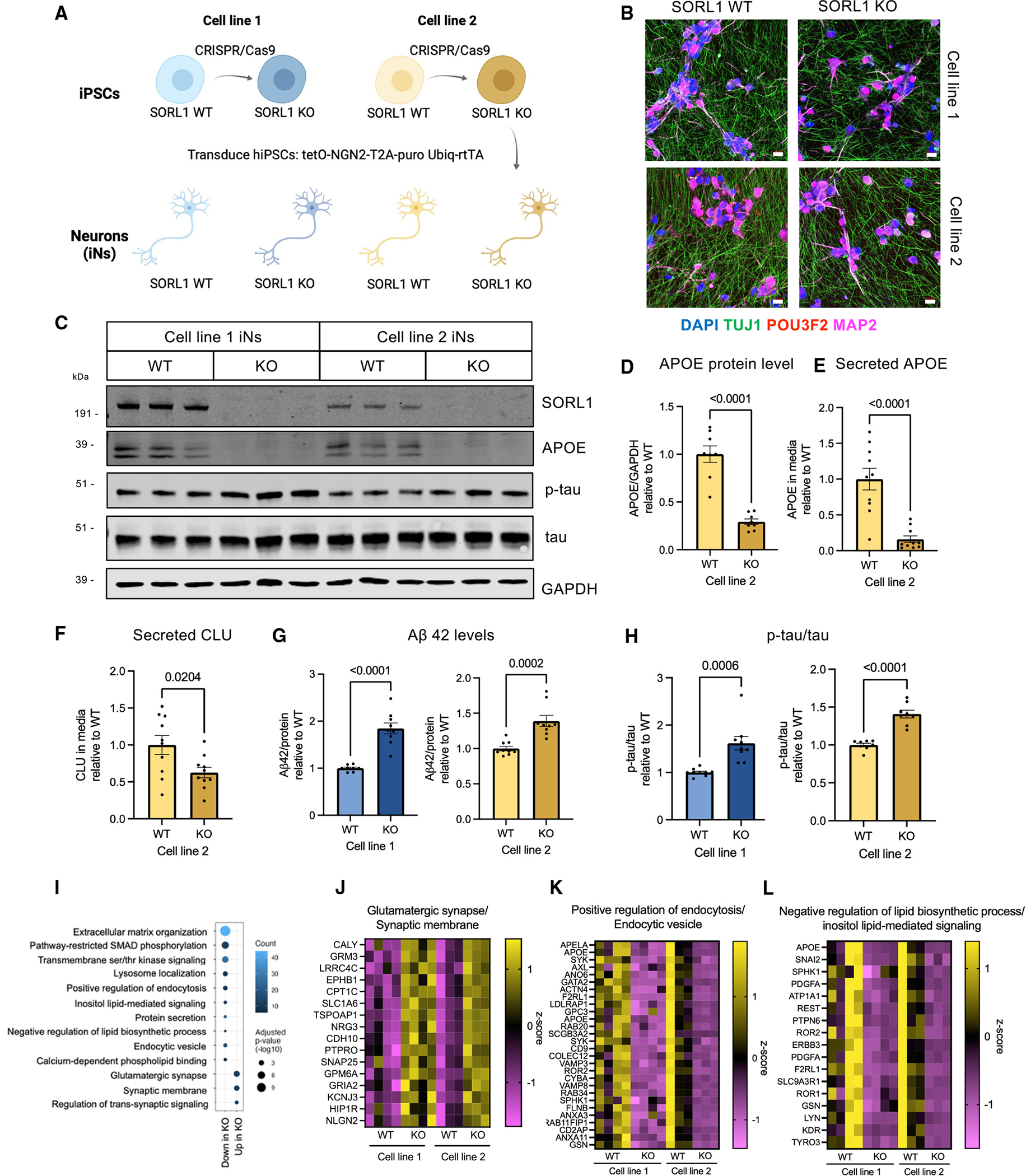
The retromer complex is a multiprotein assembly that is responsible for trafficking cargo from the endosome to the trans-Golgi network and recycling back to the plasma membrane. Recently, it has been shown that, in iPSC-derived neurons, SORL1 regulates retromer-dependent endosomal recycling in iPSC-derived neurons.41 To address whether the retromer complex is involved in driving the elevated p-tau or reduced APOE, iNs were treated with R33, a compound that stabilizes the retromer complex by putatively acting as a pharmacological chaperone.42 It previously has been shown that R33 treatment lowers Aβ generation and phosphorylated tau levels in sporadic AD neurons.43 Here, treatment with R33 at 10 μM for 72 h rescued elevated p-tau/tau in SORL1 KO iNs without altering the p-tau/tau levels of WT iNs. At a higher concentration (20 μM), R33 lowered p-tau in both WT and KO iNs (Figures 3A and 3B). Furthermore, 20 μM R33 treatment partially rescued Aβ levels in SORL1 KO iNs with no change in Aβ42/40 (Figures S6A and S6B), indicating that Aβ and p-tau phenotypes in SORL1 KO iNs are connected to the role SORL1 plays in regulating the retromer complex. However, the same R33 treatments were not sufficient to rescue the decreased APOE levels in SORL1 KO iNs, suggesting that the SORL1 KO APOE phenotype is separable from its Aβ and p-tau phenotypes (Figure 3C).
Figure 3. Enhancement of retromer function or autophagy rescues Aβ and tau phenotypes, while modulation of SMAD signaling regulates APOE levels in a SORL1-dependent manner.
(A–C) Line #1 iNs were treated with R33, a retromer stabilizer, at either 10 or 20 μM for 72 h and harvested at D21 of differentiation. A representative western blot of lysates from these SORL1 WT and KO iNs is shown in (A). Quantification shown of the protein expression of p-tau/tau (p202/205) (B) and APOE/GAPDH (C).
(D–F) iNs were treated with trehalose, a putative enhancer of autophagy, at 100 mM for 72 h and harvested at D21 of differentiation. A representative western blot of these lysate is shown in (D). Quantifications shown for p-tau/tau (p202/205) (E) and APOE/GAPDH (F).
(G–I) iNs were treated with 5 μM chloroquine (CQ) (to inhibit autophagic vesicle fusion with the lysosome) for 72 h and harvested at D21 of differentiation. A representative western blot of lysates is shown (G). Quantifications shown for LC3b-II/GAPDH (H) and APOE/GAPDH (I).
(J) SORL1 WT and KO iNs were treated with vehicle, TGF-β, or SB-431542 for 72 h. Lysates were collected, RNA purified, and qPCR performed for APOE and GAPDH (J, left) or else cells were lysed and western blotting was performed for APOE and GAPDH (J, right). For all quantifications in (B)–(J), data show mean ± SEM from three independent differentiations. For each round of differentiation, three replicates were included for each group. Values are normalized to WT iNs treated with vehicle. One-way ANOVA with Tukey’s multiple comparisons test.
(K) Schematic showing the intracellular life cycle of APOE, including a speculative model for the regulation of APOE by TGF-β signaling in neurons, with activation of TGF-β signaling resulting in repression of APOE transcription.
Modulating autophagy via chloroquine or trehalose rescues elevated p-tau but does not rescue APOE levels
Autophagy-related pathways were associated with SORL1 loss in the analyses performed herein, and a recent study reported that SORL1 KO neurons show a defect in autophagic flux,17 raising the possibility that dysregulated autophagy is mediating the effects observed on APOE levels. Alternatively, APOE levels may be regulated by loss of SORL1 via an increase in trafficking of APOE toward lysosomes and the degradation pathway. iNs were treated with 100 mM trehalose (a putative augmenter of autophagy) or 5 μM chloroquine (CQ; blocks fusion of autophagic vesicles with the lysosome) at D18. After 72 h of treatment, trehalose reduced the elevated p-tau levels but did not affect levels of Aβ or APOE in SORL1 KO iNs (Figures 3D–3F, S6C, and S6D). Similarly, while CQ treatment successfully blocked autophagy as measured by an increase in LC3b-II protein levels (Figures 3G and 3H), it did not significantly alter APOE levels in SORL1 WT or KO iNs (Figure 3I). Thus, modulation of autophagy by SORL1 loss does not appear to strongly contribute to APOE protein levels in iNs.
Modulation of SMAD signaling regulates APOE levels in neurons in a SORL1-dependent manner
TGF-β and SMAD signaling pathways were associated with SORL1 through multiple analyses (Figures 1E, 2I, and S1F), and were replicated in an independently generated RNA-seq dataset of SORL1 KO iPSC-derived neurons41 (Table S3). This association was of particular interest given the observation that SORL1 KO affected APOE RNA levels (Figure S2F) as well as protein levels (Figure 1H). SMAD proteins themselves were dysregulated in SORL1 KO iNs and inspection of the APOE genomic locus using the JASPAR database revealed putative SMAD2, SMAD3, and SMAD4 binding sites in the 3-kb region upstream of the APOE transcriptional start site as well as in the first intron and 3′ UTR (Table S4). TGF-β treatment reduced both RNA and protein levels of APOE, while treatment with an inhibitor of TGF-β receptor-mediated SMAD phosphorylation (SB-431542) increased levels of APOE RNA and protein levels in WT neurons (Figure 3J). This modulation of APOE was SORL1 dependent, as the same treatments had no effects on APOE levels in SORL1 KO iNs (Figure 3J). The results presented here suggest that TGF-β/SMAD signaling acts in concert with other factors to repress APOE transcription in iNs in a SORL1-dependent manner (Figure 3K).
SORL1 shows a strong positive association with APOE and CLU in neurons from the ROSMAP aging cohorts
While the KO vs. WT experimental system is valuable for uncovering cellular roles of SORL1, complete loss of SORL1 does not accurately represent a state that occurs in the human brain. To probe the relationship between SORL1 and APOE and other apolipoproteins in a system that captures genetically encoded variation in SORL1 levels in human neurons, we utilized a cohort of >50 iPSC lines derived from the ROSMAP cohorts.32 Previously, we reported the generation of these iPSC lines and showed that iNs from these individuals reflect significant features of the protein networks and neuropathology present in the brain tissue of the same individuals from whom the cells were derived.32 Here, we utilized this system to interrogate putative connections between apolipoproteins and SORL1. Individuals were selected to reflect varying degrees of pathological and clinical diagnosis, then iPSCs from these individuals were differentiated into neuron and astrocyte fates (Figures 4A, S1A, and S1C).
Figure 4. SORL1 levels show a strong positive correlation with APOE and CLU levels in iPSC-derived neurons from the ROSMAP cohort.
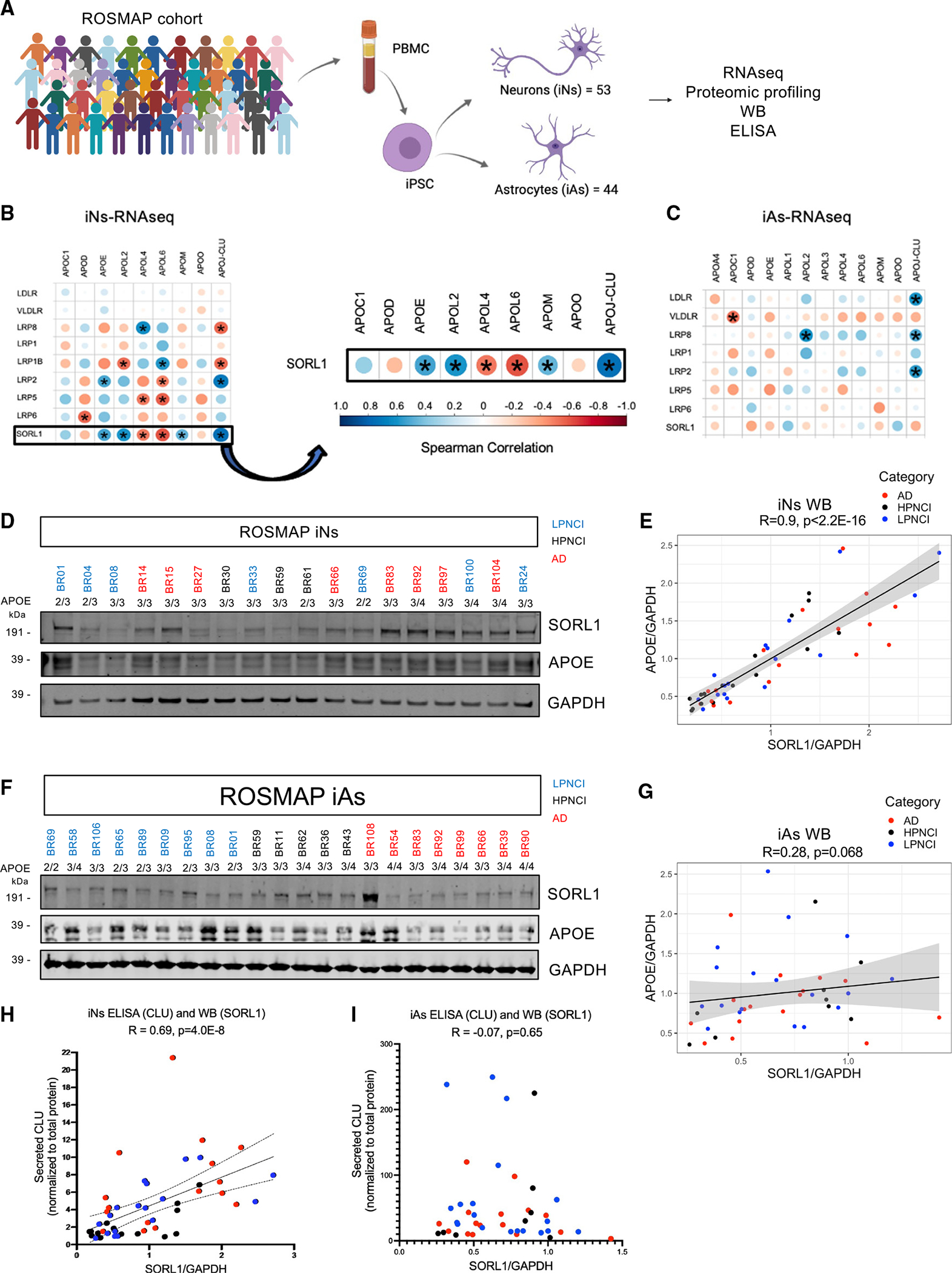
(A) Peripheral blood mononuclear cells (PBMCs) were collected from ROSMAP cohort participants, which were converted to iPSCs (previously published in Lagomarsino et al.32) and differentiated into neurons (iNs) and astrocytes (iAs). RNA-seq and proteomic profiling were then performed.
(B and C) RNA expression levels of SORL1 and other members of the LDL receptor family were each compared to APOE and members of the apolipoprotein family in both iNs (B) and iAs (C). Correlation dot plots are used to represent Spearman correlations for each comparison, with the size of the dot increasing with lower p values (asterisks indicate p < 0.001) and color indicating R value.
(D) A representative western blot image of SORL1, APOE, and GAPDH levels of ROSMAP iN protein lysates. Individuals have a unique ID that is colored based on their AD diagnosis status. LPNCI, low AD neuropathology not cognitively impaired; HPNCI, high AD neuropathology not cognitively impaired; AD, clinical and pathological diagnosis of Alzheimer’s disease and dementia.
(E) Spearman correlation between SORL1/GAPDH and APOE/GAPDH levels in iN lysates.
(F) Representative western blot image of SORL1, APOE, and GAPDH expression in ROSMAP iA protein lysates.
(G) Spearman correlation between SORL1/GAPDH and APOE/GAPDH levels in iA lysates. All western blots can be found in Figure S7.
(H and I) Conditioned media from the same cells were analyzed via ELISA to measure CLU levels. Shown are correlations between SORL1/GAPDH and CLU in iNs (H, R = 0.69, p = 4.0E−08) and iAs (I, R = −0.07, p = 0.65).
Analysis of RNA-seq data across these ROSMAP iNs and iAs revealed a high correlation between APOE and CLU with SORL1 in neurons but not astrocytes (Figures 4B and 4C). In addition, SORL1 levels were significantly positively correlated with a subset of other apolipoproteins in neurons (APOL2 and APOM). However, SORL1 levels were negatively correlated with APOL4 and APOL6 levels (Figure 4B). In addition, the association of lipoprotein receptors with APOE and CLU was specific to SORL1 and LRP2 in neurons, with other related lipoprotein receptors showing no such association (Figure 4B). CLU and APOE also showed robust positive correlations with SORL1 in iNs at the protein level, as measured by western blot (Figures 4D, 4E, S7A, and S7D) and ELISA of the conditioned media (Figure 4H). Concordant with the RNA-seq data, western blot and ELISA analyses of iAs derived from ROSMAP iPSCs showed no correlation between APOE or CLU and SORL1 (Figures 4F, 4G, 4I, and S7G). The iN experimental system captures the genetics of the individuals from whom the cells are derived, and varying levels of SORL1 and APOE are encoded at least in part within the genome of the individuals. It is important to note that, while AD diagnosis, SORL1 5′ haplotypes, and APOE genotype may contribute to their RNA and protein levels across iNs, they do not fully explain the variance in levels of SORL1 and APOE (Figures S7A–S7O). These data suggest that other genetic variants are contributing in a complex manner to affect levels of SORL1, which in turn affects APOE levels, and future studies with greater power will be required to uncover the genetic variants that mediate the expression levels of SORL1.
Unbiased analyses of RNA-seq and proteomic data from ROSMAP iNs identify genes and pathways associated with genetically encoded natural variation in SORL1 levels
To globally examine the individual genes and pathways associated with SORL1 in ROSMAP iNs in an unbiased manner, we calculated the correlation coefficients between SORL1 and all factors detected within both the RNA-seq and proteomics (tandem mass tag [TMT] mass spectrometry [MS]) datasets (Figure 5A; Table S5). This analysis allowed us to identify genes that were consistently associated with SORL1 at both the RNA and protein level across independent differentiations. While RNA and protein expression do not always agree given the important contributions of post-transcriptional and post-translational regulation, there was clear overall concordance between RNA and protein associations with SORL1 for a subset of genes (Figure 5A). First, we confirmed that SORL1 shows a strong positive correlation with APOE and CLU in both RNA and protein datasets (Figure 5B). Several genes of interest to SORL1 biology were identified in this association analysis, including the LOAD GWAS gene BIN1 and the retromer protein VPS26B (Figure 5C). Of note, BIN1 levels also were elevated with SORL1 KO in iNs (Figure 5D). In accord with observations in SORL1 KO iNs, pathway analyses revealed significant associations between SORL1 levels and intracellular transport pathways (Figure 5D). Elevated levels of proteins associated with tau protein kinase activity and cellular response to amyloid beta also were identified with lower SORL1 levels (Figure 5D), consistent with the observed increase in Aβ and p-tau observed in SORL1 KO iNs (Figure 3). Gene concept network analyses illustrate that SORL1-associated endosomal transport and autophagy pathways are linked to one another via VPS26B, CLEC16A, and TBC1D14, and to synaptic pathways via DNM1L and other genes (Figure 5E). Similarly, the SORL1-associated pathways relating to WNT and SMAD signaling were linked to one another via transcription co-regulator activity (Figure 5F). These datasets and analyses provide a framework in which to further probe the cellular consequences of loss of neuronal SORL1.
Figure 5. Unbiased RNA-seq and proteomics of ROSMAP iNs identify genes and pathways associated with genetically encoded natural variation in SORL1 levels.
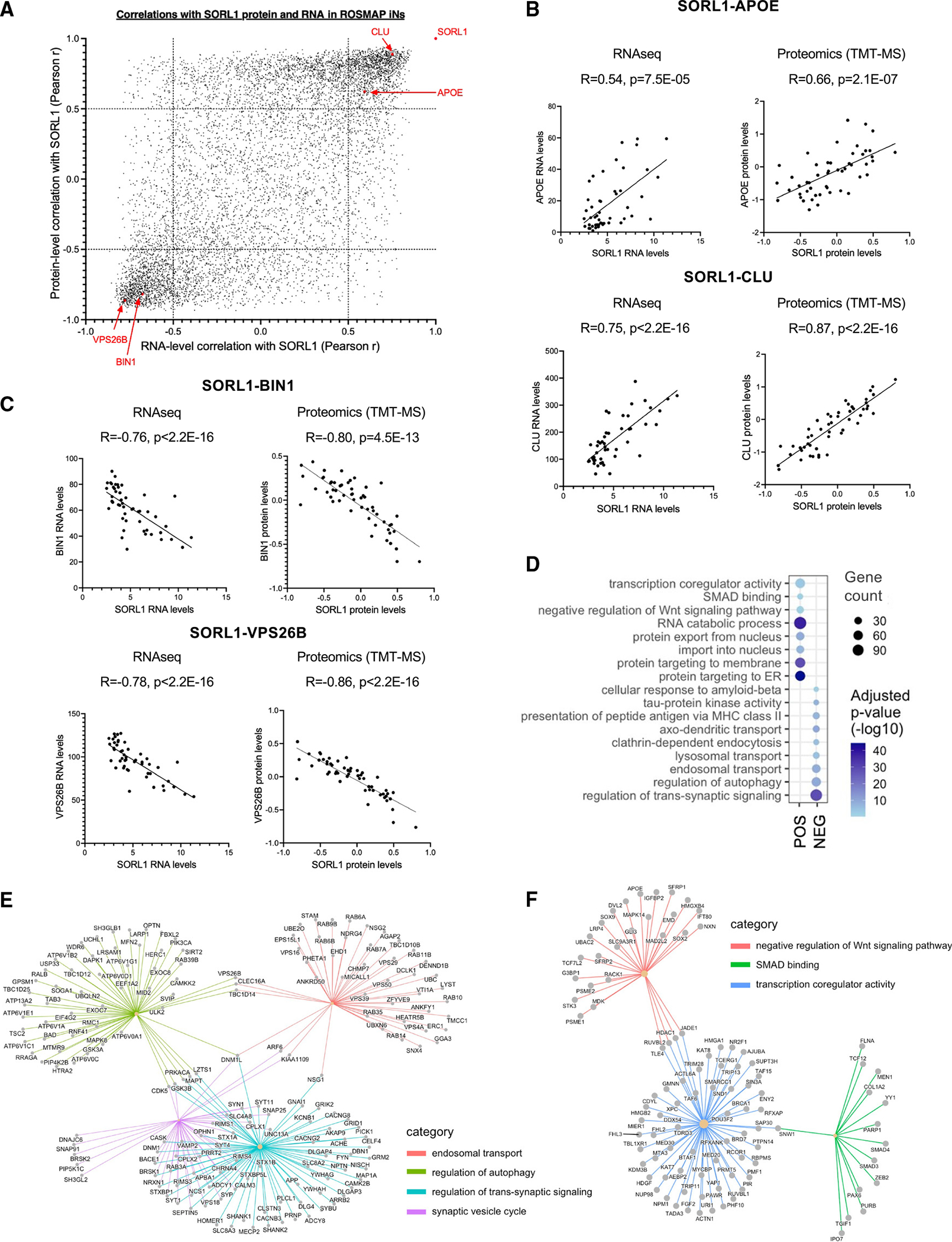
(A) ROSMAP iPSC lines were differentiated to neuron fates, and RNA-seq and TMT-MS proteomic profiling was performed. Pearson correlation coefficients between SORL1 and all other genes detected within both the RNA-seq and proteomics datasets are plotted. Each dot represents data for a single gene. High-density areas of the plot highlight genes that are either positively or negatively correlated with SORL1 in both the RNA-seq and the proteomics data.
(B) Correlation between SORL1 and APOE and CLU in ROSMAP iN RNA-seq and proteomics data.
(C) Correlation between SORL1 and BIN1 and VPS26B in ROSMAP iN RNA-seq and proteomics data. For (B) and (C), Each dot represents data from iNs derived from one individual.
(D) Pathway enrichment dot plot representing biological gene cohorts that are significantly overrepresented in either the upper right (POS) or lower left (NEG) sections of the SORL1 correlation plot in (A). Dot size represents the number of genes passing a correlation cutoff of p < −0.05 or p > 0.5 within the gene set, while color indicates q value. All pathways shown had q < 0.02.
(E and F) Gene concept network plots representing genes that belong to the pathways identified in (D) whose levels are associated with SORL1 expression.
snRNA-seq of postmortem brain tissue validates an association between SORL1 and both APOE and CLU in excitatory neurons
To assess if this SORL1-APOE-CLU relationship also exists in the aged human brain, we examined RNA-seq data from the medial frontal cortex from the same ROSMAP individuals for whom we derived iPSC lines (Figure 6A). At bulk level in brain tissue, there was no association between SORL1 and APOE, but there was a positive association between SORL1 and CLU (Figures S8A and S8B). The APOE result is not surprising given the cell-type specificity in the cell models compared to the mixed cell types in brain tissue. Pathway analysis of genes associated with SORL1 in both the brain data and the iN data (>0.5 R value < −0.5) highlighted a set of genes involved in intracellular transport, endosomal transport, and the nuclear pore complex that were concordantly associated with SORL1 expression in the brain and iNs (Figure 6B). Intriguingly, two of these genes, BIN1 and TMEM106B, are LOAD GWAS genes (Figure 6B).
Figure 6. snRNA-seq of postmortem brain tissue validates an association between SORL1 and both APOE and CLU in excitatory neurons.
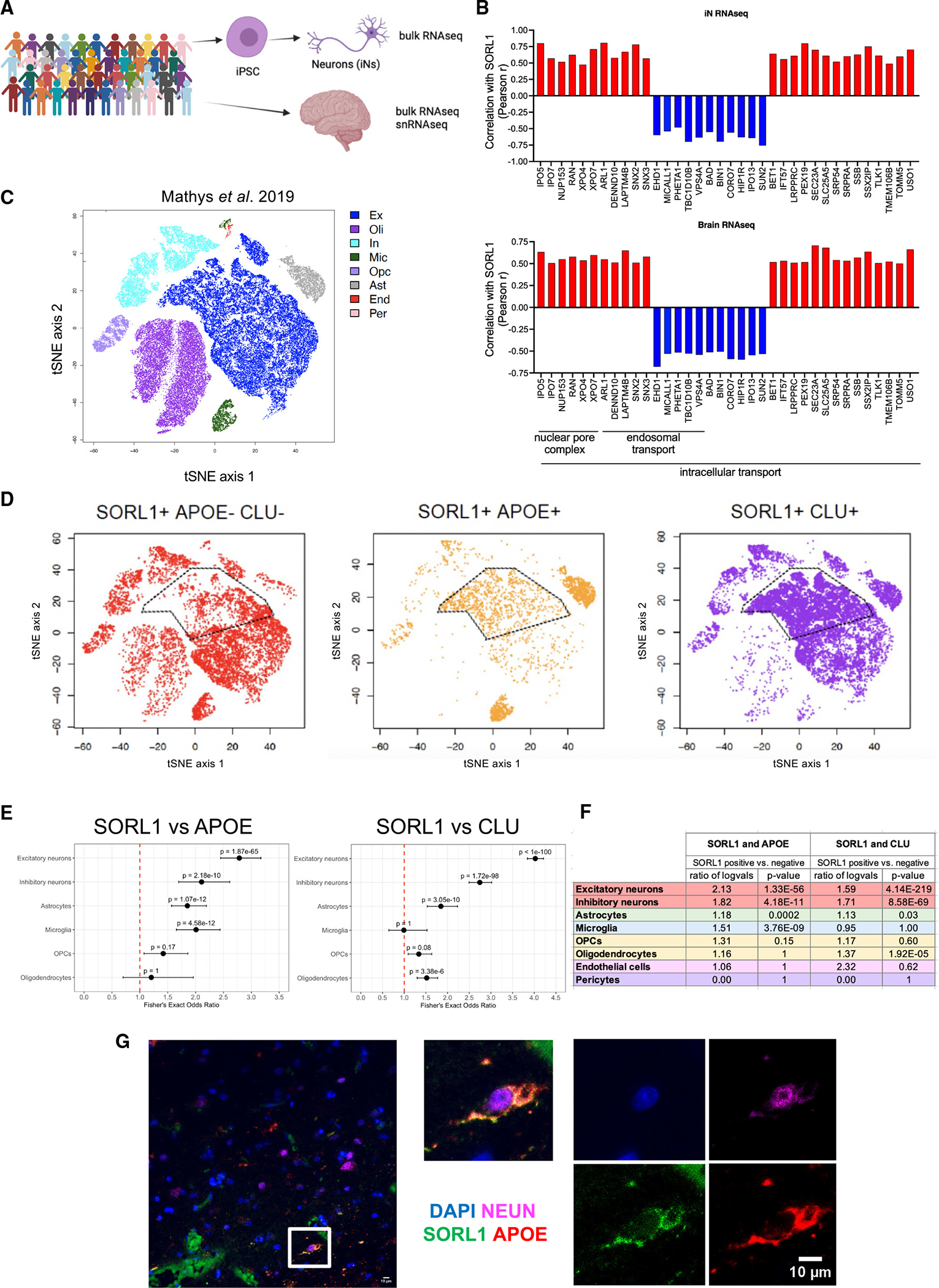
(A) iN bulk RNA-seq and postmortem brain bulk RNA-seq as well as snRNA-seq dataset (medial frontal cortex) were generated from the ROSMAP cohort.
(B) Correlation coefficient values (Pearson r) were calculated between SORL1 and other genes in bulk RNA-seq data from iNs and brain tissue. Shown are examples of concordant associations of SORL1 with genes in intracellular transport, endosomal transport, and nuclear pore complex gene sets within both iN and brain RNA-seq datasets.
(C) TSNE plot of snRNA-seq data derived from the ROSMAP cohort, colored by cell type, as determined by marker expression (data from Mathys et al.44). Data are from 70,634 cells from 48 individuals.
(D) TSNE plots with cells colored by SORL1, APOE, and/or CLU detection, as labeled. A high degree of overlap is observed between SORL1 expression and APOE and CLU expression in a subset of excitatory neurons, as well as in microglia and astrocytes.
(E) Fisher’s exact test results (adjusted p value and odds ratio) for the detection overlap of SORL1, APOE, and CLU within nuclei, separated by cell type.
(F) Differential expression results of APOE and CLU between SORL1+ and SORL1− nuclei using a Wilcoxon rank-sum test, separated by cell type. See also Table S6.
(G) Representative immunocytochemistry image of human brain section showing co-localization of DAPI, NEUN, SORL1, and APOE in a subset of cells. Scale bars, 10 μm.
Finally, to examine cell-type-specific relationships between APOE and SORL1, we examined previously reported snRNA-seq data derived from the ROSMAP cohort.44 This dataset is generated from 48 individuals with a median age of 87 years, 24 of whom had a diagnosis of AD. A total of 70,634 cells were used for the analysis (Figure 6C). Visual inspection of tSNE plots of the data showed SORL1 expression across virtually all cell clusters. Within the excitatory neuron clusters (Figure 6C, navy blue), a large proportion appeared to be co-expressing SORL1, APOE, and CLU (Figure 6D). To quantify this observation, we performed two types of analysis. First, we performed a Fisher’s exact test to determine whether the percentage of cells expressing SORL1 that express APOE or CLU in each cell type cluster is greater or less than expected by chance (Figure 6E; Table S6). For both APOE and CLU, excitatory neurons demonstrated the highest odds ratio, indicating that this cell type is the most enriched for double-positive (SORL1+/APOE+ or SORL1+/CLU+) cells. Next, we compared the ratio of log-transformed means for APOE or CLU expression in SORL1-positive versus -negative cells. Differential expression analyses show that APOE and CLU expression is higher in SORL1+ excitatory neurons (Figure 6F; Table S6). We then grouped the ROSMAP cohort by AD diagnosis and APOE genotype to determine whether AD or APOE genotype affects this relationship. Across all groups, excitatory neurons consistently showed the highest enrichment for double-positive (SORL1+/APOE+ or SORL1+/CLU+) cells (Table S6). Immunocytochemical staining of human postmortem brain for APOE and SORL1 confirmed that a subset of neurons co-express SORL1 and APOE protein at high levels (Figure 6G).
SORL1 KO neurons show altered lipid profiles and elevated LD accumulation
APOE is a component of several types of lipoprotein particles and has a well-established role in lipid transport. APOE delivers cholesterol to neurons via cell surface receptor-mediated uptake. Thus, we predicted that the reduced APOE levels in SORL1 KO neurons would result in changes in lipid profiles. We differentiated SORL1 KO and isogenic control iPSCs to D21 iNs and performed lipidomic profiling (Figure 7A; see Table S7 for full datasets). The levels of several lipid classes and species were differentially detected in SORL1 WT versus KO neurons (Figures 7B–7D, 7F, and 7G). For example, diacylglycerol (DG), methyl-dehydrocholesteryl ester (methyl-DE), and trihexosylcermide (Hex3Cer) were reduced in KO neurons (Figures 7B, 7C, 7F), while monohexosylceramide (HexCer) was significantly elevated in KO neurons (Figure 7G). In addition to these class-level changes, specific lipid species that are outside of these classes also were altered. For example, several species of phosphatidylinositol (PI) were upregulated, and a species of GM3 ganglioside (GM3(d18:1/24:0)) was downregulated (Figure 7D), among others. Interestingly, multiple lipid classes with altered levels in SORL1 KO neurons also were shown to be concordantly altered with APOE loss of function in other studies. Lipidomic profiling of APOE KO organoids revealed a reduction in triacylglycerol (TG) levels, an upregulation of phosphatidylcholine, and altered sphingolipid profiles,27 similar to what is observed here with SORL1 KO.
Figure 7. Dysregulation of lipid metabolism with SORL1 mutation.
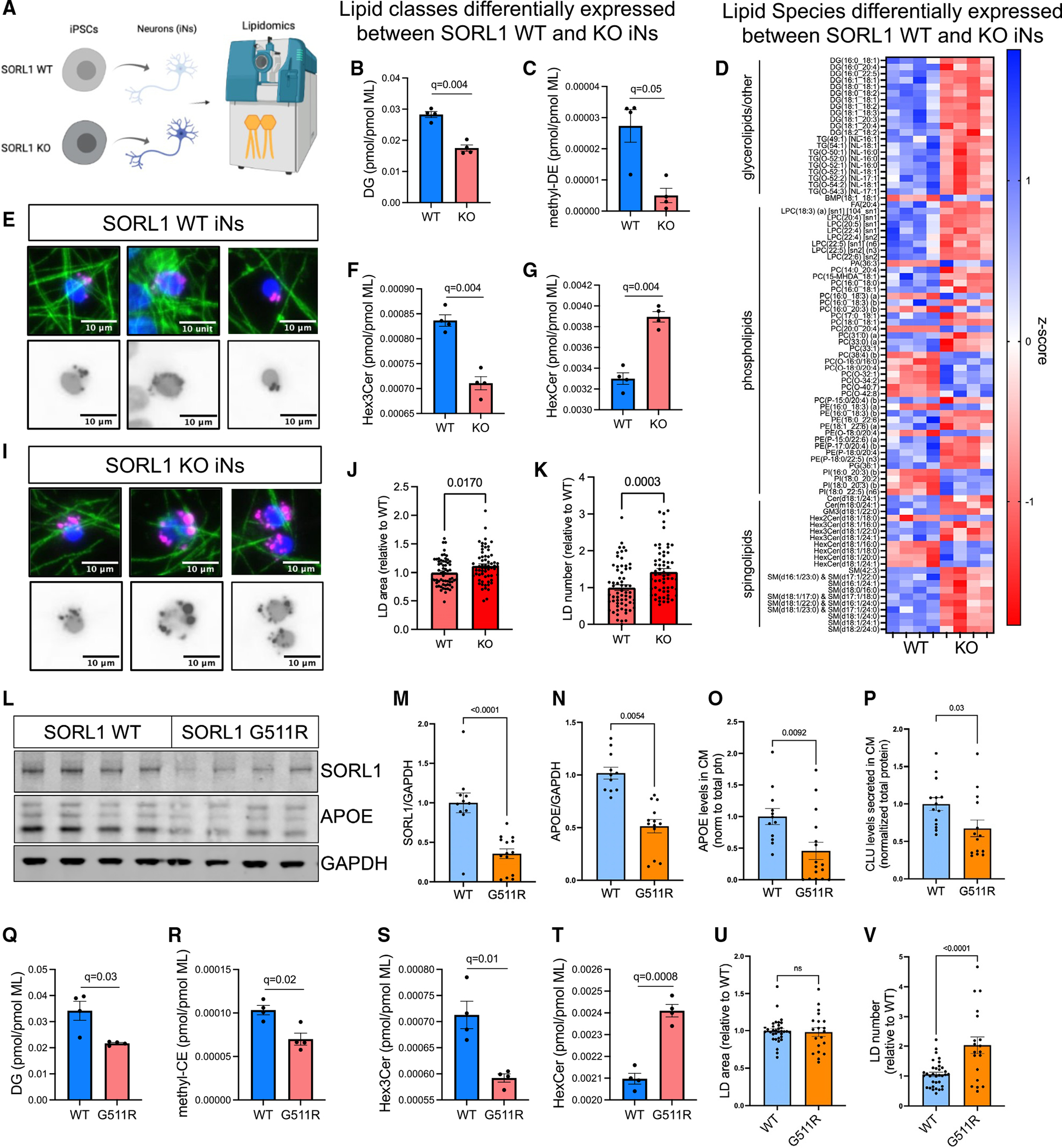
(A) SORL1 WT and KO neurons were harvested for lipidomic analysis at D21, and 813 lipid species were quantified representing 47 different classes. Values for each species and class were normalized by total membrane lipid content within each sample to control for subtle differences in cell density and efficiency of lipid extraction. Then t tests were performed between WT and KO, with multiple comparisons testing using two-stage step-up (Benjamini-Hochberg), FDR 5%. Data showing four biological replicates.
(B, C, F, and G) Eight lipid classes show dysregulation in SORL1 KO iNs including diacylglycerol (DG, B), methyl-dehydrocholesteryl ester (me-DE, C), trihexosylceramide (“ex3Cer, F), and monohexosylceramide (HexCer, G). Data show mean ± SEM.
(D) Heatmap of Z scores of all lipid species that were dysregulated (q < 0.05).
(E and I–K) Representative images of LDs, stained using LipidSpot (E and I). Quantification of LD area (J) and LD number (K) per cell normalized to WT iNs. Data show mean ± SEM from three independent differentiations, three wells per differentiation.
(L–V) iNs derived from SORL1 G511R mutation iPSC lines45 and their isogenic WT paired iPSC line were analyzed. Representative WB (L) and quantification of SORL1 (M) and APOE (N) protein levels, normalized to GAPDH. Levels of APOE (O) and CLU (P) present in the media of the same cells were quantified via ELISA. Lipidomic analyses revealed 17 classes of lipids that were differentially present. Box and whisker plots (mean with error bars from minimum to maximum) of four of these classes are shown (Q–T). The complete datasets for all lipidomic analyses can be found in Table S7. Quantification of LD area (J) and LD number (K) per cell normalized to WT iNs. Data in (Q)–(T) show four biological replicates; data in (M)–(P), (U), and (V) show mean ± SEM from three independent differentiations, three or four replicates per group.
To complement our lipidomic analysis, we next stained for LDs, which play a role in lipid homeostasis and have been shown to accumulate in the AD brain. SORL1 KO neuronal cultures displayed an elevation in the area and the number of LDs per cell, which further supports the potential role of SORL1 in lipid metabolism in neurons (Figures 7E and 7I–7K). These findings are concordant with previous studies that showed increased LD accumulation in APOE KO organoids and in cells expressing APOE4 relative to those expressing APOE3.27,28
Neurons harboring an early-onset AD mutation in SORL1 display reduced APOE and CLU levels, lipid dyshomeostasis, and an accumulation of LDs
We next examined iPSC-derived neurons that harbor a SORL1 mutation, Gly511Arg, originally identified in a mother and daughter who each had early-onset AD3 along with paired isogenic SORL1 WT neurons.45 As in SORL1 KO iNs, heterozygous G115R mutation of SORL1 was sufficient to induce a reduction in APOE and CLU in neurons (Figures 7L–7P). In addition, lipidomic profiling of G511R iNs revealed three classes of lipids that showed concordant dysregulation with SORL1 KO iNs: DG, HexCer, and Hex3Cer (Figures 7Q–7T). In addition, while KO iNs displayed a significant increase in methyl-DE ester (Figure 7C), G511R iNs showed a significant increase in the closely related species methyl-cholesteryl ester (methyl-CE; Figure 7R). Last, while G511R iNs did not show an elevation in area of LDs, they did contain a higher number of LDs (Figures 7U and 7V), similar to SORL1 KO iNs. Taken together, these results reveal that both loss of SORL1 and an EOAD mutation in SORL1 result in lipid dysregulation and a reduction in APOE and CLU levels.
DISCUSSION
SORL1 functional studies to date have primarily focused on its intracellular trafficking role. Here, we began our study with an unbiased assessment of the consequences of SORL1 loss of function across multiple brain cell types. We chose to use the iPSC experimental system for this screen to have a genetically manipulatable human system that can generate an unlimited supply of consistent cell populations. While we found that SORL1 expression was highest in microglia at both the RNA and protein level, the highest numbers of DEGs with SORL1 KO were identified in astrocytes and neurons. While microglia showed many fewer DEGs, this does not preclude an important role for SORL1 in microglia. Indeed, BIN1, a gene consistently associated with LOAD through GWAS, was elevated in SORL1 KO microglia (and neurons), as were MHC-II genes and other inflammatory response genes (Figure S2D; Table S1). Interestingly, a recent study reported that SORL1 KO microglia display higher APOE levels.46 We did not observe a change in APOE levels in our SORL1 KO microglia, and this may be due to differences in genetic backgrounds between the two studies and/or to the differences in the experimental system.
Most intriguing to us was the finding that SORL1 plays a neuron-specific role in the regulation of APOE and CLU levels. This finding is of particular interest given that APOE and CLU variants also are high-confidence genetic risk factors for AD, in addition to SORL1. Little is known about the potential role of SORL1 as a lipoprotein receptor, and the regulation of APOE and CLU levels through SORL1 may be related to this role. In 2004, Scherzer et al. first showed reduced SORL1 expression in the AD brain and speculated that this was connected to its role as a neuronal APOE receptor.7 Findings of reduced SORL1 in the AD brain have since been replicated,8,47 but there have been few studies of the connection between SORL1 and APOE. These few studies used exogenously overexpressed SORL1 and APOE to identify their ability to interact or the ability of SORL1 to mediate uptake of APOE in heterologous immortalized cell lines.25,26 Here, with SORL1 KO, APOE levels are reduced at both the protein and RNA levels in neurons but not in astrocytes or microglia.
In addition to protein-level changes, APOE and CLU RNA levels were reduced in SORL1 KO iNs, and APOE and CLU RNA levels also were correlated with SORL1 expression across the ROSMAP iNs, suggesting that reductions in APOE and CLU protein levels were mediated at least in part at the level of transcription or RNA stability (Figures S2F and 4B). Previous studies have examined transcriptional regulation of APOE in macrophages, hepatocytes, adipocytes, and astrocytes, but little is known about transcriptional regulation of APOE in neurons. Intriguingly, multiple pathway analyses performed herein indicated that TGF-β-mediated SMAD signaling was affected by SORL1 modulation. Components of this pathway were found to be differentially expressed at both the RNA and protein level with SORL1 KO (Figures 1 and 2I) and correlated with SORL1 RNA and protein levels in the ROSMAP iN cohort (Figure 5D). TGF-β has been shown to increase APOE expression in macrophages and monocytes.48 Conversely, here we see that TGF-β treatment of iPSC-derived neurons decreases APOE expression, while SMAD inhibition increases APOE levels. SMAD-mediated regulation of transcription is highly complex, and cell-type-specific effects on induction and suppression of transcription at the same genomic locus across different cell types have been reported,49,50 which may contribute to the neuron-specific effects on APOE RNA levels observed here. In this context, in iNs, SMADs appear to be acting to actively repress APOE transcription in neuronal cells, as TGF-β treatment of WT neurons repressed APOE transcription while inhibition of SMAD signaling elevated APOE RNA levels (Figure 3J).
The roles of astrocytic and microglial APOE in neurodegeneration and AD neuropathology have been deeply investigated by many groups, but much less is known about the consequences of neuronal APOE expression. APOE is expressed at higher levels in astrocytes and microglia, and thus questions arise regarding the potential implications of SORL1-mediated regulation of neuronal APOE. Neuronal APOE4 has been reported to cause an impairment of synaptic function,51 an increase in phosphorylation of tau and Aβ levels,23 and impairment of endolysosomal trafficking when it accumulates in endosomes.52 A recent study suggests that neuronally derived APOE also may have a role in regulating inflammation.24 In that study, human brain snRNA-seq data revealed that neurons with low APOE expression have lower levels of MHC-I. Here, we find that neurons with either SORL1 loss-of-function mutations or a SORL1 EOAD mutation both show reduced APOE and CLU levels, as well as perturbed lipid homeostasis and the accumulation of LDs. The lipid phenotypes observed in SORL1 mutant iNs may well be related to the reduction in APOE, as they phenocopy a subset of changes in lipid profiles in APOE KO organoids.27 Specifically, both loss of SORL1 in neurons and loss of APOE in organoids result in disrupted membrane trafficking, the accumulation of LDs, a reduction in TG levels, an upregulation of phosphatidylcholine, and altered sphingolipid profiles. These findings indicate a potential role of the neuronal SORL1-APOE axis in regulating neuronal lipid metabolism, the maintenance of which is well established to play a critical role in synaptic biology.
While speculative, it is worth noting that the reduction in neuronal APOE levels induced by SORL1 mutations may represent a protective mechanism. Data from the Herz lab and others suggest that APOE4 may fail to dissociate from its receptors within acidic endosomes due to its elevated isoelectric point relative to other APOE isoforms and that the subsequent accumulation of APOE may lead to impaired trafficking within neurons and underly the neuronal endosomal swelling observed in the AD brain.52,53 Thus, the accumulation of APOE in endosomes and the loss of function of SORL1 both result in endosomal trafficking defects. With loss of SORL1, there is an elevated risk for APOE endosomal accumulation and thus the reduction in APOE transcription and resultant lowering of APOE protein levels may represent a protective mechanism in neurons to prevent the accumulation of APOE in endosomes. This reduction would be expected to be particularly important in APOE ε4 carriers. Aligned with this possibility, the iPSC line used to generate neurons that have an APOE ε3/ε4 genotype had twice as many DEGs with SORL1 KO than the ε3/ε3 line (Tables S1 and S2). Future studies are warranted to interrogate the consequences of SORL1 mutation in neurons of different APOE genotypes in a genetically controlled manner.
Limitations of the study
The iPSC systems employed herein are reductionist systems that allow us to study the contribution of genetic mutations and natural variation of purified cell populations of defined fates. Here, we chose to study specific cell types in isolation to determine the cell-autonomous roles of SORL1. However, using this simplified system, we do not capture cell-nonautonomous interactions that are certainly contributing to disease mechanisms. In addition, other factors that play an important role in AD, such as aging, are not captured. Last, while the differentiation protocols are continuously improving, iPSC-derived cell types have not fully recapitulated the expression profiles of cells present in vivo in the brain. Despite these limitations, iPSC systems provide a well-controlled manipulatable system that allows for the identification of cell biological processes affected by modulation of specific genes of interest. Hypotheses generated by iPSC studies can then be supported or challenged by data from complementary analyses of brain tissue. Here, our findings of a role for SORL1 in regulating levels of the apolipoproteins APOE and CLU in neurons are supported by data from human brain tissue and genetic studies of AD. The rich datasets generated in this study are provided as a set of supplemental tables that can be interrogated to further study the biology of SORL1 in human brain cells.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tracy Young-Pearse, (tpearse@bwh.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement (MTA). ROSMAP iPSC lines are available from the New York Stem Cell Foundation through the NYSCF Repository (repository@nyscf.org) with completed MTA to obtain cohort data and samples (radc.rush.edu).
Data and code availability
Expression matrices and meta data for RNAseq, proteomic profiling and lipidomic profiling can be found in the supplemental tables, along with the gene-level and pathways-level output of each analysis performed using these data. RNA-seq data have been deposited at GEO and the AMP-AD Knowledge Portal and are publicly available as of the date of publication. Accession numbers and ids are listed in the key resources table. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and to the AMP-AD Knowledge Portal and are publicly available as of the date of publication. Accession ids are listed in the key resources table. Lipidomics data are deposited in Metabolights and are publicly available as of the date of publication. Accession ids are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
The results published here are in part based on data obtained from the AMP-AD Knowledge Portal (https://doi.org/10.7303/syn2580853). ROSMAP Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Goat anti-APOE | Milipore | Cat#178479; RRID:AB_10682965 |
| Rabbit anti-Amyloid Precursor Protein | Sigma-Aldrich | Cat# A8717, RRID:AB_258409 |
| Mouse anti-Phospho-Tau (Ser202, Thr205) | Thermo Fisher Scientific | Cat# MN1020, RRID:AB_223647 |
| Mouse anti-GAPDH | Proteintech | Cat# 60004-1-Ig, RRID:AB_2107436 |
| Chicken anti-GFAP | Abcam | Cat# ab4674, RRID:AB_304558 |
| Goat anti-AIF1 | Abcam | Cat# ab5076, RRID:AB_2224402 |
| Rabbit anti- LC3 | MBL International | Cat# PM036, RRID:AB_2274121 |
| Chicken anti-MAP2 | Abcam | Cat# ab5392, RRID:AB_2138153 |
| Mouse anti-LR11 (SORL1) | BD Biosciences | Cat# 611860, RRID:AB_399340 |
| Rabbit anti-Synapsin 1 | Millipore | Cat# 574777-10UG, RRID:AB_10683065 |
| Rabbit anti-Tau | Agilent | Cat# A0024, RRID:AB_10013724 |
| Mouse anti-ZO-1 | Thermo Fisher Scientific | Cat# MA3-39100-A555, RRID:AB_2663168 |
| Mouse anti-Tubulin, beta III isoform | Millipore | Cat# MAB1637, RRID:AB_2210524 |
| Mouse anti-vimentin | Millipore | Cat# CBL202, RRID:AB_93387 |
| Rabbit anti-clusterin | Abcam | Cat# ab92548, RRID:AB_10585132 |
| APOE | Invitrogen | PA5-18361, RRID:AB_10979861 |
| Mouse anti-SORL1 (Clone 525122) | R and D Systems | Cat# MAB5699, RRID:AB_2239592 |
| Rabbit anti-NeuN | Abcam | Cat# ab190565, RRID:AB_2732785 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pTet-O-NGN2-puro | Zhang et al. 201329 | Addgene plasmid #52047 |
| Tet-O-FUW-EGFP | Zhang et al. 201329 | Addgene plasmid #30130 |
| FUdeltaGW-rtTA | Zhang et al. 201329 | Addgene plasmid #19780 |
| Tet-O-SOX9-puro | Canals et al. 201830 | Addgene plasmid #117269 |
| Tet-O-NFIB-hygro | Canals et al. 201830 | Addgene plasmid #117271 |
| APOE shRNA construct1 | Sigma Aldrich | TRCN0000371278 |
| APOE shRNA construct2 | Sigma Aldrich | TRCN0000377711 |
|
| ||
| Biological samples | ||
|
| ||
| Postmortem brain tissues | New York Brain Bank | see Supplemental Tables |
| Human brain samples MPFC | RUSH RADC Brain Bank | http://www.radc.rush.edu/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| R33 | MedKoo Biosciences | 504211 |
| Trehalose dihydrate | Sigma Aldrich | 625625 |
| Chloroquine | Tocris | 4109 |
| TGF-β | Sigma Aldrich | T7039 |
| SB431542 | Calbiochem | 616464 |
|
| ||
| Critical commercial assays | ||
|
| ||
| LipidSpotTM 610 | Biotium | 70069 |
| V-PLEX Aβ Peptide Panel 1 (6E10) Kit | Meso Scale Discovery | cat. #K15200G-1 |
| R-PLEX Human ApoE Assay | Meso Scale Discovery | cat. #K1512IR-2 |
| R-PLEX Human Clusterin Assay | Meso Scale Discovery | cat. #K151YLR-2 |
|
| ||
| Deposited data | ||
|
| ||
| RNAseq data | This paper | NCBI GEO database, GEO: GSE238013 |
| Lagomarsino et al. 202132 | AMP-AD Knowledge Portal; SynID: syn2580853 and SynID: syn3219045 For quick visualization of SORL1 KO and WT data, please also see https://youngpearselab.shinyapps.io/sorl1_namei_cells/. | |
| Lipidomics data | This paper | Metabolights, study ID: MTBLS8242 |
| Mass spectrometry proteomics data | This paper | ProteomeXchange Consortium via the PRIDE partner repository PRIDE: PXD044093 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human iPSC line: Wildtype clone A7 (“line #1”, >90% European ancestry) | Knupp et al. 202016 | N/A |
| Human iPSC line: SORL1 Knockout clone E4 (“line #1” >90% European ancestry) | Knupp et al. 202016 | N/A |
| Human iPSC line: SORL1 G511R clone C6 (“line #1”, >90% European ancestry) | Mishra et al. 202245 | N/A |
| Human iPSC line: Wildtype clone 5 (“line #2”, >90% European ancestry) | This paper | N/A |
| Human iPSC line: SORL1 Knockout clone 4 (“line #2”, >90% European ancestry) | This paper | N/A |
| HUMAN IPSC LINE: BR01, 99% European ancestry | Lagomarsino et al. 202132 | BR01, AJ0006 |
| HUMAN IPSC LINE: BR04, 98% European ancestry | Lagomarsino et al. 202132 | BR04, AJ0056 |
| HUMAN IPSC LINE: BR08, 94% European ancestry | Lagomarsino et al. 202132 | BR08, AJ0044 |
| HUMAN IPSC LINE: BR09, 92% European ancestry | Lagomarsino et al. 202132 | BR09, AJ0038 |
| HUMAN IPSC LINE: BR103, 94% European ancestry | Lagomarsino et al. 202132 | BR103, AJ0121 |
| HUMAN IPSC LINE: BR104, 95% European ancestry | Lagomarsino et al. 202132 | BR104, AJ0113 |
| HUMAN IPSC LINE: BR108, 82% European ancestry | Lagomarsino et al. 202132 | BR108, AJ0117 |
| HUMAN IPSC LINE: BR11, 99% European ancestry | Lagomarsino et al. 202132 | BR11, AJ0002 |
| HUMAN IPSC LINE: BR13, 92% European ancestry | Lagomarsino et al. 202132 | BR13, AJ0039 |
| HUMAN IPSC LINE: BR14, 99% European ancestry | Lagomarsino et al. 202132 | BR14, AJ0003 |
| HUMAN IPSC LINE: BR15, 93% European ancestry | Lagomarsino et al. 202132 | BR15, AJ0008 |
| HUMAN IPSC LINE: BR21, 92% European ancestry | Lagomarsino et al. 202132 | BR21, AJ0046 |
| HUMAN IPSC LINE: BR22, 93% European ancestry | Lagomarsino et al. 202132 | BR22, AJ0021 |
| HUMAN IPSC LINE: BR24, 93% European ancestry | Lagomarsino et al. 202132 | BR24, AJ0040 |
| HUMAN IPSC LINE: BR26, 94% European ancestry | Lagomarsino et al. 202132 | BR26, AJ0043 |
| HUMAN IPSC LINE: BR27, 97% European ancestry | Lagomarsino et al. 202132 | BR26, AJ0001 |
| HUMAN IPSC LINE: BR28, 93% European ancestry | Lagomarsino et al. 202132 | BR28, AJ0029 |
| HUMAN IPSC LINE: BR29, 95% European ancestry | Lagomarsino et al. 202132 | BR29, AJ0030 |
| HUMAN IPSC LINE: BR30, 94% European ancestry | Lagomarsino et al. 202132 | BR30, AJ0042 |
| HUMAN IPSC LINE: BR33, 91% European ancestry | Lagomarsino et al. 202132 | BR33, AJ0047 |
| HUMAN IPSC LINE: BR36, 95% European ancestry | Lagomarsino et al. 202132 | BR36, AJ0022 |
| HUMAN IPSC LINE: BR37, 93% European ancestry | Lagomarsino et al. 202132 | BR37, AJ0031 |
| HUMAN IPSC LINE: BR39, 98% European ancestry | Lagomarsino et al. 202132 | BR39, AJ0005 |
| HUMAN IPSC LINE: BR40, 94% European ancestry | Lagomarsino et al. 202132 | BR40, AJ0045 |
| HUMAN IPSC LINE: BR41, 95% European ancestry | Lagomarsino et al. 202132 | BR41, AJ0024 |
| HUMAN IPSC LINE: BR43, 91% European ancestry | Lagomarsino et al. 202132 | BR43, AJ0020 |
| HUMAN IPSC LINE: BR46, 98% European ancestry | Lagomarsino et al. 202132 | BR46, AJ0004 |
| HUMAN IPSC LINE: BR48, 91% European ancestry | Lagomarsino et al. 202132 | BR48, AJ0028 |
| HUMAN IPSC LINE: BR50, 94% European ancestry | Lagomarsino et al. 202132 | BR50, AJ0041 |
| HUMAN IPSC LINE: BR54, 91% European ancestry | Lagomarsino et al. 202132 | BR54, AJ0048 |
| HUMAN IPSC LINE: BR57, 93% European ancestry | Lagomarsino et al. 202132 | BR57, AJ0073 |
| HUMAN IPSC LINE: BR58, 89% European ancestry | Lagomarsino et al. 202132 | BR58, AJ0068 |
| HUMAN IPSC LINE: BR59, 93% European ancestry | Lagomarsino et al. 202132 | BR59, AJ0072 |
| HUMAN IPSC LINE: BR60, 89% European ancestry | Lagomarsino et al. 202132 | BR60, AJ0067 |
| HUMAN IPSC LINE: BR61, 93% European ancestry | Lagomarsino et al. 202132 | BR61, AJ0076 |
| HUMAN IPSC LINE: BR62, 93% European ancestry | Lagomarsino et al. 202132 | BR62, AJ0069 |
| HUMAN IPSC LINE: BR63, 94% European ancestry | Lagomarsino et al. 202132 | BR63, AJ0070 |
| HUMAN IPSC LINE: BR64, 94% European ancestry | Lagomarsino et al. 202132 | BR64, AJ0066 |
| HUMAN IPSC LINE: BR65, 86% European ancestry | Lagomarsino et al. 202132 | BR65, AJ0080 |
| HUMAN IPSC LINE: BR66, 93% European ancestry | Lagomarsino et al. 202132 | BR66, AJ0094 |
| HUMAN IPSC LINE: BR67, 94% European ancestry | Lagomarsino et al. 202132 | BR67, AJ0099 |
| HUMAN IPSC LINE: BR68, 92% European ancestry | Lagomarsino et al. 202132 | BR68, AJ0095 |
| HUMAN IPSC LINE: BR69, 94% European ancestry | Lagomarsino et al. 202132 | BR69, AJ0103 |
| HUMAN IPSC LINE: BR72, 91% European ancestry | Lagomarsino et al. 202132 | BR72, AJ0090 |
| HUMAN IPSC LINE: BR83, 94% European ancestry | Lagomarsino et al. 202132 | BR83, AJ0083 |
| HUMAN IPSC LINE: BR89, 93% European ancestry | Lagomarsino et al. 202132 | BR89, AJ0089 |
| HUMAN IPSC LINE: BR91, 91% European ancestry | Lagomarsino et al. 202132 | BR91, AJ0115 |
| HUMAN IPSC LINE: BR92, 92% European ancestry | Lagomarsino et al. 202132 | BR92, AJ0116 |
| HUMAN IPSC LINE: BR93, 94% European ancestry | Lagomarsino et al. 202132 | BR93, AJ0114 |
| HUMAN IPSC LINE: BR95, 93% European ancestry | Lagomarsino et al. 202132 | BR95, AJ0119 |
| HUMAN IPSC LINE: BR97. 90% European ancestry | Lagomarsino et al. 202132 | BR97, AJ0123 |
| HUMAN IPSC LINE: BR98, 96% European ancestry | Lagomarsino et al. 202132 | BR98, AJ0109 |
| HUMAN IPSC LINE: BR99, 95% European ancestry | Lagomarsino et al. 202132 | BR99, AJ0107 |
| Human iPS Cell Line (Episomal, CD34+, ApoE Knockout) | Alstem cell advancement | iPS36 |
| Human iPS Cell Line (Episomal, CD34+, ApoE3) | Alstem cell advancement | iPS26 |
| Human iPS Cell Line (Episomal, CD34+) | Alstem cell advancement | iPS16 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SORL1 KO line#1 gRNA: ‘ATTGAACGACATG AACCCTC’ | Knupp et al. 202016 | N/A |
| SORL1 KO line#1 ssODN: ‘GGGAATTGATCC CTATGACAAACCAAATACCATCTACATTGAACGACA TGAACCCTCTGGCTACTCCACGTCTTCCGA AG TACAGATTTCTTCCAGTCCCGGGAAAACCAGGAAG’ | Knupp et al. 202016 | N/A |
| SORL1 G511R gRNA: ‘CTCTTGCATTTTAGGCTCAG’ | Mishra et al. 202245 | N/A |
| SORL1 G511R ssODN: ‘CTGACATATTCTTGAAA TTAAAAATAATTATTTCTCTTGCATTTTAGGCTCA GTGCGAAAGAACTTGGCTAGCAAGACAAACGTG TACATCTCTAGCAGTGCTGGAGCCAGGTGGCG’ | Mishra et al. 202245 | N/A |
| SORL1 KO line#2 gRNA: ‘ATGTTCCTGAATCATGATCC’ | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| R Studio, v3.6.1 of R; v1.2.5019 of R Studio | R Core Team, 202054 | https://www.rstudio.com |
| Sleuth, v0.30.0 | Pimentel et al., 201740 | https://www.rdocumentation.org/packages/sleuth/versions/0.30.0 |
| Cell Profiler | McQuin et al., 201855 | https://cellprofiler.org/ |
| All original code used in this paper | This paper |
https://github.com/genejockey33000/typGumbo. Zenodo; https://doi.org/10.5281/zenodo.8183364 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Induced pluripotent stem cell lines
iPSCs were generated from cryopreserved peripheral blood mononuclear cell (PBMC) samples from autopsied participants from either the Religious Order Study (ROS) or Rush Memory and Aging Project (MAP). ROS and MAP are cohort studies of aging and dementia. Each participant signed an informed consent, Anatomic Gift Act, and Repository Consent to allow their data to be repurposed, following approval by an Institutional Review Board of Rush University Medical Center. iPSCs were generated using Sendai reprogramming method.32 iPSCs undergo a rigorous quality procedure that includes a sterility check, mycoplasma testing, karyotyping, and pluripotency assays performed by the New York Stem Cell Foundation (NYSCF). iPSC cell line used to generate line 1 SORL1 KO cells was previously published.56 iPSC cell line used to generate line 2 has been previously published,31,57 generated in collaboration with the Harvard Stem Cell Institute. The parental iPSC line (HVRDi002-A) harbored a APPV717I mutation, which was corrected using CRISPR/Cas technology.31 Stem Cell Technologies hPSC Genetic Analysis kit (#07550) was used for the isogenic pairs (iPSCs) to check for karyotype abnormalities. APOE ε3/ε3 (cat#iPS26), APOE ε4/ε4 (cat#iPS16), and APOE KO (cat#iPS36) iPSCs were from obtained from Alstem. All three lines are isogenic, sharing the same genetic background, and APOE ε4/ε4 (cat#iPS16) was used as a parental line to edit APOE sequence. iPSCs were maintained using StemFlex Medium (Thermo Fisher Scientific). All cell lines were routinely tested for mycoplasma using PCR kit (MP0035–1KT) and STR profiling to prevent potential contamination or alteration to the cell lines.
CRISPR/Cas9 to generate SORL1 KO and SORL1 variant (G511R) iPSCs
Description and characterization of SORL1 WT/KO iPSCs in line 1 have been previously published.16 gRNA ‘ATTGAACGACATGAACCCTC’ was used to target exon 6 at the VPS10p domain. Description and characterization of SORL1 variant iPSCs containing heterozygous G511R variant have been previously published.45 gRNA ‘CTCTTGCATTTTAGGCTCAG’ and G511R ssODN: CTGACATATTCTTGAAATTAAAAATAATTATTTCTCTTGCATTTTAGGCTCAGTGCGAAAGAACTTGGCTAGCAAGACAAACGTGTACATCTCTAGCAGTGCTGGAGCCAGGTGGCG was used to target VPS10p domain in iPSC line 1, which is the same iPSCs used to generate SORL1 KO iPSCs. The Zhang Lab CRISPR Design website (crispr.mit.edu) was used to generate guide RNAs (gRNAs) with minimal off-target effects. gRNAs were cloned into the px458 vector that also expresses GFP and the Cas-9 nuclease. hiPSCs were electroporated with plasmids, sorted by flow cytometry for GFP expression, and plated at a clonal density of ~1×10^4 cells per 10cm plate. After approximately two weeks of growth, colonies were picked and split into identical sets. To generate SORL1 WT/KO iPSCs in line 2, gRNA was chosen from Genescript’s website, which designs the gRNAs based on the algorithms by the Broad Institute. The gRNA sequence ‘ATGTTCCTGAATCATGATCC’ was used to target exon 5 at the VPS10p domain. IDT CRISPR Cas9 guide RNA design checker was used to check for any potential off-target effects.
iPSC-derived neuron differentiation
iPSC-derived neurons (iNs) were differentiated following a previously published paper29 with minor modifications,31,32 iPSCs were plated at a density of 95k cells/cm2 on plates coated with growth factor reduced Matrigel one day prior to virus transduction (Corning #354230). Then, iPSCs were transduced with three lentiviruses – pTet-O-NGN2-puro (Addgene plasmid #52047, a gift from Marius Wernig), Tet-O-FUW-EGFP (Addgene plasmid #30130, a gift from Marius Wernig), and FUdeltaGW-rtTA (Addgene plasmid #19780, a gift from Konrad Hochedlinger). The cells were then replated at 200,000 cells/cm2 using StemFlex Medium (Thermo Fisher Scientific) and ROCK inhibitor (10μM) (D0). The media was changed to KSR media (D1), 1:1 of KSR and N2B media (D2) and N2B media (D3). On day 4, cells were dissociated using Accutase, and plated at 50,000 cells/cm2 using iN D4 media (NBM media +1:50 B27 + BDNF, GDNF, CNTF (10 ng/mL, Peprotech). Doxycycline (2ug/ml, Sigma) was added from D1 to the end of the differentiation, and puromycin (5 μg/ml, Gibco) was added from D2 to the end of the differentiation. On D3, B27 supplement (1:100) (Life Technologies) was added. From D4 to the end of differentiation D21, cells were cultured iN D4 media and fed every 2–3 days.
Induced neuron protocol media
KSR media: Knockout DMEM, 15% KOSR, 1x MEM-NEAA, 55 μM beta-mercaptoethanol, 1x GlutaMAX (Life Technologies).
N2B media: DMEM/F12, 1x GlutaMAX (Life Technologies), 1x N2 supplement B (Stemcell Technologies), 0.3% dextrose (D-(+)-glucose, Sigma).
NBM media: Neurobasal medium, 0.5x MEM-NEAA, 1x GlutaMAX (Life Technologies), 0.3% dextrose (D-(+)-glucose, Sigma).
iPSC-derived astrocyte differentiation
iPSC-derived astrocytes (iAs) were differentiated following a previously published paper30 with minor modifications.32 iPSCs were plated at 95k cells/cm2 on growth factor reduced Matrigel (Corning #354230) coated plates prior to virus transduction. Then, iPSCs were transduced with three lentiviruses – Tet-O-SOX9-puro (Addgene plasmid #117269), Tet-O-NFIB-hygro (Addgene plasmid #117271), and FUdeltaGW-rtTA (Addgene plasmid #19780). The cells were then replated at 200,000 cells/cm2 using StemFlex Medium (Thermo Fisher Scientific) and ROCK inhibitor (10μM) (D0). The media was changed daily with Expansion Media (EM) from D1 to 3, and gradually switched from EM to FGF media from D4 to D7. On day 8, cells were dissociated using Accutase, and plated at 84,000 cells/cm2 using FGF media. Doxycycline (2.5ug/ml, Sigma) was added from D1 to the end of the differentiation, puromycin (1.25μg/ml, Gibco) was added on D3 of the differentiation, and hygromycin (100μg/ml, InvivoGen # ant-hg-1) was added from D4-D6 of the differentiation. From D8 to the end of differentiation D21, cells were cultured with maturation media and fed every 2–3 days.
Induced astrocyte protocol media
Expansion Media: DMEM/F12 (Thermo Fisher Scientific), 10% FBS, 1% N2 Supplement (Stemcell Technologies), 1% GlutaMAX (Life Technologies)
FGF Media: Neurobasal media, 2% B27, 1% NEAA, 1% GlutaMAX, 1% FBS, 8 ng/ml FGF, 5 ng/ml CNTF, 10 ng/ml BMP4
Maturation Media: 1:1 DMEM/F12 and neurobasal media, 1% N2, 1% GlutaMAX, 1% Sodium Pyruvate, 5ug/ml N-1% N2, 1% GlutaMAX, 1% Sodium Pyruvate, 5ug/ml N-N-acetyl cysteine, 5 ng/ml heparin-binding EGF-like GF, 10 ng/ml CNTF, 10 ng/ml BMP4, 500ug/ml dbcAMP
iPSC-derived microglia-like cells differentiation
iPSC-derived microglia-like cells (iMGLs) were differentiated following a previously published paper35,36 with minor modifications.32,58 iPSCs were plated on growth factor reduced Matrigel (Corning #354230) using StemFlex Medium (Thermo Fisher Scientific) and ROCK inhibitor (10μM). From D0 to D12, StemDiff Hematopoietic Kit (Stemcell Technologies) was used to generated hematopoietic precursor cells (HPCs)At D12, cells were replated at 100,000 cells per 35mm well in iMGL media supplemented with 3 cytokines (IL-34 (100 ng/mL, Peprotech), TGF-β1 (50 ng/mL, Militenyi Biotech)M-CSF (25 ng/mL, Thermo Fisher Scientific)). From D12 to D24, iMGL media with freshly added cytokines were added to the culture every other day. On D24, cells were replated at 100,000 cells per 15.6mm well with 1:1 mixture of old media and fresh iMGL media with 3 cytokines. From D24 to D37, iMGL media with freshly added 3 cytokines were added to the culture every other dayOn D37, cells are resuspended in iMGL media with five cytokines, supplemented every other day until D40
iPSC-derived microglia-like cells protocol media
iMGL media: DMEM/F12, 2X insulin-transferrin-selenite, 2X B27, 0.5X N2, 1X GlutaMAX, 1X non-essential amino acids, 400μM monothioglycerol, 5 μg/mL insulin
3 cytokines: 100 ng/mL IL-34 (Peprotech), 50 ng/mL TGF-β1 (Miltenyi Biotech), and 25 ng/mL M-CSF (ThermoFisher Scientific)
5 cytokines: 100 ng/mL IL-34, 50 ng/mL TGF-β1, 25 ng/mL M-CSF, 100 ng/mL, CD200 (Novoprotein) and 100 ng/mL CX3CL1 (Peprotech)
iPSC-derived brain microvascular endothelial cells differentiation
iPSC-derived brain microvascular endothelial cells (iECs) were differentiated following previously published papers37–39 with minor modifications.58 iPSCs were plated on growth factor reduced Matrigel (Corning #354230) using StemFlex Medium (Thermo Fisher Scientific) and ROCK inhibitor (10μM) at a density of 180,000 cells per 35mm well (D-2). From D0 to D4, the media was changed daily with E6 medium (ThermoFisher) using hypoxia condition (5%CO2–5%O2-N2). From D5, the media was changed to EC media supplemented with 20 ng/ml FGF and 10uM retinoic acid. On D7, the media is replaced with new EC media supplemented with fresh FGF and retinoic acid. From D8-D14, the media was changed daily with EC medium until harvest.
Endothelial media (EC): hESFM (Thermo Fisher, Cat#: 11111044), 50X B27 supplement (Thermo Fisher, Cat#: 17504044), and (0.5%) pen/strep (Thermo Fisher, Cat#: 15140163)
METHOD DETAILS
Western blotting
Cells were lysed with the RIPA lysis buffer (Thermo Fisher Scientific #89900) or in a buffer containing 1%NP40, 10mM EDTA, 150mM NaCl, 50mM Tris, with the protease inhibitor (cOmpleteTM mini protease inhibitor, Roche) and phosphatase inhibitor (phosphoSTOP, Roche) added freshly before the lysis. Using BCA Protein Assay Kit (Pierce), protein concentrations were measured, and equal amount of protein were loaded onto 4–12% Bis-Tris NuPAGE gels (Life Technologies). Gels were then transferred to nitrocellulose membranes, blocked with LI-COR TBS blocking buffer for 1 h, then probed with primary antibodies overnight. After washing three times in TBST (0.05% Tween in TBS), the secondary antibodies were added for 1hr rocking in RT, followed by three more washes in TBST. Blots were imaged using Odyssey Clx system (LI-COR) and quantified using ImageStudio (LICOR).
Antibodies for immunocytochemistry and western blot
| Antigen | Host | ICC/WB | Dilution | Vendor | Catalog # |
|---|---|---|---|---|---|
|
| |||||
| APOE | Goat | ICC and WB | 1:1000 | Sigma | 178479 |
| APP | Rabbit | WB | 1:1000 | Sigma | A8717 |
| P202, 205 AT8 (MAPT) | Mouse | WB | 1:250 | ThermoFisher | MN1020 |
| GAPDH | Mouse | WB | 1:10,000 | ProteinTech | 60004 |
| GFAP | Chicken | ICC | 1:1000 | Abcam | ab4672 |
| IBA1 | Goat | ICC | 1:500 | Abcam | Ab5076 |
| LC3 | Rabbit | WB | 1:1000 | MBL | PM036 |
| MAP2 | Chicken | ICC | 1:5000 | Abcam | ab5392 |
| SORL1 (LR11) | Mouse | WB | 1:1000 | BD Biosciences | 611860 |
| SYN1 | Rabbit | ICC | 1:1000 | Calbiochem | 574777 |
| TAU (MAPT) | Rabbit | WB | 1:2000 | Dako | A0024 |
| TJP1 | Mouse | ICC | 1:1000 | Invitrogen | 339100 |
| TUJ1 | Mouse | ICC | 1:1000 | Milipore | MAB1637 |
| VIMENTIN | Mouse | ICC | 1:500 | Milipore | CBL202 |
| LipidSpot-610 | ICC | 1:1000 | Biotium | 70069 | |
| CLU | Rabbit | WB | 1:500 | Abcam | Ab92548 |
qPCR
At iN d21, cells were harvested and RNA was purified using Purelink RNA Mini kit (Invitrogen). cDNA was generated using SuperScript II (Invitrogen). qPCR was performed using Power SYBRTM Green Master Mix and run on ViiA7 system (Applied Biosystems).
Primers
| Target | Species | F/R | Sequence |
|---|---|---|---|
|
| |||
| APOE | Human | F | GTTGCTGGTCACATTCCTGG |
| APOE | Human | R | GCAGGTAATCCCAAAAGCGAC |
| GAPDH | Human | F | GGGAGCCAAAAGGGTCATC |
| GAPDH | Human | R | TGGTTCACACCCATGACGAA |
ELISAs
48hr conditioned media was collected from d19 to d21 before harvest. Extracellular Aβ 38, 40, and 42 levels were measured using MSD V-PLEX Aβ Peptide Panel 1 (6E10) Kit (cat. #K15200G-1). Extracellular APOE and CLU were measured using MSD R-PLEX Human ApoE Assay (cat. #K1512IR-2) and MSD R-PLEX Human Clusterin Assay (cat. #K151YLR-2).
Immunocytochemistry
At iN d21, iA d21, iMGL d40, and iEC d14, cells were washed once with PBS, then was fixed in 4% paraformaldehyde solution for 15 min at RT. After completing the fixation, cells were incubated in blocking buffer (2% donkey serum with 0.1% Triton X-100) for 1 h rocking at room temperature followed by overnight incubation in primary antibody at 4C. Then, cells are washed with PBS three times, incubated with secondary antibodies, and then washed with PBS three times. DAPI (1:1000) and LipidSpot (Lipid Droplet, 1:1000) staining was performed during the second wash with PBS. Then, the cells were imaged using LSM710 confocal microscopy, analyzed using CellProfiler.55
Drug treatments
Prior to harvest, cells were fed with iN media containing the following drugs: 10uM or 20uM R33 (MedKoo Biosciences), 100mM Trehalose dihydrate (Sigma), and 5nM Chloroquine (Tocris) for 72hrs; 10 ng/ml TGF-β (T7039) and 10uM SB-431542 (616464) for 48hrs (Western blot) and 24hrs (qPCR). The vehicles were 1:1 mix of water and DMSO for R33, ethanol for SB-431542, and water for chloroquine and TGF-β. Trehalose was prepared directly in iN media. For trehalose and chloroquine, cells were treated one more time on D20 before final harvest on d21.
Lentiviral transduction (shRNA)
Lentivirus expressing pLKO.1 vector was used to generate two APOE shRNA constructs. TRCN0000371278, referred to ‘APOE1’ in this study and TRCN0000377711, referred to ‘APOE2’ in this study were obtained from Sigma. iNs were transduced at D17 with either empty virus, APOE1, or APOE2, at MOI of 3. Following 18hrs of incubation, the media was replaced with fresh media and incubated for additional 72hr. At d21, media and cells were collected for further analysis.
Bulk RNA sequencing
High quality total RNA (eRIN >9.0) was harvested from differentiated iNs, iAs, iMGLs, iECs, or from undifferentiated iPSCs. Libraries were generated and sequenced to a depth of >20 million read pairs using the GeneWiz next-gen sequencing service. RNAseq reads were quality trimmed to remove end base called with phred33 scores below 25 and eliminate resulting reads that are shorter than 30 bases. Trimmed reads are quantified using the Kalliso (v 0.43.1) pseudoalignment algorithm with 50 bootstraps using a GRCh38 cDNA+ncRNA ref.59.
Genomic data analyses
TPM Normalized expression matrices and differential expression statistics were generated using the Sleuth (v 0.30) package in R (v 4.0.3). Expression differences were tested using a Wald test after controlling for differentiation batch. Significant hits were defined by q value (FDR) < .05 and a b value >.5 Gene Ontology enrichments were tested using the clusterProfiler package (v 3.18.1)60 in R. Enrichment dot plots were generated using ggplot2 (v 3.3.5)61 while the gene concept network analyses were generated using the enrichplot (v 1.12.2)62 package. Correlations were analyzed using custom R scripts and base correlation functions.
Protein digestion
Each cell pellet was individually homogenized in 300 μL of urea lysis buffer (8 M urea in 10 mM Tris, 100 mM NaH2PO4 buffer, pH = 8.5), including 5 μL (100x stock) HALT protease and phosphatase inhibitor cocktail (Pierce). All homogenization was performed using a Bullet Blender (Next Advance) according to manufacturer protocols. Briefly, each tissue piECe was added to Urea lysis buffer in a 1.5 mL Rino tube (Next Advance) harboring 750 mg stainless steel beads (0.9–2 mm in diameter) and blended twice for 5 min intervals in the cold room (4°C). Protein supernatants were transferred to 1.5 mL Eppendorf tubes and sonicated (Sonic Dismembrator, Fisher Scientific) 3 times for 5 s with 15 s intervals of rest at 30% amplitude to disrupt nucleic acids and subsequently vortexed. Protein concentration was determined by the bicinchoninic acid (BCA) method, and samples were frozen in aliquots at −80°C. Protein homogenates (50μg) treated with 1 mM dithiothreitol (DTT) at 25°C for 30 min, followed by 5 mM iodoacetimide (IAA) at 25°C for 30 min in the dark. Protein mixture was digested overnight with 1:100 (w/w) lysyl endopeptidase (Wako) at room temperature. The samples were then diluted with 50 mM NH4HCO3 to a final concentration of less than 2M urea and then and further digested overnight with 1:50 (w/w) trypsin (Promega) at 25°C. Resulting peptides were desalted with a Sep-Pak C18 column (Waters) and dried under vacuum.
Tandem mass tag (TMT) labeling
Peptides were reconstituted in 100ul of 100mM triethyl ammonium bicarbonate (TEAB) and labeling performed using TMTPro isobaric tags (Thermofisher Scientific, A44520). Briefly, the TMT labeling reagents were equilibrated to room temperature, and anhydrous ACN (200 μL) was added to each reagent channel. Each channel was gently vortexed for 5 min, and then 20 μL from each TMT channel was transferred to the peptide solutions and allowed to incubate for 1 h at room temperature. The reaction was quenched with 5% (v/v) hydroxylamine (5 μL) (Pierce). All 16 channels were then combined and dried by SpeedVac (LabConco) to approximately 100 μL and diluted with 1 mL of 0.1% (v/v) TFA, then acidified to a final concentration of 1% (v/v) FA and 0.1% (v/v) TFA. Peptides were desalted with a 60 mg HLB plate (Waters). The eluates were then dried to completeness. High pH fractionation was performed essentially as described (3) with slight modification. Dried samples were re-suspended in high pH loading buffer (0.07% v/v NH4OH, 0.045% v/v FA, 2% v/v ACN) and loaded onto a Water’s BEH (2.1 mm × 150 mm with 1.7 μm beads). An Thermo Vanquish UPLC system was used to carry out the fractionation. Solvent A consisted of 0.0175% (v/v) NH4OH, 0.01125% (v/v) FA, and 2% (v/v) ACN; solvent B consisted of 0.0175% (v/v) NH4OH, 0.01125% (v/v) FA, and 90% (v/v) ACN. The sample elution was performed over a 25 min gradient with a flow rate of 0.6 mL/min with a gradient from 0 to 50% B. A total of 96 individual equal volume fractions were collected across the gradient and dried to completeness using a vacuum centrifugation.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS)
All samples were analyzed on the Evosep One system using an in-house packed 15 cm, 75 μm i.d. capillary column with 1.9 μm Reprosil-Pur C18 beads (Dr. Maisch, Ammerbuch, Germany) using the pre-programmed 21 min gradient (60 samples per day) essentially as described (4). Mass spectrometry was performed with a high-field asymmetric waveform ion mobility spectrometry (FAIMS) Pro equipped Orbitrap Eclipse (Thermo) in positive ion mode using data-dependent acquisition with 2 s top speed cycles. Each cycle consisted of one full MS scan followed by as many MS/MS events that could fit within the given 2 s cycle time limit. MS scans were collected at a resolution of 120,000 (410–1600 m/z range, 4×10^5 AGC, 50 ms maximum ion injection time, FAIMS compensation voltage of −45). All higher energy collision-induced dissociation (HCD) MS/MS spectra were acquired at a resolution of 30,000 (0.7 m/z isolation width, 35% collision energy, 1.25 × 10^5 AGC target, 54 ms maximum ion time, TurboTMT on). Dynamic exclusion was set to exclude previously sequenced peaks for 20 s within a 10-ppm isolation window.
Database searching and protein quantification
All raw files were searched using Thermo’s Proteome Discoverer suite (version 2.4.1.15) with Sequest HT. The spectra were searched against a human Uniprot database downloaded August 2020 (86395 target sequences). Search parameters included 10ppm precursor mass window, 0.05 Da product mass window, dynamic modifications methionine (+15.995 Da), deamidated asparagine and glutamine (+0.984 Da), phosphorylated serine, threonine, and tyrosine (+79.966 Da), and static modifications for carbamidomethyl cysteines (+57.021 Da) and N-terminal and Lysine-tagged TMT (+304.207 Da). Percolator was used filter PSMs to 0.1%. Peptides were group using strict parsimony and only razor and unique peptides were used for protein level quantitation. Reporter ions were quantified from MS2 scans using an integration tolerance of 20 ppm with the most confident centroid setting. Only unique and razor (i.e., parsimonious) peptides were considered for quantification. All RAW FILES are available on PRIDE.
Lipidomics
Sample preparation: At iN d21, cells (500,000 cells per sample) were washed twice with ice-cold PBS, then the cell pellets were collected by centrifuging at 300g for 5 min. After removing the supernatant, the cells were resuspended in 200ul PBS followed by sonication using probe sonifier for 5–10 s at 25% amplification. BCA protein assay was performed to measure the protein concentration in each sample.
Experiment summary: Lipidomic profiling was performed on 16 iPSC-derived neuronal cell samples and 3 PBS media samples. The samples were randomized before lipid extraction. The lipids were then extracted in one batch with quality control samples spaced throughout. These included pooled plasma QC (PQC) (1 in every 5 samples), and 3 NIST1950 samples. The samples were extracted and run through our routine lipidomics platform established by the Metabolomics Laboratory.
Quality control: There are 3 types of QC samples used throughout this study. Technical QCs (TQC) were used to monitor the overall performance of the mass spectrometer. PQCs were used to monitor any experimental issues during the extraction process. NIST1950 were included as they represent the “population” lipidomic profile and can be used to align this cohort with other cohorts that utilize NIST1950 samples. The QC samples show a consistent run.
Data analysis: In total, 813 lipid species from 47 classes were measured with their peaks integrated using vendor software (Masshunter, Agilent) and their areas were exported. There are two tabs of results provided for each type of sample - one corresponding to the individual lipid species and one for their subsequent lipid classes. Concentrations were calculated and the final concentration data is presented as pmol/pmol total membrane lipid content. We considered lipid species to be differentially expressed between SORL1 WT and KO neurons if they showed association with multiple comparisons testing using two-stage step-up (Benjamini-Hochbergi); FDR 5%.
snRNAseq
Raw counts data from the Mathys et al. (2019)44 study were converted to Counts Per Million (CPM) by dividing counts for each nucleus by the sum of counts for that nucleus and then multiplying by 10^6. From this normalized matrix, the following analyses were carried out:
1. Differential expression of APOE among SORL1+ versus SORL1-nuclei. For each major cell class, nuclei were separated into SORL1+ (with expression>0) and SORL1− (with expression = 0) groups, and a Mann-Whitney test was performed (using the wilcox.test function in R) to determine differential expression of APOE. Raw p values were adjusted with Bonferroni correction (p value*number of tests performed). The fold change was calculated as the ratio of the log-transformed (log10(CPM+1)) mean expression of APOE in the SORL1+ group divided by the log-transformed mean expression of APOE in the SORL1-group. This same analysis was performed for differential expression of CLU between the two groups as well.
2. Significance of overlap in detection of APOE among SORL1+ nuclei. For each major cell class, a Fisher’s exact test was performed to determine whether the number of double-positive nuclei (APOE+/SORL1+) was greater or less than expected by chance. Odds ratio (with upper and lower bounds) were extracted from these results and graphed. Raw p values were adjusted with Bonferroni correction (p value*number of tests performed). This same analysis was performed to assess the significance of overlap in detection of CLU among SORL1+ nuclei.
Brain immunocytochemistry
Formalin-fixed postmortem brain tissues were obtained from New York Brain Bank. 6μm sections of formalin-fixed paraffin-embedded (FFPE) tissue from the dorsolateral prefrontal cortex were stained with APOE (Invitrogen, PA5–18361), SORL1 (Novus Biological, 525122) and NeuN (abcam, 190565). Heated-induced epitope retrieval was performed using citrate (pH = 6) using a microwave (800W, 30% power setting) for 25 min. The sections were blocked with blocking medium (3% BSA) for 30 min at Room Temperature, then incubated with primary antibody anti-APOE and anti-SORL1 prepared in 1% BSA for overnight at 4°C. Sections were washed three times with PBS and incubated with fluochrome conjugated secondary antibodies (Thermo Fisher) for 1 h at RT. Sections were then incubated with Alexa Fluor 647 anti-NeuN for 1 h and half at RT. Anti-fading reagent with Dapi (P36931, Life technology) was used for coverslipping. The images were acquired using Nikon Eclipse Ni-E fluorescence microscope at magnification ×40.
Data visualization
Schematics were generated using Biorender. The five-way venn diagram demonstrating overlap of common DEGs between cell types in Figure 1 was generated using nvenn.63 Graphs and heat maps were generated using R Studio54 or GraphPad Prism 9.
QUANTIFICATION AND STATISTICAL ANALYSIS
Information regarding statistical analyses can be found in the figure legends. All statistical tests were performed using GraphPad Prism 9 or R Studio. All data is shown as mean ± SEM. Comparisons between two groups were using student’s t test, and comparisons between more than two groups were analyzed using one-way ANOVA followed by Tukey’s post-hoc test. Spearman correlation method (rank-order based) and Pearson correlation method (linear relationship) was used for correlation analyses.
Supplementary Material
Highlights.
Loss of SORL1 induces a reduction of APOE and CLU levels in neurons
APOE levels and CLU levels are associated with SORL1 levels in human brain in neurons
Modulation of SMAD signaling regulates APOE levels in neurons in a SORL1-dependent manner
SORL1 KO and SORL1 G511R neurons display altered lipid profiles
ACKNOWLEDGMENTS
We thank the AMP-AD consortium for valuable feedback and data sharing (see also “Data and code availability”) and the NeuroTechnology Studio at Brigham and Women’s Hospital for providing Zeiss LSM710 confocal instrument access and consultation on data acquisition and data analysis; iPSC NeuroHub at Brigham and Women’s Hospital for technical assistance with iN differentiation; Saranna Fanning for providing reagents and guidance on lipid droplet analysis; Charles Jennings, Scott Small, and Gregory Petsko for manuscript feedback; Thomas Schwarz, Dennis Selkoe, and Francisco Quintana for their Dissertation Advisory Committee guidance; and to members of the Young-Pearse lab for their input. This work was supported by NIH grants F31AG063399, U01AG072572, U01AG061356, U01AG061359, RF1NS117446, and R01AG055909.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112994.
REFERENCES
- 1.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. (2013). Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 45, 1452–1458. 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, et al. (2022). New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias. Nat. Genet. 54, 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, et al. ; PHRC GMAJ Collaborators (2012). High Frequency of Potentially Pathogenic SORL1 Mutations in Autosomal Dominant Early-Onset Alzheimer Disease. Mol. Psychiatr. 17, 875–879. 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- 4.Vardarajan BN, Zhang Y, Lee JH, Cheng R, Bohm C, Ghani M, Reitz C, Reyes-Dumeyer D, Shen Y, Rogaeva E, et al. (2015). Coding Mutations in SORL 1 and Alzheimer Disease: SORL1 Variants and AD. Ann. Neurol. 77, 215–227. 10.1002/ana.24305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holstege H, van der Lee SJ, Hulsman M, Wong TH, van Rooij JG, Weiss M, Louwersheimer E, Wolters FJ, Amin N, Uitterlinden AG, et al. (2017). Characterization of Pathogenic SORL1 Genetic Variants for Association with Alzheimer’s Disease: A Clinical Interpretation Strategy. Eur. J. Hum. Genet. 25, 973–981. 10.1038/ejhg.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raghavan NS, Brickman AM, Andrews H, Manly JJ, Schupf N, Lantigua R, Wolock CJ, Kamalakaran S, Petrovski S, Tosto G, et al. ; Alzheimer’s Disease Sequencing Project (2018). The Alzheimer’s Disease Sequencing Project. Whole-Exome Sequencing in 20,197 Persons for Rare Variants in Alzheimer’s Disease. Ann. Clin. Transl. Neurol. 5, 832–842. 10.1002/acn3.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, Schaller C, Bujo H, Levey AI, and Lah JJ (2004). Loss of Apolipoprotein E Receptor LR11 in Alzheimer Disease. Arch. Neurol. 61, 1200–1205. 10.1001/archneur.61.8.1200. [DOI] [PubMed] [Google Scholar]
- 8.Dodson SE, Gearing M, Lippa CF, Montine TJ, Levey AI, and Lah JJ (2006). LR11/SorLA Expression Is Reduced in Sporadic Alzheimer Disease but Not in Familial Alzheimer Disease. J. Neuropathol. Exp. Neurol. 65, 866–872. 10.1097/01.jnen.0000228205.19915.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Q-L, Galasko DR, Ringman JM, Vinters HV, Edland SD, Pomakian J, Ubeda OJ, Rosario ER, Teter B, Frautschy SA, and Cole GM (2009). Reduction of SorLA/LR11, a Sorting Protein Limiting β-Amyloid Production, in Alzheimer Disease Cerebrospinal Fluid. Arch. Neurol. 66, 448–457. 10.1001/archneurol.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, and van der Flier WM (2021). Alzheimer’s Disease. Lancet 397, 1577–1590. 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Small SA, and Gandy S (2006). Sorting through the Cell Biology of Alzheimer’s Disease: Intracellular Pathways to Pathogenesis. Neuron 52, 15–31. 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CAF, Breiderhoff T, Jansen P, Wu X, et al. (2005). Neuronal Sorting Protein-Related Receptor SorLA/LR11 Regulates Processing of the Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 102, 13461–13466. 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, and Lah JJ (2006). The Lipoprotein Receptor LR11 Regulates Amyloid β Production and Amyloid Precursor Protein Traffic in Endosomal Compartments. J. Neurosci. 26, 1596–1603. 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young JE The Alzheimer’s Gene SORL1 Is a Key Regulator of Endosomal Recycling in Human Neurons. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen OM, Rudolph I-M, and Willnow TE (2016). Risk Factor SORL1: From Genetic Association to Functional Validation in Alzheimer’s Disease. Acta Neuropathol. 132, 653–665. 10.1007/s00401-016-1615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knupp A, Mishra S, Martinez R, Braggin JE, Szabo M, Kinoshita C, Hailey DW, Small SA, Jayadev S, and Young JE (2020). Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 31, 107719. 10.1016/j.celrep.2020.107719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung C, Tuck E, Stubbs V, van der Lee SJ, Aalfs C, van Spaendonk R, Scheltens P, Hardy J, Holstege H, and Livesey FJ (2021). SORL1 Deficiency in Human Excitatory Neurons Causes APP-Dependent Defects in the Endolysosome-Autophagy Network. Cell Rep. 35, 109259. 10.1016/j.celrep.2021.109259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodson SE, Andersen OM, Karmali V, Fritz JJ, Cheng D, Peng J, Levey AI, Willnow TE, and Lah JJ (2008). Loss of LR11/SORLA Enhances Early Pathology in a Mouse Model of Amyloidosis: Evidence for a Proximal Role in Alzheimer’s Disease. J. Neurosci. 28, 12877–12886. 10.1523/JNEUROSCI.4582-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, and Roses AD (1993). Binding of Human Apolipoprotein E to Synthetic Amyloid Beta Peptide: Isoform-Specific Effects and Implications for Late-Onset Alzheimer Disease. Proc. Natl. Acad. Sci. USA 90, 8098–8102. 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtzman DM, Herz J, and Bu G (2012). Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2, a006312. 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butt OH, Meeker KL, Wisch JK, Schindler SE, Fagan AM, Benzinger TLS, Cruchaga C, Holtzman DM, Morris JC, and Ances BM (2022). Network Dysfunction in Cognitively Normal APOE E4 Carriers Is Related to Subclinical Tau. Alzheimers Dement. 18, 116–126. 10.1002/alz.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martens YA, Zhao N, Liu C-C, Kanekiyo T, Yang AJ, Goate AM, Holtzman DM, and Bu G (2022). ApoE Cascade Hypothesis in the Pathogenesis of Alzheimer’s Disease and Related Dementias. Neuron 110, 1304–1317, S089662732200229X. 10.1016/j.neuron.2022.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, et al. (2018). Gain of Toxic Apolipoprotein E4 Effects in Human IPSC-Derived Neurons Is Ameliorated by a Small-Molecule Structure Corrector. Nat. Med. 24, 647–657. 10.1038/s41591-018-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zalocusky KA, Najm R, Taubes AL, Hao Y, Yoon SY, Koutsodendris N, Nelson MR, Rao A, Bennett DA, Bant J, et al. (2021). Neuronal ApoE Upregulates MHC-I Expression to Drive Selective Neurodegeneration in Alzheimer’s Disease. Nat. Neurosci. 24, 786–798. 10.1038/s41593-021-00851-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taira K, Bujo H, Hirayama S, Yamazaki H, Kanaki T, Takahashi K, Ishii I, Miida T, Schneider WJ, and Saito Y (2001). LR11, a Mosaic LDL Receptor Family Member, Mediates the Uptake of ApoE-Rich Lipoproteins In Vitro. ATVB 21, 1501–1506. 10.1161/hq0901.094500. [DOI] [PubMed] [Google Scholar]
- 26.Yajima R, Tokutake T, Koyama A, Kasuga K, Tezuka T, Nishizawa M, and Ikeuchi T (2015). ApoE-Isoform-Dependent Cellular Uptake of Amyloid-β Is Mediated by Lipoprotein Receptor LR11/SorLA. Biochem. Biophys. Res. Commun. 456, 482–488. 10.1016/j.bbrc.2014.11.111. [DOI] [PubMed] [Google Scholar]
- 27.Zhao J, Lu W, Ren Y, Fu Y, Martens YA, Shue F, Davis MD, Wang X, Chen K, Li F, et al. (2021). Apolipoprotein E Regulates Lipid Metabolism and α-Synuclein Pathology in Human IPSC-Derived Cerebral Organoids. Acta Neuropathol. 142, 807–825. 10.1007/s00401-021-02361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, Ralvenius WT, Akay L, Lockshin E, He L, et al. (2021). APOE4 Disrupts Intracellular Lipid Homeostasis in Human IPSC-Derived Glia. Sci. Transl. Med. 13, eaaz4564. 10.1126/scitranslmed.aaz4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canals I, Ginisty A, Quist E, Timmerman R, Fritze J, Miskinyte G, Monni E, Hansen MG, Hidalgo I, Bryder D, et al. (2018). Rapid and Efficient Induction of Functional Astrocytes from Human Pluripotent Stem Cells. Nat. Methods 15, 693–696. 10.1038/s41592-018-0103-2. [DOI] [PubMed] [Google Scholar]
- 31.Muratore CR, Zhou C, Liao M, Fernandez MA, Taylor WM, Lagomarsino VN, Pearse RV, Rice HC, Negri JM, He A, et al. (2017). Cell-Type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Rep. 9, 1868–1884. 10.1016/j.stemcr.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagomarsino VN, Pearse RV, Liu L, Hsieh Y-C, Fernandez MA, Vinton EA, Paull D, Felsky D, Tasaki S, Gaiteri C, et al. (2021). Stem Cell-Derived Neurons Reflect Features of Protein Networks, Neuropathology, and Cognitive Outcome of Their Aged Human Donors. Neuron 109, 3402–3420.e9. 10.1016/j.neuron.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao L, Zhang Z, Lu J, and Pei G (2019). Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell. Neurosci. 13, 316. 10.3389/fncel.2019.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobrindt K, Zhang H, Das D, Abdollahi S, Prorok T, Ghosh S, Weintraub S, Genovese G, Powell SK, Lund A, et al. (2021). Publicly Available HiPSC Lines with Extreme Polygenic Risk Scores for Modeling Schizophrenia. Complex Psychiatry 6, 68–82. 10.1159/000512716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. (2017). IPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 94, 278–293.e9. 10.1016/j.neuron.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, and Blurton-Jones M (2018). Development and Validation of a Simplified Method to Generate Human Microglia from Pluripotent Stem Cells. Mol. Neurodegener. 13, 67. 10.1186/s13024-018-0297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, and Shusta EV (2012). Derivation of Blood-Brain Barrier Endothelial Cells from Human Pluripotent Stem Cells. Nat. Biotechnol. 30, 783–791. 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, and Shusta EV (2014). A Retinoic Acid-Enhanced, Multicellular Human Blood-Brain Barrier Model Derived from Stem Cell Sources. Sci. Rep. 4, 4160. 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park T-E, Mustafaoglu N, Herland A, Hasselkus R, Mannix R, Fitz-Gerald EA, Prantil-Baun R, Watters A, Henry O, Benz M, et al. (2019). Hypoxia-Enhanced Blood-Brain Barrier Chip Recapitulates Human Barrier Function and Shuttling of Drugs and Antibodies. Nat. Commun. 10, 2621. 10.1038/s41467-019-10588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential Analysis of RNA-Seq Incorporating Quantification Uncertainty. Nat. Methods 14, 687–690. 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 41.Mishra S, Knupp A, Szabo MP, Williams CA, Kinoshita C, Hailey DW, Wang Y, Andersen OM, and Young JE (2022). The Alzheimer’s Gene SORL1 Is a Regulator of Endosomal Traffic and Recycling in Human Neurons. Cell. Mol. Life Sci. 79, 162. 10.1007/s00018-022-04182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, and Small SA (2014). Pharmacological Chaperones Stabilize Retromer to Limit APP Processing. Nat. Chem. Biol. 10, 443–449. 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, and Goldstein LSB (2018). Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Rep. 10, 1046–1058. 10.1016/j.stemcr.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mishra S; Knupp A; Kinoshita C; Martinez R; Theofilas P; Young J. Pharmacologic Stabilization of Retromer Rescues Endosomal Pathology Induced by Defects in the Alzheimer’s Gene SORL1. 10.1101/2022.07.31.502217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu T, Zhu B, Liu Y, Zhang X, Yin J, Li X, Jiang L, Hodges AP, Rosenthal SB, Zhou L, et al. (2020). Multi-Omic Comparison of Alzheimer’s Variants in Human ESC–Derived Microglia Reveals Convergence at APOE. J. Exp. Med. 217, e20200474. 10.1084/jem.20200474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sager KL, Wuu J, Leurgans SE, Rees HD, Gearing M, Mufson EJ, Levey AI, and Lah JJ (2007). Neuronal LR11/SorLA Expression Is Reduced in Mild Cognitive Impairment. Ann. Neurol. 62, 640–647. 10.1002/ana.21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh NN, and Ramji DP (2006). Transforming Growth Factor-β–Induced Expression of the Apolipoprotein E Gene Requires c-Jun N-Terminal Kinase, P38 Kinase, and Casein Kinase 2. ATVB 26, 1323–1329. 10.1161/01.ATV.0000220383.19192.55. [DOI] [PubMed] [Google Scholar]
- 49.Derynck R, and Zhang YE (2003). Smad-Dependent and Smad-Independent Pathways in TGF-β Family Signalling 425, 8. [DOI] [PubMed] [Google Scholar]
- 50.Luo K (2017). Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 9, a022137. 10.1101/cshperspect.a022137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, et al. (2018). APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 98, 1141–1154.e7. 10.1016/j.neuron.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xian X, Pohlkamp T, Durakoglugil MS, Wong CH, Beck JK, Lane-Donovan C, Plattner F, and Herz J (2018). Reversal of ApoE4-Induced Recycling Block as a Novel Prevention Approach for Alzheimer’s Disease. Elife 7, e40048. 10.7554/eLife.40048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, Ambat J, Levy E, Cookson MR, Mathews PM, and Duff KE (2017). The Endosomal–Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front. Neurosci. 11, 702. 10.3389/fnins.2017.00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.R Core Team (2020). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). Https://Www.R-Project.Org/. [Google Scholar]
- 55.McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-Generation Image Processing for Biology. PLoS Biol. 16, e2005970. 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young JE, Boulanger-Weill J, Williams DA, Woodruff G, Buen F, Revilla AC, Herrera C, Israel MA, Yuan SH, Edland SD, and Goldstein LSB (2015). Elucidating Molecular Phenotypes Caused by the SORL1 Alzheimer’s Disease Genetic Risk Factor Using Human Induced Pluripotent Stem Cells. Cell Stem Cell 16, 373–385. 10.1016/j.stem.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muratore CR, Srikanth P, Callahan DG, and Young-Pearse TL (2014). Comparison and Optimization of HiPSC Forebrain Cortical Differentiation Protocols. PLoS One 9, e105807. 10.1371/journal.pone.0105807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chou V, Fancher SB, Pearse RV, Lee H, Lam M, Seyfried NT, Bennett DA, De Jager PL, Menon V, and Young-Pearse TL (2023). INPP5D/SHIP1 Regulates Inflammasome Activation in Human Microglia; preprint. Neuroscience. 10.1101/2023.02.25.530025. [DOI] [Google Scholar]
- 59.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 60.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wickham H (2016). Ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; )978–3-319–24277-4. https://Ggplot2.Tidyverse.Org. [Google Scholar]
- 62.Yu G (2022). Enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.12.2. https://Yulab-Smu.Top/Biomedical-Knowledge-Mining-Book/. [Google Scholar]
- 63.Pérez-Silva JG, Araujo-Voces M, and Quesada V (2018). NVenn: Generalized, Quasi-Proportional Venn and Euler Diagrams. Bioinformatics 34, 2322–2324. 10.1093/bioinformatics/bty109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Expression matrices and meta data for RNAseq, proteomic profiling and lipidomic profiling can be found in the supplemental tables, along with the gene-level and pathways-level output of each analysis performed using these data. RNA-seq data have been deposited at GEO and the AMP-AD Knowledge Portal and are publicly available as of the date of publication. Accession numbers and ids are listed in the key resources table. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and to the AMP-AD Knowledge Portal and are publicly available as of the date of publication. Accession ids are listed in the key resources table. Lipidomics data are deposited in Metabolights and are publicly available as of the date of publication. Accession ids are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
The results published here are in part based on data obtained from the AMP-AD Knowledge Portal (https://doi.org/10.7303/syn2580853). ROSMAP Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Goat anti-APOE | Milipore | Cat#178479; RRID:AB_10682965 |
| Rabbit anti-Amyloid Precursor Protein | Sigma-Aldrich | Cat# A8717, RRID:AB_258409 |
| Mouse anti-Phospho-Tau (Ser202, Thr205) | Thermo Fisher Scientific | Cat# MN1020, RRID:AB_223647 |
| Mouse anti-GAPDH | Proteintech | Cat# 60004-1-Ig, RRID:AB_2107436 |
| Chicken anti-GFAP | Abcam | Cat# ab4674, RRID:AB_304558 |
| Goat anti-AIF1 | Abcam | Cat# ab5076, RRID:AB_2224402 |
| Rabbit anti- LC3 | MBL International | Cat# PM036, RRID:AB_2274121 |
| Chicken anti-MAP2 | Abcam | Cat# ab5392, RRID:AB_2138153 |
| Mouse anti-LR11 (SORL1) | BD Biosciences | Cat# 611860, RRID:AB_399340 |
| Rabbit anti-Synapsin 1 | Millipore | Cat# 574777-10UG, RRID:AB_10683065 |
| Rabbit anti-Tau | Agilent | Cat# A0024, RRID:AB_10013724 |
| Mouse anti-ZO-1 | Thermo Fisher Scientific | Cat# MA3-39100-A555, RRID:AB_2663168 |
| Mouse anti-Tubulin, beta III isoform | Millipore | Cat# MAB1637, RRID:AB_2210524 |
| Mouse anti-vimentin | Millipore | Cat# CBL202, RRID:AB_93387 |
| Rabbit anti-clusterin | Abcam | Cat# ab92548, RRID:AB_10585132 |
| APOE | Invitrogen | PA5-18361, RRID:AB_10979861 |
| Mouse anti-SORL1 (Clone 525122) | R and D Systems | Cat# MAB5699, RRID:AB_2239592 |
| Rabbit anti-NeuN | Abcam | Cat# ab190565, RRID:AB_2732785 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pTet-O-NGN2-puro | Zhang et al. 201329 | Addgene plasmid #52047 |
| Tet-O-FUW-EGFP | Zhang et al. 201329 | Addgene plasmid #30130 |
| FUdeltaGW-rtTA | Zhang et al. 201329 | Addgene plasmid #19780 |
| Tet-O-SOX9-puro | Canals et al. 201830 | Addgene plasmid #117269 |
| Tet-O-NFIB-hygro | Canals et al. 201830 | Addgene plasmid #117271 |
| APOE shRNA construct1 | Sigma Aldrich | TRCN0000371278 |
| APOE shRNA construct2 | Sigma Aldrich | TRCN0000377711 |
|
| ||
| Biological samples | ||
|
| ||
| Postmortem brain tissues | New York Brain Bank | see Supplemental Tables |
| Human brain samples MPFC | RUSH RADC Brain Bank | http://www.radc.rush.edu/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| R33 | MedKoo Biosciences | 504211 |
| Trehalose dihydrate | Sigma Aldrich | 625625 |
| Chloroquine | Tocris | 4109 |
| TGF-β | Sigma Aldrich | T7039 |
| SB431542 | Calbiochem | 616464 |
|
| ||
| Critical commercial assays | ||
|
| ||
| LipidSpotTM 610 | Biotium | 70069 |
| V-PLEX Aβ Peptide Panel 1 (6E10) Kit | Meso Scale Discovery | cat. #K15200G-1 |
| R-PLEX Human ApoE Assay | Meso Scale Discovery | cat. #K1512IR-2 |
| R-PLEX Human Clusterin Assay | Meso Scale Discovery | cat. #K151YLR-2 |
|
| ||
| Deposited data | ||
|
| ||
| RNAseq data | This paper | NCBI GEO database, GEO: GSE238013 |
| Lagomarsino et al. 202132 | AMP-AD Knowledge Portal; SynID: syn2580853 and SynID: syn3219045 For quick visualization of SORL1 KO and WT data, please also see https://youngpearselab.shinyapps.io/sorl1_namei_cells/. | |
| Lipidomics data | This paper | Metabolights, study ID: MTBLS8242 |
| Mass spectrometry proteomics data | This paper | ProteomeXchange Consortium via the PRIDE partner repository PRIDE: PXD044093 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human iPSC line: Wildtype clone A7 (“line #1”, >90% European ancestry) | Knupp et al. 202016 | N/A |
| Human iPSC line: SORL1 Knockout clone E4 (“line #1” >90% European ancestry) | Knupp et al. 202016 | N/A |
| Human iPSC line: SORL1 G511R clone C6 (“line #1”, >90% European ancestry) | Mishra et al. 202245 | N/A |
| Human iPSC line: Wildtype clone 5 (“line #2”, >90% European ancestry) | This paper | N/A |
| Human iPSC line: SORL1 Knockout clone 4 (“line #2”, >90% European ancestry) | This paper | N/A |
| HUMAN IPSC LINE: BR01, 99% European ancestry | Lagomarsino et al. 202132 | BR01, AJ0006 |
| HUMAN IPSC LINE: BR04, 98% European ancestry | Lagomarsino et al. 202132 | BR04, AJ0056 |
| HUMAN IPSC LINE: BR08, 94% European ancestry | Lagomarsino et al. 202132 | BR08, AJ0044 |
| HUMAN IPSC LINE: BR09, 92% European ancestry | Lagomarsino et al. 202132 | BR09, AJ0038 |
| HUMAN IPSC LINE: BR103, 94% European ancestry | Lagomarsino et al. 202132 | BR103, AJ0121 |
| HUMAN IPSC LINE: BR104, 95% European ancestry | Lagomarsino et al. 202132 | BR104, AJ0113 |
| HUMAN IPSC LINE: BR108, 82% European ancestry | Lagomarsino et al. 202132 | BR108, AJ0117 |
| HUMAN IPSC LINE: BR11, 99% European ancestry | Lagomarsino et al. 202132 | BR11, AJ0002 |
| HUMAN IPSC LINE: BR13, 92% European ancestry | Lagomarsino et al. 202132 | BR13, AJ0039 |
| HUMAN IPSC LINE: BR14, 99% European ancestry | Lagomarsino et al. 202132 | BR14, AJ0003 |
| HUMAN IPSC LINE: BR15, 93% European ancestry | Lagomarsino et al. 202132 | BR15, AJ0008 |
| HUMAN IPSC LINE: BR21, 92% European ancestry | Lagomarsino et al. 202132 | BR21, AJ0046 |
| HUMAN IPSC LINE: BR22, 93% European ancestry | Lagomarsino et al. 202132 | BR22, AJ0021 |
| HUMAN IPSC LINE: BR24, 93% European ancestry | Lagomarsino et al. 202132 | BR24, AJ0040 |
| HUMAN IPSC LINE: BR26, 94% European ancestry | Lagomarsino et al. 202132 | BR26, AJ0043 |
| HUMAN IPSC LINE: BR27, 97% European ancestry | Lagomarsino et al. 202132 | BR26, AJ0001 |
| HUMAN IPSC LINE: BR28, 93% European ancestry | Lagomarsino et al. 202132 | BR28, AJ0029 |
| HUMAN IPSC LINE: BR29, 95% European ancestry | Lagomarsino et al. 202132 | BR29, AJ0030 |
| HUMAN IPSC LINE: BR30, 94% European ancestry | Lagomarsino et al. 202132 | BR30, AJ0042 |
| HUMAN IPSC LINE: BR33, 91% European ancestry | Lagomarsino et al. 202132 | BR33, AJ0047 |
| HUMAN IPSC LINE: BR36, 95% European ancestry | Lagomarsino et al. 202132 | BR36, AJ0022 |
| HUMAN IPSC LINE: BR37, 93% European ancestry | Lagomarsino et al. 202132 | BR37, AJ0031 |
| HUMAN IPSC LINE: BR39, 98% European ancestry | Lagomarsino et al. 202132 | BR39, AJ0005 |
| HUMAN IPSC LINE: BR40, 94% European ancestry | Lagomarsino et al. 202132 | BR40, AJ0045 |
| HUMAN IPSC LINE: BR41, 95% European ancestry | Lagomarsino et al. 202132 | BR41, AJ0024 |
| HUMAN IPSC LINE: BR43, 91% European ancestry | Lagomarsino et al. 202132 | BR43, AJ0020 |
| HUMAN IPSC LINE: BR46, 98% European ancestry | Lagomarsino et al. 202132 | BR46, AJ0004 |
| HUMAN IPSC LINE: BR48, 91% European ancestry | Lagomarsino et al. 202132 | BR48, AJ0028 |
| HUMAN IPSC LINE: BR50, 94% European ancestry | Lagomarsino et al. 202132 | BR50, AJ0041 |
| HUMAN IPSC LINE: BR54, 91% European ancestry | Lagomarsino et al. 202132 | BR54, AJ0048 |
| HUMAN IPSC LINE: BR57, 93% European ancestry | Lagomarsino et al. 202132 | BR57, AJ0073 |
| HUMAN IPSC LINE: BR58, 89% European ancestry | Lagomarsino et al. 202132 | BR58, AJ0068 |
| HUMAN IPSC LINE: BR59, 93% European ancestry | Lagomarsino et al. 202132 | BR59, AJ0072 |
| HUMAN IPSC LINE: BR60, 89% European ancestry | Lagomarsino et al. 202132 | BR60, AJ0067 |
| HUMAN IPSC LINE: BR61, 93% European ancestry | Lagomarsino et al. 202132 | BR61, AJ0076 |
| HUMAN IPSC LINE: BR62, 93% European ancestry | Lagomarsino et al. 202132 | BR62, AJ0069 |
| HUMAN IPSC LINE: BR63, 94% European ancestry | Lagomarsino et al. 202132 | BR63, AJ0070 |
| HUMAN IPSC LINE: BR64, 94% European ancestry | Lagomarsino et al. 202132 | BR64, AJ0066 |
| HUMAN IPSC LINE: BR65, 86% European ancestry | Lagomarsino et al. 202132 | BR65, AJ0080 |
| HUMAN IPSC LINE: BR66, 93% European ancestry | Lagomarsino et al. 202132 | BR66, AJ0094 |
| HUMAN IPSC LINE: BR67, 94% European ancestry | Lagomarsino et al. 202132 | BR67, AJ0099 |
| HUMAN IPSC LINE: BR68, 92% European ancestry | Lagomarsino et al. 202132 | BR68, AJ0095 |
| HUMAN IPSC LINE: BR69, 94% European ancestry | Lagomarsino et al. 202132 | BR69, AJ0103 |
| HUMAN IPSC LINE: BR72, 91% European ancestry | Lagomarsino et al. 202132 | BR72, AJ0090 |
| HUMAN IPSC LINE: BR83, 94% European ancestry | Lagomarsino et al. 202132 | BR83, AJ0083 |
| HUMAN IPSC LINE: BR89, 93% European ancestry | Lagomarsino et al. 202132 | BR89, AJ0089 |
| HUMAN IPSC LINE: BR91, 91% European ancestry | Lagomarsino et al. 202132 | BR91, AJ0115 |
| HUMAN IPSC LINE: BR92, 92% European ancestry | Lagomarsino et al. 202132 | BR92, AJ0116 |
| HUMAN IPSC LINE: BR93, 94% European ancestry | Lagomarsino et al. 202132 | BR93, AJ0114 |
| HUMAN IPSC LINE: BR95, 93% European ancestry | Lagomarsino et al. 202132 | BR95, AJ0119 |
| HUMAN IPSC LINE: BR97. 90% European ancestry | Lagomarsino et al. 202132 | BR97, AJ0123 |
| HUMAN IPSC LINE: BR98, 96% European ancestry | Lagomarsino et al. 202132 | BR98, AJ0109 |
| HUMAN IPSC LINE: BR99, 95% European ancestry | Lagomarsino et al. 202132 | BR99, AJ0107 |
| Human iPS Cell Line (Episomal, CD34+, ApoE Knockout) | Alstem cell advancement | iPS36 |
| Human iPS Cell Line (Episomal, CD34+, ApoE3) | Alstem cell advancement | iPS26 |
| Human iPS Cell Line (Episomal, CD34+) | Alstem cell advancement | iPS16 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SORL1 KO line#1 gRNA: ‘ATTGAACGACATG AACCCTC’ | Knupp et al. 202016 | N/A |
| SORL1 KO line#1 ssODN: ‘GGGAATTGATCC CTATGACAAACCAAATACCATCTACATTGAACGACA TGAACCCTCTGGCTACTCCACGTCTTCCGA AG TACAGATTTCTTCCAGTCCCGGGAAAACCAGGAAG’ | Knupp et al. 202016 | N/A |
| SORL1 G511R gRNA: ‘CTCTTGCATTTTAGGCTCAG’ | Mishra et al. 202245 | N/A |
| SORL1 G511R ssODN: ‘CTGACATATTCTTGAAA TTAAAAATAATTATTTCTCTTGCATTTTAGGCTCA GTGCGAAAGAACTTGGCTAGCAAGACAAACGTG TACATCTCTAGCAGTGCTGGAGCCAGGTGGCG’ | Mishra et al. 202245 | N/A |
| SORL1 KO line#2 gRNA: ‘ATGTTCCTGAATCATGATCC’ | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| R Studio, v3.6.1 of R; v1.2.5019 of R Studio | R Core Team, 202054 | https://www.rstudio.com |
| Sleuth, v0.30.0 | Pimentel et al., 201740 | https://www.rdocumentation.org/packages/sleuth/versions/0.30.0 |
| Cell Profiler | McQuin et al., 201855 | https://cellprofiler.org/ |
| All original code used in this paper | This paper |
https://github.com/genejockey33000/typGumbo. Zenodo; https://doi.org/10.5281/zenodo.8183364 |