

Abstract
Checkpoint immunotherapy has yielded meaningful responses across many cancers but has shown modest efficacy in advanced prostate cancer. B7 homolog 3 protein (B7-H3/CD276) is an immune checkpoint molecule and has emerged as a promising therapeutic target. However, much remains to be understood regarding B7-H3’s role in cancer progression, predictive biomarkers for B7-H3 targeted therapy, and combinatorial strategies. Our multi-omics analyses identified B7-H3 as one of the most abundant immune checkpoints in prostate tumors containing PTEN and TP53 genetic inactivation. Here, we sought in vivo genetic evidence for, and the mechanistic understanding of, the role of B7-H3 in PTEN/TP53 deficient prostate cancer. We found that loss of PTEN and TP53 induced B7-H3 expression by activating transcriptional factor Sp1. Prostate-specific deletion of Cd276 resulted in delayed tumor progression and reversed the suppression of tumor-infiltrating T cells and NK cells in Pten/Trp53 genetically engineered mouse models. Furthermore, we tested the efficacy of the B7-H3 inhibitor in preclinical models of castration-resistant prostate cancer (CRPC). We demonstrated that enriched regulatory T cells and elevated Programmed Cell Death Ligand 1 (PD-L1) in myeloid cells hinder the therapeutic efficacy of B7-H3 inhibition in prostate tumors. Finally, we showed that B7-H3 inhibition combined with blockade of PD-L1 or Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) achieved durable anti-tumor effects and had curative potential in PTEN/TP53-deficient CRPC model. Given that B7-H3 targeted therapies have been evaluated in early clinical trials, our studies provide new insights into the potential of biomarker-driven combinatorial immunotherapy targeting B7-H3 in prostate cancer, among other malignancies.
One-sentence summary:
Immune Checkpoint B7-H3 is a Promising Therapeutic Target in PTEN/p53-deficient Prostate Cancer
INTRODUCTION
Prostate cancer is the most diagnosed cancer and the leading cause of cancer death in men worldwide. Although nearly all patients respond to androgen deprivation therapy (1), the duration of response varies from months to years, followed by progression to castration-resistant prostate cancer (CRPC), with high rates of metastasis and mortality (2). Genetic deletions and mutations in the tumor suppressor genes Phosphatase and tensin homolog (PTEN) and Tumor protein p53 (TP53) occur in ~25% of primary prostate tumors and are further enriched in > 60% of CRPCs (3–7). They are associated with metastatic disease and worse overall outcomes in patients with advanced prostate cancer (3, 5, 6, 8). TP53 and PTEN gene defects are two of the three molecular signatures that characterize “aggressive variant prostate cancers”, a clinical-biological subset of androgen-indifferent prostate tumors (5). Effective therapeutics and combinatorial strategies are urgently needed for patients with advanced prostate cancer, particularly those with PTEN and TP53 defects.
Immunotherapy has yielded meaningful responses across many cancers but has shown only modest activity in prostate cancer (9). The FDA-approved dendritic cell-based cancer vaccine sipuleucel-T has shown modest survival benefits in prostate cancer (10). Immune checkpoint inhibitors that target cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death 1 (PD-1), and its ligand (PD-L1) display minimal or no activity as single agents or in combination with androgen receptor (AR) inhibitors in advanced prostate cancer (11–16). Recently, the FDA approved treatment with the PD-1 inhibitor pembrolizumab for all solid tumors containing mismatch repair deficiency, high microsatellite instability, or high tumor mutational burden (17–19), opening a new horizon for biomarker-driven immunotherapy. There is growing recognition of the importance of identifying the molecular subtypes that better respond to certain checkpoint immunotherapies. However, a better understanding is needed of the biology of immune checkpoints as well as their interactions with oncogenic pathways and molecular features in malignancy.
Immune checkpoint B7-H3, encoded by the CD276 gene, is a member of the B7 family of immunoregulators and shares 88% amino acid identity between human and mouse homologies (20). Although B7-H3 was initially thought to stimulate T-cell responses and suppress tumor growth, the results of more recent studies have indicated that it is involved in immunosuppression and cancer progression (21–25). B7-H3 overexpression is correlated with increased risks of clinical recurrence, disease spread, and poor outcomes in patients with cancer (25–32). Distinct from many cancers expressing B7-H3 in both tumor cells and vasculature, prostate cancers express B7-H3 more often in tumor cells than in stromal cells (33). However, the roles of B7-H3 in cancer development and the tumor immune microenvironment remain unclear, partially because of the lack of tissue-specific deletion models. Although prior studies reported androgen receptor (AR) signaling might be associated with the B7-H3 expression (26, 34–36), we still have a limited understanding of genetic alterations and oncogenic pathways that contribute to B7-H3 dysregulation. These gaps in knowledge hinder the application of immunotherapy targeting B7-H3 in malignancies.
Several therapeutic approaches to targeting B7-H3 have been tested in preclinical and clinical studies (21), including monoclonal antibodies (mAbs), antibody-drug conjugates, and chimeric antigen receptor T cells (32, 33, 37–40). Enoblituzumab (MGA271), a mAb against B7-H3, is currently under phase I/II clinical investigation for refractory solid tumors, including high-risk prostate cancer (NCT02923180, NCT02982941, NCT02381314, and NCT02475213). Much remains to be understood with regard to the mechanisms of action of B7-H3 inhibitors, the rational combinatorial strategies, and identifying the patient subsets that are most likely to benefit from B7-H3 targeted therapy. Such information is critical in order to arrive at curative effects.
Here, we sought in vivo genetic evidence for, and the mechanistic understanding of, the role of B7-H3 in prostate cancer. We demonstrated that PTEN and TP53 defects induced B7-H3 overexpression in cancer cells and elucidated the underlying mechanisms. Using syngeneic models and genetically engineered mouse models (GEMMs), we uncovered the key roles of B7-H3 in promoting tumor progression and immunosuppression in PTEN/p53-deficient cancer. Furthermore, we tested the efficacy of the B7-H3 inhibitor in preclinical models of CRPC, identified compensatory mechanisms, and developed effective combinatorial strategies for advanced prostate cancers that contain PTEN and TP53 defects.
RESULTS
Expression of B7-H3 is elevated in tumors with PTEN and TP53 defects
To identify PTEN- and p53-associated immune checkpoints, we generated a 53-gene list that includes 23 inhibitory and 30 stimulatory checkpoint molecules (table S1), followed by correlation analyses with genetic deletions and mutations of PTEN and TP53 in human prostate cancer. The results revealed that B7-H3 (CD276) is the most significantly overexpressed immune checkpoint in prostate tumors that contain PTEN and TP53 defects (P < 0.0001; Fig. 1, A and B, and table S1). In addition, the elevation of B7-H3 was observed in PTEN/p53-deficient metastatic prostate cancer and invasive breast cancer (fig. S1, A and B).
Figure 1. B7-H3(CD276) expression is elevated in tumors with PTEN and TP53 defects.

(A) Correlation analyses of 53 immune checkpoints with genetic defects of PTEN or TP53 in human prostate tumors (TCGA). Differential expression of immune checkpoints in tumors with PTEN or TP53 defects were analyzed by Student’s t-test, and P values are presented on the X- or Y-axis, respectively. The color indicates the expression (Log2[RNA Seq V2 RSEM+1]) of immune checkpoints in tumors with PTEN or TP53 defects. (B) CD276 mRNA expression in prostate tumors containing PTEN or TP53 defects (TCGA dataset). One-way ANOVA with Tukey’s post hoc tests was performed using GraphPad Prism version 9.2.0..; ∗∗∗∗ P < 0.0001. (C) The CopyKat inferred copy number segmentation identified 14 luminal cell subclones in human prostate tumor samples (Single-cell transcriptional profiling data are from Chen et al., 2021 (41); n=12 patients). (D) The luminal cells were clustered by CD276 expression (left). PTEN and TP53 expression in CD276-low versus -high clusters are shown (right). Differential expression analysis was performed by the Wilcoxon rank-sum test. ∗∗∗∗ adjusted P < 0.0001. (E,F) B7-H3 expression in normal prostate (WT) and prostate tumors from PB-Cre; PtenL/L (PbP) and PB-Cre; PtenL/L; Trp53L/L (PbPP) mice, as determined by qPCR (E) and IHC (F). One-way ANOVA with Tukey’s post hoc tests were performed using GraphPad Prism version 9.2.0.; ∗∗∗∗ P < 0.0001. Scale bar, 100 µm.
To determine the expression pattern of B7-H3 in tumors at the single-cell level, we analyzed the published single-cell transcriptome dataset from 13 primary and metastatic prostate tumor tissues (41). We found that B7-H3 was predominantly expressed in a subset of luminal epithelial cells in prostate tumors (fig. S1C). Next, we subset the luminal cells from all samples and inferred the copy number alterations (CNAs) from single-cell transcriptome using CopyKat (42). Fourteen subclones were identified from the CNA profiling, including 12 aneuploid subclones and two diploid subclones (fig. S1D). Among the 12 aneuploid subclones, three (c2, c3, and c7) highly expressed B7-H3 (Fig. 1, C to E). Compared to subclones expressing low B7-H3 mRNA, those B7-H3-high subclones showed significantly lower expression of PTEN and TP53 (P < 0.0001; Fig. 1D), suggesting their negative correlations in cancer cells at single-cell resolution.
In GEMMs, Pb-Cre-driven Pten loss in the prostate (PbP) triggers non-lethal invasive tumors after a long latency, and co-deletion of Pten and Trp53 (PbPP) drives aggressive prostate cancer progression (43–45). Compared to normal prostate tissue, prostate-specific Pten deletion led to a slight increase in B7-H3 expression, whereas Pten/Trp53 double deletion caused significant elevation of B7-H3 in prostate tumors, in both mRNA and protein ( P < 0.0001; Fig. 1, E and F). Collectively, these studies in human samples and GEMMs uncovered that B7-H3 is overexpressed in tumors containing PTEN and TP53 defects.
Loss of PTEN and TP53 upregulates B7-H3 by activating transcriptional factor Sp1
Next, we determined the impact of PTEN or TP53 loss on B7-H3 expression in human prostate cancer cell lines using the Clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 system. Knockout of TP53 in LNCaP (PTEN-null; p53-intact) cells induced B7-H3 expression and activated CD276 gene transcription (Fig. 2, A and B). PTEN deletion led to increased expression and transcription of CD276 in DU145 (p53-mutated; PTEN-intact) cells (Fig. 2, C and D). Together with the above GEMMs, these results revealed the regulatory roles of PTEN and p53 tumor suppressors in B7-H3 expression.
Figure 2. Loss of PTEN and TP53 upregulates B7-H3(CD276) by activating Sp1.

(A) Western blot analysis of B7-H3 in LNCaP cells (PTEN-null) with or without TP53 knockout. (B) Dual luciferase assay of vector (Vec) or pGL4 plasmid containing CD276 promoter (CD276-reporter) in LNCaP cells with or without TP53 knockout (n=3 per group). (C) Western blot analysis of B7-H3 in DU145 cells (TP53-deficient) with or without PTEN knockout. (D) Dual luciferase assay of vector or CD276-reporter in DU145 cells with or without PTEN knockout (n=4 per group). (E) Binding of Sp1 protein to the promoter region of the CD276 gene was determined using ChIP-qPCR (n=4 per group). Locations of primers in the CD276 gene promoter region are shown. TSS: transcription start site. (F,G) PTEN/p53-deficient LNCaP cells (PTEN-null; TP53 KO) were transfected with siRNA targeting SP1 or scrambled control, followed by dual luciferase assay of CD276-reporter (F) and qPCR of CD276 (G). Relative luciferase activities were normalized to vector control samples (n=4 per group). (H) Western blot analysis of B7-H3 and Sp1 in LNCaP cells treated with different doses of AKT inhibitor MK2206. (I) Western blot analysis of Sp1 and B7-H3 in LNCaP cells upon TP53 deletion. (J) Schematics of the mechanism by which PTEN-AKT and p53 pathways regulate B7-H3 expression. Data represent the mean ± SD. One-way ANOVA with Tukey’s post hoc tests (B, D, F, and G) and student’s t-tests (E) were performed using GraphPad Prism version 9.2.0.. ∗P < 0.05, ∗∗ P < 0.01, ∗∗∗ P < 0.001, ∗∗∗∗ P < 0.0001.
To understand the regulatory mechanism of PTEN and TP53 loss on B7-H3, we searched the GeneHancer database and found 27 putative transcriptional regulators (TRs) of B7-H3 by intersecting different datasets (table S2). To identify PTEN/p53-associated TRs, we performed a protein-protein interaction network analysis of PTEN, p53, and 27 TRs using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING). Five TRs, including Specificity Protein 1 (SP1), Fos Proto-Oncogene, AP-1 Transcription Factor Subunit (FOS), Early Growth Response 2 (EGR2), IKAROS Family Zinc Finger 1 (IKZF1), and E1A Binding Protein P300 (EP300), showed associations with both PTEN and p53 (fig. S2A). When searching the literature, we found that all five TRs are involved in gene transcriptional activation, but only transcriptional factor Sp1 is negatively regulated by p53. P53 and Sp1 have negative feedback regulation of one another and play opposite roles when co-regulating the expression of target genes (46, 47). In addition, PI3K-AKT signaling was also found to activate Sp1 in cancer cells (48). Given the role of PTEN in suppressing AKT Serine/Threonine Kinase (AKT), we hypothesize that Sp1 may serve as a transcriptional factor of B7-H3 and mediate B7-H3 upregulation induced by PTEN and TP53 defects.
To test this hypothesis, we first performed Chromatin immunoprecipitation (ChIP) assays and found that the Sp1 protein bound to the promoter region of the CD276 gene (Fig. 2E). Luciferase assays showed that knocking down SP1 suppressed the transcriptional activation of the CD276 gene (Fig. 2F). In PTEN/p53-deficient prostate cancer cells, inhibition of Sp1 using siRNA or inhibitor mithramycin A led to a reduction of B7-H3 mRNA and protein (Fig. 2G, and fig. S2, B and C). Conversely, overexpression of SP1 elevated B7-H3 in PTEN/p53-deficient cells and rescued the effects of SP1 knockdown (fig. S2D). In human prostate tumor samples (29), we found that Sp1 expression is associated with CD276 mRNA expression (fig. S2E). These results indicated that B7-H3(CD276) is a direct target of Sp1, and its transcription is activated by Sp1.
Next, we treated PTEN-deficient LNCaP cells with AKT inhibitor MK2206 and found that AKT inhibition reduced both Sp1 and B7-H3 abundance in a dose-dependent manner and suppressed CD276 gene transcription (Fig. 2H and fig. S2F). In addition, we found that p53 depletion augmented the expression of Sp1 and B7-H3 (Fig. 2I), and inhibition of Sp1 impaired the upregulation of B7-H3 induced by TP53 loss (fig. S2G). Furthermore, we found that overexpression of Sp1 failed to upregulate B7-H3 in cancer cells with intact PTEN or p53 (fig. S2H), suggesting the regulatory role of Sp1 on B7-H3 expression requires the inactivation of both PTEN and p53 tumor suppressors. Altogether, these mechanistic studies demonstrated that PTEN-AKT and p53 pathways suppress the expression of B7-H3, whereas genetic inactivation of PTEN and TP53 genes causes B7-H3 overexpression by activating its transcriptional factor Sp1 (Fig. 2J).
B7-H3 promotes PTEN/p53-deficient tumors and immunosuppression
To determine the impact of B7-H3 on PTEN/p53-deficient tumors, we performed sgRNA-mediated CRISPR and knocked out Cd276 in the syngeneic prostate cancer cell line DX1 (Fig. 3A), which was derived from the PB-Cre; PtenL/L; Trp53L/L; Smad4L/L (PbPPS) GEMM (49). B7-H3 depletion suppressed the growth of PTEN/p53-deficient tumors in immunocompetent C57BL/6 mice (Fig. 3B, and fig. S3A); in contrast, it had little impact on tumor growth in immunodeficient NSG mice (Fig. 3C, and fig. S3B). These results indicated that B7-H3 plays a tumor-promoting role in PTEN/p53-deficient tumors, and the effects are largely dependent on immune cells in the tumor microenvironment (TME).
Figure 3. B7-H3 promotes PTEN/p53-deficient tumors and immunosuppression.

(A) Cd276 was knocked out in the PTEN/p53-deficient syngeneic prostate cancer cell line DX1, which was derived from the PB-Cre; PtenL/L; Trp53L/L; Smad4L/L (PbPPS) GEMM model. B7-H3 protein expression was determined using Western blot analysis. (B) Tumor growth of control and B7-H3-depleted syngeneic tumors in immunocompetent C57BL/6 mice. (C) Growth of control and B7-H3-depleted tumors in immunodeficient NSG mice. Data represent the mean ± SD. One-way ANOVA with Tukey’s post hoc tests was performed using GraphPad Prism version 9.2.0.. ∗ P < 0.05, ∗∗∗∗ P < 0.0001. ns, not significant. (D, E, F) Immunoprofiling was performed in control and B7-H3-depleted syngeneic tumors derived from (B) using mass cytometry (CyTOF). Colored viSNE plots of CD45+ immune cells are presented (D). Quantification of tumor-infiltrating lymphocytes (E) and myeloid cells (F) is shown. TIL: Tumor Infiltrating Leukocytes. Data from sgCd276 #2 and #3 tumors were combined for statistical analysis. Data represent the mean ± SD. Student’s t-tests were performed using GraphPad Prism version 9.2.0. ∗ P < 0.05, ∗∗ P < 0.01, ∗∗∗∗ P < 0.0001. ns, not significant.
To capture the impact of B7-H3 on immune components in tumors with PTEN and TP53 defects, we performed mass cytometry (CyTOF) in control and B7-H3-depleted syngeneic tumors in C57BL/6 mice. Briefly, a 28-marker panel (table S3) was used to label tumor-infiltrating immune cells, including CD4+ T cells, CD8+ T cells, regulatory T (Treg) cells, B cells, dendritic cells (DCs), natural killer (NK) cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs). In immunocompetent mice, depletion of B7-H3 in syngeneic cancer cells caused an increase in tumor-infiltrating T cells and NK cells, and meanwhile reduced the abundance of immunosuppressive MDSCs (Fig. 3, D, E, and F). To determine B7-H3’s impact on cytokine activities of lymphocytes, we designed a T cell-focused panel (table S3) and performed CyTOF analyses in control and B7-H3-depleted syngeneic tumors. As shown in fig. S3, C and D, B7-H3 depletion in PTEN/p53-deficient cancer cells enhanced the cytotoxic activities of CD8+ T and NK cells, characterized by increased Granzyme B, Interferon γ (IFNγ), and Interleukin 2 (IL2) production. In addition, both Th1-type, IFNγ and IL2, and Th2-type, IL4, cytokines were increased in CD4+ T cells upon B7-H3 depletion (fig. S3D). We also found an increase in Granzyme B-producing CD4+ T cells in B7-H3 depleted tumors (fig. S3D), suggesting that the CD4+ cytotoxic T cells may also contribute to killing cancer cells.
Furthermore, we performed multiplex immunohistochemistry (IHC) staining to visualize the spatial arrangement and cytokine activities of T cells. Consistent with our previous observations, B7-H3 depletion promoted the infiltration of CD8+ T and CD4+ T cells in syngeneic tumors and augmented their cytokine production (fig. S3, E to G). Collectively, these results indicated that elevated B7-H3 contributes to tumor growth and the immunosuppression of T cells and NK cells in PTEN/p53-deficient tumors.
Prostate-specific Cd276 deletion GEMM revealed B7-H3’s impact on cancer progression
Conventional Cd276 knockout (Cd276-null) GEMM mice have been reported in multiple studies (25, 33, 50). In 2017, Seaman et al. reported the first Cd276 conditional knockout (Cd276Loxp) mouse model (33). To further determine B7-H3’s impact on the development and progression of spontaneous tumors containing PTEN and TP53 defects, we crossed the Cd276Loxp allele onto the PbPP mice (fig. S4A) and generated the first prostate-specific Cd276 deletion prostate cancer GEMM. This new model contains the PB-Cre transgene, mTmG reporter, and conditional knockout alleles of Pten, Trp53, and Cd276 (PbPPCd276) (Fig. 4A). Compared to PbPP mice, PbPPCd276 mice showed significantly delayed prostate tumor development (P < 0.01; Fig. 4B and fig. S4B) and prolonged overall survival (P < 0.001; Fig. 4C). Histopathology analyses showed that prostate-specific Cd276 deletion suppressed the progression of PTEN/p53-deficient tumors (Fig. 4D). This newly established GEMM of PbPPCd276 provides genetic evidence supporting B7-H3’s tumor-promoting role in PTEN/p53-deficient prostate cancer.
Figure 4. Prostate-specific Cd276 deletion GEMM model demonstrates B7-H3’s impact on prostate cancer progression.

(A) GEMM model design: conditional knockout (KO) alleles of Cd276Loxp, PtenLoxp, and Trp53Loxp were crossed with prostate-specific PB-Cre and Rosa-mTmG to establish PB-Cre; PtenL/L; Trp53L/L; Cd276L/L (PbPPCd276) GEMM model. (B) Representative MRI images and tumor volume quantification of prostate tumors from PbPP and PbPPCd276 mice at four and five months of age (n ≥ 4 per group per age). Prostate tumor regions are circled with red dashed lines. (C) Kaplan–Meier survival curve of PbPP and PbPPCd276 mice. Statistical analysis by Log-rank (Mantel-Cox) test. (D) Histopathology analysis of prostate tumors from PbPP and PbPPCd276 mice at five months of age. H&E images at 1x (left) and 20x (middle) magnifications are presented. Depletion of B7-H3 was verified by IHC staining (right). Scale bars are indicated in each image. (E, F) Multiplex IHC staining and quantification of indicated CD8 T (E) and CD4 T (F) cells in prostate tumors from PbPP and PbPPCd276 mice ( n >10 individual views from 3–5 mice per group). Data represent the mean ± SD. (G) Immunofluorescence (IF) and quantification of NK (NK1.1+) cells in PbPP and PbPPCd276 tumors. Data represent the mean ± SD of >10 individual views. Student’s t-tests were performed using GraphPad Prism version 9.2.0. ∗P < 0.05, ∗∗ P < 0.01, ∗∗∗∗ P < 0.0001. ns, not significant.
Next, we characterized lymphocytes in prostate tumors from PbPP and PbPPCd276 mice using multiplex IHC staining. The results revealed that prostate-specific B7-H3 depletion led to increased infiltration of CD8+ T and CD4+ T cells in PTEN/p53-deficient spontaneous tumors and promoted their cytokine production, including Granzyme B and IFNγ (Fig. 4, E and F, and fig. S4C). In addition, we also found an increase in NK cells in prostate tumors from PbPPCd276 mice (Fig. 4G, and fig. S4D). Together with the immunoprofiling in syngeneic mouse models, these results established the biological function of B7-H3 in the suppression of tumor-killing T cells and NK cells, as well as revealed the mechanism by which B7-H3 elevation induced by PTEN and p53 inactivation contributes to cancer development and progression. These functional studies using syngeneic and GEMMs also established B7-H3 as a potential immunotherapeutic target in cancers containing PTEN and TP53 deficiencies.
Effects of B7-H3 inhibitor in preclinical models of PTEN/p53-deficient CRPC
Given that B7-H3-targeted therapies have been tested in clinical trials (32, 33, 37–40), we next sought to evaluate the anti-tumor effects of B7-H3 inhibition in preclinical models of CRPC with PTEN and TP53 defects. We previously reported that PbPPS GEMM mice, containing prostate-specific co-deletion of Pten, Trp53, and Smad4, developed castration-resistant prostate tumors after androgen deprivation therapy and showed resistance to checkpoint immunotherapy targeting PD-1 or CTLA-4 (49, 51, 52). Considering B7-H3 was found to be associated with TGFβ signaling wherein SMAD4 serves as a core component (26, 36, 53), we first determined the impact of SMAD4 on B7-H3 expression in prostate tumors. When comparing the prostate cancer GEMMs with or without Smad4 deletion, we found that SMAD4 did not affect B7-H3 expression (fig. S5A). This result indicated that high expression of B7-H3 in PbPPS tumors is induced by loss of PTEN and p53, but not SMAD4.
Next, we tested the in vivo efficacy of a B7-H3 inhibitor (MJ18 mAb against mouse B7-H3 (54)) in the CRPC preclinical model derived from PbPPS GEMM mice. After prostate tumor formation confirmed by Magnetic Resonance Imaging (MRI) imaging and tumor volume reached approximately 150mm3, PbPPS GEMM mice were treated with surgical castration and an enzalutamide (AR inhibitor)-admixed diet for three weeks (Fig. 5A). Once relapse of CRPC tumors was confirmed by MRI, the mice were randomly assigned for single or combination treatment of enzalutamide and B7-H3 inhibition (Fig. 5A). Compared to enzalutamide alone, the combination of enzalutamide and B7-H3 inhibitor suppressed the progression of CRPC tumors (Fig. 5, B to D), suggesting the therapeutic potential of targeting B7-H3 in PTEN/p53-deficient CRPC.
Figure 5. Effects of B7-H3 inhibitor in preclinical models of PTEN/p53-deficient CRPC.
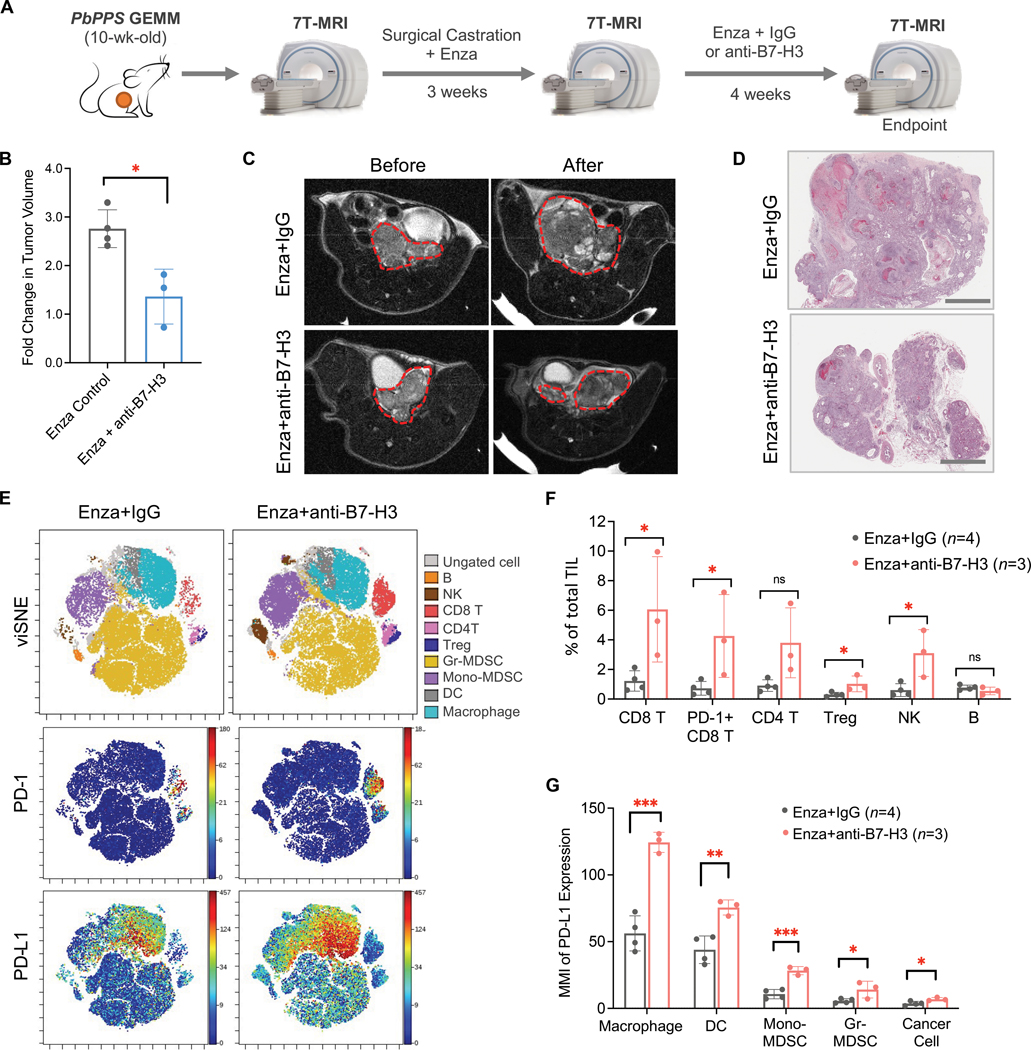
(A) Schematics of treatment design using the CRPC GEMM model. PbPPS mice were surgically castrated, followed by treatment with enzalutamide (Enza) for three weeks. Tumor relapse was monitored by 7T-MRI. Mice were then treated with Enza in combination with B7-H3 inhibitor (MJ18, 300 μg/injection) for four weeks. Isotype IgG was used in the control group. (B,C,D) Fold changes in PbPPS CRPC tumor volume (B), representative MRI images (C), and histopathology analysis (D) after treatment. Prostate tumor regions are circled with red lines. n = 4 in Enza Control group and n = 3 in Enza+anti-B7-H3 group. Scale bars = 2mm. (E,F) Immunoprofiling of syngeneic tumors treated with Enza in combination with IgG or anti-B7-H3 using CyTOF. Colored viSNE plots of CD45+ immune cells and expression patterns of PD-1 and PD-L1 are shown (E). Quantification of tumor-infiltrating lymphocytes is presented (F). TIL: Tumor Infiltrating Leukocytes. (G) Expression of PD-L1 (Median Metal Intensity, MMI) in cancer cell and myeloid components after treatment. Data represent the mean ± SD. Student’s t-tests were performed using GraphPad Prism version 9.2.0. ∗ P < 0.05, ∗∗ P < 0.01, ∗∗∗ P < 0.001. ns, not significant.
To capture the impact of B7-H3 inhibition on CRPC tumor immune landscape at the single-cell level, we performed CyTOF in DX1-derived syngeneic CRPC tumors treated with enzalutamide or in combination with MJ18. DX1 syngeneic cells were derived from the PbPPS model and showed resistance to enzalutamide in vivo, phenocopying the PbPPS CRPC model (49). The immunoprofiling results showed that pharmacological inhibition of B7-H3 augmented the infiltration of T cells and NK cells in CRPC tumors (Fig. 5, E and F); however, those CD8+ T cells highly expressed the inhibitory molecules Programmed death-ligand 1 (PD-L1), Lymphocyte-activation gene 3 (LAG-3), and T-cell immunoglobulin and mucin domain 3 (TIM-3) (Fig. 5, E and F, and fig. S5B). Similar effects were observed in CRPC GEMMs upon B7-H3 inhibition (fig. S5C). Compared to enzalutamide alone, the combination of enzalutamide and B7-H3 inhibition also increased PD-L1 expression in diverse myeloid cells, such as TAMs and DCs (Fig. 5, E and G, and fig. S5D). Although PD-L1 in cancer cells was also upregulated, the expression was much lower than that in TAMs and DCs (Fig. 5G), suggesting myeloid components play a dominant role in stimulating PD-L1/PD-1 signaling in CRPC tumors treated with a B7-H3 inhibitor. In addition, negative correlations of B7-H3 with PD-1 or PD-L1 expression were found in human prostate cancer samples (fig. S5E). These results indicated that B7-H3 inhibitor treatment led to an increase in tumor-infiltrating T and NK cells but also induced the immunoinhibitory pathway of PD-L1/PD-1 in the TME, which may dampen its anti-tumor effects in CRPC tumors.
In addition, we found that B7-H3 inhibition caused an increase in Treg in both syngeneic and GEMMs (Fig. 5F and fig. S5F), which restricted the ratio of CD8+ T cells to Tregs in CRPC tumors (fig. S5G). However, we didn’t find the percentage of Tregs in CD4+ T cells upregulated upon B7-H3 inhibition (fig. S5H), suggesting that Treg enrichment may be a consequence of overall T cell infiltration increase. Collectively, these preclinical studies demonstrated that pharmacological inhibition of B7-H3 suppresses tumor progression and augments T cells infiltration in PTEN/p53-deficient CRPC, but the therapeutic efficacy may be hindered by the upregulated PD-L1/PD-1 signaling and increased Treg in the TME.
B7-H3 inhibition combined with PD-L1 or CTLA-4 blockade achieves durable anti-tumor effects in PTEN/p53-deficient CRPC model
Based on the immunoprofiling studies, we hypothesized that inhibitors targeting PD-L1/PD-1 signaling or Treg could augment the anti-tumor effects of B7-H3 inhibition and reverse the compensatory immunosuppression in PTEN/p53-deficient CRPC. CTLA-4 is an inhibitory immune checkpoint receptor that plays a vital role in modulating T-cell responses (55, 56). Tregs constitutively express high amounts of CTLA-4, and CTLA-4 blockade impaired the suppressive activity of Tregs in several preclinical models (57–60). Hence, we determined the therapeutic effects of B7-H3 inhibition combined with antibodies against PD-1, PD-L1, or CTLA-4 in the syngeneic CRPC model (fig. S6A). Briefly, 1 × 106 DX1 cells were subcutaneously injected into male C57BL/6 mice. One week after injection, tumors formed and were measured by calipers. Then, mice were treated with enzalutamide-admixed diet food and randomly assigned to different checkpoint inhibitor treatment groups. Anti-mouse B7-H3 (MJ18 mAb, 300 μg/injection), anti-mouse PD-1 (200 μg/injection), anti-mouse PD-L1 (200 μg/injection), and anti-mouse CTLA-4 (200 μg/injection) were intraperitoneally (i.p.) administered alone or in combination every three days for six times in total. Isotype IgG was also used in control group (fig. S6A).
As shown in Fig. 6, A and B, inhibitors targeting B7-H3 or PD-L1 alone only showed modest effects, but dual blockade of B7-H3/PD-L1 rendered the CRPC tumors more sensitive to enzalutamide, prolonged the overall survival of mice (P < 0.0001), and cured 69.2% (9/13) of mice. The best therapeutic effects were found in mice treated with enzalutamide combined with dual blockade of B7-H3/CTLA-4 (Fig. 6, A and B). The combination delayed the tumor growth, significantly prolonged the survival (P < 0.0001), and caused 76.5% (13/17) of mice to be tumor-free. Both combinatorial strategies successfully protected the mice against secondary tumor rechallenge (Fig. 6, A and B), and no visible toxicity was observed (fig. S6B). In contrast, the combination of B7-H3 inhibition with PD-1 blockade only cured four in 13 mice (30.8%) and didn’t show a benefit on mice survival compared to PD-1 blockade control.
Figure 6. B7-H3 inhibition combined with PD-L1 or CTLA-4 blockade achieves synergistic effects in PTEN/p53-deficient CRPC.

(A) Growth of syngeneic CRPC tumors treated with Enza in combination with IgG, anti-B7-H3, anti-PD-1, anti-PD-L1, anti-CTLA-4, anti-B7-H3/PD-1, anti-B7-H3/PD-L1, and anti-B7-H3/CTLA-4. 1 × 106 DX1 cells were subcutaneously injected into male C57BL/6 mice. One week after injection, tumors formed and measured by calipers. Then, mice were treated with enzalutamide-admixed diet food and randomly assigned to different checkpoint inhibitor treatment groups. Monoclonal antibodies against B7-H3 (BE0124, 300 µg/injection), PD-1 (BE0273, 200 µg/injection), PD-L1 (BE0101, 200 µg/injection), and CTLA-4 (BE0131, 200 µg/injection) were administered i.p. as single agents or in combination every three days for six times in total. Isotype IgG was also used in control group. The numbers of tumor-free mice are shown. Black arrows showed the date of secondary tumor rechallenge in tumor-free mice. (B) Overall survival of mice treated with different combinations. The log-rank (Mantel-Cox) test was used for survival comparison (GraphPad Prism version 9.2.0.). Black arrows showed the date of secondary tumor rechallenge in tumor-free mice. (C,D) Multiplex IHC staining and quantification of total CD8 T (C) and the ratio of PD1+ CD8 T to total CD8 T (D) cells in tumors treated with single agents or combinations ( n = 10 individual views from two mice per group). Data represent the mean ± SD. One-way ANOVA with Tukey’s post hoc tests were performed using GraphPad Prism version 9.2.0.. ∗ P < 0.05, ∗∗ P < 0.01, ∗∗∗ P < 0.001, ∗∗∗∗ P < 0.0001. ns, not significant.
Furthermore, we performed multiplex IHC staining to compare the effects of different combinations on tumor-infiltrating T cells. The results showed that, when combined with enzalutamide, CTLA-4 or B7-H3 inhibitor led to a significant increase of CD8+ T cells (P < 0.0001), whereas PD-1 or PD-L1 blockade only showed little or modest effects (Fig. 6C). In contrast, the combinations of B7-H3 inhibitors with PD-1 (P < 0.001), PD-L1 (P < 0.0001), or CTLA-4 (P < 0.0001) blockade significantly promoted the infiltration of CD8+ T cells in CRPC tumors, compared to enzalutamide alone (Fig. 6C). Antibodies against PD-1, PD-L1, or CTLA-4 effectively reversed the exhaustion of CD8+ T cells induced by B7-H3 inhibitor (Fig. 6D and fig. S6C). CTLA-4 blockade successfully reduced the abundance of Tregs in the presence of a B7-H3 inhibitor (fig. S6D). We found that the combination of B7-H3 inhibition and PD-1 blockade caused an increase of Tregs in CRPC tumors, which may explain why B7-H3/PD-1 dual blockade had less durable anti-tumor responses than co-targeting B7-H3 and PD-L1. Altogether, these preclinical studies demonstrated that B7-H3 inhibition combined with the blockade of PD-L1 or CTLA-4 have durable anti-tumor effects and resulted in suppression of PTEN/p53-deficient CRPC by reversing the immunosuppression.
DISCUSSION
Although both B7-H3 and PD-L1, also called B7-H1, belong to B7 families, much less is known about the biology of B7-H3 signaling in cancer. A prior study crossed a conventional global Cd276 knockout strain (Cd276−/−) onto the TRAMP (transgenic adenocarcinoma of the mouse prostate) model and reported increased prostate tumor growth in the absence of B7-H3 (61). Although the authors performed a bone marrow transplant and showed the phenotype was not due to B7-H3 defect in bone marrow-derived immune cells, the global depletion of B7-H3 still hindered a direct assessment of B7-H3’s roles in prostate cancer development and the TME. Here, we report the first prostate-specific Cd276 GEMM and provide in vivo genetic evidence supporting the tumor-promoting and immunosuppressing roles of B7-H3 in prostate cancer, especially in the context of PTEN and TP53 defects. These studies advance our understanding of B7-H3 biology and lay an important foundation for targeting B7-H3 signaling in advanced prostate cancer.
B7-H3 has emerged as a promising therapeutic target, and multiple agents targeting B7-H3 are in development. However, much remains to be determined regarding biomarkers for predicting the responses to B7-H3 inhibitors and combinatorial strategies to elicit a cure in patients with cancer. Genetic defects of PTEN and TP53 are common in cancers and are correlated with disease progression and worse prognosis (3, 5, 6). Due to the nature of tumor suppressors, there are limited treatment options for targeting PTEN and TP53 defects. In the current study, we uncovered that PTEN and TP53 defects are associated with B7-H3 overexpression in human diseases, as well as elucidated the mechanism by which PTEN-AKT and p53 pathways are involved in the transcriptional regulation of B7-H3 (fig. S6C). Using approaches of genetic deletion and pharmacological inhibition, we demonstrated that B7-H3 is a promising therapeutic target in PTEN/p53-deficient tumors. Given that genetic testing of PTEN and TP53 has been used for the clinical management of prostate cancer and other cancer types, our studies provide new insights for using them as predictive markers to improve patient selection for clinical trials using B7-H3 targeted agents.
In this study, we also determined the efficacy of mAb against B7-H3 in preclinical models of CRPC and its effects on the tumor immune microenvironment. Like Cd276 genetic deletion, inhibition of B7-H3 using mAb increased the abundance of T cells and NK cells in CRPC tumors; however, it also caused compensatory immunoinhibitory effects, including upregulation of PD-L1 expression in myeloid cells and increased Treg infiltration into tumors (fig. S6E). This observation prompted us to combine B7-H3 inhibition with PD-L1 or CTLA4 blockade, and both combinatorial strategies showed curative potential in PTEN/p53-deficient CRPC preclinical models. Enoblituzumab (a mAb against B7-H3) in combination with ipilimumab (a CTLA-4 inhibitor) has recently entered clinical investigation in melanoma and non-small cell lung cancer (NCT02381314). Our studies offer new mechanistic insights into this rational combination and provide preclinical evidence supporting its potential therapeutic effects in patients with PTEN/p53-deficient CRPC. Enoblituzumab has undergone early clinical tests with PD-1 inhibitors in refractory cancers (NCT02475213, NCT04129320, and NCT04634825) (62). However, only modest synergistic effects were observed when combining B7-H3 inhibitor with PD-1 blockade in our CRPC preclinical models. In contrast, the combination of mAbs against PD-L1 and B7-H3 showed more profound tumor-suppressing effects in PTEN/p53-deficient CRPC. Further clinical studies are urgently needed to determine the therapeutic potential of B7-H3/PD-L1 dual blockade in patients with CRPC.
AR signaling plays a vital role in the development and progression of prostate cancer and is a crucial target for therapeutic interventions (2). AR inhibitors enzalutamide and apalutamide have been approved by FDA and are now in widespread clinical use. Our studies uncover the therapeutic potential of co-targeting AR and B7-H3 in PTEN/p53-deficient prostate cancer, particularly in combination with PD-L1 or CTLA-4 blockade. Recent studies found that AR signaling in immune cells also contributes to T cell suppression (63, 64), and AR inhibition promotes response to targeted therapy in melanoma and other diseases (65). Our studies in prostate cancer may provide new insights into the biomarkers and combinatorial strategies co-targeting B7-H3 and AR signaling in other cancer types. In addition to mAbs, the combinatorial strategies tested in our studies might also be applied to other B7-H3-targeting agents, such as antibody-drug conjugates or small-molecule inhibitors.
There are some limitations in our study. Our mechanistic and preclinical studies support that B7-H3 is a therapeutic target in PTEN/p53-deficient cancers. It is worthwhile to investigate the clinical relevance between the genetic status of PTEN and TP53 genes and the responsiveness to B7-H3 inhibitors in patients with advanced prostate cancer or other malignancies. The combinatorial strategies in our studies were largely based on the combination treatment with enzalutamide. The efficacies of dual blockade of B7-H3/PD-L1 or B7-H3/CTLA4, in the absence of an AR inhibitor, have yet to be tested in our preclinical models. Given the importance of AR signaling in prostate cancer, efforts have been made to study the associations between B7-H3 and AR (26, 34–36). The potential regulatory role of AR signaling on B7-H3 and whether B7-H3 mediated immunosuppression contributes to castration resistance have yet to be investigated. In addition to cytotoxic T and NK cells, we also found that B7-H3 is involved in modulating immunosuppressive Treg and myeloid cells in the TME. However, the underlying molecular mechanisms remain elusive. Future studies involving transcriptional profiling at single-cell resolution, signaling pathway identification, and clinical relevance assessment are required to answer these questions and advance our knowledge of B7-H3 biology.
In conclusion, we show here that B7-H3 was overexpressed in PTEN/p53-deficient tumors and demonstrated that PTEN-AKT signaling and the p53 pathway negatively regulate B7-H3 expression by suppressing transcriptional factor Sp1. By establishing a new prostate-specific Cd276 deletion GEMM, we provided in vivo genetic evidence for B7-H3’s role in promoting tumor progression and immunosuppression in PTEN/p53-deficient cancer. Moreover, our preclinical studies revealed that inhibition of B7-H3 using mAb suppressed the progression of CRPC tumors, but its therapeutic efficacy was dampened by elevated PD-L1 expression in myeloid cells and enriched Tregs in the TME. Finally, we show that B7-H3 inhibition combined with PD-L1 or CTLA-4 blockade has druable effects and curative potential in CRPC containing PTEN and TP53 deficiencies. Collectively, our findings demonstrate that immune checkpoint B7-H3 is a potential immunotherapy target in advanced prostate cancers that harbor PTEN and TP53 defects. Our studies advance our knowledge of B7-H3 biology, facilitate the development of biomarker-driven therapies targeting B7-H3 signaling, and inform rational biology-based combinations with curative intent for patients with CRPC and other malignancies.
MATERIALS AND METHODS
Study Design
The main objectives of this study were to determine the role of B7-H3 in cancer progression and immunosuppression, understand the impact of oncogenic signaling on B7-H3 expression, and evaluate the therapeutic potential of B7-H3 inhibition and combinations in CRPC. To this end, we analyzed tumor samples from patients with prostate cancers, performed in vitro assays (Western blot, quantitative real-time PCR, chromatin immunoprecipitation, and dual-luciferase reporter assay, multiplex IHC staining, IF, CyTOF), and in vivo experiments using syngeneic and GEMMs. For in vitro cell assays, data were collected from at least three independent cultures, as indicated in the figure legends. In vivo experiments were performed in biological replicates as indicated by n values in the figure legends. Due to the nature of prostate cancer, only male mice were used for in vivo experiments. Sample sizes were determined on the basis of previous experience for each experiment. Mice were randomly assigned to experimental groups whenever possible. No mice, outliers, or other data points were excluded. Tumor progression was evaluated by tumor size and survival rate. Tumor-bearing mice were euthanized when the tumor volumes reached approximately 2000 mm3. All experiments were performed and analyzed blinded to the genotype, group, and treatment. Statistical analysis was performed under the guidance of an expert in statistics and described in the main text and figure legends.
Experimental GEMMs
PB-Cre; PtenL/L (PbP), PB-Cre; PtenL/L; Trp53L/L (PbPP), and PB-Cre; PtenL/L; Trp53L/L; Smad4L/L (PbPPS) mice were established by the DePinho laboratory at The University of Texas MD Anderson Cancer Center (Houston, TX), as reported in previous studies (51, 52). Briefly, Cd276L/L mice were gifts from Dr. B.St. Croix at the National Cancer Institute (Frederick, MD). The knockout vector was designed to remove exon 2 containing the start codon and signal peptide in the presence of Cre (33). The prostate-specific Cd276 deletion model of PB-Cre; PtenL/L; Trp53L/L; Cd276L/L (PbPPCd276) mice were generated in this study by crossing the Cd276L/L allele onto the PbPP model. Prostate tumor development in PbPP and PbPPCd276 male mice was monitored by Bruker 7T MRI (Bruker Biospin MRI) monthly at the MD Anderson Small Animal Imaging Facility. Tumor volume was calculated using ImageJ (Fiji) software (version 2.1.0/1.53c, National Institutes of Health). Mice survival was determined by Kaplan-Meier analysis. Mice from each cohort were sacrificed at 5 months of age for tumor collection and histopathology analysis. All the GEMM mice in this study were genotyped regularly. Only male mice were used due to the prostate cancer study. All mice were interbred and maintained at MD Anderson, monitored for signs of ill health every day, and euthanized and necropsied when moribund. All operations were performed according to the protocol after being reviewed and approved by MD Anderson’s Institutional Animal Care and Use Committee (protocol #00001955).
Tumor implantation and treatment in syngeneic models
The syngeneic prostate cancer cell line DX1 was generated from the PB-Cre; PtenL/L; Trp53L/L; Smad4L/L; mTmG (PbPPS) GEMM, as reported in our prior study (49). Briefly, the prostate tumors of PbPPS mice were isolated and dissociated into single-cell suspension, followed by sorting of GFP-positive cancer cells and cultured in DMEM medium (Corning, 10-013-CV) with 10% FBS (Thermo Scientific, 10082147) and 1% penicillin-streptomycin (Thermo Scientific, 15140163). Every 10 passages, genetic co-deletions of Pten, Trp53 and Smad4 in DX1 cells were determined by genotyping and western blot. Monthly mycoplasma tests were performed using the MycoAlert PLUS detection kit (Lonza, LT07-710). To determine the impact of B7-H3 on tumor growth in immunocompetent and immunodeficient mice, 1 × 106 DX1 cells, with or without Cd276 knockout, were subcutaneously injected into one flank of five-week-old male C57BL/6 mice or two flanks of six-week-old male NSG mice. Tumor size was measured every three days using calipers. Tumor volume was calculated (width × width × length/2). At the endpoint, tumors were collected for histopathologic analyses. C57BL/6 and NSG mice were purchased from Taconic or the Experimental Radiation Oncology animal facility at MD Anderson.
For the CRPC syngeneic model and combination treatment, 1 × 106 DX1 cells were subcutaneously injected into one flank of male C57BL/6 mice. One week later, tumor volumes were measured by calipers, and mice were treated with enzalutamide-admixed diet food (MedKOO Biosciences, 201821) and then randomly assigned to checkpoint inhibitor treatment. Anti-mouse B7-H3 (300 μg/injection, Bio X Cell, BE0124), anti-mouse PD-1 (200 μg/injection, Bio X Cell, BE0273), anti-mouse PD-L1 (200 μg/injection, Bio X Cell, BE0101), and anti-mouse CTLA-4 (200 μg/injection, Bio X Cell, BE0131) were i.p. administered alone or in combination every three days for six times in total. Isotype IgG control (Bio X Cell, BE0089 and BE0088) was also used. The detailed information on antibodies used throughout the study is listed in table S4. The body weights of the mice were measured weekly. Tumor growth was measured every three days using calipers. Tumor volume was calculated (width × width × length/2). For secondary tumor rechallenge, DX1 (1 × 106) cells were subcutaneously injected into another flank of cured mice on day 60 after treatment. Mice were monitored for 102 days, and mice survival was determined by Kaplan-Meier analysis. At the endpoint, tumors were collected for histopathologic analyses or immunoprofiling using CyTOF or Flow.
PbPPS CRPC model and treatment
As described in our prior studies (49, 51, 52), tumor development of PbPPS male mice was monitored by 7T-MRI, as described above. PbPPS mice at 10 weeks old were surgically castrated, followed by treatment with the enzalutamide-admixed diet for three weeks. Tumor relapse was monitored by 7T-MRI. The mice were then randomized and treated with enzalutamide, alone or in combination with IgG control or anti-B7-H3 antibody (300 μg/injection) every three days. After four-week treatments, CRPC tumor volume was determined by 7T-MRI. Tumors were collected for histopathologic analyses.
Cell lines
22RV1 and LNCaP cells were cultured in RMPI-1640 medium (Corning, 10-040-CV) with 10% FBS (Thermo Scientific, 10082147) and 1% penicillin-streptomycin (Thermo Scientific, 15140163). DU145 was cultured in EMEM medium (ATCC, 30-2003) with 10% FBS (Thermo Scientific, 10082147) and 1% penicillin-streptomycin (Thermo Scientific, 15140163). HEK293T and DX1 cells were cultured in DMEM medium (Corning, 10-013-CV) with 10% FBS (Thermo Scientific, 10082147) and 1% penicillin-streptomycin (Thermo Scientific, 15140163). All cells were maintained at 37 °C in 5% CO2. All cell lines were confirmed by MD Anderson’s Cytogenetic and Cell Authentication Core and were negative for mycoplasma using the MycoAlert PLUS detection kit (Lonza, LT07-710), according to the manufacturer’s instructions.
Immune checkpoint expression pattern analyses
Transcriptional profiling, copy number alteration, and mutation datasets from the TCGA prostate cancer study (7) were downloaded from cbioportal (www.cbioportal.org). Tumor samples containing deep deletion or mutations of PTEN were grouped as “PTEN defects”, and the remaining samples were grouped as “PTEN unaltered”. The mRNA expression of 53 immune checkpoint genes in two groups was compared using the unpaired Student’s t-test (table S1). The statistical analysis was performed using GraphPad Prism version 9.2.0. The same method was used to analyze TP53- associated immune checkpoints. Expression of immune checkpoints in prostate tumors containing PTEN or TP53 defects was calculated (Log2[RNA Seq V2 RSEM+1]) (table S1). The bubble plot was generated using GraphPad Prism version 9.2.0.
To assess the expression pattern of CD276 in prostate and breast cancers, transcriptional profiling, copy number alteration, and mutation datasets of prostate cancer TCGA (7), metastatic prostate cancer SU2C/PCF (8), and invasive breast cancer TCGA (66) studies were downloaded from cbioportal (www.cbioportal.org). Tumor samples containing deep deletion or mutations of PTEN were grouped as “PTEN defects”, and those containing deep deletion or mutations of TP53 were grouped as “TP53 defects”; the remaining samples were grouped as “unaltered”. mRNA expression of CD276 in the above groups was compared using the unpaired Student’s t-test. The statistical analysis was performed using GraphPad Prism version 9.2.0. Pearson correlation analyses between CD276 and SP1 mRNA expression in prostate cancer (Brady dataset) (29) were performed using GraphPad Prism version 9.2.0. Pearson correlations of B7-H3 with PD-1 or PD-L1 in protein (Brady dataset) were analyzed using GraphPad Prism version 9.2.0.
Single-cell transcriptome analyses
The single-cell RNA-seq data was obtained from GSE141445 (41). The cells were clustered and annotated using Seurat 4.1.0 (67) in R 4.1.2. The gene expression data was normalized following “LogNormalize” method with “scale.factor = 10000”. The top 2000 most variable expressed genes are used for the subsequent linear dimensional reduction and cell clustering. Cell type is annotated by using the identified markers (41). Copy number alterations is inferred by CopyKat (42) with parameters: “ngene.chr”=5, “win.size”=25, “KS.cut”=0.1. 22,495 luminal cells were subset from the whole dataset to identify aneuploid cells and investigate gene expression of CD276, PTEN, and TP53. The published workflow (68) is used to identify the subclones from the CopyKat inferred segmentation data. In brief, the inferred copy number segmentation data was embedded in two dimensions using UMAP with R package ‘uwot’ (v.0.1.8) with following parameters: min dist = 0, n_neighbours = 35, seed = 55. The subclones were clustered and identified with hdbscan algorithm (dbscan v1.1-5). The hdbscan assigned outlier cells are inferred to the closest non-outlier cluster according to Euclidean distance. To compare the expression of CD276, PTEN, and TP53 between CD276-high and CD276-low clusters, the luminal cells with at least one of the three genes (CD276, PTEN, TP53) showing expression > 0 were subset. The differential expression analysis is performed by Seurat “FindMarkers” function with Wilcoxon rank sum test.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
Mouse tissues were paraffin-embedded. H&E staining of mouse tissue was performed. Antigen retrieval was performed with citrate-based antigen unmasking solution (Vector Laboratories, H-3300-250) at 95 °C for 20 min and cooled down to room temperature. Sections were blocked with 10% normal donkey serum ([NDS], Sigma-Aldrich, 566460-5ML) for 1 hour. For immunohistochemistry, primary antibodies (table S4) were prepared in 10% NDS and incubated with slides at 4 °C overnight. Sections were immersed in 3% H2O2 solution for 30 min and incubated with secondary antibodies (5 µg/ml) for 1 hour. Horseradish peroxidase streptavidin (Vector Laboratories, SA-5004-1) was diluted at a ratio of 1:200 with 10% NDS and stained for 1 hour. Colorimetric signals were developed with DAB (Vector Laboratories, SK-4100), and the nuclei were stained with hematoxylin (Fisher Scientific, 1.05174.2500). The slides were scanned by Aperio CS2 Scanscope (Leica). For immunofluorescence staining, primary antibodies (table S4) were prepared in antibody dilution buffer (1% BSA [Fisher Scientific, BP9703100] in DPBS [Corning, 21-030-CV]) supplemented with 0.3% TritonX-100 (Sigma-Aldrich, X100-500ML), followed by incubation with slides at 4 °C overnight. Secondary antibodies (1:200) in antibody dilution buffer were incubated with slides for 1 hour. Slides were stained with DAPI (0.5 µg/mL, Sigma-Aldrich, D9542-1MG) for 10 min, followed by scanning by Vectra Polaris (Akoya Biosciences). IF was performed in 3–5 tumors per cohort or group throughout the study. At least five views (20× magnification) per tumor were captured in a blinded manner using Phenochart 1.1.0.. Quantification plots represent the mean ± standard deviation of >10 individual views. Statistical analyses were performed using GraphPad Prism version 9.2.0.
Multiplex IHC
Multiplex IHC staining was performed with Opal Polaris 7 Color Manual IHC Detection Kit (Akoya Biosciences, NEL861001KT) as per manufacturer’s instruction. Paraffin-embedded mouse tissues were dewaxed, rehydrated, fixed in 10% neutral buffered formalin for 20 min, and then immersed in antigen retrieving buffer and treated in the microwave oven to retrieve antigens. After antigen retrieving, the cooled slides were blocked by blocking buffer for 10 min at room temperature and incubated with primary antibody (table S4) overnight at 4 °C. To introduce Opal polymer HRP, slides were incubated in Opal polymer HRP Ms+Rb for 10 min at room temperature. For goat- or rat-derived primary antibodies, Goat-on-Rodent HRP-Polymer (Biocare Medical, GHP516H) or Rat-on-Mouse HRP-Polymer (Biocare Medical, RT517H) kits were utilized to obtain HRP. To generate Opal signals, Opal Working Solutions were dropped onto the tissues, and slides were incubated for 10 min at room temperature. Once all the staining cycles were completed, slides were stained with DAPI working solution (supplied by the kit), followed by scanning by Vectra Polaris (Akoya Biosciences). Multiplex IHC were performed in 3–5 tumors per cohort or group throughout the study, except for Fig. 6, C and D and fig. S6, C and D (two tumors per treatment group were used). At least five views (20× magnification) per tumor were captured in a blinded manner using Phenochart 1.1.0.. In each quantification plot, data represent the mean ± standard deviation of >10 individual views. Statistical analyses were performed using GraphPad Prism version 9.2.0.
Western blot analysis
Cell pellets were lysed in 1x Laemmli sample buffer (Bio-Rad Laboratories, 1610747), premixed with 2-mercaptoethanol (Bio-Rad Laboratories, 1610710), at 99 °C for 15 min. Proteins were separated with 4%−15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad, 4561086) and transferred to the nitrocellulose membrane using the Trans-Blot Turbo RTA Mini 0.2-µm nitrocellulose transfer kit (Bio-Rad Laboratories, 1704270), and incubated with primary antibodies (table S4) overnight at 4 °C. HRP-conjugated secondary antibodies were incubated with membranes for 1 hour. Colorimetric and chemiluminescence signals were developed by Western ECL substrates (Bio-Rad Laboratories, 1705060) and captured using the ChemiDoc Imaging System (Bio-Rad Laboratories). The uncropped raw data images were shown in Data File S2.
Quantitative real-time PCR
Total RNAs were extracted from cell samples with the RNeasy Mini Kit (Qiagen, Inc., 74106) and cDNA was synthesized through reverse-transcriptional reaction with a High-Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368813) according to the standard manufacturer’s protocol. Quantitative real-time PCR (qPCR) was applied by QS3 Real-Time PCR Instruments (Thermo Fisher Scientific) using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25778), according to the manufacturer’s instructions. Gene relative expression was quantitatively determined using the specific primers (hCD276-F: 5’-AGCACTGTGGTTCTGCCTCACA-3’, hCD276-R: 5’-CACCAGCTGTTTGGTATCTGTCAG-3’, mCd276-F: 5’-ATGCTTCGAGGATGGGGTG-3’, mCd276-R: 5’-CCAGGCTCTGGGGAAAAGG-3’, hSP1-F: 5’-TGGCAGCAGTACCAATGGC-3’, and hSP1-R: 5’- CCAGGTAGTCCTGTCAGAACTT-3’). The relative mRNA expression of genes was normalized to that of ACTB. All of the data were analyzed by GraphPad Prism version 9.2.0.
Chromatin immunoprecipitation
Cells were fixed with 1% formaldehyde (Thermo Scientific, 28908) for 10 min, followed by quenching with glycine (2.5 M) for 5 min. Cells were washed with cold PBS and lysed with cell lysis buffer (12 mM Tris-HCl [pH 8.0], 10% PBS, 6 mM EDTA [pH 8.0], 12.5% FBS, 0.5% SDS, 1x protease, and phosphatase inhibitor [Thermo Scientific, 78442]) on ice for 15 min, followed by sonication using Bioruptor Pico (Diagenode). The DNA fragment was immunoprecipitated with IgG or Sp1 (Cell Signaling Technology, 9389S) antibody at 4 °C overnight. Target-bound DNA fragments were de-crosslinked and purified with phenol:chloroform:isoamyl alcohol (Invitrogen, 15593031) and chloroform:isoamyl alcohol (Sigma-Aldrich, C0549). The concentration of DNA was measured using a Qubit 4 Fluorometer (Invitrogen, Q33226) and Qubit 1X dsDNA High Sensitivity Assay Kit (Invitrogen, Q33230), according to the manufacturer’s instructions. DNA was analyzed by qPCR using the CD276 promoter primers (hCD276pro-F1: 5’-CCAAGACTGGGGTTGGACAG-3’, hCD276pro-R1: 5’-TTCCCACACTTCCAAGAGCC-3’, hCD276pro-F2: 5’-AGTGGAAATTGTCCTGCGGT-3’, hCD276pro-R2: 5’-TGGAATCCTGCTGTCCAACC-3’, hCD276pro-F3: 5’-CGTCCCTGAGTCCCAGAGT-3’, hCD276pro-R3: 5’-GGTTCCCGGGACTCCTGT-3’). The means and standard deviations of the normalized triplicate values were plotted using GraphPad Prism version 9.2.0.
Transient transfection and lentiviral transduction
Transient transfection of siRNA or plasmids was performed using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, 11668019), according to the manufacturer’s instructions. After 48 h, the cells were used for further analyses. For lentiviral transduction, the lentiviral constructs psPAX2 and pMD2.G were transfected into 293T cells using Lipofectamine 2000. After 48 h, viral supernatants were harvested and filtered. Transduced cells were incubated with viral supernatants with polybrene (Sigma-Aldrich, TR-1003-G) and selected using puromycin (Gibco, A1113803). Commercial siRNAs targeting SP1 (siSP1 #1, SASI_Hs01_00070994; siSP1 #2, SASI_Hs01_00070995) and Universal negative control (SIC001) were purchased from Sigma-Aldrich. Human SP1 overexpression plasmid was purchased from Genscript (OHu18205D).
CRISPR-mediated gene knockout
To generate TP53 or PTEN knockout cells, plasmids containing sgRNA targeting TP53 (Santa Cruz, sc-416469) or PTEN (Santa Cruz, sc-400103) were transfected into LNCaP, DU145, or 22Rv1 cell lines using Lipofectamine 2000 (Invitrogen, 11668019). After 72 hours, GFP+ cells were sorted into 96-well tissue culture plates (10 cells/well) by BD FACSAria II (BD Biosciences). Single clones were expended, followed by the determination of p53 and PTEN expression using Western blot analysis. To knock out Cd276 in syngeneic DX1 cells, sgRNAs targeting the mouse Cd276 gene (sgCd276 #2: 5’-GCGCGTCCGAGTAACCGACG-3’ and sgCd276 #3: 5’-ATCGAACAAGCCCCGCTCGT-3’) were designed using CHOPCHOP (https://chopchop.cbu.uib.no) and cloned into pLentiCRISPRv2 (Addgene, 52961), followed by lentiviral preparation and transduction.
Dual-luciferase reporter assay
The CD276-promoter region with putative Sp1-binding sites (~1.2 kb) was cloned into the pGL4 plasmid by Genewiz and verified by sequencing. pGL4-CD276pro and Renilla control plasmid (pRL, a gift from DePinho laboratory) were co-transfected into LNCaP cells. After 48 hours, dual-luciferase reporter assays were performed using the Dual-LuciferaseReporter Assay System kit (Promega, E1980), following the manufacturer’s protocol. Firefly and Renilla luciferase activity were read by the Synergy 2 Multi-Mode Microplate reader (BioTek). Firefly luciferase activity was normalized to the Renilla luciferase activity. Experiments were performed at least in triplicate, and statistical analyses were performed using GraphPad Prism version 9.2.0.
In vitro inhibitor treatment
For Sp1 inhibition, p53-depleted LNCaP cells were treated with DMSO or 20 nM mithramycin A (Sigma-Aldrich, 5303100001) for 48 hours. For AKT inhibition, LNCaP cells were treated with DMSO or MK-2206 2HCl (Selleck Chemicals, S1078) at serial concentrations for 48 hours, followed by Western blot analysis.
Mass cytometry (CyTOF)
Fresh tumor tissue (~50 mg) was digested in 1 ml of collagenase type II solution (5 mg/ml, Life Technologies, 17101-015) at 37 °C for 1 hour, followed by secondary digestion with TrypLE (Life Technologies, 12605028) at 37 °C for 15 min. Single cells were collected and incubated with 1x RBC lysis buffer (BioLegend, 420301) at room temperature for 10 min. As described previously (49, 51), single cells were blocked with purified rat anti-mouse CD16/CD32 (BD Biosciences, 553142) and incubated with cisplatin (1:2000, Fluidigm, 201064), followed by surface marker staining at 4 °C for 30 min. Cells were then fixed and permeabilized using the Foxp3/transcription factor staining buffer set (eBioscience, 00-5523-00) for 1 hour, followed by intracellular marker staining for 1 hour in the dark. Cells were further intercalated with Ir (Fluidigm, 201192A) in MaxPar Fix/Perm Buffer (Fluidigm, 201067). For T cell-specific panel, fresh tumor tissue was digested in collagenase type II solution (5 mg/ml, supplying 1:1000 diluted Brefeldin A [BioLegend, 420601] and Monensin [BioLegend, 420701]) at 37 °C for 1 hour. Secondary digestion, red cell lysing, FcR blocking, and cisplatin and surface marker staining were performed as described above. Cells were then fixed and permeabilized using the Maxpar Fix I Buffer (Fluidigm, 201065) and Maxpar Perm-S Buffer (Fluidigm, 201066) for 30 min, followed by intracellular marker staining for 30 min in the dark. Antibody-labeled cells were further fixed in 1.6% formaldehyde (VWR, PI28908) solution and intercalated with Ir (Fluidigm, 201192A) in MaxPar Fix/Perm Buffer (Fluidigm, 201067). Mass cytometry (Helios, Fluidigm) was performed at the MD Anderson Flow Cytometry and Cellular Imaging Core Facility. CyTOF data were analyzed using Cytobank (Beckman Coulter LS). Lists of antibodies against surface and intracellular markers are shown in table S3.
Flow cytometry analysis
Single cells from the digested mouse tumor tissues were blocked by purified rat anti-mouse CD16/CD32 (BD Biosciences, 553142) and stained with surface antibodies (table S4) for 15 min on ice. Dead cells were excluded by staining with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Invitrogen, L34964) and incubating for 30 min on ice. Fluorescence signals were determined by Gallios Flow Cytometer (Beckman Coulter) and analyzed with FlowJo 10.7.1 software (Becton Dickinson & Company).
Statistical analyses
All raw, individual-level data for experiments where n < 20 are presented in data file S1. The number of samples is shown in figures and described in figure legends. All data are presented as the mean ± standard deviation of at least triplicate experiments, unless otherwise stated. the Comparisons between the two groups were performed using an unpaired two-tailed Student t-test or Wilcoxon rank-sum test. One-way analysis of variance (ANOVA) with Tukey’s post hoc tests were performed to analyze data from three or more groups, as indicated in the figure legends. Statistical analysis was performed using GraphPad Prism version 9.2.0. Mice survival was determined by Kaplan-Meier analysis with Log-rank test. The hazards ratio is estimated by Cox proportional hazards model. Correlations of gene expression were performed using Pearson correlation. For all experiments, statistical significance is defined as follows: ****P < 0.0001, *** P < 0.001, ** P < 0.01, * P < 0.05. ns, not significant.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. R. A. DePinho at The University of Texas MD Anderson Cancer Center for sharing prostate cancer GEMMs of PbPP and PbPPS and the pGL4 plasmid for Dual-luciferase reporter assay. The authors thank A. Suttonin and Editing Services of Research Medical Library at MD Anderson for editing the article. The authors also thank D. H. Mak and V. Van at the MD Anderson Core facilities and their funders for their help with flow cytometry, CyTOF and Animal imaging (MRI).
Funding:
This study was partially supported by Prostate Cancer Foundation Young Investigator Award 21YOUN13 (to Y.Z.) and the MD Anderson Odyssey Postdoctoral Fellowship (to Y.Z.). This study was supported by NIH Pathway to Independence Award-NCI R00 CA226360 (to D.Z.), CPRIT Recruitment of First-Time Tenure-Track Faculty Award RR190021 (to D.Z., a CPRIT Scholar in Cancer Research), NIH/NCI R01 CA275990 (to D.Z.), and Prostate Cancer Foundation Challenge Award FP00016492 (to D.Z.).
Footnotes
Competing Interests
A. Aparicio has performed consulting or advisory work for Sanofi Genzyme, Janssen Biotech, Inc., and Bristol-Myers Squibb. All other authors declare that they have no competing interests.
Data and Materials Availability
All data associated with this study are present in the paper or supplementary materials. Source data are provided in Data Files S1 and S2. All biological resources, antibodies, and model organisms and tools are either available through commercial sources or may be requested from D.Z. and can be shared with the scientific community through a completed materials transfer agreement (MTA).
REFERENCES
- 1.Huggins C, Stevens RE Jr & Hodges CV, Studies on prostatic cancer II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg 43, 209–223 (1941). [Google Scholar]
- 2.Watson PA, Arora VK, Sawyers CL, Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15, 701–711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM, Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL, Integrative genomic profiling of human prostate cancer. Cancer cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, Wu G, Wang X, Troncoso P, Corn P, Thompson TC, Broom B, Baggerly K, Maity SN, Logothetis CJ, Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin Cancer Res 22, 1520–1530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA, The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer N. Genome Atlas Research, The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, Sboner A, Fedrizzi T, Mosquera JM, Robinson BD, De Sarkar N, Kunju LP, Tomlins S, Wu YM, Nava Rodrigues D, Loda M, Gopalan A, Reuter VE, Pritchard CC, Mateo J, Bianchini D, Miranda S, Carreira S, Rescigno P, Filipenko J, Vinson J, Montgomery RB, Beltran H, Heath EI, Scher HI, Kantoff PW, Taplin ME, Schultz N, deBono JS, Demichelis F, Nelson PS, Rubin MA, Chinnaiyan AM, Sawyers CL, Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 116, 11428–11436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cha HR, Lee JH, Ponnazhagan S, Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res 80, 1615–1623 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS, Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363, 411–422 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Maia MC, Hansen AR, A comprehensive review of immunotherapies in prostate cancer. Crit Rev Oncol Hematol 113, 292–303 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, Piulats JM, Gonzalez Mella P, Ng SS, Jaeger D, Parnis FX, Franke FA, Puente J, Carvajal R, Sengelov L, McHenry MB, Varma A, van den Eertwegh AJ, Gerritsen W, Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J Clin Oncol 35, 40–47 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, Ng S, Maio M, Franke FA, Sundar S, Agarwal N, Bergman AM, Ciuleanu TE, Korbenfeld E, Sengelov L, Hansen S, Logothetis C, Beer TM, McHenry MB, Gagnier P, Liu D, Gerritsen WR, Investigators CA, Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15, 700–712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graff JN, Alumkal JJ, Drake CG, Thomas GV, Redmond WL, Farhad M, Cetnar JP, Ey FS, Bergan RC, Slottke R, Beer TM, Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 7, 52810–52817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powles T, Yuen KC, Gillessen S, Kadel EE 3rd, Rathkopf D, Matsubara N, Drake CG, Fizazi K, Piulats JM, Wysocki PJ, Buchschacher GL Jr., Alekseev B, Mellado B, Karaszewska B, Doss JF, Rasuo G, Datye A, Mariathasan S, Williams P, Sweeney CJ, Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat Med 28, 144–153 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subudhi SK, Siddiqui BA, Aparicio AM, Yadav SS, Basu S, Chen H, Jindal S, Tidwell RSS, Varma A, Logothetis CJ, Allison JP, Corn PG, Sharma P, Combined CTLA-4 and PD-L1 blockade in patients with chemotherapy-naive metastatic castration-resistant prostate cancer is associated with increased myeloid and neutrophil immune subsets in the bone microenvironment. J Immunother Cancer 9, e002919 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr., PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA Jr., Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, de la Fouchardiere C, Rivera F, Elez E, Bendell J, Le DT, Yoshino T, Van Cutsem E, Yang P, Farooqui MZH, Marinello P, Diaz LA Jr., K.−. Investigators, Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med 383, 2207–2218 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Kontos F, Michelakos T, Kurokawa T, Sadagopan A, Schwab JH, Ferrone CR, Ferrone S, B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin Cancer Res 27, 1227–1235 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picarda E, Ohaegbulam KC, Zang X, Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin Cancer Res 22, 3425–3431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad DVR, Nguyen T, Li ZX, Yang Y, Duong J, Wang Y, Dong C, Murine B7-H3 is a negative regulator of T cells. J Immunol 173, 2500–2506 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Leitner J, Klauser C, Pickl WF, Stockl J, Majdic O, Bardet AF, Kreil DP, Dong C, Yamazaki T, Zlabinger G, Pfistershammer K, Steinberger P, B7-H3 is a potent inhibitor of human T-cell activation: No evidence for B7-H3 and TREML2 interaction. Eur J Immunol 39, 1754–1764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashiguchi M, Kobori H, Ritprajak P, Kamimura Y, Kozono H, Azuma M, Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses (vol 105, pg 10495, 2008). P Natl Acad Sci USA 105, 14744–14744 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, Duncan GS, Bukczynski J, Plyte S, Elia A, Wakeham A, Itie A, Chung S, Da Costa J, Arya S, Horan T, Campbell P, Gaida K, Ohashi PS, Watts TH, Yoshinaga SK, Bray MR, Jordana M, Mak TW, The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol 4, 899–906 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Benzon B, Zhao SG, Haffner MC, Takhar M, Erho N, Yousefi K, Hurley P, Bishop JL, Tosoian J, Ghabili K, Alshalalfa M, Glavaris S, Simons BW, Tran P, Davicioni E, Karnes RJ, Boudadi K, Antonarakis ES, Schaeffer EM, Drake CG, Feng F, Ross AE, Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: an expression-based analysis. Prostate Cancer Prostatic Dis 20, 28–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zang X, Thompson RH, Al-Ahmadie HA, Serio AM, Reuter VE, Eastham JA, Scardino PT, Sharma P, Allison JP, B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc Natl Acad Sci U S A 104, 19458–19463 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X, Inman BA, Krambeck AE, McKenney ME, Karnes RJ, Blute ML, Cheville JC, Sebo TJ, Kwon ED, B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res 67, 7893–7900 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, Gulati R, Plymate SR, Zhou Z, Birditt B, Meredith R, Geiss G, Hoang M, Beechem J, Nelson PS, Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun 12, 1426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arigami T, Narita N, Mizuno R, Nguyen L, Ye X, Chung A, Giuliano AE, Hoon DS, B7-h3 ligand expression by primary breast cancer and associated with regional nodal metastasis. Ann Surg 252, 1044–1051 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Mao Y, Li W, Chen K, Xie Y, Liu Q, Yao M, Duan W, Zhou X, Liang R, Tao M, B7-H1 and B7-H3 are independent predictors of poor prognosis in patients with non-small cell lung cancer. Oncotarget 6, 3452–3461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N, Takahama T, Tanaka K, Hayashi H, Takeda M, Kato S, Maenishi O, Sakai K, Chiba Y, Okabe T, Kudo K, Hasegawa Y, Kaneda H, Yamato M, Hirotani K, Miyazawa M, Nishio K, Nakagawa K, B7-H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clinical Cancer Research 24, 2653–2664 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, Morris K, Szot C, Morris H, Swing DA, Tessarollo L, Smith SW, Degrado S, Borkin D, Jain N, Scheiermann J, Feng Y, Wang Y, Li J, Welsch D, DeCrescenzo G, Chaudhary A, Zudaire E, Klarmann KD, Keller JR, Dimitrov DS, St Croix B, Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer cell 31, 501–515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo C, Figueiredo I, Gurel B, Neeb A, Seed G, Crespo M, Carreira S, Rekowski J, Buroni L, Welti J, Bogdan D, Gallagher L, Sharp A, Fenor de la Maza MD, Rescigno P, Westaby D, Chandran K, Riisnaes R, Ferreira A, Miranda S, Cali B, Alimonti A, Bressan S, Nguyen AHT, Shen MM, Hawley JE, Obradovic A, Drake CG, Bertan C, Baker C, Tunariu N, Yuan W, de Bono JS, B7-H3 as a Therapeutic Target in Advanced Prostate Cancer. Eur Urol 83, 224–238 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendes AA, Lu J, Kaur HB, Zheng SL, Xu J, Hicks J, Weiner AB, Schaeffer EM, Ross AE, Balk SP, Taplin ME, Lack NA, Tekoglu E, Maynard JP, De Marzo AM, Antonarakis ES, Sfanos KS, Joshu CE, Shenderov E, Lotan TL, Association of B7-H3 expression with racial ancestry, immune cell density, and androgen receptor activation in prostate cancer. Cancer 128, 2269–2280 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi X, Day A, Bergom HE, Tape S, Baca SC, Sychev ZE, Larson G, Bozicevich A, Drake JM, Zorko N, Wang J, Ryan CJ, Antonarakis ES, Hwang J, Integrative molecular analyses define correlates of high B7-H3 expression in metastatic castrate-resistant prostate cancer. NPJ Precis Oncol 6, 80 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, Burke S, Ciccarone V, Li H, Yang Y, Son T, Chen Y, Easton AN, Li JC, Rillema JR, Licea M, Fieger C, Liang TW, Mather JP, Koenig S, Stewart SJ, Johnson S, Bonvini E, Moore PA, Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res 18, 3834–3845 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Kendsersky NM, Lindsay J, Kolb EA, Smith MA, Teicher BA, Erickson SW, Earley EJ, Mosse YP, Martinez D, Pogoriler J, Krytska K, Patel K, Groff D, Tsang M, Ghilu S, Wang YF, Seaman S, Feng Y, St Croix B, Gorlick R, Kurmasheva R, Houghton PJ, Maris JM, The B7-H3-Targeting Antibody-Drug Conjugate m276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models. Clinical Cancer Research 27, 2938–2946 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, Labanieh L, Dhingra S, Leruste A, Majzner RG, Xu P, Mueller S, Yecies DW, Finetti MA, Williamson D, Johann PD, Kool M, Pfister S, Hasselblatt M, Fruhwald MC, Delattre O, Surdez D, Bourdeaut F, Puget S, Zaidi S, Mitra SS, Cheshier S, Sorensen PH, Monje M, Mackall CL, Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 26, 712–719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, Tiruthani K, Mirlekar B, Michaud D, Greene K, Herrera SG, Xu Y, Sun C, Chen Y, Ma X, Ferrone CR, Pylayeva-Gupta Y, Yeh JJ, Liu R, Savoldo B, Ferrone S, Dotti G, Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer cell 35, 221–237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen S, Zhu G, Yang Y, Wang F, Xiao YT, Zhang N, Bian X, Zhu Y, Yu Y, Liu F, Dong K, Mariscal J, Liu Y, Soares F, Loo Yau H, Zhang B, Chen W, Wang C, Chen D, Guo Q, Yi Z, Liu M, Fraser M, De Carvalho DD, Boutros PC, Di Vizio D, Jiang Z, van der Kwast T, Berlin A, Wu S, Wang J, He HH, Ren S, Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat Cell Biol 23, 87–98 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, Kumar T, Hu M, Sei E, Davis A, Wang F, Shaitelman SF, Wang JR, Chen K, Moulder S, Lai SY, Navin NE, Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol 39, 599–608 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP, Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, Hoshida Y, Hiller D, Hu B, Jiang S, Zheng H, Stegh AH, Scott KL, Signoretti S, Bardeesy N, Wang YA, Hill DE, Golub TR, Stampfer MJ, Wong WH, Loda M, Mucci L, Chin L, DePinho RA, SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H, Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer cell 4, 209–221 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, Lu YY, Tang YA, Yang YC, Yang PC, Wang YC, Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res 70, 5807–5817 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Li H, Zhang Y, Strose A, Tedesco D, Gurova K, Selivanova G, Integrated high-throughput analysis identifies Sp1 as a crucial determinant of p53-mediated apoptosis. Cell Death Differ 21, 1493–1502 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin P, Zhao C, Li Z, Mei C, Yao W, Liu Y, Li N, Qi J, Wang L, Shi Y, Qiu S, Fan J, Zha X, Sp1 is involved in regulation of cystathionine gamma-lyase gene expression and biological function by PI3K/Akt pathway in human hepatocellular carcinoma cell lines. Cell Signal 24, 1229–1240 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Zhao D, Cai L, Lu X, Liang X, Li J, Chen P, Ittmann M, Shang X, Jiang S, Li H, Meng C, Flores I, Song JH, Horner JW, Lan Z, Wu CJ, Li J, Chang Q, Chen KC, Wang G, Deng P, Spring DJ, Wang YA, DePinho RA, Chromatin Regulator, CHD1, Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Deficient Prostate Cancer. Cancer Discov 10, 1374–1387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo L, Zhu G, Xu H, Yao S, Zhou G, Zhu Y, Tamada K, Huang L, Flies AD, Broadwater M, Ruff W, van Deursen JM, Melero I, Zhu Z, Chen L, B7-H3 Promotes Pathogenesis of Autoimmune Disease and Inflammation by Regulating the Activity of Different T Cell Subsets. PLoS One 10, e0130126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, Jiang S, Chang Q, Spring DJ, Sharma P, Zebala JA, Maeda DY, Wang YA, DePinho RA, Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 543, 728–732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu GC, Wang G, Lu X, Labrot ES, Hu J, Wang W, Xiao Y, Zhang H, Zhang J, Zhang J, Gan B, Perry SR, Jiang S, Li L, Horner JW, Wang YA, Chin L, DePinho RA, Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 148, 896–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou X, Mao Y, Zhu J, Meng F, Chen Q, Tao L, Li R, Fu F, Liu C, Hu Y, Wang W, Zhang H, Hua D, Chen W, Zhang X, TGF-beta1 promotes colorectal cancer immune escape by elevating B7-H3 and B7-H4 via the miR-155/miR-143 axis. Oncotarget 7, 67196–67211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagashima O, Harada N, Usui Y, Yamazaki T, Yagita H, Okumura K, Takahashi K, Akiba H, B7-H3 contributes to the development of pathogenic Th2 cells in a murine model of asthma. J Immunol 181, 4062–4071 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA, CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413 (1994). [DOI] [PubMed] [Google Scholar]
- 56.Krummel MF, Allison JP, CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182, 459–465 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S, CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F, Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol 177, 4376–4383 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S, Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med 192, 303–310 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kavanagh B, O’Brien S, Lee D, Hou Y, Weinberg V, Rini B, Allison JP, Small EJ, Fong L, CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood 112, 1175–1183 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kreymborg K, Haak S, Murali R, Wei J, Waitz R, Gasteiger G, Savage PA, van den Brink MR, Allison JP, Ablation of B7-H3 but Not B7-H4 Results in Highly Increased Tumor Burden in a Murine Model of Spontaneous Prostate Cancer. Cancer Immunol Res 3, 849–854 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aggarwal C, Prawira A, Antonia S, Rahma O, Tolcher A, Cohen RB, Lou Y, Hauke R, Vogelzang N, P. Z. D, Kalebasty AR, Atkinson V, Adjei AA, Seetharam M, Birnbaum A, Weickhardt A, Ganju V, Joshua AM, Cavallo R, Peng L, Zhang X, Kaul S, Baughman J, Bonvini E, Moore PA, Goldberg SM, Arnaldez FI, Ferris RL, Lakhani NJ, Dual checkpoint targeting of B7-H3 and PD-1 with enoblituzumab and pembrolizumab in advanced solid tumors: interim results from a multicenter phase I/II trial. J Immunother Cancer 10, e004424 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang C, Jin J, Yang Y, Sun H, Wu L, Shen M, Hong X, Li W, Lu L, Cao D, Wang X, Sun J, Ye Y, Su B, Deng L, Androgen receptor-mediated CD8(+) T cell stemness programs drive sex differences in antitumor immunity. Immunity 55, 1268–1283 (2022). [DOI] [PubMed] [Google Scholar]
- 64.Guan X, Polesso F, Wang C, Sehrawat A, Hawkins RM, Murray SE, Thomas GV, Caruso B, Thompson RF, Wood MA, Hipfinger C, Hammond SA, Graff JN, Xia Z, Moran AE, Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 606, 791–796 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vellano CP, White MG, Andrews MC, Chelvanambi M, Witt RG, Daniele JR, Titus M, McQuade JL, Conforti F, Burton EM, Lastrapes MJ, Ologun G, Cogdill AP, Morad G, Prieto P, Lazar AJ, Chu Y, Han G, Khan MAW, Helmink B, Davies MA, Amaria RN, Kovacs JJ, Woodman SE, Patel S, Hwu P, Peoples M, Lee JE, Cooper ZA, Zhu H, Gao G, Banerjee H, Lau M, Gershenwald JE, Lucci A, Keung EZ, Ross MI, Pala L, Pagan E, Segura RL, Liu Q, Borthwick MS, Lau E, Yates MS, Westin SN, Wani K, Tetzlaff MT, Haydu LE, Mahendra M, Ma X, Logothetis C, Kulstad Z, Johnson S, Hudgens CW, Feng N, Federico L, Long GV, Futreal PA, Arur S, Tawbi HA, Moran AE, Wang L, Heffernan TP, Marszalek JR, Wargo JA, Androgen receptor blockade promotes response to BRAF/MEK-targeted therapy. Nature 606, 797–803 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cancer Genome Atlas N, Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Minussi DC, Nicholson MD, Ye H, Davis A, Wang K, Baker T, Tarabichi M, Sei E, Du H, Rabbani M, Peng C, Hu M, Bai S, Lin YW, Schalck A, Multani A, Ma J, McDonald TO, Casasent A, Barrera A, Chen H, Lim B, Arun B, Meric-Bernstam F, Van Loo P, Michor F, Navin NE, Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 592, 302–308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McInnes L, Healy J, Astels S, Hdbscan: hierarchical density based clustering. Journal of Open Source Software 2, 205 (2017) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or supplementary materials. Source data are provided in Data Files S1 and S2. All biological resources, antibodies, and model organisms and tools are either available through commercial sources or may be requested from D.Z. and can be shared with the scientific community through a completed materials transfer agreement (MTA).