


Abstract
Implementation of therapeutic in vivo gene editing using CRISPR/Cas relies on potent delivery of gene editing tools. Administration of ribonucleoprotein (RNP) complexes consisting of Cas protein and single guide RNA (sgRNA) offers short-lived editing activity and safety advantages over conventional viral and non-viral gene and RNA delivery approaches. By engineering lentivirus-derived nanoparticles (LVNPs) to facilitate RNP delivery, we demonstrate effective administration of SpCas9 as well as SpCas9-derived base and prime editors (BE/PE) leading to gene editing in recipient cells. Unique Gag/GagPol protein fusion strategies facilitate RNP packaging in LVNPs, and refinement of LVNP stoichiometry supports optimized LVNP yield and incorporation of therapeutic payload. We demonstrate near instantaneous target DNA cleavage and complete RNP turnover within 4 days. As a result, LVNPs provide high on-target DNA cleavage and lower levels of off-target cleavage activity compared to standard RNP nucleofection in cultured cells. LVNPs accommodate BE/sgRNA and PE/epegRNA RNPs leading to base editing with reduced bystander editing and prime editing without detectable indel formation. Notably, in the mouse eye, we provide the first proof-of-concept for LVNP-directed in vivo gene disruption. Our findings establish LVNPs as promising vehicles for delivery of RNPs facilitating donor-free base and prime editing without formation of double-stranded DNA breaks.
Graphical Abstract
Graphical Abstract.
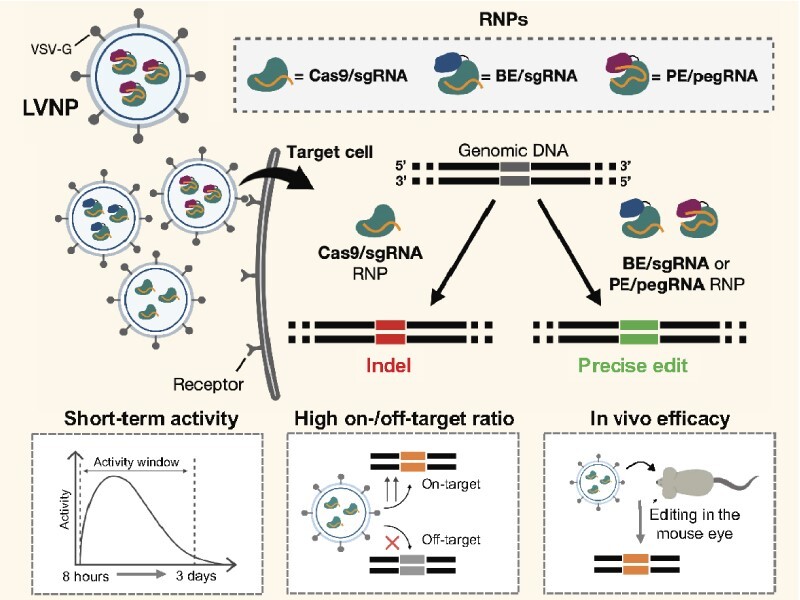
INTRODUCTION
CRISPR/Cas9-based genome editing has the potential to transform the lives of patients suffering from severe genetic diseases (1). Editing of disease-causing genes is based on targeted DNA cleavage mediated by the Cas9 endonuclease directed by a single guide RNA (sgRNA) to a predetermined locus. Repair of the DNA double-stranded break (DSB) by non-homologous end joining (NHEJ) leads to indel formation, whereas repair by homology-directed repair (HDR) supports precise editing (2), but suffers from low efficacy in primary cells as well as the need for co-administering a DNA repair template. Genotoxicity is a significant concern due to the formation of DSBs in both on- and off-target genomic loci (3). Therefore, effective genome editing ideally relies on delivery tools supporting short-lived activity of Cas9/sgRNA complexes to restrict unintended modifications of the genome. Contemporary virus-based delivery approaches, including lentiviral (LV) and adeno-associated virus (AAV)-derived vectors, facilitate delivery of Cas9 and sgRNA gene expression cassettes, although limited packaging capacity of AAV restricts the flexibility of vector designs. Importantly, delivery of gene cassettes potentially causes prolonged cellular exposure to active Cas9/sgRNA complexes, which may lead to genotoxicity (4) including formation of large deletions (5,6) and translocations (7,8), chromothripsis (9), and unwanted genomic integration (10,11). Also, high intracellular levels of Cas9 may cause depletion of gene-corrected cells (12).
To comply with the major challenges of conventional Cas9-based gene editing, alternative genome editing approaches involving neither formation of DSBs nor repair using a donor sequence have been developed. Base editing involves deamination of a predetermined target nucleotide using the base editor (BE), an engineered protein consisting of a Cas9 nickase fused to a deaminase (13,14). A potential risk of base editing is unwanted ‘bystander’ editing of nucleotides within the range of the deaminase (15). Prime editing facilitates genomic incorporation of the edit sequence copied from the prime editing guide RNA (pegRNA) (16). This procedure is facilitated by the prime editor (PE), a fusion protein consisting of a Cas9 nickase fused to the reverse transcriptase derived from Moloney murine leukaemia virus (M-MLV) (16). As both base and prime editing rely on the administration of larger fusion proteins, therapeutic intervention is challenged by the restricted packaging capacity of conventional gene vehicles (17).
Efficient ex vivo genome editing can be achieved by DNA-free delivery of CRISPR/Cas9 ribonucleoprotein (RNP) complexes consisting of recombinant Cas9 protein complexed with synthetic sgRNAs by nucleofection (18). Others and we have engineered retro- and lentivirus-derived virus particles to accommodate and transiently deliver foreign proteins, including DNA transposases (19–21), zinc-finger nucleases (ZFN), TAL-effector nucleases (22,23), Cas9 (24–27) and base editors (28). Such vehicles combine receptor-directed uptake of retroviral particles in recipient cells with the ability of such particles to transport engineered protein and protein-RNA complexes. Transient Cas9/sgRNA RNP delivery may offer substantial benefits compared to delivery of gene expression cassettes, the latter which may support prolonged protein activity leading to increased risk of genotoxicity (29,30).
Here, we report Cas9/sgRNA RNP delivery in engineered lentivirus-derived particles (LVNPs) leading to potent gene editing in recipient cells. We establish and optimize LVNP production for delivery of RNP complexes consisting of Streptococcus pyogenes Cas9 and sgRNA, implement scaffold-optimized sgRNAs for improved performance, achieve gene editing within 12 hours of delivery, demonstrate high on/off-target ratios relative to state-of-the-art nucleofection, and show targeted gene modification in vivo in the mouse eye. We further refine the LVNP technology for BE/sgRNA and PE/epegRNA delivery and show efficient base editing with markedly reduced bystander editing and prime editing without detectable indel formation. These data mark the first examples of in vivo gene modification and potent BE and PE delivery by exploiting enveloped particles based on HIV-1 biology for RNP delivery in mammalian cells and expand the options for effective and safe delivery of CRISPR therapeutics.
MATERIALS AND METHODS
Cell lines
All cells were cultured in DMEM (Sigma-Aldrich) supplemented with 5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Thermo Fischer Scientific). Transgenic cell lines were maintained in puromycin (1 ug/ml) (Thermo Fisher Scientific) or blasticidin (5 ug/ml) (Thermo Fisher Scientific). Cells were maintained at 60–90% confluence at 37°C with 5% carbon dioxide and tested negative for mycoplasma (Eurofins Genomics).
Plasmid construction
Construction of all plasmids is described in the Supplementary material. Plasmids were constructed using NEBuilder® HiFi DNA Assembly Master Mix (New England BioLabs) and has been deposited to Addgene. In brief, the plasmid backbone was linearized by digestion with the relevant restriction enzyme (Thermo Fischer Scientific) and insert(s) were PCR amplified from plasmid DNA and/or synthetic DNA (TWIST Bioscience). The resulting fragments were combined using NEBuilder® HiFi DNA Assembly Master Mix (New England BioLabs) according to manufacturer's instructions. All sgRNAs were designed using Synthego software or CRISPOR (31) and pegRNAs by PegIT (32) (Supplementary Table S2).
Nuclease activity assay
The assay for monitoring nuclease activity of SpCas9 delivered by LVNP2.2 was carried out by seeding 5 × 104 HEK293T cells in a 24-well plate format (Sarstedt). Following 24h of incubation, the cells were transduced with 60 ng LVNP2.2 loaded with SpCas9 and a sgRNA targeting d2eGFP. The LVNP2.2-tranduced cells were subsequently co-transduced with LV/PGK-d2eGFP-IRES-Puro at a MOI of 1 in 24 h intervals (0–7 days) after LVNP2.2 transduction. 24h after the final LV-transduction (8 days after LVNP2.2 transduction), the LV-transduced cells were subjected to 1 week of puromycin selection at a concentration of 1 μg/ml to discard non-transduced cells. After 1 week of puromycin selection, the cells were analysed on a NovoCyte flow cytometer (ACEA Bioscience). The SpCas9 activity was quantified by calculating the KO efficiency according to the formula:
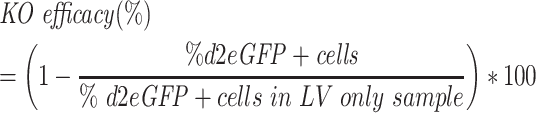 |
LV, LVNP and eVLP production
Both LV, IDLV, LVNP and eVLP were produced by transient transfection of Lenti-X (Takara Bio). Cells were seeded at a density of 3.75–4.00 × 106 in 10-cm dishes and incubated overnight. A calcium phosphate solution (33) containing relevant plasmid (Supplementary Table S1) was dropwise added to 10-cm dishes and incubated overnight. The medium was replenished after 24 h, and the supernatant was harvested 48 and 72 h post-transfection. The supernatant was passed through a 0.45 um filter and concentrated 300× by ultracentrifugation in a cushion of 20% (w/v) sucrose and PBS unless otherwise stated. Ultracentrifugation was performed at 25 000 RPM at 4°C for 2 h in a Beckman SW27 or SW28 rotor. Pellets were resuspended in 85 ul PBS overnight (4°C), pooled (first and second harvest), and centrifuged at 1200 RPM to pellet residual debris. For in vivo experiments, LVNP3.0 and eVLP was pooled from 9 × 10-cm dishes and additionally concentrated by Amicon filtration (100 kDa, Sigma-Aldrich). The viral concentration was quantified by p24 ELISA (XpressBio) according to the manufacturer's protocol and stored at −80C in aliquots until use.
Titer determination
The functional titer was estimated by limiting dilution. 1 × 105 HEK293T were seeded in 6-well plates and transduced with serial dilutions of virus in polybrene (8 μg/ml). After three days, cells were analysed for eGFP expression by flow cytometry on a NovoCyte Flow Cytometer (ACEA Biosciences). Dilutions resulting in 5–20% eGFP positive cells were used to calculate the functional titer:
 |
Flow cytometry
Cells were washed with PBS, detached by trypsinization, and resuspended in FACS buffer (1% BSA, 2.5 mM EDTA, 25 mM HEPES dissolved in PBS). Cells were analysed on a NovoCyte Flow Cytometer (ACEA Bioscience). Data was analysed in Novo Express v1.5.6 or FlowJo v10.1.
Western blotting
LVNP and IDLV was produced either in the presence or absence of 2 μM saquinavir (SQV). Ultracentrifuged particles (90 ng p24) were lysed in RIPA buffer (Thermo Fisher Scientific) supplemented with 10 mM NaF and 1× complete protease inhibitor cocktail (Roche). The lysate was denatured in XT Sample Buffer supplemented with XT Reducing Agent (Bio-Rad), separated by SDS-PAGE, and transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% skimmed milk dissolved in TBS/0.05% Tween-20 for 1 h and incubated overnight with a FLAG antibody (Sigma-Aldrich). The membrane was washed and incubated with anti-mouse secondary antibodies (Dako) and visualized by chemiluminescence using Clarity Western ECL Substrate (Bio-Rad). The antibodies were removed with stripping buffer (Thermo Fisher Scientific), and the membrane was incubated overnight with a p24 antibody (R&D Systems) followed by anti-rabbit secondary antibodies.
Transduction procedures
Cells were plated in 24-well plates (Sarstedt) at a density of 5 × 104 cells/well (HEK293T, HEK293T-Vegfa, or 3 × 104 cells/well (AML12) and incubated overnight unless stated otherwise. A mastermix containing the indicated amount of LVNP (p24 normalized) and DMEM containing polybrene (8 μg/ml) was prepared separately for each condition. For the 24-well format, 500 ul mastermix was applied to replica wells and incubated overnight. Cells were harvested 3 days post-transduction and used for downstream analysis.
Genomic DNA extraction and PCR
Genomic DNA was isolated by NaCl/EtOH precipitation as previously described (20). Following resuspension in TE-buffer or ddH2O, 1 μl (1–20 ng) was used for PCR amplification of the target region using Phusion Master Mix (Thermo Fisher Scientific). PCR products were purified by gel extraction (Omega Bio-tek) or PCR clean-up (SAP/EXO). A solution of 0.5 μl FastAP, 0.5 μl Exo1, 9 μl PCR product, and H2O to a final volume of 18 μl was incubated at 37°C for 15 min followed by inactivation at 85°C for 15 min in a thermocycler. The resulting amplicon was sequenced by Eurofins Genomics. The resulting indel frequencies were deconvoluted by ICE analysis (34) or DECODR (35), EditR for base editing (36) and CRISPResso2 (37) for prime editing. Primers are listed in Supplementary Table S3.
Determination of sgRNA abundance using ddPCR
Total RNA from ultracentrifuged LVNPs was extracted using Roche High Pure miRNA Isolation Kit (Roche Applied Science) and treated with DNase I (Thermo Fisher Scientific) to remove any residual plasmid DNA as previous described (38,39). Both yield and purity were evaluated on a DeNovix DS-11 Spectrophotometer. Equal amounts of input RNA were used for cDNA synthesis using Maxima H Minus cDNA Synthesis Master mix (Thermo Fisher Scientific). The cDNA was diluted 2 times (recipient cells) or 512 times (LVNPs) and quantitative droplet digital PCR (ddPCR) was performed on a QX200TM Droplet DigitalTM PCR System with ddPCR Supermix for Probes (No dUTP) (BioRad) according to the manufacturer. Primers and probes are listed in Supplementary Table S4.
Plasmid transfection
Plasmids were purified using NucleoBond Xtra Midi (AH Diagnostics) with endotoxin removal. HEK293T were plated for transfection in 24-well plates (Corning) at a density of 5 × 104 cells/well and allowed to adhere overnight. Cells were transfected with 1 μl TurboFect and 1μg of plasmid DNA. Unless stated otherwise, 750 ng of base editor and 250 ng of sgRNA expression plasmid were co-transfected per well. The medium was replenished after 24 h. The cells were processed for downstream analysis 3 days post-transfection.
Nucleofection
To evaluate on/off-target disruption of the Pcsk9 locus in AML12, the indicated amount of chemically modified (2′-O-methyl at 3 first and last bases, 3′ phosphorothioate bonds between first 3 and last 2 bases) sgRNA (Synthego) and indicated amount of Cas9 protein (Alt-R SpCas9 Nuclease V3, IDT) were incubated at 25°C for 15 min. The RNP solution was applied to 2 × 105 AML12 or HEK293T cells in 20 μl OptiMEM. Cells were nucleofected using a 4D-nucleofector device (Lonza, Switzerland) in 20 μl Nucleocuvette strips (Lonza) using the program CM-138 set to P3 buffer. Cells were reseeded in 24-well plates (Corning) in DMEM until downstream analysis. Genomic DNA was extracted using QuickExtract (Lucigen) Primers are listed in Supplementary Table S3
ChIP-qPCR
ChIP-qPCR against MRE11 was carried out using a scaled down version of the DISCOVER-seq protocol (40). In brief, 5 × 105 AML12 cells were seeded (day 0) and transduced (day 1) using 180 ng p24 LVNP2.2 for each time point. Cells were harvested, crosslinked in 1% formaldehyde, washed in PBS, and stored at −80°C until use. Crosslinked cells were lysed using 1 ml LB1 followed by 1 ml LB2, and lastly 100ul LB3. Lysed nuclear extract was sonicated for 15 minutes in 30 second pulses on high using a Bioruptor Standard (Diagenode) and mixed with 185 μl LB3 and 15 μl 20% Triton X-100 (Sigma-Aldrich). 5 μl lysed nuclear extract was stored as input DNA. The remaining solution was incubated overnight rotating at 4°C with Dynabeads Protein A (Thermo Fisher Scientific) prepared from 10 μl stock bead slurry bound to 1 μg anti-MRE11 (Abcam, ab208020) per sample, the beads were blocked with 0.5% BSA before addition of sample. Beads were washed 5× in RIPA buffer, 1× in TBS before the crosslinking was reversed in 200 μl elution buffer, while rotating overnight at 65°C in a hybridization oven alongside the input DNA (5 μl) diluted in 195 μl elution buffer. The eluate was treated with 8 μl 10 mg/ml RNAse A (Thermo Fisher Scientific) for 30 minutes at 37°C followed by 4 μl of 20 mg/ml Proteinase K (Thermo Fisher Scientific) treatment for 1 hour at 55°C. Hereafter, DNA was purified from the samples using a MiniElute PCR Purification kit (Qiagen) according to the manufacturer's protocol and eluted in 33 μl nuclease free water. Purified DNA (4.8 μl) was used as template in a 10 μl qPCR reaction using the RealQ Plus 2× Master Mix Green without ROX (Ampliqon). Samples were denatured at 95°C for 15 min followed by 40 cycles of (95°C for 10 s; 60°C for 20 s and 72°C for 30 s). A melt curve gradient from 65°C to 97°C was applied with a ramp rate at 0.11°C/s in technical triplicates. The enrichment was calculated as: Enrichment = 2ΔCt(on-target region)/2ΔCt(control-region), where ΔCt = Ct(Input DNA) − Ct(ChIP DNA) (40). Primers are listed in Supplementary Table S3.
In vivo experiments
Mice were kept on a 12 h/12 h light/dark cycle at the Animal Facilities at the Department of Biomedicine, Aarhus University, Denmark. Mice had ad libitum access to Altromin maintenance feed (Altromin) and water. Animals were handled in accordance with the ‘Statement for the Use of Animals in Ophthalmic and Vision Research’ from the Association for Research in Vision and Ophthalmology (ARVO). All animal experiments were performed under the approval of The Danish Animal Inspectorate (Case# 2020-15-0201-00556).
Subretinal injection
8-week-old, male C57Bl/6J mice were purchased from Janvier Labs and allowed to acclimate for a week. Mice (n = 17) were anaesthetized by medetomidine hydrochloride 0.5–1 mg/kg (Cepetor) and ketamine 60–100 mg/kg (Ketador). One drop of 1% tropicamide solution (Mydriacyl) was used for pupil dilation, and carbomer eye gel (2 mg/g, Viscotears) was used to lubricate the eyes during sedation. For LVNP2.2, the subretinal space was accessed via a posterior transscleral approach using an OPMI 1 FR PRO Surgical microscope (Zeiss), and mice received an unilateral injection of 2 μl (16 ng p24) of LVNP2.2 (encoding a sgRNA targeting Vegfa and a transfer vector encoding eGFP) as previously described (41). For LVNP3.0 and eVLP, the eye was perforated posterior to the limbus with a 30-gauge needle. A 34-gauge blunt-ended needle (World Precision Instruments, NF34BL-2) connected to a microinjection syringe (World Precision Instruments, NANOFIL) with Silifilex tubing (World Precision Instruments, SILFLEX-2) was then inserted through the opening and advanced through the vitreous cavity. Finally, the retina was perforated to access the subretinal space and the compound slowly injected. Each mouse received bilateral injections of 1.6 μl (48 ng p24) of LVNP3.0 (encoding a sgRNA targeting Vegfa). Anaesthesia was reversed with Atipamezole hydrochloride 0.5–1 mg/kg (Antisedan). Mice were kept warm on a heating pad and transferred back into their cages when mobile. Mice received subcutaneous injections of carprofen 5 mg/kg (Norodyl) prior to the subretinal injection and during the subsequent 3 days via their drinking water (3.33 mg/100 ml).
Fundus photography and optical coherence tomography (OCT)
To detect eGFP expression in vivo, and to inspect retinal structures following subretinal injection of LVNP2.2, fundus fluorescence imaging and OCT was carried out 3 days post-injection according to established protocols (42) using a commercial imaging device for rodents, the OCT2 integrated with the Micron IV retinal imaging system and the accompanying Reveal software (Phoenix Research Labs).
RPE/choroid/sclera flat mounts
5 days post-injection, mice treated with LVNP2.2 were sacrificed (n = 3), eyes enucleated, and flat mounts were prepared as previously described (43). In brief, eyes were cleaned and fixed in fresh 4% paraformaldehyde at room temperature for 2 h. The cornea, lens, and neuroretina were removed, and 8 incisions from the periphery to the optic nerve enabled flat mounting of the tissue with the retinal pigment epithelial (RPE) cells facing upward on a SuperFrost®Plus glass slide (Menzel-Glaser). Cover glass was mounted using ProLong® Gold antifade reagent (Invitrogen). Flatmounts were analysed for eGFP expression by fluorescence microscopy using a Leica DM IRBE (Leica Microsystems). Images were captured with a Leica DFC 360 FX camera and associated software (Leica Application Suite v3).
Collection of RPE cells and FACS
RPE cells were collected and pooled from LVNP2.2-injected mice (n = 6) and from naïve (non-injected) eyes (n = 3) according to established protocols (44). One eye was lost during dissection of the eye cup. In brief, hyaluronidase was used to detach the neural retina from the RPE layer followed by enzymatic digestion using trypsin combined with shaking of the eyecup to gently detach the RPE cells from the Bruch's membrane. Following the last centrifugation step, RPE cells were resuspended in FACS buffer (1% BSA, 2.5 mM EDTA, 25 mM HEPES dissolved in PBS) and the RPE cell solution was transferred to a 100 μm cell strainer (Thermo Fisher Scientific). Cells were kept on ice and sorted immediately after collection. FACS was performed using a three-laser FACS Aria III cell sorter (FACS Core Facility, Department of Biomedicine, Aarhus University). The gating strategy was defined in RPE cells isolated from non-injected eyes. Following FACS, the indel frequency was evaluated in both eGFP-negative and eGFP-positive populations isolated from both naïve and LVNP2.2-injected eyes. Primers are listed in Supplementary Table S3.
Collection of RPE cells for indel analysis
Mouse eyes treated with LVNP3.0-SpCas9 or eVLP were dissected under light microscope to separate the posterior eyecup from the anterior segment, lens, and retina. The eyecup was transferred to 350 μl of RLT Plus tissue lysis buffer (Qiagen) and incubated for 1 min. The RPE cells were detached and lysed by gentle pipetting before the remaining eyecup was removed. DNA was extracted from the RPE cells using the AllPrep DNA/RNA Mini Kit (Qiagen; 80284) according to manufactures instructions.
Targeted next-generation sequencing
Library preparation was performed using a two-step protocol. First, the genomic region (HEK3-CTTins) was PCR amplified using primers with TruSeq dual-index adapter overhangs (IDT; Supplementary Table S6) in 2X Phusion Plus PCR Master Mix (Thermo Fischer Scientific). PCR1 was performed as follows: 98°C for 30 s; 25 cycles of (94°C for 30 s, 63°C for 10 s and 72°C for 30 s); 72°C for 5 min. The amplicon was purified using HighPrep™ PCR Clean-up beads (MAGBIO Genomics) and quantified using a Qubit flex fluorometer (Thermo Fischer Scientific). A secondary PCR was run with indexing primers (IDT; Supplementary Table S6): 25 uL 2X Phusion Plus PCR Master Mix, 1 ul indexing primers (5 uM), 4 ng PCR1 amplicon, and milli-Q water. PCR2 was performed as follows: 98°C for 30 s; 8 cycles of (94°C for 30 s, 60°C for 10 s and 72°C for 30s); 72°C for 5 min. The amplicon was purified and measured as described above and diluted to a final concentration of 1 nM in RSB buffer (10 mM Tris–Cl, pH 8.5). Paired-end libraries (150 bp) were sequenced on iSeq100 (Illumina) according to the manufacturer's instructions. The level of genome editing was quantified using CRISPResso2 (37) using standard settings. Oligonucleotides for NGS are listed in Supplementary Table S5.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Significant P-values were determined by the Mann–Whitney U-test. Sample size and the statistical tests used are described in the figure legends. All statistical analysis was performed using GraphPad Prism v9.
RESULTS
Targeted DNA cleavage by cas9/sgRNA RNPs incorporated in LVNPs by C-terminal fusion to GagPol
Lentiviruses, including human immunodeficiency virus type 1 (HIV-1), assemble through multimerization of Gag and GagPol polypeptides at the plasma membrane. In conjugation with a dimeric RNA genome, Gag and GagPol polypeptides are embedded in and accumulate at a segment of the plasma membrane leading to budding of enveloped virus particles from virus-producing cells. Released virus particles are immature and undergo maturation triggered by cleavage of the polypeptides by the viral protease. To incorporate Streptococcus pyogenes Cas9 (SpCas9) in LVNPs (Figure 1A, B), we first fused FLAG-tagged SpCas9 to the integrase protein in the C-terminus of GagPol (pGagPol-IntSpCas9), based on the rationale that this strategy (referred to as LVNP1.0) was less likely to interfere with virus function due to the restricted number of GagPol molecules, relative to Gag, in the particles (45). To facilitate protease-directed release of SpCas9 from GagPol during virion maturation, a protease cleavage site (PCS) was incorporated between integrase (C-terminal end of Pol) and SpCas9 (Figure 1C), and the presence of 160-kDa SpCas9 protein in LVNP1.0, indicative of effective SpCas9 incorporation and release from GagPol during particle maturation, was confirmed (Supplementary Figure S1a). Next, by co-packaging eGFP-encoding vector RNA, we observed that the transductional titer of LVNP1.0 was markedly reduced compared to a standard integrase-defective lentiviral vector (IDLV) without SpCas9 protein (Figure 1D), suggesting that the yield of virus particles capable of transferring the eGFP reporter gene was restricted. To study functional LVNP transfer of SpCas9, we investigated two routes for sgRNA delivery based on (i) packaging of a sgRNA-encoding vector genome or (ii) overexpression of sgRNA in LVNP-producing cells. By packaging a vector containing a U6-driven expression cassette encoding a sgRNA targeting the AFF1 gene (LentiGuide-Puro; Supplementary Figure S1b), modest indel frequencies (∼8%) were observed in recipient HEK293T cells without selection (Supplementary Figure S1c). In contrast, by co-transfection of a standard sgRNA expression plasmid (U6-sgRNA-CBh-eGFP; Supplementary Figure S1d) in LVNP-producing cells together with the remaining plasmids required for LVNP1.0 production (Supplementary Table S1), we observed disruption of the AFF1 locus in a dose-dependent manner in LVNP-treated cells, with a peak indel frequency at ∼38% in the bulk population of HEK293T cells without selection (Figure 1E). Notably, indels were not observed following transduction of LVNP1.0 without vesicular stomatitis virus glycoprotein (VSV-G) envelope protein (Supplementary Figure S1e), suggesting that gene disruption occurred after uptake of LVNP1.0 by VSV-G-directed endocytosis. Collectively, we demonstrate robust LVNP1.0-directed targeted DNA cleavage following plasmid-based sgRNA overexpression in the LVNP-producing cells.
Figure 1.

Incorporation of SpCas9 into lentivirus-derived nanoparticles (LVNPs). (A) Schematic representation of LVNP and (B) LVNP loaded with a vector genome. (C) Schematics of pGagPol-IntSpCas9 composed of FLAG-tagged SpCas9 fused to the C-terminus of the integrase domain (encoded by Pol) and flanked by a PCS for HIV-1 proteolytic release. (D) Functional titers of IDLV/PGK-eGFP and LVNP1.0/PGK-eGFP was evaluated by flow cytometry. (E) Indel frequencies in the AFF1 locus after transduction with increasing dosages of LVNP1.0. (F) Schematics of pGagPol-MatSpCas9 composed of FLAG-tagged SpCas9 fused to the N-terminal of Gag harbouring an intervening phospholipase C-δ1 pleckstrin homology (PH) domain. (G) Western Blot analysis of FLAG-tagged SpCas9 (FLAG antibody) and p24 loading control of purified LVNP2.0 (90 ng p24) and IDLV (90 ng p24) produced in the presence/absence of the HIV-1 protease inhibitor saquinavir (SQV). (H) Estimation of functional titers of LVNP2.0 with/without titration of increasing amounts of pGagPol-D64V. (I) Indel frequencies in the AFF1 locus after transduction with increasing dosages of LVNP2.0 (a 1:3 ratio of pGagPol-D64V:pGagPol-MatSpCas9 was used). (J) Indel frequency after transduction of LVNP2.0 with (+) and without (−) VSV-G pseudotyping. Significant P-values (Mann–Whitney U-test) are marked by *P< 0.05, **P< 0.01, ***P< 0.005. All data is presented as ±SD of triplicates. n.s: non-significant.
Enhanced efficacy of SpCas9 fused to N-terminus of gag/GagPol-D64V
Next, to optimize incorporation of SpCas9 in LVNPs, we fused FLAG-tagged SpCas9 to the N-terminus of Gag/GagPol harbouring an intervening phospholipase C-δ1 pleckstrin homology (PH) domain thought to serve as the membrane anchoring motif (46). This fusion protein, which mimicked the strategy that we previously used for incorporation of zinc finger nucleases (ZFNs) and TAL-effector nucleases into lentiviral particles (22), was expressed from pGagPol-MatSpCas9 (Figure 1F). Using this configuration, referred to as LVNP2.0, we confirmed effective SpCas9 incorporation and release from Gag during maturation (Figure 1G; Supplementary Figure S2a). In addition, when LVNP2.0 was produced in the presence of saquinavir (SQV), an inhibitor of the protease, release of both p24 and SpCas9 was restricted, confirming that liberation of SpCas9 was directed by HIV-1 protease (Figure 1G; Supplementary Figure S2a). Notably, the gene transfer capacity of LVNP2.0 was markedly restricted (Figure 1H), but gene transfer could be reconstituted in a dose-dependent manner by adding packaging plasmid encoding normal Gag/GagPol-D64V during LVNP2.0 production (Figure 1H), as we previously described for LVNPs packaging transposases and zinc-finger nucleases (19). We then produced SpCas9-loaded LVNP2.0 carrying the LentiGuide-Puro vector encoding AFF1-targeting sgRNA and observed markedly higher levels of gene disruption in recipient cells relative to the LVNP1.0 configuration (26% versus 8% indels; Supplementary Figure S2b). Intriguingly, in HEK293T cells exposed to LVNP2.0 incorporating AFF1-targeted sgRNA that was transiently expressed from pU6-sgRNA-CBh-eGFP in the producer cells, we observed a dose-dependent increase in AFF1 disruption leading to >50% indel formation in the bulk cell population with a dose corresponding to 90 ng p24 (Figure 1I). In control experiments, we found that the uptake of LVNP2.0, resulting in 82% indel formation in the AFF1 locus, was dependent on endocytosis mediated by the VSV-G pseudotype (Figure 1J). Together, these findings demonstrate effective co-incorporation of SpCas9 and sgRNA in LVNP2.0, supporting effective delivery and targeted gene disruption in recipient cells.
Modified sgRNA scaffolds enhance LVNP2.0-directed gene disruption
We hypothesized that LVNP2.0-directed DNA cleavage could be further enhanced by incorporating sgRNAs with improved stability. This could potentially favour the interaction between SpCas9 and sgRNA and reduce sgRNA degradation during LVNP2.0 biogenesis. For LVNP2.0 targeting three different genes (Pcsk9, Vegfa and SERPING1), we compared two scaffold-optimized sgRNAs (OptScf1 (47) and OptScf2 (48)) with the chimeric sgRNA (ChimScf (2)) (Figure 2A–C). For Pcsk9 and Vegfa, LVNP2.0-directed gene disruption was investigated in murine AML12 hepatocytes and HEK293T cells carrying an inserted murine Vegfa gene cassette (49,50), respectively, resulting in complete gene disruption (Figure 2D, E). In contrast, only 25% indel formation was observed in the endogenous SERPING1 locus in HEK293T (Figure 2F), possibly reflecting low accessibility due to low transcriptional activity in this locus in HEK293T cells (according to the RNAseq database in the Human Protein Atlas). In all cases, indel formation improved with increasing dosages of LVNP2.0. To discriminate performance between sgRNA scaffolds, the dosing was reduced to 7.5 ng p24 to avoid complete disruption of Pcsk9 in murine AML12 hepatocytes (Figure 2D), whereas differences were most evident at 60 ng for Vegfa in transgenic HEK293T-Vegfa cells (Figure 2E). In both cases, and in particular at lower doses, the OptScf2 scaffold performed best (Figure 2D, E), whereas no significant differences were observed for SERPING1 (Figure 2F). The difference in sgRNA performance leading to variable editing efficiencies may be attributable to genomic accessibility and sgRNA design. To determine whether the discrepancy in scaffold performance was a result of improved sgRNA incorporation, we performed digital droplet PCR (ddPCR) on RNA isolated from p24-normalized amounts of LVNP2.0. For LVNP2.0 loaded with OptScf2 for both Pcsk9 and Vegfa (Figure 2F, H), but not SERPING1 (Figure 2I), we observed a significant increase in sgRNA abundance. These results demonstrate increased sgRNA incorporation and LVNP2.0 efficacy using the OptScf2 scaffold in VSV-G-pseudotyped LVNPs. We refer to this scaffold-optimized configuration as LVNP2.1.
Figure 2.

Incorporation of scaffold-modified sgRNAs in LVNPs. (A–C) Schematics of the chimeric scaffold (ChimScf), optimized scaffold 1 (OptScf1) and optimized Scaffold 2 (OptScf2). The modifications are highlighted in red. (D–F) Heatmaps representing the indel frequency corresponding to the indicated p24 dose for ChimScf, OptScf1 and OptScf2 for (D) Pcsk9 in AML12 hepatocytes, (E) Vegfa in transgenic HEK293T-Vegfa cells, and (F) SERPING1 in HEK293T. (G–I) The sgRNA abundance was determined by digital droplet PCR (ddPCR) in purified LVNP2.0 loaded with indicated scaffold for (G) Pcsk9, (H) Vegfa and (I) SERPING1. (J) The sgRNA abundance in purified LVNP2.1 produced with/without inclusion of the U6 promoter in the sgRNA expression backbone and in LVNPs loaded with Zink Finger Nucleases (ZFN) instead of SpCas9 (n = 1). Significant P-values (Mann–Whitney U-test) are marked by *P< 0.05. All data is except panel j is presented as ±SD of triplicates. Significant P-values (Mann–Whitney U-test) are marked by *P< 0.05. n.s: non-significant.
SpCas9-dependent sgRNA incorporation in LVNP2.1
To elucidate whether incorporation of sgRNA was dependent on co-packaging of SpCas9 protein or a result of random incorporation due to overexpression in the producer cells, we measured the sgRNA abundance in LVNP2.1. As negative control, we used LVNPs loaded with ZFNs fused to the N-terminus of Gag/GagPol, for which we have previously demonstrated effective protein packaging (22). We observed SpCas9-dependent incorporation of sgRNA in LVNP2.1, whereas only background levels were observed in ZFN-loaded LVNPs (Figure 2J). To rule out background signals from plasmid DNA, we included LVNP2.1 produced by co-transfection with a sgRNA expression plasmid lacking the U6 promoter. Only background levels of sgRNA were observed, consistent with SpCas9-dependent sgRNA incorporation (Figure 2J). These results corroborate that pre-assembled SpCas9/sgRNA RNP complexes are incorporated during assembly of LVNP2.1.
Adjustment of LVNP stoichiometry enhances LVNP2.1 performance
To maximize LVNP2.1 yield without compromising efficacy, we explored the plasmid stoichiometry during LVNP production by adjusting the ratio of transfected plasmid DNA (Supplementary Table S1). We tested 11 different ratios of the packaging plasmids pGagPol-MatSpCas9 and pGagPol-D64V during production. Increasing p24 yield was observed with increasing amount of pGagPol-D64V plasmid (Figure 3A), suggesting that the SpCas9 fusion domain had an overall negative impact on LVNP2.1 production. However, the indel frequencies in transgenic HEK293T-Vegfa cells ranged from 75 to 98% in cells treated with LVNP2.1 corresponding to 60 ng p24 and from 25 to 46% with a lower dose (15 ng p24) (Figure 3B). This suggested that equal amounts of LVNP2.1 (based on p24 measurements) supported comparable levels of gene disruption, thus favouring LVNP2.1 compositions with high yield. We chose the 70/30 (pGagPol-D64V/pGagPol-MatSpCas9) composition as it retained full activity with a negligible drop in yield. To ensure maximum packaging of sgRNA in LVNP2.1, we employed ddPCR to determine the saturating amount of sgRNA expression plasmid. The sgRNA abundance in LVNP2.1 was largely unaffected (350–400 sgRNA copies/μl) by the ratio between transfer vector plasmid (pCCL-PGK-eGFP) and sgRNA-encoding plasmid (pU6-sgRNA-CBh-eGFP), although a significant drop was evident when the sgRNA expression plasmid was reduced to 20% (Figure 3C). Whereas the indel frequencies were comparable for the six tested configurations (Figure 3D), we found a positive correlation between the percentage of eGFP-positive cells and the amount of pCCL-PGK-eGFP in the production (Figure 3E; Supplementary Figure S3). We chose the 60/40 ratio to maintain a high rate of vector transfer capacity without compromising Cas9/sgRNA RNP saturation. To consolidate the optimized configuration (referred to as LVNP2.2), we targeted four genes, Pcsk9, Vegfa, Fah, and AFF1, in appropriate cell lines and showed high indel frequencies above 75% in all these loci (Figure 3F). In summary, we find that LVNP efficacy can be optimized by modulating stoichiometry of plasmids during production.
Figure 3.

Impact of ratio of LVNP production plasmids on yield and efficacy. (A) The optimal LVNP2.1 stoichiometry was evaluated by titration of increasing amounts of pGagPol-D64V versus pGagPol-MatSpCas9 (13 μg plasmid in total) in producer cells to maximize LVNP production. The concentration (μg/ml) was determined by p24 ELISA after purification and resuspension in equal volumes of buffer (n = 1). (B) Corresponding Vegfa indel frequencies following administration to transgenic HEK293T-Vegfa cells (60 and 15 ng p24) (n = 1). (C) Determination of sgRNA abundance in purified LVNP2.1 produced by titration of increasing amounts of pCCL-PGK-eGFP (transfer vector) versus pU6-sgRNA-CBh-eGFP (13 μg plasmid in total) (n = 3). (D) The corresponding Vegfa indel frequencies by Sanger sequencing, and (E) percentage of eGFP-positive HEK293T-Vegfa cells by flow cytometry after transduction of the in (C) produced LVNP2.1 (60 ng and 15 ng p24) (n = 1). (F) Performance of LVNP2.2 across multiple loci including Pcsk9 (AML12), Vegfa (HEK293T-Vegfa), Fah (Fah reporter cells), AFF1 (HEK293T) and scrambled control (HEK293T) determined by indel frequencies (black columns) and transduction efficacy estimated by flow cytometry (purple columns) (n = 3). Panel C and F are presented as the mean ± SD of triplicates. NTC: non-transduced control, Scr: scrambled sgRNA, –RT: minus reverse transcriptase.
Transient LVNP-based RNP delivery reduces off-target DNA cleavage activity
To investigate the kinetics of LVNP2.2-directed genome editing, we first performed ChIP-qPCR against the double-strand break repair protein MRE11 at different time points after delivery. This analysis demonstrated a ∼10-fold enrichment of MRE11-bound on-target DNA 8 hours after LVNP treatment, compared to background, and >20-fold enrichment after 12 hours (Figure 4A). Accordingly, indels appeared after 8 h, reaching >40% indels after 12 h and a plateau at >85% indels after 24 h (Figure 4B). To investigate the durability of RNP activity after LVNP delivery, we developed a dual-fluorescent reporter system monitoring endonuclease activity after administration (Figure 4C). HEK293T cells were first transduced with LVNP2.2 (loaded with sgRNAs targeting d2eGFP and a transfer vector encoding mCherry) and subsequently with LV/PGK-d2eGFP-IRES-Puro at different time points, allowing the longevity of Cas9/sgRNA RNPs after administration to be evaluated by measuring d2eGFP expression by flow cytometry (Supplementary Figure S4A–E). Following co-transduction at day 0, the knockout efficacy reached >90% confirming the high potency of pre-assembled RNP complexes (Figure 4D). The efficacy decreased in a stepwise and time-dependent manner, with no detectable RNP activity 4 days after LVNP2.2 transduction (Figure 4D).
Figure 4.

Transient DNA cleavage activity and reduced genotoxicity by LVNP-directed genome editing. (A) The emergence of double-stranded DNA breaks in Pcsk9 was evaluated over the course of 24 h in MRE11 ChIP-qPCR experiments. Fold enrichment refers to the ratio of MRE11-bound on-target DNA compared to background, as measured by qPCR. NTC: Non-transduced control. (B) The indel frequency was measured continuously in the Pcsk9 locus in AML12 hepatocytes after LVNP2.2 transduction. (C) Schematic presentation of the dual fluorescence assay to monitor RNP kinetics. (D) HEK293T cells were transduced by LVNP2.2/PGK-mCherry-sgRNA-d2eGFP and co-transduced with LV/PGK-d2eGFP-IRES-Puro at the indicated time points. Using flow cytometry, we calculated the level of d2eGFP knockout at each time point to evaluate the kinetics of transient LVNP delivery. (E) Monitoring of on/off-target events in the Pcsk9 locus and a well-characterized off-target locus (Off-target 1) following nucleofection (6 μg Cas9, 3.2 μg sgRNA) or LVNP2.2 transduction (60 ng p24) in murine AML12 hepatocytes. (F) AML12 hepatocytes was transduced with increasing amount of LVNP2.2 loaded with a sgRNA targeting Pcsk9. The emergence of indels in off-target site 1 was evaluated by Sanger sequencing 3 days after transduction. (G) Identification of additional off-target sites following nucleofection (6 μg Cas9, 3.2 μg sgRNA) or LVNP2.2 (15 ng p24). (H) Same as (D) following nucleofection at non-saturation conditions (2 μg Cas9, 0.7 μg sgRNA). (I) Indel frequencies in AML12 hepatocytes transduced with LVNP2.2 (5 ng p24) or decreasing amounts of Cas9/sgRNA RNP nucleofection (red bars: 6 ug Cas9 + 3.2 ug sgRNA; green bars: 6 ug Cas9 + 0.64 ug sgRNA; blue bars: 2 ug Cas9 + 0.64 ug sgRNA; purple bars: 2 ug Cas9 + 0.46 ug sgRNA). All data is presented as ± SD of triplicates. n.d.: not detected.
Based on the immediate and short-term activity of RNPs after LVNP delivery, we speculated that this delivery approach would support high on/off-target DNA cleavage rates and compared with standard RNP nucleofection. To determine the ′time-to-event′ in an endogenous locus, we targeted the Pcsk9 gene in AML12 cells and measured the on/off-target kinetics over the course of 14 days using a promiscuous sgRNA with known off-target sites (51). We evaluated the level of off-target disruption in off-target locus 1 (Off-target 1; Supplementary Figure S5a) following administration by (i) nucleofection of recombinant SpCas9 complexed with synthetic sgRNA targeting Pcsk9 and (ii) LVNP2.2 loaded with the same sgRNA (encoded by plasmid) targeting Pcsk9. Accompanying molecular analysis revealed >90% on-target and >70% off-target events within 72 h after SpCas9/sgRNA RNP nucleofection without further accumulation over time (Figure 4E). In contrast, we found no detectable off-target events in AML12 hepatocytes exposed to LVNP2.2 (15 ng p24; Figure 4E), although the level of on-target disruption was comparable to RNP nucleofection (Figure 4E). To examine whether off-target cleavage was detectable after LVNP treatment, we treated the cells with increasing dosages of LVNP2.2, most facilitating full on-target activity (Figure 4F). Indel formation in Off-target 1 was indeed detectable at dosages of 60 ng p24 and higher (Figure 4F), corresponding to a >5-fold increase in dosing needed to obtain complete on-target gene disruption. Notably, the incidence plateaued at approximately 5% after 72 h (Figure 4F) with no further accumulation over time (Supplementary Figure S5b, c) consistent with the decay of LVNP2.2-delivered RNPs (Figure 4D). To further explore the relationship between on/off-target abundancy, we reduced the dosing of nucleofected RNPs to non-saturating conditions (2 μg Cas9; 0.7 μg sgRNA) providing indel rates at 90% (Figure 4G; Supplementary Figure S5d, e) and expanded the analysis to include additional known off-target loci (Off-target sites 2–4) (Figure 4G). With this dose, cleavage in Off-target 1 was not evident suggesting that indel formation in this locus was dependent on the amount of nucleofected RNP. However, among the additional sites, we found high off-target activity in Off-target 4 after nucleofection (Figure 4G). Interestingly, DNA cleavage leading to indel formation was not detected in this site after LVNP treatment (Figure 4G). Using the same RNP nucleofection dose (2 μg Cas9; 0.7 sgRNA) in a dual-fluorescence reporter assay (similar to the setup used for LVNP in Figure 3C), we found evidence of prolonged RNP activity with detectable levels of gene disruption 6 days after RNP nucleofection (Figure 4H). Finally, we directly compared on- and off-target cleavage between a LVNP dose resulting in very high on-target indel formation (>95%) and decreasing dosages of nucleofected RNP. Notably, whereas cleavage in Off-target 1 vanished with lower dosages of RNP, cleavage in Off-target 4 remained evident with all nucleofected RNP dosages (Figure 4I). In contrast, indel formation was not observed in any of the two off-target sites with LVNP treatment, despite the higher level of on-target activity (Figure 4I). Collectively, these findings are consistent with a model suggesting that low-abundant LVNP-based RNP delivery is sufficient to produce high levels of gene disruption and may reduce the level of genotoxicity typically associated with prolonged and high-abundant RNP delivery.
In vivo gene disruption in mouse retina using LVNP as a vehicle of RNPs
To determine the effectiveness of in vivo genome editing, we chose to target Vegfa in retinal pigment epithelial (RPE) cells in the murine eye (Figure 5A). First, LVNP2.2 (loaded with SpCas9 and sgRNAs targeting Vegfa as well as vector RNA encoding eGFP) was administered by subretinal injection (Figure 5B) to the left eye (∼16 ng p24), whereas the right eye served as negative control (n = 10 mice). Each mouse was examined by optical coherence tomography (OCT) and fundus photography to confirm reattachment of the neuroretina and monitor for eGFP expression (Figure 5C). No eGFP expression was detectable by in vivo fluorescence imaging. However, following sacrifice of the mice and enucleation, we found eGFP expression in two out of three retinal flatmounts consistent with LVNP2.2-directed eGFP gene transfer (Figure 5D). To evaluate the level of Vegfa knockout, the RPE cells were isolated and pooled (n = 6) and separated into eGFP-positive or eGFP-negative populations by FACS (Supplementary Figure S6a). Accompanying molecular analysis revealed 17% indel formation in the targeted Vegfa locus in eGFP-positive cells (Figure 5E) without any detectable off-target events (Figure 5F).
Figure 5.

Gene disruption in the murine eye. (A) Time course for in vivo evaluation of LVNP2.2-directed Vegfa disruption. (B) Administration of 2 μl (16 ng p24) LVNP2.2 (encoding a sgRNA targeting Vegfa and an eGFP-encoding transfer vector) by injection into the subretinal space in 8-week-old, male C57Bl/6J mice (n = 10). (C) Representative fundus and OCT images of the murine retina from two mice injected with LVNP2.2. (D) LVNP2.2 transduction of murine RPE cells was confirmed in retinal flat mounts by fluorescence microscopy. Upper: Visualization of eGFP expression (green channel) and autofluorescence (Texas Red channel) in representative sections. Lower: close-up of the red square. Scale bars: 50 μM (upper) and 20 μM (lower). (E) The frequency of ′on-target′ disruption of Vegfa in eGFP positive FACS sorted cells, and (F) for two off-target loci. (G) Schematics of the pGagPro-SpCas9 packaging constructs. (H) Comparison of the performance of LVNP2.2 and LVNP3.0, both carrying SpCas9 and sgRNA targeting Fah and Pcsk9 (both 7.5 ng p24) in Fah reporter cells and AML12 hepatocytes, respectively. Significant P-values (Mann–Whitney U-test) are marked by *P< 0.05 and presented as ±SD of triplicates. (I) Indel frequencies in transgenic HEK293T-Vegfa cells following transduction of LVNP3.0 or eVLPs loaded with a sgRNA targeting Vegfa. (J) Administration of 1.6 μl (48 ng p24) LVNP3.0 or 1.6 μl eVLP by injection into the subretinal space in 8-week-old, male C57Bl/6J mice (n = 6 eyes for each condition +2 controls). (K) The level of Vegfa disruption in RPE cells isolated from the murine eye cup. Significant P-values (Mann–Whitney U-test) are marked by *P< 0.05, **P< 0.01. All data is presented as ±SD of at least triplicates. n.s: non-significant.
To further improve in vivo editing rates, we engineered particles that could potentially better accommodate large fusion proteins. In the LVNP3.0 configuration, we deleted regions encoding reverse transcriptase and integrase from the pol gene, allowing production of a polypeptide containing Gag, the viral protease (Pro), and the protein of interest (POI), the latter fused to the C-terminus of Gag through a linker containing a PCS, 3xFLAG, and an additional NLS to enhance nuclear localization (52) (referred to as pGagPro). This configuration was validated by fusing SpCas9 to the C-terminus of GagPro (Figure 5G). Using LVNP3.0-SpCas9, we found slightly improved levels of gene disruption of two loci, Fah and Pcsk9, as compared to the LVNP2.2 configuration (Figure 5H).
Based on the LVNP3.0 configuration, we also optimized the LVNP production by including an additional purification step by Amicon filtration (Supplementary Figure S6b). Moreover, to benchmark the LVNP platform, we compared the performance of LVNP3.0 with engineered virus-like particles (eVLPs) derived from murine leukemia virus (MLV). eVLPs were recently shown to facilitate delivery of BE and sgRNA in the mouse eye (28). Here, we produced LVNPs and eVLPs loaded with SpCas9 and a sgRNA targeting Vegfa. As eVLPs were previously produced without vector RNA, we chose to produce both types of particles without vector RNA. As a result, transductional titers could not be determined, and therefore we used the same volume of LVNP and eVLP that were produced, concentrated, and purified in parallel. Following administration to HEK293T-Vegfa cells, we found equal levels of Vegfa disruption at approximately 56% (2 μl) for both LVNP and eVLP (Figure 5I). For in vivo experiments, we delivered LVNPs or eVLPs by subretinal injection (1.6 μl, corresponding to 48 ng p24) (Figure 5J). After 4 days, the mice were sacrificed, and the level of Vegfa disruption was determined in RPE cells. Intriguingly, we found up to 32% Vegfa disruption for eyes treated with LVNPs (median 22.2%) and up to 36% for eVLP-treated eyes (median 20.5%) in the bulk population of cells without FACS (Figure 5K). Collectively, these findings provide evidence for LVNP-directed in vivo genome editing and suggest that this platform supports in vivo targeted DNA cleavage at levels that are comparable with an MLV-based eVLP configuration.
LVNP-directed base editing with reduced bystander editing and prime editing without indel formation
To demonstrate LVNP-directed gene editing without creating double-stranded breaks, we first incorporated the FLAG-tagged adenine base editor ABE7.10 (14) in LVNP2.2-F7.10 (pGagPol-MatF7.10; Supplementary Figure S7a). By incorporation of OptScf2-type sgRNA displaying high performance in transfection-based assays (Supplementary Figure S7b), we found LVNP2.2-F7.10-directed base editing in 17% of the HEK293T cell genomic site 2 (hereafter referred to as ‘Site 2′) alleles (14) in a VSV-G dependent manner (Figure 6A). Based on the LVNP3.0 configuration, we then engineered pGagPro-ABE8e (53) (Figure 6B) and found robust levels of base editing in Site 2 ranging between 18 and 45% in a dose-dependent manner (Figure 6c-d). LVNP delivery almost matched the level of base editing observed with DNA transfection in HEK293T cells (Figure 6C, D). Notably, when evaluating the level of bystander editing at two adenines (A2 and A8) amenable to bystander editing within the sgRNA target region in Site 2 (position of the adenines), the percentage of bystander editing relative to on-target editing was markedly reduced at position A2 and A8 in cells treated with LVNP3.0-ABE8e compared to plasmid-transfected cells (Figure 6e; Supplementary Figure S7c).
Figure 6.

LVNP-directed base and prime editing. (A) The conversion rate at ′site 2′ after transduction of VSV-G-pseudotyped LVNP2.2-F7.10 (90 ng p24) in HEK293T. (B) Schematics of the pGagPro-ABE8e, pGagPro-PEmax-RRE and pGagPro-PEmaxΔRH-RRE fused to the C-terminus of GagPro packaging constructs. (C) Representative example of on-target and bystander base editing rates in the site 2 locus after delivery of ABE8e and sgRNA by LVNP3.0-ABE8e and plasmid DNA transfection. For comparison of bystander editing, conditions leading to higher on-target editing with LVNP3.0-ABE8e are shown. (D) Same as (C) for LVNP3.0-ABE8e in a dose-escalation manner. (E) The percentage of bystander editing relative to ′on-target′ conversion rate at position A2 and A8 for LVNP3.0-ABE8e and plasmid DNA transfection. (F) The percentage of intended (correct editing) and unwanted editing (indels) in the HEK3 locus 3 days after transduction in HEK293T (500 ng p24; 6-well plate). Significant P-values (Mann–Whitney U-test) are marked by *P< 0.05, **P< 0.01, ***P< 0.005. All data is presented as ± SD of triplicates. n.s: non-significant.
To investigate the capacity of LVNPs to accommodate and deliver prime editors, we constructed LVNP3.0-PEmax (PEmax described in (54)) and LVNP3.0-PEmaxΔRH, the latter which ferries a truncated version of PEmax containing an MLV-RT domain devoid of the RNase H domain (55) (Figure 6B). To support RNP stability, we expressed and incorporated engineered pegRNAs (epegRNAs) containing a structural motif (tevopreQ1) at the 3′ end to minimize degradation (56). By exposing HEK293T cells to LVNPs loaded with PEmax and an epegRNA targeting HEK3 (CTTins), we found robust levels of CTT insertion (6%) in the HEK3 locus without detectable indel formation (<0.01%) using targeted next-generation sequencing (Figure 6F; Supplementary Figure S8a, b). For PEmaxΔRH, we found comparable editing rates (5%) without indel formation (Figure 6F; Supplementary Figure S9a, b). Collectively, our data demonstrate the capacity of LVNP to deliver larger genome editing toolkits including prime editing RNPs consisting of PEmax (with a size of 230 kDa) complexed with epegRNA. These findings may pave the way for the development of LVNP-based formulations for therapeutic delivery of base and prime editors allowing gene editing without DSB formation and the need of an HDR donor sequence.
DISCUSSION
By engineering of lentivirus-derived nanoparticles to ferry RNP complexes, we have demonstrated potent gene knockout, base editing, and prime editing in a DNA-free fashion that is compatible with no transfer of genetic material except for sgRNAs or epegRNAs. We showed that LVNP yield is negatively affected by incorporation of Cas fusions and that both yield and titer may be reconstituted by titration of unfused Gag/GagPol-D64V, allowing assembly of chimeric virus particles consistent with previous studies (19,27). LVNP efficacy was improved further by incorporation of scaffold-modified sgRNAs (LVNP2.1) and optimal stoichiometry of packaging plasmids during production balancing high yields and activity in recipient cells (LVNP2.2). This is consistent with two recent studies, in which the authors showed that balancing the amount of RNP incorporated in eVLPs (gag-cargo versus gag-pro-pol) (28) and Cas9-VLPs (gag-cargo versus gagpol) (27) was a key determinant for viral maturation and genome editing efficacy.
Using the LVNP2.2 configuration for delivery of a promiscuous sgRNA, we found very high on/off-target ratio even at high LVNP doses (>10-fold higher than needed for complete gene knockout) compared to RNP nucleofection. We speculate that this difference partially reflects the low abundance of Cas9/sgRNA RNPs following LVNP delivery. In previous studies addressing virus-based protein delivery, we found relatively low levels of transferred protein and rapid turnover in recipient cells (19,23). Moreover, another study investigating different fusions of GFP to retroviral Gag/GagPol reported increased nuclear localization of GFP protein carrying an added nuclear localization signal (57). This could potentially suggest that NLS-tagged RNPs packaged into engineered LVNPs are released into the cytoplasm upon endocytosis and imported into the nucleus by intracellular protein trafficking. Also possible is that RNPs are ferried through the cytoplasm and into the nucleus as part of the viral core, and that the effect of added NLS domains is supporting import of RNPs that are being exported from the nucleus. Additional studies are needed to clarify this, but one may speculate that active, directed transport of viral core proteins, potentially involving transport on microtubules, would explain how low abundant NLS-tagged RNPs support high levels of gene editing with minimal off-target activities. This notion is supported by earlier studies showing high cellular activity of LVNP-delivered DNA-modifying transposases and nucleases (19,23,28). This would argue that specific biological properties of the viral configuration are crucial for directed intracellular transport of RNPs and may also potentially be involved in targeted delivery of cargo at or near transcriptionally active regions of the genome. If this is correct, such properties may help explain how low RNP levels support potent targeted DNA cleavage with limited off-target activities. This may also support the notion that ‘more’ is not necessarily better, and that attempts should be made to develop delivery technologies that do not overload cells with Cas9/sgRNA complexes, but allow these to accumulate near desired regions of the genome. Along those lines, Banskota and co-workers recently demonstrated that AAV-based delivery of the genes encoding the base editor and the accompanying sgRNA to the mouse liver resulted in detectable off-target editing, which was not detectable for eVLPs (28).
Based on the assumption that larger fusion proteins interfere with particle production and function due to their larger size and steric hindrance, we engineered a configuration without integrase and reverse transcriptase (LVNP3.0). A single subretinal injection of LVNP3.0 resulted in potent Vegfa knockout (22.2%) in the mouse retina, which was comparable with the efficacy of eVLP-directed RNP delivery (20.5%). Subretinal injections are micro-precise processes with inherent variability between injections mainly due to uneven distribution of the injected solution as well as variation in the amount of backflow to the vitreous body. This leads to differences in the size of the transduced area, which is consistent with our previous investigations of cellular transduction following subretinal injection of lentiviral vectors encoding eGFP (58). The present study is the first to demonstrate in vivo genome editing following subretinal delivery of HIV1-derived LVNPs loaded with SpCas9/sgRNA RNPs in the absence of a vector genome. Recently, nonviral delivery based on lipid nanoparticles (LNP) co-formulated with Cas9 mRNA and sgRNA has been adapted for effective in vivo gene knockout in e.g. mouse liver (59,60) and muscle (61). Although LNP-mediated delivery of mRNA is relatively short-lived, each mRNA copy will generate functional RNPs until degradation of the mRNA (62). Delivery of pre-assembled RNPs offers an even shorter window of action which may potentially reduce risk of genotoxicity. Furthermore, insertional mutagenesis remains a significant concern for lentiviral and AAV-mediated delivery as recently demonstrated by the development of AAV-induced hepatocellular carcinoma in newborn mice (63) and non-malignant neoplasms in long-term AAV dog studies (64).
A configuration like LVNP3.0 was recently published by Hamilton et al. (27) for concurrent delivery of Cas9-RNPs and a transgene to produce chimeric antigen receptor (CAR) primary human T cells (CAR-T). In contrast to LVNPs, Cas9-VLPs are produced by titration of integrase-competent Gag/GagPol to allow the transgene to be integrated into the host genome after reverse transcription. Whereas LVNP3.0 is engineered to circumvent the risk of insertional mutagenesis, this feature may be reconstituted by including normal Gag and GagPol in the particles. This gives the LVNP modality extensive flexibility, as demonstrated by tailoring the delivery platform for administration of Cas9/sgRNA and BE/sgRNA as well as PEmax/epegRNA RNPs. If required for a certain application, vector RNA encoding for example a reporter gene can be delivered along with editing toolkits.
To the best of our knowledge, our study is the first to incorporate ‘all-in-one’ PE/epegRNA RNPs into engineered particles for potent delivery. As proof-of-concept, we showed LVNP-directed prime editing in the HEK3 locus resulting in gene editing without indel formation using epegRNAs (56). We found no significant difference in the performance between PEmax and a truncated variant of PEmax without the RNaseH domain, potentially suggesting that the LVNP3.0 architecture can incorporate even larger fusion proteins. Alternative viral strategies, like dual-AAV systems based on split-intein fusion of the N- and C-terminal domain, have been employed to deliver PE in vitro and in vivo (52,65–67). However, dual-AAV systems require co-transduction of recipient cells, trans-splicing of the two intein fragments, and complex formation with (e)pegRNA before gene editing may occur. Our data demonstrate incorporation and transfer of PE/epegRNA RNP complexes in LVNP3.0, leading to gene editing in recipient cells. Despite removal of the reverse transcriptase and integrase, the LVNP3.0 architecture is amenable to conventional LV pseudotyping allowing specific cell types to be targeted. The LVNP modality evades the packaging limit by incorporation of RNP complexes, abrogates the risk of insertional mutagenesis, maintains the optional inclusion of a vector genome, and allows surface engineering for cell type-specific gene editing. Collectively, our results establish LVNPs as a modular platform for in vivo RNP delivery of gene editing agents.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Charlotte Thornild Møller for excellent technical support in the laboratory. Furthermore, we acknowledge the FACS Core Facility at Aarhus University for assistance with flow cytometry and Kamilla Zahll Hornbek from the Animal Facility at Department of Biomedicine, Aarhus University, for excellent technical support.
Notes
Present address: Sofie Andersen, RNA and Gene Therapies, Research & Early development, Novo Nordisk A/S, Måløv, Denmark.
Contributor Information
Jakob Haldrup, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Sofie Andersen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Alexander Rafael LaVilla Labial, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Jonas Holst Wolff, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Frederik Plum Frandsen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Thomas Wisbech Skov, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Anne Bruun Rovsing, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Ian Nielsen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Thomas Stax Jakobsen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark; Department of Ophthalmology, Aarhus University Hospital, Aarhus N, Denmark.
Anne Louise Askou, Department of Biomedicine, Aarhus University, Aarhus C, Denmark; Department of Ophthalmology, Aarhus University Hospital, Aarhus N, Denmark.
Martin K Thomsen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Thomas J Corydon, Department of Biomedicine, Aarhus University, Aarhus C, Denmark; Department of Ophthalmology, Aarhus University Hospital, Aarhus N, Denmark.
Emil Aagaard Thomsen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
Jacob Giehm Mikkelsen, Department of Biomedicine, Aarhus University, Aarhus C, Denmark.
DATA AVAILABILITY
NGS data have been deposited at the National Center of Biotechnology Information's Sequence Read Archive database at (ID: 974390). Plasmids have been deposited at Addgene for distribution.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Lundbeck Foundation [R324-2019-1832]; Innovation Fund Denmark [PASCAL-MID, 8056-00010A]; Synoptik Foundation; Fight for Sight Denmark; VELUX Foundation [GET-AMD-CURE, 38189]. Funding for open access charge: External funding.
Conflict of interest statement. J.G.M. is a member of the Scientific Advisory Board of Nvelop Therapeutics. The company was not involved in the present study. The remaining authors declare no competing interests.
REFERENCES
- 1. Doudna J.A. The promise and challenge of therapeutic genome editing. Nature. 2020; 578:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sheridan C. CRISPR therapies march into clinic, but genotoxicity concerns linger. Nat. Biotechnol. 2021; 39:897–899. [DOI] [PubMed] [Google Scholar]
- 4. Wang D., Zhang F., Gao G.. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020; 181:136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Song Y., Liu Z., Zhang Y., Chen M., Sui T., Lai L., Li Z.. Large-fragment deletions induced by Cas9 cleavage while not in the BEs system. Mol. Ther. Nucleic Acids. 2020; 21:523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kosicki M., Tomberg K., Bradley A.. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018; 36:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F.et al.. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020; 367:eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giannoukos G., Ciulla D.M., Marco E., Abdulkerim H.S., Barrera L.A., Bothmer A., Dhanapal V., Gloskowski S.W., Jayaram H., Maeder M.L.et al.. UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018; 19:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D.. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021; 53:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E.et al.. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019; 10:4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandler R.J., Sands M.S., Venditti C.P.. Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum. Gene Ther. 2017; 28:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner D.L., Amini L., Wendering D.J., Burkhardt L.M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M.. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019; 25:242–248. [DOI] [PubMed] [Google Scholar]
- 13. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R.. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017; 551:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arbab M., Shen M.W., Mok B., Wilson C., Matuszek Ż., Cassa C.A., Liu D.R.. Determinants of base editing outcomes from target library analysis and machine learning. Cell. 2020; 182:463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A.et al.. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019; 576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Haasteren J., Li J., Scheideler O.J., Murthy N., Schaffer D.V.. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020; 38:845–855. [DOI] [PubMed] [Google Scholar]
- 18. Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S.et al.. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016; 539:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai Y., Bak R.O., Krogh L.B., Staunstrup N.H., Moldt B., Corydon T.J., Schrøder L.D., Mikkelsen J.G.. DNA transposition by protein transduction of the piggyBac transposase from lentiviral Gag precursors. Nucleic Acids Res. 2014; 42:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skipper K.A., Nielsen M.G., Andersen S., Ryo L.B., Bak R.O., Mikkelsen J.G.. Time-restricted PiggyBac DNA transposition by transposase protein delivery using lentivirus-derived nanoparticles. Mol. Ther. Nucleic Acids. 2018; 11:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomsen E.A., Skipper K.A., Andersen S., Haslund D., Skov T.W., Mikkelsen J.G.. CRISPR-Cas9-directed gene tagging using a single integrase-defective lentiviral vector carrying a transposase-based Cas9 off switch. Mol. Ther. Nucleic Acids. 2022; 29:563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cai Y., Bak R.O., Mikkelsen J.G.. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife. 2014; 3:e01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai Y., Laustsen A., Zhou Y., Sun C., Anderson M.V., Li S., Uldbjerg N., Luo Y., Jakobsen M.R., Mikkelsen J.G.. Targeted, homology-driven gene insertion in stem cells by ZFN-loaded ‘all-in-one’ lentiviral vectors. Elife. 2016; 5:e12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi J.G., Dang Y., Abraham S., Ma H., Zhang J., Guo H., Cai Y., Mikkelsen J.G., Wu H., Shankar P.et al.. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016; 23:627–633. [DOI] [PubMed] [Google Scholar]
- 25. Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massouridès E., Sohier T.J.M., Corbin A.et al.. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019; 10:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lyu P., Javidi-Parsijani P., Atala A., Lu B.. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘hit-and-run’ genome editing. Nucleic Acids Res. 2019; 47:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamilton J.R., Tsuchida C.A., Nguyen D.N., Shy B.R., McGarrigle E.R., Sandoval Espinoza C.R., Carr D., Blaeschke F., Marson A., Doudna J.A.. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. 2021; 35:109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banskota S., Raguram A., Suh S., Du S.W., Davis J.R., Choi E.H., Wang X., Nielsen S.C., Newby G.A., Randolph P.B.et al.. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022; 185:250–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Doman J.L., Raguram A., Newby G.A., Liu D.R.. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020; 38:620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rees H.A., Komor A.C., Yeh W.H., Caetano-Lopes J., Warman M., Edge A.S.B., Liu D.R.. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017; 8:15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Concordet J.P., Haeussler M.. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anderson M.V., Haldrup J., Thomsen E.A., Wolff J.H., Mikkelsen J.G.. pegIT - a web-based design tool for prime editing. Nucleic Acids Res. 2021; 49:W505–W509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graham F.L., van der Eb A.J.. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973; 52:456–467. [DOI] [PubMed] [Google Scholar]
- 34. Conant D., Hsiau T., Rossi N., Oki J., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K.et al.. Inference of CRISPR edits from Sanger trace data. CRISPR J. 2022; 5:123–130. [DOI] [PubMed] [Google Scholar]
- 35. Bloh K., Kanchana R., Bialk P., Banas K., Zhang Z., Yoo B.C., Kmiec E.B.. Deconvolution of complex DNA repair (DECODR): establishing a novel Deconvolution algorithm for comprehensive analysis of CRISPR-edited sanger sequencing data. CRISPR J. 2021; 4:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S.. EditR: a method to quantify base editing from sanger sequencing. CRISPR J. 2018; 1:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E.et al.. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019; 37:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomsen E.A., Rovsing A.B., Anderson M.V., Due H., Huang J., Luo Y., Dybkaer K., Mikkelsen J.G.. Identification of BLNK and BTK as mediators of rituximab-induced programmed cell death by CRISPR screens in GCB-subtype diffuse large B-cell lymphoma. Mol. Oncol. 2020; 14:1978–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thomsen E.A., Andersen S., Marqvorsen M.H.S., Skipper K.A., Paludan S.R., Mikkelsen J.G.. Single-cell monitoring of activated innate immune signaling by a d2eGFP-based reporter mimicking time-restricted activation of IFNB1 expression. Front. Cell. Infect. Microbiol. 2021; 11:784762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wienert B., Wyman S.K., Yeh C.D., Conklin B.R., Corn J.E.. CRISPR off-target detection with DISCOVER-seq. Nat. Protoc. 2020; 15:1775–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Askou A.L., Alsing S., Benckendorff J.N.E., Holmgaard A., Mikkelsen J.G., Aagaard L., Bek T., Corydon T.J.. Suppression of choroidal neovascularization by AAV-based dual-acting antiangiogenic gene therapy. Mol. Ther. Nucleic Acids. 2019; 16:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holmgaard A.B., Askou A.L., Jensen E.G., Alsing S., Bak R.O., Mikkelsen J.G., Corydon T.J.. Targeted knockout of the vegfa gene in the retina by subretinal injection of RNP complexes containing Cas9 protein and modified sgRNAs. Mol. Ther. 2021; 29:191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Askou A.L., Benckendorff J.N.E., Holmgaard A., Storm T., Aagaard L., Bek T., Mikkelsen J.G., Corydon T.J.. Suppression of choroidal neovascularization in mice by subretinal delivery of multigenic lentiviral vectors encoding anti-angiogenic MicroRNAs. Hum. Gene Ther. Methods. 2017; 28:222–233. [DOI] [PubMed] [Google Scholar]
- 44. Alsing S., Doktor T.K., Askou A.L., Jensen E.G., Ahmadov U., Kristensen L.S., Andresen B.S., Aagaard L., Corydon T.J.. VEGFA-targeting miR-agshRNAs combine efficacy with specificity and safety for retinal gene therapy. Mol. Ther. Nucleic Acids. 2022; 28:58–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jacks T., Power M.D., Masiarz F.R., Luciw P.A., Barr P.J., Varmus H.E.. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988; 331:280–283. [DOI] [PubMed] [Google Scholar]
- 46. Urano E., Aoki T., Futahashi Y., Murakami T., Morikawa Y., Yamamoto N., Komano J.. Substitution of the myristoylation signal of human immunodeficiency virus type 1 Pr55Gag with the phospholipase C-delta1 pleckstrin homology domain results in infectious pseudovirion production. J. Gen. Virol. 2008; 89:3144–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S.et al.. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013; 155:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grevet J.D., Lan X., Hamagami N., Edwards C.R., Sankaranarayanan L., Ji X., Bhardwaj S.K., Face C.J., Posocco D.F., Abdulmalik O.et al.. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science. 2018; 361:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holmgaard A., Askou A.L., Benckendorff J.N.E., Thomsen E.A., Cai Y., Bek T., Mikkelsen J.G., Corydon T.J.. In vivo knockout of the Vegfa gene by lentiviral delivery of CRISPR/Cas9 in mouse retinal pigment epithelium cells. Mol. Ther. Nucleic Acids. 2017; 9:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pihlmann M., Askou A.L., Aagaard L., Bruun G.H., Svalgaard J.D., Holm-Nielsen M.H., Dagnaes-Hansen F., Bek T., Mikkelsen J.G., Jensen T.G.et al.. Adeno-associated virus-delivered polycistronic microRNA-clusters for knockdown of vascular endothelial growth factor in vivo. J. Gene Med. 2012; 14:328–338. [DOI] [PubMed] [Google Scholar]
- 51. Akcakaya P., Bobbin M.L., Guo J.A., Malagon-Lopez J., Clement K., Garcia S.P., Fellows M.D., Porritt M.J., Firth M.A., Carreras A.et al.. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018; 561:416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu P., Liang S.-Q., Zheng C., Mintzer E., Zhao Y.G., Ponnienselvan K., Mir A., Sontheimer E.J., Gao G., Flotte T.R.et al.. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021; 12:2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E.et al.. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020; 38:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen P.J., Hussmann J.A., Yan J., Knipping F., Ravisankar P., Chen P.F., Chen C., Nelson J.W., Newby G.A., Sahin M.et al.. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021; 184:5635–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gao Z., Ravendran S., Mikkelsen N.S., Haldrup J., Cai H., Ding X., Paludan S.R., Thomsen M.K., Mikkelsen J.G., Bak R.O.. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol. Ther. 2022; 30:2942–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nelson J.W., Randolph P.B., Shen S.P., Everette K.A., Chen P.J., Anzalone A.V., An M., Newby G.A., Chen J.C., Hsu A.et al.. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022; 40:402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robert M.A., Lytvyn V., Deforet F., Gilbert R., Gaillet B.. Virus-like particles derived from HIV-1 for delivery of nuclear proteins: improvement of production and activity by protein engineering. Mol. Biotechnol. 2017; 59:9–23. [DOI] [PubMed] [Google Scholar]
- 58. Alsing S., Lindholm A.B., Haldrup J., Jensen E.G., Mikkelsen J.G., Aagaard L., Askou A.L., Corydon T.. Simple autofluorescence-restrictive sorting of eGFP+ RPE cells allows reliable assessment of targeted retinal gene therapy. Front. Drug Deliv. 2022; 2:898568. [Google Scholar]
- 59. Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J.et al.. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent In vivo genome editing. Cell Rep. 2018; 22:2227–2235. [DOI] [PubMed] [Google Scholar]
- 60. Qiu M., Glass Z., Chen J., Haas M., Jin X., Zhao X., Rui X., Ye Z., Li Y., Zhang F.et al.. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kenjo E., Hozumi H., Makita Y., Iwabuchi K.A., Fujimoto N., Matsumoto S., Kimura M., Amano Y., Ifuku M., Naoe Y.et al.. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat. Commun. 2021; 12:7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Newby G.A., Yen J.S., Woodard K.J., Mayuranathan T., Lazzarotto C.R., Li Y., Sheppard-Tillman H., Porter S.N., Yao Y., Mayberry K.et al.. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021; 595:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rosas L.E., Grieves J.L., Zaraspe K., La Perle K.M., Fu H., McCarty D.M.. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther. 2012; 20:2098–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.-A., Merricks E.P., Long C.T.et al.. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021; 39:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zheng C., Liang S.Q., Liu B., Liu P., Kwan S.Y., Wolfe S.A., Xue W.. A flexible split prime editor using truncated reverse transcriptase improves dual-AAV delivery in mouse liver. Mol. Ther. 2022; 30:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhi S., Chen Y., Wu G., Wen J., Wu J., Liu Q., Li Y., Kang R., Hu S., Wang J.et al.. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2022; 30:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Böck D., Rothgangl T., Villiger L., Schmidheini L., Matsushita M., Mathis N., Ioannidi E., Rimann N., Grisch-Chan H.M., Kreutzer S.et al.. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med. 2022; 14:eabl9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NGS data have been deposited at the National Center of Biotechnology Information's Sequence Read Archive database at (ID: 974390). Plasmids have been deposited at Addgene for distribution.