

Abstract
Earth abundant metal catalysts hold advantages in cost, environmental burden and chemoselectivity over precious metal catalysts. Differences in reactivity for a given metal center result from ligand field strength, which can promote reaction through either open- or closed-shell carbon intermediates. Herein we report a simple protocol for cobalt-catalyzed alkene reduction. Instead of using an oxidative turnover mechanism that requires stoichiometric hydride, we find a reductive turnover mechanism that requires stoichiometric proton. The reaction mechanism appears to involve coordination and hydrocobaltation of terminal alkenes.
Keywords: Cobalt, Hydrogenation, Reduction, Alkene, Manganese
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
Introduction
Hydrogenation of alkenes finds use across scales to introduce saturation and stereocenters.1 Precious metal catalysts (Rh, Pt, Pd) and hydrogen gas can be limited, however, by catalyst cost or availability, ignition of hydrogen/oxygen mixtures, and high-pressure requirements. The canonical Horiuti–Polanyi mechanism for precious metal-catalyzed hydrogenation2 involves alkene coordination (adsorption), metal-hydride migratory insertion and reductive elimination: traditional inner-sphere elementary steps (Fig. 1A). In contrast, hydride complexes of earth abundant metals like manganese, iron and cobalt can undergo an outer-sphere elementary step of metal-hydride hydrogen atom transfer (MHAT).3 During attempts to induce reductive turnover of MHAT hydrogenation (Fig. 1B),4 we discovered a simple alkene reduction protocol that uses cheap reagents, avoids hydrogen gas and operates at standard pressure and temperature. In contrast to our recent investigations of Co- and Mn-catalyzed alkene hydrofunctionalization, this reaction likely involves an alkene coordination step.
Figure 1.

Reaction design.
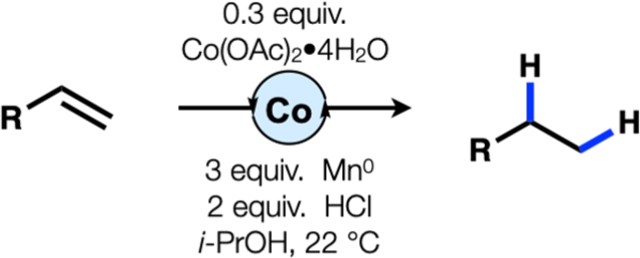
Our work originated in reports by Drago that cobalt salen complexes in ethanol at reflux under an aerobic atmosphere could catalyze alkene hydration.5 This hydration system was adapted by Mukaiyama to a more practical variant by replacement of salens with β-diketonate ligands and substitution of isopropanol with a more reactive silane hydride donor. We, in turn, adapted Mukaiyama’s modification to effect a hydrogenation of alkenes by replacement of O2 with TBHP as a stoichiometric oxidant. An unusual paradigm underlies this hydrogenation: a stoichiometric hydride source and an oxidant are necessary for catalyst turnover and therefore must be mutually compatible (Fig. 1B). This dual requirement of a stoichiometric reductant and stoichiometric oxidant can complicate the merger of these MHAT cycles with cross-coupling cycles.6 Therefore, we sought to replace the [H–]/[O] combination with an [e−]/[H+] combination, so that metal hydride might be formed by protonation of a low valent metal complex (Fig. 1B). Herein we report a method that takes advantage of this reaction design, albeit to effect a coordinative hydrogenation of terminal olefins (Fig. 1C).
Results and Discussion
Screening for reductive hydrogenation initiated with manganese(III) complexes in combination with stoichiometric manganese(0) reductant in a variety of solvents, utilizing 4-phenylbutene (1a) as substrate. These efforts failed to deliver product and our attention turned to cobalt complexes. The Co(II)(salen) class was initially examined for its hydrogenation activity in the presence of stoichiometric reductants such as titanium(III) salts, manganese(0), zinc(0), and magnesium(0). None of these conditions provided product either. Lewis acids were considered to enhance ligand lability and facilitate catalyst reduction. A brief screen of inexpensive aluminum salts revealed AlCl3 as a suitable Lewis acid to effect reduction, but reaction only occurred in protic solvent. Along with a Co(II)(salen) catalyst and manganese(0) in isopropanol, this combination of reagents proved able to effect the reaction at ambient temperature and pressure, providing 84% hydrogenation of 1a (as observed by 1H NMR spectroscopy against an internal standard). Other reductants failed to deliver product under these conditions. A brief screen of other cobalt salts revealed that Co(OAc)2•4H2O served as a superior catalyst, providing 2a with an isolated yield of 96%.
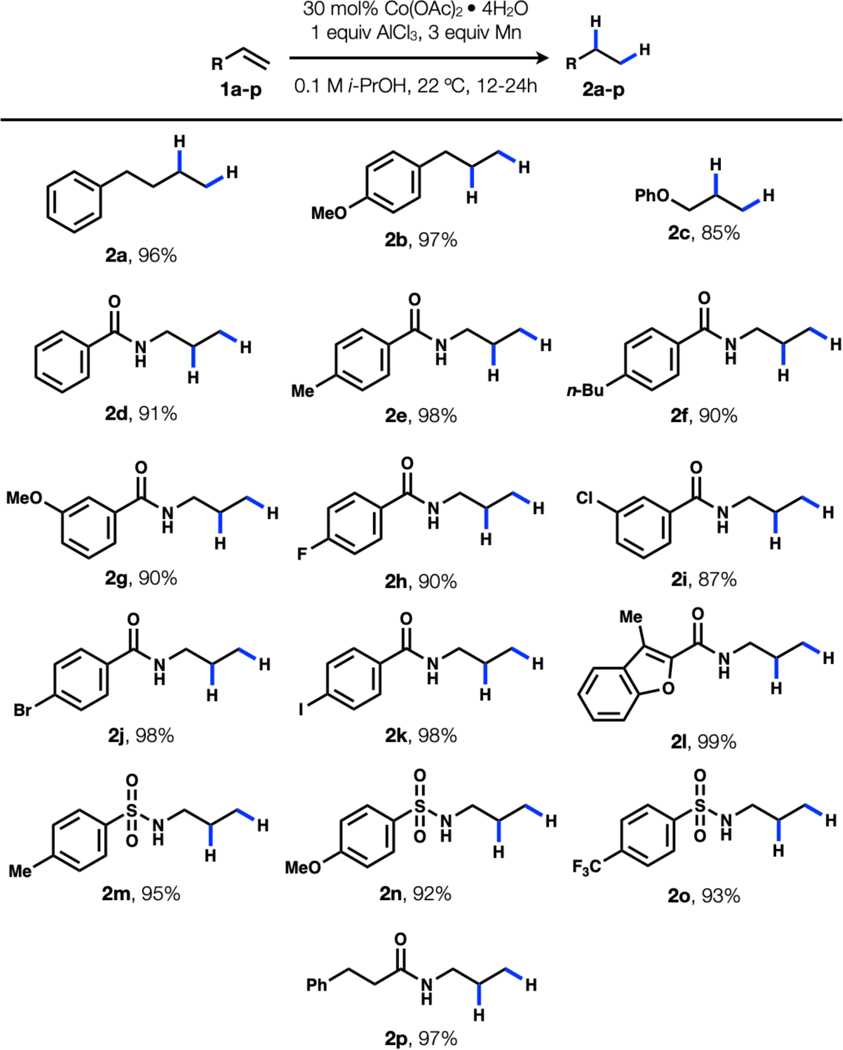
With conditions in hand, we next examined the scope of the reaction (Table 1). Allylbenzene 1b and allylether 1c could be hydrogenated to the corresponding products (2b-c). A range of N-allylbenzamides could also be hydrogenated under the reaction conditions (2d-l). Halides that are prone to reduction with heterogenous palladium(0) catalysts remain untouched over the course of this reaction (2j-k). N-Allylsulfonamides 2m-o are also reduced, as are N-allylalkylamides such as 2q. There are limitations to the reaction. For example, the reaction fails to hydrogenate some allylethers, allylamides, and styrenes. 1,1Disubstiuted and internal olefins are unaffected by the reaction. The reaction also deprotects silyl ethers. Further details can be found in the ESI. We noted that gas evolution occurs at the beginning of the reaction and hypothesized that the HCl generated by reaction of AlCl3 with alcohol was reduced by Mn powder. Indeed, we found that AlCl3 could be replaced with
Table 1.
Substrate scope of the reaction.a
|
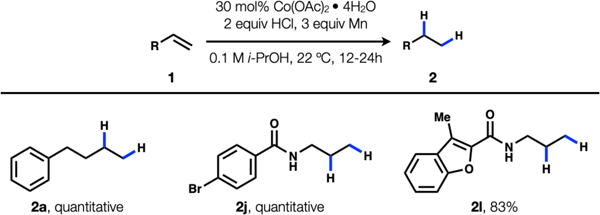
Reagents and conditions: 0.2 mmol 1, 30 mol% Co(OAc)2•4H2O, 1 equiv. AlCl3, 3 equiv. Mn powder (−325 mesh), 2 mL i-PrOH, 22 °C, 12–24 h, isolated yield reported. only 2 equivalents of HCl and led to similar yields for representative substrates (1a, 1j, and 1l) (Table 2).
We then turned our attention to the mechanism of this reaction. A few observations guided hypothesis and experiment. First, no reaction occurred in the absence of added acid. Second, no reaction occurred in the absence of Co salt or Mn reductant. Third, Mn powder could be replaced with Zn powder, but Zn caused significant variations in yield between runs. Fourth, different cobalt salts were competent in the reaction and likely formed CoCl2 in situ [based on the formation of a blue alcoholic solution].
In light of these observations, several hypotheses seemed plausible:
Solvent (isopropanol) coordinates to Co(II) and undergoes β-hydride elimination to afford a Co(II)–H. Migratory insertion into the olefin substrate forms an alkylcobalt(II) complex. The Co(II)–C bond is then protonated to afford hydrogenation product (Scheme 1). (excluded)
Co(II) is reduced by Mn(0) to Co(I). The Co(I) species is protonated to Co(III)–H. This species undergoes MHAT to the olefin to generate the free radical and a Co(II) species. The alkyl radical abstracts a hydrogen atom from Co(III)–H or solvent to afford the hydrogenation product (Scheme 2). (excluded)
Co(II) is reduced by Mn(0) to Co(0 or I). The cobalt(0 or I) species is protonated to Co(II or III)–H. This species undergoes migratory insertion into the olefin to afford a Co(II or III)–alkyl species. Protonation of the Co–C bond affords hydrogenation product and a Co(II or III) species that is reduced by Mn(0) to close the catalytic cycle (Scheme 3).
Co(II) precatalysts are reduced in situ by Mn(0) to afford Co(0 or I) capable of undergoing oxidative addition with hydrogen gas generated by reaction of solvent with AlCl3, generating HCl that goes on to react with Mn(0), affording H2 and MnCl2. The Co(II or III) dihydride undergoes 1,2-insertion and reductive elimination sequence to afford hydrogenated product (Scheme 4). (excluded)
Mn(0) reacts with the generated HCl to afford Cl–Mn(II)–H. This species undergoes ligand exchange with Co(II) to afford a Co(II)–H that undergoes 1,2-insertion with the olefin substrate. Subsequent protonation of the Co(II)–C bond affords product (Scheme 5). (excluded)
Scheme 1.

Scheme 2.

Scheme 3.

Scheme 4.

Scheme 5.

Subsequent experiments refute a number of these mechanisms. Exclusion of manganese powder led to complete arrest of the reaction, so mechanism 1 was excluded since this mechanism had no obvious role for manganese. Substrate 4 failed to undergo any cyclization (only product 5 is observed by GCMS) (Fig. 2A). Therefore, either a radical never formed or the radical-metal cage pair exhibited very high fractional cage efficiency (Fc). We recently found that Co(salen) complexes undergo HAT to generate radical pairs with apparently low Fc, leading to efficient radical cyclization onto arenes. For this reason, the fastest reacting substrates tended to be 1,1-disubstituted alkenes, which form tertiary radicals and resist collapse to an organometallic. In contrast, these reductions with Co(II) and Mn(0) only react with terminal alkenes, inconsistent with an MHAT mechanism.7 Thus, mechanism 2 may be ruled out. When argon is bubbled through the reaction to remove hydrogen gas before addition of olefin, product is still observed (Fig. 2B). The reaction also fails to proceed in the presence of catalytic manganese and hydrogen atmosphere (Fig. 2C). Therefore, mechanism 4 may be ruled out. When MnCl2 is used in combination with PhSiH3 instead of Mn(0)/HCl, the reaction fails to proceed (Fig. 2D). On the basis of this observation and the ability to replace Mn(0) with Zn(0), mechanism (5) may be excluded from consideration (it is possible that the Si–H/Mn–Cl ligand exchange is endergonic and does not proceed under the reaction conditions). Put together, these experiments leave mechanism 3 as a plausible pathway for this reduction. Further work is required to elucidate the complete details, but this rough sketch may provide guidance for future work in reductive protonation to LnCo-H complexes, especially those with weak-field ligands.8
Figure 2.

Mechanistic studies.
Conclusion
We have disclosed a method for the hydrogenation of terminal olefins catalyzed by cobalt and mediated by a stoichiometric reductant and Brønsted acid. The cost of goods makes such a system appealing: Mn(0) (Alfa Aesar): 0.1 $/g @ 1 kg; Co(OAc)2•4H2O (VWR/ChemImpex) : 0.085 $/g @ 1 kg; i-PrOH (anhydrous) (Acros): 0.02 $/g @ 20 L; 1 N HCl (Acros) 0.02 $/mL @10L. Our current understanding of the mechanism of this reaction is limited and requires further interrogation. Nonetheless, this reaction represents a valuable entry to the field of olefin hydrogenation considering the abundant, inexpensive nature of the reactants, and its chemoselectivity over halides.
Supplementary Material
Table 2.
Examples with HCl instead of AlCl3.a
|
Reaction conditions: 0.1 mmol 1, 30 mol% Co(OAc)2•4H2O, 2 equiv HCl/i-PrOH solution (1–2 M), 3 equiv Mn powder (−325 mesh), 1 mL i-PrOH, 22 °C, 18h, 1H NMR yield reported.
Acknowledgments
We are grateful for support by the NSF (CHE 1955922) and the NIH (R35 GM122606) (R.A.S.), the Fulbright Scholarship Program (R.O.M.) and the Skaggs Graduate School (V.v.d.P.).
Footnotes
Dedication
This paper is dedicated to Dale Boger, a trusted colleague, mentor, friend and recipient of the 2020 Tetrahedron Prize.
Supplementary Material
Experimental procedures and characterization for new compounds can be found in the Supplementary Material.
Declaration of Competing Interest
No competing financial or personal interests have influenced the work reported in this paper.
References and notes
- 1. (a). Liu W; Sahoo B; Junge K; Beller M.“Cobalt Complexes as an Emerging Class of Catalysts for Homogeneous Hydrogenations. Acc. Chem. Res 2018, 51, 1858–1869. [DOI] [PubMed] [Google Scholar]; (b) Ai W; Zhong R; Liu X; Liu Q.“Hydride Transfer Reactions Catalyzed by Cobalt Complexes.” Chem. Rev 2019, 119, 1876–2953. [DOI] [PubMed] [Google Scholar]; (c) Alig L; Fritz M; Schneider S.“First-Row Transition Metal (De)Hydrogenation Catalysis Based On Functional Pincer Ligands.” Chem. Rev 2019, 119, 2681–2751. [DOI] [PubMed] [Google Scholar]; (d) Seo CSG; Morris RH “Catalytic Homogeneous Asymmetric Hydrogenation: Successes and Opportunities.” Organometallics 2019, 38, 417–65. [Google Scholar]; (e) Mukherjee A; Milstein D.“Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes.” ACS Catalysis. 2018, 8, 11435–11469. [Google Scholar]; (f) Johnson NB; Lennon IC; Moran PH; Ramsden JA “Industrial-Scale Synthesis and Applications of Asymmetric Hydrogenation Catalysis.” Acc. Chem. Res 2007, 40, 1291–1299. [DOI] [PubMed] [Google Scholar]; (g) Saudan LA “Hydrogenation Processes in the Synthesis of Perfumery Ingredients.” Acc. Chem. Res 2007, 40, 1309–1319. [DOI] [PubMed] [Google Scholar]; (h) Schultz CS; Krska SW “Unlocking the Potential of Asymmetic Hydrogenation at Merck.” Acc. Chem. Res 2007, 40, 1320–1326. [DOI] [PubMed] [Google Scholar]; (i) Minnaard AJ; Feringa BL; Lefort L; De Vries JG “Asymmetric Hydrogenation Using Monodentate Phosphoramidite Ligands.” Acc. Chem. Res 2007, 40, 1267–1277. [DOI] [PubMed] [Google Scholar]; (j) Cui X; Burgess K.“Catalytic Asymmetric Hydrogenations of Largely Unfunctionalized Alkenes.” Chem. Rev 2005, 105, 3272–3296. [DOI] [PubMed] [Google Scholar]
- 2. (a). Polanyi M; Horiuti J.“Exchange reactions of hydrogen on metallic catalysts” Trans. Faraday Soc 1934, 30, 1164; [Google Scholar]; (b) Mattson B; Foster W; Greimann J; Hoette T; Le N; Wankum S; Cabri A; Reichenbacher C; Schwanke E.J. Chem. Educ 2013, 90, 5, 613–619 [Google Scholar]
- 3. (a). Crossley SWM; Obradors C; Martinez RM; Shenvi RA “Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins.” Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shenvi RA; Matos JLM; Green SA Hydrofunctionalization of Alkenes by Hydrogen-Atom Transfer. In Organic Reactions, Vol. 100. John Wiley & Sons, Inc., 2020. [Google Scholar]
- 4. (a). Shey J; McGinley CM; McCauley KM; Dearth AS; Young BT, van der Donk WA “Mechanistic Investigation of a Novel Vitamin B12-Catalyzed Carbon–Carbon Bond Forming Reaction, the Reductive Dimerization of Arylalkenes.” J. Org. Chem 2002, 67, 837–846. [DOI] [PubMed] [Google Scholar]; (b) Kamei Y; Seino Y; Yamaguchi Y; Yoshino T; Maeda S; Kojima M; Matusnaga S.“Silane- and peroxide-free hydrogen atom transfer hydrogenation using adcorbic acid and cobalt-photoredox dual catalysis.” Nature Commun. 2021, 12, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zombeck A; Hamilton DE; Drago RS “Novel Catalytic Oxidations of Terminal Olefins by Cobalt(II)-Schiff Base Complexes.” J. Am. Chem. Soc 1982, 104, 6782–6784. [Google Scholar]
- 6. (a). Green SA; Matos JLM; Yagi A; Shenvi RA Branch-Selective Hydroarylation: Iodoarene-Olefin Cross Coupling. J. Am. Chem. Soc 2016, 138, 12779–12782. [DOI] [PubMed] [Google Scholar]; (b) Green SA; Vásquez-Céspedes S; Shenvi RA Iron-Nickel Dual-Catalysis: A New Engine for Olefin Functionalization. J. Am. Chem. Soc 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Green SA; Huffman TR; McCourt RO; van der Puyl VA; Shenvi RA Hydroalkylation of Olefins to form Quaternary Carbons. J. Am. Chem. Soc 2019, 141, 7709–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crossley SWM; Barabé F; Shenvi RA “Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc 2014, 136, 16788–16791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shevick SL; Wilson CV; Kotesova S; Kim D; Holland PL; Shenvi RA “Catalytic hydrogen atom transfer to alkenes: a roadmap for metal hydrides and radicals” Chem. Sci 2020, 11, 12401–12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.